

Abstract
Objective – To review the human and veterinary literature on the biology of apoptosis in health and disease.
Data Sources – Data were examined from the human and veterinary literature identified through Pubmed and references listed in appropriate articles pertaining to apoptosis.
Human Data Synthesis – The role of apoptosis in health and disease is a rapidly growing area of research in human medicine. Apoptosis has been identified as a component of human autoimmune diseases, Alzheimer's disease, cancer, and sepsis.
Veterinary Data Synthesis – Research data available from the veterinary literature pertaining to apoptosis and its role in diseases of small animal species is still in its infancy. The majority of veterinary studies focus on oncologic therapy. Most of the basic science and human clinical research studies use human blood and tissue samples and murine models. The results from these studies may be applicable to small animal species.
Conclusions – Apoptosis is the complex physiologic process of programmed cell death. The pathophysiology of apoptosis and disease is only now being closely evaluated in human medicine. Knowledge of the physiologic mechanisms by which tissues regulate their size and composition is leading researchers to investigate the role of apoptosis in human diseases such as cancer, autoimmune disease and sepsis. Because it is a multifaceted process, apoptosis is difficult to target or manipulate therapeutically. Future studies may reveal methods to regulate or manipulate apoptosis and improve patient outcome.
Keywords: cancer, caspases, cell death, intrinsic pathway, sepsis
Introduction
All tissues must be able to tightly control cell numbers and tissue size and to protect themselves from rogue cells that threaten homeostasis. In the early 1970s, Kerr et al, 1 observed a single‐cell‐death phenomenon that occurred in the dying cells of healthy tissues, as well as in cells associated with teratogenesis, neoplasia, tumor regression, atrophy, and involution. The term apoptosis, from Greek origins (apo=for, ptosis=falling), was chosen to describe the cellular process of programmed cell death. 1 , 2 , 3 Apoptosis is a tightly regulated intracellular program in which cells destined to die activate enzymes that degrade the cell's DNA and nuclear and cytoplasmic proteins. 4 Programmed cell death eliminates unwanted cells or potentially reactive cell lines either before or after maturation. This process is vital to fetal and embryonic development and to tissue remodeling. 3 Cell populations that normally have a high rate of proliferation, such as the intestinal epithelium, depend upon apoptosis to maintain the necessary number of cells. 5 The number of activated immune cells must be controlled to contain the inflammatory response. 6 , 7 Hormone‐dependent apoptosis occurs during estrus and causes prostatic atrophy after castration. 8
Kerr et al 1 observed that apoptotic cells share many morphologic features distinct from those in necrotic cells. Cells undergoing apoptosis exhibit 1 or more of the following: cell shrinkage, chromatin condensation and nucleosomal fragmentation, and bubbling of the plasma membrane (blebbing). Biochemical features of apoptosis include DNA fragmentation, protein cleavage at specific locations, increased mitochondrial membrane permeability, and the appearance of phosphatidylserine on the cell membrane surface. 3 , 9 There is an increase in mitochondrial permeability leading to the release of pro‐apoptotic proteins and subsequent formation of apoptotic bodies. The resulting membrane‐bound apoptotic bodies are consumed by neighboring cells or by macrophages. Apoptosis is a single‐cell event, and does not induce an inflammatory reaction.
Apoptosis must be distinguished from necrosis, which is also a form of cellular death. In contrast to apoptosis, necrosis is not a genetically programmed function, it affects groups of neighboring cells, and produces an inflammatory response. 10 The death of a cell by necrosis leads to the release of alarm signal molecules that stimulate 1 or more pattern‐recognition receptors on macrophages, dendritic cells, and natural killer cells. 11 The presence of necrotic cells in a tissue is frequently interpreted by the immune system as dangerous and therefore acts as a signal to initiate an immune response. 11 Unlike apoptosis, with necrosis there is cellular swelling with loss of cell membrane integrity, organelle swelling, and lysosomal leakage. The degradation of DNA is random and lysed cells are ingested by macrophages. 12
Whether a cell survives or dies by apoptosis is determined by the balance between pro‐apoptotic (stress or death) signals and anti‐apoptotic (mitogenic or survival) signals within and around the cell (see 1, 2). Cell injury via oxygen deprivation, heat stress, chemical agents, radiation, infectious agents, genetic derangements, nutritional imbalances, immunologic reactions (eg, anaphylaxis), and other types of severe cell stress will initiate the pro‐apoptotic pathways. 4 Dysregulation of apoptosis can affect the equilibrium between cell growth and cell death, resulting in organ dysfunction.
Table 1.
Pro‐apoptotic mediators
| Agent | Mechanism |
|---|---|
| Extrinsic pathway | |
| TRAIL | Ligand binds to TNF‐α receptor |
| TNF‐α | Binds to TNF‐α receptor; breakdown of sphingomyelin to ceramide |
| FasL | Binds to Fas receptor; breakdown of sphingomyelin to ceramide |
| DISC | Activation of caspase‐8,‐10; recruits c‐FLIP; cleaves tBid to increase MOMP |
| TRADD, FADD | Adaptor proteins; death domain grouping recruits procasapase‐8, ‐10 |
| TWEAK | Ligand that binds to pro‐apoptotic receptor |
| NGF | Ligand that binds to pro‐apoptotic receptor |
| Intrinsic mitochondrial pathway | |
| Bcl‐2 family proteins Group II, III | |
| BH3‐only | Mediates diverse death stimuli from environment and from within cell; inactivates Bcl‐2, Bcl‐xL, triggering Bax/Bak |
| Bim | Binds and inhibits Bcl‐2; may inactivate Bax/Bak |
| Bmf | Binds and inhibits Bcl‐2 |
| Bik, Bad | Binds and inhibits Bcl‐2 |
| Bid | Cleaved by caspase‐8 to form tBid, which causes conformational change in Bax to allow it to insert into mitochondrial membrane inducing channel formation; may also inactive Bcl‐2 |
| PUMA, NOXA | Activates Bax to increase MOMP |
| BH1, 2, 3 or multi‐domain | |
| Bax, Bak | Mitochondrial damage or changes cause cytochrome c release and ER depletion of calcium and caspase‐12 activation |
| Mitochondrial substances | |
| AIF | Induces caspase‐independent chromatin condensation and DNA fragmentation |
| Endo G | Breaks up DNA; caspase‐independent |
| Smac/DIABLO, HtrA2/Omi | Binds and neutralizes IAPs |
| Procaspases‐2,‐3,‐9 | Initiates caspase cascade |
| Cytochrome c | Can cause reduction in mitochondrial membrane potential; binds with procaspase‐9 and Apaf‐1 to form apoptosome |
| ER pathway | Dissociation of TRAF2 and activation of caspase‐12; release of cytochrome c |
| Caspases (effector) | |
| ‐3, ‐6, ‐7 | Cleave cell membrane proteins, nucleus and cytoplasm |
| Alternate substances | |
| Granzyme B | Activates effector and activator caspases |
| Ceramide | Accumulates on mitochondrial membrane inhibiting Bcl‐2; causes cytochrome c release; activates caspase‐9 and effector caspases; activates Bax; releases cathepsins |
| p53 | Suppresses the transcription of Bcl‐2; induces the manufacture of Bax, insulin growth factor binding protein‐3; upregulates the Fas receptor |
| Cathepsin D | Activates procaspase‐3,‐9; cleavage of Bid |
| c‐Abl tyrosine kinase | Release of cytochrome c from the mitochondria |
TRAIL, tumor necrosis factor‐related apoptosis inducing ligand; FasL, Fas ligand; DISC, death‐inducing signaling complex; c‐FLIP, FLICE‐like inhibitory protein; MOMP, mitochondrial outer membrane permeability; FADD, Fas‐associated death domain; TRADD, TNFR1‐associated death domain; TWEAK, TNF‐like WEAK inducer of apoptosis; NGF, nerve growth factor; Bcl‐2, B‐cell lymphoma 2; BH3, Bcl‐homology‐3; tBid, truncated Bid; Bax, Bcl‐2‐associated protein x; Bak, Bcl‐2‐associated protein k; Apaf‐1, apoptotsis activating factor‐1 – activates procaspase 9; AIF, apoptosis inducing factor; Endo G, endonuclease G; IAPs, inibitor of apoptosis proteins; Smac/DIABLO, second mitochondrial‐derived activator of caspases – director inhibitor of apoptosis‐binding protein with LOw pI; TRAF2, TNF receptor associated factor 2.
Table 2.
Anti‐apoptotic mediators
| Agent | Mechanism |
|---|---|
| Intrinsic pathway | |
| Bcl‐2 (Group I), Bcl‐xL | Controls mitochondrial permeability |
| Inhibits Bax, Bak, granzyme B, p53 | |
| Extrinsic pathway | |
| c‐FLIP | Prevents caspase‐8 binding to various death receptors |
| NF‐κB | Upregulates anti‐apoptotic mediators c‐FLIP, IAPs; accelerates growth; activates anti‐apoptotic gene regulator p65 |
| Alternate pathways | |
| IAP | Mimics Bcl‐2; inhibits caspase‐9 activity |
| Survivin | Regulates cell cycle mitosis; inhibits caspases‐3,‐7 |
| XIAP | Inhibits caspase‐3,‐7,‐9; activates NF‐κB |
| Cytokine receptors | |
| JAK, STAT | Cytokine receptor interaction induces survival gene production through activation of NF‐κB |
| MAPK | Translocates to nucleus; induces genetic production of anti‐apoptotic factors |
| PKR | Phosphorylation of the protein initiation factor 2 alpha and IκB kinase complex; delays apoptosis |
| CDKs, cyclins, | Controls cell cycle machinery |
| CDK inhibitors | |
Bcl‐2, B‐cell lymphoma 2; Bcl‐xL, Bcl‐2‐associated protein xL; Bax, Bcl‐2‐associated protein x; Bak, Bcl‐2‐associated protein k; c‐FLIP, FLICE‐like inhibitory protein; NF‐κB, nuclear factor‐κB; IκB, inhibitory‐κB; IAPs, inibitor of apoptosis proteins; XIAP, X‐linked inhibitor of apoptosis protein; JAK, Janus kinase; STAT, signal transducers and activators of transcription; MAPK, mitogen‐activated protein kinase; PKR, protein kinase R; CDK, cyclin‐dependent kinase.
Apoptosis in health is a finely balanced process. Too much or too little apoptosis contributes to disease. Apoptosis of infected cells is part of the host's defense mechanism. Some viruses and bacteria, however, have developed the ability to inhibit the infected cell's apoptotic mechanisms and protect their environment. 13 Inhibition of apoptosis is linked to uncontrolled cell growth and the formation of many types of cancer. In humans, excessive apoptosis is linked to stroke and Alzheimer's disease. 14 The activation or restoration of apoptosis is emerging as a key strategy for treatment of cancer and other diseases. 15 , 16 , 17 , 18 , 19 , 20
Our aim is to provide a basic review of the literature regarding the mechanisms and regulation of apoptosis. Extracellular ligand‐directed or intracellular stress‐induced stimuli can activate this highly regulated process. Caspases play a central role by initiating and executing the intracellular cascade of events that result in protein and nucleic acid cleavage, and ultimately, cell death. Many of the key apoptotic proteins have been identified, however there is still much to learn regarding the molecular mechanisms of action or activation of these proteins. Knowledge of the pro‐apoptotic and anti‐apoptotic cell pathways is important to understanding the mechanisms of many life‐altering diseases in humans and animals and realizing the potential for novel therapeutics.
Pathways of Apoptosis
Apoptosis can be genetically encoded or can occur in response to cellular or external stimuli. There are 3 features that characterize apoptosis: protein cleavage or hydrolysis, breakdown of nuclear DNA, and recognition of the apoptotic cell by phagocytic cells. 4 The cleavage of proteins primarily occurs with the activation of a family of cysteine proteases called caspases (Cysteine ASPartate‐Specific ProteASEs). 8 Caspases are synthesized in an inactive form and activated by specific initiation mechanisms. 10 Programmed cell death can also result from caspase‐independent mechanisms triggered by cell membrane receptor‐ligand binding or damage to cell organelles. 21 , 22 , 23 , 24 , 25 , 26
Initiation of Caspase Cascades
There are 3 known pathways that initiate the activation of caspase cascades and the programmed death of a cell. The route utilized is dependent on the initial death signal, the cell type involved, and the balance between pro‐apoptotic and anti‐apoptotic signals. 10 One initiating path may lead to another with cross‐talk between them possible. Two of the pathways, the death receptor (DR) (extrinsic) and mitochondrial (intrinsic), have been detailed in the literature. 2 , 14 , 27 , 28 , 29 The third is an intrinsic pathway involving the endoplasmic reticulum (ER) and is the least understood. 30 , 31 , 32 , 33 , 34 This pathway is believed to be a pathologically relevant form of apoptosis occurring in response to cellular stress. 10 , 35
Extrinsic (DR) pathway: The extrinsic pathway (see Figure 1) begins with pro‐apoptotic receptors on the cell's surface activated by a pro‐apoptotic molecule or ligands specific for that receptor. These cell DRs belong to the tumor necrosis factor (TNF) receptor superfamily, with the Fas receptor and TNFR1 as the most intensely studied members. 15 Fas is present on a variety of cell types including activated B cells and T cells. 36 Ligands that activate pro‐apoptotic receptors include the Fas ligand (FasL) and TNF‐α 37 , 38 , 39 , 40 , 41 , 42 (Table 1). FasL is expressed by a variety of cell types, including activated T cells and natural killer cells. 36 TNF‐α is produced predominantly by activated monocyte/macrophages and lymphocytes. 26
Figure 1.
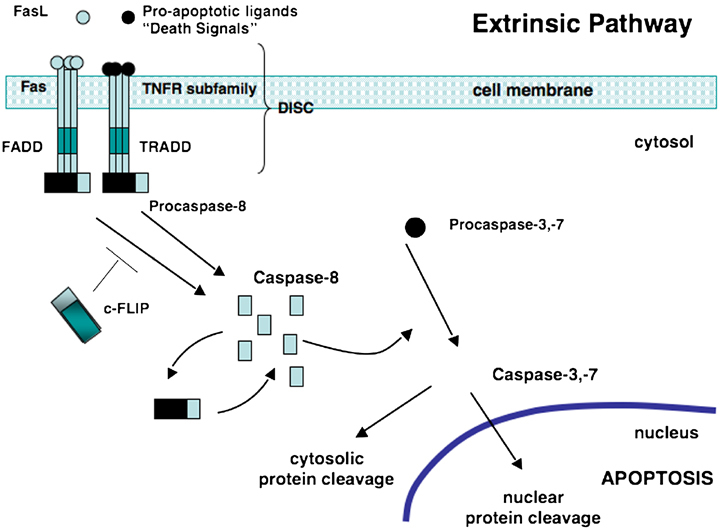
The extrinsic or death receptor (DR) pathway. Pro‐apoptotic ligands, death signals, and Fas bind to Fas or TNFRs. The intracellular portion of the DR is known as the death domain (DD). Bunching of the receptor‐ligand complexes groups their DDs and a binding site for an adaptor protein is formed. This ligand‐receptor‐adaptor protein complex is called the death‐inducing signaling complex (DISC). It recruits and assembles initiator caspase‐8 that releases active caspase enzyme molecules into the cytosol. Here, they activate the effector caspases‐3 and ‐7, resulting in nuclear protein cleavage and the initiation of apoptosis. FasL, Fas ligand; TNFR tumor necrosis factor receptor; FADD, Fas‐associated death domain; TRADD, TNF‐associated death domain; c‐FLIP, FLICE‐like inhibitory protein; DISC, death‐inducing signaling complex.
The intracellular portion of the DR is known as the death domain (DD). Once 3 or more DR‐ligand complexes bunch, their DDs are brought into close proximity and a binding site for an adaptor protein is formed. The adaptor protein is specific for that receptor (eg, Fas‐associated DD [FADD] or TNF receptor‐associated DD [TRADD]). This complex of ligand‐receptor‐adaptor protein is called the death‐inducing signaling complex (DISC), leading to the recruitment and assembly of initiator caspases 8 and 10. 43 , 44 , 45 , 46 These caspases can now undergo self‐processing and release active caspase enzyme molecules into the cytosol. Here, they activate the effector caspases 3, 6, and 7. 15 , 47 , 48 , Figure 1 illustrates the sequence of events that trigger the extrinsic pathway.
Intrinsic mitochondrial pathway: The intrinsic mitochondrial pathway (see Figure 2) is initiated from within the cell in response to cellular stresses such as DNA damage, radical oxygen species, radiation, hormone or growth‐factor deprivation, chemotherapeutic agents, cytokines, and glucocorticoids. 30 Initiation of this pathway eventually results in the release of pro‐apoptotic proteins from the mitochondria that will activate caspase enzymes and trigger apoptosis. 49 , 50 , 51 , 52 The success of the pathway in inducing apoptosis depends on the balance of activity between pro‐apoptotic and anti‐apoptotic members of the B‐cell lymphoma‐2 (Bcl‐2) superfamily of proteins (Table 1).
Figure 2.
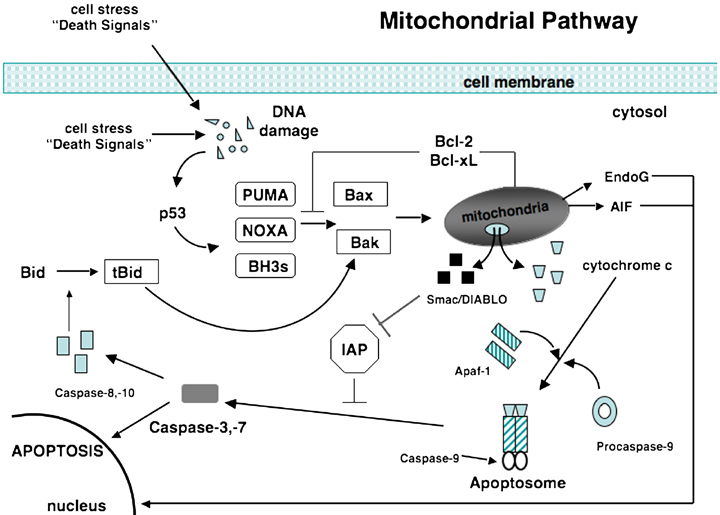
The mitochondrial or intrinsic pathway. Activation of the pro‐apoptotic proteins Bax and Bak occurs through conversion of Bid to tBid by caspase‐8 or‐10 and through activation of PUMA, Noxa, or other BH3 initiator proteins when p53 is induced by DNA damage. Activated Bax and Bak oligomerize at the mitochondrial membrane and cause the release of several mitochondrial factors. Cytochrome c combines with Apaf‐1 and procaspase‐9 forming an apoptosome. Also released from the mitochondria are Smac/DIABLO, proteins that inactivate IAPs. Activated caspase‐9 then is able to activate caspase‐3 or ‐7 allowing apoptosis to proceed. Also released from the mitochondria are EndoG and AIF that stimulate apoptosis independent of caspases. Bcl‐2 and Bcl‐xL block the activation of Bax and Bak. Bcl‐2, B‐cell lymphoma‐2; IAP, inhibitor of apoptosis protein; Apaf‐1, apoptosis‐activating factor‐1; Smac, second mitochondrial‐derived activator of caspases; DIABLO, director inhibitor of apoptosis‐binding protein with LOw pI; BH3, Bcl‐homology‐3; tBid, truncated Bid; EndoG, endonuclease G; AIF, apoptosis‐inducing factor; Bax, Bcl‐2‐associated protein x; Bak, Bcl‐2‐associated protein k.
Bcl‐2 superfamily of proteins derives its name as the second member of a range of proteins found in follicular lymphoma. 4 , 53 All of the Bcl‐2 family members are present on the outer mitochondrial membranes as dimers where they control membrane permeability in ion channel fashion or through the creation of pores. 53 The permeability of the mitochondrial outer membrane determines whether or not there is release of the pro‐apoptogenic substances from the mitochondria.
This Bcl‐2 family of proteins is subdivided into 3 groups based on structural similarities and functional criteria. Group I possess anti‐apoptotic activity while groups II and III promote cell death. 2 The Bcl‐2 family share 1 or more of 4 characteristic domains of homology crucial for function. The anti‐apoptotic Bcl‐2 family proteins, such as Bcl‐2 and Bcl‐xL, contain all 4 domains and exert their control of mitochondrial permeability by stimulating ADP/ATP exchange, stabilizing the mitochondrial inner transmembrane potential, and preventing the opening of a permeability transition pore. 54 Overexpression of Bcl‐2 and Bcl‐xL is known to be associated with a number of human malignancies 49 , 55 , 56 (Table 2). These proteins also act by inhibiting the action of the pro‐apoptotic proteins, Bax and Bak.
Pro‐apoptotic proteins of the Bcl‐2 family initiate apoptosis by blocking the anti‐apoptotic activity of Bcl‐2 and Bcl‐xL by binding to their mitochondrial binding sites or by triggering the activation of pro‐apoptotic Bax/Bak. 57 A third type of pro‐apoptotic activity is through the cytoplasmic protein, Bid. This molecule is found in the cytoplasm in an inactive form. When cleaved by activated caspase‐8 from the extrinsic pathway, Bid (once activated, referred to as t‐Bid) causes a structural change to Bax making it similar to the structure of the anti‐apoptotic molecule, Bcl‐2, allowing Bax to translocate to the mitochondria. 58 , 59 , 60 , 61 This is but one method of cross‐talk that occurs between the intrinsic and extrinsic pathways.
Each Bcl‐2 family member can interact with other Bcl‐2 members, so that large numbers of heterodimer combinations within a cell are possible. Cells with more pro‐death proteins are sensitive to death and cells with an excess of protective family members are usually apoptosis‐resistant. 2
There are at least 3 current theories describing the exact mechanism by which the Bcl‐2 pro‐apoptotic proteins lead to increased mitochondrial permeability. The first theory describes the insertion of Bcl‐2 proteins into the mitochondrial membrane and directly forming a channel. 2 , 62 A second theory explains the Bcl‐2 pro‐apoptotic proteins interacting with other mitochondrial membrane proteins, possibly voltage‐dependent anion channel, to form large pores. 63 The size of the voltage‐dependent anion channel is too small for proteins to pass, so this model assumes that there is a conformation change with Bcl‐2 binding. 2 The third theory describes the Bcl‐2 proteins modulating the mitochondrial proteins resulting in the formation of a permeability transition pore and loss of membrane potential, organelle swelling, and loss of cytochrome c from the pore. 64 , 65 Once the permeability of the membrane has been compromised, cytochrome c is released and combines with a cytosolic molecule called apoptosis activating factor‐1 (Apaf‐1). Cytochrome c and Apaf‐1 combine with procaspase‐9 for activation of this caspase (Figure 2). The binding of these 3 substances forms an apoptosome, which then activates procaspase‐3.
Alternate substances can initiate the intrinsic and extrinsic pathways (Table 1). Phosphoprotein p53 is a transcription factor that regulates the cell cycle and functions as a cell stress sensor molecule capable of inducing the intrinsic mitochondrial pathway. Factors that damage DNA, such as ionizing radiation, genotoxic drugs, and free radicals, activate p53. Activated p53 promotes apoptosis primarily through its ability to suppress the transcription of anti‐apoptotic factors like Bcl‐2 or induce the manufacture of pro‐apoptotic factors like Bax, insulin growth factor binding protein‐3 and upregulation of the Fas receptor. 10 , 66 Bcl‐xL can inhibit p53, 64 and inactivation or loss of p53 is a common abnormality of many human cancers. 66 , 67 One veterinary study showed an increased risk in cats, diagnosed with oral squamous cell carcinoma and exposed to secondhand tobacco smoke, to overexpress p53. In human cancer, p53 is the most commonly disrupted gene and is also the most frequently mutated gene in human oral cancer. 68
Intrinsic ER pathway: The third and least understood pathway is referred to as the endoplasmic reticulum or ER pathway. 30 It involves caspase‐12 and is said to be able to function independently of the mitochondria. 32 Cellular stresses such as hypoxia, glucose starvation, disturbances in calcium homeostasis, and exposure to free radicals injure the ER, resulting in the unfolding of proteins and reduction in protein synthesis. In normal cells, an adaptor protein, TNF receptor‐associated factor 2 (TRAF2), is bound to procaspase‐12, rendering it inactive. Stress of the ER leads to the dissociation of TRAF2, activation of caspase‐12. 34 , 35 , 69 Once activated, caspase‐12 cleaves procaspase‐9, which then cleaves procaspase‐3. This mechanism is independent of the mitochondria although there is evidence that caspase 12 can cause the release of cytochrome c from the mitochondria and thus stimulate the intrinsic pathway 35 (Table 1).
There are other mechanisms of cross‐talk between the intrinsic pathways that initiate apoptosis. Ito et al, 69 demonstrated that ER stress can cause activation of a positive apoptosis regulator, c‐Abl tyrosine kinase, known for its tumorogenic characteristics and results in the release of cytochrome c from the mitochondria. 25 , 69 In addition, the mitochondrial pathway pro‐apoptotic Bcl‐2 family protein, Bak, has been implicated in causing ER depletion of calcium, which can induce caspase‐12 activation 70 (Table 1).
Caspase cascade
The central executioners of apoptosis are part of a large protein family known as the caspases. 2 , 71 To date, 14 caspases have been identified. Specific caspases are found in relatively large amounts as inactive precursors called procaspases within the cytoplasm. Procaspases can be activated by 1 of 3 methods: (1) exposure to another activated caspase, (2) autocatalysis, or (3) association with an activator protein, such as caspase‐9, Apaf‐1, and cytochrome c. 2
The caspases involved in apoptosis are subdivided into initiator caspases (2, 8, 9, 10) and effector (or executioner) caspases (3, 6, 7). The initiator caspases are activated by adaptor‐mediated self‐cleavage. The specific interaction of caspases and activator protein promotes the formation of a multimeric complex that is necessary to bring 2 caspase precursors together to activate each other and produce an active tetramer. 72 , 73 This caspase cascade strategy of activation is used by the initiator caspases to cleave and activate the effector caspases. 65
When activated, the effector caspases selectively cleave a restricted set of target proteins that follow an aspartate residue. In most cases, this results in inactivation of the target protein. However, they can also activate proteins, either directly by cleaving off a negative regulatory domain, or indirectly, by inactivating a regulatory subunit. 2 , 74 Effector caspases cause cytoskeletal filament aggregation, clumping of ribosomal particles and rearrangement of rough ER to form a series of concentric whorls as seen on electron microscopy (Table 1). 10 , 65 Caspases are also responsible for cleaving nuclear lamins required for nuclear shrinking and budding, and for loss of cellular shape and membrane blebbing. 2 , 75 , 76
Caspases will activate caspase‐activated DNAases (CAD) that breakdown nuclear DNA, the second feature of apoptosis. CAD exists in an inactive form (ICAD) in the nucleus when it is bound to a subunit. Once the effector caspase‐3 is activated it migrates to the nucleus and cleaves the inhibitory subunit thus activating CAD. 77 CAD is the nuclease responsible for breaking down the DNA into 50–300‐kb pieces that are then cleaved into 180–200‐bp fragments by endonucleases. It is these fragments that compose the DNA ladders visualized by agarose gel electrophoresis, a biochemical hallmark of apoptosis. 10
Alternate pathways
Granzyme B: There is an accessory method of triggering apoptosis by the serine protease granzyme B, a lymphocyte granular enzyme expressed by activated cytotoxic T lymphocytes and natural killer cells. Granzyme B will bind to its cell surface receptor, an insulin‐like growth factor II receptor, causing endocytosis of the protease. It remains in the endocytic vesicle until stimulation by an activated cytotoxic T cell. 78 Activation of almost all of the activator and effector caspases can occur through the action of the granzyme B pathway. A number of caspase sensitive targets, as well as other unique proteins not normally cleaved by caspases, can be cleaved directly by granzyme B. 74 , 79 This mechanism is dependent on mitochondrial disruption, as overexpression of anti‐apoptotic Bcl‐2 will halt this process. Granzyme‐mediated apoptosis is integral to the immune surveillance machinery that rid the body of virally infected or malignantly transformed cells 26 (Table 1).
Mitochondrial factors: Increased mitochondrial outer membrane permeability can result in the release of mitochondrial pro‐death substances in addition to cytochrome c, such as apoptosis inducing factor, 67 Smac/DIABLO 80 , 81 , 82 (Second Mitochondrial‐derived Activator of Caspases/Direct Inhibitor of Apoptosis‐Binding protein with LOw pI), endonuclease G, HtrA2/Omi, and several procaspases 83 (eg, procaspase‐2, ‐3, and ‐9). Smac/DIABLO and HtrA2/Omi bind to cytoplasmic inhibitor of apoptosis proteins (IAPs), neutralizing their anti‐apoptotic activity. The migration of apoptosis inducing factor from the mitochondria to the nucleus induces caspase‐independent chromatin condensation and DNA fragmentation. Endonuclease G can also directly cause the break up of DNA independent of caspases 84 (Table 1, Figure 2).
Ceramide: The ceramide/sphingomyelin pathway can proceed with or without caspase interaction. Ceramides are sphingolipid‐signaling mediators involved in the regulation of differentiation, growth suppression, cell aging, stress responses, and apoptosis. Various stresses, such as ultraviolet radiation, radical oxygen species, chemotherapeutics, IL‐1β, TNF‐α, or Fas activation, initiate this pathway. 21 Sphingomyelinases are activated by binding to TNF receptor family DD, FADD, and TRADD and cleave sphingomyelin, a member of the phospholipid bilayer, into ceramide. 22 Ceramide can also be generated on lysosomes, ER, and mitochondrial membranes, when stimulated by a variety of cytotoxic agents. 85 , 86 , 87
Increased levels of ceramide initiate an apoptotic program involving mitochondrial membrane disruption (Table 1). Ceramide‐induced apoptosis is mediated through the mitochondria when ceramide accumulates in the mitochondrial membrane. Elevation of ceramide and sphingosine result in increased mitochondrial membrane permeability, cyctochrome c release, and activation of caspase‐9. 26 The susceptibility of a cell for ceramide‐induced apoptosis is reliant on the cell's Bax to Bcl‐2 ratio. 23 Ceramide production, DNA damage, and radical oxygen species also stimulate cellular lysosomal release of cathepsins, which leads to release of mitochondrial factors, activation of procaspase‐9 and ‐3, and activation of Bid. 24 , 25
Inhibitors of apoptosis
The most well‐known anti‐apoptotic factors are members of the Bcl‐2 family (Table 2). There is a dynamic equilibrium between the anti‐apoptotic members and pro‐apoptotic members of the Bcl‐2 family. Additional inhibitors include IAPs, FLICE‐like inhibitory protein (c‐FLIP) and nuclear factor‐κB (NF‐κB).
IAPs: A failsafe inhibitory mechanism exists in the intrinsic pathway. Pro‐apoptotic activity is counterbalanced by a family of at least 8 proteins, known as IAPs (eg, survivin and XIAP). The IAPs compromise the effector phase of apoptosis through blocking or inactivating caspases 88 , 89 , 90 , 91 , 92 , 93 (see Table 2). They have been found associated with the activated TNF receptors 88 where they block the activation of caspase‐8 and are upregulated by NF‐κB activation. 89 , 90
IAPs also act downstream of the mitochondrial release of cytochrome c to prevent activation of caspase‐9. 94 , 95 Caspase‐9 uses a peptide from one of its end subunits to attract an IAP family molecule. This binding allows caspase‐9 to remain dormant even though the initial steps for activation have taken place. 96 , 97 The overexpression of IAPs is associated with a drug‐resistant phenotype of cancer cells. 98
c‐FLIP: c‐FLIP is a protein‐deficient caspase homolog. This inhibitor prevents both the binding of caspase‐8 to various DRs and its activation. 99 The ratio of c‐FLIP to caspase‐8 is critical for the assembly of the DISC. Formation of the DISC recruits c‐FLIP to bind to its target molecule. 57 This works as a built‐in safety system and alludes to the integral balance between pro‐apoptotic and anti‐apoptotic mediators.
Upregulation of c‐FLIP has been associated with diverse hematologic cancer cell lines. 100 , 101 The sensitization of many cancer cells to death ligand‐mediated apoptosis appears to be mediated by c‐FLIP downregulation. 102 , 103 There does not appear to be inactivation of caspase‐9 mechanisms because c‐FLIP does not prevent apoptosis induced by granzyme B or by chemotherapeutic drugs and irradiation. 104
NF‐κB: NF‐κB is a transcription factor and an anti‐apoptotic gene regulator composed of a p50/p65 heterodimer. The p65 subunit provides the gene regulatory function. NF‐κB is kept quiescent in the cytoplasm as a dimer bound to its repressor, inhibitors of NF‐κB (IκB) family. Phosphorylation of IκB by upstream kinases frees NF‐κB, which then translocates to the nucleus. 105 The p65 subunit is eventually released from the DNA and binds to newly synthesized IκB α, which complexes with NF‐κB. This complex translocates back into the cytoplasm. Stimulation of NF‐κB activation has been associated with accelerated growth, resistance to apoptosis, and propensity to form metastases. Inversely, inhibition of NF‐κB activation produces an increase in apoptosis, indicating that the balance of cell viability versus cell death is maintained in some degree to NF‐κB activation. 106
Recognition of apoptotic cells
The third characteristic of apoptosis is recognition of the dying cells by phagocytic cells. The apoptotic cell expresses markers on their membrane that are recognized by phagocytes. 4 Through internal cellular signals phosphatidylserine, a phospholipid component, is shifted from the inner to the outer layer of the plasma membrane. 10 This allows for early recognition and removal of a dying cell without release of pro‐inflammatory mediators, as occurs with necrosis. 4
Regulation of apoptosis
Control of whether the pro‐apoptotic or anti‐apoptotic pathway is chosen is subject to positive and negative genetic and environmental regulators. Pro‐apoptotic gene activation will lead to cell death while deactivation of the gene will block apoptotic pathways. Genetic regulation can also be modified by exogenous stimuli from the cell's immediate environment. A cell destined or started on a death pathway can receive a survival signal that can save the cell from apoptosis. Genetic regulators (mostly pro‐apoptotic) include the c‐Myc gene family, the p53 tumor suppressor gene, DRs and the caspase family.
The c‐Myc gene is a member of the Janus kinase or JAK family, and is involved in both cell proliferation and apoptosis. In the presence of anti‐apoptotic cytokines (eg, insulin‐like growth factor‐1) or negative regulators of apoptosis (eg Bcl‐2), c‐Myc drives cellular proliferation. In the absence of these factors, the c‐Myc family promotes apoptosis. 10
Survival signals, such as growth factors and other soluble mediators, are often released by neighboring cells. Hematopoietic cell lines and differentiated cells are dependent on survival factors like granulocyte macrophage colony stimulating factor, granulocyte colony stimulating factor, IL‐3, and erythropoietin. 10 T and B lymphocytes are dependent on certain interleukins, such as IL‐7 and IL‐2, to mature properly or lead to cellular differentiation. 57 The role of environmental survival substances leads to the possibility of dysregulation of apoptosis in the presence of inappropriate survival signals, as observed in tumors and sepsis. 12 , 107 , 108
Apoptosis in disease
Dysregulated apoptosis best describes pathologic disease states that induce or inhibit cell death inappropriately. In humans, excessive apoptosis is linked to stroke and Alzheimer's disease and reduced apoptosis to cancer and autoimmune disease. 14 , 28 , 109 Apoptosis also plays a key role in sepsis. A review of the current literature in companion animal medicine yields research pertaining to apoptosis in the fields of oncology, orthopedics, and virology, as well as in other disease states. Table 3 lists some of the articles of veterinary origin that investigate apoptosis in disease.
Table 3.
Veterinary studies pertaining to apoptosis from 2005 to March 2008
| Author | Journal | Topic |
|---|---|---|
| Oncology | ||
| 1. Scase, TJ et al | J Vet Intern Med 2006:20(1):151–8 | Canine mast cell tumors and markers of apoptosis and cell proliferation |
| 2. Johnson, ME et al | Vet Pathol 2004;41(6):599–607 | Review of survivin, an IAP, and its role in cancer |
| 3. Greissmayr, PC et al | J Vet Intern Med 2007;21(6):1409–12 | Mushroom‐derived Maitake PET fraction in the treatment of canine LSA |
| 4. Fan, TM | Vet Clin North Am Small Anim Pract 2007;37(6):1091–110 | Biphosphonates and cancer management |
| 5. Lee, JJ et al | Am J Vet Res 2007;68(4):411–22 | VP3 gene of chicken anemia virus causing apoptosis in K9 mammary tumor cells |
| 6. Kano, R et al | Vet Clin Pathol 2008;37(1):57–60 | Bcl‐2 expression in feline lymphoma cell lines |
| 7. Sano, J et al | Res Vet Sci 2005;79(3):197–201 | Antineoplastic drug effects on Bcl‐2 and Bcl‐xL genes in feline T‐cell leukemia |
| Orthopedics | ||
| 8. Lim, S et al | Vet Res Commun 2008;32(3):243–53 | Enrofloxacin causes apoptosis in canine tendons and chondrocytes |
| 9. Gyger, O et al | Vet J 2007;174(2):371–7 | Apoptosis in normal and diseased canine cruciate ligaments |
| 10. Echigo, R et al | J Vet Med Sci 2006;68(8):899–902 | Hyaluronan decreases chondrocyte apoptosis in dogs with ACL injury |
| Viral | ||
| 11. Ruggieri, A et al | Vet Microbiol 2007;121(1–2):64–72 | Canine coronavirus induces apoptosis through caspase‐3 activation |
| 12. Takano, T et al | Vet Microbiol 2007;119(2–4):121–31 | TNF‐α produced by FIP‐infected macrophages induces and apoptosis in uninfected T cells |
| 13. Natoni, A et al | J Gen Virol 2006;87(Pt 2):357–61 | Mitochondrial pathway triggered during feline calicivirus infection |
| Miscellaneous | ||
| 14. Reusch, CE et al | Vet Rec 2007;160(7):219–24 | Trilostane causing adrenal gland apoptosis and necrosis |
| 15. Ammersbach, MA et al | J Vet Intern Med 2006;20(5):1166–71 | Glucocorticoid immunosuppressive effects on canine lymphocytes may involve apoptosis |
Apoptosis in Cancer
A feature characteristic of cancer cells is the uncoupling of cell division and cell death; cells that should have died were not properly signaled to do so. 3 Oncologic studies are investigating the enhancement of apoptosis to arrest the growth of tumor cells. One mechanism through which normal cellular numbers are maintained is through p53 and its control over‐apoptosis. Deficiencies in p53 can lead to reduced apoptosis and tumor development. 66 , 110 Some cancers harbor mutations in the p53 genome or disrupt normal p53 functions whereas others increase or overexpress Bcl‐2 proteins leading to a cessation of the normal cellular death program. 78
Apoptosis in sepsis
Cellular demise in sepsis can occur through apoptosis as well as necrosis. In 1996, Bone first proposed that apoptosis contributes to the multiple organ dysfunction in sepsis. 10 , 111 Most treatments had been aimed at blunting the over‐reactive or pro‐inflammatory response. Bone proposed that the anergic or hypoimmune aspect of sepsis, when apoptosis becomes most critical, must also be addressed, 111 Apoptotic loss of B cells, T cells, and dendritic cells in sepsis decreases antibody production, macrophage activation, and antigen presentation, respectively. 112
Leukocytes are responsible for opsonization and phagocytosis of infected cells and antigens at the site of inflammation. Neutrophils produce highly toxic and unstable reactive oxygen species and release bactericidal substances. A hallmark of sepsis is the loss of normal apoptosis of neutrophils. This prolonged life produces neighboring cell damage and contributes to activation of pro‐inflammatory cytokines. Normally, pro‐inflammatory mediators (TNF‐α, IL‐1β, IL‐6, IL‐8, and IFN‐γ) released from macrophages and neutrophils have overlapping effects and function to limit damage, combat pathogenic organisms, eliminate foreign antigens, and promote repair. 113 Anti‐inflammatory cytokines (IL‐4, IL‐10, TGF‐β, soluble receptors and receptor antagonists) are also quickly released to try to reduce and locally contain the inflammatory response. 114 Many of these inflammatory components are the key factors responsible for the dysregulated apoptosis of immune cells in sepsis.
Key cells involved in the inflammatory process (neutrophils, macrophages, dendritic cells, and lymphocytes) are also cells targeted for apoptosis. Apoptosis of immune cells is normally not a pathologic process because inflammatory cells must be eliminated so that inflammation does not continue unabated. 115 , 116 However, in sepsis or other overwhelming inflammatory processes (like trauma 117 and severe burns) there is extensive cell death of lymphocytes 118 and dendritic cells and delayed cell death of neutrophils. This leads to a blunted immune response coinciding with increased cellular damage.
Neutrophils are one of the first cells to migrate to the site of inflammation with an average half life of 6–12 hours when unstimulated. 119 , 120 Once a neutrophil is released into circulation, its apoptotic program has been activated. Studies have shown that sepsis can shorten as well as prolong the life span of the activated neutrophils (early or delayed apoptosis, respectively). 121 , 122 , 123 , 124
Early apoptosis of neutrophils dampens respiratory burst activity and may lessen secondary tissue injury. 125 Delayed apoptosis of neutrophils contributes to increased cellular damage, especially in the lung, liver, kidneys, and gastrointestinal tract. Acute respiratory distress syndrome (ARDS) is marked by significant pulmonary accumulation of neutrophils, 126 , 127 and is considered to be a direct effect of neutrophil‐induced injury. Cells retrieved from the lungs of septic patients show reduced rates of neutrophil apoptosis with the degree of inhibition paralleling the severity of sepsis. 128 , 129 Additional studies have shown that increased apoptosis (via Fas/FasL‐dependent mechanism) of pulmonary epithelial cells will lead to permeability changes characteristic of ARDS. 130 , 131
The cytokines produced by activated neutrophils summon macrophages to the area of inflammation; cytokines are also produced by tissue macrophages in response to foreign invasion. Macrophages are antigen‐presenting cells (APC) capable of engulfing foreign material, infected cells, and apoptotic cells through recognition of specific cell surface molecules. Dendritic cells, another type of APC, are viewed as the sentinels of the immune system. 132 Like macrophages, mature dendritic cells are able to activate lymphocytes through the presentation of antigen.
Lymphocytes need 2 signals to stimulate differentiation and initiation of the immune response. The first signal is the presentation of antigen thus accounting for the specificity of the response. The second signal involves costimulatory molecules on APCs or APC secretion of cytokines. The costimulatory molecules interact with specific T cell sites producing a pro‐apoptotic or anti‐apoptotic state. Failure of the appropriate second signal after interaction with an APC results in anergy or apoptosis of the lymphocyte. 133 , 134 Anergy is a state of unresponsiveness to antigen. 112 This is also the mechanism for self‐tolerance. 135
Immature dendritic cells are capable of ingesting apoptotic cells but this will render them incapable of maturing and stimulating T cells. 136 Macrophages and dendritic cells will secrete IL‐10 after engulfing apoptotic cells. IL‐10, considered an anti‐inflammatory cytokine, selectively blocks the maturation of dendritic cells. 137 This has been shown to suppress the phagocytic activity as well as pro‐inflammatory cytokine production of alveolar macrophages. 138 IL‐6 is also secreted by dendritic cells after ingesting apoptotic cells, leading to autocrine blockage of maturation. 139 This lack of maturation leads to a tolerogenic state with no further stimulation of the immune system.
After ingesting apoptotic cells, dendritic cells can mature only when there are danger signals expressed by the apoptotic cell or when dendritic cells are engulfing an excessive number of apoptotic cells. This leads to secretion of pro‐inflammatory cytokines IL‐1β and TNF‐α by these signaled dendritic cells. 137 Inadequate clearance of surplus apoptotic cells results in these cells becoming necrotic and inducing a pro‐inflammatory response. 140
Studies have demonstrated apoptosis of intestinal and splenic B cells, CD4 T cells and dendritic cells in sepsis. 141 , 142 , 143 Overwhelming infection should lead to massive clonal expansion of B and T lymphocytes 142 but instead there is significant loss of these cell lines in sepsis. Lack of stimulation by APC cells experiencing apoptosis leads to poor B cell and T cell stimulation. These unstimulated T cells are removed by apoptosis.
In as many as 30% of bacteremic patients who die from systemic inflammatory response syndrome or multiple organ dysfunction syndrome, no focus of infection is identified. Premature B cell and intestinal epithelial cell death through apoptosis in the intestines is one theory to explain intestinal bacterial translocation and loss of the first line of intestinal defense. 10
Treatment strategies
Current human treatment strategies for manipulating apoptosis focus mainly on cancer and sepsis. A Pubmed literature search at the time of writing revealed a total of 645 citations for apoptosis and veterinary. Most of the studies are experimental at this point but many oncologic studies are manipulating apoptosis in the treatment of their patients (Table 3).
In human cancer research, peptidomimetics are in the early stages of experimental study. They are synthetic peptides that are resistant to enzymatic degradation and are being used for their pro‐apoptotic effects on Bid as well as their antagonism of IAPs. 144 , 145 In cancer treatments, there are trials attempting to block the overexpression of Bcl‐2 because it is the cessation of normal apoptosis that leads to the growth of tumors. In addition to the direct effects on the apoptotic programs, discoveries are being made that allow chemotherapeutics to act synergistically with various anti‐apoptotic therapeutics. 78
Treatment strategies for sepsis had previously targeted the hyperimmune phase rather than the hypoimmune phase. Addressing apoptosis can be therapeutically challenging because targeted blockade of apoptosis in lymphocyte populations must be specific enough to primarily target those cell populations undergoing increased apoptosis and to be sufficiently transient to prevent the risk of malignant transformation associated with prolonged blockade of apoptosis. 146 This poses a challenge to try to target specific pathways. Attempts at blocking the circulating mediators and cytokines that induce apoptosis have not been successful because of the inherent redundancy and fail‐safe mechanisms of the apoptotic pathways. 119 Caspase inhibitors show promise because caspases are common factors to many apoptotic pathways. Broad‐spectrum caspase inhibitors have been shown to prevent lymphocyte apoptosis and improve survival in animal models of sepsis. 13 , 147 , 148 Caution must be expressed, though, because increased dosages of caspase inhibitors can cause cytotoxicity and TNF‐α induced injury. 149 , 150
Gene therapy studies have shown that overexpression of Bcl‐2 delays or blocks apoptosis and improves survival in septic mice. 10 , 151 , 152 Other therapies target Akt, a regulator of cell proliferation and death. Mice overexpressing Akt have reduced lymphocyte apoptosis and increased survival after cecal ligation and puncture. 153 Fas fusion proteins, and attempts at altering gene expression of members of the DR pathways, are also areas of ongoing apoptotic research. 147 , 154 , 155 , 156 , 157 Recombinant human activated protein C, a product being used successfully in some septic patients, 158 , 159 may counteract the induction of apoptosis in monocyte 160 and endothelial cell lines, modulating the inflammatory and coagulation cascades during sepsis. 161 , 162 , 163 , 164 , 165 , 166 It also helps to attenuate the levels of pro‐ and anti‐apoptotic proteins in favor of survival.
In contrast to anti‐apoptotic strategies, recent studies have addressed the hypothesis that apoptosis in sepsis may in some cases be beneficial by downregulating the inflammatory response. Earlier onset of apoptosis may, in fact, favor survival. Giarmarellos‐Bourboulis et al 167 have associated monocyte apoptosis at the onset of sepsis to a favorable outcome due to a decrease in the amount of pro‐inflammatory cytokines produced.
Summary
Apoptosis is a normal biologic process necessary to maintain cellular homeostasis. There are characteristic pathways that lead to this form of cell death continually influenced by local cellular events, growth factors, and neighboring stresses. This complicated system has numerous built‐in avenues and failsafe mechanisms, including pro‐apoptotic and anti‐apoptotic factors. During sepsis and cancer, just 2 of the many diseases causing dysregulation of the apoptotic process, cells are either killed too quickly or survive too long. Newer therapies, designed to manipulate apoptosis depending on the pathology involved, can promote or delay this form of cellular demise. Although an extremely complicated process, understanding the relationship between sepsis and apoptosis will undoubtedly lead to new treatment modalities.
References
- 1. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972; 26:239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hengartner MO. The biochemistry of apoptosis. Nature 2000; 407 (6805):770–776. [DOI] [PubMed] [Google Scholar]
- 3. Perl M, Chung CS, Ayala A. Apoptosis. Crit Care Med 2005; 33 (12, suppl):S526–S529. [DOI] [PubMed] [Google Scholar]
- 4. Robbins SL CR. Robbins and Cotran pathologic basis of disease, 7th edn Philadelphia: Elsevier Saunders; 2005. [Google Scholar]
- 5. Hall PA, Coates PJ, Ansari B, et al Regulationan of cell number in the mammalian gastrointestinal tract; the importance of apoptosis. J Cell Sci 1994; 107:3569–3577. [DOI] [PubMed] [Google Scholar]
- 6. Cohen JJ, Duke RC. Apoptosis and programmed cell death in immunity. Annu Rev Immunol 1992; 10:267–293. [DOI] [PubMed] [Google Scholar]
- 7. Ekert PG, Vaux DL. Apoptosis and the immune system. Br Med Bull 1997; 53 (3):591–603. [DOI] [PubMed] [Google Scholar]
- 8. Gerschenson LE, Rotello RJ. Apoptosis: a different type of cell death. FASEB J 1992; 6:2450–2455. [DOI] [PubMed] [Google Scholar]
- 9. Ziegler U, Groscurth P. Morphological features of cell death. News Physiol Sci 2004; 19:124–128. [DOI] [PubMed] [Google Scholar]
- 10. Power C, Fanning N, Redmond HP. Cellular apoptosis and organ injury in sepsis: a review. Shock 2002; 18 (3):197–211. [DOI] [PubMed] [Google Scholar]
- 11. Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev 2008; 9:231–241. [DOI] [PubMed] [Google Scholar]
- 12. Proskuryakov S, Gabai V, Konoplyannikov A, et al Immunology of apoptosis and necrosis. Biochemistry (Moscow) 2005; 70 (12):1310–1320. [DOI] [PubMed] [Google Scholar]
- 13. Hotchkiss RS, Tinsley KW, Karl IE. Role of apoptotic cell death in sepsis. Scand J Infect Dis 2003; 35 (9):585–592. [DOI] [PubMed] [Google Scholar]
- 14. Finkel E. The mitochondrion: Is it central to apoptosis. Science 2001; 292 (5517):624–626. [DOI] [PubMed] [Google Scholar]
- 15. Ghobrial IM, Witzig TE, Adjel AA. Targeting apoptosis pathways in cancer. CA Cancer J Clin 2005; 55:178–194. [DOI] [PubMed] [Google Scholar]
- 16. Rowinsky EK. Targeted induction of apoptosis in cancer management: the emerging role of necrosis factor-related apoptosis-inducing ligand receptor activating agents. J Clin Oncol 2005; 23:9394–9307. [DOI] [PubMed] [Google Scholar]
- 17. Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer 2005; 5:876–885. [DOI] [PubMed] [Google Scholar]
- 18. Fischer U, Schultze‐Osthoff K. New approaches and therapuetics targeting apoptosis in disease. Pharmacol Rev 2005; 57:187–215. [DOI] [PubMed] [Google Scholar]
- 19. Reed JC. Drug insight: cancer therapy strategies based on restoration of endogenous cell death mechanisms. Nat Clin Pract Oncol 2006; 3:388–398. [DOI] [PubMed] [Google Scholar]
- 20. Reed JC, Pellecchia M. Apoptosis‐based therapies for hematologic malignancies. Blood 2005; 106:408–418. [DOI] [PubMed] [Google Scholar]
- 21. Grullich C, Sullards M, Fuks Z, et al CD95 (Fas/Apo‐1) signals ceramide generation independent of effector stage of apoptosis. J Biol Chem 2000; 275 (12):8650–8656. [DOI] [PubMed] [Google Scholar]
- 22. Kee T, Vit P, Melendez A. Sphingosine kinase signalling in immune cells. Clin Exp Pharmacol Physiol 2005; 32:153–161. [DOI] [PubMed] [Google Scholar]
- 23. Von Haifen C, T W, Gillissen B, et al Activation of bax by ceramide is independent of caspases. Oncogene 2002; 21:4009–4019. [DOI] [PubMed] [Google Scholar]
- 24. Minarowska A, Miarowski L, Karwowski A, et al Regulatory role of cathepsin D in apoptosis. Folia Histochem Cytobiol 2007; 45 (3):159–163. [PubMed] [Google Scholar]
- 25. Kim R, Emi M, Tanabe K. Role of mitochondria as the gardens of cell death. Cancer Chemother Pharmacol 2006; 57:545–553. [DOI] [PubMed] [Google Scholar]
- 26. Klener P Jr, Andera L, Klener P, et al Cell death signalling pathways in the pathogenesis and therapy of haematologic malignancies: overview of apoptotic pathways. Folia Biol (Praha) 2006; 52:34–44. [DOI] [PubMed] [Google Scholar]
- 27. Srinivasula SM, Hegde R, Saleh A, et al A conserved XIAP‐interaction motif in caspase‐9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 2001; 410:112–116. [DOI] [PubMed] [Google Scholar]
- 28. Krammer PH. CD95's deadly mission in the immune system. Nature 2000; 407:789–795. [DOI] [PubMed] [Google Scholar]
- 29. Adams JM. Ways of dying: multiple pathways to apoptosis. Genes Dev 2003; 17:2481–2495. [DOI] [PubMed] [Google Scholar]
- 30. Grutkoski PSCC, Albina J, Biffl W, et al Apoptosis in the critically ill, In: Fink MPAE, Vincent JL, Kochanek PM, eds. Textbook of Critical Care, 5th ed 2005, pp. 195–201. [Google Scholar]
- 31. Szegezdi E, Logue SE, Gorman AM, et al Mediators of endoplasmic reticulum stress‐induced apoptosis. EMBO Rep 2006; 7:880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Szegezdi E, Fitzgerald U, Samali A. Caspase‐12 and ER‐stress‐mediated apoptosis: the story so far. Ann NY Acad Sci 2003; 1010:186–194. [DOI] [PubMed] [Google Scholar]
- 33. Rao RV, Castro‐Obregon S, et al Coupling endoplasmic reticulum stress to the cell death program. J Biol Chem 2002; 277 (24):21836–21842. [DOI] [PubMed] [Google Scholar]
- 34. Yoneda TIK, Oono K, Yui D, et al Activation of caspase‐12, an endoplasmic reticulum (ER) resident caspase, through tumor necrosis factor receptor associated factor 2‐dependent mechanism in response to er stress. J Biol Chem 2001; 276 (17):13935–13940. [DOI] [PubMed] [Google Scholar]
- 35. Morishima NNK, Takenouchi H, Shibata T, et al An endoplasmic reticulum stress‐specific caspase cascade in apoptosis. J Biol Chem 2002; 277 (37):34287–34294. [DOI] [PubMed] [Google Scholar]
- 36. Watanabe N, Ikuta K, Nisitani S, et al Activation and differentiation of autoreactive b‐1 cells by interleukin 10 induce autoimmune hemolytic anemia in Fas‐deficient antierythrocyte immunoglobulin transgenic mice. J Exp Med 2002; 196 (1):141–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Golks A, Brenner D, Schmitz I, et al The role of CAP3 in CD95 signaling: new insights into the mechanism of procaspase-8 activation. Cell Death Differ 2006; 13:489–498. [DOI] [PubMed] [Google Scholar]
- 38. Gomez‐Angelats M, Cidlowski JA. Protein kinase C regulates FADD recruitment and death‐inducing signaling complex formation in Fas/CD95‐induced apoptosis. J Biol Chem 2001; 27:44944–44952. [DOI] [PubMed] [Google Scholar]
- 39. Imtiyaz HZ, Zhang Y, Zhang J, et al Structural requirements for signal‐induced target binding of FADD determined by functional reconstitution of FADD deficiency. J Biol Chem 2005; 280:31360. [DOI] [PubMed] [Google Scholar]
- 40. Sandu C, Gavathiotis E, Huang J, et al A mechanism for death receptor discrimination by death adaptors. J Biol Chem 2005; 280:31974–31980. [DOI] [PubMed] [Google Scholar]
- 41. Wajant H. Death receptors. Essays Biochem 2003; 39:53–71. [DOI] [PubMed] [Google Scholar]
- 42. Lavrik IN, Golks A, Krammer PH. Death receptor signaling. J Cell Sci 2005; 118:265–267. [DOI] [PubMed] [Google Scholar]
- 43. Boldin MP, Varfolomeev EE, Pancer Z, et al A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J Biol Chem 1995; 270:7795–7798. [DOI] [PubMed] [Google Scholar]
- 44. Chinnaiyan AM, O'Rourke K, Tewari M, et al FADD, a novel death domain‐containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995; 81:505–512. [DOI] [PubMed] [Google Scholar]
- 45. Kischkel FC, Hellbardt S, Behrmann I, et al Cytotoxicity‐dependent APO‐1 (Fas/CD95)‐associated proteins form a death‐inducing signaling complex (DISC) with the receptor. EMBO J 1995; 14:5579–5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang J, Chun HJ, Wong W, et al Caspase‐10 is an initiator caspase in death receptor signaling. Proc Natl Acad Sci USA 2001; 98:13884–13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ashkenazi A. Targeting death and decoy receptors of the tumor‐necrosis factor superfamily. Nat Rev Cancer 2002; 2:420–430. [DOI] [PubMed] [Google Scholar]
- 48. Lavrik IN, Golks A, Krammer PH. Caspases: pharmacological manipulation of cell death. J Clin Invest 2005; 115:2665–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Coultas L, Stasser A. The role of the Bcl‐2 protein family in cancer. Semin Cancer Biol 2003; 13:115–123. [DOI] [PubMed] [Google Scholar]
- 50. Letai A. Pharmacological manipulation of Bcl‐2 family members to control cell death. J Clin Invest 2005; 115:2648–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Thornberry NA, Lazebnik Y. Caspases: enemies within. Science 1998; 281:1312–1316. [DOI] [PubMed] [Google Scholar]
- 52. Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006; 25:4798–4811. [DOI] [PubMed] [Google Scholar]
- 53. Minn AJ, Velez P, Schendel SL, et al Bcl‐x(L) forms an ion channel in synthetic lipid membranes. Nature 1997; 385:353–357. [DOI] [PubMed] [Google Scholar]
- 54. Adams JM, Cory S. The Bcl‐2 protein family: arbiters of cell survival. Science 1998; 281:1322–1326. [DOI] [PubMed] [Google Scholar]
- 55. Bush JA, Li G. The role of Bcl‐2 family members in the progression of cutaneous melanoma. Clin Exp Metastasis 2001; 20:531–539. [DOI] [PubMed] [Google Scholar]
- 56. Igney FH, Krammer PH. Death and anti‐death: tumour resistance to apoptosis. Nat Rev Cancer 2002; 2:277–288. [DOI] [PubMed] [Google Scholar]
- 57. Opferman J. Apoptosis in the development of the immune system. Cell Death Differ 2007; 1–9. [DOI] [PubMed] [Google Scholar]
- 58. Luo X, Budihardjo I, Zou H, et al Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998; 94 (4):481–490. [DOI] [PubMed] [Google Scholar]
- 59. Li H, Zhu H, Xu CJ, et al Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998; 94 (4):491–501. [DOI] [PubMed] [Google Scholar]
- 60. Gross A, Yin XM, Wang K, et al Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL‐XL prevents this release but not tumor necrosis factor‐R1/Fas death. J Biol Chem 1999; 274 (2):1156–1163. [DOI] [PubMed] [Google Scholar]
- 61. Ruffolo SC, Breckenridge DG, Nguyen M, et al BID‐dependent and BID‐independent pathways for BAX insertion into mitochondria. Cell Death Differ 2000; 7:1101–1108. [DOI] [PubMed] [Google Scholar]
- 62. Reed JC. Double identity for proteins of the Bcl‐2 family. Nature 1997; 387:773–776. [DOI] [PubMed] [Google Scholar]
- 63. Shimizu S, Narita M, Tsujimoto Y. Bcl‐2 family proteins regulate the release of apoptogenic chytochrome c by the mitochondrial channel VDAC. Nature 1999; 399:483–487. [DOI] [PubMed] [Google Scholar]
- 64. Ertster S, Mihara M, Kim RH, et al In vivo mitochondrial p53 translocation triggers a rapid first wave of cell death in response to DNA damage that can precede p53 target gene activation. Mol Cell Biol 2004; 24:6728–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Padanilam BJ. Cell death induced by acute renal injury: a perspective on the contributions of apoptosis and necrosis. Am J Physiol Renal Physiol 2003; 284:F608–F627. [DOI] [PubMed] [Google Scholar]
- 66. Bauer JH, Helfand SL. New tricks of an old molecule: lifespan regulation by p53. Aging Cell 2006; 5:437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lorenzo H, Susin S, Penninger JM, et al Apoptosis inducing factor (AIF): a phyologenticially old, caspase-independent effector of cell death. Cell Death Differ 1999; 6:516–524. [DOI] [PubMed] [Google Scholar]
- 68. Snyder L, Bertone E, Jakowski R, et al p53 Expression and environmental tobacco smoke exposure in feline oral squamous cell carcinoma. Vet Pathol 2004; 41 (3):209–214. [DOI] [PubMed] [Google Scholar]
- 69. Ito Y, Pandey P, Mishra N, et al Targeting of the c‐Abl Tyrosine kinase to mitochondria in endoplasmic reticulum stress‐induced apoptosis. Mol Cell Biol 2001; 21 (18):6233–6242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zong W, Li C, Hatzivassiliou G, et al Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol 2003; 162 (1):59–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Alnemri ES, Livingston DJ, Nicholson DW, et al Human ICE/CED‐3 protease nomenclature. Cell 1996; 87:171. [DOI] [PubMed] [Google Scholar]
- 72. Yang X, Chang HY, Baltimore D. Autoproteolytic activation of pro‐caspases by oligomerization. Mol Cell 1998; 1 (2):319–325. [DOI] [PubMed] [Google Scholar]
- 73. Martin DA, Siegel RM, Zheng L, et al Membrane oligomerization and cleavage activates the caspase‐8 (FLICE/MACHalpha1) death signal. J Biol Chem 1998; 273 (8):4345–4349. [DOI] [PubMed] [Google Scholar]
- 74. Andrade F, Roy S, Nicholson D, et al Granzyme B directly and efficiently cleaves several downstream caspase substrates: implications for CTL-induced apoptosis. Immunity 1998; 8 (4):451–460. [DOI] [PubMed] [Google Scholar]
- 75. Rao L, Perez D, White E. Lamin proteolysis facilitates nuclear events during apoptosis. J Cell Biol 1996; 135:1441–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Buendia B, Santa‐Maria A, Courvalin JC. Caspase‐dependent proteolysis of integral and peripheral proteins of nuclear membranes and nuclear pore complex proteins during apoptosis. J Cell Sci 1999; 112:1743–1753. [DOI] [PubMed] [Google Scholar]
- 77. Nagata S. Apoptotic DNA fragmentation. Exp Cell Res 2000; 256:12–18. [DOI] [PubMed] [Google Scholar]
- 78. Fadeel B, Orrenius S. Apoptosis: a basic biological phenomenon with wide-ranging implications in human disease. J Int Med 2005; 258 (6):479–517. [DOI] [PubMed] [Google Scholar]
- 79. Rosen A, Casciola‐Rosen L. Autoantigens as substrates for apoptotic proteases: implications for the pathogenesis of systematic autoimmune disease. Cell Death Differ 1999; 6 (1):6–12. [DOI] [PubMed] [Google Scholar]
- 80. Verhagen AM, Ekert PG, Pakusch M, et al Identification of DIABLO, a mammalian protein that promotes apoptosis by bindingn to and antagonizing IAP proteins. Cell 2000; 102:43–53. [DOI] [PubMed] [Google Scholar]
- 81. Du C, Fang M, Li Y, et al Smac, a mitochondrial protein that promotes cytochrome c‐dependent caspase activation by eliminating IAP inhibition. Cell 2000; 102:33–42. [DOI] [PubMed] [Google Scholar]
- 82. Ravagnan L, Roumier T, Kroemer G. Mitochondria, the killer organelles and their weapons. J Cell Physiol 2002; 192:131–137. [DOI] [PubMed] [Google Scholar]
- 83. Loeffler M, Kroemer G. The mitochondrion in cell death control: certainties and incognita. Exp Cell Res 2000; 256:19–26. [DOI] [PubMed] [Google Scholar]
- 84. Lorenzo H, Susin S. Mitochondrial effectors in caspase‐independent cell death. FEBS Lett 2004; 557:14–20. [DOI] [PubMed] [Google Scholar]
- 85. Birbes H, Luberto C, Hsu YT, et al A mitochondrial pool of sphingomyelin is involved in TNFalpha‐induced Bax translocation to mitochondria. Biochem J 2005; 86:445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ruvolo PP, Deng X, Ito T, et al Ceramide induces Bcl2 dephosphorylation via a mechanism involving mitochondrial PP2A. J Biol Chem 1999; 274:20296–20300. [DOI] [PubMed] [Google Scholar]
- 87. Von Haefen C, Wieder T, Gillissen B, et al Ceramide induces mitochondrial activation and apoptosis via a Bax‐dependent pathway in human carcinoma cells. Oncogene 2002; 21:4009–4019. [DOI] [PubMed] [Google Scholar]
- 88. Rothe M, Pan MG, Hazel WJ, et al The TNFR2‐TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell 1995; 83 (7):1243–1252. [DOI] [PubMed] [Google Scholar]
- 89. Chu ZL, McKensey TA, Liu L, et al Suppression of tumor necrosis factor‐induced cell death by inhibitor of apoptosis c‐IAP2 is under NF‐kappaB control. Proc Natl Acad Sci USA 1997; 94 (19):10057–10062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wang CY, Mayo MW, Korneluk RG, et al NF‐kappaB antiapoptosis: induction of TRAF1 and TRAF2 adn c-IAP1 and c-IAP2 to suppress caspase-8 ativation. Science 1998; 281:1680–1683. [DOI] [PubMed] [Google Scholar]
- 91. Deveraux QL, Roy N, Stennicke HR, et al IAPs block apoptotic events induced by caspase‐8 and cytochrome c by direct inhibition of distinct caspases. EMBO J 1998; 17 (8):2215–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Duckett CS, Li F, Wang Y, et al Human IAP‐like protein regulates programmed cell death downstream of Bcl‐xL and cytochrome c . Mol Cell Biol 1998; 18 (1):608–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Shi Y. Caspase activation, inhibition and reactivation: a mechanistic view. Protein Sci 2004; 13:1979–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Finucane DM, Bossy‐Wetzel E, Waterhouse NJ, et al Bax‐induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl‐xL. J Biol Chem 1999; 274 (4):2225–2233. [DOI] [PubMed] [Google Scholar]
- 95. Huang DC, Cory S, Strasser A. Bcl‐2, Bcl‐XL and adenovirus protein E1B19kD are functionally equivalent in their ability to inhibit cell death. Oncogene 1997; 14 (4):405–414. [DOI] [PubMed] [Google Scholar]
- 96. Svrinivasula S, et al A conserved XIAP‐interaction motif in caspase‐9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 2001; 410:112–116. [DOI] [PubMed] [Google Scholar]
- 97. Nicholson DW. Baiting death inhibitors. Nature 2001; 410:33–34. [DOI] [PubMed] [Google Scholar]
- 98. Klener PJ, Andera L, Klener P, et al Cell death signalling pathways in the pathogenesis and therapy of haematologic malignancies: overview of apoptotic pathways. Folia Biol (Praha) 2006; 52:34–44. [DOI] [PubMed] [Google Scholar]
- 99. Tschopp J, Irmler M, Thome M. Inhibition of fas death signals by FLIPs. Curr Opin Immunol 1998; 10 (5):552–558. [DOI] [PubMed] [Google Scholar]
- 100. Shain KH, Landowski TH, Dalton WS. Adhesion‐mediated intracellular redistribution of c‐Fas‐associated death domain‐like IL‐1‐converting enzyme‐like inhibitory protein‐long confers resistance to CD95‐induced apoptosis in hematopoietic cancer cell lines. J Immunol 2002; 168:2544–2553. [DOI] [PubMed] [Google Scholar]
- 101. Thomas RK, Kallenborn A, Wickenhauser C, et al Constitutive expression of c‐FLIP in Hodgkin and Reed‐Sternberg cells. Am J Pathol 2002; 160:1521–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kim Y, Suh N, Sporn M, et al An inducible pathway for degradation of FLIP protein sensitizes tumor cells to TRAIL‐induced apoptosis. J Biol Chem 2002; 277:22320–22329. [DOI] [PubMed] [Google Scholar]
- 103. Siegmund D, Hadwiger P, Pfizenmaier K, et al Selective inhibition of FLICE‐like inhibitory protein expression with small interfering RNA oligonucleotides is sufficient to sensitize tumor cells for TRAIL‐induced apoptosis. Mol Med 2002; 8:725–732. [PMC free article] [PubMed] [Google Scholar]
- 104. Kataoka T, Schroter M, Hahne M, et al FLIP prevents apoptosis induced by death receptors but not by perforin/granzyme B, chemotherapeutic drug, and gamma irradiation. J Immunol 1998; 161:3936–3942. [PubMed] [Google Scholar]
- 105. Siebenlist U, Franzoso G, Brown K. Structure, regulation and funtion of NF‐kappa B. Annu Rev Cell Biol 1994; 10:405–455. [DOI] [PubMed] [Google Scholar]
- 106. Beg AA, Baltimore D. An essential role for NF‐kappaB in preventing TNF‐alpha‐induced cell death. Science 1996; 274:782–784. [DOI] [PubMed] [Google Scholar]
- 107. Bellone M. Apoptosis, cross‐presentation, and the fate of the antigen specific immune response. Apoptosis 2000; 5:307–314. [DOI] [PubMed] [Google Scholar]
- 108. Martindale J, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol 2002; 192:1–15. [DOI] [PubMed] [Google Scholar]
- 109. Krysko DV, D'Herde K, Vandenabeele P. Clearance of apoptotic and necrotic cells and its immunologic consequences. Apoptosis 2006; 11:1709–1726. [DOI] [PubMed] [Google Scholar]
- 110. Hussain SP, Harris CC. p53 Biological Network: at the crossroads of the cellular-stress response pathway and molecular carcinogenesis. J Nippon Med Sch 2006; 73 (2):54–64. [DOI] [PubMed] [Google Scholar]
- 111. Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med 1996; 24 (7):1125–1128. [DOI] [PubMed] [Google Scholar]
- 112. Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med 2003; 348 (2):138–150. [DOI] [PubMed] [Google Scholar]
- 113. Dinarello CA. Proinflammatory cytokines. Chest 2000; 118:503–508. [DOI] [PubMed] [Google Scholar]
- 114. Opal SM, DePalo VA. Anti‐inflammatory cytokines. Chest 2000; 117:1162–1172. [DOI] [PubMed] [Google Scholar]
- 115. Opferman J, Korsmeyer SJ. Apoptosis in the development and maintenance of the immune system. Nat Immunol 2003; 4 (5):410–415. [DOI] [PubMed] [Google Scholar]
- 116. Palmer E. Negative selection – clearing out the bad apples from the T‐cell repertoire. Nature Rev/Immunol 2003; 3:383–391. [DOI] [PubMed] [Google Scholar]
- 117. Hietbrink F, Koenderman L, Rijkers GT, et al Trauma: the role of the innate immune system. World J Emerg Surg 2006; 1 (15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Hotchkiss RS, Osmon SB, Chang KC, et al Accelerated lymphocyte death in sepsis occurs by both the death receptor and mitochondrial pathways. J Immunol 2005; 174 (8):5110–5118. [DOI] [PubMed] [Google Scholar]
- 119. Oberholzer C, Oberholzer A, Clare‐Salzler M, et al Apoptosis in sepsis: a new target for therapeutic exploration. FASEB J 2001; 15 (6):879–892. [DOI] [PubMed] [Google Scholar]
- 120. Ayala A, Lomas JL, Grutkoski PS, et al Pathological aspects of apoptosis in severe sepsis and shock? Int J Biochem Cell Biol 2003; 35 (1):7–15. [DOI] [PubMed] [Google Scholar]
- 121. Jimenez MF, Watson RW, Parodo J, et al Dysregulated expression of neutrophil apoptosis in the systemic inflammatory response syndrome. Arch Surg 1997; 132 (12):1263–1269; discussion 1269–1270. [DOI] [PubMed] [Google Scholar]
- 122. Ertel W, Keel M, Infanger M, et al Circulating mediators in serum of injured patients with septic complications inhibit neutrophil apoptosis through up‐regulation of protein‐tyrosine phosphorylation. J Trauma 1998; 44 (5):767–775; discussion 775–766. [DOI] [PubMed] [Google Scholar]
- 123. Keel M, Ungethum U, Steckholzer U, et al Interleukin‐10 counterregulates proinflammatory cytokine‐induced inhibition of neutrophil apoptosis during severe sepsis. Blood 1997; 90 (9):3356–3363. [PubMed] [Google Scholar]
- 124. Martins PS, Kallas EG, Neto MC, et al Upregulation of reactive oxygen species generation and phagocytosis, and increased apoptosis in human neutrophils during severe sepsis and septic shock. Shock 2003; 20 (3):208–212. [DOI] [PubMed] [Google Scholar]
- 125. Whyte MK, Meagher LC, MacDermot J, et al Impairment of function in aging neutrophils is associated with apoptosis. J Immunol 1993; 150:5124–5134. [PubMed] [Google Scholar]
- 126. Marshall JC. Neutrophils in the pathogenesis of sepsis. Crit Care Med 2005; 33 (12 suppl):S502–S505. [DOI] [PubMed] [Google Scholar]
- 127. Ware L, Matthay M. The acute respiratory distress syndrome. N Engl J Med 2000; 342:1334–1349. [DOI] [PubMed] [Google Scholar]
- 128. Feterowski C, Weighardt H, Emmanuilidis K, et al Immune protection against septic peritonitis in endotoxin‐primed mice is related to reduced neutrophil apoptosis. Eur J Immunol 2001; 31 (4):1268–1277. [DOI] [PubMed] [Google Scholar]
- 129. Matute‐Bello G, Liles WC. Neutrophil apoptosis in acute respiratory distress syndrome. Am Rev Respir Crit Care Med 1997; 156:1969–1977. [DOI] [PubMed] [Google Scholar]
- 130. Matute‐Bello G, Liles WC, Steinberg KP, et al Soluble Fas ligand induces epithelial cell apoptosis in humans with acute lung injury (ARDS). J Immunol 1999; 163:2217–2225. [PubMed] [Google Scholar]
- 131. Perl M, Chung CS, Lomas‐Neira J, et al Silencing of Fas, but not caspase‐8, in lung epithelial cells ameliorates pulmonary apoptosis, inflammation, and neutrophil influx after hemorrhagic shock and sepsis. Am J Pathol 2005; 167 (6):1545–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Moser M, Murphy K. Dendritic cell regulation of TH1‐TH2 development. Nat Immunol 2000; 1 (3):199–205. [DOI] [PubMed] [Google Scholar]
- 133. Mahidhara R, Billiar TR. Apoptosis in sepsis. Crit Care Med 2000; 28 (4 suppl):N105–N113. [DOI] [PubMed] [Google Scholar]
- 134. Van Parijs L, Abbas A. Homeostasis and self‐tolerance of the immune system: turning lymphocytes off. Science 1998; 280:243–248. [DOI] [PubMed] [Google Scholar]
- 135. Van Parijs L, Abbas A. Homeostasis and self‐tolerance in the immune system: turning lymphocytes off. Science 1998; 280 (5361):243–248. [DOI] [PubMed] [Google Scholar]
- 136. Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med 1999; 5 (11):1249–1255. [DOI] [PubMed] [Google Scholar]
- 137. Rovere P, Vallinoto C, Bondanza A, et al Cutting edge: bystander apoptosis triggers dendritic cell maturation and antigen-presenting function. J Immunol 1998; 161:4467–4471. [PubMed] [Google Scholar]
- 138. Reddy RC, Chen GH, Newstead MW, et al Alveolar macrophage deactivation in murine septic peritonitis: role of interleukin 10. Infect Immun 2001; 69 (3):1394–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Wallet M, Sen P, Tisch R. Immunoregulation of dendritic cells. Clin Med Res 3 (3):166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Liu G, Wu C, Zhao EY. Phagocytosis of apoptotic cells and immune regulation. Scand J Immunol 2006; 64:1–9. [DOI] [PubMed] [Google Scholar]
- 141. Tinsley KW, Grayson MH, Swanson PE, et al Sepsis induces apoptosis and profound depletion of splenic interdigitating and follicular dendritic cells. J Immunol 2003; 171 (2):909–914. [DOI] [PubMed] [Google Scholar]
- 142. Hotchkiss RS, Tinsley KW, Swanson PE, et al Sepsis‐induced apoptosis causes progressive profound depletion of B and CD4+T lymphocytes in humans. J Immunol 2001; 166 (11):6952–6963. [DOI] [PubMed] [Google Scholar]
- 143. Hotchkiss RS, Tinsley KW, Swanson PE, et al Depletion of dendritic cells, but not macrophages, in patients with sepsis. J Immunol 2002; 168 (5):2493–2500. [DOI] [PubMed] [Google Scholar]
- 144. Walensky LD, Kung AL, Escher I, et al Activation of apoptosis in vivo by a hydrocarbon‐stapled BH3 helix. Science 2004; 305:1466–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Li L, Thomas RM, Suzuki H, et al A small molecule Smac mimic potentiates TRAIL‐ and TNFalpha‐mediated cell death. Science 2004; 305 (5689):1411–1413. [DOI] [PubMed] [Google Scholar]
- 146. Oberholzer A, Oberholzer C, Minter RM, et al Considering immunomodulatory therapies in the septic patient: should apoptosis be a potential therapeutic target? Immunol Lett 2001; 75 (3):221–224. [DOI] [PubMed] [Google Scholar]
- 147. Ayala A, Wesche‐Soldato DE, Perl M, et al Blockade of apoptosis as a rational threapeutic strategy for the treatment of sepsis. Novartis Found Symp 2007; 280:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Caserta TM, Smith AN, Gultice AD, et al Q‐VD‐OPh, a broad spectrum caspase inhibitor with potent anti‐apoptotic properties. Apoptosis 2003; 8:345–352. [DOI] [PubMed] [Google Scholar]
- 149. Perfettini J, Kroemer G. Caspase activation is not death. Nat Immunol 2003; 4 (4):308–310. [DOI] [PubMed] [Google Scholar]
- 150. Cauwels A, Janssen B, Waeytens A, et al Caspase inhibition causes hyperacute tumor necrosis factor‐induced shock via oxidative stress and phospholipase A2. Nat Immunol 2003; 4 (4):387–393. [DOI] [PubMed] [Google Scholar]
- 151. Hotchkiss RS, Swanson PE, Knudson CM, et al Overexpression of Bcl‐2 in transgenic mice decreases apoptosis and improves survival in sepsis. J Immunol 1999; 162 (7):4148–4156. [PubMed] [Google Scholar]
- 152. Adrie C, Bachelet M, Vayssier‐Taussat M, et al Mitochondrial membrane potential and apoptosis peripheral blood monocytes in severe human sepsis. Am J Respir Crit Care Med 2001; 164 (3):389–395. [DOI] [PubMed] [Google Scholar]
- 153. Bommhardt U, Chang KC, Swanson PE, et al Akt decreases lymphocyte apoptosis and improves survival in sepsis. J Immunol 2004; 172:7583–7591. [DOI] [PubMed] [Google Scholar]
- 154. Chung CS, Song GY, Lomas J, et al Inhibition of Fas/Fas ligand signaling improves septic survival: differential effects on macrophage apoptotic and functional capacity. J Leukoc Biol 2003; 74 (3):344–351. [DOI] [PubMed] [Google Scholar]
- 155. Chung CS, Yang S, Song GY, et al Inhibition of Fas signaling prevents hepatic injury and improves organ blood flow during sepsis. Surgery 2001; 130 (2):339–345. [DOI] [PubMed] [Google Scholar]
- 156. Wesche‐Soldato DE, Chung CS, Lomas‐Neira J, et al In vivo delivery of caspase‐8 or Fas siRNA improves the survival of septic mice. Blood 2005; 106 (7):2295–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Wesche DE, Lomas‐Neira JL, Perl M, et al Leukocyte apoptosis and its significance in sepsis and shock. J Leukoc Biol 2005; 78 (2):325–337. [DOI] [PubMed] [Google Scholar]
- 158. Laterre PF, Levy H, Clermont G, et al Hospital mortality and resource use in subgroups of the Recombinant Human Activated Protein C Worldwide Evaluation in Severe Sepsis (PROWESS) trial. Crit Care Med 2004; 32:2207–2218. [DOI] [PubMed] [Google Scholar]
- 159. Angus DC, Laterre PF, Helterbrand J, et al The effect of drotrecogin alfa (activated) on long‐term survival after severe sepsis. Crit Care Med 2004; 32:2199–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Bilbault P, Lavaux T, Launoy A, et al Inuence of drotrecogin alpha (activated) infusion on the variation of Bax/Bcl‐2 and Bax/Bcl‐xl ratios in circulating mononuclear cells: a cohort study in septic shock patients. Crit Care Med 2007; 35:69–75. [DOI] [PubMed] [Google Scholar]
- 161. Wesche‐Soldato DE, Swan R, Chung CS, et al The apoptotic pathway as a therapeutic target in sepsis. Curr Drug Targets 2007; 8 (4):493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Joyce DE, Gelbert L, Ciaccia A, et al Gene expression profile of antithrombotic protein c defines new mechanisms modulating inflammation and apoptosis. J Biol Chem 2001; 276 (14):11199–11203. [DOI] [PubMed] [Google Scholar]
- 163. Joyce DE, Grinnell BW. Recombinant human activated protein C attenuates the inflammatory response in endothelium and monocytes by modulating nuclear factor‐kappaB. Crit Care Med 2002; 30 (5 suppl):S288–S293. [DOI] [PubMed] [Google Scholar]
- 164. Joyce D, Nelson DR, Grinnell BW. Leukocyte and endothelial cell interactions in sepsis: relevance of the protein C pathway. Crit Care Med 2004; 32:S280–S286. [DOI] [PubMed] [Google Scholar]
- 165. Cheng T, Liu D, Griffin JH, et al Activated protein C blocks p53‐mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med 2003; 9 (3):338–342. [DOI] [PubMed] [Google Scholar]
- 166. Alam JJ. Apoptosis: target for novel drugs. Trends Biotechnol 2003; 21 (11):479–483. [DOI] [PubMed] [Google Scholar]
- 167. Giamarellos‐Bourboulis EJ, Routsi C, Plachouras D, et al Early apoptosis of blood monocytes in the septic host: Is it a mechanism of protection in the event of septic shock? Crit Care 2006; 10 (3):R76. [DOI] [PMC free article] [PubMed] [Google Scholar]