

Dear Editor,
Distal deletion of the short arm of chromosome 4, including 4p16.3, causes Wolf-Hirschhorn syndrome (WHS; OMIM 194190), which is a rare congenital disorder characterized by intellectual disability, seizures, specific facial deformities (Greek warrior helmet appearance), and multiple congenital anomalies [1]. In contrast, more proximal interstitial deletion of chromosome 4p, including 4p12–p16, has been reported more rarely than WHS and is often termed proximal 4p deletion syndrome; it is distinct from WHS [2]. The phenotypic manifestation of proximal 4p deletion syndrome is generally less severe than that of WHS. Although several cases of WHS have been reported in Korea [3, 4], proximal 4p deletion syndrome is yet to be reported. As this type of deletion is not well known among healthcare professionals and confers a broad spectrum of congenital abnormalities, it may go undiagnosed until the affected person is older. We present the first case of proximal 4p deletion syndrome in a Korean patient with developmental delay. The genetic analysis was conducted after obtaining informed consent from the parents for the clinical analysis. Reporting of this case without informed consent was approved by the Institutional Review Board of Soonchunhyang University Bucheon Hospital, Bucheon, Korea (2019-10-016).
Between June 2016 and April 2017, a 5-yr-old boy visited Soonchunhyang University Bucheon Hospital for psychomotor retardation evaluation. At birth (41 weeks of gestation), he weighed 2.56 kg (3rd percentile). Neither of his parents had intellectual disabilities or epilepsy. On physical examination, the patient’s growth parameters, including weight (16.5 kg, 25th percentile) and height (109.5 cm, 75th percentile), were within normal ranges; however, his developmental milestones were delayed. His head control was delayed by four to five months of age, and he started to walk without assistance only at 18 months of age. His ability to understand and express language was low, and his pronunciation was inaccurate. Formal intellectual assessments found that his intelligence quotient was very low (full-scale intelligence quotient 61). He had no apparent facial dysmorphism, except for hypertelorism, and no limb defects or seizure episodes. However, he had bilateral cryptorchidism and an atrial septal defect. The electroencephalogram results were normal for his age. We compared the clinical features of our patient with those reported in a previous study on 4p interstitial deletion (Table 1) [2].
Table 1.
Comparison of clinical features of the patient in this study and patients in a previous study on 4p interstitial deletion
| Phenotype | This study | Total n/N* (%) for Alesi et al. [2] |
|---|---|---|
| Deleted region | 4p15.1–15.31 | 4p14–16.3 |
| Mental retardation | + | 15/16 (94) |
| Hypotonia | − | 13/16 (81) |
| Large nose | − | 11/15 (73) |
| Epicanthal folds | − | 10/15 (67) |
| Micrognathia/retrognathia | − | 10/16 (63) |
| Tall thin body habitus | − | 8/13 (62) |
| Long face | − | 9/15 (60) |
| High palate | − | 9/15 (60) |
| Cryptorchidism | + | 4/7 (57) |
| Midface hypoplasia | − | 7/13 (54) |
| Upslanting palpebral fissures | − | 8/15 (53) |
| Ptosis | − | 5/16 (31) |
| Hypertelorism | + | 4/15 (27) |
| Broad short neck | − | 4/15 (27) |
| Craniosynostosis | − | 1/16 (6) |
| Congenital heart disease† | + | 1/16 (6) |
n/N indicates the ratio of the number of positive patients and the total number of examined patients;
Atrial septal defect.
Conventional G-banding chromosome analysis showed a deletion of the short arm of chromosome 4, 46,XY,del(4)(p15. 2p15.3) in all metaphases analyzed (Fig. 1A); parental karyotypes were normal. A commercially available FISH probe for the WHS critical region mapping to 4p16.3 was used according to the manufacturer’s protocol (Abbott, Rungis, France). The FISH results revealed that the deletion observed in the chromosome study did not include the WHS critical regions on chromosome 4p16.3 (Fig. 1B). Next, we used the CytoScan 750K chromosomal microarray (Affymetrix, Santa Clara, CA, USA) for high-resolution genome-wide DNA copy number analysis. The analysis was performed according to the manufacturer’s instructions, and data were analyzed according to human genome assembly 19. The results revealed an interstitial 12 Mb deletion at 4p15.31p15.1 (Fig. 1C). The final karyotype was 46,XY,del(4)(p15.2p15.3).arr[GRCh37] 4p15.31p15.1(19,766,331-31,541, 260)X1.
Fig. 1.
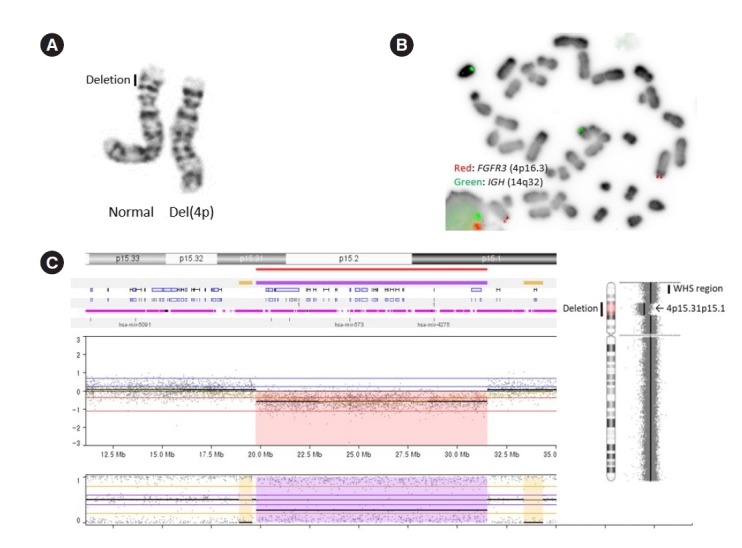
Genetic study of a patient with developmental delay. (A) An interstitial deletion in 4p was revealed by conventional G-banding chromosome analysis. (B) FISH analysis revealed two copies of FGFR3 (red) on 4p16.3. The green signal indicates IGH (14q32). (C) Chromosomal microarray showed a 12-Mb deletion at 4p15.31p15.1. The deletion is proximal to the typical WHS region.
Abbreviations: FISH, Fluorescence in situ hybridization; WHS, Wolf-Hirschhorn syndrome.
We report a rare de novo interstitial deletion of 4p15.1–15.31 in a patient with developmental delay. The deleted region does not overlap with WHS regions and encompasses 19 genes: SLIT2, MIR218-1, KCNIP4, ADGRA3, GBA3, PPARGC1A, DHX15, SOD3, LGI2, SEPSECS, PI4K2B, ZCCHC4, ANAPC4, SLC34A2, SMIM20, RBPJ, CCKAR, STIM2, and PCDH7. Several of these genes are of special interest regarding brain development. SLIT2 regulates neurodevelopmental processes, such as migration and axon guidance, by binding to roundabout receptors [5]. KCNIP4 is highly expressed in brain tissue and modulates the activity of voltage-gated potassium channels that contribute to the frequency of slow repetitive firing and back-propagation of action potentials in neurons [6]. LGI2 is a member of the leucine-rich glioma-inactivated (LGI) family [7]. Human LGI proteins play an important role in nervous system development, and variants are related to epilepsy and amyelination [7]. RBPJ is a transcription regulator of Notch signaling, which plays an important role in neurodevelopment [8]. Hassed et al. [9] reported two missense mutations in RBPJ in two independent families affected by Adams-Oliver syndrome 3 (AOS3; OMIM 614814) characterized by vertex scalp defects and terminal limb defects. Interestingly, our patient exhibited haploinsufficiency of RBPJ but did not manifest the characteristic severe vascular and limb defects seen in AOS3. This report will contribute to improve the understanding of the genetic background of proximal 4p deletion syndrome and facilitate accurate genetic counselling, including recurrence risks and prenatal testing options.
ACKNOWLEDGEMENTS
None.
Footnotes
CONFLICTS OF INTEREST
No potential conflicts of interest relevant to this article are reported.
AUTHOR CONTRIBUTIONS
PS and JMA designed the study and wrote the manuscript. JBR and LYK conducted chromosome/FISH experiments and analysis. KCS carried out chromosomal microarray analysis. All authors reviewed the manuscript and approved the final manuscript.
RESEARCH FUNDING
This work was supported by the Soonchunhyang University Research Fund.
REFERENCES
- 1.Battaglia A, Carey JC, South ST. Wolf-Hirschhorn syndrome: a review and update. Am J Med Genet C Semin Med Genet. 2015;169:216–23. doi: 10.1002/ajmg.c.31449. [DOI] [PubMed] [Google Scholar]
- 2.Alesi V, Barrano G, Morara S, Darelli D, Petrilli K, Capalbo A, et al. A previously undescribed de novo 4p15 deletion in a patient with apparently isolated metopic craniosynostosis. Am J Med Genet A. 2011;155A:2543–51. doi: 10.1002/ajmg.a.34201. [DOI] [PubMed] [Google Scholar]
- 3.Lee JS, Kang ES, Huh JW, Yoo JH, Lee YK, Cho HC, et al. A Case of Wolf-Hirschhorn Syndrome with del (4) (p15.2) Korean J Clin Pathol. 1999;19:137–40. [Google Scholar]
- 4.Park SH, Chi JG. Oligomeganephronia associated with 4p deletion type chromosomal anomaly. Pediatr Pathol. 1993;13:731–40. doi: 10.3109/15513819309048260. [DOI] [PubMed] [Google Scholar]
- 5.Unni DK, Piper M, Moldrich RX, Gobius I, Liu S, Fothergill T, et al. Multiple Slits regulate the development of midline glial populations and the corpus callosum. Dev Biol. 2012;365:36–49. doi: 10.1016/j.ydbio.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 6.Holmqvist MH, Cao J, Hernandez-Pineda R, Jacobson MD, Carroll KI, Sung MA, et al. Elimination of fast inactivation in Kv4 A-type potassium channels by an auxiliary subunit domain. Proc Natl Acad Sci U S A. 2002;99:1035–40. doi: 10.1073/pnas.022509299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kegel L, Jaegle M, Driegen S, Aunin E, Leslie K, Fukata Y, et al. Functional phylogenetic analysis of LGI proteins identifies an interaction motif crucial for myelination. Development. 2014;141:1749–56. doi: 10.1242/dev.107995. [DOI] [PubMed] [Google Scholar]
- 8.Imayoshi I, Sakamoto M, Yamaguchi M, Mori K, Kageyama R. Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains. J Neurosci. 2010;30:3489–98. doi: 10.1523/JNEUROSCI.4987-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hassed SJ, Wiley GB, Wang S, Lee JY, Li S, Xu W, et al. RBPJ mutations identified in two families affected by Adams-Oliver syndrome. Am J Hum Genet. 2012;91:391–5. doi: 10.1016/j.ajhg.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]