

Abstract
The ability to control cellular and viral gene expression, either globally or selectively, is central to a successful viral infection, and it is also crucial for the host to respond and eradicate pathogens. In eukaryotes, regulation of message stability contributes significantly to the control of gene expression and plays a prominent role in the normal physiology of a cell as well as in its response to environmental and pathogenic stresses. Not surprisingly, emerging evidence indicates that there are significant interactions between the eukaryotic RNA turnover machinery and a wide variety of viruses. Interestingly, in many cases viruses have evolved mechanisms not only to evade eradication by these pathways, but also to manipulate them for enhanced viral replication and gene expression. Given our incomplete understanding of how many of these pathways are normally regulated, viruses should be powerful tools to help deconstruct the complex networks and events governing eukaryotic RNA stability. Copyright © 2010 John Wiley & Sons, Ltd.
This article is categorized under:
-
1
RNA Turnover and Surveillance > Turnover/Surveillance Mechanisms
-
2
RNA in Disease and Development > RNA in Disease
The virus–host interaction is akin to an evolutionary arms race; viruses continually evolve and fine‐tune mechanisms to exploit the cellular machinery for their own replication, while the host counter‐attacks using a multitude of strategies to detect and destroy foreign invaders. Many such ‘molecular battles’ are waged to control gene expression, as commandeering this machinery is crucial to the success of all viral infections. Given that the regulation of cellular gene expression is orchestrated in large measure by changes in messenger RNA (mRNA) stability, viruses must interface with the cellular RNA decay pathways to control the levels of cellular and viral RNAs. In addition, the cellular RNA destruction machinery likely represents a formidable obstacle to viral genomic and/or mRNA accumulation and integrity. Therefore, while viral RNAs can be targeted for turnover by the host machinery, in many cases viruses have evolved to circumvent and even subvert these pathways to facilitate their own replication. This review will highlight both these themes in relation to the variety of cellular regulators and effectors controlling the ultimate fate of RNA in an infected cell. We will focus on how viruses are influenced by the basal mRNA decay machinery, as well as pathways governing mRNA quality control. Although an extensive body of literature also exists for microRNA and small interfering RNA‐mediated regulation of viral and cellular gene expression during infection, this topic is reviewed elsewhere in this journal and is thus not covered herein.
DEADENYLATION
Cellular mRNAs are protected at their termini by a 5′ 7‐methylguanosine cap and a 3′ non‐templated poly(A) tail, which facilitate translation and restrict access of exonucleases. Within the nucleus, deadenylases trim poly(A) tails for mRNA export and participate in the destruction of non‐coding or aberrant transcripts.1 In the cytoplasm, poly(A) tail removal is the first and often the rate‐limiting step of general mRNA turnover and is usually required both for subsequent decapping and degradation of the message body2 (Figure 1). Several deadenylase complexes have been identified, including Ccr4‐Caf1‐Not1, PAN2‐PAN3, and PARN.3 While to date no viruses have been shown to directly interfere with specific deadenylases, several examples exist of viral RNAs evading the general deadenylation process.
Figure 1.
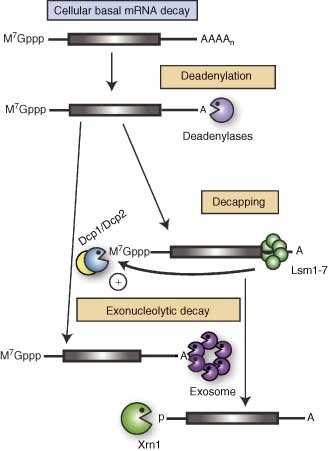
Basal pathways of cellular mRNA decay. Degradation of normal cellular mRNA initiates with removal of the poly(A) tail by one of the cellular deadenylases. Subsequently, the Lsm1–7 protein complex binds the 3′ untranslated region (UTR) of the deadenylated messages and stimulates decapping by the Dcp2 enzyme and its activator protein Dcp1. The message body is subject to 3′→5′ exonucleolytic decay by the exosome or 5′→3′ decay by Xrn1.
Alphaviruses such as Sindbis virus (SINV) and Venezuelan equine encephalitis virus (VEEV) are positive‐sense single‐stranded RNA [(+)RNA] viruses whose genomic RNAs are both capped and polyadenylated, resembling cellular mRNAs. Consequently, to avoid degradation, SINV and VEEV RNAs have evolved sequences that can stall deadenylation4 (Figure 2(a)). A combination of conserved sequences in the 3′ untranslated region (UTR) of the viral genomes act in cis to repress deadenylation in mosquito cell extracts, presumably by mediating interaction with a protective cellular factor.4 A 38‐kDa mosquito protein binds both the SINV and the VEEV 3′ UTRs and can be specifically titrated away in the presence of excess viral UTR sequences, leading to decreased viral RNA stability. Interestingly, removal of the 38‐kDa protein from SINV RNA leads to increased binding of a 32‐kDa mosquito protein. This suggests a mechanism whereby the viral UTRs may be stabilized by displacing a deadenylation‐promoting factor via preferential binding to a protective factor. The identity of these two cellular proteins and their conservation across species is currently unknown. However, in the future their characterization will likely provide insight both into how both cellular mRNA deadenylation and viral RNA stabilization are orchestrated.
Figure 2.
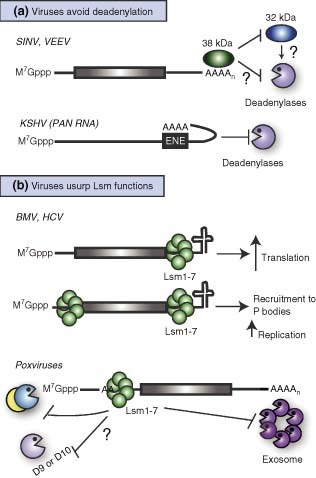
Viral circumvention or utilization of cellular deadenylation and decapping pathways. (a) The RNAs of Sindbis virus (SINV) and Venezuelan equine encephalitis virus (VEEV) contains elements in the 3′ UTR that recruit a cellular 38‐kDa protein and block deadenylation, presumably by preventing binding of a 32‐kDa deadenylation‐promoting factor. The PAN RNA of Kaposi's sarcoma‐associated herpesvirus contains an element (ENE) that interacts in cis with the poly(A) tail thus preventing deadenylase access. (b) The RNA genomes of Brome mosaic virus (BMV) and hepatitis C virus (HCV) can be bound by Lsm proteins in their 3′ and/or 5′ UTRs. This enhances their translation, as well as facilitates replication, possibly through recruitment of the RNAs to P bodies. Poxviruses transcripts have polyadenine sequences within their 5′ UTRs, which bind Lsm1–7. Lsm binding prevents decapping (and presumably 5′→3′ decay), as well as 3′→5′ degradation of the messages.
A second mechanism of protection from deadenylation is used by a highly abundant non‐coding nuclear RNA of Kaposi's sarcoma‐associated herpesvirus (KSHV), termed PAN (Figure 2(a)). PAN contains a 79‐nt element (ENE) near its 3′ end that acts post‐transcriptionally to stabilize the RNA and promote its nuclear retention.5 The ENE forms a stem‐loop structure containing a U‐rich loop. This loop makes intramolecular contacts with the PAN poly(A) tail and blocks deadenylation, presumably in part by preventing access of deadenylases to the RNA.6 However, a simple U‐rich stretch is not sufficient to recapitulate the effects of PAN‐ENE,6 and mutations in the stem region of PAN‐ENE can also abolish its function.7 These observations suggest that the entire structure is required for efficient interaction and potentially for the recruitment of additional protective factors. As the function of PAN remains unknown, at present the importance of the PAN‐ENE in KSHV infection cannot be assessed. The fact that the decay kinetics of PAN are different from those of cytoplasmic polyadenylated RNAs has led to the speculation that PAN RNA degradation, particularly at the stage of deadenylation, may be carried out by a dedicated nuclear pathway that normally disposes of incorrectly processed RNAs.6 Thus, the study of PAN RNA protection from nuclear degradation may provide information as to how polyadenylation and deadenylation are carried out in the nucleus of mammalian cells and how they contribute to nuclear RNA quality control.
THE Lsm COMPLEX
As deadenylation is completed, a ring‐shaped heptameric complex of Sm‐like proteins (Lsm1–7) binds to the 3′ end of the message and recruits the cellular decapping enzymes Dcp1 and Dcp2, which promote cap hydrolysis8, 9, 10, 11 (Figure 1). The Lsm complex is highly conserved in eukaryotes, and in yeast it has been shown to associate with several additional components of the RNA decay machinery, including decapping activators and the 5′→3′ exonuclease Xrn1.10, 11, 12, 13, 14 While the role of the Lsm proteins in eukaryotes is closely linked to mRNA degradation, several viruses use this complex instead to facilitate viral replication and, surprisingly, to enhance viral RNA stability.
The Lsm complex has been shown to participate in both viral replication and translation of Brome mosaic virus (BMV)15, 16, 17 and hepatitis C virus (HCV)18 (Figure 2(b)). Like those of all other (+)RNA viruses, the genomes of BMV and HCV are used first as mRNAs for protein production, and subsequently as templates for replication. These two processes are considered mutually exclusive and often require different cis‐acting sequences. The genome of BMV consists of three RNA segments that are 5′ capped, but terminate in a 3′ tRNA‐like structure rather than a poly(A) tail. Although plants are its natural host, BMV can also replicate in yeast, which has greatly facilitated genetic dissection of host factors involved in its replication cycle. A mutagenesis screen in yeast identified Lsm1p as a host factor required for BMV RNA replication.15 Subsequent studies showed that Lsm1p, together with other components of the Lsm complex, is also required for efficient BMV genomic RNA translation elongation, but not initiation.17 While Lsm‐stimulated BMV translation requires multiple regions of the viral RNAs, the effect of Lsm on BMV RNA replication is specifically dependent on 3′ UTR elements.16,17 Thus, partially overlapping signals appear to participate in the seemingly opposing activities of translational enhancement and redirection of RNAs from the translational pool into replication complexes. Notably, Lsm proteins do not regulate the translation of the BMV coat protein from a fourth subgenomic RNA segment, RNA4, which is generated during BMV replication, suggesting that the effects of Lsm proteins on BMV RNA translation and replication are tightly linked.17
As mentioned before, the Lsm complex is similarly important for the HCV lifecycle, suggesting that the observations on the role of Lsm in BMV replication and translation may generalize to many (+)RNA viruses. Indeed, siRNA‐mediated knockdown of Lsm1 and its co‐activators PatL1 and Rck/p54 decreases HCV intracellular RNA levels, translation, and viral particle production.18 Using gel shift assays, the Lsm1–7 ring was shown to interact with select HCV 5′ and 3′ UTR sequences necessary for translation and replication, suggesting that it may play a direct role in modulating these processes during infection.
It has been suggested that the Lsm proteins may direct BMV RNAs to replication complexes by recruiting them to P bodies, which are sites of RNA storage and/or decay19 (Figure 2(b)). Because P bodies are thought to hold mRNAs in a translationally repressed state,20,21 in principle they could be ideal sites for viral genome replication.19 Surprisingly, when the BMV and HCV 3′ tRNA‐like structure are replaced with a poly(A) tail, the Lsm complex is no longer required for viral RNA accumulation and translation.16, 17, 18 One hypothesis is that these viruses co‐opt Lsm to compensate for the RNA stabilizing functions normally provided by polyadenylation and its associated poly(A) binding protein (PABP). However, association with PABP should not result in RNA recruitment to P bodies, as PABP is not a component of P bodies.22 In addition, the mRNAs associated with P bodies are generally believed to be deadenylated.23 Therefore, it is not clear that PABP and Lsm association could substitute for each other in this system. Further studies are therefore needed to delineate how and why the Lsm complex is required for viral RNA replication only in the absence of polyadenylation.
Cellular Lsm proteins may also play a role in the stability of mRNAs of poxviruses, which are large DNA viruses that replicate in the cytoplasm (Figure 2(b)). Transcripts from orthopoxviridae, particularly those expressed later in infection, often contain non‐templated adenosine stretches of 5–40 nucleotides within their 5′ UTRs, thought to be generated by polymerase stuttering.24 5′ adenosine tracts of at least 10 nt were shown to promote mRNA stabilization in cells and in cytoplasmic extracts by inhibiting both decapping and 3′→5′ exonucleolytic decay of the RNA.25 Although these adenosine stretches bind PABP, titrating away PABP with excess poly(A) RNA does not affect the stability of the 5′ adenosine tract‐containing transcripts. Their stabilization instead correlates with direct binding of the Lsm1–7 complex.25 Poxviruses encode two decapping proteins, D9 and D10, which contribute to the global cellular mRNA turnover that occurs during poxvirus infection. However, D9/D10 can also target viral transcripts and may restrict expression of early viral genes.26, 27, 28 Thus, Lsm proteins may participate in the protection of late viral messages from the viral decapping proteins, although it has yet to be directly proven that this complex protects viral messages during infection or that it blocks D9/D10 decapping activity.25
It is notable that the virus‐associated roles of the Lsm complex in mRNA stabilization, enhanced translation, and replication are at odds with its established cellular function in activating mRNA decapping to facilitate message degradation. While it is formally possible that these viruses are manipulating the Lsm complex to induce novel activities, a more likely explanation is that the Lsm proteins have multifaceted roles in cellular gene expression, which are being revealed and exploited by viruses.
5′→3′ mRNA DECAY
Decapping exposes the mRNA 5′ end, triggering rapid exonucleolytic destruction of the message body predominantly via the 5′→3′ nuclease Xrn1 (Figure 1). Viruses lacking a 5′ cap might therefore be particularly susceptible to Xrn1‐mediated degradation. Indeed, Xrn1 was initially discovered in the yeast Saccharomyces cerevisiae for its ‘Superkiller’ (Ski) phenotype: loss of Xrn1 resulted in cell death as a consequence of increased levels of the toxin‐encoding uncapped M satellite double‐stranded RNA (dsRNA) of the L‐A virus.29 In addition, both in yeast30 and in the plant Nicotiana benthamiana,31 Xrn1 and its homolog Xrn4 prevent the genomic RNA of Tomato bushy stunt virus from accumulating (Figure 3(a)). These exonucleases also reduce the levels of viral RNA intermediates necessary for recombination, a process that some RNA viruses exploit to repair their genomes and increase diversity.32
Figure 3.
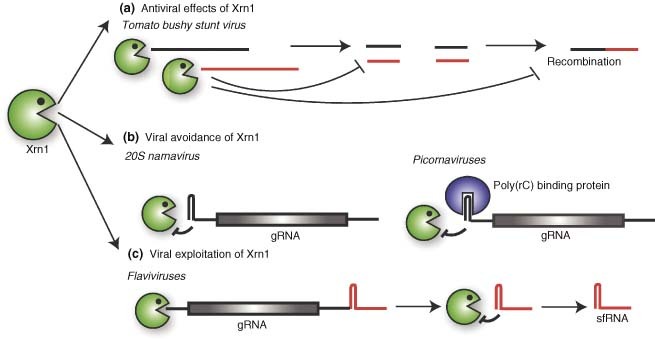
Viral interactions with Xrn1. (a) During infection with Tomato bushy stunt virus, Xrn1 can prevent the accumulation of viral RNA and of viral RNA intermediates that serve as substrates for recombination. (b) Some viruses prevent Xrn1 degradation of their uncapped genomic RNAs (gRNA) by having highly structured sequences in the 5′ UTR, as in the case of 20S narnavirus, or by recruiting protein complexes to this region, as in the case of Picornaviruses. (c) Flaviviral RNAs contain Xrn1 blocking sequences in their 3′ UTR. Xrn1‐mediated degradation of the RNAs up to the blocking sequence results in the generation of a functional subgenomic RNA, sfRNA.
How do RNA viruses avoid Xrn1‐mediated degradation? Some protect their RNAs by encoding cis elements that can physically block Xrn1. The yeast 20S RNA narnavirus, for example, has a highly structured G‐rich motif in its 5′ region that can block Xrn1 activity, thereby likely enhancing the 20S virus persistence in yeast laboratory strains33 (Figure 3(b)). In contrast, poliovirus (PV) recruits a ribonucleoprotein (RNP) complex including poly(rC) binding proteins to a 5′ cloverleaf RNA structure34 (Figure 3(b)). Mutations in this structure and consequent loss of the RNP adversely affect PV RNA stability, possibly as a consequence of increasing susceptibility to Xrn1‐mediated degradation.35 This strategy is probably common to many other members of the Picornaviridae family, as the 5′ cloverleaf structure is highly conserved.36
Interestingly, some viruses instead exploit the activity of Xrn1 (Figure 3(c)). Flaviviruses such as West Nile and Dengue viruses may appropriate Xrn1 to produce a 3′ subgenomic RNA (termed sfRNA) that plays an essential role in viral pathogenicity in both cell culture and mice.37 These viruses have a highly structured sequence (stem loop II or SLII) within the 3′ UTR of their genome that is necessary for the production of the sfRNA. Depletion of Xrn1 prevents production of sfRNA, suggesting that sfRNA is a product of Xrn1‐mediated degradation of the viral genomic RNA upstream of SLII. In turn, this indicates that the SLII element can stall Xrn1 activity. Because no other Xrn1‐blocking element has been described in mammalian cells, the SLII element will likely prove a valuable tool to study Xrn1‐mediated degradation both in the context of viral infections and in uninfected cells.
3′→5′ mRNA DECAY
The exosome, a multisubunit protein complex with 3′→5′ exonucleolytic activity, is the major driver of RNA degradation from the 3′ end (Figure 1). In the cytoplasm, this complex participates in mRNA turnover after deadenylation, whereas in the nucleus it plays an essential role in RNA processing and quality control.38 Little is currently known about the interaction of the exosome with viruses. However, the zinc‐finger antiviral protein (ZAP), an important mediator of cellular response to retroviruses,39 alphaviruses,40 and filoviruses,41 has been shown to bind the hRrp46 component of the exosome and to recruit the complex to viral mRNAs to promote their degradation.42 Depletion of the exosome using siRNAs directed against the hRrp41 and hRrp46 subunits reduces turnover of reporter RNAs by ZAP, suggesting that exosome activity is an integral component of ZAP function. ZAP binds viral RNAs in a sequence‐specific manner, perhaps explaining why this protein is not a broad‐spectrum viral inhibitor.43 However, the absence of significant homology between the various viral sequences bound by ZAP implicates RNA structure as the primary determinant for recognition. An important next step will be to directly test whether exosome depletion rescues viral replication defects in ZAP‐expressing cells.
These findings suggest a role for the exosome in controlling viral infection. In contrast, no evidence currently exists for viruses directly co‐opting this complex. Nonetheless, given the widespread roles for the exosome in RNA quality control and turnover, viruses presumably activate this complex at least indirectly when triggering turnover of cellular mRNAs. For example, infection by select herpes and coronaviruses results in a global destruction of host messages.44, 45, 46, 47, 48 The exosome, as well as other RNA turnover pathways, may contribute to degradation of full‐length or partially degraded mRNAs in these cases.
mRNA QUALITY CONTROL
Cells are equipped with specific mechanisms for the recognition and disposal of defective mRNAs generated by errors introduced during transcription and RNA processing or as a result of mutations in the DNA sequence. The best characterized of these pathways is nonsense‐mediated decay (NMD), which targets mRNAs harboring a premature stop codon (PTC) (Figure 4). The core NMD effectors are the Upf proteins, which associate with prematurely terminating mRNAs and trigger their rapid destruction. Current models for NMD activation in human cells suggest that Upf1 is recruited to these aberrant mRNAs during the initial round of translation via interactions with the eRF1 and eRF3 translation termination factors, whereupon other effectors of NMD (including Upf2, Upf3, and Smg proteins) join the complex.49 The aberrant mRNA is then cleaved endonucleolytically near the PTC by the NMD factor Smg6, and the message body is degraded by cellular exonucleases50,51 (Figure 4). Two features can mark mRNAs for destruction via the NMD pathway. In human cells, one important feature is the presence of exon–exon junctions downstream of the termination codon, as determined by the retention of exon‐junction complexes (EJCs) at these sites. Normally EJCs, which are deposited during splicing, are displaced from the mRNA by translating ribosomes.52, 53, 54 However, NMD can also occur in the absence of splicing, suggesting that downstream EJCs are not always required for activation of NMD.49 Indeed, a second determinant is the length of the 3′ UTR, which is monitored by the proximity of the termination codon to the poly(A) tail. Messages bearing PTCs possess longer 3′ UTRs than their wild‐type counterparts and this appears to decrease the efficiency of translation termination and facilitate Upf1 recruitment, even in the absence of downstream EJCs.55, 56, 57
Figure 4.
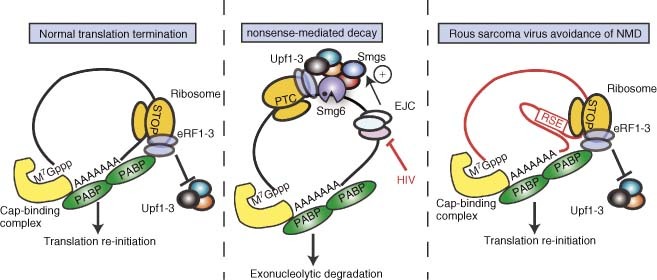
Nonsense‐mediated mRNA decay and mechanisms of viral avoidance. (a) When the translation machinery reaches the stop codon of a normal mRNA, the eukaryotic release factors eRF1 and eRF3 are recruited to the ribosome, in part via interactions with PABP. This results in efficient translation termination and re‐initiation. (b) When mRNAs containing a premature stop codon (PTC) are translated, the presence of EJCs downstream of the PTC, coupled with inefficient interactions between PABP and the termination factors, triggers nonsense‐mediated decay (NMD). NMD is induced by recruitment of Upf1–3 and the Smg proteins; cleavage by the endonuclease Smg6 triggers degradation of the mRNA fragments. Viruses such as HIV‐1 regulate the alterative splicing of their genomic RNAs to avoid the presence of EJCs downstream of stop codons. (c) The unspliced RNA of Rous sarcoma virus has a very long 3′ UTR, which is stabilized by the RSE element that directly inhibits NMD. This element may artificially ‘shorten’ the 3′ UTR by making contacts with sequences close to the poly(A) tail.
NMD represents a potentially formidable obstacle for viruses, many of which encode polycistronic transcripts that would presumably be viewed by the cellular NMD machinery as having aberrantly long 3′ UTRs. Additionally, viruses frequently use alternative splicing to maximize coding potential, possibly creating spliced variants with EJC components remaining downstream of the stop codon used by the 5′ ORF. One way viruses may escape NMD is by tightly regulating their splicing pattern, as is the case with human immunodeficiency virus 1 (HIV‐1).58 The sole HIV‐1 transcript is alternatively spliced to generate more than 30 mRNAs. The genes are always spliced out sequentially in a 5′→3′ order, so as to avoid generating transcripts where splicing of 3′ genes leads to deposition of EJCs downstream of the termination codons of the 5′ genes. However, a consequence of this strategy is that the 5′‐most genes are generally translated from completely unspliced mRNAs, which have very long UTRs between the stop codon of the first gene and the poly(A) tail. Thus, some viruses specifically avoid NMD by encoding protective elements within their long 3′ UTRs. This is best illustrated by the avian retrovirus Rous sarcoma virus (RSV), which contains a sequence element (RSE) downstream of the Gag gene stop codon that prevents the unspliced mRNA from being targeted for degradation.59 Deletion of this sequence causes a dramatic shortening of the half‐life of unspliced RSV mRNA in a translation‐ and Upf1‐dependent manner, whereas insertion of the RSE protects PTC‐containing NMD substrates from degradation. Analyses of the RSE structure using SHAPE chemistry and partial RNase digestion indicate the presence of an AU‐rich stretch and stem‐loop elements that are conserved amongst 20 different avian retroviruses.60 While the mechanisms by which the RSE blocks NMD remain unknown, an intriguing preliminary observation suggests that it may form an RNA–RNA interaction with a region just upstream of the RSV poly(A) site.60 Given the correlation between the proximity of the stop codon and poly(A) tail for NMD activation, such an interaction could facilitate translation termination by bringing the poly(A) tail closer to the Gag stop codon, thereby decreasing Upf1 recruitment (Figure 4). Alternatively, the RSE may bind proteins that influence the stability of the mRNA.
It will be of interest to examine whether similar, perhaps structurally related elements exist within other viral or cellular mRNAs with unusually long 3′ UTRs, as this may shed light on how this important pathway is regulated normally in cells and in response to infection. For example, both coronavirus and SINV have long stretches of coding sequence at their 3′ end that are not translated from the genomic mRNA, but instead are expressed from subgenomic transcripts.61,62 During the translation of genes at the 5′ end of these viral genomes such sequences would appear as very long 3′ UTRs, which might be predicted to trigger NMD even in the absence of nuclear splicing and EJC deposition. Thus, a protective mechanism may be required to actively stabilize the RNA genomes in such cases. A similar situation could arise during infection with some DNA viruses such as those of the gamma‐herpesvirus subfamily, which transcribe a number of potentially polycistronic messages.63
Additional, NMD‐independent roles in viral infection have been proposed for Upf1. For example, Upf1 associates with HIV‐1 RNA, and its presence stabilizes the viral genome and facilitates translation of the Gag precursor.64 These functions of Upf1 are genetically separable from its role in NMD, as they are observed even with mutants of the Upf1 RNA helicase domain that have dominant‐negative effects on NMD.
An intriguing idea is that cells may specifically transform viral mRNAs into targets of quality control pathways as an antiviral measure. Evidence for this comes from studies with the pokeweed antiviral protein (PAP), a ribosome‐inactivating factor isolated from the pokeweed plant Phytolacca americana that functions via RNA depurination.65, 66, 67 This protein has a broad‐spectrum antiviral activity against both plant and animal viruses, including HIV,68 and has generated interest as a potential antiviral treatment.69 Although initially antiviral activity was ascribed to the ribosome‐inactivating function of PAP, it was subsequently found that some of the PAP mutants that are unable to depurinate ribosomal RNA (rRNA) can still selectively inhibit viral replication.70 PAP was subsequently shown to depurinate capped mRNAs, and the aforementioned mutants were shown to retain the ability to depurinate mRNAs.66 Recent studies in yeast using BMV have shown that BMV RNAs become depurinated upon PAP expression, leading to ribosome stalling and enhanced RNA turnover71 (Figure 5). Interestingly, PAP‐induced turnover of BMV RNA did not occur in yeast defective in the Dom34p endonuclease, a key component of a quality control pathway termed No‐go decay (NGD). NGD is a surveillance mechanism that targets mRNAs with translation‐elongation stalls, for example as a consequence of rare codons or strong secondary structures. Following endonucleolytic cleavage by Dom34p, the resulting RNA fragments are degraded from the 5′ end by the exonuclease Xrn1 and from the 3′ end by the exosome72 (Figure 5). As expected for NGD, yeast mutants defective in Xrn1 or exosome activity accumulate cleaved BMV RNA intermediates.
Figure 5.
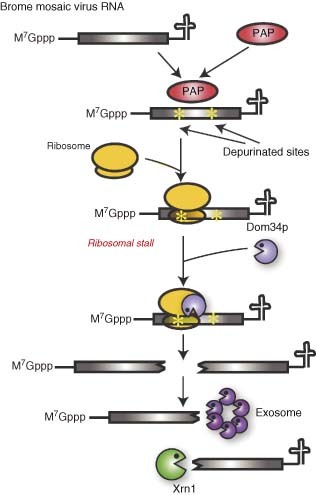
No‐go decay (NGD) as an antiviral pathway. In yeast ectopically expressing the plant protein PAP, the RNAs of Brome mosaic virus are depurinated. This causes translating ribosomes to stall, thereby triggering NGD via recruitment of the endonuclease Dom34p to the translation complex. After Dom34p cleaves the RNA close to the site of stalling, the resulting fragments become targets for exonucleases such as Xrn1 and the exosome.
While NGD has been demonstrated in yeast, an analogous surveillance pathway in mammalian cells has yet to be identified. Thus, it will be especially interesting to determine whether PAP antiviral activity against mammalian viruses similarly involves depurination followed by translation‐dependent RNA turnover, as this would provide compelling evidence for NGD in humans. Along these lines, a recent study found a reduction in the RNA levels of human T‐cell leukemia virus type I (HTLV‐I), as well as viral RNA depurination, in mammalian cells that ectopically express PAP.73 However, unlike the situation in yeast with BMV, HTLV‐I RNAs were not destabilized, arguing against an NGD‐like mechanism of suppression. It is noteworthy that whereas a number of N‐glycosidases that depurinate rRNA have been described in plants,74 no such protein has been reported in animal cells. In addition, it will be important to elucidate which features differentiate PAP‐susceptible versus non‐susceptible RNAs in light of potential antiviral uses for PAP.
RNase L
In eukaryotic cells, endonucleases are critical components of multiple cytoplasmic quality control pathways but may not be major participants in basal mRNA decay. However, an interferon‐inducible riboendonuclease, RNase L, has important roles in the innate immune response to pathogens. RNase L is activated in the presence of dsRNA, a non‐self species often present in virally infected cells (Figure 6) (reviewed in Ref 75). dsRNAs stimulate the activity of 2′,5′‐oligoadenylate synthetase (OAS), which produces the allosteric activator for RNase L, 2′‐5′‐oligoadenylate (2–5A). Activated RNase L cleaves at UU and UA dinucleotides and contributes to the host defense by destroying not only viral RNAs, but also cellular mRNAs and rRNAs that are needed for viral replication, and by stimulating apoptosis. In addition, cleavage by RNase L liberates small dsRNA species that activate the pattern recognition receptors RIG‐I and Mda‐5, leading to IFN‐β production and amplification of the antiviral interferon response.76
Figure 6.
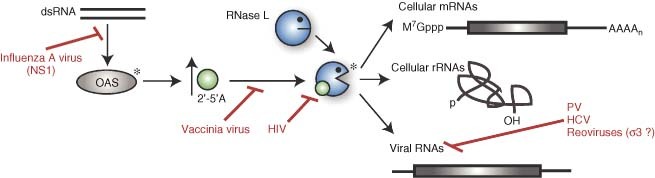
Viruses interfere with several stages of the RNase L pathway. RNase L is indirectly activated by the presence of double‐stranded RNA (dsRNA) species in the cytoplasm of infected cells. dsRNA activates 2′,5′‐oligoadenylate synthetase (OAS), which produces 2′‐5′‐oligoadenylate (2–5A), an allosteric activator for RNase L (asterisks indicate active enzymes). The NS1 protein of influenza virus A prevents OAS activation, whereas vaccinia virus and HIV‐1 block activation of RNase L by 2–5A, the latter by decreasing the affinity of RNase L for its activator. Once activated, RNase L can cleave viral RNAs and also cellular mRNAs and rRNAs. Several viruses specifically protect their RNAs from degradation, including poliovirus (PV), hepatitis C virus (HCV), and possibly reoviruses, using their σ 3 protein. Reoviruses also exploit RNase L‐mediated destruction of cellular messages to decrease competition for gene expression machinery.
dsRNA is most often found upon infection with RNA viruses that produce dsRNA intermediates during genome replication, although dsDNA viruses can also produce dsRNA due to annealing of complementary RNAs transcribed from opposite strands of the genome. RNase L‐deficient cells and mice thus exhibit increased susceptibility to infection by several viruses (reviewed in Ref 75) including encephalomyocarditis virus,77 SINV,78 and Coxsackievirus B4.79 It has also been proposed that a human reduction‐of‐function mutation in RNase L renders individuals more susceptible to xenotropic murine leukemia‐related virus, a retrovirus that has been linked to prostate cancer and chronic fatigue syndrome.80,81 A recent report has shown that in the absence of RNase L, mice infected with a neurotropic demyelinating strain of mouse hepatitis virus (MHV) succumb to CNS encephalomyelitis.82 Notably, the infected RNase L‐/‐ mice did not exhibit increased neuronal infection, interferon induction, or viral titers; mortality upon infection correlated instead with advanced demyelination and CNS tissue damage. Thus, RNase L somehow prevents extended and sustained CNS tropism of MHV. The dramatic effects of such subtle tropism alterations highlight both the diversity of RNase L functions as well as its complex interactions with infecting viruses.
Not surprisingly, many viruses have evolved mechanisms to blunt the activation of RNase L, and such inactivation occurs at a variety of different steps in the RNase L pathway (Figure 6). The NS1 protein of influenza A virus, for example, acts at a very early step by coating dsRNA and thereby preventing these species from activating OAS.83,84 Infection with vaccinia virus (VV) or HIV‐1, on the other hand, potently induces 2–5A production, but subsequent RNase L activation is blocked.75,85,86 In the case of HIV‐1, it appears that this is accomplished by somehow decreasing the affinity of RNase L for 2–5A (reviewed in Ref 85).
Some viruses, like HCV and PV, do not block RNase L activation, but their RNAs still escape degradation by this endonuclease. It has been shown that select genotypes of HCV avoid RNase L cleavage by suppressing the frequency of UU and UA sequences in their genome, perhaps as a consequence of selective evolutionary pressure.87 HCV genotypes in the human population that are less sensitive to RNase L also show increased interferon resistance.87 Conversely, the PV genome is very UU/UA rich, but is still surprisingly resistant to RNase L cleavage because of a structural element present within the PV RNA2 ORF.88 This element functions as a competitive inhibitor of RNase L thereby directly blocking its enzymatic activity.89 The role of RNase L inhibition during PV infection remains unclear, however, as PV carrying mutations that prevent the formation of the inhibitory structural element replicate to wild‐type levels in cell culture, and do not exhibit increased sensitivity to interferon.88
Although the above examples highlight RNase L as an antiviral protein, in some cases it may facilitate viral replication and/or dissemination. For example, PV forms larger plaques if cultured in cells overexpressing wild‐type RNase L, and smaller plaques in cells overexpressing a dominant‐negative form of the protein.88 It has been hypothesized that PV selectively inhibits RNase L early in infection, but then uses RNase L‐induced apoptosis to enhance viral release and spread. Mammalian reoviruses may subvert the host shutoff activity of RNase L to dispose of cellular mRNAs and clear the gene expression machinery for viral use.90 Replication of both the Jones and Dearing reovirus strains was reduced by ∼1 log in RNase L knockout relative to wild‐type mouse embryo fibroblasts, supporting the idea that the virus partially co‐opts this pathway to facilitate infection.90 Interestingly, minimal degradation of rRNAs was observed, suggesting that RNase L may not act indiscriminately as originally thought, but rather target specific sequences.91, 92, 93 The mechanism of viral escape from these pathways remains unknown, although the reovirus σ3 protein has been proposed to coat viral dsRNAs, thereby selectively inhibiting RNase L activation in cytoplasmic locales where viral RNA replication occurs, i.e., ‘replication factories’.94 However, it should be noted that the results mentioned above contrast with earlier findings implicating RNase L in restricting reovirus replication, which may be due to the use of different cell types in these studies.95,96
PERSPECTIVES
In addition to the examples highlighted herein, in many other cases viral infection presumably leads to robust engagement of cellular mRNA turnover machinery. For example, several viruses remove the 7‐methylguanosine caps from cellular mRNAs, prevent appropriate cellular mRNA splicing, 3′ processing, or polyadenylation, or directly cleave cellular messages.27,46,97, 98, 99, 100 Presumably these events lead to destruction of the impaired cellular transcripts through both basal and quality control machinery operating in the nucleus and in the cytoplasm. Such elevated global RNA degradation may overload the RNA turnover machinery, potentially indirectly stabilizing viral transcripts. Aside from affecting global RNA levels, viruses can also selectively alter the stability of specific messages through the induction of cellular signaling pathways. For example, the activation of the MK2/p38 pathway by the kaposin B protein of KSHV leads to stabilization of cytokine mRNAs bearing AU‐rich elements in their 3′ UTRs.101 In these cases, viral manipulation of select transcript stability likely has important consequences for overall pathogenesis.
Our understanding of the multitude of ways in which viruses either commandeer or are controlled by cellular mRNA turnover pathways is constantly evolving, and far from complete. Moreover, many of the pathways governing cellular mRNA decay remain incompletely understood even in uninfected cells, and new components and mechanisms are constantly described. Thus, as in many other fields of biology, viruses will continue to be invaluable tools for revealing key modulators of mRNA stability and destruction both in normal and pathogenic states.
Acknowledgements
We thank all members of the Glaunsinger lab for helpful discussions and critical reading of the manuscript. Funding to B.G. was provided by a Burroughs Wellcome Foundation Investigators in the Pathogenesis of Infectious Disease award.
RELATED WIREs ARTICLES
https://doi.org/10.1002/wrna.45
https://doi.org/10.1002/wrna.40
https://doi.org/10.1002/wrna.78
https://doi.org/10.1002/wrna.13
https://doi.org/10.1002/wrna.17
https://doi.org/10.1002/wrna.25
https://doi.org/10.1002/wrna.26
REFERENCES
- 1. Doma MK, Parker R. RNA quality control in eukaryotes. Cell 2007, 131: 660–668. [DOI] [PubMed] [Google Scholar]
- 2. Goldstrohm AC, Wickens M. Multifunctional deadenylase complexes diversify mRNA control. Nat Rev Mol Cell Biol 2008, 9: 337–344. [DOI] [PubMed] [Google Scholar]
- 3. Parker R, Song H. The enzymes and control of eukaryotic mRNA turnover. Nat Struct Mol Biol 2004, 11: 121–127. [DOI] [PubMed] [Google Scholar]
- 4. Garneau NL, Sokoloski KJ Opyrchal M, Neff CP, Wilusz CJ, et al. The 3′ untranslated region of sindbis virus represses deadenylation of viral transcripts in mosquito and Mammalian cells. J Virol 2008, 82: 880–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Conrad NK, Steitz JA. A Kaposi's sarcoma virus RNA element that increases the nuclear abundance of intronless transcripts. Embo J 2005, 24: 1831–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Conrad NK, Mili S, Marshall EL, Shu MD, Steitz JA. Identification of a rapid mammalian deadenylation‐dependent decay pathway and its inhibition by a viral RNA element. Mol Cell 2006, 24: 943–953. [DOI] [PubMed] [Google Scholar]
- 7. Conrad NK, Shu MD, Uyhazi KE, Steitz JA. Mutational analysis of a viral RNA element that counteracts rapid RNA decay by interaction with the polyadenylate tail. Proc Natl Acad Sci U S A 2007, 104: 10412–10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beelman CA, Stevens A, Caponigro G, LaGrandeur TE, Hatfield L, et al. An essential component of the decapping enzyme required for normal rates of mRNA turnover. Nature 1996, 382: 642–646. [DOI] [PubMed] [Google Scholar]
- 9. Dunckley T, Parker R. The DCP2 protein is required for mRNA decapping in Saccharomyces cerevisiae and contains a functional MutT motif. Embo J 1999, 18: 5411–5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tharun S, He W, Mayes AE, Lennertz P, Beggs JD, et al. Yeast Sm‐like proteins function in mRNA decapping and decay. Nature 2000, 404: 515–518. [DOI] [PubMed] [Google Scholar]
- 11. Tharun S, Parker R. Targeting an mRNA for decapping: displacement of translation factors and association of the Lsm1p‐7p complex on deadenylated yeast mRNAs. Mol Cell 2001, 8: 1075–1083. [DOI] [PubMed] [Google Scholar]
- 12. Bonnerot C, Boeck R, Lapeyre B. The two proteins Pat1p (Mrt1p) and Spb8p interact in vivo, are required for mRNA decay, and are functionally linked to Pab1p. Mol Cell Biol 2000, 20: 5939–5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bouveret E, Rigaut G, Shevchenko A, Wilm M, Seraphin B. A Sm‐like protein complex that participates in mRNA degradation. Embo J 2000, 19: 1661–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Coller JM, Tucker M, Sheth U, Valencia‐Sanchez MA, Parker R. The DEAD box helicase, Dhh1p, functions in mRNA decapping and interacts with both the decapping and deadenylase complexes. RNA 2001, 7: 1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Diez J, Ishikawa M, Kaido M, Ahlquist P. Identification and characterization of a host protein required for efficient template selection in viral RNA replication. Proc Natl Acad Sci U S A 2000, 97: 3913–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mas A, Alves‐Rodrigues I, Noueiry A, Ahlquist P, Diez J. Host deadenylation‐dependent mRNA decapping factors are required for a key step in brome mosaic virus RNA replication. J Virol 2006, 80: 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Noueiry AO, Diez J, Falk SP, Chen J, Ahlquist P. Yeast Lsm1p‐7p/Pat1p dead‐enylation‐dependent mRNA‐decapping factors are required for brome mosaic virus genomic RNA translation. Mol Cell Biol 2003, 23: 4094–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Scheller N, Mina LB, Galao RP, Chari A, Gimenez‐Barcons M, et al. Translation and replication of hepatitis C virus genomic RNA depends on ancient cellular proteins that control mRNA fates. Proc Natl Acad Sci U S A 2009, 106: 13517–13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beckham CJ, Light HR, Nissan TA, Ahlquist P, Parker R, et al. Interactions between brome mosaic virus RNAs and cytoplasmic processing bodies. J Virol 2007, 81: 9759–9768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eulalio A, Behm‐Ansmant I, Izaurralde E. P bodies: at the crossroads of post‐transcriptional pathways. Nat Rev Mol Cell Biol 2007, 8: 9–22. [DOI] [PubMed] [Google Scholar]
- 21. Parker R, Sheth U. P bodies and the control of mRNA translation and degradation. Mol Cell 2007, 25: 635–646. [DOI] [PubMed] [Google Scholar]
- 22. Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke‐Andersen J, et al. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol 2005, 169: 871–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zheng D, Ezzeddine N, Chen CY, Zhu W, He X, et al. Deadenylation is prerequisite for P‐body formation and mRNA decay in mammalian cells. J Cell Biol 2008, 182: 89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schwer B, Visca P, Vos JC, Stunnenberg HG. Discontinuous transcription or RNA processing of vaccinia virus late messengers results in a 5′ poly(A) leader. Cell 1987, 50: 163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bergman N, Moraes KC, Anderson JR, Zaric B, Kambach C, et al. Lsm proteins bind and stabilize RNAs containing 5′ poly(A) tracts. Nat Struct Mol Biol 2007, 14: 824–831. [DOI] [PubMed] [Google Scholar]
- 26. Parrish S, Moss B. Characterization of a vaccinia virus mutant with a deletion of the D10R gene encoding a putative negative regulator of gene expression. J Virol 2006, 80: 553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Parrish S, Resch W, Moss B. Vaccinia virus D10 protein has mRNA decapping activity, providing a mechanism for control of host and viral gene expression. Proc Natl Acad Sci U S A 2007, 104: 2139–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shors T, Keck JG, Moss B. Down regulation of gene expression by the vaccinia virus D10 protein. J Virol 1999, 73: 791–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Toh EA, Guerry P, Wickner RB. Chromosomal superkiller mutants of Saccharomyces cerevisiae . J Bacteriol 1978, 136: 1002–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheng CP, Serviene E, Nagy PD. Suppression of viral RNA recombination by a host exoribonuclease. J Virol 2006, 80: 2631–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jaag HM, Nagy PD. Silencing of Nicotiana benthamiana Xrn4p exoribonuclease promotes tombusvirus RNA accumulation and recombination. Virology 2009, 386: 344–352. [DOI] [PubMed] [Google Scholar]
- 32. Domingo E. Virus evolution In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, et al., eds. Fields Virology. 5th ed. Philadelphia: Lippincott Williams and Wilkins; 2007; 389–421. [Google Scholar]
- 33. Esteban R, Vega L, Fujimura T. 20S RNA narnavirus defies the antiviral activity of SKI1/XRN1 in Saccharomyces cerevisiae . J Biol Chem 2008, 283: 25812–25820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Murray KE, Roberts AW, Barton DJ. Poly(rC) binding proteins mediate poliovirus mRNA stability. RNA 2001, 7: 1126–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kempf BJ, Barton DJ. Poly(rC) binding proteins and the 5′ cloverleaf of uncapped poliovirus mRNA function during de novo assembly of polysomes. J Virol 2008, 82: 5835–5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zell R, Stelzner A. Application of genome sequence information to the classification of bovine entero‐ viruses: the importance of 5′‐ and 3′‐nontranslated regions. Virus Res 1997, 51: 213–229. [DOI] [PubMed] [Google Scholar]
- 37. Pijlman GP, Funk A, Kondratieva N, Leung J, Torres S, et al. A highly structured, nuclease‐resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 2008, 4: 579–591. [DOI] [PubMed] [Google Scholar]
- 38. Schmid M, Jensen TH. The exosome: a multipurpose RNA‐decay machine. Trends Biochem Sci 2008, 33: 501–510. [DOI] [PubMed] [Google Scholar]
- 39. Gao G, Guo X, Goff SP. Inhibition of retroviral RNA production by ZAP, a CCCH‐type zinc finger protein. Science 2002, 297: 1703–1706. [DOI] [PubMed] [Google Scholar]
- 40. Bick MJ, Carroll JW, Gao G, Goff SP, Rice CM, et al. Expression of the zinc‐finger antiviral protein inhibits alphavirus replication. J Virol 2003, 77: 11555–11562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Muller S, Moller P, Bick MJ, Wurr S, Becker S, et al. Inhibition of filovirus replication by the zinc finger antiviral protein. J Virol 2007, 81: 2391–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guo X, Ma J, Sun J, Gao G. The zinc‐finger antiviral protein recruits the RNA processing exosome to degrade the target mRNA. Proc Natl Acad Sci U S A 2007, 104: 151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guo X, Carroll JW, Macdonald MR, Goff SP, Gao G, et al. The zinc finger antiviral protein directly binds to specific viral mRNAs through the CCCH zinc finger motifs. J Virol 2004, 78: 12781–12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Glaunsinger B, Ganem D. Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol Cell 2004, 13: 713–723. [DOI] [PubMed] [Google Scholar]
- 45. Kwong AD, Frenkel N. Herpes simplex virus‐infected cells contain a function(s) that destabilizes both host and viral mRNAs. Proc Natl Acad Sci U S A 1987, 84: 1926–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee YJ, Glaunsinger BA. Aberrant herpesvirus‐induced polyadenylation correlates with cellular messenger RNA destruction. PLoS Biol 2009, 7: e1000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Narayanan K, Huang C, Lokugamage K, Kamitani W, Ikegami T, et al. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J Virol 2008, 82: 4471–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tohya Y, Narayanan K, Kamitani W, Huang C, Lokugamage K, et al. Suppression of host gene expression by nsp1 proteins of group 2 bat coronaviruses. J Virol 2009, 83: 5282–5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Muhlemann O, Eberle AB, Stalder L, Zamudio R. Recognition and elimination of nonsense mRNA. Biochim Biophys Acta 2008, 1779: 538–549. [DOI] [PubMed] [Google Scholar]
- 50. Eberle AB, Lykke‐Andersen S, Muhlemann O, Jensen TH. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat Struct Mol Biol 2009, 16: 49–55. [DOI] [PubMed] [Google Scholar]
- 51. Huntzinger E, Kashima I, Fauser M, Sauliere J, Izaurralde E. SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA 2008, 14: 2609–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Isken O, Maquat LE. Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev 2007, 21: 1833–1856. [DOI] [PubMed] [Google Scholar]
- 53. Zhang J, Sun X, Qian Y, LaDuca JP , Maquat LE, et al. At least one intron is required for the nonsense‐mediated decay of triosephosphate isomerase mRNA: a possible link between nuclear splicing and cytoplasmic translation. Mol Cell Biol 1998, 18: 5272–5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang J, Sun X, Qian Y, Maquat LE. Intron function in the nonsense‐mediated decay of beta‐globin mRNA: indications that pre‐mRNA splicing in the nucleus can influence mRNA translation in the cytoplasm. RNA 1998, 4: 801–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Eberle AB, Stalder L, Mathys H, Orozco RZ, Muhlemann O, et al. Posttranscriptional gene regulation by spatial rearrangement of the 3′ untranslated region. PLoS Biol 2008, 6: e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ivanov PV, Gehring NH, Kunz JB, Hentze MW, Kulozik AE, et al. Interactions between UPF1, eRFs, PABP and the exon junction complex suggest an integrated model for mammalian NMD pathways. Embo J 2008, 27: 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Singh G, Rebbapragada I, Lykke‐Andersen J. A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense‐mediated mRNA decay. PLoS Biol 2008, 6: e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bohne J, Wodrich H, Krausslich HG. Splicing of human immunodeficiency virus RNA is position‐dependent suggesting sequential removal of introns from the 5′ end. Nucleic Acids Res 2005, 33: 825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Weil JE, Beemon KL. A 3′ UTR sequence stabilizes termination codons in the unspliced RNA of Rous sarcoma virus. RNA 2006, 12: 102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Weil JE, Hadjithomas M, Beemon KL. Structural characterization of the Rous sarcoma virus RNA stability element. J Virol 2009, 83: 2119–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kuhn RJ. Togaviridiae: the viruses and their replication In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, et al., eds. Fields Virology. 5th ed. Philadelphia: Lippincott Williams and Wilkins; 2007; 1002–1022. [Google Scholar]
- 62. Lai MC, Perlman S, Anderson LJ. Coronaviridiae In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, et al., eds. Fields Virology. 5th ed. Philadephia: Lippincott Williams and Wilkins; 2007; 1305–1335. [Google Scholar]
- 63. Ganem D. Kaposi's Sarcoma‐associated herpesvirus In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, et al., eds. Fields Virology. 5th ed. Philadelphia: Lippincott Williams and Wilkins; 2007; 2847–2888. [Google Scholar]
- 64. Ajamian L, Abrahamyan L, Milev M, Ivanov PV, Kulozik AE, et al. Unexpected roles for UPF1 in HIV‐1 RNA metabolism and translation. RNA 2008, 14: 914–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Endo Y, Tsurugi K. RNA N‐glycosidase activity of ricin A‐chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J Biol Chem 1987, 262: 8128–8130. [PubMed] [Google Scholar]
- 66. Hudak KA, Wang P, Tumer NE. A novel mechanism for inhibition of translation by pokeweed antiviral protein: depurination of the capped RNA template. RNA 2000, 6: 369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Irvin JD. Purification and partial characterization of the antiviral protein from Phytolacca americana which inhibits eukaryotic protein synthesis. Arch Biochem Biophys 1975, 169: 522–528. [DOI] [PubMed] [Google Scholar]
- 68. Zarling JM, Moran PA, Haffar O, Diegel M, Myers DE, et al. Inhibition of HIV‐1 replication in seropositive patients' CD4+ T‐cells by pokeweed antiviral protein‐monoclonal antibody conjugates. Int J Immunopharmacol 1991, 13(Suppl 1): 63–68. [DOI] [PubMed] [Google Scholar]
- 69. Parikh BA, Tumer NE. Antiviral activity of ribosome inactivating proteins in medicine. Mini Rev Med Chem 2004, 4: 523–543. [DOI] [PubMed] [Google Scholar]
- 70. Tumer NE, Hwang DJ, Bonness M. C‐terminal deletion mutant of pokeweed antiviral protein inhibits viral infection but does not depurinate host ribosomes. Proc Natl Acad Sci U S A 1997, 94: 3866–3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gandhi R, Manzoor M, Hudak KA. Depurination of Brome mosaic virus RNA3 in vivo results in translation‐dependent accelerated degradation of the viral RNA. J Biol Chem 2008, 283: 32218–32228. [DOI] [PubMed] [Google Scholar]
- 72. Doma MK, Parker R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature 2006, 440: 561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mansouri S, Choudhary G, Sarzala PM, Ratner L, Hudak KA, et al. Suppression of human T‐cell leukemia virus I gene expression by pokeweed antiviral protein. J Biol Chem 2009, 284: 31453–31462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Park SW, Prithiviraj B, Vepachedu R, Vivanco JM. Isolation and purification of ribosome‐inactivating proteins. Methods Mol Biol 2006, 318: 335–347. [DOI] [PubMed] [Google Scholar]
- 75. Silverman RH. Viral encounters with 2′,5′‐oligoadenylate synthetase and RNase L during the interferon antiviral response. J Virol 2007, 81: 12720–12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Malathi K, Dong B, Gale M Jr, Silverman RH. Small self‐RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448: 816–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhou A, Paranjape J, Brown TL, Nie H, Naik S, et al. Interferon action and apoptosis are defective in mice devoid of 2′,5′‐oligoadenylate‐dependent RNase L. Embo J 1997, 16: 6355–6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sawicki DL, Silverman RH, Williams BR, Sawicki SG. Alphavirus minus‐strand synthesis and persistence in mouse embryo fibroblasts derived from mice lacking RNase L and protein kinase R. J Virol 2003, 77: 1801–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Flodstrom‐Tullberg M, Hultcrantz M, Stotland A, Maday A, Tsai D, et al. RNase L and double‐stranded RNA‐dependent protein kinase exert complementary roles in islet cell defense during coxsackievirus infection. J Immunol 2005, 174: 1171–1177. [DOI] [PubMed] [Google Scholar]
- 80. Lombardi VC, Ruscetti FW, Das Gupta J, Pfost MA, Hagen KS, et al. Detection of an infectious retrovirus, XMRV, in blood cells of patients with chronic fatigue syndrome. Science 2009, 326: 585–589. [DOI] [PubMed] [Google Scholar]
- 81. Urisman A, Molinaro RJ, Fischer N, Plummer SJ Casey G, et al. Identification of a novel Gammaretrovirus in prostate tumors of patients homozygous for R462Q RNASEL variant. PLoS Pathog 2006, 2: e25. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 82. Ireland DD, Stohlman SA, Hinton DR, Kapil P, Silverman RH, et al. RNase L mediated protection from virus induced demyelination. PLoS Pathog 2009, 5: e1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hale BG, Randall RE, Ortin J, Jackson D. The multifunctional NS1 protein of influenza A viruses. J Gen Virol 2008, 89: 2359–2376. [DOI] [PubMed] [Google Scholar]
- 84. Min JY, Krug RM. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: inhibiting the 2′‐5′ oligo (A) synthetase/RNase L pathway. Proc Natl Acad Sci U S A 2006, 103: 7100–7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bolinger C, Boris‐Lawrie K. Mechanisms employed by retroviruses to exploit host factors for translational control of a complicated proteome. Retrovirology 2009, 6: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Rice AP, Roberts WK, Kerr IM. 2–5A accumulates to high levels in interferon‐treated, vaccinia virus‐infected cells in the absence of any inhibition of virus replication. J Virol 1984, 50: 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Han JQ, Barton DJ. Activation and evasion of the antiviral 2′‐5′ oligoadenylate synthetase/ribonuclease L pathway by hepatitis C virus mRNA. RNA 2002, 8: 512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Han JQ, Townsend HL, Jha BK, Paranjape JM, Silverman RH, et al. A phylogenetically conserved RNA structure in the poliovirus open reading frame inhibits the antiviral endoribonuclease RNase L. J Virol 2007, 81: 5561–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Townsend HL, Jha BK, Han JQ, Maluf NK, Silverman RH, et al. A viral RNA competitively inhibits the antiviral endoribonuclease domain of RNase L. RNA 2008, 14: 1026–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Smith JA, Schmechel SC, Williams BR, Silverman RH, Schiff LA, et al. Involvement of the interferon‐regulated antiviral proteins PKR and RNase L in reovirus‐induced shutoff of cellular translation. J Virol 2005, 79: 2240–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bisbal C, Silhol M, Laubenthal H, Kaluza T, Carnac G, et al. The 2′‐5′ oligoadenylate/RNase L/RNase L inhibitor pathway regulates both MyoD mRNA stability and muscle cell differentiation. Mol Cell Biol 2000, 20: 4959–4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Khabar KS, Siddiqui YM, al‐Zoghaibi F, al‐Haj L, Dhalla M, et al. RNase L mediates transient control of the interferon response through modulation of the double‐stranded RNA‐dependent protein kinase PKR. J Biol Chem 2003, 278: 20124–20132. [DOI] [PubMed] [Google Scholar]
- 93. Li XL, Blackford JA, Judge CS, Liu M, Xiao W, et al. RNase‐L‐dependent destabilization of interferon‐induced mRNAs. A role for the 2–5A system in attenuation of the interferon response. J Biol Chem 2000, 275: 8880–8888. [DOI] [PubMed] [Google Scholar]
- 94. Schmechel S, Chute M, Skinner P, Anderson R, Schiff L, et al. Preferential translation of reovirus mRNA by a sigma3‐dependent mechanism. Virology 1997, 232: 62–73. [DOI] [PubMed] [Google Scholar]
- 95. Nilsen TW, Maroney PA, Baglioni C. Synthesis of (2′‐5′)oligoadenylate and activation of an endoribonuclease in interferon‐treated HeLa cells infected with reovirus. J Virol 1982, 42: 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. Embo J 1998, 17: 3351–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Chen Z, Li Y, Krug RM. Influenza A virus NS1 protein targets poly(A)‐binding protein II of the cellular 3′‐end processing machinery. Embo J 1999, 18: 2273–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Dias A, Bouvier D, Crepin T, McCarthy AA, Hart DJ, et al. The cap‐snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 2009, 458: 914–918. [DOI] [PubMed] [Google Scholar]
- 99. Kamitani W, Huang C, Narayanan K, Lokugamage KG, Makino S. A two‐pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat Struct Mol Biol 2009, 16: 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lai MC, Peng TY, Tarn WY. Functional interplay between viral and cellular SR proteins in control of post‐transcriptional gene regulation. FEBS J 2009, 276: 1517–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. McCormick C, Ganem D. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science 2005, 307: 739–741. [DOI] [PubMed] [Google Scholar]