

Summary
This study applied molecular‐based method to investigate the presence of porcine deltacoronavirus (PDCoV) in 59 commercial pig farms in South Korea. The results of RT‐PCR screening on a relatively large collection of faeces samples (n = 681) from January 2013 to March 2015 did not reveal the presence of PDCoV until the end of 2014. However, on March 2015, PDCoV‐positive samples (SL2, SL5) were detected from SL swine farm in Gyeongbuk province. The phylogenetic trees based on the complete spike‐ and nucleocapsid protein‐coding genes showed that SL2 and SL5 closely related to the US PDCoV strains rather than those in China. Thought Korean strains of PDCoV isolated in 2014 (KNU14.04) and in 2015 (SL2 and SL5) grouped within US PDCoV cluster, the reconstruction of ancestral amino acid changes suggested that they are different.
Keywords: porcine deltacoronavirus, swine, South Korea
Introduction
Coronaviruses are single‐stranded, positive‐sense enveloped RNA viruses belonging to the Coronaviridae family and are divided into 4 genera (Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus) (Woo et al., 2012). Until 2014, three members of the Alphacoronavirus genus such as porcine epidemic diarrhoea virus (PEDV), transmissible gastroenteritis virus (TGEV) and porcine respiratory coronavirus (PRCV) are known to cause enteric and respiratory diseases of swine. More recently, a novel emerging porcine deltacoronavirus (PDCoV) was demonstrated to be enteropathogenic and causes severe diarrhoea resemble those of PEDV and TGEV infections (Chen et al., 2015; Jung et al., 2015), and mild interstitial pneumonia (Ma et al., 2015). Since the first report of PDCoV in Hong Kong in 2012 (Woo et al., 2012), the virus is identified in the United States (Wang et al., 2014a,b), South Korea (Lee and Lee, 2014) and China (Song et al., 2015). In this study, we further report the presence and genetic characterization of PDCoV from cases showing symptoms of diarrhoea in Korean swine farms.
Materials and Methods
Molecular detection
In this study, faecal samples of pigs showing signs of diarrhoea (n = 681) collected from January 2013 to March 2015 were screened for the presence of porcine deltacoronavirus (PDCoV). The sampling locations were given in the Fig. S1. Total RNA was extracted using Trizol LS (Invitrogen, USA) following the manufacturer's instructions. The RNA was then converted into cDNA with the use of random hexamers and commercial RNA to cDNA EcoDry Premix kit (Clontech, Otsu, Japan) following the manufacturer's protocol. To enhance the specificity, two pairs of PDCoV primer were utilized. The first method designed primer set of reference (Woo et al., 2012). The other PDCoV‐specific primers were designed in this study, targeting a region of 587 bp of the nucleocapsid protein‐coding gene (PDCoV‐587F 5′‐CCCAGCTCAAGGTTTCAGAG‐3′, PDCoV‐587R 5′‐CCCAATCCTGTTTGTCTGCT‐3′). The thermal profile was initial denaturation at 94°C for 5 min, followed by 38 cycles of 94°C for 30 s, 56°C for 30 s, 72°C for 30 s and a final extension at 72°C for 7 min. The screening for other porcine enteric viruses was performed with pathogen‐specific primers using AccuPower® ProFi Taq PCR PreMix (Bioneer Ltd., Daejeon, Korea). The detection of Kobuvirus and group A rotavirus was following the previous studies (Reuter et al., 2009; Lee et al., 2013). For porcine epidemic diarrhoea virus (PEDV) and transmissible gastroenteritis virus (TGEV), we used i‐TGEV/PEDV Detection kit (iNtRON Ltd., Daejeon, Korea).
Nucleotide sequencing and phylogenetic analysis
For sequencing of genes encoded spike protein (S) and nucleocapsid protein (N), we followed the protocol described in the previous study (Hu et al., 2015). PDCoV‐positive samples were amplified with primer sets (PDCoV‐SF2, PDCoV‐SR2 and PDCoV‐NF1, PDCoV‐NR1). The specific PCR bands were purified by QIAquick Gel Extraction Kit (Qiagen, Daejeon, Germany), cloned utilizing TA cloning kit (Topcloner TA kit; Enzynomics, Daejeon, Korea) and subsequently transformed into competent Escherichia coli cells (DH5α). The purified recombinant plasmids were sequenced by Macrogen Inc (Seoul, Korea). New sequences of PDCoV generated in this study were addressed in GenBank accession no. KR060082–KR060085. The genetic relationship of two PDCoV strains (SL2, SL5) with other PDCoVs was inferred from a codon‐based alignment of 31 sequences of complete S gene (3483 bases) and 31 sequences of complete N gene (1029 bases). The details of the data set are summarized in Table S1. The phylogenetic tree was reconstructed by the maximum likelihood model with 1000 bootstrap replicates implemented in IQ‐TREE version 1.3.8 (Nguyen et al., 2015). The best‐fitting nucleotide substitution model for each alignment was determined automatically by specifying ‘‐m TEST’ option.
Inferring ancestral amino acid changes
Amino acid changes on the evolutionary path of PDCoV (based on S and N genes) were inferred using the codeml program implemented in PAML 4.8 (Yang, 2007). Substitutions occurred on a given node of a phylogeny were annotated by treesub program (Tamuri, 2013).
Results and Discussions
The screening results by RT‐PCR carried out on 681 samples of 59 swine farms (Table 1) showed that until the end of 2014 all of tests were negative for nucleic acid of PDCoV. It was on March 2015, PDCoV‐positive samples were detected in a 600‐scale sow farm (SL farm) in Gyeongbuk province. This farm was reported to be infected by PEDV in 2014 and had severe diarrhoea with 100% mortality in piglets. In early 2015, it was observed that up to 20% pigs of all ages had diarrhoea and 10% died. The diagnosis of porcine enteric viruses (Table 2) revealed the dual infection of PDCoV and PEDV, while TGEV, group A rotavirus and Kobuvirus were not detected. In the literature, it was reported that PDCoV co‐infected with others enteric viruses, such as: group C rotavirus (Marthaler et al., 2014), TGEV (Dong et al., 2015) and PEDV (Song et al., 2015). Combining the detection results of this study with the above‐mentioned reports, it could be inferred that PEDV was the most frequent co‐infected viruses.
Table 1.
Results of retrospective detection of PDCoV in NINE provinces from 2013 to March 2015
| Sampling sites | Sample collection year | |||||
|---|---|---|---|---|---|---|
| 2013 | 2014 | 2015a | ||||
| n | (+) | n | (+) | n | (+) | |
| Gyeonggi | 78 | 0 | 43 | 0 | 14 | 0 |
| Gangwon | 46 | 0 | 22 | 0 | 11 | 0 |
| Chungnam | 46 | 0 | 31 | 0 | 13 | 0 |
| Chungbuk | 49 | 0 | 22 | 0 | 10 | 0 |
| Jeonbuk | 38 | 0 | 26 | 0 | 8 | 0 |
| Jeonnam | 32 | 0 | 18 | 0 | 10 | 0 |
| Gyeongbuk | 38 | 0 | 22 | 0 | 18 | 2 |
| Gyeongnam | 22 | 0 | 24 | 0 | 12 | 0 |
| Jeju | 11 | 0 | 13 | 0 | 4 | 0 |
| Total | 360 | 0 | 221 | 0 | 100 | 2 |
n, number of faecal samples; +, number of positive samples.
Until March 2015.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Table 2.
Detection of porcine enteric viruses in diarrhoeal intestinal/faecal samples from pigs of SL farm in March 2015
| Name of samples/Specimens | Clinical symptoms | Pig groupa | Collection date | PDCoV | PEDV | TGEV | Group A rotavirus | Kobuvirus |
|---|---|---|---|---|---|---|---|---|
| SL1/Faeces | Diarrhoea | Sow | 25 March 2015 | − | + | − | − | − |
| SL2/Faeces | Diarrhoea, wasted | Finisher | 25 March 2015 | + | + | − | − | − |
| SL3/Faeces | Diarrhoea, wasted | Finisher | 25 March 2015 | − | + | − | − | − |
| SL4/Faeces | Diarrhoea, wasted | Finisher | 25 March 2015 | − | + | − | − | − |
| SL5/Intestine | Acute watery diarrhoea | Suckling | 31 March 2015 | + | + | − | − | − |
| SL6/Intestine | Diarrhoea | Suckling | 31 March 2015 | − | + | − | − | − |
Pigs were classified into six groups of sow, suckling pigs (<30 days), weaner (30–60 days), grower (60–90 days) and finisher (≥90 days).
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
For the genetic characterization, the maximum likelihood phylogenetic trees reconstructed from the S and N genes (Fig. 1a, b) showed a clear separation between Chinese and US strains of PDCoV and is similar to the previous studies (Marthaler et al., 2014; Wang et al., 2016). Of which, Korean strains of PDCoV isolated in 2014 (KNU14.04) and in 2015 (SL2 and SL5) were grouped within US PDCoV cluster; however, they located at different branches (highlights, Fig. 1a, b). Based on the S gene, the inferred ancestral amino acid changes along the nodes of the phylogeny (Fig. 2a) showed that the branches leading to Korean PDCoV isolates in 2014 and in 2015 shared 1 back substitution (node #40: Q106L, node #37: L106Q) and four unique substitutions (node #39: S697A, node #38: V550A, I669L and node #37: I1014V). However, the branch that leaded to 2015 isolates (SL2 and SL6) had further 2 mutations locating near the tip of the phylogeny (node #59: I110V, T582A). Based on the N gene, it was observed only amino acid mutations (six changes) near the tip of the phylogeny, on the node leading to SL2 and SL5 (Fig. 2b). The details of non‐synonymous substitutions at every node of the phylogeny can be found in Tables S2, S3. At present, the significance of these substitutions is almost obscured. Of the all, the phylogenetic analyses suggested that the PDCoVs strains (SL2, SL5) detected in early 2015 are different with the previously emerged virus (KNU14.04).
Figure 1.
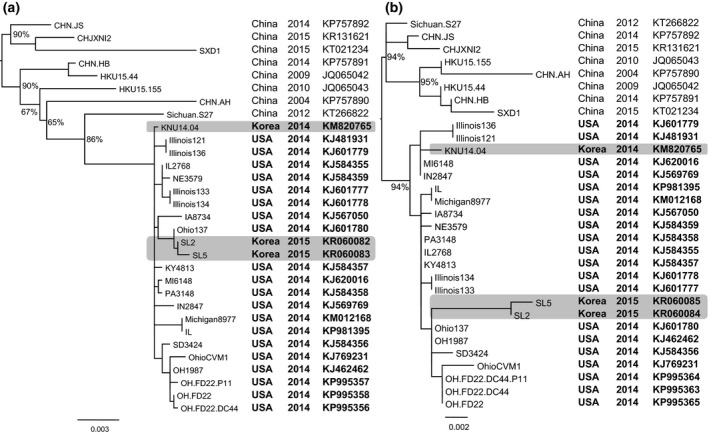
Maximum likelihood phylogeny of PDCoVs based on the spike protein‐coding gene (a) and the nucleocapsid protein‐coding gene (b). The numbers at the nodes of the phylogenies denote the bootstrap values to which they belong (for clarity, labels of some terminal nodes were omitted). The phylogenetic trees showed that Korean PCDoV isolates in 2014 (KNU14.04) and in 2015 (SL2, SL5) were grouped within US PDCoV cluster, but they located at different branches (highlights).
Figure 2.
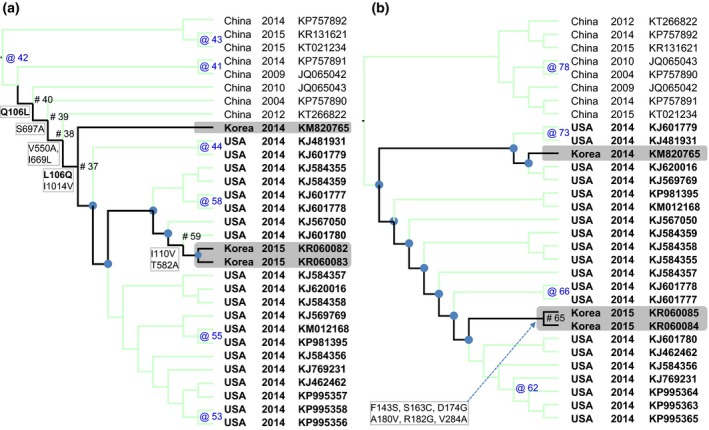
The maximum likelihood trees based on the S gene (a) and the N gene (b) with reconstructed non‐synonymous substitutions were mapped to the nodes of the phylogeny. For clarity, only branches leading to Korean PDCoV isolates were highlighted (black lines). The nodes where non‐synonymous substitutions occurred were indicated by # (for the highlighted branches) and by @ (for the others). The nodes without non‐synonymous substitutions were marked by ● (for the highlighted branches) and were not marked (for the others). It was observed that the branch which leaded to 2015 isolates (SL2, SL5) accumulated further mutations in comparing to the branch which leaded to 2014 isolate (KNU14.04).
In summary, by screening the samples collected from January 2013 to March 2015, this study confirmed the presence PDCoV in Korean swine farms. The phylogenetic analyses suggested that the Korean PDCoV isolated in 2014 and in 2015 are closely related to US strains of PDCoV, but they are different.
Conflict of interest
The authors declare that there are no conflict of interests.
Supporting information
Figure S1. Sampling sites for retrospective detection of PDCoV in 9 provinces from 2013 to March 2015.
Table S1. List of sequences used in this study.
Table S2. List of non‐synonymous substitutions at the nodes of the PDCoV phylogeny based on the spike protein coding gene (shown in Figure 2A).
Table S3. List of non‐synonymous substitutions at the nodes of the PDCoV phylogeny based on the nucleocapsid‐protein coding gene (shown in Figure 2B).
Acknowledgements
This study was supported by a grant (No. PJ011184) from BioGreen 21 Program, Rural Development Administration.
References
- Chen, Q. , Gauger P., Stafne M., Thomas J., Arruda P., Burrough E., Madson D., Brodie J., Magstadt D., Derscheid R., Welch M., and Zhang J., 2015: Pathogenicity and pathogenesis of a United States porcine deltacoronavirus cell culture isolate in 5‐day‐old neonatal piglets. Virology 482, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, N. , Fang L., Zeng S., Sun Q., Chen H., and Xiao S., 2015: Porcine deltacoronavirus in mainland China. Emerg. Infect. Dis. 21, 2254–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, H. , Jung K., Vlasova A. N., Chepngeno J., Lu Z., Wang Q., and Saif L. J., 2015: Isolation and characterization of porcine deltacoronavirus from pigs with diarrhea in the United States. J. Clin. Microbiol. 53, 1537–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, K. , Hu H., Eyerly B., Lu Z., Chepngeno J., and Saif L. J., 2015: Pathogenicity of 2 porcine deltacoronavirus strains in gnotobiotic pigs. Emerg. Infect. Dis. 21, 650–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. , and Lee C., 2014: Complete genome characterization of Korean porcine deltacoronavirus strain KOR/KNU14‐04/2014. Genome Announc. 2, e01191–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M.‐H. , Jeoung H.‐Y., Park H.‐R., Lim J.‐A., Song J.‐Y., and An D.‐J., 2013: Phylogenetic analysis of porcine astrovirus in domestic pigs and wild boars in South Korea. Virus Genes 46, 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Y. , Zhang Y., Liang X., Lou F., Oglesbee M., Krakowka S., and Li J., 2015: Origin, evolution, and virulence of porcine deltacoronaviruses in the United States. mBio, 6, e00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthaler, D. , Raymond L., Jiang Y., Collins J., Rossow K., and Rovira A., 2014: Rapid detection, complete genome sequencing, and phylogenetic analysis of porcine deltacoronavirus. Emerg. Infect. Dis. 20, 1347–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, L. T. , Schmidt H. A., von Haeseler A., and Minh B. Q., 2015: IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter, G. , Boldizsár Á., and Pankovics P., 2009: Complete nucleotide and amino acid sequences and genetic organization of porcine kobuvirus, a member of a new species in the genus Kobuvirus, family Picornaviridae. Arch. Virol. 154, 101–108. [DOI] [PubMed] [Google Scholar]
- Song, D. , Zhou X., Peng Q., Chen Y., Zhang F., Huang T., Zhang T., Li A., Huang D., Wu Q., He H., and Tang Y., 2015: Newly emerged porcine deltacoronavirus associated with diarrhoea in swine in China: identification, prevalence and full‐length genome sequence analysis. Transbound Emerg. Dis., 62, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamuri, A. U. , 2013: Treesub: annotating ancestral substitution on a tree. Available at: https://github.com/tamuri/treesub (accessed January 28, 2013).
- Wang, L. , Byrum B., and Zhang Y., 2014a: Detection and genetic characterization of deltacoronavirus in pigs, Ohio, USA, 2014. Emerg. Infect. Dis. 20, 1227–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Byrum B., and Zhang Y., 2014b: Porcine coronavirus HKU15 detected in 9 US states, 2014. Emerg. Infect. Dis. 20, 1594–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Hayes J., Sarver C., Byrum B., and Zhang Y., 2016: Porcine deltacoronavirus: histological lesions and genetic characterization. Arch. Virol., 161, 171–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P. C. , Lau S. K., Lam C. S., Lau C. C., Tsang A. K., Lau J. H., Bai R., Teng J. L., Tsang C. C., and Wang M., 2012: Discovery of seven novel mammalian and avian coronaviruses in Deltacoronavirus supports bat coronaviruses as the gene source of Alphacoronavirus and Betacoronavirus and avian coronaviruses as the gene source of Gammacoronavirus and Deltacoronavirus. J. Virol., 86, 3995–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. , 2007: PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Sampling sites for retrospective detection of PDCoV in 9 provinces from 2013 to March 2015.
Table S1. List of sequences used in this study.
Table S2. List of non‐synonymous substitutions at the nodes of the PDCoV phylogeny based on the spike protein coding gene (shown in Figure 2A).
Table S3. List of non‐synonymous substitutions at the nodes of the PDCoV phylogeny based on the nucleocapsid‐protein coding gene (shown in Figure 2B).