

Abstract
H‐bonding, as a non covalent stabilizing interaction of diverse nature, has a central role in the structure, function and dynamics of chemical and biological processes, pivotal to molecular recognition and eventually to drug design. Types of conventional and non conventional (H−H, dihydrogen, H‐ π, CH‐ π, anti‐ , proton coordination and H−S) H‐bonding interactions are discussed as well as features emerging from their interplay, such as cooperativity (σ‐ and π‐) effects and allostery.
Its utility in many applications is described. Catalysis, proton and electron transfer processes in various materials or supramolecular architectures of preorganized hosts for guest binding, are front‐line technology.
The H‐bond–related concept of proton transfer (PT) addresses energy issues or deciphering the mechanism of many natural and synthetic processes. PT is also of paramount importance in the functions of cells and is assisted by large complex proteins embedded in membranes. Both intermolecular and intramolecular PT in H‐bonded systems has received attention, theoretically and experimentally, using prototype molecules. It is found in rearrangement reactions, protein functions, and enzyme reactions or across proton channels and pumps.
Investigations on the competition between intra‐ and intermolecular H bonding are discussed. Of particular interest is the H‐bond furcation, a common phenomenon in protein‐ligand binding. Multiple H‐bonding (H‐bond furcation) is observed in supramolecular structures.
Keywords: H-bonds, catalysis, crystal engineering, proteins, tautomerism
In H‐bond we trust. H‐bonding, as a highly decorated non covalent interaction has established itself as a major contributor towards not only sustaining life but also improving its quality through diverse and rapidly expanding applications in Chemistry, Biology and Materials Science. The H‐bonding virtues are succinctly described in important Chemistry and Biology topics.
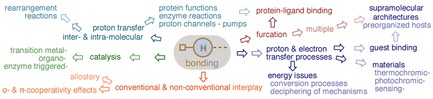
Introduction
Early advances in either Chemistry or Biology progressed independently among the disciplines, thus, without the benefit of a mutual impact. Today, however, boundaries of (Organic) Chemistry and Biology have been blurred as knowledge and techniques are quickly interchanged and adopted. Natural Product Chemists analyze newly sequenced genomes for clues to previously undiscovered bioactive molecules or biosynthetic pathways. Biologists identify molecules, which regulate signaling events, during cell development and control interactions among species. Synthetic Organic Chemists rely on biological assays to guide the design of molecules to be used as ligands tightly binding to enzymes and protein receptors. Various techniques of Physical Organic Chemistry and Molecular Biology are applied to enzymes and other biomolecules to gain some understanding of their structures, functions and mechanisms of action at levels earlier accessible only to small molecules.1
This flow of intellect, so to speak, between the disciplines, would not have been realized, had both of them not been in partnership. Chemical Biology, i. e. the application of Chemistry to elucidate problems of Biology at the molecular level, was, thus, born. The significance and power of this partnership has been amply demonstrated in the post‐genomics era.
Understanding the physiology of diseases is vital to drug design and molecular mechanisms of diseases are a core theme.[2] There is a move from high‐throughput to high‐output screening to select disease‐relevant targets for synthesis. Data from structure, function and post‐genomics technologies on biological targets are invaluable in identifying compounds with unique profiles. When such information is not available, chemists generate small molecule leads binding to the target. Furthermore, they attempt to mimic nature by identifying compounds to fit the structural constraints of the target's binding site(s).
Selective binding plays a key role in enzyme–substrate interactions. Artificial non protein‐based enzymes, incorporating a binding site along with catalytic groups, can achieve high rates and selectivity.
Biological membranes, on the other hand, have inspired attempts towards suitable structure modifications to facilitate transport of molecules through them.3
All biological processes are regulated and mediated by various interactions. Prominent among them is H‐bonding, a key‐ interaction in the world, as we know it. The unusual and complex properties of water, the ability of proteins to fold into stable 3D structures, the fidelity of DNA base pairing and the binding of ligands to receptors, are among the manifestations of this ubiquitous non covalent interaction.
It has been the subject of scrutiny for decades and has gradually developed and expanded into a field in its own merit. Today, it has reached a stage of maturity such that it gives rise to philosophical quests as to its nature or identity. Expertly written reviews, monographs, books or treatises have appeared covering the massive work produced so far and are cited in the various sections. However, these works treat the subject within the Chemistry or Biology domains separately.
Certain features of this remarkable interaction, evident in a couple of ongoing projects, have motivated the authors into shaping the present account. This is, thus, intended to be an interdisciplinary extensive “executive summary”, so to speak, highlighting, critically in parts, major features and concepts of H‐bonding and its significance, spanning through important overlapping fields of Chemistry and Biology, eventually demonstrating its role as a bridge between the two disciplines.
It is hoped that through these lines it will become apparent that (a) the partnership of these disciplines is a sine qua non to the progress of both either as individual domains or more importantly as joined forces and (b) activity in deciphering the full potential of this remarkable interaction will thrive unabated into a bright future.
1. Non Covalent Bonding Interactions
Non covalent interactions are stabilizing bonding interactions, pivotal to molecular recognition and eventually to drug design. The total stabilization energy of these interactions is partitioned into various contributions. Electrostatic, charge transfer (CT) and dispersion are the dominant ones.[4] H‐bonding stands prominently among them, having a central role in the structure, function and dynamics of chemical and biological systems.
1.1. Conventional H‐bonding
H‐bond Profile
A conventional H‐bond is formed between a polar species XH and a lone pair carrier Y:
Xδ−−Η⋅⋅⋅⋅Yδ+ (X=O, N, F & Y=electron rich atom).
In general, an H‐bond is characterized4, 5, 6, 7, 8, 9 by:
a weak to medium interaction energy (ca.10‐40KJmol−1),
a substantial overlap of electron clouds of the heteroatoms,
an electron transfer between the heteroatoms,
a preferred geometry (most frequently observed) of 1.6 Å>d<3.2 Å
Å>d< 2.6 Å) and an angle ϕ>90° (ideally ϕ>130°).
What is the driving force behind the geometry and spectral features of H‐bonding? Natural Bond Orbital (NBO) Analysis has shown[10] that there is a charge transfer (CT) from the electron donor to the antibonding orbitals of the electron acceptor (proton donor). Accumulation of electron density in those orbitals causes elongation of the latter and a red shift of its IR stretching frequency. According to a recent report,11 a Natural Bond‐Bond Polarizability (NBBP) index, within the NBO framework, at the ab initio and DFT levels, may describe delocalization phenomena, H‐bonding interactions among them.
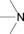
The H donor/acceptor groups commonly found, particularly in biomolecules, are shown in Table 1.
Table 1.
Common H‐bonding sites in biomolecules.
|
Acceptor sites |
Donor sites |
||
|---|---|---|---|
|
|
amines |
|
water, alcohols, phenols, carboxylic acids |
|
|
imines |
|
amines, amides, azoles |
|
|
nitriles |
|
ammonium ions |
|
|
alcohols, ether, water |
|
|
|
|
amides, esters, ketones |
|
|
|
|
P (phosphine‐) N (amine‐)oxides S (sulfo‐) |
|
|
|
|
Fluorine |
|
|
|
Anions |
RCO2 − : carboxylates RSO3 − : sulfonates ROPO3 2−: phosphates Hal−: halide (fluoride, chloride) |
|
|
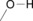
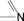




This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
H‐bonding is an interaction of diverse nature, no longer limited to the long‐known conventional interactions. As an attractive interaction between an H donor and an H acceptor, it can be either in the same (Figure 1, type a) or in different molecules (Figure 1, type b).
Figure 1.
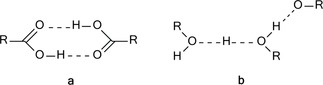
Intermolecular H‐bonding interactions between same or different molecules.
Arrangements of types A or B describe an intermolecular H‐bonding. Type B is labile, occurs randomly, thus, it is difficult to determine.12
If the donor and acceptor sites within a molecule are close enough, then an equilibrium is usually set up between pseudo‐ring and open conformations.13 The former conformation facilitates and triggers an intramolecular H‐bond while the latter one forces the polar groups to be exposed to the surroundings (e.g solvent) (Figure 2).
Figure 2.
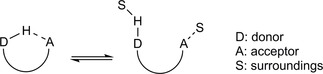
H‐bonding sites in “closed” (pseudo ring) and “open” conformations.
Intramolecular H‐bonding is essential in chemical and biological processes. It may be strong enough, in many molecular or supramolecular structures. In water, it can stabilize the secondary or tertiary structure of biological molecules, interfering in their conformation dynamics or transition state energetics.
Its utility in many applications is a major research theme. Catalysis, particularly organocatalysis, proton and electron transfer processes (excited state proton transfer included) in thermochromic, photochromic and sensing materials or supramolecular preorganized hosts for guest binding, are some front‐line applications (see later sections). Its activating effect in organocatalysis14 and basicity of amine bases,15 are two important and practically useful features.
There have been investigations on the competition between intra‐ and intermolecular H‐bonding [16]. The former is preferred among 5‐ or 6‐membered rings.16 Strong and very strong H‐bonds have been classified [17] into +/‐ charge‐assisted (+/‐CAHBs) and resonance‐assisted (RAHBs). Of moderate strength are the polarization‐assisted H‐bonds (PAHBs) of ⋅⋅⋅⋅O−H⋅⋅⋅⋅O−H⋅⋅⋅⋅ groups observed in water, alcohols or phenols.
The stability of the intramolecular H‐bond is attributed to resonance assistance giving extended π‐delocalization. In this context, the impact of intramolecular H‐bonding on substituent properties or intramolecular cyclizations has been investigated.18, 19
Intramolecular H‐bonding has also been investigated by 1H and 17O NMR spectroscopy in many applications.20, 21 A special issue on some aspects indicative of its significance has recently been devoted to it.22
Multiple H‐bonding (H‐bond furcation) is observed in supramolecular structures.[23] 3‐Centered (bifurcated) are the common ones (Figure 3) but 4‐centered (trifurcated) are also found in biomolecules.
Figure 3.
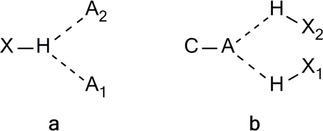
Furcated H‐bonding types: (a) bifurcated donor, and (b) bifurcated acceptor.
Bifurcated H‐bonds have been measured by DFT calculations and isotope‐edited FTIR spectroscopy in transmembrane (influenza A M2 protein and Severe Acute Respiratory Syndrome coronavirus [SARS] E protein)24 or in complex RAHB environments.25 In a modern context, furcation includes all kinds of strong (O−H⋅⋅⋅⋅O, N−H⋅⋅⋅⋅O) and weak (C−H⋅⋅⋅⋅O) H‐bonds.26, 27 More than 25 % of O−H⋅⋅⋅⋅O H‐bonds in carbohydrates, and more so in amino acids and proteins, are multifurcated.28 A special issue on the spectroscopy of H‐bonded systems, describing excited state dynamic interactions has been published recently.29
Symmetry is of significance for an H‐bond.30 H‐bonded sites of the same proton affinity raise the question of an H‐bond equidistant from both ends, i. e. either midway or closer to one of them, hopping in rapid equilibrium. These possibilities correspond to double‐well or single‐well potentials.
Double–well potentials are associated with strong H‐bonds whereas the single‐well H‐bond potentials are among the strongest, e. g. [F⋅⋅⋅⋅H⋅⋅⋅⋅F]−. All H‐bonds are described as resonance hybrids reaching maximum stabilization when the resonance forms have identical energies as in a single‐well potential. The majority, however, of H‐bonds are asymmetrical, i. e. H lies closer to one of the atoms, even if they are the same
The nature of H‐bonding has been recently revisited31 with reference to bond critical point (bcp) and its corresponding chemical bond relationship. The study has been based on homologous diols in their ring‐like conformation (i. e., a 5‐, 6‐ & a 7‐ring conformation) (Figure 4).
Figure 4.
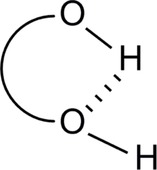
Revisiting intramolecular version of H‐bonding.
Geometry and IR spectroscopy secure an intramolecular H‐bond. It has, thus, been suggested that a bcp is only one of the criteria to assess the existence of a bond. The question of the nature of H‐bond as a true bond or just a weak interaction, in the form of an energy continuum, remains a strongly debated issue.32, 33, 34 Imaging methods such as scanning tunneling microscopy (STM) and atomic force microscopy (AFM) have been called in to “see” H‐bonding.35 Not only the “revered” O−H⋅⋅⋅⋅OH bonds have been unveiled but also C−H⋅⋅⋅⋅O and C−H⋅⋅⋅⋅N ones with C−H groups as donors. In this context, a “carbon bond” has recently been proposed by analogy to H and halogen bonds.36
Proton transfer (PT) Phenomena
An H‐bond between XH and Y is a proton sharing between the two ends X and Y, thus, it is, in fact, a step prior to Proton Transfer (PT).
The correlation between H‐bonding and PT has been pointed out by Bürgi and Dunnitz [37] and this relationship has been a research theme ever since (see also Tautomerism, section 3.4).38
Its fundamental importance in chemistry and biology38 has been recognized. Both intermolecular and intramolecular PTs in H‐bonded systems have received much attention, theoretically and experimentally,12, 39, 40 either as a single PT41, 42 or a multiple one,43, 44, 45, 46 using prototype molecules. It is found in rearrangement reactions, protein functions, enzyme reactions or across proton channels and pumps. Many experimental and theoretical reports, as well as book chapters and reviews, have been devoted to PT along H‐bonds.47 Tunneling (a quantum effect of going through and not over an energy reaction barrier),[48] transport in water molecules[49] or hopping (or “hop‐turn” or Grotthuss mechanism)[50] are the currently accepted proton relay modes. Interestingly, examples of PT, occurring without engaging H‐bonds, have recently been encountered.51 Investigations have also focused on PT processes in the excited state (ESIPT).52, 53
The concept of proton (PT) and electron transfer (ET) is of growing interest in addressing energy issues such as conversion processes54 or even deciphering the mechanism of many natural and synthetic processes.55 What is more important is that the PT and ET may be a coupled process (PCET). Ongoing research focuses on the possibility that the two transfers proceed concertedly (CPET) instead of following a stepwise pathway.56
H‐bond Acidity and Basicity
During a PT process, surroundings are reorganized to adjust to a new equilibrium state. This is accompanied by charge,[56] as well as bonding57 redistribution. PT basicity and H‐bond basicity are important in biological systems.
The H‐bond may be looked at as an arrested intermediate in a deprotonation event. This implies that there is an acid‐base component to its behavior.58 The impact of intramolecular H‐bonding on the O−H dissociation enthalpy has been studied in this respect.59 H‐bond participants act as, either H acceptors (Brönsted bases) and H donors (Brönsted acids) or as electron donors (Lewis bases) and electron acceptors (Lewis acids).
The H‐bond is measured by its binding energy not directly and accurately but usually calculated from the thermodynamic parameters ΔG, ΔH and ΔS of the complex X−H⋅⋅⋅⋅Y.
Many physical properties such as hydrophobic, electronic and steric substituent constants, as well as biological activities, are related to ΔG.
H‐bond acidity is the ability of a molecule to act as an H‐bond donor. In some cases, this property appears to follow the acidity concept.60 Halogen acids, however, are an example where the two concepts oppose each other. Acidity increases with halogen size while H‐bond acidity decreases.61 Ring strain (3‐or 4‐membered rings) has been found to increase H‐bond acidity.62
Several experimental scales for H‐bond acidity and basicity have been developed.63, 64 Intramolecular H‐bonding markedly reduces acidity while basicity remains essentially unaffected.65 Lipophilicity, on the other hand, increases and this has been observed in ortho‐substituted phenols.66
Hydrophobic Interactions
Hydrophobic interactions (HI) between apolar molecules or apolar parts of molecules in water have been studied as early as end of 19th century. HI are different from all other non covalent interactions in solution.67 They do not arise directly from intermolecular interactions but are rather driven by the H‐bonded aggregation tendency of water molecules, minimizing the contact area between water and non polar entities. Their mechanism is still not clear though it has been under scrutiny for a long time.
Hydrophobicity is an important factor, in the structures of proteins and nucleic acids, surfactant aggregations, phospholipid or lipid components in biomembranes, binding of enzymes to substrates and binding of antigens to antibodies.
Some time ago, it was found68 that organic reactions can be carried out under hydrophobic control with high rates and selectivities, particularly if assisted by salting‐in or salting‐out agents (e. g. LiCl, LiClO4) or so‐called water‐like solvents such as β‐cyclodextrin, urea, formamide.
Strong H‐bonding between H2 O and non polar molecules points to HI. Currently, there are two competing views for its origin.69 The classical one suggests that interactions among H2 O molecules are much stronger than those between H2 O and non polar solute molecules. The “heretic” one, on the other hand, suggests that H2 O molecules rearrange to create a cavity to accommodate the solute, clearly at an entropy cost. The use of water as solvent in synthetic chemistry has eventually, emerged as a primary concern.70, 71, 72
1.2. Non Conventional H‐bonding
This interaction, being ubiquitous in macromolecules, has attracted great attention in chemistry and structural biology.5
H‐H Interactions
This type of H‐bonding (close to a non bonding or Van der Waals contact) exists between identically or similarly charged H atoms. It has a stabilizing contribution to the bonding energy in (un)saturated hydrocarbons, even in cases where there are steric non bonding repulsions, originating from the approach of the H atoms within their Van der Waals radii.73
Dihydrogen Interactions
This type of interaction, termed as “dihydrogen bond”,74, 75 is dominated by electrostatic forces between H atoms of opposite charge of the type H+⋅⋅⋅⋅H (Figure 5). In that, it is to be distinguished from an H−H‐bond.
Figure 5.
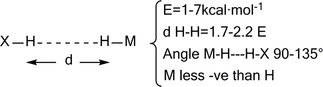
A typical dihydrogen interaction.
Geometry and energy features are similar to those of the conventional H‐bond and find applications in catalysis and crystal engineering.
H⋅⋅⋅π Interactions
This is the bond‐forming interaction between double/triple bonds, aromatic or cyclopropane rings and X⋅⋅⋅⋅H H‐donors.75 Several structures have been studied and their X‐ rays have revealed the operation of H⋅⋅⋅π interactions intra‐76 or inter‐molecularly.77, 78
C‐H⋅⋅⋅π Interactions
This is the weakest of the H contacts that occurs between a soft acid, C⋅⋅⋅⋅H and a soft base, the π‐system.79
The CH/π interaction is characteristic of a relatively large contribution from a π to a σ* charge transfer and dispersive interaction compared to normal H‐bonding. Unlike the typical hard acid‐hard base H‐bonding, CH/π interaction may operate in both polar and non polar media and is hardly disturbed by water. This is important for molecular interactions in biological environments.
Groups that can participate in CH/π interactions are methyl, isopropyl, long chain alkyls or CHs in aromatic rings whereas unsaturated bonds (conjugated or isolated), aromatic rings of amino acids, nucleic acid bases, porphyrins etc. make up the π‐part (Figure 6).
Figure 6.
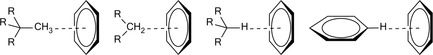
Examples of C−H–π H‐bond interactions.
Its importance has been recognized in various fields of chemistry and biochemistry,4, 80 as well as biomacromolecules, such as peptides and proteins81 and studied by ab initio MO calculations.82
Improper or anti‐H‐bond Interactions (blue shifting)
This is a C/H π interaction observed in C⋅⋅⋅⋅H⋅⋅⋅⋅benzene complexes. Its conception is opposite to that of an ordinary H‐bond. [83, 84] Accordingly, the charge transfer from the electron donor to the acceptor is directed to a remote part of the latter, accompanied by a re‐structuring of its geometry.
Proton Coordination Interactions
Besides the known H‐bond interactions, already described, much less known is the ability of H δ+ to coordinate to two or more atoms in bonding modes.[85] Agostic interactions, H‐bridges, bi (multi) furcated H atoms are all facets of its coordination power. Agostic, in particular, is a covalent contact86 of the types shown (Figure 7), similar to the well‐known H‐bridges (Figures 8 & 9), where the H atom is simultaneously bonded to both C and M.
Figure 7.
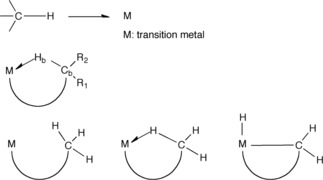
General types of agostic contacts.
Figure 8.
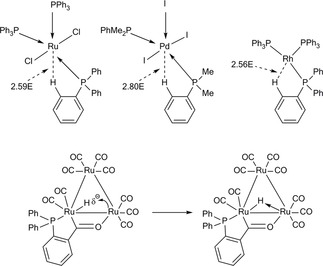
Examples of H‐bridges.
Figure 9.
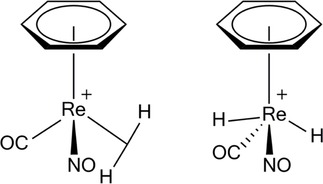
Examples of dihydrogen contacts.
Charge density topological analysis and 1H/13C NMR spectra have been used to identify agostic interactions, usually observed in metal σ‐complexes, along with dihydrogen contacts (Figure 9).
H⋅⋅⋅S Bonds
In spite of its larger size than O and a more diffuse electron density, S can and does engage in H‐bonding.[87] An H‐bond is observed if H⋅⋅⋅⋅S d ≤ 2.9 Å. For O−H⋅⋅⋅⋅S and N−H⋅⋅⋅⋅S interactions the acceptor is more frequently found in the ligand than in the protein. Intramolecular O−H⋅⋅⋅⋅S and O−H⋅⋅⋅⋅O H‐bonds have been compared in resonance‐assisted H‐bonded (RAHB) environments.88
π⋅⋅⋅π Interactions
These interactions, also known as Aromatic‐Aromatic or Arene‐Arene interactions (AI), are non covalent interactions, ubiquitous in Nature. They are thought to confer stability to duplex DNA,89 they have been proposed to contribute to the unique properties of thermophilic proteins,90 they engage in aggregation of amyloid β in AD91 and they are found in biomolecular recognition.
It has been estimated that ca. 60 % of aromatic side chains in proteins are engaged in pairs, 80 % of them form networks of ≥3 interacting aromatic rings.92 It is believed that AI consists of Van der Waals, hydrophobic and electrostatic forces.93, 94 Substituent effects can determine the geometry of interaction (e.g edge‐to‐face or face‐to‐face) and if they are substantial, they can modify it.[95, 96] The effect of heteroatoms has also been a research theme in a series of nucleic acids.[97]
Cooperativity –Allostery
The interplay of various types of H‐bonding interactions within the same structure gives rise to cooperativity effects.7 This H‐bond multiplicity has been studied experimentally[98] and theoretically.99 It is known100, 101 that in arrays of conventional H‐bonds, such as O−H⋅⋅⋅⋅O and N−H⋅⋅⋅⋅O, the strength of the individual substituents is enhanced by cooperativity effects. σ‐Cooperativity is the best known, observed in Oδ−−Hδ+⋅⋅⋅⋅Oδ−−Hδ+⋅⋅⋅⋅Oδ−−, where individual H‐bonds are strengthened by mutual polarization of the OH groups.
Complex systems, accommodating σ‐ or π‐cooperativity for inter‐ and intra‐molecular H‐bonding interactions, have been modeled.102 If the induced σ‐ or π‐polarization, is relayed in the same direction, along an H‐bonded chain, the cooperativity is homodromic whereas in the opposite direction it is antidromic or anti‐cooperativity. Coupling of non‐linearly relayed tandem associations is a common feature in biological systems. π‐ Cooperativity in heterocyclic H‐bonded systems103 has been of particular interest due to the extensive ring π– delocalization, giving rise to RAHB‐stabilized interactions.
Cooperativity refers to self‐organization events whereas allostery applies to entities of substrate‐receptor binding sites and any information relay among binding subunits. Allostery prevails in naturally found molecular architectures and holds a key‐role in the regulation of enzymatic processes.
2. Applications of H‐bonding
H‐Bonding, over its very long history, through an intensive and extensive scrutiny, has been established as an interaction of crucial significance to both chemical and biological processes. Deciphering its salient features fosters, at an impressive pace, the development of diverse applications, some of which are described in the following sections.
2.1. Catalysis
In Nature, catalysis is a complex operation of various processes the participants of which perform distinct and essential functions. For a long time transition metal (TM) complexes and enzymes have been the main efficient catalysts in every day laboratory use. The former have seen an impressive growth during the past two decades, catalyzing various transformations. In fact, Organic Synthesis has been historically dominated by transition metal catalysis.104 The vitality of the field can be witnessed by a diverse array of catalysts of broad scope and applicability, as eloquently described in many reviews, [105–110] books or series.111 Then a knowledge, as old as the early 70’s, was re‐born and structured into the concept of organocatalysis.112 This has gradually flourished into a synthetic strategy, over the last decade and has, now, reached enough maturity to stand as a research field in its own merit.
Asymmetric catalysis has long been dominated and still is by TM complexes. However, the organic catalysts have been picking up pace, offering many advantages over their metal‐based competitors, such as operational simplicity and ready availability, low cost, bench‐ stability and comparably higher robustness than enzymes or antibodies.
The use of organocatalysts in multi‐component, tandem or domino‐multi‐step reactions,113, 114, 115 as well as total synthesis,116 allows the stereo‐controlled assembly of architectures of increasing complexity while introducing “greenness” in the transformations. Nowadays, its constantly expanding potential can cover enantioselectivity, aerobic and moist reaction conditions. The advances in the field have been expertly reviewed.117, 118, 119, 120, 121, 122, 123
In addition to its primacy as a structural determinant, H‐bonding has a crucial role in catalysis. Its potential as a catalyst has been relatively recently realized and appreciated in H donor organocatalysts. A broad range of this type of catalysts has been developed.124, 125, 126, 127, 128, 129, 130
Transition Metal Catalysis
Extensive efforts and remarkable achievements have been reported over the years and still are, at an unabated rate, on the properties and reactivity patterns of metals pertaining to their application in diverse transformations. Relevant advances in transition metal catalysis, as well as its H‐bonding activity in many metal complexes, have been efficiently reviewed.131, 132
A few comments are included, here, merely to pinpoint the significance of H‐bonding in metal complex‐mediated reactions. H‐bond donors or acceptors in metal complexes are metal‐bound, ligand‐based or external to the complex. Interactions may occur either within a metal complex or with surrounding molecules or ions through these types. Many of these complexes form quite strong H‐bonds, the chemistry of which has been extensively studied in biological systems.
Organocatalysis
Organocatalysis offers many advantages to synthetic organic chemistry.133 Organocatalysts are stable to air and water, easily handled, relatively nontoxic and readily separable from a reaction mixture. The use of enantiomerically pure organocatalysts, easily accessed from natural sources or by synthesis, is extremely important in the synthesis of enantiomers of the target structures, frequently both enantiomers. The induced enantioselectivity is crucial to the efficiency/potential of a structure as a drug candidate.
Useful review articles134 detail the literature on the combined effects of (transition) metal and organic catalysts as a powerful approach in asymmetric catalysis.
A range of organocatalysts function through H‐bonding, usually intermolecularly. The intramolecular variant is less common. Nonetheless, its activating effect, through a phenol OH group135 in bifunctional organocatalysts, has been recently reviewed.14
Various ureas and thioureas have been reported as either double136, 137, 138, 139, 140, 141 or bifunctional120, 142, 143, 144 H‐bond donor catalysts in asymmetric reactions. Solvent‐free thiourea organocatalysis of asymmetric Michael additions, under ball milling conditions, has been mediated by H‐bonding.145 In their bifunctional mode, they mimic natural enzymes, allowing scope for enhanced catalyst activity and higher degree of stereo‐control.
All delineated transformations engage either the HOMO or the LUMO of the participants. A distinct departure from this trend, known as SOMO catalysis, has been developed by MacMillan.146, 147, 148, 149, 150, 151
Molecular recognition, of paramount importance to biocatalysis (i. e. enzyme catalysis, see also below Enzyme & Cocktail Catalysis), rests upon anion sensing,[152] binding153 and stabilization.153 Non covalent interactions, H‐bonding in particular,154 play a major role in these life‐sustaining biological process. Their significance and potential to the catalytic activity of enzymes has been appreciated relatively recently.155, 156, 157
Enzyme Catalysis
H‐bonding to an electrophile decreases the electron density of that species, activating it towards nucleophilic attack. This principle is employed frequently by enzymes in the acceleration of a wide range of chemical processes.
Enzyme catalysis has long been considered to rival intramolecularity.158 The focus of that correlation has been on the high reaction rates observed in both types of processes (up to 108‐1010‐1018!). Proximity of reacting centers in either intramolecular reactions via covalent forces or enzymic reactions via non covalent interactions, has been the core theme of a long debated discordance on these similarities. Fredric Menger159 and Thomas C. Bruice160 have elegantly laid out the relevant arguments. The combined effects of enzymes and organocatalysts have been eloquently reviewed.161
Organic Chemists have always viewed enzymes as organic catalysts. That is certainly true and in the words of J. Knowles “…enzymes are no different from other catalysts, only better…” The statement, put in such a captivatingly simple way, reflects the proficiency of enzymes to effect transformations, smoothly, complex ones among them, stereoselectively, fast, under mild conditions and quite often in high yields.
Enzymic action proceeds, first through binding to the substrate, stabilizes the TS, achieving selectivity and that then reacts further to the final product (Figure 10). Examples with GSH and its complexes with transferase isoenzymes (A1‐ 1, P1‐ 1 or M1‐ 1) have been reported.162
Figure 10.
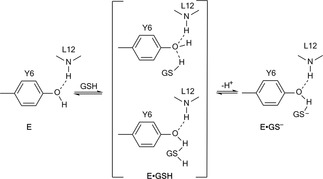
Schematic representation of the GSH‐Enzyme mechanism of action.
Several studies have been reported on the kinetic and energetic aspects of enzymic activity. Low Barrier H‐bonds (LBHBs) have been considered as a major factor.163 The dominant view, nowadays, is that of a multi‐factorial regime of interactions.163, 164 Nuclear quantum tunneling and dynamics have been invoked in enzyme–catalyzed H transfer reactions.164 Many redox processes are accompanied by proton content changes at enzyme active sites. Getting a grasp of such a reactivity path is a current chemical challenge.165
It is interesting to note that changes in ▵G≠ have been correlated to Near‐Attack Conformers (NACs) [166], a parameter defining the required conformation of reactants to enter the TS and that is yet another feature linking enzyme‐catalysed reactions to intramolecular ones.
Cocktail Catalysis
In recent years, the idea of organocatalysis, enzyme catalysis and transition metal catalysis joining forces, has been born and rapidly grown.167 The ultimate goal behind this quest is to discover and develop efficient approaches towards the synthesis of complex molecules with satisfactory chemo‐ and stereoselectivity not attainable by each of the methods applied individually.
2.2. Biomolecule‐Ligand Complexes
DNA and RNA structures, peptide and protein secondary structures, such as α‐helices, β‐sheets, β‐and γ‐loops, as well as their tertiary protein structures, have H‐bonds as major contributors (enthalpy component) and hydrophobic contacts (entropy component).
The 3D protein architecture is largely stabilized by H‐bonds. Their strength endows them with specificity in structure and function and a conserved orientation.169 Their weakness, entails fragility, during complexation, folding and conformation change.170
A protein's secondary structure is held by weak H‐bonds. A puzzling question is, thus, what gives that protein strength, elasticity and resilience? Studies on β‐sheet proteins have revealed171 that the strength of protein‐based materials may lie in the geometric configuration of the β‐strands making up the β‐sheets via cooperative H‐bond clusters. Molecular simulations have been called in to explore and decipher the geometry requirements for the observed strength.
Geometries of macromolecules are complicated and the overall complexity increases further by the various types of H donors and acceptors at the protein‐ligand interface.59, 172, 173
During the formation of a protein‐ligand complex, water molecules compete with binding. The latter prevails if the H‐bond energy in the complex and the entropy gain from water release is more favorable than the free energy contributed by the H‐bonded binding partners and water molecules (Figure 11).174, 175, 176
Figure 11.
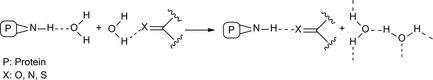
Ligand‐protein complex in presence of water molecules.
Ligands interacting with functionalities of a binding site, through H‐bonds, bind in a specific orientation. The H‐bonding interactions demand optimum distances and angles.173, 177, 178, 179
The backbone H‐bonding of amino acid side chains in their binding interactions, is still a non clarified issue. Perturbation of this backbone pattern has been recently described as a means to assess the contribution of H‐bonding to binding interactions.180
Of particular interest is the H‐bond furcation,[181] a common phenomenon in protein‐ligand binding, observed in the active sites of their complexes, ranging from bi‐ to hexa‐furcated geometries (see also sections 2.1 & 2.2).
H/D exchange has been applied to protein folding, for a long time, as an effective means for locating the amide H atoms in the interior or exterior of a protein [182]. Recently, H/ D exchange has found increasing use in small molecule design.183 Proton transfer (PT) (see section 2.1) is also of paramount importance in the functions of cells and is assisted by large complex proteins embedded in membranes.184
A recent survey of protein and nucleic acid structures has unveiled halogen bonds, stabilizing inter‐ and intra‐molecular interactions affecting ligand binding.185, 186, 187 A unified approach to describe H– and halogen– bonding interactions has been reported.188
Enzymes and receptors often show high specificities for their substrates and agonists, respectively.189 Exceptions are some metabolic enzymes, e. g. cytochromes, oxidizing a number of drugs, and some transporters.
It is known that similar ligand interactions with a receptor‐binding site exhibit similar affinities even if their structures are different. This is an important issue in drug design (Figure 12).190 Shape similarity is also important in ligand binding, sometimes more so than H‐bonding.191, 192
Figure 12.
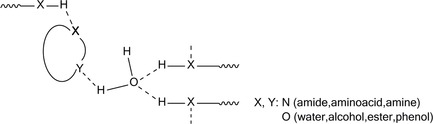
H‐bonding in receptor‐ligand binding interactions.
H‐bonding networks incorporating water molecules are important in the ligand binding sites of many protein‐ligand 3D structures (Figure 12).193, 194, 195 In protein environments, it is known that PTs occur along polar or charged residues and isolated water molecules. [196] Proton shifts, as a sequence of slow PTs, is crucial to the energetics of a cell. Recent findings, however, seem to be compatible to a rather fast diffusion through the membrane's hydration layer.197 H‐bonding also contributes to membrane protein folding and stability. Experiment has shown that the H‐bonding contribution to stability in water‐soluble proteins is only ca. 1 kcalmol−1.195 Double‐mutant cycle studies have indicated that H‐bonding is also weak in membrane proteins too. In any case, the individual H‐bonds may be weak but their overall effect may be sound.
2.3. Crystal Packing/ Engineering
In Nature, nano‐objects are assembled from macromolecular precursors. Bio‐macromolecules, such as proteins or DNA, exhibit structural homogeneity, possess exceptional mechanical properties, perform impressive catalytic functions (e. g. cytochrome P450) and have high information storage capability (e. g. DNA).
A major theme in the development of organization of matter is selectivity. In this context, molecular self‐assembly has emerged as a powerful technology for the synthesis of nanomaterials. Technologies to induce predefined secondary structural motifs by means of self‐assembly of oligo‐ and polymeric precursors allow the design of abiotic devices with capabilities superior to those displayed by their natural counterparts.
Intermolecular interactions, which, through packing, allow the construction of materials with defined physical and chemical properties, give rise to crystal engineering as proposed by Desiraju.198 H‐bond–directed organization of molecular precursors has garnered much success. H‐bonded ensembles are, generally, kinetically labile, under ambient conditions. A reversibility‐ or irreversibility‐triggering event will hamper or favour selectivity, respectively.199
Just as covalent bond formation is characterized by chemo‐ , regio‐ , stereo‐ and enantio‐selectivity, a similar classification applies to its non covalent variant. The selectivity types we shall refer to will be the H‐mediated ones.200
Chemoselectivity
This refers to the specificity guiding a functional group aggregation. For example, carboxylic acids and amides embody self‐complementary recognition groups defined by H‐bond donor and acceptor pairs. In molecules incorporating these groups, one can distinguish both homomeric or heteromeric assembly modes.
The H‐bond strength, in both modes, is of the same order of magnitude as their energy difference, thus, hindering selectivity.200 The acid‐base features of H‐bonding, on the other hand, could be called in to drive selectivity by pKα adjustments.201
Regioselectivity
Regioselective association of molecular components may give rise to alternative super‐structures. Sterically directed or preorganized molecular components can, under thermodynamic control, trigger regioselectivity.202, 203
Stereoselectivity
Association of two chiral precursors may form a diastereomeric aggregate. Chiral‐chiral or achiral‐achiral molecular associations may also exhibit diastereoselectivity.
Molecular recognition and host‐guest chemistry have been extensively studied,204, 205 particularly in amino acid / peptide receptors.206 Numerous chiral receptors, acting through H‐bonds to bind racemic guests, have been described.
Enantioselectivity
To preserve induction of enantiomeric excess in a stereochemical transformation demands that the chiral product be kinetically inert. Resolution of chiral super‐structures, arising from H‐bond associations, is viable, under following conditions: (a) crystal nuclei formed are infrequent and slow, (b) crystal growth, once initiated, is rapid and (c) enantiomer interconversion in solution is fast.
Tubular Structures
Channel‐like structures have captured the interest of chemists for a long time. Most prominent ones are ionic channels,207 zeolites208 and carbon nanotubes.209 Ionic channels, formed by proteins, are responsible for the transport of Na and K ions through the lipophilic cell walls. Both ends are surrounded by –vely charged amino acids, giving rise to a local high concentration of cations. The tubular structure, thus formed, is kept intact by H‐bonds among the amino acids.
Zeolites (silicates with regular rigid 3D frameworks made of Si, Al) form a covalently bound framework containing cavities and channels. The interior is covered with –vely charged O centers and OH groups, providing a hydrophilic environment similar to ionic channels. Inclusion of metal ions or larger molecules is, thus, allowed, anchored by H‐bonds or dipole‐dipole interactions.
Carbon nanotubes are formed from graphite sheets in which C atoms are connected / held by covalent bonds. Mechanical, optical and electronic properties of these frameworks are of particular significance to technology applications.
2.4. Tautomerism
This is an important type of isomerism, found in organic chemistry, biochemistry, medicinal chemistry, pharmacology and molecular biology. It is, in fact, a major component in organic reactions [210] and biochemical processes,211 including those that involve specific interactions with proteins, enzymes and receptors.
It is observed in bioamines (e.g histamine),212 amino acids (e.g histidine and arginine),213 pyrimidine bases (e.g cytosine, thymine, uracil),214 purine bases (adenine, guanine)215 and porphyrins.216
Tautomerism has hampered, at times, deciphering of issues, such as chemical reactivity, biological activity or structure assignment. Having to choose the biologically active tautomer among those of varying thermodynamic stability is frequently a daunting task. A less stable one is, more often than not, the active intermediate in a biotransformation, dictating its reaction course.
Proton transfer (PT) (see section 2.1), intramolecularly, in structures exhibiting tautomerism or intermolecularly, between neutral or ionic species, is a crucial step and quite often the rate‐determining one in such processes.2, 217, 218 Tunneling (see section 2.1),47, 219 in this context, has a key role in enzyme dynamics and catalytic activity (see section 3.1).220
The stabilizing effect of H‐bonding in nucleic acid or protein complexes is strongly related to proton transfer between identical221 (e.g homodimers) or distinct[222] (e.g heterodimers) tautomers of the same molecule. H‐bonding is an important factor in controlling tautomeric equilibria, typical cases being those of keto‐enol in aliphatic or aromatic carbonyl compounds,223 Schiff bases, (keto)oxime‐nitroso(enol)224 (e.g quinone oxime‐nitrosophenol) or enamine‐imine,225 to name a few.
A feature of significance, common to all these H‐bond‐based tautomeric regimes, is their resonance‐assisted strong H‐bonds (RAHBs) (see also, section 2.1).2, 225
H‐bonding has also been correlated to the aromaticity of heterocycles, as a major determinant of their tautomeric forms and corresponding reactivity.226
3. Summary and Outlook
H‐Bonding has travelled a long way and established itself as a non covalent stabilizing interaction with a decisive role on the structure, reactivity or function of molecular frameworks. Despite its long age and many applications in chemistry, biology, materials and their interface, it still has a long way to go, unraveling its potential in areas either yet unexplored or re‐assessable.
As a bridge of these disciplines, H‐bonding potential and perspectives are essentially unlimited. In fields as diverse as functional materials, crystal engineering, drug design (host‐guest chemistry, protein functions and modeling), H‐bonding, in concert or in contest with other “non conventional” bonding types of promising future, entering the scene, such as carbon, halogen and pnicogen interactions, are expected to thrive in the future.
Biographical Information
Petros G. Tsoungas, born, Thessaloniki, Greece. BSc (Hons) Chemistry, North London University (1978). MSc, PhD, Organic Chemistry, UEA, UK (1981). National Military Service (1981‐1984). Research associate, University Thessaloniki (1982‐1984). NIH post doctoral fellowship, University Alberta, Canada (1984‐1985). Head of Hygiene/Safety, EKO National Refinery (1989‐1992). Government Science Policy Department (1992‐2008). Senior Researcher, Hellenic Marine Research Centre, then Hellenic Pasteur Institute to date. Research interests: fused 5‐/ 6‐membered N, O heterocycles and ab initio‐and DFT‐addressed relevant issues.

Biographical Information
George N. Pairas, born 1961, Athens, Greece. BSc Pharmacy (1982), PhD Medicinal Chemistry (1987), University of Patras, Greece. Military National Service (1985‐1986). Abbott Laboratories Hellas (1987‐1989). Lecturer (1989), Assist. Prof. (1998), Assoc. Prof. (2015‐) ‐ Medicinal Chemistry, Department of Pharmacy, University of Patras, Greece. Research interests focus on Design, Synthesis & Conformational Studies of Bioactive Compounds (steroid derivatives with anti‐tumour activity, heterocyclic compounds as building blocks in drug synthesis, bioactive peptides, drugs against infections).

Acknowledgements
Authors are indebted to Mrs. Christina Foka for her invaluable contribution to the preparation of the manuscript.
G. N. Pairas, P. G. Tsoungas, ChemistrySelect 2016, 1, 4520.
References
- 1. Poulter C.D., J Org Chem. 2009, 74, 2631–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Edwards S., Chemistry in Britain. RSC Publishing, Cambridge, 2002. [Google Scholar]
- 3. Breslow R., Chemistry and Biology 1998, 5, R27-R28. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Hobza P., Acc Chem. Res. 2012, 45, 663–672; [DOI] [PubMed] [Google Scholar]
- 4b. Jeffrey G.A., An Introduction to Hydrogen Bonding (Topics in Physical Chemistry), Oxford University Press, New York, 1997. [Google Scholar]
- 5. Desiraju G.S., Steiner T., The Weak Hydrogen Bond in Structural Chemistry and Biology, 1st edn., Oxford University Press, New York, 1999. [Google Scholar]
- 6. Arunan E., Desiraju G.R, Klein R. A., Sadlej J., Scheiner S., Alkorta I., Clary D. C., Crabtree R. H., Dannanberg J. J., Hobza P., Kjaergaard H. G., Legon A. C., Mennucci B., Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar]
- 7. Gilli G., Gilli P., The Nature of the Hydrogen Bond: Outline of a Comprehensive H-Bond Theory, Oxford University Press, New York, 2009. [Google Scholar]
- 8. Pihko P.M., H-Bonding in Organic Synthesis, 1st edn., Wiley-VCH Verlag GmbH & KGaA, Weinheim, 2009. [Google Scholar]
- 9. Desiraju G.R., Angew. Chem. Int. Ed. 2011, 50, 52–59. [DOI] [PubMed] [Google Scholar]
- 10. Reed A., Curtiss L.A., Weihold F., Chem. Rev. 1988, 88, 899–926. [Google Scholar]
- 11. Zimmerman H.E., Weinhold F., J. Org. Chem. 2013, 78, 1844–1850. [DOI] [PubMed] [Google Scholar]
- 12. Koch U., Popelier P.L.A., J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar]
- 13. Kuhn B., Mohr P., Stahl M., J. Med. Chem. 2010, 53, 2601–2611. [DOI] [PubMed] [Google Scholar]
- 14. Chauhan P., Chimni S.S., RSC Adv. 2012, 2, 737–758. [Google Scholar]
- 15. Barić D., Dragičević I., Kovačević B., J. Org. Chem. 2013, 78, 4075–4082. [DOI] [PubMed] [Google Scholar]
- 16. Nagy P., Int. J. Mol. Sci. 2014, 15, 19562–19633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Krygowski T.M., Zachara-Horeglad J.E., Paluciak M., J. Org. Chem. 2010, 75, 4944–4949. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Gilli P., Bertolasi V., Ferretti V., Gilli G., J. Am. Chem. Soc. 1994. , 116, 909–915; [Google Scholar]
- 18b. Gilli G., Gilli P., J. Mol. Struct. 2000, 552, 1–15. [Google Scholar]
- 19. Liu X., Zhang N., Yang J., Liang Y., Zhang R., Dong D., J. Org. Chem. 2013, 78, 3323–3328. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Gerothanasis I., Prog. Nucl. Magn. Res. Spec, 2010, 56, 95–197; [DOI] [PubMed] [Google Scholar]
- 20b. Gerothanasis I., Prog. Nucl .Magn. Res. Spec, 2010, 57, 1–110; [DOI] [PubMed] [Google Scholar]
- 20c. Jaccard G., Lauterwein J, Helv. Chim. Acta, 1986, 69, 1469–1485. [Google Scholar]
- 21. Infantes L., Motherwell W.D.S., Kristallogr, 2005, 220, 333–339. [Google Scholar]
- 22. Castellano R.K., Molecules, 2014, 19, 15783–15785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rozas I., Alkorta I., Elguero J., J. Phys. Chem. A, 1998, 102, 9925–9932. [Google Scholar]
- 24. Feldblum E.S., Arkin I.T., PNAS, 2014, 111, 4085–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nowroozi A., Hajiabadi H., Struct. 2014, 25, 215–220. [Google Scholar]
- 26. Desiraju G.R., Steiner T., in The weak hydrogen bond in structural chemistry and Biology, Oxford University Press, Oxford, 1999, pp. 1–28. [Google Scholar]
- 27. Horowitz S., Rievel R.C., J. Biol. Chem. 2012, 287, 41576–41582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Panigrahi S.K., Desiraju G.R., Proteins: Structure Function and Bioinformatics 2007, 67, 128–141. [DOI] [PubMed] [Google Scholar]
- 29.M.J. Wóczik, P. Blaise, J. Sadlej, H. Flakus, J. Atom. Molec. Optic. Phys 2012, Article ID 174236 1–1
- 30. Perrin C.L., Pure Appl. Chem. 2009, 81, 571–583. [Google Scholar]
- 31. Lane J.R., Contreras-Garcia J., Piquemal J.-P., Miller B.J., Kjaegaard H.G., J. Chem. Theory Comput. 2013, 9, 3263–3266. [DOI] [PubMed] [Google Scholar]
- 32. Bachrach S.M., Computational Organic Chemistry, 2nd edn., John Wiley & Sons, Hoboken, New Jersey, 2013. [Google Scholar]
- 33. Arunan E., Mani D., Faraday Discuss. 2015, 177, 51–64. [DOI] [PubMed] [Google Scholar]
- 34. Ayoub A.T., Tuszynski J., Klodukowski M., Theoret. Chem. Acc. 2014, 133, 1520–1526. [Google Scholar]
- 35. Zhang J., Chen P., Yuan B., Ji W., Cheng Z., Qiu X., Science, 2013, 342, 611–614. [DOI] [PubMed] [Google Scholar]
- 36. Mani D., Arunan E., Phys. Chem. Chem. Phys. 2013, 15, 14377–14383. [DOI] [PubMed] [Google Scholar]
- 37.
- 37a. Bürghi H.-B., Dunnitz J.D., Acc. Chem. Res. 1983, 16, 153–161; [Google Scholar]
- 37b. Alkorta I., Rozas I., Mo O., Yañez M., Elguero J., J. Phys. Chem. A 2001, 105, 7481–7485. [Google Scholar]
- 38. Gamiz-Hernandez A., Magomedov A., Hummer G., Kaila V.R., J. Phys. Chem. B 2015, 119, 2611–2619. [DOI] [PubMed] [Google Scholar]
- 39. Braun J., Limbach H.-H., Williams P.G., Morimoto H., Wemmer D.E., J. Am. Chem. Soc. 1996, 118, 7231–7232. [Google Scholar]
- 40. Braun J., Koecher M., Schlabach M., Wehrle B., Limbach H.-H., Vogel E., J. Am. Chem. Soc. 1994, 116, 6593–6604. [Google Scholar]
- 41. Shida N., Barbara P.F., Almlof J.E., J. Chem. Phys. 1989, 91, 4061–4072. [Google Scholar]
- 42. Tuckerman M. E., Marx D., Phys Rev Lett 2001, 86, 4946–4949. [DOI] [PubMed] [Google Scholar]
- 43. Bertran J., Oliva A., Rodriguez-Santiago L., Sodupe M., J. Am. Chem. Soc. 1998, 120, 8159–8167. [Google Scholar]
- 44. Florian J., Hrouda V., Hobza P., J. Am. Chem. Soc. 1994, 116, 1457–1460. [Google Scholar]
- 45. Simperler A., Mikenda W., Schwarz K., Chem. Eur. J. 2001, 7, 1606–1613. [DOI] [PubMed] [Google Scholar]
- 46. Wu Y., Wang H., Lin Y., Gao S., Zhang F., Can. J. Chem. 2013, 91, 992–998. [Google Scholar]
- 47. Alkorta I., Elguero J., Org. Biomol. Chem. 2006, 4, 3096–3101 & references cited therein. [DOI] [PubMed] [Google Scholar]
- 48.
- 48a. Johannisen L.O., Hay S., Scrutton N.S., Phys. Chem. Chem. Phys. 2015, 17, 30775–30782; [DOI] [PubMed] [Google Scholar]
- 48b. Layfield J.P., Hammes-Schiffer S., Chem. Rev. 2014, 114, 3466–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schmitt U.W., J. Phys. Chem. 1998, 102, 5547–5551. [Google Scholar]
- 50. Voth G.A., Acc. Chem. Res. 2006, 39,143-150 & references cited therein. [DOI] [PubMed] [Google Scholar]
- 51. Golan A., Bravaya K.B., Kudirka R., Kostko O., Leone S.R., Krylov A.I., Ahmed M., Nat. Chem. 2012, 4, 323–329. [DOI] [PubMed] [Google Scholar]
- 52. Zhao G.-J., Han K.-L., Acc. Chem. Res. 2012, 45(3), 404–413 & references cited therein. [DOI] [PubMed] [Google Scholar]
- 53. Uzhinov B.M., Khimich M.N., Russ. Chem. Rev. 2011, 80, 553–577. [Google Scholar]
- 54. Reese S.Y., Nocera D.G., Annu. Rev. Biochem. 2009, 78, 673–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bonin J., Costentin C., Robert M., Savéant J.-M., Tard C., Acc. Chem. Res. 2012, 45, 372–381 & references cited therein. [DOI] [PubMed] [Google Scholar]
- 56. Savéant J.-M., Ann. Rev. Anal. Chem. 2014, 7, 537–560. [DOI] [PubMed] [Google Scholar]
- 57. Tse J.S., Ann. Rev. Phys. Chem. 2002, 53, 249–290. [DOI] [PubMed] [Google Scholar]
- 58. Krokidis X., Goncalves V., Savin A., Silvi B., J. Phys. Chem .A, 1998, 102, 5065–5073. [Google Scholar]
- 59. Chen J., McAllister M.A., Lee J. K., Houk K. N., J. Org. Chem. 1998, 63, 4611–4619. [Google Scholar]
- 60. Nazarparvar E., Zahedi M., Klein E., J. Org. Chem. 2012, 77, 10093–10104. [DOI] [PubMed] [Google Scholar]
- 61. Alkorta I., Campillo N., Rozas I., Elguero J., J. Org. Chem. 1998, 63, 7759–7763. [Google Scholar]
- 62. Legon A., Chem. Soc. Rev. 1993, 22, 153–163. [Google Scholar]
- 63. Abraham M.H., Frellier P.L., Prior D.V., Duce P.P., Morris J.J., Taylor P.J., J. Chem. Soc. Perkin Trans. 2, 1989, 6, 699–711. [Google Scholar]
- 64. Abraham M.H., Frellier P.L., Prior D.V., Morris J.J., Taylor P.J., J. Chem. Soc. Perkin Trans. 2, 1990, 4, 521–529. [Google Scholar]
- 65. Huque F.T.T., Platts J. A., Org. Biomol. Chem. 2003, 1, 1419–1424. [DOI] [PubMed] [Google Scholar]
- 66. Abraham M. H., Du C. M., Platts J. A., J. Org. Chem. 2000, 65, 7114–7118. [DOI] [PubMed] [Google Scholar]
- 67. Sijbren O., Engberts J.B.F.N., Org. Biomol. Chem. 2003. 1, 2809–2820. [DOI] [PubMed] [Google Scholar]
- 68. Breslow R., Acc. Chem. Res. 1991, 24, 159–164. [Google Scholar]
- 69. Lazarides Th., Acc. Chem. Res. 2001, 34, 931–937. [DOI] [PubMed] [Google Scholar]
- 70. Butler R.N., Coyne A.G., Chem. Rev. 2010, 110, 6302–6337. [DOI] [PubMed] [Google Scholar]
- 71. Li C.J., Trost B.M., PNAS, 2008, 105, 13197–13202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Li C.J., Chem. Rev. 2005, 105, 3095–3166. [DOI] [PubMed] [Google Scholar]
- 73. Alkorta I., Elguero J., Grabowski S.J., J. Phys. Chem. A 2008, 112, 2721–2727. [DOI] [PubMed] [Google Scholar]
- 74. Custelcean R., Jackson J.E., Chem. Rev. 2001, 101, 1963–1980. [DOI] [PubMed] [Google Scholar]
- 75. Rozas I., Alkorta I., Elguero J., J. Phys. Chem. 1997, 101, 9457–9463. [Google Scholar]
- 76. Steiner T., Starikov E.B., Tamm M., J. Chem.Soc. Perkin Trans. 2, 1996, 1, 67–71. [Google Scholar]
- 77. Steiner T., J. Chem. Soc. Chem. Commun. 1995, 1, 95–96. [Google Scholar]
- 78. Viswamitra M. A., Radhakrishnan R., Bandekar J., Desiraju G. R., J. Am. Chem. Soc. 1993, 115, 4868–4869. [Google Scholar]
- 79. Takahashi H., Suboyama T., Umezawa Y., Honda K., Nishio M., Tetrahedron, 2000, 56, 6185–6191. [Google Scholar]
- 80. Brandl M., Weiss M. S., Jabs A., Sühnel J., Hilgenfeld R., J. Mol. Biol. 2001, 307, 357–377. [DOI] [PubMed] [Google Scholar]
- 81. Umezawa Y., Nishio M., Bioorg. Med. Chem. 1998, 6, 2507–2515. [DOI] [PubMed] [Google Scholar]
- 82. Takahashi O., Yamazaki K., Kohno Y., Ueda K., Suezawa H., Nishio M., Tetrahedron 2009, 65, 3525–3528 [Google Scholar]
- 83. Kolandaivel P., Nirmala V., J. Mol. Struct. 2004, 694, 33–38. [Google Scholar]
- 84. Alabugin I., Manoharan M., Peabody S., Weinhold F., J. Am. Chem. Soc. 2003, 125, 5973–5987. [DOI] [PubMed] [Google Scholar]
- 85. Kühl O., Chem. Soc. Rev. 2011, 40, 1235–1246. [DOI] [PubMed] [Google Scholar]
- 86. Kojelka J., Non Covalent Forces, vol.19, chapter. 6, in Challenges and Advances in Computational Chemistry and Physics (Ed.: S. Scheiner), 2015, pp 129–158. [Google Scholar]
- 87. Gregoret L.M., Rader S.D., Fletterick R.J., Cohen F.E., Proteins, 1991, 9, 99–107. [DOI] [PubMed] [Google Scholar]
- 88. Hansen P.E., Molecules, 2015, 20, 2405–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kool E.T., Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 1–22. [DOI] [PubMed] [Google Scholar]
- 90. Kannan N., Vishveshwara S., Protein Eng. 2000, 13, 753–761. [DOI] [PubMed] [Google Scholar]
- 91. Gazit E., The FASEB Journal, 2002, 16, 77–83. [DOI] [PubMed] [Google Scholar]
- 92. Subrayashastry A., Narayanaswamy S., Chittaranjan D., Arumugam S., Karle I.S., Balaram P., J. Am. Chem. Soc. 2003, 125, 5308–5315. [DOI] [PubMed] [Google Scholar]
- 93. Hunter C. A., Lawson K. R., Perkins J., Urch C. J., J. Chem. Soc. Perkin Trans. 2, 2001, 5, 651–669. [Google Scholar]
- 94. Sindkhedkar M. D., Mulla H. R., Cammers-Goodwin A., J. Am. Chem. Soc. 2000, 122, 9271–9277. [Google Scholar]
- 95. Carver F.J., Hunter C.A., Seward E.M., Chem. Commun. 1998, 7, 775–776. [Google Scholar]
- 96. Butterfield S. M., Patel P. R., Waters M. L. , ibid. 2002, 124, 9751–9755. [DOI] [PubMed] [Google Scholar]
- 97. Guckian K.M., Schweitzer B.A., Ren R.X.F., Sheils C.J., Tahmassebi D.C., Kool E.T., J. Am. Chem. Soc. 2000, 122, 2213–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hunter C.A., Ihekwaba N., Misuraca M.C., Segarra-Maset M.D., Turega S.M., Chem. Commun. 2009, 26, 3964–3966. [DOI] [PubMed] [Google Scholar]
- 99. Li Q., Liu Z., Cheng J., Li W., Gong B., Sun J., J. Mol. Struct. THEOCHEM 2009, 896, 112–115. [Google Scholar]
- 100. Alkorta I., Blanco F., Deya P. M., Elguero J., Estarellas C., Frontera A., Quiñonero D., Theoret. Chem. Acc. 2010, 126, 1–14. [Google Scholar]
- 101. Allen F.H., Howard J.A.K., Hoy V.J., Desiraju G.R., Reddy D.S., Wilson C.C., J. Am. Chem. Soc. 1996, 118, 4081–4084. [Google Scholar]
- 102. Missopolinou D., Panayiotou C., J. Phys. Chem. A, 1998, 102, 3574–3581. [Google Scholar]
- 103. Kawahara S.-I., Taira K., Uchimaru T., Chem. Phys. 2003, 290, 79–83. [Google Scholar]
- 104. Beller M., Bolm C., Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals, 2nd edn., vols 1 and 2, Wiley-VCH, Weinheim, 2004. [Google Scholar]
- 105. Ojima I., Tzamarioudaki M., Li Z., Donovan R.J., Chem. Rev. 1996, 96, 635–662. [DOI] [PubMed] [Google Scholar]
- 106. Haughton L., Williams J.M.J., J. Chem. Soc. Perkin Trans. 1, 2000, 20, 3335–3349. [Google Scholar]
- 107. Fletcher A.J., Christie S.D.R., J. Chem. Soc. Perkin Trans.1, 2001, 1, 1–13. [Google Scholar]
- 108. Antos J., Francis M.B., Curr. Opin. Chem.Biol. 2006, 10, 253–262. [DOI] [PubMed] [Google Scholar]
- 109. Reetz M., Angew. Chem. Int. Ed. 2008, 47, 2556–2588. [DOI] [PubMed] [Google Scholar]
- 110. Chauder B., Green L., Snieckus V., Pure Appl. Chem. 1999, 71, 1521–1529. [Google Scholar]
- 111. Frenking G., Theoretical Aspects of Transition Metal Catalysis: Topics in Organometallic Chemistry, Vol 12, Springer Verlag, Berlin/Heidelberg, 2005. [Google Scholar]
- 112. MacMillan D.W.C., Nature, 2008, 455, 304–308. [DOI] [PubMed] [Google Scholar]
- 113. Seayad J., List B., Catalytic Asymmetric Multi-component Reactions in Multi-component Reactions (Eds.: J. Zhu, H. Bienaymé), Wiley-VCH, Weinheim, 2004, pp. 199–223. [Google Scholar]
- 114. Ramachary D.B., Kishov M., Reddy G.B., Org. Biomol. Chem. 2006, 4, 1641–1646. [DOI] [PubMed] [Google Scholar]
- 115. Guo H.-C., Ma J.-A., Angew. Chem. Int. Ed. 2006, 45, 354–366. [DOI] [PubMed] [Google Scholar]
- 116. Grondal C., Jeanty M., Enders D., Nature Chemistry 2010, 2, 167–178. [DOI] [PubMed] [Google Scholar]
- 117. Akiyama T., Itoh J., Fuchibe K., Adv. Synth. Catal. 2006, 348, 999–1010. [Google Scholar]
- 118. Taylor M.S., Jacobsen E.N., Angew. Chem. Int. Ed. 2006, 45, 1520–1543. [DOI] [PubMed] [Google Scholar]
- 119. Jacobsen E.N., Doyle M., Chem. Rev. 2007, 107, 5713–5743. [DOI] [PubMed] [Google Scholar]
- 120. Miyabe H., Takemoto Y., Bull. Chem. Soc. Jpn. 2008, 81, 785–795. [Google Scholar]
- 121. Dalko P.I., Enantioselective Organocatalysis: Reactions and Experimental Procedures, Wiley-VCH Verlag GmbH & Co KGaA, Weinheim, 2007. [Google Scholar]
- 122. Dalko P.I., Moisan L., Angew. Chem. Int. Ed. 2004, 43, 5138–5175. [DOI] [PubMed] [Google Scholar]
- 123. Dalko P.I., Moisan L. Adv. Synth. Catal. 2004, 346, 1007–1012. [Google Scholar]
- 124. Martin N.J.A., Cheng X., List B., J. Am. Chem. Soc. 2008, 130, 13862–13863. [DOI] [PubMed] [Google Scholar]
- 125. Bui T., Syed S., BarbasIII C.F., J. Am. Chem. Soc. 2009, 131, 8758–8759. [DOI] [PubMed] [Google Scholar]
- 126. Rabalakos C., Wulff W.D., J. Am. Chem. Soc. 2008, 130, 13524–13525. [DOI] [PubMed] [Google Scholar]
- 127. Reisman E., Doyle A.G., Jacobsen E.N., J. Am. Chem. Soc. 2008, 130, 7198–7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Li L., Ganesh M., Seidel D., J. Am. Chem. Soc. 2009, 131, 11648–11649. [DOI] [PubMed] [Google Scholar]
- 129. Crisostomo F. R. Pinacho, Lledo A., Shenoy S. R., Wasawa T. I., Rebek J. Jr, J. Am. Chem. Soc. 2009, 131, 7402–7410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.
- 130a. Yoon T.P., Jacobsen E. N., Angew. Chem. Int. Ed. 2005, 44, 466–468; [DOI] [PubMed] [Google Scholar]
- 130b. Gaunt M.J., Johansson C.C.C., McNally A., Vo N.T. Drug Discov. Today, 2007, 12, 8–27. [DOI] [PubMed] [Google Scholar]
- 131.
- 131a. D'Souza D.M., Müller T. J.J., Chem. Soc. Rev. 2007, 36, 1095–1108. [DOI] [PubMed] [Google Scholar]
- 131b. Gonzalez S. Diez, Marion N., Nolan S.P., Chem. Rev. 2009, 109, 3612–3676. [DOI] [PubMed] [Google Scholar]
- 132. Natale D., Mareque-Rivas J.C., Chem. Commun. 2008, 4, 425–437. [DOI] [PubMed] [Google Scholar]
- 133.
- 133a. Jacobsen E.N., MacMillan D.W.C. , PNAS 2010, 107, 20678–20685;20956302 [Google Scholar]
- 133b. Jeffrey J.L., Terrett J.A., MacMillan D.W.C., Science, 2015, 349, 1532–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.
- 134a. Shao Z., Zhang H., Chem. Soc. Rev. 2009, 38, 2745–2755; [DOI] [PubMed] [Google Scholar]
- 134b. Zhong C., Shi X., Eur. J. Org. Chem. 2010, 16, 2999–3025. [Google Scholar]
- 135. Assimomytis N., Tsoungas P.G., Sariyannis Y., Stavropoulos G., Varvounis G., Cordopatis P., Synlett, 2009, 17, 2777–2782. [Google Scholar]
- 136. Taylor M.S., Jacobsen E.N., J. Am. Chem. Soc. 2004, 126, 10558–10559. [DOI] [PubMed] [Google Scholar]
- 137. Joly G.D., Jacobsen E.N., J. Am. Chem. Soc. 2004, 126, 4102–4103. [DOI] [PubMed] [Google Scholar]
- 138. Wenzel A.G., Jacobsen E.N., J. Am. Chem. Soc. 2002, 124, 12964–12965. [DOI] [PubMed] [Google Scholar]
- 139. Vachal P., Jacobsen E. N., J. Am. Chem. Soc. 2002, 124, 10012–10014. [DOI] [PubMed] [Google Scholar]
- 140. Vachal P., Jacobsen E.N., Org. Lett. 2000, 2, 867–870. [DOI] [PubMed] [Google Scholar]
- 141. Sigman M.S., Jacobsen E.N., J. Am. Chem. Soc. 1998, 120, 4901–4902. [Google Scholar]
- 142. Kanai M., Kato N., Ichikawa E., Shibasaki M., Synlett. 2005, 10, 1491–1508. [Google Scholar]
- 143. Ma J.A., Cahard D., Angew. Chem. Int. Ed. 2004, 43, 4566–4583. [DOI] [PubMed] [Google Scholar]
- 144. Schneider J.F., Lauber M.B., Muhr V., Kratzer D., Paradies J. , Org. Biomol. Chem. 2011, 9, 4323–4327. [DOI] [PubMed] [Google Scholar]
- 145.
- 145a. Jörres M., Mersmann S., Raabe G., Bolm C., Green Chem. 2013, 1, 612–616; [Google Scholar]
- 145b. Fang X., Wang C.-J., Chem. Commun. 2015, 51, 1185–1197. [DOI] [PubMed] [Google Scholar]
- 146. MacMillan D.W.C., Nature, 2008, 455, 304–308. [DOI] [PubMed] [Google Scholar]
- 147. Jang H., Hong J., MacMillan D.W.C., J. Am. Chem. Soc. 2007, 129, 7004–7005. [DOI] [PubMed] [Google Scholar]
- 148. Kim H., MacMillan D.W.C., J. Am. Chem. Soc. 2008, 130, 398–399. [DOI] [PubMed] [Google Scholar]
- 149. Lelais G., MacMillan D.W.C., Aldrichimica Acta, 2006, 39, 79–87. [Google Scholar]
- 150. MacMillan D.W.C. , Science, 2008, 322, 77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Rendler S., MacMillan D.W.C., J. Am. Chem. Soc. 2010, 132, 5027–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Singh A.S., Sun S.-S., J.Org.Chem. 2012, 77, 1880–1890 & references cited therein. [DOI] [PubMed] [Google Scholar]
- 153. Moyano A., Rios R., Chem. Rev. 2011, 111, 4703–4832. [DOI] [PubMed] [Google Scholar]
- 154. Yu X., Wang W., Chem. Asian J. 2008, 3, 516–532 and references cited therein. [DOI] [PubMed] [Google Scholar]
- 155. Brière J., Oudeyer S., Dalla V., Levacher V., Chem. Soc. Rev. 2012, 41, 1696–1707 & references cited therein. [DOI] [PubMed] [Google Scholar]
- 156. Connon S.J., Synlett, 2009, 3, 0354–0376 & references cited therein. [Google Scholar]
- 157. Zhang Z., Schreiner P.R., Chem. Soc. Rev. 2009, 38, 1187–1198 & references cited therein. [DOI] [PubMed] [Google Scholar]
- 158. Menger F.M., Acc. Chem. Res. 1985, 18, 128–134. [Google Scholar]
- 159. Menger F.M., Acc. Chem. Res. 1993, 26, 206–212. [Google Scholar]
- 160. Bruice T.C., Lightstone F.C., Acc. Chem. Res. 1999, 32, 127–136. [Google Scholar]
- 161. Pamies O., Bäckvall J.-E., Chem. Rev. 2003, 108, 3247–3262. [DOI] [PubMed] [Google Scholar]
- 162. Pouliou F.M., Thireou T.N., Eliopoulos E.E., Tsoungas P.G., Labrou N.E., Clonis Y.D., Chem. Biol. Drug Des. 2015, 86, 1055–1063. [DOI] [PubMed] [Google Scholar]
- 163. Schiøtt B., Iversen B.B., Madsen G.K., Larsen F.K., PNAS, 1998, 95, 12799–12802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Cleland W.W., Biochem. Biophys. 2000, 382, 1–5. [DOI] [PubMed] [Google Scholar]
- 165. Warren J.J., Mayer J.M., Biochemistry, 2015, 54, 1863–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Bruice T.C.A., Acc. Chem. Res. 2002, 35, 139–148. [DOI] [PubMed] [Google Scholar]
- 167. Du Z., Shao Z., Chem. Soc. Rev. 2013, 42, 1337–1378 & references cited therein. [DOI] [PubMed] [Google Scholar]
- 168. Usui I., Schmidt S., Breit B., Org. Lett. 2009, 11, 1453–1456. [DOI] [PubMed] [Google Scholar]
- 169. Kortemme T., Morozov A.V., Baker D. J. Mol. Biol. 2003, 326, 1239–1259. [DOI] [PubMed] [Google Scholar]
- 170. Allocati N., Masulli M., Pietracupa M., Federici L., Ilio C. Di, Biochem. J. 2006, 394(Pt 1), 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Nova A., Keten S., Pugno N.M., Redaelli A., Buehler M.J., Nano Letters 2010, 10, 2626–2634. [DOI] [PubMed] [Google Scholar]
- 172. Chen D., Oezguen N., Urvil P., Ferguson C., Dann S.M., Savidge T.C., Sci. Adv. 2016, 2(3), e1501240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Kubinyi H., Hydrogen Bonding: The Last Mystery in Drug Design? in Biological Physico-Chemical and Computational Strategies: Pharmacokinetic Optimization in Drug Research (Eds.: B. Testa, H. van de Waterbeend, G. Folkers, R. Guy), Helvetica Chimica Acta & Wiley-VCH, Zürich, 2001, pp 513–524. [Google Scholar]
- 174. Böhm H.-J., Klebe G., Angew. Chem. Int. Ed. 1996, 35, 2588–2614. [Google Scholar]
- 175. Babine R.E., Bender S.L., Chem. Rev. 1997, 97, 1359–1472. [DOI] [PubMed] [Google Scholar]
- 176. Davis A.M., Teague S.J., Angew. Chem. Int. Ed. 1999, 38, 736–749. [DOI] [PubMed] [Google Scholar]
- 177. Lommerse J.P.M., Price S.L., Taylor R., J. Comput. Chem. 1997, 18, 757–774. [Google Scholar]
- 178. Pihan E., Delgadillo R.F., Tonkin M.L., Pugnière M., Lebrun M., Boulanger M.J., Douguet D., J. Comput. Aided Mol. Des. 2015, 29, 525–539. [DOI] [PubMed] [Google Scholar]
- 179.
- 179a. Gardikis Y., Tsoungas P.G., Potamitis C., Zervou M., Cordopatis P., Heterocycles 2011, 83, 1077–1091; [Google Scholar]
- 179b. Gardikis Y., Tsoungas P.G., Potamitis C., Zervou M., Pairas G., Cordopatis P., Heterocycles, 2011, 83, 1291–1302. [Google Scholar]
- 180. Eildal J.N.N., Hulqvist G., Balle T., Stuhr-Hansen N., Padrah S., Gianni S., Strømgaard K., Jemth P., J. Am. Chem. Soc. 2013, 135, 12998–13007. [DOI] [PubMed] [Google Scholar]
- 181. Torshin I.Y., Weber I.T., Harrison R.W., Protein Eng. 2002, 15, 359–363. [DOI] [PubMed] [Google Scholar]
- 182. Konermann L., Liu J. Pan„ Y.-H., Chem. Soc. Rev. 2011, 40, 1224–1234. [DOI] [PubMed] [Google Scholar]
- 183. Patgiri A., Joy S.T., Arora P.S., J. Am. Chem. Soc. 2012, 134, 11495–11502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Sapronova A., Brystof V.S., Green M.E., Front. Biosci. 2003, 8, 1356–1370. [Google Scholar]
- 185. Auffinger P., Hays F.A., Westhof E., Ho P.S., PNAS 2004, 101, 16789–16794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Domagala M., Paluciak M., Comp. Theoret. Chem. 2014, 1027, 173–178. [Google Scholar]
- 187. Aakeröy C. B., Panikkattu S., Chopade P .D., Desper J., Cryst. Eng. Commun. 2013, 15, 3125–3136. [Google Scholar]
- 188. Politzer P., Murray J.S., A Unified View of Halogen Bonding, Hydrogen Bonding and Other σ-Hole Interactions in Noncovalent Forces Challenges and Advances in Computational Chemistry (Ed. S. Scheiner), Springer International Publishing, Switzerland, 2015, pp. 291–321. [Google Scholar]
- 189. Wallimann P., Marti T., Fürer A., Diederich F., Chem. Rev. 1997, 97, 1567–1608 [DOI] [PubMed] [Google Scholar]
- 190. Chen J.M., Xu S. L., Wawrzack Z., Basarab G.S., Jordan D.B., Biochemistry 1998, 37, 17735–17744. [DOI] [PubMed] [Google Scholar]
- 191. Moran S., Ren R.X.-F., Rumney S., Kool E.T., J. Am. Chem. Soc. 1997, 119, 2056–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Moran S., Ren R.X.-F., Kool E.T., PNAS 1997, 94, 10506–10511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Veerapandian P., Structure-Based Drug Design, Marcel Dekker, New York, 1997. [Google Scholar]
- 194. Ladbury J.E., Chem. Biol. 1996, 3, 973–980. [DOI] [PubMed] [Google Scholar]
- 195. Bowie J.U., Curr. Opin. Struct. Biol. 2011, 21, 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.
- 196a. Ishikita H., Saito K., J. R. Soc. Interface 2014, 11, 20130518; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196b. Agmon N., Gutman M., Nature Chem. 2011, 3, 840–842. [DOI] [PubMed] [Google Scholar]
- 197. Mikol V., Papageorgiou C., Borer X., J. Med. Chem. 1995, 38, 3361–3367. [DOI] [PubMed] [Google Scholar]
- 198. Desiraju G.R., Crystal Engineering: The Design of Organic Solids, Elsevier, Amsterdam, 1989. [Google Scholar]
- 199. Anderson H.L., Sanders J.K.M., Acc. Chem. Res. 1993, 26, 469–475. [Google Scholar]
- 200. Archer E.A., Gong H., Krische M.J., Tetrahedron, 2001, 57, 1139–1159. [Google Scholar]
- 201. Etter M.C., Adsmond D.A., J. Chem. Soc. Chem. Commun. 1990, 8, 589–591. [Google Scholar]
- 202. Zerkowski J A., Seto C.T., Wierda D.A., Whitesides G.M., J. Am. Chem. Soc. 1990, 112, 9025–9026. [Google Scholar]
- 203. Drain C.M., Russel K.C., Lehn J.-M., J. Chem. Soc. Chem. Commun. 1996, 3, 337–338. [Google Scholar]
- 204. Zhang D.-W., Wang W.-K., Li Z.-T., Chem. Rec. 2015, 15, 233–251. [DOI] [PubMed] [Google Scholar]
- 205. Cram D.J., Angew. Chem. Int. Ed. Engl. 1986, 25, 1039–1057. [Google Scholar]
- 206. Cabrele C., Martinek T.A., Reiser O., Berlicki Ł., J. Med. Chem. 2014, 57, 9718–9739 and references cited therein. [DOI] [PubMed] [Google Scholar]
- 207. Langley P. J., Hulliger J., Chem. Soc. Rev. 1999, 28, 279–291. [Google Scholar]
- 208. Calzaferri G., Pauchard M., Maas H., Huber S., Khatyr A., Schaafsma T. , J. Mater. Chem. 2002, 12, 1–13. [Google Scholar]
- 209. Davies A.G., Thompson J.M.T., Nanoengineering: Electronics, Materials and Assembly in Phil. Trans RSC A, Royal Society Series on Advanced Science Vol. 3, Imperial College Press, 2007. [Google Scholar]
- 210. Elguero J., Katritzky A.R., Denisko O.V., Adv. Heterocycl. Chem. 2000, 76, 1–84. [Google Scholar]
- 211. Singh A., Hespanha J.P., Philos. Trans. R. Soc. A, 2010, 368, 4995–5011. [DOI] [PubMed] [Google Scholar]
- 212. Raczyńska E.D., Kosińska W., Osmialowski B., Gawinecki R., Chem. Rev. 2005, 105, 3561–3612. [DOI] [PubMed] [Google Scholar]
- 213. Day R.M., Thalhauser C.J., Sudmeier J.L., Vincent M.P., Torchilin E.V., Sanford D.G., Bachovchin C.W., Bachovchin W.W., Protein Sci. 2003, 12, 794–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Kabelac M., Kratochvil M., Sponer J., Hobza P., J. Biomol. Struct. Dyn. 2000, 17, 1077–1086. [DOI] [PubMed] [Google Scholar]
- 215. Kadish K.M., Smith K.M., Guilard R., The Porphyrin Handbook Vol 2, Academic Press, San Diego, 1999. [Google Scholar]
- 216. Zundel G., Hydrogen Bonds with Large Proton Polarizability and Proton Transfer Processes in Electrochemistry and Biology in Advances in Chemical Physics (Eds.: I. Prigogine, S.A. Rice), Volume 111, John Wiley & Sons, Hoboken, NJ, 1999. [Google Scholar]
- 217. Kohen A., Klinman J.P., Acc. Chem. Res. 1998, 31, 397–404. [Google Scholar]
- 218. Schreiner P.R., Reisenauer H.P., Ley D., Gerbig D., Wu C.-H., Allen W.D., Science 2011, 332, 1300–1303. [DOI] [PubMed] [Google Scholar]
- 219. Sutcliffe M.J., Scrutton N.S., Eur. J. Biochem. 2002, 269(13), 3095–3102. [DOI] [PubMed] [Google Scholar]
- 220. Söntjens H.M., Sijbesma R.P., Genderen M.H.P. van, Meijer E.W., J. Am. Chem. Soc. 2000, 122, 7487–7493. [Google Scholar]
- 221. Zimmerman S.C., J. Org. Chem. 1993, 58, 6625–6628. [Google Scholar]
- 222. Iglesias E ., J. Org. Chem. 2003, 68, 2680–2688. [DOI] [PubMed] [Google Scholar]
- 223. Sanz D., Perona A., Claramunt R. M., Elguero J., Tetrahedron, 2005, 61, 145–154. [Google Scholar]
- 224. Raczyńska E.D., TM. Krygowski, J. Zachara, B. Ośmiatofski, R. Gawinecki, J Phys Org Chem 2005, 18, 892–897. [Google Scholar]
- 225. Garro J.C., Manzanares G.D., Zamarbide G.N., Ponce C.A., Estrada M.R., Jáuregui E.A., J. Mol. Struct. THEOCHEM 2001, 545, 17–27. [Google Scholar]
- 226. Zubatyuk R.I., Volovenko Y.M., Shishkin O.V., Gorb L., Leszczynski J., J. Org. Chem. 2007, 72, 725 [DOI] [PubMed] [Google Scholar]