

Summary
Bat species around the world have recently been recognized as major reservoirs of several zoonotic viruses, such as severe acute respiratory syndrome coronavirus (SARS‐CoV), Middle East respiratory syndrome coronavirus (MERS‐CoV), Nipah virus and Hendra virus. In this study, consensus primer‐based reverse transcriptase polymerase chain reactions (RT‐PCRs) and high‐throughput sequencing were performed to investigate viruses in bat faecal samples collected at 11 natural bat habitat sites from July to December 2015 in Korea. Diverse coronaviruses were first detected in Korean bat faeces, including alphacoronaviruses, SARS‐CoV‐like and MERS‐CoV‐like betacoronaviruses. In addition, we identified a novel bat rotavirus belonging to group H rotavirus which has only been described in human and pigs until now. Therefore, our results suggest the need for continuing surveillance and additional virological studies in domestic bat.
Keywords: bat, coronavirus, Middle East respiratory syndrome, severe acute respiratory syndrome, group H rotavirus, Korea
Introduction
Bats are considered reservoirs of several emerging infectious disease. Recently emerging human viruses, such as severe acute respiratory syndrome coronavirus (SARS‐CoV), Middle East respiratory syndrome coronavirus (MERS‐CoV), Nipah virus, Hendra virus and Ebola virus, are thought be bat‐borne viruses (Han et al., 2015). In addition, new species of influenza A viruses, lyssaviruses, paramyxoviruses and coronaviruses continue to be discovered in bats around the world (Tong et al., 2013; Banyard et al., 2014; Yang et al., 2014; Mortlock et al., 2015). Reassortant group A rotavirus was also detected in the faeces of a straw‐coloured fruit bat (Esona et al., 2010). Therefore, viruses that originate in bats may be a source of additional spill over from wildlife into domestic animals and humans (Plowright et al., 2015).
Since the outbreak of MERS‐CoV in Korea in 2015, the prediction and control of newly emerging viruses has gained urgency. However, in Korea, there have been a few studies on viruses in domestic bat species. Serological evidence of Japanese encephalitis virus (JEV) was reported in the early 1990s (Lee and Lee, 1992). In 1995, there was a report on the isolation and genetic characterization on a hantavirus from bats (Jung and Kim, 1995). More information of viruses in bats is needed to clarify the role of these animals in infectious diseases epidemiology.
Therefore, in this study, we investigated viruses in bat species in Korea, using 49 faecal samples collected from July to December 2015 in 11 sites in natural bat habitats.
Materials and Methods
Samples
From July to December in 2015, 49 faecal samples were collected at 11 sites in natural bat habitats, such as caves, an abandoned mine and under a bridge (Table 1). Fresh bat faeces were placed into transport medium in a 10% suspension and were transported to the laboratory for further laboratory analysis.
Table 1.
Information on bat faecal samples
| Cases | Collected date | Region | Site name | Major bat species | No. of samples |
|---|---|---|---|---|---|
| 1 | 24 July 2015 | Danyang | Under HSA Bridge | Unknown | 6c |
| 2 | 4 August 2015 | Wonju | HO cave |
Rhinolophus ferrumequinum, Myotis macrodactylus |
4c |
| 3 | 14 August 2015 | Taebaek | YY cavea |
Myotis aurascens Kuzyakin Myotis petax Rhinolophus ferrumequinum |
5c |
| 4 | 16 August 2015 | Uljin | SL caveb | Unknown | 1c |
| 5 | 9 October 2015 | Jeongsun | PG cave | Rhinolophus ferrumequinum | 4 |
| 6 | 14 October 2015 | Inje | BM abandoned mine | Rhinolophus ferrumequinum | 6 |
| 7 | 18 October 2015 | Taebaek | YY cavea |
Myotis aurascens Kuzyakin Myotis petax Rhinolophus ferrumequinum |
3 |
| 8 | 25 October 2015 | Bonghwa | DRN cave |
Rhinolophus ferrumequinum Myotis macrodactylus |
3 |
| 9 | 18 November 2015 | Taebaek | YY cavea |
Myotis aurascens Kuzyakin Myotis petax Rhinolophus ferrumequinum |
1 |
| 10 | 19 November 2015 | Uljin | SL caveb | Unknown | 1 |
| 11 | 1 December 2015 | Hapcheon | BT cave | Unknown | 3 |
| 12 | 1 December 2015 | Munkyung | BGS cave | Miniopterus schreibersii | 4 |
| 13 | 1 December 2015 | Yeongwol | YJ cave | Rhinolophus ferrumequinum | 4 |
| 14 | 1 December 2015 | Miwon | CS cave | Rhinolophus ferrumequinum | 4 |
collected at the same site (YY cave) on a different date.
collected at the same site (SL cave) on a different date.
pooled and sequenced by high‐throughput sequencing.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
The major bat species at the collection sites were determined based on morphology and on previous data from bats' roosting sites (Han et al., 2012). In this study, the major bat species in the collecting sites were Rhinolophus ferrumequinum, Myotis macrodactylus, Myotis aurascens Kuzyakin, Myotis petax and Miniopterus schreibersii.
Viral detection
Consensus primer‐based reverse transcriptase polymerase chain reactions (RT‐PCRs) were performed to detect influenza A viruses, coronaviruses, lyssaviruses and paramyxoviruses (Fouchier et al., 2000; Poon et al., 2005; Vázquez‐Morón et al., 2006; Tong et al., 2008). The detailed information on the RT‐PCRs is in Table S1. A total of 49 bat faecal samples were tested. The amplicons from positive RT‐PCRs were sequenced using target‐specific forward and reverse primers synthesized by Cosmogenetech Co. Ltd (Daejeon, Korea). The nucleotide data obtained from Sanger sequencing of the PCR fragments were further analysed with related sequences in GenBank using BioEdit (Hall, 1999) and MEGA version 6 (Tamura et al., 2013).
High‐throughput sequencing
In total, 16 bat faecal samples that were collected in four sites were used for high‐throughput sequencing (Table 1). The supernatants from suspensions of one, four, five and six faecal samples collected in Danyang, Wonju, Taebaek and Uljin, respectively, were pooled by sampling site. Each of the four pooled samples was filtered through a 0.2‐μm filter and ultracentrifuged at 252 000 g for 1 h. Each pellet was resuspended in 500 μl of 1× digestion buffer (Turbo DNA Free Kit; Ambion, Darmstadt, Germany), and a total of 10 μl of Turbo DNase was added. After incubating for 30 min at 37°C, suspensions were transferred to a 1.5‐ml reaction tube and centrifuged at 1500 g for 3 min. The supernatants were used for RNA extraction. RNA was extracted using Trizol LS (Invitrogen Corp., Carlsbad, CA, USA), following the manufacturer's manual. The extracted RNA was submitted to Macrogen (Seoul, Korea) for high‐throughput sequencing in a HiSeq 2000 sequencing system based on the transcriptome de novo sequencing platform.
The obtained viral contigs were analysed and annotated by the MG‐RAST server (Meyer et al., 2008). The cut‐off for the annotation was 10−5, 60% and 15 for maximum e‐value, minimum percentage identity and minimum alignment length, respectively. The contigs annotated as mammalian viruses were validated through BLASTn (http://www.ncbi.nlm.nih.gov). The GenBank accession numbers for the nucleotide sequences in this study are KU528584–KU528594.
Results
PCR‐based detection of influenza A viruses, coronaviruses, lyssaviruses and paramyxoviruses in bat faeces
There were no samples positive for influenza A viruses, lyssaviruses or paramyxoviruses.
Among 14 cases of the submitted samples, three cases were positive for coronavirus by RT‐PCR, showing the expected 440 nt target. These included the following: (Case no. 1) one of four samples from HO cave in Wonju where Rhinolophus ferrumequinum and Myotis macrodactylus were the major species present, (Case no. 6) one of six samples BM abandoned mine in Inje where Rhinolophus ferrumequinum was the major species found and (Case no. 12) two of four samples BGS (Case no. 12) cave in Munkyung where Miniopterus schreibersii was found to be the major species (Table 1).
In the HO cave, one of four faecal samples (B15‐8) was positive by coronavirus‐specific RT‐PCR. When the PCR‐positive fragment was sequenced, it was found to be a bat coronavirus (Bat CoV) belonging to the genus alphacoronavirus and showing 89% nucleotide sequence identity with Bat CoV A620 (GenBank accession no. DQ648828) detected in China in 2005.
In the BM abandoned mine, one of four faecal samples (B15‐21) was found to be a Bat CoV belonging to the genus betacoronavirus and showing 98% nucleotide identity with SARS‐like Bat CoV Lingbao‐331 (GenBank accession no. KF294456) detected in China in 2012.
In the BGS cave, two of four faecal samples (B15‐40 and B15‐41) were found to be Bat CoVs belonging to the genus alphacoronavirus and showing 99% nucleotide identity with Bat CoV A895 (GenBank accession no. DQ648841) and 98% nucleotide identity with Bat CoV A910 (GenBank accession no. DQ648849), both of which were detected in China in 2005.
High‐throughput sequencing from the pooled bat faecal samples
As shown in Table 1, cases 1, 2, 3 and 4 were further analysed by high‐throughput sequencing. By Hiseq 2000 sequencing, 4341, 162 559, 7690 and 16 657 contigs were obtained from cases 1, 2, 3 and 4, respectively. Based on the annotation by the MG‐RAST system, several viral sequences that are related to mammalian viruses, insect viruses, plant viruses, fungal viruses and bacteriophages were identified.
The mammalian viral sequences annotated by the MG‐RAST system were further validated by BLASTn (http://www.ncbi.nlm.nih.gov). The BLASTn result is presented in Table 2. Fifteen viral sequences from case no. 1 were similar to porcine rotavirus SKA‐1 (69–73% nucleotide identity), Bat CoV SC2013 (70–91% nucleotide identity), Bat CoV HKU5‐5 (82% nucleotide identity), Alphacoronavirus Eptesicus fuscus‐related strains (95–96% nucleotide identity) and Rhinolophus pearsoni bunyavirus Shaanxi 2011 (72% nucleotide identity). Three viral sequences from case no. 2 were similar to Banna virus 02VN078b (90% nucleotide identity), Bat CoV CDPHE15 (80% nucleotide identity) and Bat CoV Neixiang‐31 (82% nucleotide identity). There was no mammalian viral sequences in cases 3 and 4.
Table 2.
Mammalian viral sequences matched with contigs from high‐throughput sequencing
| Contigsa | Size (bases) | BLASTn search parameters | Matched strain | ||
|---|---|---|---|---|---|
| E‐value | % Nucleotide identity | Accession No. | Strain name | ||
| C6091 | 404 | 2e‐37 | 73 | AB576634 | Porcine rotavirus SKA‐1 |
| C6719 | 438 | 1e‐21 | 69 | AB576631 | Porcine rotavirus SKA‐1 |
| C7554 | 211 | 9e‐19 | 73 | AB576625 | Porcine rotavirus SKA‐1 |
| C9980 | 215 | 5e‐22 | 73 | AB576629 | Porcine rotavirus SKA‐1 |
| C10008 | 285 | 2e‐35 | 73 | AB576629 | Porcine rotavirus SKA‐1 |
| C10298 | 355 | 2e‐48 | 73 | AB576631 | Porcine rotavirus SKA‐1 |
| C3339 | 230 | 1e‐55 | 83 | KJ473821 | BtVs‐BetaCoV/SC2013 |
| C6171 | 203 | 1e‐16 | 70 | KJ473821 | BtVs‐BetaCoV/SC2013 |
| C10151 | 219 | 1e‐36 | 82 | EF065512 | Bat coronavirus HKU5‐5 |
| C10579 | 207 | 3e‐67 | 91 | KJ473821 | BtVs‐BetaCoV/SC2013 |
| C6073 | 243 | 6e‐98 | 95 | HQ585081 | Alphacoronavirus Eptesicus fuscus/Appalachian Ridge/P1‐C1148/IT/USA/2009 |
| C8820 | 417 | 0.0 | 96 | JX537911 | Alphacoronavirus Eptesicus fuscus strain NeCoV isolate Ffrag_SAGA_bigbrown_2010 |
| C8271 | 270 | 2e‐61 | 95 | HQ585082 | Alphacoronavirus Eptesicus fuscus/Appalachian Ridge/P1‐C837/IT/USA/2009 |
| C7729 | 209 | 2e‐21 | 76 | KF294382 | Anlong Ms bat coronavirus isolate Neixiang‐32 |
| C5770 | 313 | 2e‐34 | 72 | KC154063 | Rhinolophus pearsoni bunyavirus isolate Bat Rp‐BunyaV/Shaanxi2011 |
| C700585 | 207 | 8e‐70 | 90 | EU265697 | Banna virus isolate 02VN078b |
| C817114 | 201 | 6e‐40 | 80 | KF430219 | Bat coronavirus CDPHE15/USA/2006 |
| C695165 | 226 | 6e‐53 | 82 | KF294381 | Neixiang Md bat coronavirus isolate Neixiang‐31 |
Contigs obtained from case no. 1 and case no 2. No mammalian viral sequences were detected in case no. 3 and case no 4.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Genetic characteristics of bat coronaviruses in Korea
Partial RNA‐dependent RNA polymerase (RDRP) sequences obtained from B15‐8 (sample in case no. 2), B15‐21 (sample in case no. 6), B15‐40 and B15‐41 (sample in case no. 12) were phylogenetically compared with other reference sequences (Fig. 1a). Three of these sequences clustered with the alphacoronaviruses (B15‐8, 40 and 41), and the other one clustered with the betacoronaviruses (B15‐21). Bat CoV B15‐8 grouped with bat CoVs found in China, showing 85.7–88.8% nucleotide identities, and was found to be closely related with Bat CoV‐512 and porcine epidemic diarrhoea virus (PEDV)‐CV777, showing 78.6% and 74.6% nucleotide identities, respectively. Bat CoV B15‐21 grouped with SARS‐CoV‐like Bat CoVs, such as Bat CoV Rf1 and Bat CoV HKU3 with 90.3–97.7% nucleotide identities, and was found to be closely related to human SARS‐CoVs (SARS‐CoV Tor2, MA15, ExoN1 strains), showing 89.1% nucleotide identity. Bat CoV B15‐40 and B15‐41 grouped with Bat CoV 1A and 1B, and nucleotide identities within the group were 92.3–97.1%.
Figure 1.
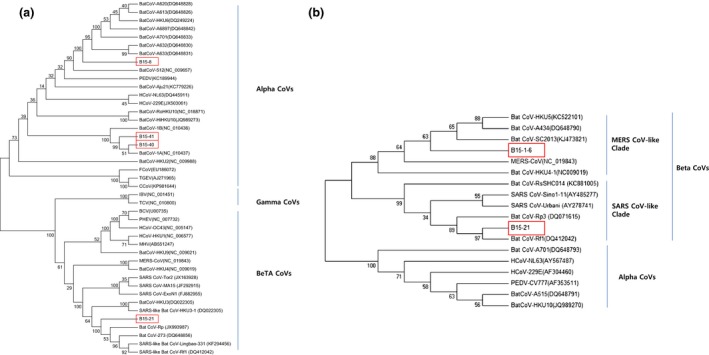
Phylogenetic trees constructed from bat coronavirus sequences detected in this study and from other coronaviruses. (a) bootstrap consensus tree inferred from 1000 replicates with the UPGMA method (evolutionary distance parameter: maximum composite likelihood method) based on 351 bases of the RNA‐dependent RNA polymerase gene, (b) bootstrap consensus tree inferred from 1000 replicates using the neighbour‐joining method (evolutionary distance parameter: maximum composite likelihood method) based on 163 bases of the spike gene. The box indicates the bat CoV sequences obtained in this study.
Because an RDRP sequence from betacoronavirus‐related Bat CoV B15‐1‐6 (detected from the pooled sample in case no. 1) was not obtained, a partial spike gene sequence (c10151) was compared with the spike genes of reference betacoronaviruses (Fig. 1b). In addition, the full spike gene sequence obtained from Bat CoV B15‐21 was also analysed. Bat CoV B15‐1‐6 grouped with MERS‐CoV‐like Bat CoVs (Bat CoV strains HKU5, A434, SC2013 and HKU4), showing 80.9–82.2% nucleotide identities, and was found to be closely related to MERS‐CoV EMC, showing 76.6% of nucleotide identity.
The amino acids sequence encoded by the full spike gene of B15‐21 was aligned and compared with human SARS‐CoV and SARS‐CoV‐like Bat CoV sequences (Figure S1). The S2 domain is more conserved than the S1 domain in the spike protein. The receptor binding domain (RBD) of the SARS‐CoV‐like Bat CoVs, except for RaSHC014, had two major deletion sites, TGNYN (446–450) and PFSPDGKPCTPPA (472–484), compared to human SARS coronaviruses sequences. The receptor binding motif (RBM) and human ACE2 (hACE2)‐interacting residue were highly variable between human SARS‐CoVs and SARS‐CoV‐like Bat CoVs but RaSHC014 strain.
Phylogenetic characterization of Group H bat rotavirus in Korea
Porcine rotavirus‐related sequences from pooled bat faeces in case no. 1 were analysed with reference sequences of rotaviruses in human and animals (Fig. 2). Based on the partial sequences of the VP1, VP3 and VP4 regions, the rotavirus‐related sequences were found to belong to rotavirus group H. The group H bat rotavirus in this study showed 69.4–72.7%, 71.1–72.9% and 67.3–72.2% nucleotide identities with the other group H rotaviruses, based on VP1, VP3 and VP4 partial sequences, respectively.
Figure 2.
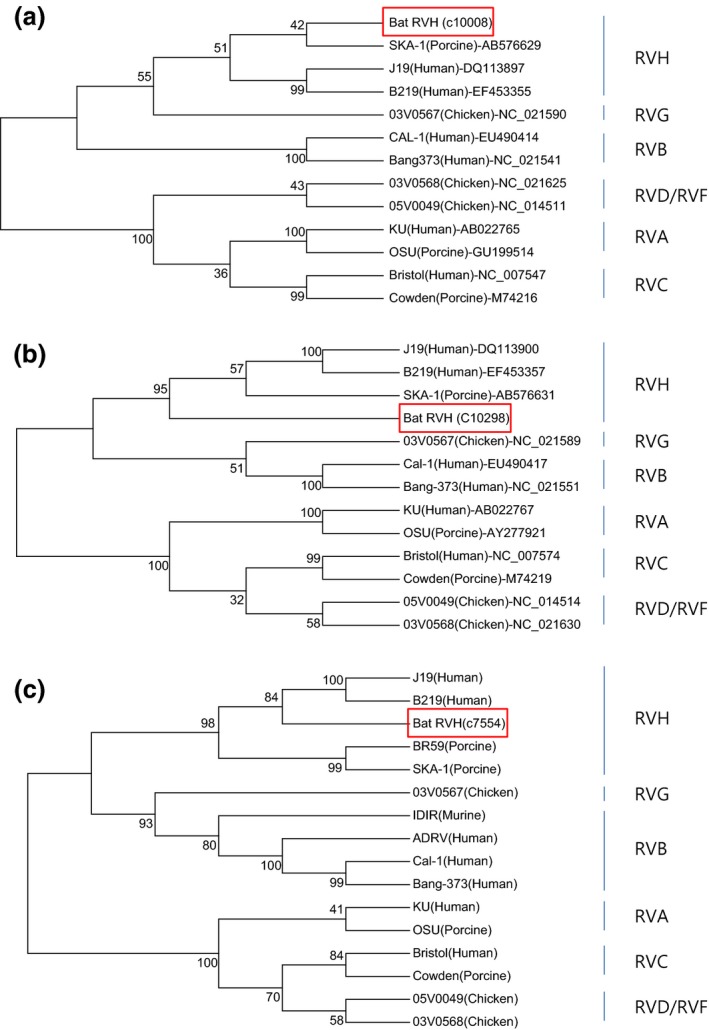
Phylogenetic trees constructed from Group H rotavirus‐related sequences detected in bat faeces in this study and sequences of other, representative rotavirus strains. The bootstrap consensus tree inferred from 1000 replicates using the neighbour‐joining method (evolutionary distances were computed using the Kimura 2‐parameter method). (a) 275 bases of the VP1 region, (b) 347 bases of the VP3 region, (c) 202 bases of the VP4 region. The box indicates the group H bat rotavirus sequences obtained in this study.
Discussion
According to a recent report, there are 23 species of bats in Korea (Han et al., 2012). Most Korean bats are thought to be insectivores, as no fruit bats have been found in Korea to date, except for some fruit bats as pets or indoor exhibitions (Han et al., 2010). Through the target‐specific RT‐PCR, partial RDRP sequences of three alphacoronaviruses (B15‐8, 40 and 41) and one betacoronavirus (B15‐21) were detected in the faeces of Korean bats. Bat CoV B15‐8 was detected in faeces collected at the HO cave where Rhinolophus ferrumequinum and Myotis macrodactylus were the major species. This virus was found to be closely related with Bat CoVs recently found in China and PEDV, a porcine diarrhoeic virus around world. On the other hand, Bat CoVs B15‐40 and 41 were detected at the BGS cave, where Miniopterus schreibersii was the major species. These sequences were closely related with Bat CoV 1A and 1B, which were reported to have a clear host species restriction to Miniopterus magnate and Miniopterus pusillus in Hong Kong (Chu et al., 2008). As Bat CoVs B15‐40 and 41 were also found in faeces in a cave where Miniopterus schreibersii was dominant, Bat CoV strains in the same lineage with Bat CoV 1A and 1B may be adapted to the Miniopterus spp.
It was interesting that Bat CoV B15‐21 found in this study was closely related to SARS‐CoV‐like Bat CoVs, such as Bat CoV Rf1, based on the partial RDRP and S genes, respectively. The newly identified Bat CoV B15‐21 clustered with the SARS‐CoV‐like Bat CoV group in the phylogenetic analysis. The major bat species in the site where Bat CoV B15‐21 was collected was Rhinolophus ferrumequinum, which is consistent with a previous report that SARS‐CoV‐like bat CoVs were mostly found in Rhinolophus spp. (Yuan et al., 2010). The amino acids of its full spike gene showed that the B15‐21 strain is closely related to SARS‐CoV‐like Bat CoVs Rp3, Rf1, HKU3‐2 and 273 rather than human SARS‐CoVs and Bat CoV RaSHC014, which was reported to have high potential for human emergence (Menachery et al., 2015) and showed two specific deletion sites in the RBD of the spike protein. One additional betacoronavirus (B15‐1‐6), which was not detected by the consensus primer‐based RDRP‐specific RT‐PCR (Poon et al., 2005), was detected by high‐throughput sequencing as shown in Table 2. Three contigs were similar to Bat CoV SC2013, which is a MERS‐related betacoronavirus that was found in bats in China (Yang et al., 2014). One contig was also similar to Bat CoV HKU5‐5, which clustered with MERS‐CoV (Woo et al., 2007; Annan et al., 2013). Phylogenetic analysis based on partial spike gene sequences of Bat CoV B15‐1‐6 showed that it belonged to a MERS‐CoV‐like clade with previously known MERS‐CoV and MERS‐CoV‐like Bat CoVs.
Although information of the associated bat species was not clearly available, this study first report that SARS‐CoV and MERS‐CoV‐like bat CoVs exist in Korea. This may imply that other strains or types of those coronaviruses may circulate in bat species in Korea. Therefore, we cannot ignore appearance of novel or variant coronavirus through local emergence in Korea, like SARS and MERS in China and Middle East, respectively.
In addition to coronaviruses, this study is the first to determine the existence of group H rotaviruses in bat faeces collected in Korea. Group H rotavirus is a recently proposed group of rotaviruses that include strains ADRV‐N and B219, which infect human adults (Alam et al., 2007; Matthijnssens et al., 2012). So far, group H rotaviruses have only been reported in human and pigs (Molinari et al., 2015), but this study provides evidence that bat species may be a host of group H RVs. To confirm that, there should be follow‐up studies including virus isolation and characterization, genomic analysis, continuous surveillance and VP6‐based classification (Matthijnssens et al., 2012) to find its prevalence, epidemiology and zoonotic potential.
Localized emergences of zoonotic diseases are usually due to expansion of the wildlife–human interface (Morse et al., 2012). The human–bat interface in Korea has not been fully described. People usually recognize that bats are flying and roosting in the deep forest or caves. However, the sites where the bat faecal samples were collected showed some evidence of human–bat contact (Figure S2). Some caves or abandoned mine where bats are roosting are located near human habitats. People living there frequently visit into the caves or the abandoned mine to take a rest or to ferment foods. Some caves were also used for some religious ceremony by Korean traditional shamans.
In this study, SARS‐CoV‐like and MERS‐CoV‐like bat CoVs and group H rotavirus were detected for this first time in Korea, which may be of interest because of their zoonosis potential. Several kinds of evidence for human–bat contact were also found. Therefore, the potential for novel or variant human viruses from bats in Korea should not be ignored. Continuous surveillance and additional virological research should be a priority to prevent future emergences of zoonotic viral diseases.
Supporting information
Table S1. Information on targets and primers for viral detection.
Figure S1. Amino acid‐based multiple alignment of spike proteins from B15‐21 and other reference strains.
Figure S2. Possible evidence of human‐bat interaction in the bat habitats in this study.
Acknowledgement
This work was supported by grants of the KRIBB Initiative programme (Grant No. KGM4691511) and by the BioNano Health‐Guard Research Center funded by the Ministry of Science, ICT & Future Planning (MSIP) of Korea as a Global Frontier Project (Grant No. H‐GUARD 2013M3A6B2078954). This research was also supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (Grant No. HI15C3036).
References
- Alam, M. M. , Kobayashi N., Ishino M., Ahmed M. S., Ahmed M. U., Paul S. K., Muzumdar B. K., Hussain Z., Wang Y. H., and Naik T. N., 2007: Genetic analysis of an ADRV‐N‐like novel rotavirus strain B219 detected in a sporadic case of adult diarrhea in Bangladesh. Arch. Virol. 152, 199–208. [DOI] [PubMed] [Google Scholar]
- Annan, A. , Baldwin H. J., Corman V. M., Klose S. M., Owusu M., Nkrumah E. E., Badu E. K., Anti P., Agbenyega O., Meyer B., Oppong S., Sarkodie Y. A., Kalko E. K. V., Lina P. H. C., Godlevska E. V., Reusken C., Seebens A., Gloza‐Rausch F., Vallo P., Tschapka M., Drosten C., and Drexler J. F., 2013: Human betacoronavirus 2c EMC/2012–related viruses in bats, Ghana and Europe. Emerg. Infect. Dis. 19, 456–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banyard, A. C. , Evans J. S., Luo T. R., and Fooks A. R., 2014: Lyssaviruses and bats: emergence and zoonotic threat. Viruses 6, 2974–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu, D. K. W. , Peiris J. S. M., Chen H., Guan Y., and Poon L. L. M., 2008: Genomic characterizations of bat coronaviruses (1A, 1B and HKU8) and evidence for co‐infections in Miniopterus bats. J. Gen. Virol. 89, 1282–1287. [DOI] [PubMed] [Google Scholar]
- Esona, M. D. , Mijatovic‐Rustempasic S., Conrardy C., Tong S., Kuzmin I. V., Agwanda B., Breiman R. F., Banyai K., Niezgoda M., Rupprecht C. E., Gentsch J. R., and Bowen M. D., 2010: Reassortant group A rotavirus from straw‐colored fruit bat (Eidolon helvum). Emerg. Infect. Dis. 16, 1844–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouchier, R. A. , Bestebroer T. M., Herfst S., Van Der Kemp L., Rimmelzwaan G. F., and Osterhaus A. D., 2000: Detection of influenza A viruses from different species by PCR amplification of conserved sequences in the matrix gene. J. Clin. Microbiol. 38, 4096–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, T. A. , 1999: BioEdit: a user‐friendly biological sequence alignment editor and analysis program for Windows 95/98/NT Nucl. Nucleic Acids Symp. Ser. 41, 95–98. [Google Scholar]
- Han, J. E. , Gomez D. K., Kim J. H., Choresca C. H. Jr, Shin S. P., and Park S. C., 2010: Isolation of a zoonotic pathogen Kluyvera ascorbata from Egyptian fruit‐bat Rousettus aegyptiacus . J. Vet. Med. Sci. 72, 85–87. [DOI] [PubMed] [Google Scholar]
- Han, S. , Jung C. W., Choi Y. G., and Kim S. S., 2012: Sounds of the Bats in Korea. National Institute of Biological Resources Press, Incheon. [Google Scholar]
- Han, H.‐J. , Wen H.‐L., Zhou C.‐M., Chen F.‐F., Luo L.‐M., Liu J.‐W., and Yu X.‐J., 2015: Bats as reservoirs of severe emerging infectious diseases. Virus Res. 205, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, Y. , and Kim G. R., 1995: Genomic characterization of M and S RNA segments of hantaviruses isolated from bats. Acta Virol. 39, 231–233. [PubMed] [Google Scholar]
- Lee, J. , and Lee Y. T., 1992: Detection of antibodies in Korean bats to Japanese encephalitis virus. Korean J. Microbiol. 30, 119–123. [Google Scholar]
- Matthijnssens, J. , Otto P., Ciarlet M., Desselberger U., Van Ranst M., and Johne R., 2012: VP6‐sequence‐based cutoff values as a criterion for rotavirus species demarcation. Arch. Virol. 157, 1177–1182. [DOI] [PubMed] [Google Scholar]
- Menachery, V. D. , Yount B. L. Jr, Debbink K., Agnihothram S., Gralinski L. E., Plante J. A., Graham R. L., Scobey T., Ge X.‐Y., Donaldson E. F., Randell S. H., Lanzavecchia A., Marasco W. A., Shi Z.‐L., and Baric R. S., 2015: A SARS‐like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 21, 1508–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, F. , Paarmann D., D'Souza M., Olson R., Glass E. M., Kubal M., Paczian T., Rodriguez A., Stevens R., Wilke A., Wilkening J., and Edwards R. A., 2008: The metagenomics RAST server – a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari, B. L. D. , Alfieri A. F., and Alfieri A. A., 2015: Genetic variability of VP6, VP7, VP4, and NSP4 genes of porcine rotavirus group H detected in Brazil. Virus Res. 197, 48–53. [DOI] [PubMed] [Google Scholar]
- Morse, S. S. , Mazet J. A. K., Woolhouse M., Parrish C. R., Carroll D., Karesh W. B., Zambrana‐Torrelio C., Lipkin W. I., and Daszak P., 2012: Prediction and prevention of the next pandemic zoonosis. Lancet 380, 1956–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortlock, M. , Kuzmin I. V., Weyer J., Gilbert A. T., Agwanda B., Rupprecht C. E., Nel L. H., Kearney T., Malekani J. M., and Markotter W., 2015: Novel paramyxoviruses in bats from Sub‐Saharan Africa, 2007–2012. Emerg. Infect. Dis. 21, 1840–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowright, R. K. , Eby P., Hudson P. J., Smith I. L., Westcott D., Bryden W. L., Middleton D., Reid P. A., McFarlane R. A., Martin G., Tabor G. M., Skerratt L. F., Anderson D. L., Crameri G., Quammen D., Jordan D., Freeman P., Wang L.‐F., Epstein J. H., Marsh G. A., Kung N. Y., and McCallum H., 2015: Ecological dynamics of emerging bat virus spillover. Proc. R. Soc. B. Biol. Sci. 282, 20142124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon, L. L. M. , Chu D. K. W., Chan K. H., Wong O. K., Ellis T. M., Leung Y. H. C., Lau S. K. P., Woo P. C. Y., Suen K. Y., Yuen K. Y., Guan Y., and Peiris J. S. M., 2005: Identification of a novel coronavirus in bats. J. Virol. 79, 2001–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. , Stecher G., Peterson D., Filipski A., and Kumar S., 2013: MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, S. , Chern S.‐W. W., Li Y., Pallansch M. A., and Anderson L. J., 2008: Sensitive and broadly reactive reverse transcription‐PCR assays to detect novel paramyxoviruses. J. Clin. Microbiol. 46, 2652–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, S. , Zhu X., Li Y., Shi M., Zhang J., Bourgeois M., Yang H., Chen X., Recuenco S., Gomez J., Chen L.‐M., Johnson A., Tao Y., Dreyfus C., Yu W., McBride R., Carney P. J., Gilbert A. T., Chang J., Guo Z., Davis C. T., Paulson J. C., Stevens J., Rupprecht C. E., Holmes E. C., Wilson I. A., and Donis R. O., 2013: New world bats harbor diverse influenza a viruses. PLoS Pathog. 9, e1003657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vázquez‐Morón, S. , Avellón A., and Echevarría J. E., 2006: RT‐PCR for detection of all seven genotypes of Lyssavirus genus. J. Virol. Methods 135, 281–287. [DOI] [PubMed] [Google Scholar]
- Woo, P. C. Y. , Wang M., Lau S. K. P., Xu H., Poon R. W. S., Guo R., Wong B. H. L., Gao K., Tsoi H.‐W., Huang Y., Li K. S. M., Lam C. S. F., Chan K.‐H., Zheng B.‐J., and Yuen K.‐Y., 2007: Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J. Virol. 81, 1574–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, L. , Wu Z., Ren X., Yang F., Zhang J., He G., Dong J., Sun L., Zhu Y., Zhang S., and Jin Q., 2014: MERS–related betacoronavirus in Vespertilio superans bats, China. Emerg. Infect. Dis. 20, 1260–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, J. , Hon C.‐C., Li Y., Wang D., Xu G., Zhang H., Zhou P., Poon L. L. M., Lam T. T.‐Y., Leung F. C.‐C., and Shi Z., 2010: Intraspecies diversity of SARS‐like coronaviruses in Rhinolophus sinicus and its implications for the origin of SARS coronaviruses in humans. J. Gen. Virol. 91, 1058–1062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Information on targets and primers for viral detection.
Figure S1. Amino acid‐based multiple alignment of spike proteins from B15‐21 and other reference strains.
Figure S2. Possible evidence of human‐bat interaction in the bat habitats in this study.