

Summary
Porcine deltacoronavirus (PDCoV) is a newly emerged enterotropic swine coronavirus that causes enteritis and diarrhoea in piglets. Here, a nested reverse transcription (RT)‐PCR approach for the detection of PDCoV was developed to identify and characterize aetiologic agent(s) associated with diarrhoeal diseases in piglets in South Korea. A PCR‐based method was applied to investigate the presence of PDCoV in 683 diarrhoeic samples collected from 449 commercial pig farms in South Korea from January 2014 to December 2016. The molecular‐based survey indicated a relatively high prevalence of PDCoV (19.03%) in South Korea. Among those, the monoinfection of PDCoV (9.66%) and co‐infection of PDCoV (6.30%) with porcine epidemic diarrhoea (PEDV) were predominant in diarrhoeal samples. The full‐length genomes or the complete spike genes of the most recent strains identified in 2016 (KNU16‐07, KNU16‐08 and KNU16‐11) were sequenced and analysed to characterize PDCoV currently prevalent in South Korea. We found a single insertion‐deletion signature and dozens of genetic changes in the spike (S) genes of the KNU16 isolates. Phylogenetic analysis based on the entire genome and spike protein sequences of these strains indicated that they are most closely related to other Korean isolates grouped with the US strains. However, Korean PDCoV strains formed different branches within the same cluster, implying continuous evolution in the field. Our data will advance the understanding of the molecular epidemiology and evolutionary characteristics of PDCoV circulating in South Korea.
Keywords: full‐length genome, nested RT‐PCR, PDCoV, phylogenetic analysis, prevalence
1. INTRODUCTION
Porcine deltacoronavirus (PDCoV) is a newly emerging enterotropic swine coronavirus that causes acute enteritis in nursing piglets (Jung et al., 2015; Woo et al., 2012). PDCoV infection is characterized by marked villous atrophy in the small intestine, which results in watery diarrhoea and vomiting, leading to dehydration and death in newborn piglets (Jung et al., 2015; Ma et al., 2015). This disease is symptomatically comparable to, but reportedly milder than, other porcine enteric coronavirus diseases caused by transmissible gastroenteritis virus (TGEV) and porcine epidemic diarrhoea virus (PEDV), with lower mortality rates in affected neonatal piglets (Hu et al., 2015). PDCoV belongs to the genus Deltacoronavirus within the family Coronaviridae of the order Nidovirales. PDCoV is a large, enveloped virus possessing a single‐stranded, positive‐sense RNA genome of approximately 25.4‐kb long with a 5′ cap and a 3′ polyadenylated tail, which is the smallest genome size among porcine coronaviruses (de Groot et al., 2011; Woo et al., 2012). The PDCoV genome consists of only six canonical coronaviral genes, except for open reading frame (ORF) 3, in the following conserved order: 5′ untranslated region (UTR)‐ORF1a‐ORF1b‐S‐E‐M‐N‐3′ UTR. ORF1a and 1b occupy the 5′‐proximal two‐thirds of the genome encoding two overlapping viral replicase polyproteins, 1a and 1ab, which are proteolytically processed into mature non‐structural proteins (nsps). The production of polyproteins requires a −1 ribosomal frame shift, which C‐terminally extends polyprotein 1a into polyprotein 1ab during translation of the genomic RNA. The 3′‐proximal last third of the genome codes for the four structural proteins, spike (S), envelope (E), membrane (M) and nucleocapsid (N), as well as two accessory genes, non‐structural gene 6 (NS6) and NS7, located between M and N, and within N, respectively (Lai, Perlman, & Anderson, 2007; Lee & Lee, 2014; Li et al., 2014; Marthaler, Jiang, Collins, & Rossow, 2014; Woo et al., 2012).
PDCoV was first discovered in Hong Kong, China in a territorial surveillance study of coronaviruses in mammals and birds in 2012 (Woo et al., 2012). A PCR‐based survey of diarrhoea samples from pigs in mainland China revealed the prevalence of PDCoV across the country since 2012. This study reported that the monoinfection of PDCoV and coninfection of PDCoV with PEDV are most common in pig herds in China (Song et al., 2015). In February 2014, the presence of PDCoV was first announced in Ohio, United States, in conjunction with diarrhoea outbreaks without other aetiologic agents. Molecular surveillance indicated that this novel coronavirus was present in 20 US states and nearly 80% of the tested samples corresponded to cases of co‐infection of PDCoV with other enteric viral pathogens such as a rotavirus or PEDV (Li et al., 2014; Ma et al., 2015; Marthaler, Jiang, et al., 2014; Marthaler, Raymond, et al., 2014; Wang, Byrum, & Zhang, 2014). Although the origin of PDCoV in the US remains unclear, sequence analyses suggest the possible introduction of a Chinese PDCoV strain into US swine (Li et al., 2014; Marthaler, Jiang, et al., 2014; Wang et al., 2014). However, recent retrospective evaluation revealed that PDCoV antibodies could be detected in archival serum samples collected in 2010, suggesting that the virus may have already existed as early as 2010 (Thachil, Gerber, Xiao, Huang, & Opriessnig, 2015). Shortly after its emergence in the US, PDCoV was also detected in South Korea from two diarrhoea samples independently collected in April and June 2014, which were positive for porcine rotavirus (PRV) and negative for other enteric viruses, respectively (Lee & Lee, 2014). Genetic and phylogenetic analyses showed that the Korean strains are more closely related to the US strains than to the Hong Kong HKU15 strains, suggesting that Korean PDCoV originated from the US (Lee & Lee, 2014). In this study, we aimed to further investigate the prevalence and full‐length genome sequence analysis of PDCoV from clinical cases associated with diarrhoea from Korean swine farms.
2. MATERIALS AND METHODS
2.1. Clinical sample collection
Small intestine or stool specimens (n = 683) were collected from piglets showing acute enteritis and watery diarrhoea from January 2014 through December 2016 (Table S1). A list of the sampling provinces is present in Table S1. Intestinal homogenates were prepared as 10% (wt/vol) suspensions in phosphate‐buffered saline (PBS) using a MagNA Lyser (Roche Diagnostics, Mannheim, Germany) by three repetitions of 15 s at a speed of 8,000 g. Faecal samples were also diluted with PBS to be 10% (wt/vol) suspensions. The suspensions were then vortexed and centrifuged for 10 min at 4,500 × g (Hanil Centrifuge FLETA5, Incheon, South Korea). The clarified supernatants were stored at −80°C for RNA extraction until use.
2.2. RNA extraction and nested reverse transcription (RT)‐PCR
To screen the presence of PDCoV from diarrhoeal faecal or intestinal samples, a nested RT‐PCR approach was developed with the following external and internal primer sets, designed to target the N gene of the PDCoV KNU14‐04 strain (GenBank accession no. KM820765; Lee & Lee, 2014): PDCoV‐N504‐Fwd (5′‐CATCAGCTGCTACCTCTCCG‐3′) and PDCoV‐N504‐Rev (5′‐GGTCTGATTAACGACCGTAT‐3′) for the first round; and PDCoV‐N504‐2nd‐Fwd (5′‐GCCCAGCTCAAGGTTTCAGAGTTG‐3′) and PDCoV‐N504‐2nd‐Rev (5′‐ATGCGAGGATCAGCCATACC‐3′) for the second round. The expected sizes of the amplicons from the primer sets are 504 and 289 bp, respectively. Total RNA was extracted using the TGE/PED detection kit (iNtRON Biotechnology, Seongnam, South Korea) according to the manufacturer's instructions. The concentrations of the extracted RNA were measured using a NanoVue spectrophotometer (GE Healthcare, Piscataway, NJ, USA). The first round of RT‐PCR was performed using 1 μg of RNA, external primers and the HyQ one‐step RT‐PCR kit (SNC, Hanam, South Korea) according to the manufacturer's protocol under the following conditions: reverse transcription at 50°C for 30 min, an initial PCR activation step at 95°C for 15 min, 30 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s and extension at 72°C for 40 s, followed by a final extension step at 72°C for 7 min. For nested PCR, the second round of amplification was conducted using 1 μl of initial PCR product, internal primers and TaKaRa Ex Taq DNA polymerase (TaKaRa, Otsu, Japan) under the following conditions: denaturation at 94°C for 5 min, 30 cycles of denaturation at 94°C for 30 s, annealing at 58°C for 30 s and extension at 72°C for 30 s, followed by a final extension step at 72°C for 10 min.
2.3. Nucleotide sequence analysis
The complete genomes of representative PDCoV field strains, designated KOR/KNU16‐07/2016 and KOR/KNU16‐11/2016, and the entire S gene of KOR/KNU16‐08/2016 were sequenced by the traditional Sanger method. To determine the full‐length genomic sequence of the Korean PDCoV isolates, KNU16‐07 and KNU16‐11, oligonucleotide primers were designed based on the PDCoV KNU14‐04 strain (GenBank accession no. KM820765) to obtain RT‐PCR fragments. The primers were further synthesized based on KNU14‐04 sequences for rapid amplification of cDNA ends (RACE) experiments and nucleotide sequencing (Table S2). Nine overlapping cDNA fragments spanning the entire viral genome were RT‐PCR amplified using gene‐specific primer sets as described above. The 5′ and 3′ ends of the PDCoV genome were determined by RACE as described previously (Lee & Lee, 2013). The individual PCR amplicons were gel‐purified, cloned into the pGEM‐T easy vector (Promega, Madison, WI, USA), and sequenced in both directions using primers for the T7 and SP6 promoters, as well as PDCoV‐specific primers. General procedures for DNA manipulation and cloning were performed according to standard procedures (Sambrook & Russell, 2001). The complete genomic sequences of the KNU16‐07 and KNU16‐11 viruses and the S gene sequence of KNU16‐08 were deposited in the GenBank database under accession numbers KY364365 and KY926512, and KY926511, respectively.
2.4. Multiple alignments and phylogenetic analyses
The sequences of the 38 fully sequenced S genes and 33 complete genomes of PDCoV isolates were independently used in sequence alignments and phylogenetic analyses. The multiple sequence alignments were generated with the ClustalX 2.0 program (Thompson, Gibson, Plewniak, Jeanmougin, & Higgins, 1997), and the percentages of the nucleotide sequence divergences were further assessed using the same software program. Phylogenetic trees were constructed from the aligned nucleotide or amino acid sequences using the neighbour‐joining method and subsequently subjected to bootstrap analysis with 1,000 replicates in order to determine the percentage reliability values of each internal node of the tree (Saitou & Nei, 1987). All figures involving phylogenetic trees were generated using the Mega 4.0 software (Tamura, Dudley, Nei, & Kumar, 2007).
3. RESULTS
3.1. Prevalence of PDCoV in clinical samples from diarrhoeic pigs
Although PDCoV was initially identified in South Korea in 2014, the prevalence of this novel virus in our nation not yet been studied. Moreover, the infection patterns of PDCoV in Korean swine farms currently remain to be determined. To accomplish this, a nested RT‐PCR for detection of PDCoV was developed and employed to screen 684 diarrhoeal samples of pigs collected from 449 Korean swine farms from January 2014 to December 2016. Clinical samples that tested positive for PDCoV by RT‐PCR were further subjected to sequencing analysis. The results of our BLAST search (http://blast.ncbi.nlm.nih.gov/Blast.cgi ) for the nested RT‐PCR amplicons showed that the sequences obtained in this study have 100% nucleotide (nt) identity with reference strains, confirming the detection of PDCoV.
Of the 684 porcine faecal/intestinal samples tested in our PDCoV prevalence survey, 130 (19.03%) were PDCoV positive; 66 of 684 (9.66%) samples that were negative for TGEV, PEDV, and PRV were confirmed as PDCoV monoinfections (Table 1). PDCoV/PEDV co‐infections were most common (6.30%); 43 of 130 PDCoV‐detected samples were found to be positive for PEDV. PDCoV/PRV co‐infections were also detected (2.78%); PRV was identified in 19 of 130 PDCoV‐positive samples. Two of the 130 PDCoV‐positive samples were confirmed as a multiple‐infection case with both PEDV and PRV (PDCoV/PEDV/PRV). None of the PDCoV‐positive samples were positive for TGEV.
Table 1.
Detection rates of Porcine deltacoronavirus (PDCoV) in diarrhoeal faecal/intestinal samples from pigs in South Korea, 2014–2016
| Year | Virus | Sample | Positive | Positive rate (%) |
|---|---|---|---|---|
| 2014 | PDCoV | 229 | 12 | 5.24 |
| PDCoV+PEDVa | 229 | 17 | 7.42 | |
| PDCoV+PRV | 229 | 4 | 1.75 | |
| PDCoV+PEDV+PRV | 229 | 1 | 0.44 | |
| 2015 | PDCoV | 215 | 27 | 12.56 |
| PDCoV+PEDV | 215 | 19 | 8.84 | |
| PDCoV+PRV | 215 | 8 | 3.72 | |
| PDCoV+PEDV+PRV | 215 | 1 | 0.47 | |
| 2016 | PDCoV | 239 | 27 | 11.3 |
| PDCoV+PEDV | 239 | 7 | 2.93 | |
| PDCoV+PRV | 239 | 7 | 2.93 | |
| PDCoV+PEDV+PRV | 239 | 0 | 0 | |
| 2014–2016 | PDCoV | 683 | 66 | 9.66 |
| PDCoV+PEDV | 683 | 43 | 6.3 | |
| PDCoV+PRV | 683 | 19 | 2.78 | |
| PDCoV+PEDV+PRV | 683 | 2 | 0.29 |
Porcine enteric viruses (TGEV, PEDV and PRV) were detected by TGE/PED and PRV RT‐PCR Kits (iNtRON Biotechnology).
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
3.2. Complete genomic characterizations of Korean PDCoV strains
The full‐length genome sequences of KOR/KNU16‐07/2016 and KOR/KNU16‐11/2016, representative epidemic strains from an intestinal sample collected in 2016, were determined to investigate their molecular characteristics. The entire genomic sequence of KNU16‐07 is 25,422 nt in length, excluding the 3′ poly(A) tail and has a typical PDCoV organization consisting of a 539‐nt 5′ UTR, a 18,803‐nt replicase gene (nt 540–11,414 for 1a and nt 11,414–19,342 for 1b), a 3,483‐nt S gene (nt 19,324–22,806), a 252‐nt E gene (nt 22,800–23,051), a 654‐nt M gene (nt 23,044–23,697), a 285‐nt NS6 (nt 23,697–23,981), a 1,029‐nt N gene (nt 24,002–25,030), a 603‐nt NS7 gene (nt 24,096–24,698) and a 392‐nt 3′ UTR. The complete genome sequence and organization of KNU16‐11 were nearly identical to that of the KNU16‐07 strain, except for the presence of a 3‐nt deletion in the S gene.
Sequence analysis showed that the KNU16 isolates are most closely related to the Korean KNU14‐04 strain and the US strains, sharing nucleotide identities of 99.3%–99.8% with 35–161 nucleotide differences at the complete genome level (Table 2; Table S3). In contrast, the full‐length genomes of the PDCoV KNU16 isolates had the lowest nucleotide identity to Chinese strains including two Hong Kong HKU15 strains, ranging from 98.4% to 99.2% (Table 2; Table S3). Comparing the complete genomes of the Korean KNU16 series to the Hong Kong HKU15 strains, all contained additional seven nucleotides (ACATGGG) at position 1, corresponding to the 5′ UTR (Fig. S1). This insertion was reported in other global strains, including Chinese strains (Lee & Lee, 2014). The Korean KNU16 strains possessed a 3‐nt insertion (IN) signature in both the 3′ UTR (nt positions 25,049–25,051) and the S gene (at nt positions 19,476–19,478 or amino acid [aa] position 52 in S) compared to those of the Hong Kong strain HKU15‐155, which is commonly present in the Korean KNU14‐04 and all US strains (Figs. S2 and S3; Lee & Lee, 2014). In addition to the IN pattern, the S genes of two KNU16‐08 and KNU16‐11 strains had a unique a 3‐nt (1‐aa) deletion (DEL) at nt positions 22,752–22,754 (aa position 1,114 in S) compared to the sequences of other reference strains, and dozens of aa mutations were identified in a putative S ectodomain and endodomain of either or both strains (Fig. S3). These genetic drifts led to a relatively low aa identity to other Korean strains, ranging from 97.8% to 98.7% (Table 2).
Table 2.
Pairwise comparisons of the nucleotide sequences of the full‐length genomes and the S protein genes of the KNU16 and reference Porcine deltacoronavirus (PDCoV) strains
| Strain | Nucleotide/amino acid identity (%) (No. of nucleotide/amino acid differences) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HKU15‐155/China/2012 | HKU15‐44/China/2012 | JS/China/2014 | AH2004/China/2015 | CHJXNI2/China/2015 | NH/China/2016 | IL2768/USA/2014 | IN2847/USA/2014 | KY4813/USA/2014 | NE3579/USA/2014 | KNU14‐04/Korea/2014 | KNU16‐07/Korea/2016 | KNU16‐08/Korea/2016 | KNU16‐11/Korea/2016 | |
| HKU15‐155/China/2012 | 99 (230) | 99.3 (174) | 99.1 (220) | 99 (238) | 99.1 (213) | 99.1 (212) | 99.1 (212) | 99.1 (206) | 99.1 (210) | 99.1 (219) | 99.1 (221) | ND | 98.6 (337) | |
| HKU15‐44/China/2012 | 99 (11) | 99 (247) | 99.1 (216) | 98.7 (314) | 98.8 (285) | 98.9 (272) | 98.9 (277) | 98.9 (268) | 98.9 (271) | 98.8 (281) | 98.8 (283) | ND | 98.4 (400) | |
| JS/China/2014 | 99.3 (8) | 99.2 (9) | 99 (250) | 99.3 (153) | 99.2 (203) | 99.2 (186) | 99.2 (189) | 99.2 (182) | 99.2 (187) | 99.2 (193) | 99.2 (197) | ND | 98.7 (311) | |
| AH2004/China/2015 | 98.7 (14) | 99 (11) | 98.7 (14) | 98.7 (315) | 98.8 (300) | 98.9 (264) | 98.9 (265) | 98.9 (258) | 98.9 (265) | 98.9 (269) | 98.9 (273) | ND | 98.4 (392) | |
| CHJXNI2/China/2015 | 98.9 (12) | 98.8 (13) | 99.3 (8) | 98.5 (17) | 98.9 (267) | 98.9 (255) | 98.9 (260) | 99 (252) | 98.9 (258) | 98.9 (265) | 98.9 (269) | ND | 98.5 (373) | |
| NH/China/2016 | 99.3 (8) | 99.2 (9) | 99.4 (6) | 98.8 (13) | 99.1 (10) | 99 (249) | 99 (250) | 99 (241) | 99 (250) | 99 (254) | 98.9 (256) | ND | 98.5 (373) | |
| IL2768/USA/2014 | 98.9 (12) | 99.3 (7) | 99.3 (8) | 99.2 (9) | 98.9 (12) | 99.1 (10) | 99.8 (35) | 99.8 (26) | 99.9 (25) | 99.8 (37) | 99.8 (39) | ND | 99.3 (160) | |
| IN2847/USA/2014 | 98.7 (15) | 99.1 (10) | 99 (11) | 98.9 (12) | 98.7 (15) | 98.8 (13) | 99.1 (3) | 99.8 (31) | 99.8 (28) | 99.8 (40) | 99.8 (44) | ND | 99.3 (161) | |
| KY4813/USA/2014 | 98.7 (14) | 99.2 (9) | 99.1 (10) | 99 (11) | 98.7 (14) | 98.9 (12) | 99.8 (2) | 99.5 (5) | 99.8 (27) | 99.8 (35) | 99.8 (35) | ND | 99.3 (154) | |
| NE3579/USA/2014 | 98.8 (13) | 99.3 (8) | 99.2 (9) | 99.1 (10) | 98.8 (13) | 99 (11) | 99.9 (1) | 99.6 (4) | 99.7 (3) | 99.8 (40) | 99.8 (40) | ND | 99.3 (161) | |
| KNU14‐04/Korea/2014 | 98.9 (12) | 99.3 (7) | 99.3 (8) | 99.2 (9) | 98.9 (12) | 99.1 (10) | 100 (0) | 99.7 (3) | 99.8 (2) | 99.9 (1) | 99.8 (48) | ND | 99.3 (157) | |
| KNU16‐07/Korea/2016 | 98.2 (20) | 98.7 (15) | 98.6 (16) | 98.5 (17) | 98.2 (20) | 98.4 (18) | 99.3 (8) | 99 (11) | 99.1 (10) | 99.2 (9) | 99.3 (8) | ND | 99.3 (171) | |
| KNU16‐08/Korea/2016 | 97.7 (26) | 98.3 (19) | 98.1 (22) | 98 (23) | 97.7 (26) | 97.9 (24) | 98.7 (14) | 98.7 (15) | 98.6 (16) | 98.7 (15) | 98.7 (14) | 98.1 (22) | ND | |
| KNU16‐11/Korea/2016 | 97.6 (27) | 98.1 (22) | 97.8 (25) | 97.7 (26) | 97.5 (29) | 97.6 (27) | 98.5 (17) | 98.6 (16) | 98.3 (19) | 98.4 (18) | 98.5 (17) | 97.8 (25) | 99.5 (5) | |
The per cent full‐length genome identity (nucleotides) was shown in the upper right, and the per cent of S protein identity (amino acid) was presented in the lower left.
ND, Not determined.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
3.3. Phylogenetic analysis
For studies to establish the genetic relationships involved, phylogenetic analyses were carried out using the nucleotide sequences of the full‐length genome and the S gene of the KNU16 isolates, which were determined in this study and those available from GenBank, together with selected coronavirus sequences from other genera (Figure 1). The phylogeny based on the complete genome sequence indicated that the KNU16 strains were clustered into the deltacoronavirus group, forming a clade with nine other swine‐origin PDCoV strains, which was distinct from deltacoronaviruses of avian‐origin (Figure 1a).
Figure 1.
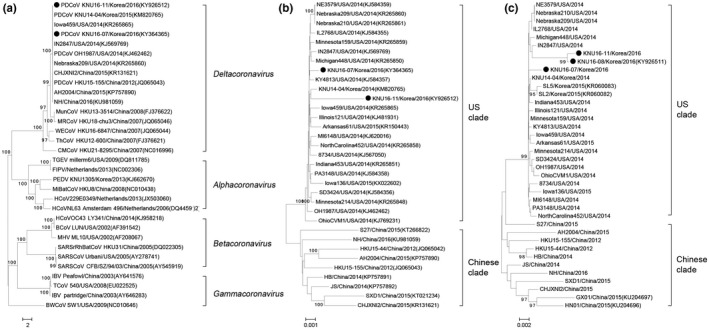
Phylogenetic analysis based on the full‐length genome sequences of four coronavirus genera (Alphacoronavirus, Betacoronavirus, Gammacoronavirus and Deltacoronavirus) (a) and the nucleotide sequences of the complete genomes (b) and the S genes (c) of PDCoV strains. Multiple sequence alignments were performed using the ClustalX program, and the phylogenetic tree was constructed from the aligned nucleotide sequences using the neighbour‐joining method. Numbers at each branch represent bootstrap values greater than 50% of 1000 replicates. Names of the strains, countries, years of isolation, GenBank accession numbers and clades proposed in this study are shown. The porcine deltacoronavirus (PDCoV) isolates identified in this study are indicated by solid circles. Scale bars indicate nucleotide substitutions per site
In agreement with previous studies (Marthaler, Raymond, et al., 2014; Wang, Hayes, Sarver, Byrum, & Zhang, 2016), phylogenetic analysis based on the PDCoV complete genomes exhibited a clear separation between US and Chinese strains. Of which, all Korean PDCoV strains identified in 2014 (KNU14‐04) and in 2016 (KNU16‐07 and KNU16‐11) were grouped within the US PDCoV clade; however, they were situated in different branches (Figure 1b). Furthermore, a subsequent phylogenetic tree reconstructed from the full‐length S gene showed the same grouping structure as the complete genome‐based tree (Figure 1c). Similarly, the KNU16 strains still belonged to the US clade but were located on a separate branch with the Korean prototype strain KNU14‐04 and the other isolates (SL2 and SL5) reported in 2015. Altogether, the phylogenetic data suggest that the current strains detected in 2016 differ from the first emergent virus in South Korea.
4. DISCUSSION
The family Coronaviridae is included in the order Nidovirales along with the families Arteriviridae, Roniviridae and Mesoniviridae, infecting a broad range of species including humans, other mammals, rodents and birds (Cavanagh, 1997; Mayo, 2002; Siddell & Snijder, 2008; Spaan et al., 2005). Coronaviruses are divided into four genera: Alpha‐, Beta‐, Gamma‐ and Deltacoronavirus, based on the phylogenetic distances of highly conserved domains (Woo et al., 2012). Although birds are the primary reservoirs for gamma‐ and deltacoronaviruses, these avian‐origin coronaviruses may jump and adapt to some mammalian species including pigs. Indeed, a study to investigate the presence of deltacoronavirus identified a novel PDCoV genome from the faecal samples of pigs in Hong Kong, China in 2012 (Woo et al., 2012). In early 2014, PDCoV was detected and reported in the US and South Korea (Lee & Lee, 2014; Wang et al., 2014). Soon thereafter, the pathogenicity and pathogenesis of this novel virus were elucidated in gnotobiotic and conventional piglets under different experimental conditions (Chen et al., 2015; Jung et al., 2015; Ma et al., 2015). In South Korea, pigs have been prone to various diarrhoeic diseases for years, predominantly by PEDV (Lee, 2015; Lee & Lee, 2014). However, little is known regarding whether PDCoV has been involved with outbreaks of diarrhoea at domestic pig farms since its emergence. To provide insight into the epidemiological status of PDCoV in South Korea, it is necessary to develop assays for the detection of this newly emerged enteric virus. In this study, therefore, a nested RT‐PCR method for PDCoV detection was established and utilized for a prevalence survey of PDCoV‐associated diarrhoeic diseases.
The high prevalence of PDCoV infections in diarrhoeal samples from pigs was confirmed in South Korea, which is similar to the results of two independent studies from the US and China (Marthaler, Raymond, et al., 2014; Song et al., 2015). RT‐PCR screening of pig diarrhoeal samples collected from January 2014 to December 2016 revealed the presence of PDCoV monoinfection (9.66%) as well as co‐infection with PDCoV/PEDV (6.30%) and PDCoV/PRV (2.78%). Like China, PDCoV/PEDV co‐infection was the most common type of co‐infection detected in South Korea, rather than PDCoV/PRV co‐infection, which was more prevalent in US pig herds (Marthaler, Raymond, et al., 2014). Further study is needed to elucidate the interactions between PDCoV and PEDV or PDCoV and PRV. In the US, PDCoV infection was reported to be associated with significant mortality rates in the field (Ma et al., 2015; Marthaler, Jiang, et al., 2014; Song et al., 2015; Wang et al., 2014). Similarly, a recent survey study revealed that a high number of piglet deaths occurring during an outbreak in China involved PDCoV infections and symptoms of severe diarrhoea (Song et al., 2015). In contrast to the US and China, PDCoV infection appears to be less prevalent in South Korea, and PDCoV strains circulating in South Korea may not typically cause the severity and mortality of PDCoV‐associated disease. Because PED is the most common enteric disease in South Korea and PDCoV causes clinical signs indistinguishable from those of PEDV, it is possible that PDCoV‐induced disease might be neglected and draw less attention for investigation. Some coronaviruses can retain some potential to infect different animal species, and subsequently can be adapted and maintained in the host by exploiting or sharing various cell surface components or other undefined factors (Su et al., 2016). As PDCoV has a non‐swine ancestor and is not swine‐borne, a potential interspecies transmission of deltacoronavirus may have occurred between wild birds or small mammals and domestic pigs (Ma et al., 2015; Woo et al., 2012). Although pigs may have initially served as a susceptible host, PDCoV may not yet be fully adapted to pigs and appears to continue to undergo genetic drift to become more completely adapted to pigs (Jung, Hu, & Saif, 2017). Intriguingly, the recently identified Korean PDCoV strains contained a unique S INDEL signature at the N‐terminus and C‐terminus of S, respectively, and several amino acid substitutions have emerged at the external and cytosolic internal domains of the S protein. Moreover, the KNU16 strains were found to be phylogenetically different from other Korean strains, implying that potential adaptation to the natural host may be ongoing.
In summary, a nested RT‐PCR assay was designed and applied for the detection of PDCoV in diarrhoeic samples from pigs. As PCR development is not the objective of this study, and PCR is not evaluated, it is not suggested be stated as a stand‐alone point in conclusion. A PCR‐based survey indicated that PDCoV monoinfection and co‐infection with PEDV or PRV were almost equally present in diarrhoeal pigs from South Korea. However, the detection rate of PDCoV in South Korea was lower than that reported in the US and China. Genetic and phylogenetic analyses indicate that the Korean isolates have a close evolutionary relationship with US strains and that the virus continues to evolve and adapt to its host in the field.
Supporting information
ACKNOWLEDGEMENT
This research was supported by a fund (Project Code No. Z‐1543056‐2016‐17‐01) by Research of Animal and Plant Quarantine Agency, South Korea.
Jang G, Lee K‐K, Kim S‐H, Lee C. Prevalence, complete genome sequencing and phylogenetic analysis of porcine deltacoronavirus in South Korea, 2014–2016. Transbound Emerg Dis. 2017;64:1364–1370. 10.1111/tbed.12690
REFERENCES
- Cavanagh, D. (1997). Nidovirales: A new order comprising Coronaviridae and Arteriviridae. Archives of Virology, 142, 629–633. [PubMed] [Google Scholar]
- Chen, Q. , Gauger, P. , Stafne, M. , Thomas, J. , Arruda, P. , Burrough, E. , … Zhang, J. (2015). Pathogenicity and pathogenesis of a United States porcine deltacoronavirus cell culture isolate in 5‐day‐old neonatal piglets. Virology, 482, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot, R. J. , Baker, S. C. , Baric, R. , Enjuanes, L. , Gorbalenya, A. E. , Holmes, K. V. , … Ziebuhr, J. (2011). Coronaviridae In King A. M. Q., Adams M. J., Carstens E. B., & Lefkowitz E. J. (Eds.), Virus taxonomy: Ninth report of the international committee on taxonomy of viruses (pp. 806–828). Oxford: Elsevier. [Google Scholar]
- Hu, H. , Jung, K. , Vlasova, A. N. , Chepngeno, J. , Lu, Z. , Wang, Q. , & Saif, L. J. (2015). Isolation and characterization of porcine deltacoronavirus from pigs with diarrhea in the United States. Journal of Clinical Microbiology, 53, 1537–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, K. , Hu, H. , Eyerly, B. , Lu, Z. , Chepngeno, J. , & Saif, L. J. (2015). Pathogenicity of 2 porcine deltacoronavirus strains in gnotobiotic pigs. Emerging Infectious Diseases, 21, 650–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, K. , Hu, H. , & Saif, L. J. (2017). Calves are susceptible to infection with the newly emerged porcine deltacoronavirus, but not with the swine enteric alphacoronavirus, porcine epidemic diarrhea virus. Archives of Virology, 162, 2357–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, M. C. , Perlman, S. , & Anderson, L. J. 2007: Coronaviridae In Knipe D. M., Howley P. M., Griffin D. E., Martin M. A., Lamb R. A., Roizman B. & Straus S. E. (Eds.), Fields virology (pp. 1305–1336). 5th ed Philadelphia, PA, USA: Williams, Lippincott, & Wilkins. [Google Scholar]
- Lee, C. (2015). Porcine epidemic diarrhea virus: An emerging and re‐emerging epizootic swine virus. Virology Journal, 12, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y. N. , & Lee, C. (2013). Complete genome sequence of a novel porcine parainfluenza virus 5 isolate in Korea. Archives of Virology, 158, 1765–1772. [DOI] [PubMed] [Google Scholar]
- Lee, S. , & Lee, C. (2014). Complete genome characterization of Korean porcine deltacoronavirus strain KOR/KNU14‐04/2014. Genome Announcements, 2, e01191–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, G. , Chen, Q. , Harmon, K. M. , Yoon, K. J. , Schwartz, K. J. , Hoogland, M. J. , … Zhang, J. (2014). Full‐length genome sequence of porcine deltacoronavirus strain USA/IA/2014/8734. Genome Announcements, 2, e00278–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Y. , Zhang, Y. , Liang, X. , Lou, F. , Oglesbee, M. , Krakowka, S. , & Li, J. (2015). Origin, evolution, and virulence of porcine deltacoronaviruses in the United States. MBio, 6, e00064–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthaler, D. , Jiang, Y. , Collins, J. , & Rossow, K. (2014). Complete genome sequence of strain SDCV/USA/Illinois121/2014, a porcine deltacoronavirus from the United States. Genome Announcements, 2, e00218–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthaler, D. , Raymond, L. , Jiang, Y. , Collins, J. , Rossow, K. , & Rovira, A. (2014). Rapid detection, complete genome sequencing, and phylogenetic analysis of porcine deltacoronavirus. Emerging Infectious Diseases, 20, 1347–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo, M. A. (2002). A summary of taxonomic changes recently approved by ICTV. Archives of Virology, 147, 1655–1656. [DOI] [PubMed] [Google Scholar]
- Saitou, N. , & Nei, M. (1987). The neighbor‐joining method: A new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 4, 406–425. [DOI] [PubMed] [Google Scholar]
- Sambrook, J. , & Russell, D. W. (2001). Molecular cloning: A laboratory manual, 3rd ed Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Siddell, S. , & Snijder, E. J. (2008). An introduction to nidovirus In Perlman S., Gallagher T., & Snijder E. J. (Eds.), Nidoviruses (pp. 1–13). Washington, DC: ASM Press. [Google Scholar]
- Song, D. , Zhou, X. , Peng, Q. , Chen, Y. , Zhang, F. , Huang, T. , … Tang, Y. (2015). Newly emerged porcine deltacoronavirus associated with diarrhoea in swine in China: Identification, prevalence and full‐length genome sequence analysis. Transboundary and Emerging Diseases, 62, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaan, W. J. M. , Cavanagh, D. , deGroot, R. J. , Enjuanes, L. , Gorbalenya, A. E. , Snijder, E. J. , & Walker, P. J. 2005: Nidovirales In Virus taxonomy. Classification and nomenclature of viruses. Fauquet C. M., Mayo M. A., Maniloff J., Desselberger U. & Ball L. A. (Eds.), San Diego, CA: Academic Press. [Google Scholar]
- Su, S. , Wong, G. , Shi, W. , Liu, J. , Lai, A. C. , Zhou, J. , … Gao, G. F. (2016). Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends in Microbiology, 24, 490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. , Dudley, J. , Nei, M. , & Kumar, S. (2007). MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Molecular Biology and Evolution, 24, 1596–1599. [DOI] [PubMed] [Google Scholar]
- Thachil, A. , Gerber, P. F. , Xiao, C. T. , Huang, Y. W. , & Opriessnig, T. (2015). Development and application of an ELISA for the detection of porcine deltacoronavirus IgG antibodies. PLoS ONE, 10, e0124363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J. D. , Gibson, T. J. , Plewniak, F. , Jeanmougin, F. , & Higgins, D. G. (1997). The ClustalX windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research, 25, 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Byrum, B. , & Zhang, Y. (2014). Detection and genetic characterization of deltacoronavirus in pigs, Ohio, USA, 2014. Emerging Infectious Diseases, 20, 1227–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Hayes, J. , Sarver, C. , Byrum, B. , & Zhang, Y. (2016). Porcine deltacoronavirus: Histological lesions and genetic characterization. Archives of Virology, 161, 171–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P. C. , Lau, S. K. , Lam, C. S. , Lau, C. C. , Tsang, A. K. , Lau, J. H. , … Yuen, K. Y. (2012). Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. Journal of Virology, 86, 3995–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials