

Abstract
Viral genomes are compact and encode a limited number of proteins. Because they do not encode components of the translational machinery, viruses exhibit an absolute dependence on the host ribosome and factors for viral messenger RNA (mRNA) translation. In order to recruit the host ribosome, viruses have evolved unique strategies to either outcompete cellular transcripts that are efficiently translated by the canonical translation pathway or to reroute translation factors and ribosomes to the viral genome. Furthermore, viruses must evade host antiviral responses and escape immune surveillance. This review focuses on some recent major findings that have revealed unconventional strategies that viruses utilize, which include usurping the host translational machinery, modulating canonical translation initiation factors to specifically enhance or repress overall translation for the purpose of viral production, and increasing viral coding capacity. The discovery of these diverse viral strategies has provided insights into additional translational control mechanisms and into the viral host interactions that ensure viral protein synthesis and replication. WIREs RNA 2014, 5:779–801. doi: 10.1002/wrna.1246
This article is categorized under:
-
1
Translation > Translation Mechanisms
-
2
Translation > Translation Regulation
INTRODUCTION
Viruses are obligate intracellular pathogens that rely on the host machinery to mediate viral protein synthesis and to replicate their genomes. Because viral genomes are compact, they do not encode the necessary components of the translational machinery and thus, the virus must usurp the available cellular resources and divert them toward viral translation. However, the virus must contend with cellular transcripts that are being translated through the canonical translation initiation pathway, and evade host innate immune responses that act to restrict viral spread by inhibiting virus translation and replication. As such, viruses have evolved various noncanonical mechanisms that confer the ability to preferentially engage the host ribosome during infection and to counter antiviral mechanisms. In this review, there will be an emphasis on viral strategies to hijack the canonical translation pathway, modulation of host factors and the use of alternate translation initiation factors, and viral recoding mechanisms. The examples of viral translation strategies presented herein are not all‐encompassing, but rather, they are illustrative of novel strategies that have emerged in recent literature, with references to classical examples of which the mechanisms have been well characterized. Comprehensive reviews on these translational mechanisms have been described.1, 2 These strategies all confer a selective advantage for viruses to compete for the ribosome and in some cases, allow the viral RNA to be exclusively translated. The strategies described focus on viral mechanisms in metazoa. Due to space limitations, the diverse translational mechanisms found in plant viruses will not be discussed and readers are directed to several excellent reviews.3, 4, 5 Before delving into specific examples, the canonical translation initiation pathway will be briefly reviewed to develop a greater appreciation for these alternate mechanisms of translation.
CANONICAL TRANSLATION INITIATION
Translation, which includes the processes of initiation, elongation, termination, and ribosome recycling, is a tightly regulated process that involves decoding of the genetic information stored within a messenger RNA (mRNA) into a functional polypeptide sequence. During elongation, the ribosome translocates codon by codon as aminoacyl‐transfer RNAs (tRNAs) are sampled to ensure the delivery of the cognate amino acid into the nascent polyprotein chain. In eukaryotes, the majority of mRNAs bear a 5′ 7‐methyl‐guanosine cap and 3′ poly(A) tail, both of which mediate translation initiation via a cap‐dependent mechanism (Figure 1). The cap moiety binds the cap‐binding protein, eukaryotic initiation factor (eIF) 4E, which is in complex with eIF4G and eIF4A to form eIF4F. eIF4A facilitates unwinding of RNA secondary structures during scanning. eIF4G acts as a scaffold that recruits factors to the 5′ end, and through interaction with the poly(A) binding protein (PABP) bound to the 3′ poly(A) tail, mediates circularization of the mRNA. The 43S preinitiation complex comprised of the 40S ribosomal subunit, the ternary complex eIF2·Met‐tRNAi·GTP, eIF3, eIF1, eIF1A, and eIF5 is delivered to the 5′ end of the transcript via interaction of eIF3 with eIF4G. In a process that is facilitated by eIF1 and 1A, the complex subsequently undergoes directional scanning to locate the authentic AUG codon positioned within a favorable context. Start codon recognition by the initiator Met‐tRNAi within the ribosomal P site results in hydrolysis of the eIF2‐bound GTP, release of the Pi, and dissociation of initiation factors. Hydrolysis of GTP and subsequent release of eIF5B facilitate 60S joining and the formation of an elongation‐competent ribosome. For a comprehensive review of the canonical translation pathway, please see Refs 7, 8.
Figure 1.
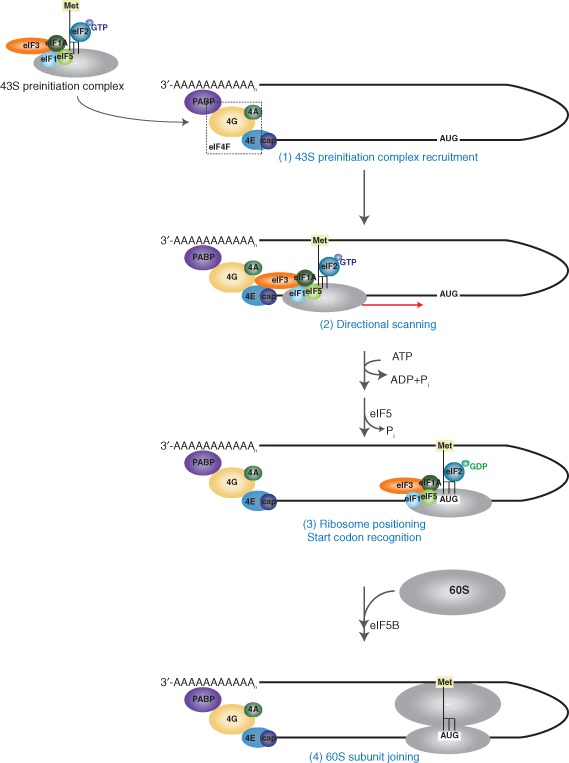
Eukaryotic translation initiation. The 5′ 7‐methyl‐guanosine cap of cellular mRNA is bound by the cap‐binding complex eIF4F, which consists of the cap‐binding protein 4E, the helicase 4A, and the scaffold protein 4G. eIF4G facilitates recruitment of the 43S preinitiation complex (1) and circularization of the mRNA through interaction with poly(A) binding proteins (PABP) bound to the 3′ poly(A) tail. Following 43S recruitment, the complex undergoes ATP‐dependent directional scanning (2) to locate the AUG start codon within a favorable context. (3) Start codon recognition and anticodon:codon pairing results in hydrolysis of the eIF2‐bound GTP in a process mediated by eIF5. Subsequently, eIF5B mediates joining of the 60S ribosomal subunit to form an elongation‐competent 80S ribosome (4). (Reprinted with permission from Ref 6. Copyright 2004 Nature Publishing Group)
NONCANONICAL TRANSLATION INITIATION
Usurping the Host Translation Machinery
Internal Ribosome Entry Sites: Picornavirus
In contrast to cap‐dependent translation, some viruses utilize a noncanonical mode of translation termed internal ribosome entry. This translation mechanism involves a cis‐acting, generally structured RNA element called an internal ribosome entry site (IRES) which is often found in the untranslated region (UTR) of a viral genome.9 IRESs recruit the ribosome internally within the viral RNA in a 5′ end‐independent manner and afford the viral genome the ability to hijack the translational machinery for viral protein synthesis during infection. IRESs were first described in picornaviruses, where the 5′‐UTRs of poliovirus (PV) and encephalomyocarditis virus (EMCV) mediate internal ribosome recruitment and initiate translation within the context of bicistronic reporter constructs.10, 11 Subsequent analyses through the concerted efforts of various groups have demonstrated that other picornaviral IRESs exist in the genomes of foot‐and‐mouth disease virus (FMDV),12, 13 human rhinoviruses,14 and hepatitis A virus (HAV).15, 16 These IRES elements share a common dependence on many canonical initiation factors including the ternary complex eIF2·Met‐tRNAi·GTP, eIF4A, eIF3, and the C‐terminal domain of eIF4G (for review see Ref 17). The fact that IRESs can mediate internal ribosome recruitment was irrevocably demonstrated through the discovery that an artificially synthesized circularized RNA harboring the EMCV IRES can initiate translation, proving that this mechanism of initiation does not depend on linear scanning of the translational apparatus.18 In general, IRESs utilize only a subset of the canonical translation factors and may also use auxiliary proteins called IRES trans‐acting factors (ITAFs), which are proteins that are not normally involved in translation but are usurped for IRES‐mediated translation. Because of the reduced requirement for initiation factors, IRES translation is active under conditions such as virus infection and cellular stress when the activities of specific targeted translation initiation factors are compromised. How does a viral IRES gain advantage over cap‐dependent translation? In the case of PV, the viral proteases 2Apro and 3Cpro not only process the viral polyprotein but also target and cleave host translation eIFs, including the scaffold protein eIF4G and PABP, which in effect leads to the shutoff of host protein synthesis and an increase in the availability of ribosomes and translation factors for viral RNA translation.19, 20, 21, 22 However, because of the limited factor requirement for PV IRES translation, the viral proteins are preferentially translated during infection (see below). Since these initial findings, additional IRES elements have been identified in other viruses as well as in a subset of cellular mRNAs.23 Thus, viral IRESs allow hijacking of the host translational machinery to mediate preferential, and in some cases like PV infection, an exclusive switch from cap‐dependent to viral IRES‐dependent protein synthesis.
IRESs can be classified into four primary classes based on the requirement for canonical initiation factors, initiator Met‐tRNAi and ITAFs.9 The picornavirus IRESs are further divided into several subgroups. Types I and II picornavirus IRESs, exemplified by the PV and EMCV IRESs, respectively, have similar factor requirements. However, while the ribosome is directly recruited to the AUG start codon on the EMCV type II IRES, it must scan a short distance to the initiation codon following recruitment on the PV type I IRES.24, 25 HAV IRES is a member of a minor group designated type III, as it is mechanistically distinct from other picornaviral IRES elements.26, 27, 28 It is generally (reasonably) assumed that IRESs from the same viral family utilize a similar mechanism for translation initiation. However, recent studies into other picornaviral IRESs identified two additional subtypes: one type, best exemplified by Simian picornavirus type 9 and porcine teschovirus 1, encompasses IRES elements that bear remarkable similarities to the hepatitis C virus (HCV)‐like IRESs,29, 30, 31, 32 while another type, exemplified by Aichivirus (AV), includes members of the Kobuvirus, Salivirus, and Paraturdivirus genera.33, 34 This further demonstrates that IRESs from the same family may mediate translation via distinct mechanisms.
Similar to the type I and II IRESs, the AV IRES is stimulated by the ITAF, polypyrimidine tract binding protein; however, the AV IRES, although having a similar core structure, adopts distinct subdomains that determine its unique translational mechanism. Unlike type I and II IRESs, the AV IRES is completely dependent on DHX29,34 which is a DExH‐Box helicase shown previously to be involved in translational initiation of mRNAs with structured 5′ UTRs.35, 36 As the initiation codon of the AV IRES is buried within a stable hairpin structure, it is proposed that DHX29 is required for unwinding of the hairpin to allow access to the initiator Met‐tRNAi. 34 This finding raises the possibility of unique IRES translation mechanisms and factor requirements within the same viral family. Similarly, recent work involving the picornaviral HAV IRES suggests that the precise strategy by which IRESs mediate translation is governed by specific cellular conditions that persist during infection. Initial characterization of the 5′‐noncoding region of HAV suggested that it shares many similar structural motifs with the EMCV IRES.37 Despite this similarity, however, the HAV IRES differs substantially in its requirements for optimal activity. Most notably, while other picornaviral IRESs are resistant to or stimulated by 2Apro, the HAV IRES is strongly inhibited under the same conditions.27 The dependence of the HAV IRES on an intact eIF4G/eIF4F complex is supported by several lines of evidence. The addition of purified eIF4F complexes following protease treatment was demonstrated to rescue HAV IRES‐mediated translation.26, 28 In a related manner, methylated cap analog, eIF4E‐binding protein 1, or rotavirus nsp3 inhibit HAV IRES function by modulation of eIF4F.26, 28 Recent reports demonstrate that 2Apro and Lpro exhibit contradicting effects on HAV IRES translation; while 2Apro impairs HAV IRES function, the FMDV Lpro strongly stimulated IRES activity, even upon proteolytic cleavage of eIF4G and arsenite‐induced phosphorylation of eIF2α.38 Resistance to eIF2α phosphorylation is reminiscent of PV and EMCV IRES‐mediated translation, as translation can proceed in an eIF2‐independent fashion late in infection.39, 40 While these findings are intriguing and may help reconcile differences that are previously thought to differentiate HAV IRES from other picornavirus IRESs, they warrant further investigations to determine the cause of divergence from previously published results.
Internal Ribosome Entry Sites: Dicistrovirus
Of the IRES classes, group I IRESs, exemplified by members of the Dicistroviridae family, have the most streamlined mechanism of action. Dicistroviruses are positive‐sense RNA viruses that are primarily pathogenic to arthropods.41 Noteworthy members of this class include the Cricket paralysis virus (CrPV) and Drosophila C virus (DCV) which are infectious to a number of insect species including the genetically tractable model Drosophila melanogaster; and the honeybee viruses including the Israeli acute paralysis, Acute bee paralysis virus, and Kashmir bee virus which have been recently implicated in the colony collapse disorder of honeybees.42 The name of the family is derived from the viral genome organization, which contains two main open reading frames that are translated under the regulation of two distinct IRES elements.43 The 5′ proximal cistron is under the regulation of the 5′ UTR IRES and encodes viral nonstructural proteins, whereas the downstream cistron encoding viral structural proteins is regulated by the intergenic region (IGR) IRES (Figure 2(a)). The two ORFs are distinctly regulated during infection; the viral structural proteins are produced in molar excess of the nonstructural proteins (nsps).43, 48, 49 Studies of the 5′ UTR IRESs have been limited. It has been shown that the Rhopalosiphum padi virus (RhPV) 5′ UTR IRES, which can direct translation in a number of systems including rabbit reticulocyte lysates, insect lysates, and wheat germ extracts50, 51 uses limited translation factors and exhibits an absolute dependence on eIF1, eIF2, and eIF3 for 48S formation.52 The 5′ UTR IRESs lack sequence conservation,53 and although it remains to be investigated, it seems unlikely that other dicistroviral 5′ UTR IRESs exhibit the same factor requirement as the RhPV 5′ UTR IRES. In contrast, the IGR IRES has been extensively studied through biochemical and structural approaches to reveal an unprecedented mechanism of action. Remarkably, the IGR IRES directly recruits the 40S and 80S ribosome in the absence of initiation factors or initiator Met‐tRNAi. 54, 55, 56 Moreover, the IRES functionally mimics a tRNA to occupy the ribosomal P site and direct translation initiation from the ribosomal A site using a non‐AUG codon.55, 56, 57 The unique triple‐pseudoknotted structure of the IGR IRES allows it to adopt two functional domains, including a solvent‐inaccessible domain largely responsible for direct ribosome binding54, 58, 59 and a tRNA‐mimicry domain that bears remarkable structural similarities to an authentic codon:anticodon pairing46, 56 (Figure 2(b)–(d)). The tRNA‐mimicry domain primes the ribosome into an elongation‐like mode where the initial pseudo‐translocation step mediated by eEF2 occurs in the absence of peptide bond formation.60, 61
Figure 2.
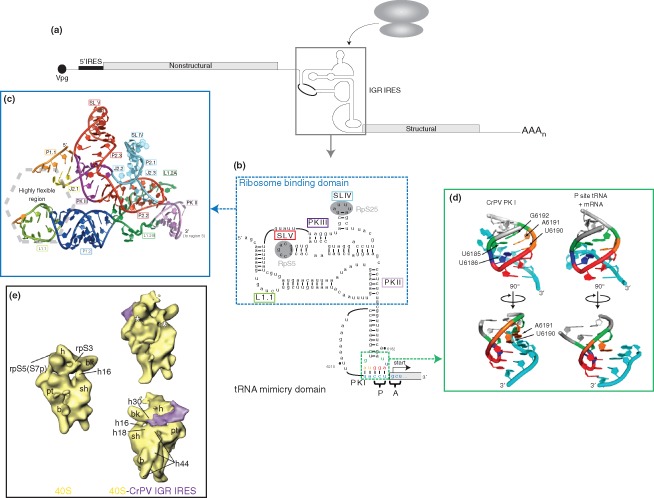
Dicistrovirus intergenic region internal ribosome entry site (IGR IRES). (a) Genome organization of members of the Dicistroviridae family. Dicistroviruses have a single‐stranded, positive‐sense RNA genome that contains the genome‐linked protein (Vpg) and poly(A) tail at the 5′‐ and 3′‐ends, respectively. The genome contains two non‐overlapping open reading frames. The upstream cistron (encoding viral nonstructural proteins) is regulated by the 5′ IRES, while the downstream cistron (encoding viral structural proteins) is expressed under the regulation of the IGR IRES (boxed). The IGR IRES can directly recruit ribosomes in the absence of canonical translation initiation factors. (b) Sequence and secondary structure of the CrPV IGR IRES. The IGR IRES adopts a triple‐pseudoknotted structure (PKI/II/III) with two independently folded domains: the ribosome binding domain (boxed in blue) and tRNA‐mimicry domain. Within the ribosome binding domain, stem‐loop (SL) V interacts with ribosomal protein (RP) S5 whereas SL IV interacts with RPS25 (shaded in gray). The conserved L1.1 bulge is thought to interact with the L1 stalk of the 60S subunit to direct 80S formation. Notable structural elements are boxed in the corresponding colors as those used in crystal structure in (c). The tRNA‐mimicry domain structurally mimics an authentic codon:anticodon interaction and establishes the translational reading frame by occupying the ribosomal P site. IRES‐mediated translation initiates from the A site and at a non‐AUG codon to direct synthesis of the viral structural proteins. The IRES codon:anticodon‐like interaction is boxed in green. Specific nucleotides within this region are depicted in the corresponding colors as the structure shown in (d). (Reprinted with permission from Ref 44. Copyright 2010 Cold Spring Harbor Laboratory Press) (c) Crystal structure of the ribosome binding domain from the Plautia stali intestinal virus. The ribosome binding domain forms a solvent‐inaccessible core that mediates contacts with the 40S ribosomal subunit. (Reprinted with permission from Ref 45. Copyright 2006 American Association for the Advancement of Science) (d) Comparison of the CrPV PKI codon:anticodon‐like interaction and an authentic P‐site mRNA:tRNA interaction. Analogous bases in both structures are highlighted in the same color. (Reprinted with permission from Ref 46. Copyright 2008 Nature Publishing Group) (e) Cryo‐EM reconstructions of the vacant human 40S ribosomal subunit (left) and the CrPV IGR IRES‐bound 40S complex (right) at 25.3 Å and 20.3 Å, respectively. The IGR IRES binds to the intersubunit space and induces conformational changes in the 40S subunit (indicated by asterisk). (Reprinted with permission from Ref 47. Copyright 2004 Cell Press)
The IGR IRES domains are modular in nature and are functionally interchangeable to generate chimeric IRESs with activities that are dictated by the ribosome binding domain.44, 62 Cryo‐EM and structural docking studies have yielded insights into the specific contacts between the IRES and the ribosome that drive IRES translation. The IGR IRES occupies the intersubunit space proximal to the decoding center, with specific contacts to the ribosomal P and E sites (Figure 2(e)).47, 63 Binding induces conformational changes in the IGR IRES, where it retracts from the A site toward the E site of the ribosome.47 Reciprocal conformational changes are also observed in the ribosome itself. IRES binding to the 40S subunit alone results in a rotation of the head relative to the body.47 An additional connection is also established between the head and the shoulder on the solvent accessible side of the subunit, which may be involved in latch closure to the mRNA entry channel to effectively anchor the 3′ end of the incoming mRNA.47 Remarkably, 60S subunit joining causes a reversion in the head rotation and opening of the mRNA channel, possibly facilitating delivery of the first incoming aminoacyl‐tRNA.47 Interestingly, binding of an unrelated IRES, the HCV IRES, to the ribosome induces similar conformational changes, suggesting that such structural rearrangements may be intrinsic to ribosome function and underlie the ability of these IRES elements to manipulate the ribosome (compare Figures 2(e) and 3(b)).47, 65
Figure 3.
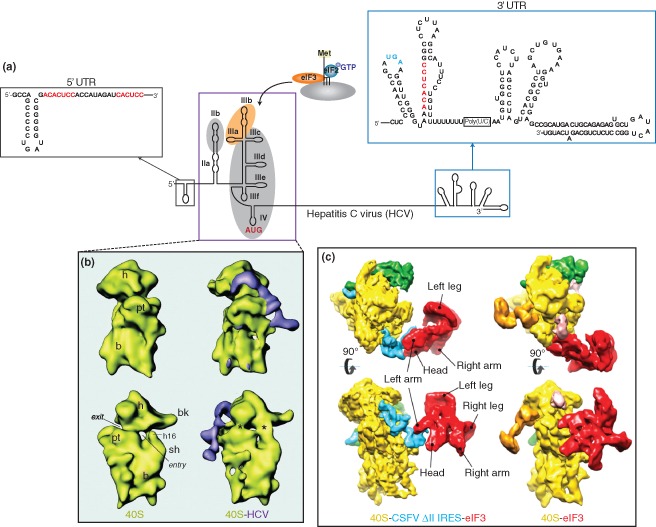
Hepatitis C virus (HCV) IRES. (a) Genome organization of the hepatitis C virus. The 5′‐untranslated region of the HCV genome contains an internal ribosome entry site (boxed in purple) that mediates ribosome recruitment using only eIF3 and the eIF2·Met‐tRNAi·GTP ternary complex. The IRES consists of domains II–IV, where the apical region of domain III interacts with eIF3 (shaded in orange), and regions within domains II, III, and IV establish contacts with the ribosome (shaded in gray). The IRES translational start site is highlighted in red. Within the 5′ and 3′ untranslated regions (UTRs) of the viral genome, three miR‐122 binding sites have been identified by base complementarity (sequences highlighted in red). The stop codon of the coding region is highlighted in blue. The structures of the HCV 5′ and 3′ UTRs are Reprinted with permission from Ref 64. Copyright 2005 American Association for the Advancement of Science. (b) Cryo‐EM structure of the vacant 40S ribosomal subunit from rabbit reticulocytes and 40S‐HCV complex at 20Å. The HCV IRES binds to the solvent accessible side of the ribosome and induces conformational changes in the 40S subunit, similar to those induced by IGR IRES‐40S binding (indicated by asterisks). (Reprinted with permission from Ref 65. Copyright 2001 American Association for the Advancement of Science). (c) Cryo‐EM structure of the 40S‐eIF3 (11.6 Å) and 40S‐CSFV ΔII IRES‐eIF3 (9.5 Å) complexes containing eIF3 and 40S from rabbit reticulocytes. In the 40S‐eIF3 complex (right), eIF3 (shown in red) interacts directly with the 40S subunit (in yellow). In contrast, eIF3 binds to the IRES (in blue) via the apical loop of domain III in the IRES‐containing complex (left), suggesting that the IRES displaces eIF3 to gain access to the ribosome. (Reprinted with permission from Ref 66. Copyright 2013 Nature Publishing Group)
Internal Ribosome Entry Sites: HCV‐like
Like the IGR IRES, the HCV IRES, which belongs to the class II IRESs, can also directly recruit the 40S subunit in the absence of translation factors. Following 40S binding, the HCV IRES then recruits eIF3 and eIF2·Met‐tRNAi to form 48S complexes that are properly positioned at the initiation site.67, 68 Interestingly, unlike the IGR IRES which binds exclusively within the mRNA cleft of the ribosome, cryo‐EM studies revealed that the HCV IRES is mostly located on the solvent side of the 40S with only domain II of the IRES occupying near the E site of the ribosome (Figure 3(b)).65, 69 The HCV IRES adopts an open RNA structure consisting of distinct domains to mediate specific functions during IRES translation. The apical region of domain III is responsible for eIF3 binding70, 71, 72 and the junction of domain III and IV forms a high‐affinity core for 40S subunit binding (Figure 3(a)).71, 73, 74 Domain II is essential for IRES translation and is thought to induce conformations within the 40S subunit to mediate translation.65, 75 At the heart of the HCV IRES, a pseudoknot within domain IV facilitates positioning of the ribosome and the ternary complex at the AUG start codon.76, 77 It is noteworthy that both the IGR IRES and HCV IRES, though using distinct strategies to manipulate the ribosome, can recruit the ribosome by binding to different regions of the 40S subunit. Moreover, both IRESs use a pseudoknot structure, which is a general feature that dictates ribosome positioning to initiate translation.
Although the precise mechanism of HCV IRES‐mediated translation is not fully understood, structural studies have provided mechanistic insight into how it hijacks the ribosome. As described above, after the HCV IRES directly recruits the 40S subunit, the eIF2 ternary complex and eIF3 play essential roles in facilitating proper ribosome positioning at the AUG start codon.67, 72 It has been proposed that eIF3 acts as a structural scaffold to facilitate 40S positioning but its mechanistic role in HCV IRES translation has not been elucidated. In a recent report, using the classical swine fever virus (CSFV) IRES, an HCV‐like IRES, cryo‐EM studies comparing the position of eIF3 within the 43S or IRES‐ribosome complexes reveal conformational discrepancies.66 In the 43S preinitiation complex, eIF3 forms extensive contacts with specific ribosomal proteins (RPs).66 In contrast, eIF3 binds exclusively to the IRES within the IRES‐ribosome complex, suggesting that the IRES (particularly domain III) functionally displaces eIF3 to gain access to the ribosome (Figure 3(c)). This finding suggests a possible strategy whereby the IRES evicts eIF3 from the 43S complex to minimize competition of cellular mRNAs for the translational machinery.66 To substantiate this finding, primer extension analysis demonstrated that 48S complex formation on β‐globin mRNA was reduced upon addition of HCV‐like IRES domain IIIabc, which alone is sufficient for eIF3 binding.66 Although it is unknown whether this represents a viable strategy to outcompete host transcripts within the context of virus infection, it nevertheless provides insight into how HCV‐like IRESs may usurp the host translational machinery. Structural analyses have similarly been useful in illuminating events downstream of initiation during HCV IRES‐mediated translation. Through the use of cryo‐EM, complemented by NMR and biochemical approaches, Kieft and colleagues demonstrated that domain IIb of the HCV IRES is responsible for promoting a switch from translation initiation to elongation and suggests that the IRES has roles beyond ribosome recruitment and positioning at the initiation site.78 Mutants bearing deletions or substitutions in this specific region could still effectively assemble 80S ribosomes and correctly position the start codon, but exhibited moderate impairment in the first translocation event.78 These findings provided the first documented example in which ribosome assembly on the HCV IRES is uncoupled from the initial translocation step. Translocation on the HCV IRES necessitates ribosomal conformational changes that are mediated by a putative interaction between the HCV IRES domain II and RPS5.65, 79 In domain IIb mutants, this essential interaction is absent, thus resulting in a defect in translocation.78 It remains to be investigated whether this property is specific for HCV IRES or a general strategy utilized by other viral IRESs. These findings add to the existing roles of domain II in HCV IRES translation including 60S joining, eIF5‐induced hydrolysis of GTP bound to eIF2, eIF3j dissociation, and configuration of the RNA in the decoding groove.68, 80, 81, 82
Internal Ribosome Entry Sites: HIV
IRES‐dependent mechanisms appear to mediate the translation of alternate isoforms of gag protein within the human immunodeficiency virus types 1 and 2 (HIV‐1 and HIV‐2, respectively),83, 84 and the related simian immunodeficiency viruses (SIVs).85, 86 These viral elements, located within the gag coding region, are translated most efficiently as leaderless RNAs, but within the context of the viral genome, are preceded by upstream 5′ UTR elements that modulate IRES activity.84, 87 Interestingly, these viral IRESs utilize a unique mechanism to recruit ribosomal complexes to an initiation site upstream of the IRES core. Using sucrose gradient centrifugation, the HIV‐2 IRES was shown to assemble three distinct translation initiation complexes, which were confirmed by toeprinting analysis to be at the authentic and internal AUG codons of the gag gene.87 RNA‐based affinity purification revealed that 48S initiation complexes formed on the HIV‐2 IRES are comprised of all the canonical initiation factors with the exception of eIF4G and eIF1.88 Intriguingly, similar to a strategy used by the dicistrovirus IGR IRES and HCV IRES, the HIV‐2 IRES can directly bind to the 40S subunit.88 eIF3 is also recruited through a conserved structural core, and can form an IRES/40S/eIF3 ternary complex that is likely a nonrate‐limiting step and a prerequisite in translation initiation.88 It is speculated that subsequent to ternary complex formation, the initiation complexes are shuttled to the alternate initiation codons to mediate expression of the gag isoforms.88 A recent report showed that the HIV‐1 gag leader IRES (to be distinguished from the coding region IRES discussed above) exhibits enhanced activity in lymphocyte cell line, suggesting that specific ITAFs found in these cells may facilitate HIV IRES translation.89 While structural conservation of the IRES suggests that it has functional implications in the HIV life cycle, further investigations will be required to identify specific ITAFs that facilitate its translation.
Direct Role of miRNA on Virus Infection
Infection by HCV, a positive‐sense RNA, is hepatotrophic in nature and is a major causative agent for chronic liver disease. Tissue tropism was initially thought to be associated with the requirement of specific cell surface receptors or the dependence of the viral life cycle on factors that are expressed exclusively in liver cells; however, this speculation was overturned by the discovery that viral RNA abundance is modulated by the liver‐specific microRNA (miRNA), miR‐122.64 Indeed, exogenous expression of miR‐122 greatly enhanced HCV abundance in nonhepatic cells.90 The HCV genome contains three potential miR‐122 binding sites identified through base complementarity with the miRNA seed sequence: one within the variable sequence of the 3′ UTR and two in the 5′ UTR (Figure 3(a), sequences in red). Sequestration of miR‐122 by antisense oligonucleotides or disruption of the miRNA seed match resulted in a decrease in viral RNA abundance.64 Expression of artificial miRNAs bearing compensatory mutations that restored the seed match ameliorated these effects, indicating a direct interaction of miR‐122 with the HCV binding sites.64, 91, 92 How does miR‐122 mediate HCV replication? Given the role of miRNAs on translation, it was proposed that miR‐122 affected translation of the HCV mRNA. Viral RNA accumulation was suggested to be a result of miR‐122‐dependent stimulation of the HCV IRES and required both binding sites within the 5′ UTR to be intact.92, 93 Additionally, the 3′ region of miR‐122 downstream of the seed sequence is essential and stimulation is facilitated by Argonaute (Ago) proteins94 and the P body protein LSm1.95 Although the contribution of miR‐122 on HCV translation is moderate, the finding that a miRNA positively affects translation is in line with other reports that miRNAs can function positively and contrasts their conventional role in translational repression.96 Furthermore, insertion of the miR‐122 binding site into the 3′ UTR of a reporter RNA negated the stimulatory effects and instead, conferred the canonical repressive function of miRNAs.92 In contrast to the modest translational effects, recent reports have proposed that the major role of miR‐122, in association with Ago2, is to prevent decay of the HCV genome, independently of its translation status.97 The exonuclease Xrn1 was shown to degrade the HCV RNA and miR‐122 binding functions to protect the HCV RNA from degradation.98 Interestingly, supplementation of miR‐122 or specific knockdown of Xrn1 has redundant and nonadditive effects on HCV RNA stability.98 However, due to the inability of Xrn1 knockdown to rescue viral replication defects from impaired miR‐122 binding, miR‐122 may have additional roles in the viral life cycle that are yet unidentified.98 Though a comprehensive understanding of miR‐122 function on the HCV life cycle is still lacking, its roles in HCV IRES stimulation and RNA stabilization suggest a multifaceted mode of regulation by miRNAs that may be prevalent in other viruses.
Specialized Ribosomes in Cap‐Independent Translation
While ribosomes may be conventionally perceived as homogenous, indiscriminatory protein‐translating apparatuses, there is emerging evidence suggesting that ribosomes are heterogeneous in nature as a result of varying RP content, unique posttranslational modifications and/or ribosomal RNA composition.99 In fact, the idea that specific constituents of the ribosome may confer a more specialized function was formally proposed as the ribosome filter hypothesis.100 It underscores the intriguing possibility that the translational activities of ribosomes are not static, but rather are selective for specific transcripts, thus representing an additional level of complexity in translational regulation. The first indication of this process came through studies of some viruses bearing IRES elements, including PV, HCV, and CrPV. A genome‐wide siRNA screen for genes involved in DCV infection of Drosophila S2 cells identified 66 ribosomal proteins that are required for DCV IGR IRES‐mediated translation and virus infection.101 Surprisingly, while knockdown of ribosomal proteins had relatively modest effects on cap‐dependent translation, IRES‐mediated translation was dramatically reduced. Furthermore, it was shown that this translational defect is not specific to insect viruses, but is observed also for PV infection.101 These initial findings suggested that the general integrity of the ribosome may be important in promoting viral growth. The notion that specific ribosomes may mediate translation of distinct classes of mRNAs was spawned by studies on the role of RPS25 on IGR IRES translation. In IGR IRES‐ribosome complexes, RPS25 crosslinks specifically with the ribosome binding domain of the IRES,102 and cryo‐EM reconstructions suggest that the IRES interacts with a protein adjacent to RPS5 that has no prokaryotic homolog.63 Additionally, RPS25‐deficient ribosomes isolated from Saccharomyces cerevisiae exhibit significantly diminished binding to the IGR IRES, and IRES translational activity is negligible in yeast translation extracts prepared from RPS25‐deficient strains.103 Interestingly, cryo‐EM studies revealed that RPS25 and the neighboring RPS5 together constitute the primary interface for ribosome‐IGR IRES binding.59, 103 In vivo characterization of the role of RPS25 in CrPV IGR IRES and HCV IRES function revealed that while global translation was only minimally affected, the loss of RPS25 yielded a significant impairment in IRES activity.104, 105 Similarly, the activities of structurally and functionally diverse IRES elements including the CSFV, EMCV, PV, and EV71 IRESs and a subset of cellular IRESs were also affected,104 suggesting that specific ribosomal proteins may have evolved to accommodate IRES translation in general. Because the IGR IRES and HCV IRESs can recruit the 40S subunit directly, it is reasonable to propose that specific ribosomal proteins such as RPS25 may interact directly with and facilitate recruitment of the IRES (Figure 4). Furthermore, it may also provide an explanation as to why these IRESs may not function in prokaryotes, which do not have RPS25. However, in the case of picornaviruses such as PV and EV71, it is not clear how RPs contribute to infection, as these IRESs require a multitude of factors to recruit the ribosome. Nevertheless, the common requirement for specific ribosomal proteins and specialized ribosomes suggest that diverse viral IRESs may in fact share more mechanistic features than initially thought.
Figure 4.
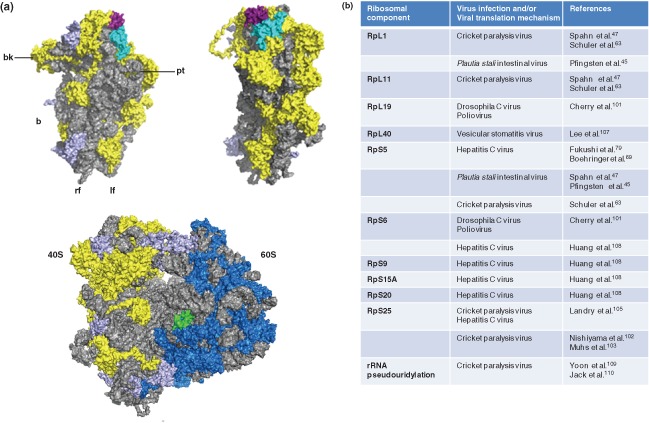
Specialized ribosomes in viral translation. (a) (Top left) The positions of ribosomal proteins S5 (in cyan) and S25 (in magenta) are shown on the 40S subunit, as viewed from the intersubunit space. Various landmarks of the 40S subunit are indicated (bk = beak, pt = platform, b = body, rf = right foot, lf = left foot). RPS5 and S25 interact with the cricket paralysis virus and hepatitis C virus IRESs. (Top right) The structure as observed after a 90° clockwise rotation. (Bottom) The location of ribosomal protein L40 (in green) within the 80S ribosome is shown. In all structures, the rRNA is shown in gray, 40S ribosomal proteins in yellow, 60S ribosomal proteins in blue, and other ribosomal proteins involved in viral translation mechanisms, as summarized in (b), are shown in purple. Structures are produced using PDB accession numbers: 3U5B, 3U5C, 3U5D, and 3U5D. (Reprinted with permission from Ref 106. Copyright 2011 American Association for the Advancement of Science) (b) Ribosomal components and/or their modifications that interact with the viral genome or that are shown to be required for viral translation based on structural and biochemical studies.
The regulatory effects of RPs may go beyond IRES translation. Through an siRNA screen of ribosomal proteins required for vesicular stomatitis virus (VSV) infection, eight ribosomal proteins, including RPL40, were shown to play an intrinsic role in viral mRNA translation but not global translation (Figure 4).107 The VSV viral genome, which is both capped and polyadenylated, is thought to be translated via a distinct cap‐dependent mechanism. During infection, host translational repression is achieved, in part, by the hypophosphorylation of eIF4E‐binding protein 1 (4E‐BP1), which effectively disrupts formation of the eIF4F complex through sequestration of eIF4E.111, 112 Despite having extensive similarities to host mRNAs, viral RNAs can escape translational shutoff and translation proceeds via an RPL40‐dependent mechanism, likely through cis‐acting elements present in the viral 5′ and 3′ UTR elements.107 RPL40 facilitates VSV translation at the level of initiation, and as a ribosomal constituent (as opposed to its potential extra‐ribosomal functions).107 Other viruses related to VSV, including the rabies virus, measles virus, and Newcastle disease virus, all have a similar sensitivity to RPL40 depletion; more intriguingly, however, is the identification of a small subset of cellular mRNAs, through sequencing polysome‐associated RNAs, that are expressed by a RPL40‐dependent pathway.107 This suggests that VSV and related viruses may have usurped an endogenous translational pathway.
It is well established that the catalytic peptidyltransferase domain of the ribosome lies in the rRNA constituent; the majority of ribosomal proteins are localized to the periphery of the ribosome and the content varies dramatically between the prokaryotic and eukaryotic ribosomes (reviewed in Ref 113). It is interesting to note that the RPs which constitute the specialized ribosomes essential for IRES‐mediated or viral translation have no prokaryotic counterparts, which may suggest that these additional RPs may facilitate ribosome selection of IRESs or in the case of VSV, viral cap‐dependent translation. Recent lines of evidence support the emerging notion that ribosomal proteins modulate translation and there is increasing interest as to how they may contribute to the differential regulation of a subset of mRNAs. Although the loss of RPs does not have a detrimental effect on cell viability or host cap‐dependent translation, it is not clear if there are certain cellular conditions that favor ribosomes with a specific composition. It is likely that the RPs are regulated via posttranslational modifications which may modulate ribosome activity and/or specificity and it remains to be investigated whether specific ribosomal proteins are regulated during virus infection to control IRES‐mediated translation.
In addition to RPs, rRNA is also subject to modifications that can regulate the translation of a subset of RNAs. In an attempt to understand how rRNA modifications contribute to the pathogenesis of X‐linked dyskeratosis congenita, a fatal disease characterized by bone marrow failure and an increased susceptibility to cancer, Ruggero and coworkers have identified a specific requirement of rRNA pseudouridylation in IRES translation.109 While global translation is not affected, the loss of pseudouridine synthase activity results in a significant downregulation of p27, XIAP, and Bcl‐xL translation. Furthermore, the effect is not exclusive to cellular IRES elements but is also observed with the CrPV IRES, suggesting an intrinsic ribosomal defect in IRES‐dependent translation.109 Further investigation into the molecular mechanism revealed that it is attributable to a decrease in affinity of pseudouridine‐deficient ribosomes for the CrPV IGR IRES, and that this defect is conserved from lower to higher eukaryotes.110 Pseudouridine‐deficient ribosomes also exhibited other translational defects including reduced translational fidelity and maintenance of translational reading frame, altered sensitivities to translational inhibitors, and reduced ribosome‐tRNA affinities.110 Because these defects are governed in part by tRNA binding and that the CrPV IGR IRES also adopts a tRNA‐like conformation, it is speculated that the loss of pseudouridylation manifests as a universal defect in ribosome‐ligand binding.110 There are 91 known pseudouridylation modification sites within human rRNA.114 Whether specific pseudouridylation modifications within the rRNA are responsible for IRES binding remains to be examined. Furthermore, these findings raise the intriguing possibility that specific nucleotides within the rRNA may be modified during virus infection. These modifications would most likely have to occur during maturation of the ribosomal subunits in order for the modifying enzymes to access sites on the rRNA.
Modulation of Translation Initiation Factors
Inactivation of the Cap‐Binding Complex
One common hallmark and dramatic consequence of some viral infections is host translational shutoff, which effectively dampens the antiviral response and diverts the cell's translational capacity toward viral production. Because translation initiation is the rate‐limiting step that is tightly regulated by the availability of canonical initiation factors, constituents of the eIF4F cap‐binding complex represent major targets for translational control. PV‐mediated translational repression provides an exemplary way in which the virus modulates canonical initiation factors for its exclusive usage while consequently rendering the host cell incapable of translation initiation. Translational repression is initiated by cleavage of eIF4GI by 2Apro, and proceeds to completion by cleavage of the more resistant functional homolog, eIF4GII.19, 20 Furthermore, the proteases 3Cpro and 2Apro lead to cleavage of PABP.21 Cleavage of eIF4G and PABP effectively inactivates the cap‐binding complex essential to canonical translation initiation. The proteolysis of eIF4G generates two cleavage products that have disparate functions: an amino‐terminal fragment containing the eIF4E binding domain115, 116 and a carboxyl‐terminal fragment containing the eIF3 and eIF4A binding sites.116, 117 While cap‐dependent translation is severely impaired by eIF4G cleavage, the C‐terminal proteolytic product of eIF4G is necessary and sufficient in supporting IRES‐mediated translation.118 A specific and direct interaction between PV IRES domain V and the central core of eIF4G is essential for ribosome recruitment.119 This interaction promotes eIF4A recruitment, and by association with eIF3 (likely as a constituent of the 43S complex), allows hijacking of the ribosome during infection.25 Interestingly, a recent report demonstrated that upregulation of miR‐141 during enterovirus EV71 infection results in the targeted inhibition of eIF4E. Transfection of antagomiR‐141 (antisense miR‐144) delayed host translational shutoff and moderately attenuated virus production.120 Furthermore, ectopic expression of miR‐141 resulted in shutoff of cap‐dependent translation concomitant with an increase in IRES translation.120 Thus, this study reveals an additional level of regulation that acts in coordination with the cleavage of eIF4G and PABP to ensure efficient shutoff of host translation and to promote the switch to IRES translation. Therefore, viral modulation of initiation factors helps to establish an environment that minimizes the ability of cellular transcripts to compete for the translational machinery to thereby promote its own preferential translation.
Stimulation of Cap‐Binding Complex
While host translational repression and a switch to viral cap‐independent translation is a hallmark of some virus infections, other viruses do not use IRES elements to recruit the ribosome. DNA viruses such as herpes simplex virus‐1 (HSV‐1) and Kaposi's sarcoma herpesvirus (KSHV) use a strategy to actively assemble eIF4F complex to mediate cap‐dependent viral protein synthesis despite suppressing host protein synthesis during virus infection.121, 122 For example, during HSV‐1 infection, the viral protein ICP6 binds to eIF4G to promote eIF4F assembly.122 In contrast, human cytomegalovirus (HCMV) infection does not lead to inhibition of host translation and thus, viral translation proceeds concomitantly with host protein synthesis.123 Insights of this mechanism have come to light. Under HCMV infection, the virus modulates the pool of translation initiation factors to actively promote eIF4F formation.124, 125, 126, 127 In particular, cellular poly(A) binding protein (PABP1) is translationally stimulated and accumulates in the cytoplasm124 (in contrast to the nuclear retention of PABP described in HSV‐1128, 129 and KSHV infections121, 130). Translational upregulation is dependent on the terminal oligopyrimidine (TOP) motif within the 5′ UTR of PABP1 and requires mTORC1 activation by the viral‐encoded UL38 mTORC1 activator.131 Preventing PABP accumulation impairs eIF4F assembly and decreased viral titer, suggesting that newly‐synthesized PABP facilitates eIF4F formation on viral RNAs.131 Thus, remodeling of translational complexes can be achieved by viral modulation of initiation factor levels, which has significant implications on the ability of the virus to mount a productive infection. Interestingly, HCMV infection coordinately increases the levels of the PABP1 repressor, Paip2, and the E3 ubiquitin ligase EDD1, which targets Paip2 for proteolytic degradation.132 This effect at first seemed paradoxical, as it neutralizes the virus' attempt to upregulate PABP1. However, the increase in Paip2 abundance has been established as a virus‐induced innate antiviral strategy in response to the upregulation of PABP1.132 The host cell precisely modulates the ratio of PABP1 and its cognate repressor in order to diminish the pool of free PABP1 available for viral translation.132 Although this mechanism of regulation remains to be elucidated under HCMV infection, Paip2 may have a more universal role in regulating PABP1 levels and thus the overall translation profile in the absence of viral infection.
Inactivation of eIF2 and Alternate Initiation Factor Usage
While a common strategy is to target the eIF4F complex (as described above), an alternate and nonexclusive way in which host translational repression can be triggered is through activation of eIF2α kinases, which results in the downstream phosphorylation and inactivation of eIF2α (Figure 5). Upon delivery of the ternary complex eIF2·Met‐tRNAi·GTP to a start codon, GTP hydrolysis occurs to allow for tRNA accommodation. For subsequent rounds of translation initiation to ensue, eIF2·GDP must be recycled to eIF2·GTP in a process mediated by the guanine nucleotide exchange factor eIF2B. The α‐subunit of eIF2 is susceptible to phosphorylation by various kinases that are activated during stress conditions as a means to repress translation. eIF2α‐P acts as a potent competitive inhibitor to eIF2B, and impedes its ability to recycle eIF2·GDP; thus targeting eIF2 serves a fundamental role in altering gene expression during different environmental stresses (for review, see Ref 133). Four eIF2α kinases exist in eukaryotes: HRI responds to fluctuations in heme:globin ratios; GCN2 is activated by UV irradiation and amino acid starvation; PERK is triggered by overload of the ER; and PKR is stimulated by dsRNAs. Perhaps the most important eIF2α kinase implicated in host antiviral immune defense is PKR, as it acts as a sensor for incoming viral dsRNAs or viral replication intermediates. GCN2 has also been identified as a regulator of the antiviral response, as it is specifically activated upon binding of two nonadjacent regions of the Sindbis virus (SV) genomic RNA to the GCN2 histidyl‐tRNA synthetase‐related domain.134 Strikingly, GCN2−/− mouse embryonic fibroblasts were highly permissive to SV (and to a lesser extent, VSV infection and Semliki forest virus), suggesting the involvement of GCN2 in an innate antiviral response pathway.134 Similarly, HIV‐1 viral RNA activates GCN2 in vitro, although GCN2 is targeted for cleavage by the HIV‐1 protease during infection.135 Conceivably, phosphorylation of eIF2α and the subsequent depletion of the available ternary complex pool is beneficial for the host cell, as incapacitating the translational machinery can restrict viral spread. The identification of viral products that counter the activation of PKR and/or eIF2α phosphorylation to maintain viral translation further points to the importance of eIF2α kinases in the antiviral response (for extensive review of such examples, see Ref 136). Alternately, viruses that utilize eIF2‐independent modes of translation may actively induce eIF2α phosphorylation to ameliorate the host antiviral response. IRESs such as the dicistrovirus IGR IRES that can recruit the ribosome independently of factors or initiator Met‐tRNAi are translationally stimulated under cellular stresses and virus infections that induce eIF2α phosphorylation.56, 137 However, there are new data to suggest that viruses may utilize other factors to recruit the Met‐tRNAi. For example, eIF5B and ligatin/eIF2D can deliver Met‐tRNAi to the 40S ribosome bound to some viral mRNAs such as HCV IRES‐like RNAs.138, 139, 140, 141 Interestingly, HCV IRES‐mediated translation is stimulated upon eIF2α phosphorylation by PKR or under conditions that decrease eIF2 activity.142, 143 The fact that the eIF2 ternary complex is dispensable for HCV translation presents an interesting conundrum, since ternary complex‐mediated positioning of the 40S at the initiator codon is a prerequisite for HCV translation.67 The significance of eIF2D during virus infection remains to be determined. In addition to the role of eIF2D on viral translation, recent studies have implicated another Met‐tRNAi‐interacting protein, eIF2A, on HCV IRES translation. In this study, eIF2A knockdown and repletion studies demonstrate that eIF2A is responsible for Met‐tRNAi delivery during stress conditions and is required for HCV proliferation.144 Upon infection, eIF2A is relocalized from the nucleus (and partially the cytoplasm) to an exclusively cytoplasmic distribution, where recruitment to the viral genome occurs by direct interaction with domain IIId of the HCV IRES.144 Since phosphorylation of eIF2α occurs under HCV infection, it is speculated that eIF2A‐dependent translation may play a physiological role during an authentic infection.144
Figure 5.
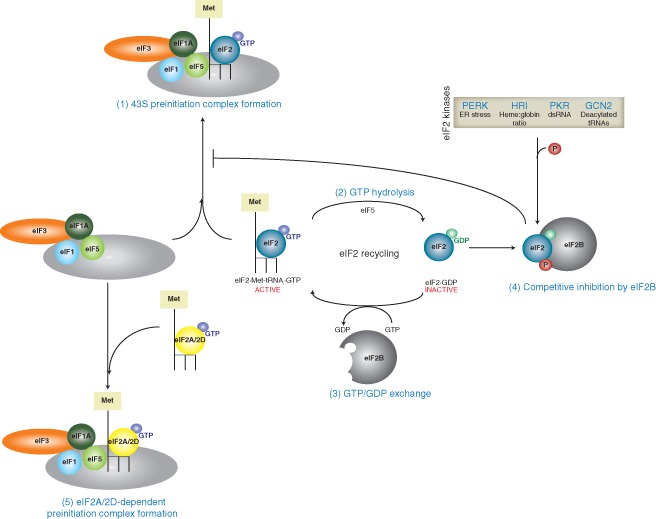
Recycling of eIF2 during translation initiation. During translation initiation, the eIF2·Met‐tRNAi·GTP ternary complex is recruited to the 43S preinitiation complex and is involved in start codon recognition (1). Upon cognate codon:anticodon pairing, eIF5 mediates hydrolysis of eIF2‐bound GTP to GDP (2), which must be recycled by the guanine nucleotide exchange factor eIF2B for subsequent rounds of initiation (3). The α‐subunit of eIF2 is susceptible to phosphorylation by various eIF2α kinases (PERK, HRI, PKR, and GCN2) in response to specific environmental triggers. The phosphorylated form of eIF2α acts as a competitive inhibitor of eIF2B (4), thus preventing recycling of the GDP‐bound eIF2 to the GTP‐bound form and decreasing the availability of the ternary complex pool for translation initiation. Interestingly, some viruses including hepatitis C virus (HCV) and alphaviruses utilize alternate initiation factors such as eIF2A and eIF2D to deliver the initiator tRNA (5). This allows viral translation to proceed when the canonical translation initiation pathway through the eIF2·Met‐tRNAi·GTP ternary complex is compromised.
The use of alternate initiation factors in viral protein synthesis is not restricted to HCV. For the alphavirus SV, translation of its capped, subgenomic 26S RNA can proceed in both an eIF2‐dependent and ‐independent manner. The specific mode of translation is dependent on the context of the experimental assay: transfection or overexpression of the 26S renders it sensitive to eIF4G cleavage and eIF2α phosphorylation whereas 26S translation is refractory to the same conditions within the context of a virally infected cell.145, 146 Resistance of 26S translation to eIF2α phosphorylation is conferred by a stable hairpin structure located ∼25 nucleotides downstream of the translation initiation site.140 Reconstitution experiments using this hairpin structure demonstrated that eIF2D, but not eIF5B, efficiently promoted translation initiation on 26S RNA.140 Interestingly, in an independent study, eIF2A was implicated in eIF2‐independent translation of 26S RNA.146 siRNA‐mediated knockdown of eIF2A results in a substantial decrease in the synthesis of SV structural proteins in PKR+/+, but not PKR0/0 cells.146 While these studies altogether provide evidence that translation of 26S subgenomic RNA during infection likely utilizes an alternate initiation factor, the precise identity of this factor is still debatable. The ability of viruses like HCV and alphaviruses to utilize alternate Met‐tRNAi delivery factors represents novel strategies to evade host translational shutoff and sustain viral translation regardless of the functional status of eIF2. More interestingly, it poses the possibility that a subset of cellular transcripts, particularly those that encode for stress inducible genes, may undergo the same initiation pathway and be preferentially expressed when general translation is compromised.
Increasing Viral Coding Capacity/Recoding Mechanisms
Because viral genomes are extremely compact with a limited sequence space, numerous strategies have evolved to effectively increase the viral coding capacity.1 Maximizing the genetic information in a viral genome by encoding multiple, commonly overlapping genes may be selectively advantageous in establishing a productive viral infection. While the ribosome must maintain a constant translational reading frame to faithfully decode the mRNA into a functional polypeptide, an elongating ribosome may be subjected to the effects of cis‐acting RNA elements that affect accuracy during decoding. A programmed ribosomal frameshift (PRF) involves a displacement in the translational reading frame of the elongating ribosome toward the 5′ end (−1 frameshift) or 3′ end (+1 frameshift) (Figure 6). The parameters constituting a −1 PRF have been extensively defined in viral genomes and involve a ‘slippery’ heptanucleotide RNA motif with the consensus X_XXY_YYZ and a downstream stimulating element (usually a pseudoknot, and in some cases, a stable hairpin) located six to eight nucleotides from the 3′ boundary of the slippery sequence (Figure 6(a)).148, 149, 150, 151 A slippery sequence alone is insufficient for frameshifting to occur152, 153; the additional stimulatory element is required to induce sufficient pausing of the elongating ribosome such that the interactions between the mRNA and the A‐ and P‐site tRNAs are disrupted to allow translocation into an alternate frame.154 Because only the wobble positions of the −1 frame codons are changed relative to the reference frame, −1 decoding involves re‐pairing of the mRNA with near‐cognate tRNAs. By reconstituting an active −1 frameshifting event using mammalian ribosomes and a variant of the coronavirus IBV frameshift signal, cryo‐EM reconstructions of the stalled complexes provided significant insight into the conformational rearrangements and mechanical tensions that occur during the decoding of a ribosomal frameshift.155 Most notably, the stimulatory pseudoknot obstructs the entrance of the mRNA channel and induces a ratchet‐like rearrangement that traps the eEF2 translocase in an orientation that precludes A‐site tRNA binding.155 Additionally, the P‐site tRNA undergoes structural deformation that results from a bending of the D‐arm, suggesting that the distortable nature of the tRNA might be essential in the frameshifting process.155 These cryo‐EM structures provide a mechanical explanation for frameshifting, wherein the ribosome attempting to undergo eEF2‐catalyzed translocation is counteracted by the blockage of the mRNA channel and occlusion of the A site.155 This resistance distorts the P‐site tRNA and places sufficient strain on the anticodon–codon interaction that causes their dissociation.155 Alleviation of this strain likely promotes realignment of the anticodon with the codon in the −1 direction and the occurrence of a frameshifting event.155
Figure 6.
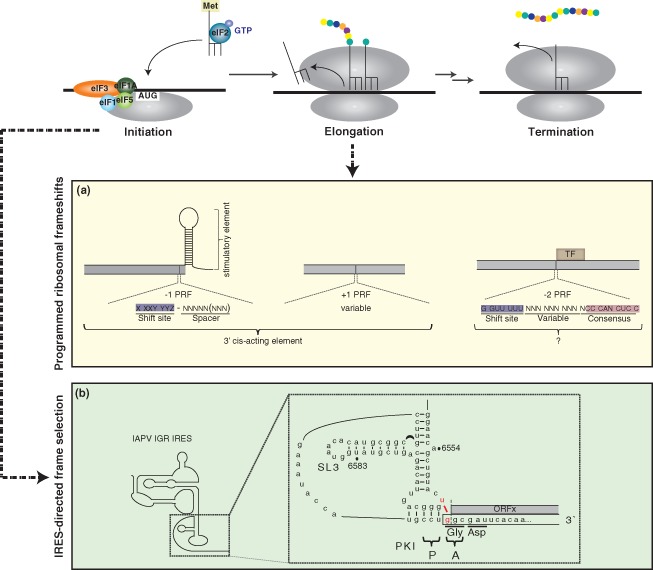
Increasing coding capacity by programmed ribosomal frameshift elements and IRES‐mediated translational reading frame selection. The translation cycle involves the steps of initiation, characterized by the recruitment and positioning of the ribosome at the translational start site; elongation, where amino acids are sequentially added to the growing polypeptide chain; and termination, where the nascent polypeptide is released from the ribosome. Programmed ribosomal frameshifts (PRFs) act on an elongating ribosome whereas translational reading frame selection mediated by the IGR IRES occurs at the level of initiation. (a) Three types of PRFs, including the −1, +1 and −2 PRFs, have been identified in various viral genomes. In the −1 PRF, the frameshift site is comprised of a heptanucleotide sequence with the consensus X_XXY_YYZ (where X represents any nucleotide, Y represents A or U, Z represents A, C, or U, and the underscores designate the codons in the 0 frame) and a downstream spacer sequence. While the consensus for −1 PRFs is well characterized, the frameshift consensus in +1 PRF is more variable. −1 PRFs require a 3′ stimulatory element and +1 PRFs depend on cis‐elements that facilitate the displacement of the ribosome into an alternate frame. The −2 PRF, recently identified in the arterivirus porcine reproductive and respiratory syndrome virus, produces a transframe fusion (TF). The −2 PRF occurs at a conserved G_GUU_UUU sequence and is stimulated by a conserved downstream CCCANCUCC motif located 11 nucleotides downstream. The mechanism of this mode of frameshift has not been fully elucidated. (b) Israeli acute paralysis virus (IAPV) IGR IRES‐mediated translation in the +1 reading frame occurs via a U:G wobble (highlighted in red) adjacent to the IRES translational start site. The first amino acid decoded in the +1 frame is alanine. Mass spectrometry analysis has identified the presence of ORFx in virally infected honeybees, although its role in virus infection is currently unknown. (Reprinted with permission from Ref 147. Copyright 2012 Elsevier Inc.)
Frameshift elements are essential in regulating the translation of proteins during the viral life cycle. In HIV‐1 and other related retroviruses, the expression of the viral proteins, including the RNA‐dependent DNA polymerase, is under the regulation of frameshift signals.156, 157, 158 Classical translation of the viral mRNA terminates at a stop codon to generate exclusively the gag protein, which represents the precursor for the viral structural proteins. Via a −1 PRF approximately 200 nucleotides upstream of the gag stop codon, the gag‐pol fusion is generated from which the viral enzymes are derived (for review, see Ref 159). The frequency of frameshift thus dictates the precise ratio of the viral structural and nonstructural proteins, which is crucial to the assembly of an infectious virion. Deviation of this ratio results in a decrease in viral yield.160 Though initially described in retroviral genomes, PRFs have since been documented in many other viruses and cellular genes and are more prevalent than originally thought. While −1 PRF appears to be the predominant type of frameshifting, examples of +1 PRF are limited. Well characterized modes of +1 PRF have been found in the yeast Ty1 and Ty3 retrotransposons which, similar to −1 PRF, are dependent on cis‐acting elements161, 162 (Figure 6(a)).
Bioinformatic algorithms have proven to be extremely powerful in identifying frameshift elements, some of which act through nonconventional mechanisms.163, 164, 165, 166, 167 For example, until recently, the utilization of −2 PRF is poorly documented in eukaryotes. Computational analysis of various genotype isolates of porcine reproductive and respiratory syndrome virus (PRRSV), a member of the Arteriviridae family, revealed a region of increased conservation in the +1 reading frame encoding viral nsp 2, designated as nsp2TF.166 Mass spectrometric and biochemical analyses demonstrated definitively that translation of nsp2TF occurs via a −2 PRF and necessitates both a G GUU UUU motif at the shift site and a downstream CCCANCUCC for efficient ribosomal frameshifting (Figure 6(a)).166 Interestingly, nsp2TF is partitioned to specific foci under infection and is excluded from replication structures where nsp2 resides.166 The observation that nsp2TF frameshift mutants severely impair virus replication further substantiates the physiological role of nsp2TF during infection. While the precise role of nsp2TF is currently unknown, the novel mode of frameshifting adds to the complexity in discerning the coding potential of a compact viral genome. It is also known that sequences downstream of the −2 PRF signal can also mediate a −1 PRF to generate the two viral replicase precursor polyproteins.168, 169 Thus, many regulatory mechanisms must be in place to ensure that appropriate partitioning of translating ribosomes occurs to allow specific viral proteins to be expressed at precise times during the viral life cycle.
A similar bioinformatics approach has been used to identify enhanced coding potential via an overlapping gene in a subset of dicistroviruses infectious to honeybees and fire ants.167, 170 The alternate gene, ORFx, is encoded in the +1 translational reading frame within the 5′ proximal region of the cistron encoding viral structural proteins.171 Its translation does not occur through a conventional ribosomal frameshift, but through a unique and novel mechanism mediated by the IGR IRES. While canonical frameshift elements act upon an elongating ribosome, the dicistrovirus IRES engages in an alternate reading frame during the initiation step. Translation initiates by a noncanonical U:G basepair adjacent to the tRNA‐mimicry domain of the IRES, which, through a mechanism that is still poorly understood, causes decoding to be displaced in the +1 frame (Figure 6(b)).171 Although the function of ORFx in viral infection is still under investigation, mass spectrometry has confirmed that it is expressed in virus‐infected bees.171 Additionally, the maintenance of the ORFx sequence under selective pressure suggests that there is a biological function during the viral life cycle.
CONCLUSION
Viruses utilize various mechanisms to harness the host translational machinery for viral propagation. While these strategies are diverse and yield differing outcomes on the translational status of the host, they provide unique means to outcompete cellular mRNAs in support of the efficient and sometimes exclusive production of viral proteins. Other mechanisms may function to increase the overall viral coding capacity, which is advantageous in the context of compact genomes. Although many noncanonical mechanisms have been identified and characterized, recent advances in bioinformatic and biochemical approaches have driven the discovery of novel viral translational strategies. For example, technologies such as SILAC (stable isotope labeling by amino acids in cell culture) in combination with Click chemistry have proven useful in the identification and quantification of newly‐synthesized proteins.172, 173 Ribosome profiling has also provided a complementary approach in examining RNAs that are translated. The subcodon resolution of this technique has made it possible to obtain genome‐wide insight into processes intrinsic to translation, including reading frame selection and initiation codon usage.174 In fact, insights into ribosomal frameshift were gleaned not only from bioinformatic approaches but also from ribosomal profiling.175 These techniques have been fundamental in expanding our understanding of the translational responses that occur upon viral infection and other cellular stresses, and will only increasingly perpetuate the discovery of novel translational mechanisms. In light of our increasing understanding of translational control mechanisms, new antiviral therapeutics can also be developed through the discovery of novel compounds that specifically target viral translational pathways176, 177 or modulate viral frameshift elements.178
ACKNOWLEDGMENTS
The authors sincerely apologize for work that was not discussed or cited due to space considerations. Funded by CIHR (MOP‐82144), NSERC, and Genome Canada. EJ is a CIHR New Investigator and MSFHR Scholar. HA is supported by an NSERC CGS D fellowship.
Conflict of interest: The authors have declared no conflicts of interest for this article.
REFERENCES
- 1. Firth AE, Brierley I. Non‐canonical translation in RNA viruses. J Gen Virol 2012, 93:1385–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Walsh D, Mathews MB, Mohr I. Tinkering with translation: protein synthesis in virus‐infected cells. Cold Spring Harb Perspect Biol 2013, 5:a012351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dreher TW, Miller WA. Translational control in positive strand RNA plant viruses. Virology 2006, 344:185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Simon AE, Miller WA. 3′ cap‐independent translation enhancers of plant viruses. Annu Rev Microbiol 2013, 67:21–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nicholson BL, White KA. 3′ Cap‐independent translation enhancers of positive‐strand RNA plant viruses. Curr Opin Virol 2011, 1:373–380. [DOI] [PubMed] [Google Scholar]
- 6. Gebauer F, Hentze MW. Molecular mechanisms of translational control. Nat Rev Mol Cell Biol 2004, 5:827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hinnebusch AG, Lorsch JR. The mechanism of eukaryotic translation initiation: new insights and challenges. Cold Spring Harb Perspect Biol 2012, 4:a011544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aitken CE, Lorsch JR. A mechanistic overview of translation initiation in eukaryotes. Nat Struct Mol Biol 2012, 19:568–576. [DOI] [PubMed] [Google Scholar]
- 9. Kieft JS. Viral IRES RNA structures and ribosome interactions. Trends Biochem Sci 2008, 33:274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jang SK, Krausslich HG, Nicklin MJ, Duke GM, Palmenberg AC, Wimmer E. A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J Virol 1988, 62:2636–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pelletier J, Sonenberg N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 1988, 334:320–325. [DOI] [PubMed] [Google Scholar]
- 12. Kuhn R, Luz N, Beck E. Functional analysis of the internal translation initiation site of foot‐and‐mouth disease virus. J Virol 1990, 64:4625–4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Belsham GJ, Brangwyn JK. A region of the 5′ noncoding region of foot‐and‐mouth disease virus RNA directs efficient internal initiation of protein synthesis within cells: involvement with the role of L protease in translational control. J Virol 1990, 64:5389–5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Borman A, Jackson RJ. Initiation of translation of human rhinovirus RNA: mapping the internal ribosome entry site. Virology 1992, 188:685–696. [DOI] [PubMed] [Google Scholar]
- 15. Brown EA, Zajac AJ, Lemon SM. In vitro characterization of an internal ribosomal entry site (IRES) present within the 5′ nontranslated region of hepatitis A virus RNA: comparison with the IRES of encephalomyocarditis virus. J Virol 1994, 68:1066–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Glass MJ, Jia XY, Summers DF. Identification of the hepatitis A virus internal ribosome entry site: in vivo and in vitro analysis of bicistronic RNAs containing the HAV 5′ noncoding region. Virology 1993, 193:842–852. [DOI] [PubMed] [Google Scholar]
- 17. Plank TD, Kieft JS. The structures of nonprotein‐coding RNAs that drive internal ribosome entry site function. Wiley Interdiscip Rev RNA 2012, 3:195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen CY, Sarnow P. Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Science 1995, 268:415–417. [DOI] [PubMed] [Google Scholar]
- 19. Etchison D, Milburn SC, Edery I, Sonenberg N, Hershey JW. Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000‐dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J Biol Chem 1982, 257:14806–14810. [PubMed] [Google Scholar]
- 20. Gradi A, Svitkin YV, Imataka H, Sonenberg N. Proteolysis of human eukaryotic translation initiation factor eIF4GII, but not eIF4GI, coincides with the shutoff of host protein synthesis after poliovirus infection. Proc Natl Acad Sci USA 1998, 95:11089–11094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Joachims M, Van Breugel PC, Lloyd RE. Cleavage of poly(A)‐binding protein by enterovirus proteases concurrent with inhibition of translation in vitro. J Virol 1999, 73:718–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuyumcu‐Martinez NM, Van Eden ME, Younan P, Lloyd RE. Cleavage of poly(A)‐binding protein by poliovirus 3C protease inhibits host cell translation: a novel mechanism for host translation shutoff. Mol Cell Biol 2004, 24:1779–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hellen CU, Sarnow P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev 2001, 15:1593–1612. [DOI] [PubMed] [Google Scholar]
- 24. Jackson RJ. The current status of vertebrate cellular mRNA IRESs. Cold Spring Harb Perspect Biol 2013, 5:a011569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sweeney TR, Abaeva IS, Pestova TV, Hellen CU. The mechanism of translation initiation on Type 1 picornavirus IRESs. EMBO J 2013, 33:76–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ali IK, McKendrick L, Morley SJ, Jackson RJ. Activity of the hepatitis A virus IRES requires association between the cap‐binding translation initiation factor (eIF4E) and eIF4G. J Virol 2001, 75:7854–7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Borman AM, Bailly JL, Girard M, Kean KM. Picornavirus internal ribosome entry segments: comparison of translation efficiency and the requirements for optimal internal initiation of translation in vitro. Nucleic Acids Res 1995, 23:3656–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Borman AM, Kean KM. Intact eukaryotic initiation factor 4G is required for hepatitis A virus internal initiation of translation. Virology 1997, 237:129–136. [DOI] [PubMed] [Google Scholar]
- 29. Pisarev AV, Chard LS, Kaku Y, Johns HL, Shatsky IN, Belsham GJ. Functional and structural similarities between the internal ribosome entry sites of hepatitis C virus and porcine teschovirus, a picornavirus. J Virol 2004, 78:4487–4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chard LS, Bordeleau ME, Pelletier J, Tanaka J, Belsham GJ. Hepatitis C virus‐related internal ribosome entry sites are found in multiple genera of the family Picornaviridae. J Gen Virol 2006, 87:927–936. [DOI] [PubMed] [Google Scholar]
- 31. Chard LS, Kaku Y, Jones B, Nayak A, Belsham GJ. Functional analyses of RNA structures shared between the internal ribosome entry sites of hepatitis C virus and the picornavirus porcine teschovirus 1 Talfan. J Virol 2006, 80:1271–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hellen CU, de Breyne S. A distinct group of hepacivirus/pestivirus‐like internal ribosomal entry sites in members of diverse picornavirus genera: evidence for modular exchange of functional noncoding RNA elements by recombination. J Virol 2007, 81:5850–5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sweeney TR, Dhote V, Yu Y, Hellen CU. A distinct class of internal ribosomal entry site in members of the Kobuvirus and proposed Salivirus and Paraturdivirus genera of the Picornaviridae. J Virol 2012, 86:1468–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yu Y, Sweeney TR, Kafasla P, Jackson RJ, Pestova TV, Hellen CU. The mechanism of translation initiation on Aichivirus RNA mediated by a novel type of picornavirus IRES. EMBO J 2011, 30:4423–4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dhote V, Sweeney TR, Kim N, Hellen CU, Pestova TV. Roles of individual domains in the function of DHX29, an essential factor required for translation of structured mammalian mRNAs. Proc Natl Acad Sci USA 2012, 109:E3150–E3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pisareva VP, Pisarev AV, Komar AA, Hellen CU, Pestova TV. Translation initiation on mammalian mRNAs with structured 5′UTRs requires DExH‐box protein DHX29. Cell 2008, 135:1237–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brown EA, Day SP, Jansen RW, Lemon SM. The 5′ nontranslated region of hepatitis A virus RNA: secondary structure and elements required for translation in vitro. J Virol 1991, 65:5828–5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Redondo N, Sanz MA, Steinberger J, Skern T, Kusov Y, Carrasco L. Translation directed by hepatitis A virus IRES in the absence of active eIF4F complex and eIF2. PLoS ONE 2012, 7:e52065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Redondo N, Sanz MA, Welnowska E, Carrasco L. Translation without eIF2 promoted by poliovirus 2A protease. PLoS ONE 2011, 6:e25699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Welnowska E, Sanz MA, Redondo N, Carrasco L. Translation of viral mRNA without active eIF2: the case of picornaviruses. PLoS ONE 2011, 6:e22230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bonning BC, Miller WA. Dicistroviruses. Annu Rev Entomol 2010, 55:129–150. [DOI] [PubMed] [Google Scholar]
- 42. Cox‐Foster DL, Conlan S, Holmes EC, Palacios G, Evans JD, Moran NA, Quan PL, Briese T, Hornig M, Geiser DM, et al. A metagenomic survey of microbes in honey bee colony collapse disorder. Science 2007, 318:283–287. [DOI] [PubMed] [Google Scholar]
- 43. Wilson JE, Powell MJ, Hoover SE, Sarnow P. Naturally occurring dicistronic cricket paralysis virus RNA is regulated by two internal ribosome entry sites. Mol Cell Biol 2000, 20:4990–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jang CJ, Jan E. Modular domains of the Dicistroviridae intergenic internal ribosome entry site. RNA 2010, 16:1182–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pfingsten JS, Costantino DA, Kieft JS. Structural basis for ribosome recruitment and manipulation by a viral IRES RNA. Science 2006, 314:1450–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Costantino DA, Pfingsten JS, Rambo RP, Kieft JS. tRNA‐mRNA mimicry drives translation initiation from a viral IRES. Nat Struct Mol Biol 2008, 15:57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Spahn CM, Jan E, Mulder A, Grassucci RA, Sarnow P, Frank J. Cryo‐EM visualization of a viral internal ribosome entry site bound to human ribosomes: the IRES functions as an RNA‐based translation factor. Cell 2004, 118:465–475. [DOI] [PubMed] [Google Scholar]
- 48. Garrey JL, Lee YY, Au HH, Bushell M, Jan E. Host and viral translational mechanisms during cricket paralysis virus infection. J Virol 2010, 84:1124–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moore NF, Kearns A, Pullin JS. Characterization of cricket paralysis virus‐induced polypeptides in Drosophila cells. J Virol 1980, 33:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Woolaway KE, Lazaridis K, Belsham GJ, Carter MJ, Roberts LO. The 5′ untranslated region of Rhopalosiphum padi virus contains an internal ribosome entry site which functions efficiently in mammalian, plant, and insect translation systems. J Virol 2001, 75:10244–10249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Royall E, Woolaway KE, Schacherl J, Kubick S, Belsham GJ, Roberts LO. The Rhopalosiphum padi virus 5′ internal ribosome entry site is functional in Spodoptera frugiperda 21 cells and in their cell‐free lysates: implications for the baculovirus expression system. J Gen Virol 2004, 85:1565–1569. [DOI] [PubMed] [Google Scholar]
- 52. Terenin IM, Dmitriev SE, Andreev DE, Royall E, Belsham GJ, Roberts LO, Shatsky IN. A cross‐kingdom internal ribosome entry site reveals a simplified mode of internal ribosome entry. Mol Cell Biol 2005, 25:7879–7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jan E. Divergent IRES elements in invertebrates. Virus Res 2006, 119:16–28. [DOI] [PubMed] [Google Scholar]
- 54. Jan E, Sarnow P. Factorless ribosome assembly on the internal ribosome entry site of cricket paralysis virus. J Mol Biol 2002, 324:889–902. [DOI] [PubMed] [Google Scholar]
- 55. Sasaki J, Nakashima N. Methionine‐independent initiation of translation in the capsid protein of an insect RNA virus. Proc Natl Acad Sci USA 2000, 97:1512–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wilson JE, Pestova TV, Hellen CU, Sarnow P. Initiation of protein synthesis from the A site of the ribosome. Cell 2000, 102:511–520. [DOI] [PubMed] [Google Scholar]
- 57. Sasaki J, Nakashima N. Translation initiation at the CUU codon is mediated by the internal ribosome entry site of an insect picorna‐like virus in vitro. J Virol 1999, 73:1219–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nishiyama T, Yamamoto H, Shibuya N, Hatakeyama Y, Hachimori A, Uchiumi T, Nakashima N. Structural elements in the internal ribosome entry site of Plautia stali intestine virus responsible for binding with ribosomes. Nucleic Acids Res 2003, 31:2434–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Costantino D, Kieft JS. A preformed compact ribosome‐binding domain in the cricket paralysis‐like virus IRES RNAs. RNA 2005, 11:332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jan E, Kinzy TG, Sarnow P. Divergent tRNA‐like element supports initiation, elongation, and termination of protein biosynthesis. Proc Natl Acad Sci USA 2003, 100:15410–15415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pestova TV, Hellen CU. Translation elongation after assembly of ribosomes on the Cricket paralysis virus internal ribosomal entry site without initiation factors or initiator tRNA. Genes Dev 2003, 17:181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hertz MI, Thompson SR. In vivo functional analysis of the Dicistroviridae intergenic region internal ribosome entry sites. Nucleic Acids Res 2011, 39:7276–7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schuler M, Connell SR, Lescoute A, Giesebrecht J, Dabrowski M, Schroeer B, Mielke T, Penczek PA, Westhof E, Spahn CM. Structure of the ribosome‐bound cricket paralysis virus IRES RNA. Nat Struct Mol Biol 2006, 13:1092–1096. [DOI] [PubMed] [Google Scholar]
- 64. Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver‐specific MicroRNA. Science 2005, 309:1577–1581. [DOI] [PubMed] [Google Scholar]
- 65. Spahn CM, Kieft JS, Grassucci RA, Penczek PA, Zhou K, Doudna JA, Frank J. Hepatitis C virus IRES RNA‐induced changes in the conformation of the 40s ribosomal subunit. Science 2001, 291:1959–1962. [DOI] [PubMed] [Google Scholar]
- 66. Hashem Y, des Georges A, Dhote V, Langlois R, Liao HY, Grassucci RA, Pestova TV, Hellen CU, Frank J. Hepatitis‐C‐virus‐like internal ribosome entry sites displace eIF3 to gain access to the 40S subunit. Nature 2013, 503:539–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pestova TV, Shatsky IN, Fletcher SP, Jackson RJ, Hellen CU. A prokaryotic‐like mode of cytoplasmic eukaryotic ribosome binding to the initiation codon during internal translation initiation of hepatitis C and classical swine fever virus RNAs. Genes Dev 1998, 12:67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Otto GA, Puglisi JD. The pathway of HCV IRES‐mediated translation initiation. Cell 2004, 119:369–380. [DOI] [PubMed] [Google Scholar]
- 69. Boehringer D, Thermann R, Ostareck‐Lederer A, Lewis JD, Stark H. Structure of the hepatitis C virus IRES bound to the human 80S ribosome: remodeling of the HCV IRES. Structure 2005, 13:1695–1706. [DOI] [PubMed] [Google Scholar]
- 70. Buratti E, Tisminetzky S, Zotti M, Baralle FE. Functional analysis of the interaction between HCV 5′UTR and putative subunits of eukaryotic translation initiation factor eIF3. Nucleic Acids Res 1998, 26:3179–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kieft JS, Zhou K, Jubin R, Doudna JA. Mechanism of ribosome recruitment by hepatitis C IRES RNA. RNA 2001, 7:194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sizova DV, Kolupaeva VG, Pestova TV, Shatsky IN, Hellen CU. Specific interaction of eukaryotic translation initiation factor 3 with the 5′ nontranslated regions of hepatitis C virus and classical swine fever virus RNAs. J Virol 1998, 72:4775–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kolupaeva VG, Pestova TV, Hellen CU. An enzymatic footprinting analysis of the interaction of 40S ribosomal subunits with the internal ribosomal entry site of hepatitis C virus. J Virol 2000, 74:6242–6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lytle JR, Wu L, Robertson HD. The ribosome binding site of hepatitis C virus mRNA. J Virol 2001, 75:7629–7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Malygin AA, Kossinova OA, Shatsky IN, Karpova GG. HCV IRES interacts with the 18S rRNA to activate the 40S ribosome for subsequent steps of translation initiation. Nucleic Acids Res 2013, 41:8706–8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Berry KE, Waghray S, Mortimer SA, Bai Y, Doudna JA. Crystal structure of the HCV IRES central domain reveals strategy for start‐codon positioning. Structure 2011, 19:1456–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Berry KE, Waghray S, Doudna JA. The HCV IRES pseudoknot positions the initiation codon on the 40S ribosomal subunit. RNA 2010, 16:1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Filbin ME, Vollmar BS, Shi D, Gonen T, Kieft JS. HCV IRES manipulates the ribosome to promote the switch from translation initiation to elongation. Nat Struct Mol Biol 2013, 20:150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fukushi S, Okada M, Stahl J, Kageyama T, Hoshino FB, Katayama K. Ribosomal protein S5 interacts with the internal ribosomal entry site of hepatitis C virus. J Biol Chem 2001, 276:20824–20826. [DOI] [PubMed] [Google Scholar]
- 80. Locker N, Easton LE, Lukavsky PJ. HCV and CSFV IRES domain II mediate eIF2 release during 80S ribosome assembly. EMBO J 2007, 26:795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fraser CS, Hershey JW, Doudna JA. The pathway of hepatitis C virus mRNA recruitment to the human ribosome. Nat Struct Mol Biol 2009, 16:397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Filbin ME, Kieft JS. HCV IRES domain IIb affects the configuration of coding RNA in the 40S subunit's decoding groove. RNA 2011, 17:1258–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Buck CB, Shen X, Egan MA, Pierson TC, Walker CM, Siliciano RF. The human immunodeficiency virus type 1 gag gene encodes an internal ribosome entry site. J Virol 2001, 75:181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Herbreteau CH, Weill L, Decimo D, Prevot D, Darlix JL, Sargueil B, Ohlmann T. HIV‐2 genomic RNA contains a novel type of IRES located downstream of its initiation codon. Nat Struct Mol Biol 2005, 12:1001–1007. [DOI] [PubMed] [Google Scholar]
- 85. Ohlmann T, Lopez‐Lastra M, Darlix JL. An internal ribosome entry segment promotes translation of the simian immunodeficiency virus genomic RNA. J Biol Chem 2000, 275:11899–11906. [DOI] [PubMed] [Google Scholar]
- 86. Nicholson MG, Rue SM, Clements JE, Barber SA. An internal ribosome entry site promotes translation of a novel SIV Pr55(Gag) isoform. Virology 2006, 349:325–334. [DOI] [PubMed] [Google Scholar]
- 87. Weill L, James L, Ulryck N, Chamond N, Herbreteau CH, Ohlmann T, Sargueil B. A new type of IRES within gag coding region recruits three initiation complexes on HIV‐2 genomic RNA. Nucleic Acids Res 2010, 38:1367–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Locker N, Chamond N, Sargueil B. A conserved structure within the HIV gag open reading frame that controls translation initiation directly recruits the 40S subunit and eIF3. Nucleic Acids Res 2011, 39:2367–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Plank TD, Whitehurst JT, Kieft JS. Cell type specificity and structural determinants of IRES activity from the 5′ leaders of different HIV‐1 transcripts. Nucleic Acids Res 2013, 41:6698–6714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chang J, Guo JT, Jiang D, Guo H, Taylor JM, Block TM. Liver‐specific microRNA miR‐122 enhances the replication of hepatitis C virus in nonhepatic cells. J Virol 2008, 82:8215–8223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Henke JI, Goergen D, Zheng J, Song Y, Schuttler CG, Fehr C, Junemann C, Niepmann M. microRNA‐122 stimulates translation of hepatitis C virus RNA. EMBO J 2008, 27:3300–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jopling CL, Schutz S, Sarnow P. Position‐dependent function for a tandem microRNA miR‐122‐binding site located in the hepatitis C virus RNA genome. Cell Host Microbe 2008, 4:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Jangra RK, Yi M, Lemon SM. Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR‐122. J Virol 2010, 84:6615–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Roberts AP, Lewis AP, Jopling CL. miR‐122 activates hepatitis C virus translation by a specialized mechanism requiring particular RNA components. Nucleic Acids Res 2011, 39:7716–7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Roberts AP, Doidge R, Tarr AW, Jopling CL. The P body protein LSm1 contributes to stimulation of hepatitis C virus translation, but not replication, by microRNA‐122. Nucleic Acids Res 2013, 42:1257–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up‐regulate translation. Science 2007, 318:1931–1934. [DOI] [PubMed] [Google Scholar]
- 97. Shimakami T, Yamane D, Jangra RK, Kempf BJ, Spaniel C, Barton DJ, Lemon SM. Stabilization of hepatitis C virus RNA by an Ago2‐miR‐122 complex. Proc Natl Acad Sci USA 2012, 109:941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Li Y, Masaki T, Yamane D, McGivern DR, Lemon SM. Competing and noncompeting activities of miR‐122 and the 5′ exonuclease Xrn1 in regulation of hepatitis C virus replication. Proc Natl Acad Sci USA 2013, 110:1881–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Xue S, Barna M. Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat Rev Mol Cell Biol 2012, 13:355–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Mauro VP, Edelman GM. The ribosome filter hypothesis. Proc Natl Acad Sci USA 2002, 99:12031–12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Cherry S, Doukas T, Armknecht S, Whelan S, Wang H, Sarnow P, Perrimon N. Genome‐wide RNAi screen reveals a specific sensitivity of IRES‐containing RNA viruses to host translation inhibition. Genes Dev 2005, 19:445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Nishiyama T, Yamamoto H, Uchiumi T, Nakashima N. Eukaryotic ribosomal protein RPS25 interacts with the conserved loop region in a dicistroviral intergenic internal ribosome entry site. Nucleic Acids Res 2007, 35:1514–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Muhs M, Yamamoto H, Ismer J, Takaku H, Nashimoto M, Uchiumi T, Nakashima N, Mielke T, Hildebrand PW, Nierhaus KH, et al. Structural basis for the binding of IRES RNAs to the head of the ribosomal 40S subunit. Nucleic Acids Res 2011, 39:5264–5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hertz MI, Landry DM, Willis AE, Luo G, Thompson SR. Ribosomal protein S25 dependency reveals a common mechanism for diverse internal ribosome entry sites and ribosome shunting. Mol Cell Biol 2013, 33:1016–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Landry DM, Hertz MI, Thompson SR. RPS25 is essential for translation initiation by the Dicistroviridae and hepatitis C viral IRESs. Genes Dev 2009, 23:2753–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ben‐Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M. The structure of the eukaryotic ribosome at 3.0 A resolution. Science 2011, 334:1524–1529. [DOI] [PubMed] [Google Scholar]
- 107. Lee AS, Burdeinick‐Kerr R, Whelan SP. A ribosome‐specialized translation initiation pathway is required for cap‐dependent translation of vesicular stomatitis virus mRNAs. Proc Natl Acad Sci USA 2013, 110:324–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Huang JY, Su WC, Jeng KS, Chang TH, Lai MM. Attenuation of 40S ribosomal subunit abundance differentially affects host and HCV translation and suppresses HCV replication. PLos Pathog 2012, 8:e1002766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Yoon A, Peng G, Brandenburger Y, Zollo O, Xu W, Rego E, Ruggero D. Impaired control of IRES‐mediated translation in X‐linked dyskeratosis congenita. Science 2006, 312:902–906. [DOI] [PubMed] [Google Scholar]
- 110. Jack K, Bellodi C, Landry DM, Niederer RO, Meskauskas A, Musalgaonkar S, Kopmar N, Krasnykh O, Dean AM, Thompson SR, et al. rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Mol Cell 2011, 44:660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Connor JH, Lyles DS. Vesicular stomatitis virus infection alters the eIF4F translation initiation complex and causes dephosphorylation of the eIF4E binding protein 4E‐BP1. J Virol 2002, 76:10177–10187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Haghighat A, Mader S, Pause A, Sonenberg N. Repression of cap‐dependent translation by 4E‐binding protein 1: competition with p220 for binding to eukaryotic initiation factor‐4E. EMBO J 1995, 14:5701–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wilson DN, Doudna Cate JH. The structure and function of the eukaryotic ribosome. Cold Spring Harb Perspect Biol 2012, 4:a011536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ofengand J. Ribosomal RNA pseudouridines and pseudouridine synthases. FEBS Lett 2002, 514:17–25. [DOI] [PubMed] [Google Scholar]
- 115. Mader S, Lee H, Pause A, Sonenberg N. The translation initiation factor eIF‐4E binds to a common motif shared by the translation factor eIF‐4 gamma and the translational repressors 4E‐binding proteins. Mol Cell Biol 1995, 15:4990–4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Lamphear BJ, Kirchweger R, Skern T, Rhoads RE. Mapping of functional domains in eukaryotic protein synthesis initiation factor 4G (eIF4G) with picornaviral proteases. Implications for cap‐dependent and cap‐independent translational initiation. J Biol Chem 1995, 270:21975–21983. [DOI] [PubMed] [Google Scholar]
- 117. Imataka H, Sonenberg N. Human eukaryotic translation initiation factor 4G (eIF4G) possesses two separate and independent binding sites for eIF4A. Mol Cell Biol 1997, 17:6940–6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Ohlmann T, Rau M, Pain VM, Morley SJ. The C‐terminal domain of eukaryotic protein synthesis initiation factor (eIF) 4G is sufficient to support cap‐independent translation in the absence of eIF4E. EMBO J 1996, 15:1371–1382. [PMC free article] [PubMed] [Google Scholar]
- 119. de Breyne S, Yu Y, Unbehaun A, Pestova TV, Hellen CU. Direct functional interaction of initiation factor eIF4G with type 1 internal ribosomal entry sites. Proc Natl Acad Sci USA 2009, 106:9197–9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Ho BC, Yu SL, Chen JJ, Chang SY, Yan BS, Hong QS, Singh S, Kao CL, Chen HY, Su KY, et al. Enterovirus‐induced miR‐141 contributes to shutoff of host protein translation by targeting the translation initiation factor eIF4E. Cell Host Microbe 2011, 9:58–69. [DOI] [PubMed] [Google Scholar]
- 121. Arias C, Walsh D, Harbell J, Wilson AC, Mohr I. Activation of host translational control pathways by a viral developmental switch. PLoS Pathog 2009, 5:e1000334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Walsh D, Mohr I. Assembly of an active translation initiation factor complex by a viral protein. Genes Dev 2006, 20:461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Stinski MF. Synthesis of proteins and glycoproteins in cells infected with human cytomegalovirus. J Virol 1977, 23:751–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Perez C, McKinney C, Chulunbaatar U, Mohr I. Translational control of the abundance of cytoplasmic poly(A) binding protein in human cytomegalovirus‐infected cells. J Virol 2011, 85:156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Walsh D, Perez C, Notary J, Mohr I. Regulation of the translation initiation factor eIF4F by multiple mechanisms in human cytomegalovirus‐infected cells. J Virol 2005, 79:8057–8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Kudchodkar SB, Yu Y, Maguire TG, Alwine JC. Human cytomegalovirus infection induces rapamycin‐insensitive phosphorylation of downstream effectors of mTOR kinase. J Virol 2004, 78:11030–11039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Isler JA, Skalet AH, Alwine JC. Human cytomegalovirus infection activates and regulates the unfolded protein response. J Virol 2005, 79:6890–6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Dobrikova E, Shveygert M, Walters R, Gromeier M. Herpes simplex virus proteins ICP27 and UL47 associate with polyadenylate‐binding protein and control its subcellular distribution. J Virol 2010, 84:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Salaun C, MacDonald AI, Larralde O, Howard L, Lochtie K, Burgess HM, Brook M, Malik P, Gray NK, Graham SV. Poly(A)‐binding protein 1 partially relocalizes to the nucleus during herpes simplex virus type 1 infection in an ICP27‐independent manner and does not inhibit virus replication. J Virol 2010, 84:8539–8548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Lee YJ, Glaunsinger BA. Aberrant herpesvirus‐induced polyadenylation correlates with cellular messenger RNA destruction. PLoS Biol 2009, 7:e1000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. McKinney C, Perez C, Mohr I. Poly(A) binding protein abundance regulates eukaryotic translation initiation factor 4F assembly in human cytomegalovirus‐infected cells. Proc Natl Acad Sci USA 2012, 109:5627–5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. McKinney C, Yu D, Mohr I. A new role for the cellular PABP repressor Paip2 as an innate restriction factor capable of limiting productive cytomegalovirus replication. Genes Dev 2013, 27:1809–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Dever TE, Dar AC, Sicheri F. The eIF2a kinases In: Mathews MB, Sonenberg N, Hershey JW, eds. Translational Control in Biology and Medicine. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2007, 319–344. [Google Scholar]
- 134. Berlanga JJ, Ventoso I, Harding HP, Deng J, Ron D, Sonenberg N, Carrasco L, de Haro C. Antiviral effect of the mammalian translation initiation factor 2alpha kinase GCN2 against RNA viruses. EMBO J 2006, 25:1730–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. del Pino J, Jimenez JL, Ventoso I, Castello A, Munoz‐Fernandez MA, de Haro C, Berlanga JJ. GCN2 has inhibitory effect on human immunodeficiency virus‐1 protein synthesis and is cleaved upon viral infection. PLoS ONE 2012, 7:e47272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Walsh D, Mohr I. Viral subversion of the host protein synthesis machinery. Nat Rev Microbiol 2011, 9:860–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Fernandez J, Yaman I, Sarnow P, Snider MD, Hatzoglou M. Regulation of internal ribosomal entry site‐mediated translation by phosphorylation of the translation initiation factor eIF2alpha. J Biol Chem 2002, 277:19198–19205. [DOI] [PubMed] [Google Scholar]
- 138. Dmitriev SE, Terenin IM, Andreev DE, Ivanov PA, Dunaevsky JE, Merrick WC, Shatsky IN. GTP‐independent tRNA delivery to the ribosomal P‐site by a novel eukaryotic translation factor. J Biol Chem 2010, 285:26779–26787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Pestova TV, de Breyne S, Pisarev AV, Abaeva IS, Hellen CU. eIF2‐dependent and eIF2‐independent modes of initiation on the CSFV IRES: a common role of domain II. EMBO J 2008, 27:1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Skabkin MA, Skabkina OV, Dhote V, Komar AA, Hellen CU, Pestova TV. Activities of ligatin and MCT‐1/DENR in eukaryotic translation initiation and ribosomal recycling. Genes Dev 2010, 24:1787–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Terenin IM, Dmitriev SE, Andreev DE, Shatsky IN. Eukaryotic translation initiation machinery can operate in a bacterial‐like mode without eIF2. Nat Struct Mol Biol 2008, 15:836–841. [DOI] [PubMed] [Google Scholar]
- 142. Rivas‐Estilla AM, Svitkin Y, Lopez Lastra M, Hatzoglou M, Sherker A, Koromilas AE. PKR‐dependent mechanisms of gene expression from a subgenomic hepatitis C virus clone. J Virol 2002, 76:10637–10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Robert F, Kapp LD, Khan SN, Acker MG, Kolitz S, Kazemi S, Kaufman RJ, Merrick WC, Koromilas AE, Lorsch JR, et al. Initiation of protein synthesis by hepatitis C virus is refractory to reduced eIF2.GTP.Met‐tRNA(i)(Met) ternary complex availability. Mol Biol Cell 2006, 17:4632–4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Kim JH, Park SM, Park JH, Keum SJ, Jang SK. eIF2A mediates translation of hepatitis C viral mRNA under stress conditions. EMBO J 2011, 30:2454–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Sanz MA, Castello A, Ventoso I, Berlanga JJ, Carrasco L. Dual mechanism for the translation of subgenomic mRNA from Sindbis virus in infected and uninfected cells. PLoS ONE 2009, 4:e4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Ventoso I, Sanz MA, Molina S, Berlanga JJ, Carrasco L, Esteban M. Translational resistance of late alphavirus mRNA to eIF2alpha phosphorylation: a strategy to overcome the antiviral effect of protein kinase PKR. Genes Dev 2006, 20:87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Wang QS, Au HH, Jan E. Methods for studying IRES‐mediated translation of positive‐strand RNA viruses. Methods 2013, 59:167–179. [DOI] [PubMed] [Google Scholar]
- 148. Kim YG, Su L, Maas S, O'Neill A, Rich A. Specific mutations in a viral RNA pseudoknot drastically change ribosomal frameshifting efficiency. Proc Natl Acad Sci USA 1999, 96:14234–14239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Brierley I, Jenner AJ, Inglis SC. Mutational analysis of the "slippery‐sequence" component of a coronavirus ribosomal frameshifting signal. J Mol Biol 1992, 227:463–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Yu CH, Noteborn MH, Pleij CW, Olsthoorn RC. Stem‐loop structures can effectively substitute for an RNA pseudoknot in ‐1 ribosomal frameshifting. Nucleic Acids Res 2011, 39:8952–8959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Dinman JD, Icho T, Wickner RB. A‐1 ribosomal frameshift in a double‐stranded RNA virus of yeast forms a gag‐pol fusion protein. Proc Natl Acad Sci USA 1991, 88:174–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Tu C, Tzeng TH, Bruenn JA. Ribosomal movement impeded at a pseudoknot required for frameshifting. Proc Natl Acad Sci USA 1992, 89:8636–8640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Kontos H, Napthine S, Brierley I. Ribosomal pausing at a frameshifter RNA pseudoknot is sensitive to reading phase but shows little correlation with frameshift efficiency. Mol Cell Biol 2001, 21:8657–8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Brierley I, Digard P, Inglis SC. Characterization of an efficient coronavirus ribosomal frameshifting signal: requirement for an RNA pseudoknot. Cell 1989, 57:537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Namy O, Moran SJ, Stuart DI, Gilbert RJ, Brierley I. A mechanical explanation of RNA pseudoknot function in programmed ribosomal frameshifting. Nature 2006, 441:244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Jacks T, Varmus HE. Expression of the Rous sarcoma virus pol gene by ribosomal frameshifting. Science 1985, 230:1237–1242. [DOI] [PubMed] [Google Scholar]
- 157. Jacks T, Power MD, Masiarz FR, Luciw PA, Barr PJ, Varmus HE. Characterization of ribosomal frameshifting in HIV‐1 gag‐pol expression. Nature 1988, 331:280–283. [DOI] [PubMed] [Google Scholar]
- 158. Jacks T, Madhani HD, Masiarz FR, Varmus HE. Signals for ribosomal frameshifting in the Rous sarcoma virus gag‐pol region. Cell 1988, 55:447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Brierley I, Dos Ramos FJ. Programmed ribosomal frameshifting in HIV‐1 and the SARS‐CoV. Virus Res 2006, 119:29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Shehu‐Xhilaga M, Crowe SM, Mak J. Maintenance of the Gag/Gag‐Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J Virol 2001, 75:1834–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Belcourt MF, Farabaugh PJ. Ribosomal frameshifting in the yeast retrotransposon Ty: tRNAs induce slippage on a 7 nucleotide minimal site. Cell 1990, 62:339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Farabaugh PJ, Zhao H, Vimaladithan A. A novel programed frameshift expresses the POL3 gene of retrotransposon Ty3 of yeast: frameshifting without tRNA slippage. Cell 1993, 74:93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Jagger BW, Wise HM, Kash JC, Walters KA, Wills NM, Xiao YL, Dunfee RL, Schwartzman LM, Ozinsky A, Bell GL, et al. An overlapping protein‐coding region in influenza A virus segment 3 modulates the host response. Science 2012, 337:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Loughran G, Firth AE, Atkins JF. Ribosomal frameshifting into an overlapping gene in the 2B‐encoding region of the cardiovirus genome. Proc Natl Acad Sci USA 2011, 108:E1111–E1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Chung BY, Miller WA, Atkins JF, Firth AE. An overlapping essential gene in the Potyviridae. Proc Natl Acad Sci USA 2008, 105:5897–5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Fang Y, Treffers EE, Li Y, Tas A, Sun Z, van der Meer Y, de Ru AH, van Veelen PA, Atkins JF, Snijder EJ, et al. Efficient ‐2 frameshifting by mammalian ribosomes to synthesize an additional arterivirus protein. Proc Natl Acad Sci USA 2012, 109:E2920–E2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Firth AE, Wang QS, Jan E, Atkins JF. Bioinformatic evidence for a stem‐loop structure 5′‐adjacent to the IGR‐IRES and for an overlapping gene in the bee paralysis dicistroviruses. Virol J 2009, 6:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. den Boon JA, Snijder EJ, Chirnside ED, de Vries AA, Horzinek MC, Spaan WJ. Equine arteritis virus is not a togavirus but belongs to the coronaviruslike superfamily. J Virol 1991, 65:2910–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Brierley I, Boursnell ME, Binns MM, Bilimoria B, Blok VC, Brown TD, Inglis SC. An efficient ribosomal frame‐shifting signal in the polymerase‐encoding region of the coronavirus IBV. EMBO J 1987, 6:3779–3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Sabath N, Price N, Graur D. A potentially novel overlapping gene in the genomes of Israeli acute paralysis virus and its relatives. Virol J 2009, 6:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Ren Q, Wang QS, Firth AE, Chan MM, Gouw JW, Guarna MM, Foster LJ, Atkins JF, Jan E. Alternative reading frame selection mediated by a tRNA‐like domain of an internal ribosome entry site. Proc Natl Acad Sci USA 2012, 109:E630–E639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 2002, 1:376–386. [DOI] [PubMed] [Google Scholar]
- 173. Somasekharan SP, Stoynov N, Rotblat B, Leprivier G, Galpin JD, Ahern CA, Foster LJ, Sorensen PH. Identification and quantification of newly synthesized proteins translationally regulated by YB‐1 using a novel Click‐SILAC approach. J Proteomics 2012, 77:e1–e10. [DOI] [PubMed] [Google Scholar]
- 174. Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome‐wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 324:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Michel AM, Choudhury KR, Firth AE, Ingolia NT, Atkins JF, Baranov PV. Observation of dually decoded regions of the human genome using ribosome profiling data. Genome Res 2012, 22:2219–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Cencic R, Hall DR, Robert F, Du Y, Min J, Li L, Qui M, Lewis I, Kurtkaya S, Dingledine R, et al. Reversing chemoresistance by small molecule inhibition of the translation initiation complex eIF4F. Proc Natl Acad Sci USA 2011, 108:1046–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, Gross JD, Degterev A, Yuan J, Chorev M, et al. Small‐molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell 2007, 128:257–267. [DOI] [PubMed] [Google Scholar]
- 178. Marcheschi RJ, Tonelli M, Kumar A, Butcher SE. Structure of the HIV‐1 frameshift site RNA bound to a small molecule inhibitor of viral replication. ACS Chem Biol 2011, 6:857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]