

Summary
Middle East respiratory syndrome coronavirus (MERS‐CoV) is caused by a novel betacoronavirus that was isolated in late 2012 in Saudi Arabia. The viral infections have been reported in more than 1700 humans, ranging from asymptomatic or mild cases to severe pneumonia with a mortality rate of 40%. It is well documented now that dromedary camels contract the infection and shed the virus without notable symptoms, and such animals had been infected by at least the early 1980s. The mechanism of camel to human transmission is still not clear, but several primary cases have been associated with camel contact. There is no approved antiviral drug or vaccine against MERS‐CoV despite the active research in this area. Vaccine candidates have been developed using various platforms and regimens and have been tested in several animal models. Here, this article reviews the published studies on MERS‐CoV vaccines with more focus on vaccines tested in large animals, including camels. It is foreseeable that the 1‐health approach could be the best way of tackling the MERS‐CoV endemic in the Arabian Peninsula, by using the mass vaccination of camels in the affected areas to block camel to human transmission. Camel vaccines can be developed in a faster time with fewer regulations and lower costs and could clear this virus from the Arabian Peninsula if accompanied by efficient public health measures.
Keywords: camel vaccine, MERS coronavirus vaccine, Middle East Respiratory Syndrome
Abbreviations used
- AdV‐hDPP4
adenoviral vector encoding human DPP4
- ChAdOx1
chimpanzee adenovirus developed at Oxford University, vector version 1
- DDD
prime boost regimen of 3 sequential DNA vaccinations
- DDP
prime boost regimen of 2 sequential DNA vaccinations followed by 1 protein vaccination
- DPP4
dipeptidyl peptidase 4, a cellular surface receptor protein for MERS‐CoV
- ELISpot
enzyme‐linked immunospot assay, used to evaluate the number of antigen‐specific cytokine‐producing cells
- MERS‐CoV
Middle East respiratory syndrome coronavirus
- MERSpp
MERS‐CoV pseudotyped viral particles
- MVA
modified vaccinia virus Ankara
- nAb
neutralizing antibody
- NHP
nonhuman primate
- PP
prime boost regimen of 2 sequential protein vaccinations
- RBD
receptor‐binding domain within the S1 subunit of spike protein
- S
MERS‐CoV spike protein
- ΔTM S
the spike protein lacking its transmembrane domain
- S1
S1 subunit of the spike protein
- S2
S2 subunit of the spike protein
- SARS‐CoV
severe acute respiratory syndrome coronavirus
- VNT
virus‐neutralizing test
1. INTRODUCTION
Almost a decade since the emergence of severe acute respiratory syndrome coronavirus (SARS‐CoV) in 2003, another novel coronavirus has emerged in humans. The Middle East respiratory syndrome coronavirus (MERS‐CoV) was isolated in 2012 from a patient experiencing severe pneumonia in the Kingdom of Saudi Arabia (KSA).1, 2 MERS‐CoV has infected more than 1700 humans, with 30% to 40% mortality rate, mainly in countries of the Arabian Peninsula.3 Dromedary camels are strongly believed to be an intermediate host and an important source of infection although the exact transmission mechanism is still unclear.2, 3, 4 Human‐to‐human transmission has been documented, although this requires a very close and lengthy contact with patients.4 In the most part, cases are mild or asymptomatic, and most fatal cases are the result of comorbidities, such as diabetes mellitus type 2, which is again very prevalent in the Arabian Peninsula.4 Asymptomatic cases are estimated to be 2.1 times higher than the reported cases5 and together with mild cases may serve to spread the virus from camels to humans. Primary cases that had no contact with infected patients are more likely to have contacted camels directly or indirectly.6 However, camels are believed to be an intermediate with speculation that bats could be the primary host.4 Dromedary camels were 80% to 90% seropositive, from samples dated back to as early as 1983, in African countries,7 Arabian Peninsula countries,8 and the Spanish Canary Islands,9 but 0% in Australian dromedary camels or Bactrian camels,10, 11 and several groups reported viral shedding from infected camels.12, 13, 14, 15, 16, 17, 18 Nevertheless, despite the impressive and quick elucidation of many issues about MERS‐CoV, there are gaps in our understanding of this emerging virus, such as the precise route of transmission of camel‐to‐human infections, the risk factors for contracting the virus, and the protective level of neutralizing antibodies (nAb). More importantly, to date, there is no specific antiviral or vaccine for treatment or prevention of MERS‐CoV infection. This article reviews the ongoing efforts on vaccine development and sheds some light on the challenges that can hinder both the progress and the speed of developing a MERS‐CoV vaccine.
2. ANIMAL MODELS FOR VACCINE EVALUATION
MERS‐CoV tropism in humans and camels is determined by the binding of its spike glycoprotein to dipeptidyl peptidase 4 (DPP4) on the surface of mammalian cells such as respiratory epithelial cells. However, the virus does not infect most small experimental animals such as mice, ferrets, rats, and hamsters, mainly because of the different DPP4 in these animals therefore posing a challenge for developing a vaccine by delaying scientific investigation on the virus infection, pathogenicity, and transmission. A rabbit model was reported to support MERS‐CoV,19 but more small animal models were required to accelerate MERS‐CoV research; therefore, susceptible mouse models were developed by different methods. The first model was developed by transducing mice with adenoviral vector encoding human DPP4 (AdV‐hDPP4) delivered intranasally to enable mouse cells in the respiratory tract to express hDPP4, allowing viral entry and infection.20 This model is easy to use and can be readily applied to a broad range of mouse strains, especially in laboratories with no transgenic animal facility available. Other models were developed by generating transgenic mice that are either expressing human DPP4 globally20 or replacing murine DPP4 with the human ortholog.21 These developments helped in evaluating vaccines and monoclonal antibody‐based therapy in small laboratory animals. Although one of the main clinical manifestations of MERS in human patients is kidney failure, supported by biochemical tests and by the virus tropism in kidney cells, MERS mouse models did not support this and did not show virus or viral RNA isolation from kidney. Large animals such as nonhuman primates (NHPs) supported MERS with common marmosets showing moderate to severe infection as compared with mild infection in rhesus macaques (reviewed by van Doremalen and Munster.22) Dromedary camels are strongly believed to be intermediate hosts and one of the sources of infection, although the mechanism of transmitting the virus to human subjects is yet to be investigated and determined. MERS‐CoV replicates in the upper respiratory tract in camels, unlike humans where MERS‐CoV infection affects mainly lungs and lower respiratory tract, and results in mild clinical manifestation with rhinorrhea. In experimental camel models, infectious viruses were detected in nasal and oropharyngeal swabs and in tissues up to 5 days postinfection, whereas viral RNA persisted in these samples for over a month.18 All these developments and studies of animal models were conducted in less than 3 years and have accelerated our understanding of and responses to MERS‐CoV. Some vaccines have already been tested in mouse, NHP, and camel models, as discussed in this section and in Figure 1 and Table 1, although there seems a long way to go before establishing a robust animal model for assessing immunogenicity and efficacy of MERS‐CoV vaccine candidates.
Figure 1.
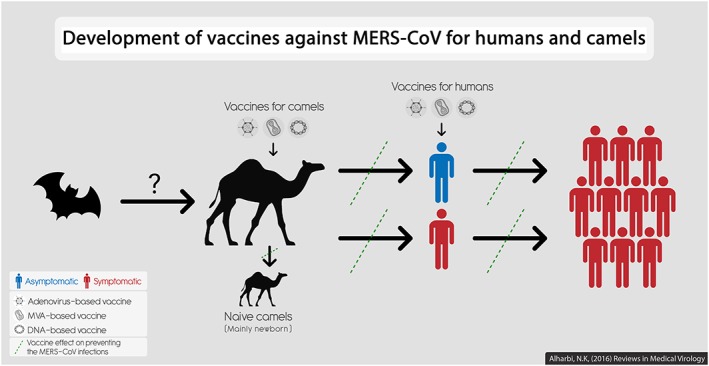
Development of MERS‐CoV vaccines for humans and camels. Three vaccines based on different vectors have been developed and tested (or planned for testing) in camels and in humans. Camel vaccination could block MERS‐CoV transmission and prevent any potential outbreaks in humans, applying the one‐health concept. Vaccinating humans, especially health care workers or individuals with comorbidity, could then further prevent MERS‐CoV infections and outbreaks. Bats are included as a suspected primary host of MERS‐CoV although this is not confirmed. MERS‐CoV can spread from infected camels to (i) naive camels or (ii) humans who may or may not show symptoms but are able to spread the virus to more susceptible individuals, causing an outbreak
Table 1.
Evaluation of vaccine candidates in large animals
| Vaccine (Ref.) | Platform, antigen, adjuvant, strain of vaccine | Animals, regimens, intervals, dose, route of immunization | Humoral immunity | Cellular immunity | Protection |
|---|---|---|---|---|---|
| Lan et al24 | Recombinant RBD367–606 residues of the S antigen. Alum adjuvant. EMC/2012 strain (GenBank: JX869059) | Rhesus macaques (n = 3), rRBD‐rRBD‐rRBD (homologous prime‐2× boost regimen), 8 wk between prime and first boost 17 wk between second and third boost, High dose: 200 μg prime and 100 μg boost with 1 mg alumLow dose: 50 μg prime and 25 μg boost with 1 mg alum, Intramuscular route | Using MERSpp NA, nAb titers were induced similarly by both doses. Sustained similar neutralization activity throughout peak (week 10, 2 wk after 1st boost) and long‐memory (week 17, at 2nd boost) stages as well as a day before challenge (week 27) | ELISpot: high‐dose induced IFN‐γ secreting PBMCs, significantly higher than the low dose | Challenge with 6.5 × 107 TCID50 of EMC/2012 strain, 27 wk postprime, intratracheally. Efficacy: reduced interstitial infiltration in lungs by x‐rayLess gross pathology and less abnormality in lung (~2‐fold). |
| Wang et al 32 | DNA and protein. S, ΔTM S, and S1 antigens. Alum adjuvant for protein. England1 strain (GenBank: AFY13307) | Rhesus macaques (n = 6).Three different regimens: DDD (S), DDP (S,S,S1), and PP (S1) 4‐wk intervals, 100 μg of S1 protein, 1 mg of S plasmid DNA, Intramuscular route |
All vaccines induced nAb in MERSpp NA, assessed 2 wk after each vaccination. Boost effect was eminent in protein involving regimens (ie, DDP and PP). Also, those regimens induced higher nAb levels than DDD. Average of nAb levels was 2.5 log10 in IC90, 10‐wk postboost in DDP and PP |
N/E | Challenged with 5 × 106 PFU of JordanN3 strain 19‐wk post boost via intratracheal route. Efficacy: 4‐ to 6‐fold reduction in the peak proportional volume of pulmonary consolidation as measured by CT scan, in PP and DDP, respectively |
| Muthumani et al35 | DNA. S antigen. Based on consensus sequence of the S gene | NHP: Rhesus macaques (n = 4)DDD 3‐wk intervals, Low (0.5 mg) and high (2 mg) doses, Intramuscular routeCamels: adult female dromedary camels (n = 3)DDD4‐wk intervals. Dose? Intramuscular route | NHP: nAb levels were similar between doses, around 100 VNT in EMC/2012 strain based microneutralization assayIn MERSpp NA: 80% neutralization activity against the vaccine based MERSpp with weak cross‐neutralization against pseudotyped viruses based on 2 other human CoVCamels: 2 wk after final immunization, nAb titers in vaccinated camels were 0, 300, and 600 VNT using EMC/2012 strain based microneutralization assay. The titers in unimmunized camels were 0–50 VNT | NHP: responses of TNF‐α and IFN‐γ (with lower IL‐2) secreting CD8+ and CD4+ T cells.Responses were not significantly different between the 2 doses.In ELISpot, some peptides were identified as immunodominant.Camels: N/E* | NHP: challenged with 7 × 106 TCID50 of the EMC/2012 strain, 4‐wk postboost, via combined intratracheal, intranasal, oral, and ocular routes. Efficacy: reduction in radiographic and histological changes in vaccinated groups.Significant reduction in viremia (viral load) in lung and bronchus tissuesLow‐dose vaccine showed better reduction in viremia level and radiographic than high dose although not significant. Camels: N/E |
| Haagmans et al38 | MVA viral vector. S antigen. EMC/2012 strain (GenBank: JX869059) | 6‐ to 8‐month‐old dromedary camels (n = 4), MVA‐MVA (homologous prime boost regimen), 4‐wk intervals. Simultaneous 2 × 108 PFU intranasal and 1 × 108 PFU intramuscular Intramuscular and intranasal routes simultaneously | Undetectable or low sera nAb at 4 wk post prime, but high levels post boost: average of 512 VNT in EMC/2012 strain based microneutralization assay. Nasal nAb titers were detected. | N/E* | Challenge with 107 TCID50 of EMC/2012 strain, 7‐wk postboost, via intranasal route.Efficacy: within 6 d postinfection, statistical reduction in viral genome and complete absent of infectious viruses (in nasal swabs), low RNA with no infectious viruses (in rectal swabs), no viral RNA or infectious viruses (in sera)In various organs, much lower or no RNA, and no infectious viruses in vaccinated camels compared with controls (NB: infectious viruses were not isolated from all organs of mock‐immunized control animals, only from nose, trachea, and some lymph node tissues). The levels of protection of this vaccine were also confirmed by immunohistochemistry and in situ hybridization |
NHP indicates nonhuman primate; S, full‐length spike antigen; ΔTM S, spike antigen lacking its transmembrane domain; S1, S1 subunit of the spike protein; RBD, receptor‐binding domain; DDD, prime boost regimen of 3 sequential DNA vaccinations; DDP, prime boost regimen of 2 sequential DNA vaccinations followed by 1 protein vaccination; PP, prime boost regimen of 2 sequential protein vaccinations; MERSpp NA, MERS‐CoV pseudotyped viral particles neutralization assay; IC90, inhibitory concentration of sera antibodies in MERSpp NA; VNT, virus neutralizing test; N/E, not evaluated; PBMC, peripheral blood monocyte; PFU, plaque‐forming unit; TCID50, 50% of tissue culture infectious dose.
Cellular immunogenicity detection reagents are not available for camels.
3. CURRENT MERS‐COV VACCINE CANDIDATES UNDER DEVELOPMENT
3.1. Recombinant DNA and protein based vaccines
Soon after the identification of MERS‐CoV, several groups developed vaccines for preclinical testing. Recombinant receptor‐binding domain (rRBD), residues 367 to 606 of MERS‐CoV spike (S) protein, has been expressed and tested in mice as a vaccine.23 It induced RBD‐specific immunoglobulin G (IgG) antibody when it was tested with various adjuvants such as alum, CpG, P(I:C), or IFA. However, only RBD with alum or with alum and CpG was able to induce nAb.23 This vaccine was then taken further and tested in Rhesus macaques at 2 different dosages, in 3 homologous vaccinations regimen, with 8‐ and 17‐week intervals. Both doses induced strong RBD‐specific antibodies 2 weeks after each vaccination, and similar nAb titers, but significantly higher than mock‐vaccinated monkeys. When compared with mock‐vaccinated control animals, both doses of vaccines reduced the interstitial infiltration and the gross pathology in lungs, after the animals were challenged with MERS‐CoV (Table 1).24 Vaccines reduced the viral RNA load in lung and trachea tissues, and in throat, nasal, and rectal swab samples. However, this reduction in viral load was statistically significant only in throat swabs from high‐dose vaccinated animals (summarized in Table 1). Both doses also reduced the infectious virus titers in the lung and trachea by around 2‐fold. Moreover, it was speculated that nAb were the underlying mechanism for this partial protection because cellular responses, as tested by IFN‐γ secreting peripheral blood monocytes, were detectable only in animals immunized with the highest dose vaccine.
A series of studies was also conducted to first define the minimum sequence of MERS‐CoV RBD to focus the immune responses.25, 26 The studies concluded that residues 377 to 588 of the S protein are the minimum RBD region that was able to induce the highest nAb levels in mice as tested by MERS‐CoV microneutralization assay. Different adjuvants were then tested with rRBD (377–588) vaccine such as Freund adjuvant, alum, MPLA, ISA51, and MF59. MF59 induced the highest RBD‐specific IgG antibodies, with IgG1 and IgG2a subtypes that neutralized the virus.27 MF59 induced higher Th2‐biased immune responses than ISA51 or Freund adjuvant, whereas alum and MPLA induced Th2‐biased immune responses. The rRBD (377–588) vaccine induced CD4+ and CD8+ T‐cell responses that were similar with or without any adjuvant.27
Subunit vaccines are more appealing because it was found that the inactivated whole SARS‐CoV used as a vaccine resulted in enhanced lung immunopathology when vaccinated animals were challenged with SARS‐CoV.28, 29, 30 This was attributed to the presence of N‐specific antibodies rather than S‐specific antibodies31; therefore, using the whole S antigen in a vaccine could be as safe as using minimum RBD. In fact, some studies reported that nAb titers were directed against other parts of the S, outside the RBD boundaries.32 A linear synthesized peptide, corresponding to 736 to 761 residues of the S protein (in the S2 subunit away from the RBD), induced nAb titers in mice. These titers were lower than using rRBD but other synthesized peptides based on immunoepitopes from the S1 subunit of the spike protein (S1) and RBD regions induced binding, but not nAb.33 This supports using the full‐length S antigen for vaccine development.
Nanoparticles of the trimerized S protein were produced as a vaccine by baculovirus expression system and used to immunize BALB/c mice with alum or matrix M adjuvants. The latter induced significantly higher nAb levels, 4‐fold higher than with alum. Moreover, there was no difference between high or low doses, in prime boost regimens.34 In a separate study, the S antigen was tested in mice in 3 different forms, the full‐length S, the S without its transmembrane domain, or the S1 subunit. All forms were tested in homologous regimens in mice, that is, prime boost regimen of 3 sequential DNA vaccinations (DDD) or prime boost regimen of 2 sequential protein vaccinations (PP), or in a heterologous regimen, that is, prime boost regimen of 2 sequential DNA vaccinations followed by 1 protein vaccination (DDP).32 The study concluded that the full‐length S DNA and S1‐based protein induced the highest nAb titers in DDP or PP regimens, but it should be noted that there was no full‐length S‐based protein vaccine in this study.32 The nAb titers cross‐protected mice against 8 distinct MERS‐CoV isolates; this could be predicted as the RBD sequence is highly conserved between the 8 isolates.
This study also showed that vaccinating with full‐length S DNA was important to achieve maximal neutralization activity; therefore, vaccines that are based on the full S or S1 antigens were then further tested in Rhesus macaques, using DDD, PP, or DDP regimens. The study used alum (ALPO4) to adjuvant the protein‐based vaccines. The DDP followed tightly by PP reduced the peak proportional volume of pulmonary consolidation, as measured by computed tomography (CT), after the animals were challenged with MERS‐CoV (Table 1). Therefore, it seemed that regimens involving protein‐based vaccine were superior to DNA‐based vaccines in driving this protection.
Another encouraging study also reported a partial protection in NHPs using only DNA vaccine based on the consensus sequence of the S gene.35 This vaccine was delivered to Rhesus macaques in 3 homologous DNA vaccinations, with 3‐week intervals. It induced high titer nAb and reduced the viral RNA load in lung tissues of vaccinated animals as compared with mock‐vaccinated controls upon MERS‐CoV challenge (Table 1). The radio examination, histology, and gross pathology were normal for 6 of the 8 vaccinated monkeys with mild infection in the other 2 (Table 1).
In mice, this vaccine had been shown to induce high S‐specific IgG levels that neutralized 3 MERS‐CoV pseudotyped viral particles (MERSpp) in neutralization assay. These 3 MERSpp were based on the S sequence from 2 clinical MERS‐CoV isolates as well as the consensus S gene. Most importantly, this vaccine was the first to undergo camel immunization. Two of 3 vaccinated dromedary camels developed S‐specific antibodies (Table 1).35 Overall, this vaccine induced high titer nAb in mice, camels, and monkeys and has now advanced to human clinical trials (discussed in the next section).
3.2. Viral vector‐based vaccines
A wide range of nonreplicating viruses have been used as vaccine vectors, encoding recombinant heterogeneous antigens, with some of them being tested in human clinical trials, including human and chimpanzee adenoviruses and attenuated poxviruses (eg, canarypox, fowlpox, and modified vaccinia virus Ankara [MVA]). Some of these vectors have been developed for MERS‐CoV vaccines.
3.2.1. Poxviral vectors
Recombinant MVA encoding the full‐length S antigen was used to immunize BALB/c mice via intramuscular or subcutaneous routes in a homologous prime boost vaccination.36 Three different doses of MVA induced comparable nAb titers in the intramuscular group, whereas in subcutaneous vaccinations, lower dose did not induced high levels nAb, especially postprime, using MERSpp neutralization assay. Comparing the routes, intramuscular route elicited higher nAb titers than subcutaneous,37 and this trend was also observed in cellular immune responses as measured by IFN‐γ enzyme‐linked immunospot assay (ELISpot). In challenge experiments, immunized mice were first transduced with AdV‐hDPP4 to render them susceptible to MERS‐CoV infection. Upon challenge with MERS‐CoV, the intramuscular route vaccination drove 100‐fold reduction or undetectable viral RNA load, and this protection was less evident in subcutaneous vaccinated mice.37
Dromedary camels, which are the most relevant animal species, were then vaccinated with MVA, given intramuscular and intranasal simultaneously, and the animals were boosted 4 weeks later with the same manner of vaccination. Postprime, only 2 of the 4 vaccinated animals developed nAb, but after the boost, nAb levels were high in all vaccinated animals. Three weeks after the boost vaccination, camels were challenged with MERS‐CoV via intranasal route (Table 1).38 As a promising result, vaccinated camels did not show rhinorrhea as compared with the control group. Significant reduction in viral genome and complete absent of infectious viruses were observed in nasal swabs. A low viral RNA load was detected, but no infectious viruses were isolated from rectal swabs. Serum samples from vaccinated camels were clear of the virus or the viral RNA. In a wide range of examined organs from vaccinated camels, there were low or undetectable RNA levels, with no infectious viruses, as compared with control animals. Looking at the control group alone, infectious MERS‐CoV was obtained only from nasal and tracheal tissues. The partial protection of this vaccine, as shown by reduced viral shedding, was also confirmed by immunohistochemistry and in situ hybridization,38 and this vaccine is now heading to a phase I clinical trial.
3.2.2. Measles viral vector
Recombinant measles virus was also developed as a vector for MERS‐CoV vaccine, using the S antigen either full‐length S antigen (expressed on infected cell surfaces) or soluble version of S antigen (secreted from the infected cells), lacking transmembrane and cytoplasmic domains.39 It was tested in immunocompromised mice, vaccinated twice with a month interval. The vaccine induced nAb and cellular immune responses (in IFN‐γ ELISpot). Mice were then transduced with AdV‐hDPP4 and subsequently challenged with MERS‐CoV. The vaccinated mice showed reduction in viral RNA loads and infectious virus titers in lung tissues. This partial protection was also confirmed by histopathology and immunohistochemistry of lung. In all experiments, soluble S was similar to nonsoluble S‐based vaccine.
3.2.3. Human adenoviral vectors
Vaccines based on adenoviral vectors were developed by 2 different groups using human adenovirus type 5 (Ad5) and Ad41. The first group used Ad5 vectors encoding either the full‐length S antigen or the S1 antigen to immunize BALB/c mice in a prime boost vaccination (intramuscular route for prime and intranasal route for the boost). Levels of IgG1 and IgG2a, which were similar for both antigens, increased over time and then were boosted to their peak level as they plateaued for 3 weeks postboost. Similarly, the nAb levels doubled after the boost, and they plateaued for 3 weeks.40 The second group used an Ad41 vector, derived from Ad41 virus that infects humans naturally via the gastrointestinal route, making it a suitable vector to induce mucosal immunity and to allow for oral immunization. Therefore, Ad41 viral vector delivering MERS S antigen was tested in comparison to Ad5 in BALB/c mice. Mice were vaccinated with Ad5 or Ad41 via intramuscular or intragastric routes, respectively. The intramuscular vaccination induced higher titers of total RBD‐specific IgG antibodies than the intragastric route with either vector. Using MERSpp neutralization assay, nAb titers followed the same trend, with Ad5 in intramuscular vaccination inducing the highest nAb titers. Although Ad41 was hypothesized to induce mucosal immunity, only the intramuscular route showed RBD‐specific IFN‐γ secreting cells in lungs as well as in spleens.41
3.2.4. Chimpanzee adenoviral vectors
Human adenoviral vectors, such as Ad5, have safety issues because highly prevalent preexisting immunity in human populations can eliminate adenoviral vectors, hindering immune responses to the encoded vaccine antigens. This may have affected the HIV vaccine efficacy in the Step clinical trial despite encouraging preclinical studies.42 It has prompted scientists to search for adenoviruses with less or no natural exposure to humans such as simian adenovirus type 63 (ChAd63) and ChAd3, both of which are safe and immunogenic when evaluated in humans.43, 44 Chimpanzee adenovirus isolate Y25 has been developed as a vaccine vector,45 called chimpanzee adenovirus developed at Oxford University, vector version 1 (ChAdOx1), and has now been evaluated in dromedary camels in Saudi Arabia for a vaccine against Rift Valley fever virus.46 ChAdOx1 required a single dose of 109 infectious units, given intramuscular, to elicit high titer nAb in camels. ChAdOx1 has also been tested in humans47 for Influenza vaccine, and in both camels and humans, ChAdOx1 showed excellent safety and immunogenicity. Therefore, a MERS‐CoV vaccine based on ChAdOx1 has been developed by Prof. Sarah Gilbert's group at the University of Oxford and induced high antibody titers in BALB/c mice (this work is being prepared for publication). The vaccine will also be tested in both dromedary camels and humans in the United Kingdom and Saudi Arabia.
3.2.5. Venezuelan equine encephalitis replicons (VRP)
Nearly all vaccines developed for MERS‐CoV were based on the S protein or its versions to induce nAb. However, eliciting cellular immunity can be achieved by vaccines that encode internal antigens such as nucleocapsid protein (N) or immunoepitopes from the N protein.
VRP encoding CD4+ T‐cell‐specific epitope from SARS‐CoV nucleocapsid protein (VRP‐SARS‐N) was developed and tested in BALB/c mouse models. VRP‐SARS‐N induced airway CD4+ T cells that were able to activate dendritic cell migration to lymph nodes, which in turn stimulated epitope‐specific cytotoxic CD8+ T cells. This resulted in the complete protection of vaccinated mice, immunized in prime boost regimen.48 The same researchers then developed VRP‐MERS‐N vaccine that is based on the N epitope analogue from MERS‐CoV, and similar results were obtained. The depletion of airway CD4+ T cells or inhibition of airway IFN‐γ significantly decreased the protection of VRP‐MERS‐N.48 This, in addition to the fact that the N epitope is a weak CD8+ T‐cell epitope, supports the role and importance of airway CD4+ T cells in controlling MERS‐CoV infection as well as in vaccine development. Mice immunized with VRP‐MERS‐N showed cross‐reactivity to several human CoVs (including SARS‐CoV, HKU4, and HKU5) with a moderate level of protection upon SARS‐CoV challenge. This study clearly supports the cellular immunity role in developing MERS‐CoV vaccine and paves the way toward a pan‐vaccine as a universal encounter against existing or potentially emerging human CoVs.
3.3. Technical variability in assessing vaccines
Studies on vaccine development for MERS‐CoV or any other pathogens show a range of technical variabilities. This may not allow for comparing results of various vaccine studies, but it shows a range of possible ways of applying vaccines. For instance, challenge experiments on immunized NHPs showed partial protection despite differences in vaccine construction, platforms, the dose and route of immunization, or the dose of challenge virus, and different MERS‐CoV isolates were also used for the challenge (summarized in Table 1).32, 35 Moreover, various S gene sequences from different MERS‐CoV strains were used to construct vaccines discussed in this article, mostly EMC/2012 strain. In addition, the assessment of nAb titers is performed by microneutralization assays based on the infectious MERS‐CoV virus (different isolates were used, but mostly EMC/2012) and reporting viral nAb titers in virus‐neutralizing test (VNT).
However, if the same experimental settings were used in this assay, sera from different sources (eg, mice, NHPs, camels, or humans) or from different vaccine platforms (DNA, proteins, viral vectors) can be compared by reporting VNT. For example, MV‐based vaccine, given twice to mice, induced lower VNT than MVA‐based vaccine, given twice to mice (1000 and 2000 VNT, respectively), when VERO cells, EMC/2012 isolate, and other settings were the same in the assay.36, 39
The MERSpp neutralization assays is also a convenient way of measuring nAb titer, based on lentivirus core that has a reporter luciferase with the MERS‐CoV spike protein displayed on the surface.49 This method is widely used mainly because it assesses humoral immune responses in BSL‐2 laboratory conditions instead of BSL‐3 because of the unavailability of the virus or a BSL‐3 laboratory. It also allows for testing a range of MERSpp based on spike proteins from various MERS‐CoV strains.32, 35
4. CHALLENGES IN DEVELOPING MERS‐COV VACCINES
In addition to challenges in finding the best small animal model, mentioned previously, developing a MERS‐CoV vaccine could face a few challenges as follow.
4.1. Funding and cost‐effectiveness of vaccines
Developing a vaccine for human use is a very long process that may take a decade to take a vaccine from bench through clinics to licensure, given that there is a productive and coherent collaboration between clinicians, scientists, regulators, and pharmaceutical industry. Yet one of the big obstacles is that a vaccine should be cost‐effective for a company to take it into further development and licensing. In the case of MERS‐CoV, although the mortality rate is already high (at least 30%), outbreaks seem sporadic, nosocomial, and zoonotic and may not pose risk as serious as global major infections such as malaria, HIV, TB, or influenza. For example, an influenza outbreak may become a global epidemic in a matter of hours because of the mass air‐travel era that we live in. MERS‐CoV outbreaks have occurred in the Arabian Peninsula, where it was first isolated, and where there is human contact with a considerable population of dromedary camels, less than the number of camels in eastern Africa. MERS‐CoV infections reported outside the Arabian Peninsula in China, Europe, or the United States did not cause any outbreaks, presumably because of stricter infection control measures, but it could be due to less camel contact or the absence of camel population in these countries. So far, the only documented MERS‐CoV outbreak outside the primary regions was the Korean outbreak in 2015, started by an index patient travelling back from the Arabian Gulf states. This outbreak had infected 186 people with mortality rate of 19% but was contained in a few months, again likely because of the strict infection control practices.50 Therefore, a pharmaceutical company may be discouraged from developing a vaccine to an infection that (i) seems to be sporadic, nosocomial, or zoonotic with no sustained transmission; (ii) might be easily contained by effective infection control practices; and (iii) might disappear for a long time if strict public health measures were taken, similar to what happened with SARS‐CoV. If industrial entities cannot be interested, then the hope is that governments, at least in the endemic areas, could consider funding vaccine development from the basic early studies through clinical trials to approval and marketing. Although the most affected countries are rich, such as KSA, Qatar, and UAE, they might look into quicker measures to contain this virus even if those measures are more expensive than vaccines, which take long and laborious efforts. For example, a tertiary hospital in Riyadh, KSA, one of the biggest in the Middle East with 1200 beds and 250,000 visits to the ER per annum, with some VIP royal clinics, was hit by a MERS‐CoV outbreak in August 2015, resulting in 130 cases, with 40% mortality rate.51 The hospital responded by several actions regardless of the cost, among them closing down the hospital for weeks, building a separate ER for flulike symptoms with several negatively pressurized rooms, and constructing a car drive‐thru screening checkpoint at the entrance of the medical city to direct flulike patients to the new ER without any contact with them. The hospital was then reopened and had to pay overtime to some staff to catch up with clinic schedules. These responses took only 7 weeks to complete and helped in clearing the MERS‐CoV from this large hospital in an astonishingly short time (personal communication with Dr. Bandar Al Knawy, CEO of King Abulaziz Medical City.51) Therefore, such rich governments, under public pressure, might prefer implementing similar actions to all other major hospitals because these are quick and efficient responses and might be favored to funding the lengthy process of vaccine development. Yet the Saudi Ministry of Health has shown interest in vaccines from early stages of development and held a special workshop in Riyadh (November 2015), inviting WHO officials, major funding bodies, and vaccinologists who are interested in MERS‐CoV vaccines, aimed at helping and accelerating such process. It discussed the science, the funds, and the plans to set up a blueprint on the subject. There were also explanations of the Saudi FDA regulations on trials, in an attempt to welcome vaccine clinical trials in the KSA.
4.2. Target populations for MERS‐CoV vaccine trials
The question of who to vaccinate is becoming more critical as scientists have conducted vaccine studies in animal models and are now approaching phase I human clinical trials. Two vaccines have begun clinical testing: the DNA‐based GLS‐5300 vaccine that was codeveloped by Inovio Pharmaceuticals and GeneOne Life Science Inc. is now being tested in humans by Walter Reed Army Institute of Research.52 The second vaccine, scheduled for a phase I safety trial, is the MVA‐based vaccine that showed promising protection results in dromedary camels.38 Phase I trials focus on the safety of a given vaccine and are tested in healthy volunteers. Therefore, it is anticipated that, because of the safety records of MVA and DNA vaccines, the vaccines could move to phase II immunogenicity and safety trials, in bigger number of subjects by the end of 2017/18 and might also move to endemic areas, provided funding is available. However, the target population to receive the vaccine would be an important question. From the epidemiological studies, the MERS‐CoV infects mainly middle‐aged male, health care workers, and people with comorbidities such as diabetes mellitus and heart diseases.4 Therefore, these factors would set the criteria for selecting participants for phase II trials. Following safety trials, efficacy trials could be considered next, where scientists generally use a curable strain of the pathogen to challenge vaccines and assess if the respective vaccine is efficacious; this is being done in malaria vaccine trials (for an example study, see Hodgson et al53). For viral infections such as HIV, scientists need to rely on natural infection as a challenge model. Therefore, for a MERS‐CoV vaccine efficacy, vaccinating health care workers, especially those in the ER and waiting for the next outbreak to appear to assess the vaccine efficacy would be 1 way of efficacy trial setting. Alternatively, vaccinating the contacts of infected people during an outbreak would be a way to assess whether these contacts can be protected as compared with unvaccinated contacts of infected people. This approach, called ring vaccination, was used during smallpox eradication campaign by the WHO vaccination program and was recently used for Ebola vaccine in Africa.54 However, this may become difficult for MERS‐CoV for 3 reasons. First, it requires a very efficient coordination between scientists, clinicians, and health officials in endemic areas to identify volunteers, to get them to consent to participate, and to vaccinate them within the right time. Second, it requires quick actions, as the peak of the infection may not last for long, 2 weeks after the onset of MERS illness.4 Third, and most importantly, it requires identifying a large number of infected people with a high number of their contacts (n = 200–2000) who are willing to participate; however, clustered MERS‐CoV outbreaks are usually less than 255 cases,50, 51, 55 making it difficult to conduct a phase III efficacy trial during an outbreak.
5. CONCLUDING REMARKS
To date, none of the tested vaccines against MERS‐CoV failed in inducing nAb, protecting mice, or partially protecting large animals (Table 1). This virus does not seem very polymorphic in the spike protein that is involved in tropism and cell entry and contains neutralizing epitopes. Therefore, vaccines could become available in 5 to 10 years if there is sufficient financial and logistical support. Because there is increasing evidence of camels as a source of infection, using 1‐health concept to vaccinate camels might decrease the human cases, especially mild or asymptomatic infection, which may be undetectable but important in circulating the virus (Figure 1). Developing vaccines for animals would require less time, less cost, fewer regulations, and no cGMP manufacturing. If governments in the affected Arabian areas are willing to contain the virus, then a mass camel screening program can identify and vaccinate seronegative camels, which should not be a large number because 80% of camels are already seropositive. Camel vaccination can also focus on immune naïve newborn (or pregnant) camels as well as seronegative imported camels.
Lastly, a response to a future emerging coronavirus would be to develop a universal vaccine that incorporates a conserved region from most coronaviruses; however, this might be difficult if coronaviruses do not share similarities in neutralizing epitopes or immunodominant epitopes that are able to induce cross‐neutralizing immunity. Therefore, the most realistic approach would be to increase the preparedness to respond rapidly to any emerging pathogen, not necessarily coronaviruses. This includes developing technology that can be easily used for any vaccine development. For example, the cGMP manufacturing of MVA viral vector (used for a MERS‐CoV vaccine) is based on chicken embryo fibroblast primary cells, which are not ideal for large scale manufacturing. Developing a cheaper and efficient cGMP cell line instead of primary cells would accelerate MVA‐based vaccines. Another example would be to develop a technology that excludes the cold‐chain transfer of vaccines that would reduce both costs and time to a great extent and make the world more prepared to distribute vaccines against the next emerging pathogen.
ACKNOWLEDGMENTS
The author thanks Prof. Sarah Gilbert, University of Oxford, for her comments on this article and Mr. Sultan Alharbi (srab4d@gmail.com) for the infographic design in Figure 1.
Alharbi NK. Vaccines against Middle East respiratory syndrome coronavirus for humans and camels. Rev Med Virol. 2017;27:e1917 10.1002/rmv.1917
REFERENCES
- 1. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814–1820. [DOI] [PubMed] [Google Scholar]
- 2. Mackay IM, Arden KE. Middle East respiratory syndrome: an emerging coronavirus infection tracked by the crowd. Virus Res. 2015;202:60–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. World Health Organization . Middle East respiratory syndrome coronavirus (MERS‐CoV) [Internet]. Geneva: World Health Organization; 2016 Sep [cited 2016 Sep 01]. Available from: http://www.who.int/emergencies/mers-cov/en/ [Google Scholar]
- 4. Mackay IM, Arden KE. MERS coronavirus: diagnostics, epidemiology and transmission. Virol J. 2015;12(1):222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lessler J, Salje H, Van Kerkhove MD, et al. Estimating the severity and subclinical burden of Middle East respiratory syndrome coronavirus infection in the Kingdom of Saudi Arabia. Am J Epidemiol. 2016;183(7):657–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alraddadi BM, Watson JT, Almarashi A, et al. Risk factors for primary Middle East respiratory syndrome coronavirus illness in humans, Saudi Arabia, 2014. Emerg Infect Dis. 2016;22(1):49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Muller MA, Corman VM, Jores J, et al. MERS coronavirus neutralizing antibodies in camels, Eastern Africa, 1983–1997. Emerg Infect Dis. 2014;20(12):2093–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hemida MG, Perera RA, Al Jassim RA, et al. Seroepidemiology of Middle East respiratory syndrome (MERS) coronavirus in Saudi Arabia (1993) and Australia (2014) and characterisation of assay specificity. Euro Surveill : bulletin Europeen sur les maladies transmissibles = European communicable disease bulletin. 2014;19(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gutierrez C, Tejedor‐Junco MT, Gonzalez M, Lattwein E, Renneker S. Presence of antibodies but no evidence for circulation of MERS‐CoV in dromedaries on the Canary Islands, 2015. Euro Surveill : bulletin Europeen sur les maladies transmissibles = European communicable disease bulletin. 2015;20(37). [DOI] [PubMed] [Google Scholar]
- 10. Shehata MM, Gomaa MR, Ali MA, Kayali G. Middle East respiratory syndrome coronavirus: a comprehensive review. Front Med. 2016;10(2):120–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miguel E, Perera RA, Baubekova A, et al. Absence of Middle East respiratory syndrome coronavirus in Camelids, Kazakhstan, 2015. Emerg Infect Dis. 2016;22(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sabir JS, Lam TT, Ahmed MM, et al. Co‐circulation of three camel coronavirus species and recombination of MERS‐CoVs in Saudi Arabia. Science. 2016;351(6268):81–84. [DOI] [PubMed] [Google Scholar]
- 13. Alagaili AN, Briese T, Mishra N, et al. Middle East respiratory syndrome coronavirus infection in dromedary camels in Saudi Arabia. MBio. 2014;5(2):e00884–e00814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Azhar EI, El‐Kafrawy SA, Farraj SA, et al. Evidence for camel‐to‐human transmission of MERS coronavirus. N Engl J Med. 2014;370(26):2499–2505. [DOI] [PubMed] [Google Scholar]
- 15. Haagmans BL, Al Dhahiry SH, Reusken CB, et al. Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect Dis. 2014;14(2):140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wernery U, Corman VM, Wong EY, et al. Acute Middle East respiratory syndrome coronavirus infection in livestock Dromedaries, Dubai, 2014. Emerg Infect Dis. 2015;21(6):1019–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Khalafalla AI, Lu X, Al‐Mubarak AI, Dalab AH, Al‐Busadah KA, Erdman DD. MERS‐CoV in upper respiratory tract and lungs of dromedary camels, Saudi Arabia, 2013–2014. Emerg Infect Dis. 2015;21(7):1153–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adney DR, van Doremalen N, Brown VR, et al. Replication and shedding of MERS‐CoV in upper respiratory tract of inoculated dromedary camels. Emerg Infect Dis. 2014;20(12):1999–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Haagmans BL, van den Brand JM, Provacia LB, et al. Asymptomatic Middle East respiratory syndrome coronavirus infection in rabbits. J Virol. 2015;89(11):6131–6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Agrawal AS, Garron T, Tao X, et al. Generation of a transgenic mouse model of Middle East respiratory syndrome coronavirus infection and disease. J Virol. 2015;89(7):3659–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pascal KE, Coleman CM, Mujica AO, et al. Pre‐ and postexposure efficacy of fully human antibodies against Spike protein in a novel humanized mouse model of MERS‐CoV infection. Proc Natl Acad Sci U S A. 2015;112(28):8738–8743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Doremalen N, Munster VJ. Animal models of Middle East respiratory syndrome coronavirus infection. Antiviral Res. 2015;122:28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lan J, Deng Y, Chen H, et al. Tailoring subunit vaccine immunity with adjuvant combinations and delivery routes using the Middle East respiratory coronavirus (MERS‐CoV) receptor‐binding domain as an antigen. PLoS One. 2014;9(11):e112602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lan J, Yao Y, Deng Y, et al. Recombinant receptor binding domain protein induces partial protective immunity in Rhesus macaques against Middle East respiratory syndrome coronavirus challenge. EBioMedicine. 2015;2(10):1438–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ma C, Wang L, Tao X, et al. Searching for an ideal vaccine candidate among different MERS coronavirus receptor‐binding fragments—the importance of immunofocusing in subunit vaccine design. Vaccine. 2014;32(46):6170–6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Du L, Zhao G, Kou Z, et al. Identification of a receptor‐binding domain in the S protein of the novel human coronavirus Middle East respiratory syndrome coronavirus as an essential target for vaccine development. J Virol. 2013;87(17):9939–9942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang N, Channappanavar R, Ma C, et al. Identification of an ideal adjuvant for receptor‐binding domain‐based subunit vaccines against Middle East respiratory syndrome coronavirus. Cell Mol Immunol. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tseng CT, Sbrana E, Iwata‐Yoshikawa N, et al. Immunization with SARS coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. PLoS One. 2012;7(4):e35421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lokugamage KG, Yoshikawa‐Iwata N, Ito N, et al. Chimeric coronavirus‐like particles carrying severe acute respiratory syndrome coronavirus (SCoV) S protein protect mice against challenge with SCoV. Vaccine. 2008;26(6):797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bolles M, Deming D, Long K, et al. A double‐inactivated severe acute respiratory syndrome coronavirus vaccine provides incomplete protection in mice and induces increased eosinophilic proinflammatory pulmonary response upon challenge. J Virol. 2011;85(23):12201–12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deming D, Sheahan T, Heise M, et al. Vaccine efficacy in senescent mice challenged with recombinant SARS‐CoV bearing epidemic and zoonotic spike variants. PLoS Med. 2006;3(12):e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang L, Shi W, Joyce MG, et al. Evaluation of candidate vaccine approaches for MERS‐CoV. Nat Commun. 2015;6:7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang Y, Deng Y, Wen B, et al. The amino acids 736–761 of the MERS‐CoV spike protein induce neutralizing antibodies: implications for the development of vaccines and antiviral agents. Viral Immunol. 2014;27(10):543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Coleman CM, Liu YV, Mu H, et al. Purified coronavirus spike protein nanoparticles induce coronavirus neutralizing antibodies in mice. Vaccine. 2014;32(26):3169–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Muthumani K, Falzarano D, Reuschel EL, et al. A synthetic consensus anti‐spike protein DNA vaccine induces protective immunity against Middle East respiratory syndrome coronavirus in nonhuman primates. Sci Transl Med. 2015;7(301):301ra132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Song F, Fux R, Provacia LB, et al. Middle East respiratory syndrome coronavirus spike protein delivered by modified vaccinia virus Ankara efficiently induces virus‐neutralizing antibodies. J Virol. 2013;87(21):11950–11954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Volz A, Kupke A, Song F, et al. Protective efficacy of recombinant modified vaccinia virus Ankara (MVA) delivering Middle East respiratory syndrome coronavirus spike glycoprotein. J Virol. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Haagmans BL, van den Brand JM, Raj VS, et al. An orthopoxvirus‐based vaccine reduces virus excretion after MERS‐CoV infection in dromedary camels. Science. 2016;351(6268):77–81. [DOI] [PubMed] [Google Scholar]
- 39. Malczyk AH, Kupke A, Prufer S, et al. A highly immunogenic and protective Middle East respiratory syndrome coronavirus vaccine based on a recombinant measles virus vaccine platform. J Virol. 2015;89(22):11654–11667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim E, Okada K, Kenniston T, et al. Immunogenicity of an adenoviral‐based Middle East respiratory syndrome coronavirus vaccine in BALB/c mice. Vaccine. 2014;32(45):5975–5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guo X, Deng Y, Chen H, et al. Systemic and mucosal immunity in mice elicited by a single immunization with human adenovirus type 5 or 41 vector‐based vaccines carrying the spike protein of Middle East respiratory syndrome coronavirus. Immunology. 2015;145(4):476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Buchbinder SP, Mehrotra DV, Duerr A, et al. Efficacy assessment of a cell‐mediated immunity HIV‐1 vaccine (the Step Study): a double‐blind, randomised, placebo‐controlled, test‐of‐concept trial. Lancet. 2008;372(9653):1881–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. de Barra E, Hodgson SH, Ewer KJ, et al. A phase Ia study to assess the safety and immunogenicity of new malaria vaccine candidates ChAd63 CS administered alone and with MVA CS. PLoS One. 2014;9(12):e115161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tapia MD, Sow SO, Lyke KE, et al. Use of ChAd3‐EBO‐Z Ebola virus vaccine in Malian and US adults, and boosting of Malian adults with MVA‐BN‐Filo: a phase 1, single‐blind, randomised trial, a phase 1b, open‐label and double‐blind, dose‐escalation trial, and a nested, randomised, double‐blind, placebo‐controlled trial. Lancet Infect Dis. 2016;16(1):31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dicks MD, Spencer AJ, Edwards NJ, et al. A novel chimpanzee adenovirus vector with low human seroprevalence: improved systems for vector derivation and comparative immunogenicity. PLoS One. 2012;7(7):e40385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Warimwe GM, Gesharisha J, Carr BV, et al. Chimpanzee adenovirus vaccine provides multispecies protection against Rift Valley fever. Sci Rep. 2016;6:20617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Antrobus RD, Coughlan L, Berthoud TK, et al. Clinical assessment of a novel recombinant simian adenovirus ChAdOx1 as a vectored vaccine expressing conserved Influenza A antigens. Mol Ther. 2014;22(3):668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhao J, Zhao J, Mangalam AK, et al. Airway memory CD4(+) T cells mediate protective immunity against emerging respiratory coronaviruses. Immunity. 2016;44(6):1379–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Grehan K, Ferrara F, Temperton N. An optimised method for the production of MERS‐CoV spike expressing viral pseudotypes. MethodsX. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Korea Centers for Disease, C. and Prevention . Middle East respiratory syndrome coronavirus Outbreak in the Republic of Korea, 2015. Osong Public Health Res Perspect. 2015;6(4):269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Balkhy HH, Alenazi TH, Alshamrani MM, et al. Notes from the field: nosocomial outbreak of Middle East respiratory syndrome in a large tertiary care hospital—Riyadh, Saudi Arabia, 2015. MMWR Morb Mortal Wkly Rep. 2016;65(6):163–164. [DOI] [PubMed] [Google Scholar]
- 52. Pellerin C. Army scientists begin first MERS vaccine clinical trial. DoD News, Defense Media Activity . 2016.
- 53. Hodgson SH, Ewer KJ, Bliss CM, et al. Evaluation of the efficacy of ChAd63‐MVA vectored vaccines expressing circumsporozoite protein and ME‐TRAP against controlled human malaria infection in malaria‐naive individuals. J Infect Dis. 2015;211(7):1076–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Henao‐Restrepo AM, Longini IM, Egger M, et al. Efficacy and effectiveness of an rVSV‐vectored vaccine expressing Ebola surface glycoprotein: interim results from the Guinea ring vaccination cluster‐randomised trial. Lancet. 2015;386(9996):857–866. [DOI] [PubMed] [Google Scholar]
- 55. Oboho IK, Tomczyk SM, Al‐Asmari AM, et al. 2014 MERS‐CoV Outbreak in Jeddah—A Link to Health Care Facilities. N Engl J Med. 2015;372(9):846–854. [DOI] [PMC free article] [PubMed] [Google Scholar]