

Abstract
Type I interferons (IFN‐I) have been known for decades for their indispensable role in curtailing viral infections. It is, however, now also increasingly recognized that IFN‐I is detrimental to the host in combating a number of bacterial infections. We have previously reported that viral infections induce partial lymphocyte activation, characterized by significant increases in the cell surface expression of CD69 and CD86, but not CD25. This systemic partial activation of lymphocytes, mediated by IFN‐I, is rapid and is followed by a period of IFN‐I unresponsiveness. Here we propose that IFN‐I exhaustion that occurs soon after a primary viral infection may be a host response protecting it from secondary bacterial infections.
The double‐edged sword of IFN‐I
Since it was first shown in 1957 that IFN‐I ‘interferes’ with viral replication within host cells 1, it has become one of the best studied cytokine. The beneficial effects of IFN‐I are well appreciated in numerous viral experimental models as inducers of antiviral state. Type I interferon is one of the few successful antiviral treatments in therapeutic clinical use, as in chronic hepatitis C infections 2. Viral infections of most somatic cells result in an early synthesis of IFN‐I production. Specialized cells called plasmacytoid dendritic cells (pDCs) are the major IFN‐I producers 3 and mediate systemic IFN‐I responses following viral infections 4. The primary role of IFN‐I is to limit initial viral replication and to facilitate subsequent adaptive immune responses. IFN‐I is a multifunctional cytokine that positively influences cells of both innate and adaptive immunity and therefore is considered as a bridge that links innate and adaptive immunity (reviewed in 5). With a few exceptions of chronic viral infections 6, 7, most studies agree that IFN‐I is protective against acute viral infections. This has been clearly demonstrated in knockout mouse studies, in which mice deficient of functional IFN‐I receptors are highly susceptible to viral infections, such as influenza virus 8, encephalitic flaviviruses 9, Schmallenberg virus 10 and lymphocytic choriomeningitis virus 11.
Besides the antiviral response, a bacterial infection also leads to the induction of IFN‐I synthesis. However, in contrast to the role of IFN‐I in response to a viral infection, the effect on the host in the case of bacteria may be either beneficial or detrimental (Table 1). The precise mechanism/s behind this dualistic effect of IFN‐I on bacteria is not fully understood, but recent studies have provided some insights into how IFN‐I can suppress antibacterial immunity. For example, Teles et al.12 reported that the in vitro induction of IFN‐I by human monocytes in response to Mycobacterium leprae promotes the production of the anti‐inflammatory cytokine IL‐10. IL‐10 together with IFN‐I synergistically limits the production of type II IFN (IFN‐γ) 12, an important effector cytokine against bacterial infections. In a mouse model of Francisella tularensis and Listeria monocytogenes infections, IFN‐I was shown to suppress gamma delta T cell/IL‐17 responses and a subsequent neutrophil recruitment 13. As both IL‐17 and neutrophils play an important role in antibacterial immunity (reviewed in 14), IFN‐I is highly detrimental to the host during F. tularemia infections. Regardless of differences in reported mechanism/s, it is clear that IFN‐I can enhance the host susceptibility to certain bacterial pathogens by suppressing the host's antibacterial immunity.
Table 1.
Benefits and adverse effects of IFN‐I.
| Protective against: | Reference | Detrimental against: | Reference |
|---|---|---|---|
| Streptococcus pneumoniae | 57 | Listeria monocytogenes | 13, 58, 59, 60, 61 |
| Bacillus anthracis | 62 | Francisella tularensis | 13 |
| Salmonella typhimurium | 63, 64, 65 | Chlamydia muridarum | 66 |
| Shigella flexneri | 65 | Yersinia pestis | 67 |
| Chlamydia trachomatis | 68 | Mycobacterium tuberculosis | 38, 39 |
| Chlamydia psittaci | 69, 70 | Staphylococcus aureus | 46, 71 |
| Legionella pneumophila | 72 | Streptococcus pneumoniae | 34, 35, 46 |
| Brucella abortus | 73 | ||
| Mycobacterium leprae | 12 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Virus‐induced partial lymphocyte activation and associated IFN‐I exhaustion
Live viral infections in a mouse model cause IFN‐I‐dependent systemic partial lymphocyte activation 5, 15, 16, characterized by increased expression of activation markers CD69 and CD86, but not CD25 (the interleukin‐2 receptor α chain) 15, 16. The vast majority of lymphocytes undergo this partial activation within 24 h of a viral infection with the cell surface marker expression returning to normal at around day 5 post‐infection 16. A recent report suggested a possible biological role for this phenomenon. It has been shown that the early activation of CD69 temporarily retains lymphocytes in secondary lymphoid organs, presumably promoting antigen‐specific interactions of lymphocytes with antigen‐presenting cells 17.
Concurrent respiratory infections are common among young children and the elderly, and epidemiological studies during the influenza pandemic of 2009 identified co‐infection with other respiratory viruses such as coronavirus, human bocavirus, respiratory syncytial virus and human rhinoviruses 18, 19, 20. Consistent with epidemiology studies, mouse models of viral diseases show enhanced susceptibility to secondary, unrelated viral episodes following primary viral infections 16, 21. While the biological role of partial lymphocyte activation during a primary infection is not yet understood, its consequence, a refractory period of an IFN‐I response to secondary infections, has attracted attention due to their clinical implications 5, 16. We showed that an unrelated secondary adenovirus infection following a primary Semliki Forest virus (SFV) infection fails to trigger partial lymphocyte activation for a duration of 5–9 days post‐primary infection due to IFN‐I exhaustion 16. We found that IFN‐I levels are below the detection limit at day 1 after a secondary viral infection, and the hosts regain its capacity to mount IFN‐I responses 9 or more days after a primary viral infection. Thus, it is likely that IFN‐I exhaustion is responsible for the heightened susceptibility to secondary viral infections.
Co‐infection models examining synergistic consequences between respiratory pathogens are predominantly concerned with combinations of viral and bacterial pathogens. This is largely due to information gained from the devastating Spanish influenza pandemic of 1918 when the majority of deaths were due to bacterial co‐infections or subsequent bacterial infections 22, 23. In the case of the 2009 Swine flu pandemic, 18~34% of influenza episodes admitted to intensive care units worldwide were due to complications caused by bacterial co‐infections 24, 25, 26, 27, 28, 29. Of these cases, Staphylococcus aureus and Streptococcus pneumoniae were the most commonly isolated bacterial pathogens. These pathogens colonize the upper respiratory tract and nasopharyngeal cavity 30, 31, and it has therefore been hypothesized that influenza infections allow outgrowth of colonized S. pneumoniae or S. aureus and result in mucosal co‐infections 32, 33, 34. Such secondary infections occur most frequently at 5–10 days after primary viral infections, thus suggesting that a transient immunosuppression may be responsible for the bacterial outgrowth. A mechanism proposed for a synergism between influenza and S. pneumoniae suggests that the antiviral IFN‐I response elicited by the primary influenza virus infection enhances the susceptibility of the host to secondary bacterial challenge via suppression of antibacterial immunity 34, 35, 36.
Recent mathematical modelling of epidemiological data from the 1918 influenza pandemic has shown a positive correlation between Mycobacterium tuberculosis and influenza death 37. M. tuberculosis is a clinically important bacterial pathogen that latently infects one‐third of the world's population. Negative effects of IFN‐I during M. Tuberculosis infection have repeatedly been shown 38, 39, 40, 41. However, with an exception of highly virulent strains 40, M. tuberculosis does not generally induce strong IFN‐I responses 42 despite possessing a Toll‐like receptor (TLR)‐9 agonist (DNA‐containing CpG motifs), which is a potent IFN‐I inducer. This phenomenon has been recently explained, namely the detection of mycobacterial lipoproteins through TLR‐2 inhibits the TLR‐9 signalling pathway 42, 43 via depletion of a signalling molecule, IL‐1R‐associated kinase 1, and thereby in turn suppresses IFN‐I production during M. tuberculosis infections. This TLR‐2‐dependent negative regulation of the IFN‐I response during M. tuberculosis infections is likely to be beneficial to the host by limiting the harmful effects of IFN‐I. This inhibitory mechanism may also play a positive role during other bacterial infections as TLR‐2 recognizes a wide range of bacterial pathogens. What is interesting is that TLR‐2 signalling impairs TLR‐7‐, TLR‐9‐ but not TLR‐3‐induced IFN‐I synthesis 42, 43. This in turn explains why influenza virus co‐infections in M. tuberculosis‐infected mice impairs bacterial control in an IFN‐I‐dependent manner 44. Influenza virus generates multiple ligands of pattern recognition receptors during the viral replication cycle, which includes dsRNA (TLR‐3 agonist) and ssRNA (TLR‐7 agonist). Thus, influenza virus infections can override TLR‐2‐dependent inhibition of IFN‐I responses in M. tuberculosis‐infected mice through TLR‐3 signalling and induce IFN‐I responses that ultimately result in outgrowth of M. tuberculosis. These findings provide answers as to why the risk of influenza death was higher among patients with tuberculosis than non‐tuberculosis patients during an influenza pandemic 37.
Hypothesis: Type I IFN exhaustion serves to increase the resistance of virally infected mice against secondary bacterial infections
Recent studies have focused on the mechanism of how primary viral infections render the host vulnerable to a sequel of bacterial infections. Severe forms of viral–bacterial co‐infections are rare and only seen when the virus itself is highly virulent such as the 1918 Spanish influenza virus 23. In fact, according to the Centre for Disease Control and Prevention, only 29% of fatal cases of patients with H1N1 influenza had bacterial co‐infection 45. When the primary viral infection is highly pathogenic, it is difficult to ascertain whether the increased susceptibility is due to suppression of antibacterial immunity or the consequence of viral pathology itself. We hypothesize that severe forms of viral–bacterial co‐infection are an exception to the rule and that in most cases, that is, with less virulent viruses, primary infections do not lead to severe secondary bacterial pathology. Thus, there have to exist immune mechanisms that limit secondary co‐infections.
Our current understanding of the biology of IFN‐I is that it is beneficial and essential to recover from most if not all acute viral infections, but may be detrimental to the host when fighting off bacterial pathogens. We also know from our previous studies 16 and reports from others 21 that IFN‐I deficiency as a consequence of exhaustion occurs after primary viral infections and the host is rendered more susceptible to secondary unrelated viral infections during this transient period of IFN‐I exhaustion. Based on these observations, we hypothesize that the host evolved a negative feedback loop mechanism to limit IFN‐I production, which is rapid but transient rendering the host less susceptible to opportunistic bacterial infections that are more prevalent than secondary unrelated virus infections. This transient deficiency in IFN‐I benefits the host as it does not lower resistance to common secondary bacterial infections (Fig. 1). In support of this hypothesis, IFN‐I exhaustion is most likely to be evolutionarily as it appears to be a consequence of all primary viral infections. We and others have shown this to be the case for adenoviruses, alphaviruses, orthomyxoviruses, murine cytomegalovirus and lymphocytic choriomeningitis virus 16, 21. From an evolutionary perspective, there must have been a strong selective advantage to transiently exhaust IFN‐I responses after primary viral infections to occur. Thus, it is reasonable to speculate the evolutionary advantage of negative feedback regulation to suppress virus‐induced immune responses that are detrimental against secondary bacterial infections.
Figure 1.
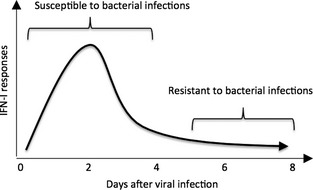
Proposed impact of virus‐induced host IFN‐I deficiency on susceptibility against secondary bacterial infections. Viral infections induce early IFN‐I responses followed by a transient period of IFN‐I exhaustion. The hosts are highly susceptible to secondary bacterial infections during the peak of IFN‐I and then become resistant during transient suppression of IFN‐I responses.
Experimental approach
It has been shown previously, exploring influenza virus/S. pneumoniae co‐infection models, that secondary challenges, with either virus or bacteria, at the peak or during the IFN‐I response, are highly lethal and the increased lethality is attributable to IFN‐I 34, 35, 36. It would be interesting to find out whether the outcome of such co‐infection experiments would differ if mice undergoing a primary virus infection were challenged with bacterial pathogens at the time of IFN‐I exhaustion, 5–9 days post‐infection. Thus, to provide evidence for the above‐outlined hypothesis, all that would be required is to establish correlates of strength of IFN‐I response and exhaustion with severity of secondary bacterial challenges. A time course of bacterial infections after primary virus infection and/or poly I:C treatment would provide an answer to this question. Poly I:C, a synthetic analogue of double‐stranded RNA, mimics RNA viral infections, but would eliminate potential unrelated viral‐induced pathologies affecting secondary bacterial pathologies. It has been shown that poly I:C‐treated mice mount IFN‐I responses that render the host transiently more susceptible to bacterial infections 41, 46. Evaluation of the severity of bacterial growth, morbidity and mortality should establish whether IFN‐I exhaustion ameliorates secondary bacterial pathology. Poly I:C‐treated experimental groups will eliminate potential unknown viral‐induced complications.
Discussion
It is somewhat surprising that the by now widely known phenomenon, that of an IFN‐I refractory period after a viral infection, has as yet not been investigated as to its consequences for the host's susceptibility to bacterial infections, given its potential clinical implications. The known detrimental consequences of the refractory period to secondary viral infections, namely heightened susceptibility, are somewhat hard to understand in evolutionary terms unless there exists an overriding host–benefit rationale. This may well turn out to be protection from potentially lethal bacterial infection, which can be controlled in the absence of IFN‐I.
What has been investigated are the causes for the impaired IFN‐I responses following viral infections. As pDCs are the principal secretors of IFN‐I, the prevailing hypothesis for IFN‐I impairment is centred on pDCs 5, 21, 47. pDCs that have been induced to produce large amounts of IFN‐I in a primary antiviral response are either depleted, through mechanisms such as NK cell‐mediated cytotoxicity 48, 49, or are induced to mature and have to be replaced by haematopoesis, or they acquire a transient state of unresponsiveness and paralysis such as that reported in experiments using in vitro stimulation after in vivo viral infections 50. Although, in our mouse model using avirulent SFV, we did not observe quantitative reduction in pDCs 16, others have reported significant decrease in numbers of pDCs soon after acute or during persistent viral infections 21, 51. Consistent with the above animal data, human patients infected with hepatitis B virus (HBV), hepatitis C virus (HCV) or HIV have decreased numbers of circulating pDCs 52, 53, 54, 55. In addition, patients with HCV infection receiving IFN‐Iα therapy exhibit decreased numbers of pDCs in blood compared with untreated controls 56. Thus, a strong negative correlation exists between the quantity of the IFN‐I response and pDC numbers. Recent study by Swiecki et al.51 has shown that pDC depletion during systemic viral infection occurs in an IFN‐I‐dependent manner through upregulation of pro‐apoptotic expressions of Bid, Bim, Noxa and Bax and downregulation of anti‐apoptotic Bcl‐xl and Bcl‐2.
Besides quantitative changes, qualitative differences in pDCs have also been documented. pDCs isolated from mice undergoing IFN‐I exhaustion are unable to produce IFN‐I in response to CpG, a TLR‐9 agonist, after treatment ex vivo 21. Interestingly, the functional defect of pDCs is limited to IFN‐I production because synthesis and secretion of other cytokines such as TNF‐α, IL‐12 and MCP‐1 are not impaired 21. Collectively, it is likely that the inability of the host to mount an IFN‐I response during the refractory period against a secondary challenge is due to both a pDC intrinsic defect in IFN‐I production and an overall reduction in pDC numbers, the consequence being a vastly reduced IFN‐I output, which may render the host less susceptible to secondary bacterial infections.
Research into viral/bacterial co‐infections has in recent years become much more fashionable due to its potential clinical significance. Most studies have focused on understanding how viral infections cause heightened susceptibility to subsequent bacterial infections. Much less attention has been directed on understanding how the host has evolved mechanisms to enhance resistance against such secondary bacterial infections. The evidence presented above supports our hypothesis that inhibition of IFN‐I production is a mechanism by the host to reduce susceptibility to bacterial infections during recovery from primary virus infections.
Acknowledgment
We would like to thank to Drs Mohammed Alsharifi (University of Adelaide, South Australia) and Herbert P. Ludewick (Albany Medical College, NY) for scientific discussions.
References
- 1. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci 1957;147:258–67. [PubMed] [Google Scholar]
- 2. Alric L, Duffaut M, Selves J et al Maintenance therapy with gradual reduction of the interferon dose over one year improves histological response in patients with chronic hepatitis C with biochemical response: results of a randomized trial. J Hepatol 2001;35:272–8. [DOI] [PubMed] [Google Scholar]
- 3. Asselin‐Paturel C, Boonstra A, Dalod M et al Mouse type I IFN‐producing cells are immature APCs with plasmacytoid morphology. Nat Immunol 2001;2:1144–50. [DOI] [PubMed] [Google Scholar]
- 4. Cella M, Facchetti F, Lanzavecchia A, Colonna M. Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nat Immunol 2000;1:305–10. [DOI] [PubMed] [Google Scholar]
- 5. Alsharifi M, Mullbacher A, Regner M. Interferon type I responses in primary and secondary infections. Immunol Cell Biol 2008;86:239–45. [DOI] [PubMed] [Google Scholar]
- 6. Teijaro JR, Ng C, Lee AM et al Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013;340:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wilson EB, Yamada DH, Elsaesser H et al Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013;340:202–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Szretter KJ, Gangappa S, Belser JA et al Early control of H5N1 influenza virus replication by the type I interferon response in mice. J Virol 2009;83:5825–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lobigs M, Mullbacher A, Wang Y, Pavy M, Lee E. Role of type I and type II interferon responses in recovery from infection with an encephalitic flavivirus. J Gen Virol 2003;84:567–72. [DOI] [PubMed] [Google Scholar]
- 10. Wernike K, Breithaupt A, Keller M, Hoffmann B, Beer M, Eschbaumer M. Schmallenberg virus infection of adult type I interferon receptor knock‐out mice. PLoS ONE 2012;7:e40380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Muller U, Steinhoff U, Reis LF et al Functional role of type I and type II interferons in antiviral defense. Science 1994;264:1918–21. [DOI] [PubMed] [Google Scholar]
- 12. Teles RM, Graeber TG, Krutzik SR et al Type I interferon suppresses type II interferon‐triggered human anti‐mycobacterial responses. Science 2013;339:1448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Henry T, Kirimanjeswara GS, Ruby T et al Type I IFN signaling constrains IL‐17A/F secretion by gammadelta T cells during bacterial infections. J Immunol 2010;184:3755–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kolls JK, Linden A. Interleukin‐17 family members and inflammation. Immunity 2004;21:467–76. [DOI] [PubMed] [Google Scholar]
- 15. Alsharifi M, Lobigs M, Regner M, Lee E, Koskinen A, Mullbacher A. Type I interferons trigger systemic, partial lymphocyte activation in response to viral infection. J Immunol 2005;175:4635–40. [DOI] [PubMed] [Google Scholar]
- 16. Alsharifi M, Regner M, Blanden R et al Exhaustion of type I interferon response following an acute viral infection. J Immunol 2006;177:3235–41. [DOI] [PubMed] [Google Scholar]
- 17. Shiow LR, Rosen DB, Brdickova N et al CD69 acts downstream of interferon‐alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 2006;440:540–4. [DOI] [PubMed] [Google Scholar]
- 18. Renois F, Talmud D, Huguenin A et al Rapid detection of respiratory tract viral infections and coinfections in patients with influenza‐like illnesses by use of reverse transcription‐PCR DNA microarray systems. J Clin Microbiol 2010;48:3836–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schnepf N, Resche‐Rigon M, Chaillon A et al High burden of non‐influenza viruses in influenza‐like illness in the early weeks of H1N1v epidemic in France. PLoS ONE 2011;6:e23514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peci A, Winter AL, Gubbay JB et al Community‐acquired respiratory viruses and co‐infection among patients of Ontario sentinel practices, April 2009 to February 2010. Influenza Other Respi Viruses 2012;7:559–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zuniga EI, Liou LY, Mack L, Mendoza M, Oldstone MB. Persistent virus infection inhibits type I interferon production by plasmacytoid dendritic cells to facilitate opportunistic infections. Cell Host Microbe 2008;4:374–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis 2008;198:962–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brundage JF, Shanks GD. Deaths from bacterial pneumonia during 1918–19 influenza pandemic. Emerg Infect Dis 2008;14:1193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Estenssoro E, Rios FG, Apezteguia C et al Pandemic 2009 influenza A in Argentina: a study of 337 patients on mechanical ventilation. Am J Respir Crit Care Med 2010;182:41–8. [DOI] [PubMed] [Google Scholar]
- 25. Farias JA, Fernandez A, Monteverde E et al Critically ill infants and children with influenza A (H1N1) in pediatric intensive care units in Argentina. Intensive Care Med 2010;36:1015–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kumar A, Zarychanski R, Pinto R et al Critically ill patients with 2009 influenza A(H1N1) infection in Canada. JAMA 2009;302:1872–9. [DOI] [PubMed] [Google Scholar]
- 27. Martin‐Loeches I, Sanchez‐Corral A, Diaz E et al Community‐acquired respiratory coinfection in critically ill patients with pandemic 2009 influenza A(H1N1) virus. Chest 2011;139:555–62. [DOI] [PubMed] [Google Scholar]
- 28. Rice TW, Rubinson L, Uyeki TM et al Critical illness from 2009 pandemic influenza A virus and bacterial coinfection in the United States. Crit Care Med 2012;40:1487–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Webb SA, Pettila V, Seppelt I et al Critical care services and 2009 H1N1 influenza in Australia and New Zealand. N Engl J Med 2009;361:1925–34. [DOI] [PubMed] [Google Scholar]
- 30. Kluytmans J, van Belkum A, Verbrugh H. Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin Microbiol Rev 1997;10:505–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kwambana BA, Barer MR, Bottomley C, Adegbola RA, Antonio M. Early acquisition and high nasopharyngeal co‐colonisation by Streptococcus pneumoniae and three respiratory pathogens amongst Gambian new‐borns and infants. BMC Infect Dis 2011;11:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Peltola VT, Boyd KL, McAuley JL, Rehg JE, McCullers JA. Bacterial sinusitis and otitis media following influenza virus infection in ferrets. Infect Immun 2006;74:2562–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Diavatopoulos DA, Short KR, Price JT et al Influenza A virus facilitates Streptococcus pneumoniae transmission and disease. FASEB J 2010;24:1789–98. [DOI] [PubMed] [Google Scholar]
- 34. Nakamura S, Davis KM, Weiser JN. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J Clin Invest 2011;121:3657–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shahangian A, Chow EK, Tian X et al Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest 2009;119:1910–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li W, Moltedo B, Moran TM. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of gammadelta T cells. J Virol 2012;86:12304–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oei W, Nishiura H. The relationship between tuberculosis and influenza death during the influenza (H1N1) pandemic from 1918‐19. Comput Math Methods Med 2012;2012:124861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Manca C, Tsenova L, Freeman S et al Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak‐Stat pathway. J Interferon Cytokine Res 2005;25:694–701. [DOI] [PubMed] [Google Scholar]
- 39. Stanley SA, Johndrow JE, Manzanillo P, Cox JS. The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX‐1‐mediated secretion and contributes to pathogenesis. J Immunol 2007;178:3143–52. [DOI] [PubMed] [Google Scholar]
- 40. Manca C, Tsenova L, Bergtold A et al Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN‐alpha/beta. Proc Natl Acad Sci USA 2001;98:5752–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Antonelli LR, Gigliotti Rothfuchs A, Goncalves R et al Intranasal Poly‐IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen‐permissive monocyte/macrophage population. J Clin Invest 2010;120:1674–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Simmons DP, Canaday DH, Liu Y et al Mycobacterium tuberculosis and TLR2 agonists inhibit induction of type I IFN and class I MHC antigen cross processing by TLR9. J Immunol 2010;185:2405–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu YC, Simmons DP, Li X, Abbott DW, Boom WH, Harding CV. TLR2 signaling depletes IRAK1 and inhibits induction of type I IFN by TLR7/9. J Immunol 2012;188:1019–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Redford PS, Mayer‐Barber KD, McNab FW et al Influenza A virus impairs control of Mycobacterium tuberculosis co‐infection through a Type I Interferon receptor dependent pathway. J Infect Dis 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Centers for Disease Control and Prevention (CDC) . Bacterial coinfections in lung tissue specimens from fatal cases of 2009 pandemic influenza A (H1N1) ‐ United States, May‐August 2009. MMWR Morb Mortal Wkly Rep 2009;58:1071–4. [PubMed] [Google Scholar]
- 46. Tian X, Xu F, Lung WY et al Poly I:C enhances susceptibility to secondary pulmonary infections by gram‐positive bacteria. PLoS ONE 2012;7:e41879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Baranek T, Dalod M. How opportunistic agents benefit from viral infections: the plasmacytoid dendritic cell connection. Cell Host Microbe 2008;4:305–7. [DOI] [PubMed] [Google Scholar]
- 48. Della Chiesa M, Vitale M, Carlomagno S, Ferlazzo G, Moretta L, Moretta A. The natural killer cell‐mediated killing of autologous dendritic cells is confined to a cell subset expressing CD94/NKG2A, but lacking inhibitory killer Ig‐like receptors. Eur J Immunol 2003;33:1657–66. [DOI] [PubMed] [Google Scholar]
- 49. Hayakawa Y, Screpanti V, Yagita H et al NK cell TRAIL eliminates immature dendritic cells in vivo and limits dendritic cell vaccination efficacy. J Immunol 2004;172:123–9. [DOI] [PubMed] [Google Scholar]
- 50. Bjorck P. Dendritic cells exposed to herpes simplex virus in vivo do not produce IFN‐alpha after rechallenge with virus in vitro and exhibit decreased T cell alloreactivity. J Immunol 2004;172:5396–404. [DOI] [PubMed] [Google Scholar]
- 51. Swiecki M, Wang Y, Vermi W, Gilfillan S, Schreiber RD, Colonna M. Type I interferon negatively controls plasmacytoid dendritic cell numbers in vivo . J Exp Med 2011;208:2367–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Altfeld M, Fadda L, Frleta D, Bhardwaj N. DCs and NK cells: critical effectors in the immune response to HIV‐1. Nat Rev Immunol 2011;11:176–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Duan XZ, Wang M, Li HW, Zhuang H, Xu D, Wang FS. Decreased frequency and function of circulating plasmocytoid dendritic cells (pDC) in hepatitis B virus infected humans. J Clin Immunol 2004;24:637–46. [DOI] [PubMed] [Google Scholar]
- 54. Finke JS, Shodell M, Shah K, Siegal FP, Steinman RM. Dendritic cell numbers in the blood of HIV‐1 infected patients before and after changes in antiretroviral therapy. J Clin Immunol 2004;24:647–52. [DOI] [PubMed] [Google Scholar]
- 55. Kanto T, Inoue M, Miyatake H et al Reduced numbers and impaired ability of myeloid and plasmacytoid dendritic cells to polarize T helper cells in chronic hepatitis C virus infection. J Infect Dis 2004;190:1919–26. [DOI] [PubMed] [Google Scholar]
- 56. Goutagny N, Vieux C, Decullier E et al Quantification and functional analysis of plasmacytoid dendritic cells in patients with chronic hepatitis C virus infection. J Infect Dis 2004;189:1646–55. [DOI] [PubMed] [Google Scholar]
- 57. Weigent DA, Huff TL, Peterson JW, Stanton GJ, Baron S. Role of interferon in streptococcal infection in the mouse. Microb Pathog 1986;1:399–407. [DOI] [PubMed] [Google Scholar]
- 58. Auerbuch V, Brockstedt DG, Meyer‐Morse N, O'Riordan M, Portnoy DA. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes . J Exp Med 2004;200:527–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stockinger S, Kastner R, Kernbauer E et al Characterization of the interferon‐producing cell in mice infected with Listeria monocytogenes . PLoS Pathog 2009;5:e1000355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Carrero JA, Calderon B, Unanue ER. Lymphocytes are detrimental during the early innate immune response against Listeria monocytogenes . J Exp Med 2006;203:933–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. O'Connell RM, Saha SK, Vaidya SA et al Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med 2004;200:437–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gold JA, Hoshino Y, Hoshino S, Jones MB, Nolan A, Weiden MD. Exogenous gamma and alpha/beta interferon rescues human macrophages from cell death induced by Bacillus anthracis . Infect Immun 2004;72:1291–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Freudenberg MA, Merlin T, Kalis C, Chvatchko Y, Stubig H, Galanos C. Cutting edge: a murine, IL‐12‐independent pathway of IFN‐gamma induction by gram‐negative bacteria based on STAT4 activation by Type I IFN and IL‐18 signaling. J Immunol 2002;169:1665–8. [DOI] [PubMed] [Google Scholar]
- 64. Bukholm G, Berdal BP, Haug C, Degre M. Mouse fibroblast interferon modifies Salmonella typhimurium infection in infant mice. Infect Immun 1984;45:62–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Niesel DW, Hess CB, Cho YJ, Klimpel KD, Klimpel GR. Natural and recombinant interferons inhibit epithelial cell invasion by Shigella spp. Infect Immun 1986;52:828–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nagarajan UM, Prantner D, Sikes JD et al Type I interferon signaling exacerbates Chlamydia muridarum genital infection in a murine model. Infect Immun 2008;76:4642–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Patel AA, Lee‐Lewis H, Hughes‐Hanks J, Lewis CA, Anderson DM. Opposing roles for interferon regulatory factor‐3 (IRF‐3) and type I interferon signaling during plague. PLoS Pathog 2012;8:e1002817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shemer‐Avni Y, Wallach D, Sarov I. Reversion of the antichlamydial effect of tumor necrosis factor by tryptophan and antibodies to beta interferon. Infect Immun 1989;57:3484–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Carlin JM, Borden EC, Byrne GI. Interferon‐induced indoleamine 2,3‐dioxygenase activity inhibits Chlamydia psittaci replication in human macrophages. J Interferon Res 1989;9:329–37. [DOI] [PubMed] [Google Scholar]
- 70. Carlin JM, Weller JB. Potentiation of interferon‐mediated inhibition of Chlamydia infection by interleukin‐1 in human macrophage cultures. Infect Immun 1995;63:1870–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Martin FJ, Gomez MI, Wetzel DM et al Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J Clin Invest 2009;119:1931–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Schiavoni G, Mauri C, Carlei D, Belardelli F, Pastoris MC, Proietti E. Type I IFN protects permissive macrophages from Legionella pneumophila infection through an IFN‐gamma‐independent pathway. J Immunol 2004;173:1266–75. [DOI] [PubMed] [Google Scholar]
- 73. de Almeida LA, Carvalho NB, Oliveira FS et al MyD88 and STING signaling pathways are required for IRF3‐mediated IFN‐beta induction in response to Brucella abortus infection. PLoS ONE 2011;6:e23135. [DOI] [PMC free article] [PubMed] [Google Scholar]