

Abstract
Delivering polynucleotides into animals has been a major challenge facing their success as therapeutic agents. Given the matured understanding of antibody‐mediated delivery techniques, it is possible to rationally design delivery vehicles that circulate in the blood stream and are specifically delivered into target organs. If the targeting moiety is designed to contain the cargo of an RNAi mediator without impacting its paratope, directed delivery can be achieved. In this article, we review the state of art in delivery technology for RNA mediators and address how this technique could soon be used to enhance the efficacy of the numerous small RNA therapeutic programs currently under evaluation. Copyright © 2010 John Wiley & Sons, Ltd.
This article is categorized under:
-
1
Regulatory RNAs/RNAi/Riboswitches > RNAi: Mechanisms of Action
-
2
RNA Methods > RNA Analyses in Cells
-
3
RNA in Disease and Development > RNA in Disease
RNA interference (RNAi) is a Nobel Prize winning phenomenon that has been developed into an invaluable technology for cell biologists to characterize gene function and is being harnessed for therapeutic development.1 An RNAi mediator is a term that encompasses any molecule capable of inducing RNAi, including short interfering (siRNA) and microRNA (miRNA). These classes of polynucleotides have the ability to target an mRNA in the cell and change its expression levels predominantly by modulating mRNA turnover or translation. Although RNAi mediators are typically active once in the correct cellular compartment, they are intrinsically difficult to get into the cell from an external source because of their susceptibility to nucleases, relatively large molecular weight, negatively charged backbone, and their inability to efficiently penetrate membranes without an aid. While technologies such as lipids and membrane penetrating polypeptides are widely available to aid delivery of RNAi mediators into cells in culture, techniques to efficiently deliver RNAi mediators into animal organs are not as easily obtainable. Delivering RNA into cells in animal tissues is much more complicated as target cells are typically deeply embedded in dense and complex microenvironments.
Although numerous techniques have been developed to systemically deliver RNAi mediators into some of the major organ systems,2, 3, 4, 5 these techniques lack efficiency in specifically targeting particular genes in cell types of interest. Using systemic delivery techniques, several reports suggest over 90% of often costly RNAi mediator is delivered to the wrong location for therapeutic indication.6 The distribution of RNAi mediators to incorrect locations can induce unintended side effects.7,8 It is therefore preferred to direct charge neutralized RNAi mediators to specific cells and organs of interest. In turn, we are focusing this article on techniques being developed for directing the delivery of RNAi mediators.
IN VIVO ORGAN DELIVERY
Delivering RNAi mediators into the liver has been a common theme among many technologies.9, 10, 11, 12, 13, 14 The liver's active filter function allows it to be easily targeted through intravenous injection and the liver is an important site for metastasis, and has several disease targets including hepatitis‐B virus, hepatitis‐C virus, and several genes involved in lipid and glucose metabolism. One of the first successful gene reductions of Apolipoprotein B in the jejunum and liver utilized cholesterol conjugated siRNA.15 Studies utilizing modified siRNA10,16,17 along with liposomal formulations18 continue to be main strategies in liver targeting. Wolfrum et al.19 found that efficient uptake of siRNA conjugates depends on interactions with lipoprotein particles, receptors and transmembrane proteins. Their study showed that high‐density lipoprotein directed siRNA into liver, gut, and kidney whereas low‐density lipoprotein targeted siRNA primarily into the liver. These results illustrate nicely that effective uptake via a common mechanism can be exploited to optimize delivery. In terms of delivery into the respiratory epithelium, unformulated siRNAs were shown to be efficacious against respiratory infections caused by respiratory syncytial virus (RSV)20 and severe acute respiratory syndrome21 in primate and mouse models. Bitko et al.20 found nasally administered siRNA at a dose of 3 mg/kg reduced RSV and Parainfluenza virus (PIV) titers by 99% without inducing an interferon response. However, combinational therapies using an excess of one siRNA over the other had an inhibitory effect, most likely a result of RNAi protein mediator competition. Using 10–40 mg/kg accumulated doses of siRNA in rhesus macaque severe acute respiratory syndrome models, Li et al. were able to reduce coronavirus induced fever, viral titer, and acute diffuse alveoli damage without any signs of siRNA induced toxicity. This study suggested that siRNA exemplifies a new means to combat emerging respiratory infections. Progressing respiratory tract therapies to the clinic, Alnylam has begun phase II trials on siRNA targeting RSV (described in detail in the following text).
Despite the unfriendly environment in the intestine for nucleic acid‐based therapeutics, several groups have developed unique delivery strategies to circumvent these limitations.22, 23, 24, 25, 26 Xiang et al. engineered Escherichia coli to transcribe shRNAs, allowing successful transfer of shRNAs into mammalian cells. Upon oral or intravenous administration, E. coli encoding shRNA against β‐catenin induced silencing in the intestinal epithelium and in human colon cancer xenografts in mice. This study suggests there is a potential for bacterial, trans‐kingdom‐mediated RNAi‐based therapies. In another study aimed at treating inflammatory bowel disease by reducing TNFα and colon inflammation, Zhang et al. directly administered liposomal complexed siRNA in cervicovaginal and rectal mucosa. Single vaginal or rectal administrations reduced corresponding mRNA levels by up to 90%. These liposomal formulations were shown to be nontoxic and did not elicit an interferon response suggesting another means for genetic engineering of the gastrointestinal tract (GI) tract in vivo.
Two significant impediments to the delivery of RNAi to treat central nervous system (CNS) diseases are systemic delivery of therapeutics across the blood–brain barrier and the targeting of drugs to specific tissues within the brain.27,28 Currently, most preclinical and clinical studies on delivery of RNAi to the CNS have utilized invasive intracerebral techniques. Small RNA therapy directed to the brain however will likely require multiple administrations, making simple IV injection the most advantageous route of delivery. There have been some studies that have successfully utilized monoclonal antibodies (mAbs) that bind endogenous transport systems such as the transferrin receptor (TfR) or insulin receptor to target antisense agents and neuropeptides through the blood–brain barrier.29 Using another approach, cationic liposomal delivery of nucleic acids30, 31, 32, 33 caused nonspecific accumulation to the lungs, with no expression in the brain; however, incorporating nucleic acids in the interior of antibody tethered, pegylated neutral liposomes enabled transport into the brain via the TfR.34 Using a similar technique, this time with an antibody against epidermal growth factor receptor (EGFR), Xia et al. saw a 90% increase in the survival time in mice with intracranial brain cancer.35
Direct delivery to the eye paved the way for the first RNAi therapeutic human clinical trials.36,37 Earlier it had been shown to be successful in the form of intravitreal injections in cynomolgus induced monkeys38 and subretinal injections in mice.39 In both studies, siRNA targeting vascular endothelial growth factors (VEGFs) were used to treat age‐related macular degeneration (AMD). This type of delivery is advantageous because a low, single dose of siRNA produces high efficacy with little or no side effects. In primates, the effect of a single siRNA dose injection persisted for 36 days38 and in mice, subretinal injection resulted in 75% reduction in neovascularization when compared with controls.39 Subconjunctival injections have also demonstrated success as 200 nM siRNA targeted to Transforming growth factor beta (TGF‐β) and complexed with a lipid formulation led to an approximately 50% decrease in inflammatory cell infiltration only 48‐h postinjection.40 Human testing have now reached Phase II clinical trials and are further described in the following text.
RNAi mediator drugs in clinical development
The development of polynucleotide technologies as therapeutic agents has led to the Food and Drug Administration (FDA) approval in 1998, of Formivirsen, a 21‐mer phosphothioate against cytomegalovirus retinitis in acquired immune deficiency syndrome patients, giving hope that other drugs with a similar mechanism of action would also be approved. Unfortunately, the ensuing years were disappointing in this regard. Nevertheless, with the advent of RNAi, the number of active clinical trials with therapeutic oligonucleotides has grown enormously. According to some estimates, 80% of all therapeutic targets are not ‘druggable’ with small molecules or protein‐based therapies making RNAi mediators very attractive. Unlike traditional small drug or protein‐based approaches, any disease‐causing gene and any cell type or tissue can potentially be targeted with RNAi mediators if delivery were not a major limitation.
On March of 2009, OPKO Health, Inc. announced that it had decided to terminate the first Phase III clinical study of Bevasiranib,41 for the treatment of wet AMD because of the recommendation of an Independent Data Monitoring Committee (not included in Table 1 for this reason). A report last year from the University of Kentucky suggested that Bevasiranib suppressed blood vessel formation by activating an immune receptor (TLR3),8,42 suggesting the drug's effect may be nonspecific. Despite this, several companies are still actively progressing RNAi technology through clinical trials. Currently, 88.2% of RNAi mediator drugs in development are in preclinical trials, 6.5% are in Phase I clinical trials, 4.3% are in Phase II clinical trials and 1.1% are in Phase III clinical trials. Of these, 88.2% are synthetic nucleic acids. Table 1 shows a listing of the 11 companies that have active RNAi mediator drug programs in clinical trials. We will review what is known on a few of these companies' efforts including Alnylam, Calando, and Santaris.
Table 1.
List of companies with active RNAi mediator drug programs.
| Generic Name | Status | Originator | Indication |
|---|---|---|---|
| NUC‐B100 | Phase I | Nucleonics | Infection, hepatitis‐B virus |
| SPC‐3649 | Phase I | Santaris Pharma | Infection, hepatitis‐C virus |
| RNAi HIV therapy (Benitec 1) | Phase I | Benitec | Infection, HIV/AIDS |
| Tenascin‐C RNAi | Phase I | Senetek | Cancer, brain |
| CALAA‐01 | Phase I | Calando | Cancer, unspecified |
| TD‐101 | Phase I | TransDerm | Pachyonychia congenita |
| ALN‐VSP02 | Phase I | Alnylam | Liver cancer |
| ALN‐RSV01 | Phase II | Alnylam | Infection, respiratory syncytial virus |
| AKIi‐5 | Phase II | Quark | Renal failure |
| RTP‐801i‐14 | Phase II | Quark | Macular degeneration, age‐related, wet |
| AGN‐211745 | Phase II | Allergan | AMD and Choroidal neovascularization (CNV) infection |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Directing Delivery into the Lungs Through Inhalation
Alnylam's siRNA against RSV has entered Phase II clinical trials, and preliminary reports are promising. Alnylam's RNAi therapeutic, ALN‐RSV01, was designed to target the conserved N gene of the RSV genome, critical for the replication of the virus. Since initiating the ALN‐RSV01 therapeutic program in 2005, Alnylam has made rapid progress. Alnylam has licensed intellectual property from the University of South Alabama,43,44 covering RNAi compositions and methods of treating pulmonary viral infections with siRNAs. The company has also formed an exclusive alliance with Kyowa Hakko to develop and commercialize ALN‐RSV01 in Japan and other major markets in Asia. In February 2008, Alnylam announced that it had achieved human proof of concept with ALN‐RSV01, the first for the industry. Results from the company's GEMINI trial with ALN‐RSV01 showed statistically significant antiviral efficacy. GEMINI was a randomized, placebo‐controlled, double‐blind Phase II trial in 88 healthy adult volunteers experimentally inoculated with RSV. Intranasal ALN‐RSV01 administered once a day for 5 days (2 days prior to and 3 days following viral infection) gave a 37.1% reduction in infection rate as measured by plaque assay, and similar significant reductions as measured by polymerase chain reaction (PCR), spin‐enhanced culture and RSV antigen assay. There was a 95% increase in the number of patients remaining free of infection (24/43 on ALN‐RSV01 vs. 12/42 on placebo). Viral load, peak viral load, duration of viral shedding, and mean daily viral load all showed consistent trends favoring an ALNRSV01 treatment effect. There were no significant differences in clinical symptoms. ALN‐RSV01 was safe and well tolerated. In April 2008, Alnylam initiated a Phase II clinical trial to assess the safety and tolerability of aerosolized ALN‐RSV01 versus placebo in adult lung transplant patients naturally infected with RSV. Those receiving ALN‐RSV01 will have the drug administered by inhalation via nebulizer, which is the expected delivery formulation for commercialization. As a secondary objective, this trial will be the first to evaluate the antiviral activity of ALN‐RSV01 in a naturally acquired RSV lower respiratory tract infection.
Liver Targeting of RNAi Mediators
Alnylam has also been actively advancing its RNAi therapeutic, ALN‐VSP02, for the treatment of solid tumors with liver involvement. ALN‐VSP02 targets two key genes each involved in the disease pathway of liver cancer: kinesin spindle protein (KSP) and VEGF. It is thought that the dual‐target approach increases the potential for a significant therapeutic benefit for cancer treatment. In preclinical studies, ALN‐VSP demonstrated the ability to silence both tumor KSP and VEGF and stopped the proliferation of cancer cells. In these studies, ALN‐VSP02 showed a striking reduction in the growth and number of significant tumor masses in the liver. Data reported at the 23rd Annual Meeting of the International Society for Biological Therapy of Cancer demonstrated robust efficacy in an orthotopic liver tumor model, including the inhibition of tumor growth as measured by serum levels of alpha‐fetoprotein (AFP) and a significant survival benefit. Specific silencing of KSP and VEGF for ALN‐VSP02 was demonstrated, and an RNAi mechanism of action toward both target gene mRNAs was confirmed. In late 2008, an investigational new drug (IND) application for ALN‐VSP02 was submitted to the U.S. FDA and accepted. A Phase I clinical trial titled ‘Dose Escalation Trial to Evaluate the Safety, Tolerability, Pharmacokinetics of Intravenous ALN‐VSP02 in patients with advanced solid tumors with liver involvements’ is enrolling an estimated 58 people (http://www.cancer.gov/drugdictionary/?CdrID=642288 ).
Directed Delivery to Transferrin Receptor Containing Cells
In June 2008, Calando Pharmaceuticals dosed the first patient with CALAA‐01, a transferrin‐targeted nanoparticle siRNA that silences the M2 subunit of ribonucleotide reductase.45 This medication consisting of four components includes a synthetic nonchemically modified siRNA, a cyclodextran polymer, a stabilizing agent and a targeting agent that form an approximately 100 nm particle. The targeting agent contains the human transferrin protein. The cationic polymer (cyclodextran) interacts electrostatically with anionic siRNA to assemble into nanocomplexes below approximately 100 nm in diameter that protect the siRNA from nuclease degradation in serum. The siRNA‐containing nanocomplexes are targeted to cells that over‐express the TfR. Upon reaching a target cell, transferrin binds to TfRs on the cell surface and the siRNA‐containing nanocomplex enters the cell by endocytosis. Inside the cell, chemistry built into the polymer achieves unpackaging of the siRNA from the nanocomplex, permitting it to function via RNA interference.46 The surface‐modifying agents have terminal adamantane groups that form inclusion complexes with the cyclodextrin and contain polyethylene glycol (PEG) to endow the particles with properties that prevent aggregation, enhance stability, and enable systemic administration. Ligands to cell surface receptors can be covalently attached to the adamantane‐PEG modifier, enabling the siRNA‐containing particles to be targeted to tissues of interest. Workers at Caltech and Children's Hospital located in Los Angeles have also used nanoparticles made with Calando's delivery system and siRNA against the EWS‐Fli1 fusion gene to prevent tumor growth in a disseminated murine model of Ewing's sarcoma.47,48 Treatments were made using female Non‐obese diabetic/severe combined immunodeficiency (NOD/SCID) mice injected with luciferase‐expressing TC71 (human Ewing's sarcoma) cells at siRNA doses of 2.5 mg/kg. Doses were administered weekly twice for 4 weeks. Each treatment group contained 10 mice. Safety evaluations are ongoing, as well as additional efficacy studies in animal models. Please refer to our list of publications for more information.
While most therapeutic microRNAs are in the preclinical stages, Santaris recently began clinical trials for SPC3649, an antisense‐based molecule against miR‐122 for the treatment of Hepatitis‐C virus. Santaris is using interspersed locked nucleic acid combinations along with a phosphorothioate backbone which acts by sequestering miRNA in the cytoplasm.49 Despite this opportunity, there will be numerous challenges that an miRNA‐based drug would have to overcome as every miRNA can target multiple genes and influence tens or hundreds of biological pathways. The number of safety concerns that will have to be addressed will be significant. Another issue will be choosing the right miRNA to target. Recently, there has been a growing body of evidence that suggest a link between the miRNA profile of a cell and its oncogenic potential. A more lucid picture of the role miRNAs play in cancer will likely come forward once efforts to examine the cancer genome reveal the number of mutations in miRNAs and their protein‐coding gene target sequences.
Targeted Delivery Technology Using Antibody Conjugates
Conjugation of siRNA molecules to a targeting molecule is a promising means of delivery.50, 51, 52, 53, 54, 55 The conjugate is usually formed through a covalent attachment of the targeting molecule to the passenger strand of the RNAi mediator, so as not to disrupt silencing activity. Conjugation is an attractive approach because of the wide variety of potential targeting moieties, including nucleic acid or peptide aptamers, cell permeable peptides, antibodies, and natural molecules such as cholesterol which would be expected to have low toxicity.52,56,57 Antibody‐based siRNA delivery is an active area of investigation. In the immunoliposome approach, siRNA‐containing liposomes are chemically coupled to antibodies to form immunoliposome‐siRNA complexes.58, 59, 60 After injection, the complexes bind to surface antigens and the liposomes fuse with the cell membrane and deliver siRNA into cells. An alternative antibody‐based approach uses modified antibody‐fusion proteins, which have distinct antigen‐ and RNA‐binding domains.61,62 In theory, after the siRNA‐fusion protein complex is injected into the animal it binds the target cells and is subsequently internalized by receptor‐mediated endocytosis. The RNAi mediator should then dissociate from the targeting delivery vehicle, exit the endosome, enter the cytoplasm and enter the RNAi silencing protein complex where it is used to induce gene silencing.63, 64, 65, 66 For all of this to take place, a fine balance of siRNA affinity for the carrier is needed so that siRNA can be released upon entering the endosome.
Cancer or Disease Targeting Using Antibody Conjugates
Current efforts at antibody‐based siRNA delivery can be classified into three categories: immunoliposomes, recombinant antibody fusions, and antibody‐carrier conjugates (targeted transport technology described below). Recombinant fusion immunoconjugates (r‐FIs) have distinct antigen‐ and RNA‐binding domains fused into one protein unit. Prior to delivery the RNAi mediator is mixed with the r‐FI and forms a non‐covalent complex with the RNA‐binding domain of the fusion complex. The siRNA‐r‐FI complex is now in a form to be injected into an animal model and bind target cells. After subsequent receptor‐mediated endocytosis, the siRNA dissociates from the r‐FI and interacts with the RNA‐induced silencing complex (RISC) assembly.61 In the research paper by Song and colleagues, a gene encoding the nucleic acid binding protein was fused to the gene encoding a Fab fragment of an antibody directed against the human immunodeficiency virus (HIV) gp160 (env) protein. The Fab‐protamine fusion protein is then loaded with siRNA and used to selectively deliver the siRNA into cells expressing the env gene. Recombinant antibody–protamine fusion to deliver siRNA into cells expressing the integrin lymphocyte function associated antigen‐1 (LFA‐1) protein67 has also shown efficacy. Using a single chain antibody specific against CD7 fused to an oligo‐9‐arginine, Shankar's group has demonstrated T cell specific siRNA delivery and effective inhibition of HIV using antiviral siRNA.62 These and other reports demonstrate the promise of directed siRNA delivery into specific blood cells.68
While a great deal of exciting progress toward in vivo delivery has been achieved using the recombinant immunoconjugate approach developed, there are still several barriers which limit its development: (1) the small size of the RNA‐binding domain limits the amount of siRNA than can associate, (2) the process for producing recombinant modified antibody fusions often provides low yields of the desired product, (3) the recombinant antibody‐fusion production process is difficult to scale‐up (elimination of required denaturation/refolding steps would improve production efficiency and scalability), and (4) the release of functional siRNA from antibodies lacks any control.
TARGETED DELIVERY TECHNOLOGY
Antibody–carrier conjugates have made all commercially available antibodies potential delivery platforms. By chemically conjugating more than one positively charged RNAi mediator binding domain onto an antibody we have increased RNAi loading capacity to at least 15 RNAi mediators per conjugate enabling larger quantities of RNAi mediators to enter the cytoplasm. As a model system for many studies Bioo Scientific has performed conjugations with the Human Epidermal growth factor receptor 2 (HER2‐erb‐B‐2) antibody. The erbB family of receptor tyrosine kinase proteins includes EGFR, HER2 (erbB2), HER3 (erbB3), and HER4 (erbB4).69,70 These cell membrane receptors are composed of an extracellular ligand‐binding domain, a transmembrane lipophilic domain, and an intracellular tyrosine kinase domain. Phosphorylation of the tyrosine kinase domain followed by homodimerization between different receptors of the same family leads to protein activation that promotes signaling cascades including ERK1/2 that promote cell growth.71 We also studied the uptake specificity of siRNA in cell culture and in an animal tumor model. In order to determine if HER2 antibody conjugate delivers siRNA into target cells we incubated the antibody conjugates with siRNA at varying molar ratios and applied the complex to cells that were plated onto cover slips. In these studies, HER2 antibody conjugates were shown to deliver siRNA into cell lines expressing HER2 only while cell lines lacking receptors for the antibody lacked siRNA cellular uptake suggesting the HER2 antibody conjugate and siRNA they carry can enter cells.
Validation of targeted delivery in an animal model system
The in vivo siRNA delivery efficacy of immunoconjugates was tested by systemic injection into NOD/SCID mice to silence Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) expression in tumor cells over‐expressing target antigens. We performed the animal tests using the HER2 antibody conjugate with tumors made with cell lines expressing HER2 (SKBR3) or a breast cancer cell line containing significantly low levels of Her2 receptor (MCF7). NOD/SCID mice injected subcutaneously with either 1 million SKBR3 or MCF7 cells into flanks were monitored postinjection for tumor formation. Once the tumors reached sufficient size, siRNA treatment using the HER2 antibody conjugate delivery vehicle was commenced by mixing HER2 antibody conjugate with either GAPDH siRNA or a negative‐control siRNA and injected tumors twice with an 8‐h interval between injections. Seventy‐two hours following the injections the mice were sacrificed; tumors were removed, processed and analyzed for knockdown using a GAPDH enzyme‐linked immunosorbent assay (ELISA). The data illustrated in Figure 1 suggests that the HER2 antibody conjugate may deliver its siRNA load and knockdown GAPDH in cells that only express HER2.
Figure 1.
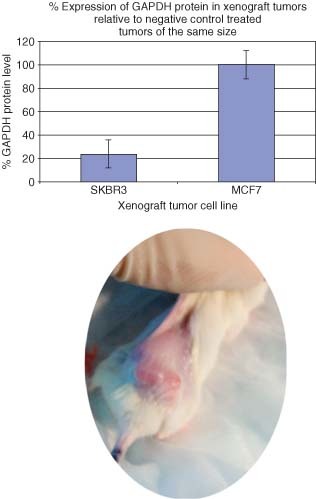
The analysis of knockdown of GAPDH in SKBR3 or MCF7 xenograft tumor cells using receptor‐mediated siRNA delivery. We added 50‐ng GAPDH siRNA or a scrambled negative‐control siRNA with 100 ng of HER2 antibody conjugate. This complex was incubated for 30 min at room temperature and then injected directly into xenograft tumors made using either SKBR3 or MCF7 cell lines. Seventy‐two hours postinjection, the animals were sacrificed, and tumors were removed and analyzed using an in‐house GAPDH ELISA assay. The relative gene reduction compared with scrambled negative controls was determined. The circular panel illustrates a xenograft tumor that was generated in NOD/SCID mice before delivery of the T3 conjugate.
Targeted Delivery Technology Using Peptides and Immuno‐liposomal Conjugates
The effective use of RNAi mediators as a therapy currently depends on the availability of a delivery vehicle that can be systemically administered and targeted to particular cells while limiting side effects. Naked RNAi mediators when delivered to the bloodstream have half‐lives in the order of minutes because of renal clearance.72 In addition, while most serum nucleases do not recognize double stranded RNA, some serum RNAses can degrade siRNA. To overcome these challenges RNAi mediators have been mixed with antibody‐containing liposomes or peptides for targeted IV delivery in animal models of cancer. To work effectively, these systemic, yet targeted siRNA delivery systems should have the following characteristics. The RNAi mediator must be protected in the blood stream to prolong half‐life, transport must occur across endothelial walls, there must be a specific binding to the cell plasma membrane, active endocytosis in the cell, and the RNAi mediator must be active in the target cell to elicit the desired biological response. While currently no available RNAi mediator delivery technology espouses all these characteristics, several studies have come close.
To develop a tumor selective delivery system, Schiffelers et al.73 attached an arginine‐glycine‐aspartic acid (RGD) peptide ligand to the distal end of PEG as a means to target tumor neovasculature‐expressing integrins. The PEGylated polyethyleneimine (PEI) was complexed with siRNA against VEGF‐R2. Tail vein injection into tumor‐bearing mice showed selective uptake and inhibition of tumor angiogenesis and growth rate. The complex was shown to improve serum stability over 12 h when compared with unformulated RNAi mediator. Using a similar approach, Kim et al.74 utilized PEI‐graft‐PEG‐folate to deliver siRNA targeting green fluorescent protein (GFP) to folate receptor over‐expressing keratin‐forming tumor cell line (KB) cells. Results demonstrated successful target reduction in the cytoplasm. Building on the premise of integrin‐targeted RNAi mediator delivery, Peer et al.75 developed liposomal based β7 integrin‐targeted siRNA vehicles to target specific leukocyte subsets in gut inflammation. Using stabilized unilamellar vesicles (ULV), they covalently attached hyaluronan to palmitoylphosphatidylethanolamine (DPPE) in the ULV, which stabilized these particles during systemic circulation and endocytosis. A monoclonal antibody against β7 integrins was covalently attached to the hyaluronan. This systemic application silenced CyD1 in leukocytes and reversed induced colitis in mice by suppressing leukocyte proliferation and T helper cell 1 cytokine expression. Covalent conjugations as performed by Peer et al. are typically performed through reactive sites carried on the head groups of phospholipids with the use of a crosslinker or other intermediary. These conjugations may be performed with homo‐bifunctional or hetero‐bifunctional crosslinking reagents, carbodiimides, reductive amination, N‐hydroxysuccinimide (NHS) esters, activation of carboxylates, or non‐covalent use of an avidin–biotin interaction. Stability and in vivo use of these complexes often depend on the size of each liposomal particle, the amount of antibody in the reaction and the mole amount of lipid in the bilayer formulation. The use of liposomal‐antibody conjugates was demonstrated by Pirollo et al.58,59 to specifically deliver siRNAs to both primary and metastatic tumors, including prostate, pancreatic, and breast tumors after systemic administration. Pirollo et al. utilized a TfRscFV‐liposomal complex to deliver an anti‐HER‐2 siRNA. The liposome encapsulated the siRNA by a cationic 1,2‐dioleoyl‐trimethylammonium‐propane (DOTAP) dioleoyl phosphatidylethanolamine (DOPE) lipid and was labeled with the anti‐TfRscFV targeting moiety. TfR levels have been shown to correlate with the proliferative nature of tumor cells and recycle during internalization in rapidly dividing cancer cells,76,77 contributing to the uptake of TfR‐targeted liposomal complexes.
Once siRNA has reached its intended target it must be released from the endosome to knockdown a gene of interest. This has often limited the efficacy of many studies. The incorporation of pH sensitive components into liposomes has been developed to try to enhance the efficiency of liposomal payload by exploiting the fact that endosomal compartments are generally acidic in nature. Destabilization of targeted liposomes following acidification has been enhanced by: (1) inclusion of (DOPE) which undergoes a lamellar to hexagonal phase transition at low pH, releasing captured liposomal components and (2) pH sensitive peptides that attach to the liposome at neutral pH and bind adjacent bilayers at acidic pH. Utilizing the latter approach, Chen et al.78 and Aoki et al.79 have developed histidine–lysine polymers that were shown to disrupt the endosomes and increase transfection efficiency over unmodified cationic liposomes. Using liposomes containing siRNA against HER2 with labeled polymers adapted from these studies, Pirollo et al.59 demonstrated that tumor specific delivery resulted in near‐complete knockdown of HER2 expression in tumors. They further showed size reduction of PANC‐1 xenograft tumors using this technology.
CONCLUSION
The field of siRNA is like no other as the rapid progression in technology has propelled it from in vitro cell culture studies to advanced clinical trials in only 6 years. Data from preclinical programs suggest that siRNA therapeutics have the potential for treating disease. Many of the preclinical studies have led to ongoing clinical trials with encouraging preliminary results. Despite this progress, delivery remains the largest obstacle for improving siRNA therapy. Understanding how RNA‐based therapies can be delivered into tissues affected by disease will be essential if they are to be widely successful in treating disease. Most current clinical trials utilize direct injection into organs or targeted administration of unmodified siRNA or siRNA with minor chemical modifications. While direct injection or targeted administrations remain the best option for siRNA delivery today, it is unlikely that siRNA‐based therapeutics will be limited to these approaches. Broad yet targeted systemic clinical treatment will likely be the future for safe and efficacious human application. To move in this direction, multiple delivery technologies will be required that will evolve based on the unique challenges each organ or disease may pose.
We propose that antibody‐mediated delivery vehicles will lead the way for directed siRNA targeting. This assumption is based on previous successes that have been obtained using antibodies in treating disease. From a practical standpoint, antibodies can be designed to target any cellular surface antigen in a specific fashion. Antibodies can also be engineered so they do not have an impact on the immune system (humanized). Antibody drug conjugates such as mylotarg are already approved by the FDA and are used to treat human disease. These factors as well as others make antibody‐based methods for drug delivery very attractive.
RELATED WIREs ARTICLES
REFERENCES
- 1. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double‐stranded RNA in Caenorhabditis elegans . Nature 1998, 391: 806–811. [DOI] [PubMed] [Google Scholar]
- 2. Han SE, Kang H, Shim GY, Kim SJ, Choi HG, Kim J, Hahn SK, Oh YK. Cationic derivatives of biocompatible hyaluronic acids for delivery of siRNA and antisense oligonucleotides. J Drug Target 2009, 17: 123–132. [DOI] [PubMed] [Google Scholar]
- 3. Howard KA. Delivery of RNA interference therapeutics using polycation‐based nanoparticles. Adv Drug Deliv Rev 2009, 61: 710–720. [DOI] [PubMed] [Google Scholar]
- 4. Tseng YC, Mozumdar S, Huang L. Lipid‐based systemic delivery of siRNA. Adv Drug Deliv Rev 2009, 61: 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang C, Newsome JT, Mewani R, Pei J, Gokhale PC, Kasid UN. Systemic delivery and pre‐clinical evaluation of nanoparticles containing antisense oligonucleotides and siRNAs. Methods Mol Biol 2009, 480: 65–83. [DOI] [PubMed] [Google Scholar]
- 6. Gao S, Dagnaes‐Hansen F, Nielsen EJ, Wengel J, Besenbacher F, Howard KA, Kjems J. The effect of chemical modification and nanoparticle formulation on stability and biodistribution of siRNA in mice. Mol Ther 2009, 17: 1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kariko K, Bhuyan P, Capodici J, Ni H, Lubinski J, Friedman H, Weissman D. Exogenous siRNA mediates sequence‐independent gene suppression by signaling through toll‐like receptor 3. Cells Tissues Organs 2004, 177: 132–138. [DOI] [PubMed] [Google Scholar]
- 8. Cho WG, Albuquerque RJ, Kleinman ME, Tarallo V, Greco A, Nozaki M, Green MG, Baffi JZ, Ambati BK, De Falco M, et al. Small interfering RNA‐induced TLR3 activation inhibits blood and lymphatic vessel growth. Proc Natl Acad Sci U S A 2009, 106: 7137–7142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Morrissey DV, Blanchard K, Shaw L, Jensen K, Lockridge JA, Dickinson B, McSwiggen JA, Vargeese C, Bowman K, Shaffer CS, et al. Activity of stabilized short interfering RNA in a mouse model of hepatitis B virus replication. Hepatology 2005, 41: 1349–1356. [DOI] [PubMed] [Google Scholar]
- 10. Morrissey DV, Lockridge JA, Shaw L, Blanchard K, Jensen K, Breen W, Hartsough K, Machemer L, Radka S, Jadhav V, et al. Potent and persistent in vivo anti‐HBV activity of chemically modified siRNAs. Nat Biotechnol 2005, 23: 1002–1007. [DOI] [PubMed] [Google Scholar]
- 11. Zimmermann TS, Lee AC, Akinc A, Bramlage B, Bumcrot D, Fedoruk MN, Harborth J, Heyes JA, Jeffs LB, John M, et al. RNAi‐mediated gene silencing in non‐human primates. Nature 2006, 441: 111–114. [DOI] [PubMed] [Google Scholar]
- 12. Alshamsan A, Haddadi A, Incani V, Samuel J, Lavasanifar A, Uludag H. Formulation and delivery of siRNA by oleic acid and stearic acid modified polyethylenimine. Mol Pharm 2009, 6: 121–133. [DOI] [PubMed] [Google Scholar]
- 13. Andersen MO, Howard KA, Kjems J. RNAi using a chitosan/siRNA nanoparticle system: in vitro and in vivo applications. Methods Mol Biol 2009, 555: 77–86. [DOI] [PubMed] [Google Scholar]
- 14. Subramanian N, Mani P, Roy S, Gnanasundram SV, Sarkar DP, Das S. Targeted delivery of hepatitis C virus‐specific short hairpin RNA in mouse liver using Sendai virosomes. J Gen Virol 2009, 90: 1812–1819. [DOI] [PubMed] [Google Scholar]
- 15. Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, Elbashir S, Geick A, Hadwiger P, Harborth J, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432: 173–178. [DOI] [PubMed] [Google Scholar]
- 16. Manoharan M. RNA interference and chemically modified siRNAs. Nucleic Acids Res Suppl 2003, 3: 115–116. [DOI] [PubMed] [Google Scholar]
- 17. Manoharan M. RNA interference and chemically modified small interfering RNAs. Curr Opin Chem Biol 2004, 8: 570–579. [DOI] [PubMed] [Google Scholar]
- 18. Akinc A, Goldberg M, Qin J, Dorkin JR, Gamba‐Vitalo C, Maier M, Jayaprakash KN, Jayaraman M, Rajeev KG, Manoharan M, et al. Development of lipidoid‐siRNA formulations for systemic delivery to the liver. Mol Ther 2009, 17: 872–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wolfrum C, Shi S, Jayaprakash KN, Jayaraman M, Wang G, Pandey RK, Rajeev KG, Nakayama T, Charrise K, Ndungo EM, et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat Biotechnol 2007, 25: 1149–1157. [DOI] [PubMed] [Google Scholar]
- 20. Bitko V, Musiyenko A, Shulyayeva O, Barik S. Inhibition of respiratory viruses by nasally administered siRNA. Nat Med 2005, 11: 50–55. [DOI] [PubMed] [Google Scholar]
- 21. Li BJ, Tang Q, Cheng D, Qin C, Xie FY, Wei Q, Xu J, Liu Y, Zheng BJ, Woodle MC, et al. Using siRNA in prophylactic and therapeutic regimens against SARS coronavirus in Rhesus macaque. Nat Med 2005, 11: 944–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grillot‐Courvalin C, Goussard S, Huetz F, Ojcius DM, Courvalin P. Functional gene transfer from intracellular bacteria to mammalian cells. Nat Biotechnol 1998, 16: 862–866. [DOI] [PubMed] [Google Scholar]
- 23. Palliser D, Chowdhury D, Wang QY, Lee SJ, Bronson RT, Knipe DM, Lieberman J. An siRNA‐based microbicide protects mice from lethal herpes simplex virus 2 infection. Nature 2006, 439: 89–94. [DOI] [PubMed] [Google Scholar]
- 24. Santel A, Aleku M, Keil O, Endruschat J, Esche V, Fisch G, Dames S, Loffler K, Fechtner, M, Arnold W, et al. A novel siRNA‐lipoplex technology for RNA interference in the mouse vascular endothelium. Gene Ther 2006, 13: 1222–1234. [DOI] [PubMed] [Google Scholar]
- 25. Xiang S, Fruehauf J, Li CJ. Short hairpin RNA‐expressing bacteria elicit RNA interference in mammals. Nat Biotechnol 2006, 24: 697–702. [DOI] [PubMed] [Google Scholar]
- 26. Yang R, Yang X, Zhang Z, Zhang Y, Wang S, Cai Z, Jia Y, Ma Y, Zheng C, Lu Y, et al. Single‐walled carbon nanotubes‐mediated in vivo and in vitro delivery of siRNA into antigen‐presenting cells. Gene Ther 2006, 13: 1714–1723. [DOI] [PubMed] [Google Scholar]
- 27. Agrawal A, Min DH, Singh N, Zhu H, Birjiniuk A, von Maltzahn G, Harris TJ, Xing D, Woolfenden SD, Sharp PA, et al. Functional delivery of siRNA in mice using dendriworms. ACS Nano 2009, 3: 2495–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mathupala SP. Delivery of small‐interfering RNA (siRNA) to the brain. Expert Opin Ther Pat 2009, 19: 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pardridge WM. Intravenous, non‐viral RNAi gene therapy of brain cancer. Expert Opin Biol Ther 2004, 4: 1103–1113. [DOI] [PubMed] [Google Scholar]
- 30. Reimer DL, Zhang Y, Kong S, Wheeler JJ, Graham RW, Bally MB. Formation of novel hydrophobic complexes between cationic lipids and plasmid DNA. Biochemistry 1995, 34: 12877–12883. [DOI] [PubMed] [Google Scholar]
- 31. Huang L, Li S. Liposomal gene delivery: a complex package. Nat Biotechnol 1997, 15: 620–621. [DOI] [PubMed] [Google Scholar]
- 32. Mahato RI, Rolland A, Tomlinson E. Cationic lipid‐based gene delivery systems: pharmaceutical perspectives. Pharm Res 1997, 14: 853–859. [DOI] [PubMed] [Google Scholar]
- 33. Matsui H, Johnson LG, Randell SH, Boucher RC. Loss of binding and entry of liposome‐DNA complexes decreases transfection efficiency in differentiated airway epithelial cells. J Biol Chem 1997, 272: 1117–1126. [DOI] [PubMed] [Google Scholar]
- 34. Huwyler J, Wu D, Pardridge WM. Brain drug delivery of small molecules using immunoliposomes. Proc Natl Acad Sci U S A 1996, 93: 14164–14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xia CF, Zhang Y, Boado RJ, Pardridge WM. Intravenous siRNA of brain cancer with receptor targeting and avidin‐biotin technology. Pharm Res 2007, 24: 2309–2316. [DOI] [PubMed] [Google Scholar]
- 36. Whelan J. Beyond PEGylation. Drug Discov Today 2005, 10: 301. [DOI] [PubMed] [Google Scholar]
- 37. Whelan J. First clinical data on RNAi. Drug Discov Today 2005, 10: 1014–1015. [DOI] [PubMed] [Google Scholar]
- 38. Tolentino MJ, Brucker AJ, Fosnot J, Ying GS, Wu IH, Malik G, Wan S, Reich SJ. Intravitreal injection of vascular endothelial growth factor small interfering RNA inhibits growth and leakage in a nonhuman primate, laser‐induced model of choroidal neovascularization. Retina 2004, 24: 660. [DOI] [PubMed] [Google Scholar]
- 39. Reich SJ, Fosnot J, Kuroki A, Tang W, Yang X, Maguire AM, Bennett J, Tolentino MJ. Small interfering RNA (siRNA) targeting VEGF effectively inhibits ocular neovascularization in a mouse model. Mol Vis 2003, 9: 210–216. [PubMed] [Google Scholar]
- 40. Nakamura H, Siddiqui SS, Shen X, Malik AB, Pulido JS, Kumar NM, Yue BY. RNA interference targeting transforming growth factor‐beta type II receptor suppresses ocular inflammation and fibrosis. Mol Vis 2004, 10: 703–711. [PubMed] [Google Scholar]
- 41. Singerman L. Combination therapy using the small interfering RNA bevasiranib. Retina 2009, 29(Suppl 6): S49–S50. [DOI] [PubMed] [Google Scholar]
- 42. Kleinman ME, Yamada K, Takeda A, Chandrasekaran V, Nozaki M, Baffi JZ, Albuquerque RJ, Yamasaki S, Itaya M, Pan Y, et al. Sequence‐ and target‐independent angiogenesis suppression by siRNA via TLR3. Nature 2008, 452: 591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barik S. Development of gene‐specific double‐stranded RNA drugs. Ann Med 2004, 36: 540–551. [DOI] [PubMed] [Google Scholar]
- 44. Barik S. Treating respiratory viral diseases with chemically modified, second generation intranasal siRNAs. Methods Mol Biol 2009, 487: 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. RNA interference targeting the M2 subunit of ribonucleotide reductase enhances pancreatic adenocarcinoma chemosensitivity to gemcitabine. Oncogene 2004, 23: 1539–1548. [DOI] [PubMed] [Google Scholar]
- 46. Xu L, Huang CC, Huang W, Tang WH, Rait A, Yin YZ, Cruz I, Xiang LM, Pirollo KF, Chang EH. Systemic tumor‐targeted gene delivery by anti‐transferrin receptor scFv‐immunoliposomes. Mol Cancer Ther 2002, 1: 337–346. [PubMed] [Google Scholar]
- 47. Kovar H, Ban J, Pospisilova S. Potentials for RNAi in sarcoma research and therapy: Ewing's sarcoma as a model. Semin Cancer Biol 2003, 13: 275–281. [DOI] [PubMed] [Google Scholar]
- 48. Davis ME. The first targeted delivery of siRNA in humans via a self‐assembling, cyclodextrin polymer‐based nanoparticle: from concept to clinic. Mol Pharm 2009, 6: 659–668. [DOI] [PubMed] [Google Scholar]
- 49. Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’ Nature 2005, 483: 685–689.. [DOI] [PubMed] [Google Scholar]
- 50. Pappas TC, Bader AG, Andruss BF, Brown D, Ford LP. Applying small RNA molecules to the directed treatment of human diseases: realizing the potential. Expert Opin Ther Targets 2008, 12: 115–127. [DOI] [PubMed] [Google Scholar]
- 51. Blackburn WH, Dickerson EB, Smith MH, McDonald JF, Lyon LA. Peptide‐functionalized nanogels for targeted siRNA delivery. Bioconjug Chem 2009, 20: 960–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jeong JH, Mok H, Oh YK, Park TG. siRNA conjugate delivery systems. Bioconjug Chem 2009, 20: 5–14. [DOI] [PubMed] [Google Scholar]
- 53. Wullner U, Neef I, Tur MK, Barth S. Targeted delivery of short interfering RNAs–strategies for in vivo delivery. Recent Pat Anticancer Drug Discov 2009, 4: 1–8. [DOI] [PubMed] [Google Scholar]
- 54. Yu B, Zhao X, Lee LJ, Lee RJ. Targeted delivery systems for oligonucleotide therapeutics. AAPS J 2009, 11: 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zheng X, Vladau C, Zhang X, Suzuki M, Ichim TE, Zhang ZX, Li M, Carrier E, Garcia B, Jevnikar AM, et al. A novel in vivo siRNA delivery system specifically targeting dendritic cells and silencing CD40 genes for immunomodulation. Blood 2009, 113: 2646–2654. [DOI] [PubMed] [Google Scholar]
- 56. Lorenz C, Hadwiger P, John M, Vornlocher HP, Unverzagt C. Steroid and lipid conjugates of siRNAs to enhance cellular uptake and gene silencing in liver cells. Bioorg Med Chem Lett 2004, 14: 4975–4977. [DOI] [PubMed] [Google Scholar]
- 57. Wang XL, Xu R, Lu ZR. A peptide‐targeted delivery system with pH‐sensitive amphiphilic cell membrane disruption for efficient receptor‐mediated siRNA delivery. J Control Release 2009, 134: 207–213. [DOI] [PubMed] [Google Scholar]
- 58. Pirollo KF, Chang EH. Targeted delivery of small interfering RNA: approaching effective cancer therapies. Cancer Res 2008, 68: 1247–1250. [DOI] [PubMed] [Google Scholar]
- 59. Pirollo KF, Rait A, Zhou Q, Hwang SH, Dagata JA, Zon G, Hogrefe RI, Palchik G, Chang EH. Materializing the potential of small interfering RNA via a tumor‐targeting nanodelivery system. Cancer Res 2007, 67: 2938–2943. [DOI] [PubMed] [Google Scholar]
- 60. Pirollo KF, Zon G, Rait A, Zhou Q, Yu W, Hogrefe R, Chang EH. Tumor‐targeting nanoimmunoliposome complex for short interfering RNA delivery. Hum Gene Ther 2006, 17: 117–124. [DOI] [PubMed] [Google Scholar]
- 61. Song E, Zhu P, Lee SK, Chowdhury D, Kussman S, Dykxhoorn DM, Feng Y, Palliser D, Weiner DB, Shankar P, et al. Antibody mediated in vivo delivery of small interfering RNAs via cell‐surface receptors. Nat Biotechnol 2005, 23: 709–717. [DOI] [PubMed] [Google Scholar]
- 62. Kumar P, Ban HS, Kim SS, Wu H, Pearson T, Greiner DL, Laouar A, Yao J, Haridas V, Habiro K, et al. T cell‐specific siRNA delivery suppresses HIV‐1 infection in humanized mice. Cell 2008, 134: 577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tuschl T, Zamore PD, Lehmann R, Bartel DP, Sharp PA. Targeted mRNA degradation by double‐stranded RNA in vitro. Genes Dev 1999, 13: 3191–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Parrish S, Fleenor J, Xu S, Mello C, Fire A. Functional anatomy of a dsRNA trigger: differential requirement for the two trigger strands in RNA interference. Mol Cell 2000, 6: 1077–1087. [DOI] [PubMed] [Google Scholar]
- 65. Zamore PD, Tuschl T, Sharp PA, Bartel DP. RNAi: double‐stranded RNA directs the ATP‐dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 2000, 101: 25–33. [DOI] [PubMed] [Google Scholar]
- 66. Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21‐ and 22‐nucleotide RNAs. Genes Dev 2001, 15: 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Peer D, Zhu P, Carman CV, Lieberman J, Shimaoka M. Selective gene silencing in activated leukocytes by targeting siRNAs to the integrin lymphocyte function‐associated antigen‐1. Proc Natl Acad Sci U S A 2007, 104: 4095–4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liu Z, Winters M, Holodniy M, Dai H. siRNA delivery into human T cells and primary cells with carbon‐nanotube transporters. Angew Chem Int Ed Engl 2007, 46: 2023–2027. [DOI] [PubMed] [Google Scholar]
- 69. Duffy MJ. Predictive markers in breast and other cancers: a review. Clin Chem 2005, 51: 494–503. [DOI] [PubMed] [Google Scholar]
- 70. Ono M, Kuwano M. Molecular mechanisms of epidermal growth factor receptor (EGFR) activation and response to gefitinib and other EGFR‐targeting drugs. Clin Cancer Res 2006, 12: 7242–7251. [DOI] [PubMed] [Google Scholar]
- 71. Meden H, Kuhn W. Overexpression of the oncogene c‐erbB‐2 (HER2/neu) in ovarian cancer: a new prognostic factor. Eur J Obstet Gynecol Reprod Biol 1997, 71: 173–179. [DOI] [PubMed] [Google Scholar]
- 72. Dykxhoorn DM, Lieberman J. The silent revolution: RNA interference as basic biology, research tool, and therapeutic. Annu Rev Med 2005, 56: 401–423. [DOI] [PubMed] [Google Scholar]
- 73. Schiffelers RM, Ansari A, Xu J, Zhou Q, Tang Q, Storm G, Molema G, Lu PY, Scaria PV, Woodle MC. Cancer siRNA therapy by tumor selective delivery with ligand‐targeted sterically stabilized nanoparticle. Nucleic Acids Res 2004, 32: e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kim SH, Mok H, Jeong JH, Kim SW, Park TG. Comparative evaluation of target‐specific GFP gene silencing efficiencies for antisense ODN, synthetic siRNA, and siRNA plasmid complexed with PEI‐PEG‐FOL conjugate. Bioconjug Chem 2006, 17: 241–244. [DOI] [PubMed] [Google Scholar]
- 75. Peer D, Park EJ, Morishita Y, Carman CV, Shimaoka M. Systemic leukocyte‐directed siRNA delivery revealing cyclin D1 as an anti‐inflammatory target. Science 2008, 319: 627–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Daniels TR, Delgado T, Helguera G, Penichet ML. The transferrin receptor part II: targeted delivery of therapeutic agents into cancer cells. Clin Immunol 2006, 121: 159–176. [DOI] [PubMed] [Google Scholar]
- 77. Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The transferrin receptor part I: biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol 2006, 121: 144–158. [DOI] [PubMed] [Google Scholar]
- 78. Chen QR, Zhang L, Luther PW, Mixson AJ. Optimal transfection with the HK polymer depends on its degree of branching and the pH of endocytic vesicles. Nucleic Acids Res 2002, 30: 1338–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Aoki Y, Hosaka S, Kawa S, Kiyosawa K. Potential tumor‐targeting peptide vector of histidylated oligolysine conjugated to a tumor‐homing RGD motif. Cancer Gene Ther 2001, 8: 783–787. [DOI] [PubMed] [Google Scholar]