

Abstract
Pathogen safety is crucial for plasma‐derived clotting factor concentrates used in the treatment of bleeding disorders. Plasma, the starting material for these products, is collected by plasmapheresis (source plasma) or derived from whole blood donations (recovered plasma). The primary measures regarding pathogen safety are selection of healthy donors donating in centers with appropriate epidemiologic data for the main blood‐transmissible viruses, screening donations for the absence of relevant infectious blood‐borne viruses, and release of plasma pools for further processing only if they are nonreactive for serologic markers and nucleic acids for these viruses. Despite this testing, pathogen inactivation and/or removal during the manufacturing process of plasma‐derived clotting factor concentrates is required to ensure prevention of transmission of infectious agents. Historically, hepatitis viruses and human immunodeficiency virus have posed the greatest threat to patients receiving plasma‐derived therapy for treatment of hemophilia or von Willebrand disease. Over the past 30 years, dedicated virus inactivation and removal steps have been integrated into factor concentrate production processes, essentially eliminating transmission of these viruses. Manufacturing steps used in the purification of factor concentrates have also proved to be successful in reducing potential prion infectivity. In this review, current techniques for inactivation and removal of pathogens from factor concentrates are discussed. Ideally, production processes should involve a combination of complementary steps for pathogen inactivation and/or removal to ensure product safety. Finally, potential batch‐to‐batch contamination is avoided by stringent cleaning and sanitization methods as part of the manufacturing process.
Abbreviations
- B19V
parvovirus B19
- BVDV
bovine viral diarrhea virus
- EMA
European Medicines Agency
- HAV
hepatitis A virus
- PARV4
human parvovirus 4
- PPV
porcine parvovirus
- PRV
pseudorabies virus
- QSEAL
Quality Standards of Excellence, Assurance, and Leadership
- SINV
Sindbis virus
- vCJD
variant Creutzfeldt‐Jakob disease
- WNV
West Nile virus
Human plasma is a source of clotting factor concentrates used for treatment of bleeding disorders caused by coagulation factor deficiencies. Such disorders include hemophilia A (Factor [F]VIII), hemophilia B (F IX), and von Willebrand disease (von Willebrand factor [VWF]); rarer disorders such as fibrinogen, FXIII, and FX deficiencies; and a number of acquired bleeding disorders. For von Willebrand disease, some rare bleeding disorders, and acquired bleeding, plasma‐derived clotting factor concentrates, but not recombinant concentrates, are available. A four‐factor prothrombin complex concentrate (Kcentra [Beriplex P/N in Europe], CSL Behring, Marburg, Germany) has been recently approved in the United States for reversal of warfarin.1
Efforts to prevent transmission of infectious agents were accelerated after the recognition of human immunodeficiency virus (HIV) transmission to hemophilia patients in 1983 and the identification of hepatitis C virus (HCV) thereafter. By the 1990s, there was substantial clinical evidence of a lack of transmission of lipid membrane‐enveloped viruses (hepatitis B [HBV], HCV, and HIV) using solvent/detergent (S/D) treatment.2 Nonenveloped viruses, including hepatitis A virus (HAV) and parvovirus B19 (B19V), presented additional challenges, as they are not inactivated by this technique.3, 4, 5 In Germany, pasteurization (heating at 60°C for 10 hours in a stabilized aqueous solution) was developed in the late 1970s for a clotting factor concentrate as a dedicated virus reduction method; this FVIII and VWF concentrate was licensed in 1981 and no proven cases of virus transmissions were reported.6
More recently, prion transmission has become a concern.7, 8, 9 Manufacturing processes have evolved to include multiple steps to prevent transmission of both known and emerging blood‐borne pathogens.10 The objective of this review is to examine current methods to prevent pathogen transmission via plasma‐derived clotting factor concentrates by state‐of‐the‐art selection of plasma, and effective pathogen inactivation and removal by the manufacturing process.
Pathogen Reduction Regulations for Plasma‐Derived Products
Clotting factor concentrates are derived from pools consisting of large numbers of donations.2 The fractionation pool size varies depending on the size of the vessel used to pool the plasma. A batch of final product can include several fractionation pools. Donor screening and testing of donations and fractionation pools are the first steps in product safety, followed by pathogen inactivation and/or removal (i.e., reduction) during manufacturing. Source plasma from apheresis commonly comes from compensated donors, while recovered plasma is from whole blood donors not compensated in cash.11, 12 The manufacturing process of clotting factor concentrates from both starting materials and their safety records are virtually identical.11, 12
The European Medicines Agency (EMA) published a guideline focusing on the safety of plasma‐derived products;10 the fourth version details measures necessary to prevent transmission of viruses to recipients of plasma‐derived products. These measures include selection of donors, screening of donations and plasma pools for markers of infection with known viruses, and a manufacturing process with a high capacity to inactivate and/or remove viruses by selected steps validated for their virus reduction capacity. The guidelines state that “effective steps for the inactivation/removal of a wide range of viruses of diverse physicochemical characteristics” are required during manufacture. Both the EMA and the World Health Organization (WHO) recommend that two distinct steps with complementary modes of action be used,10, 13 helping to ensure that any virus surviving Step 1 is inactivated and/or removed in Step 2. At least one step should inactivate or remove nonenveloped viruses. The EMA guideline further states that if a manufacturing process contains a step that reliably inactivates and/or removes a wide range of enveloped and nonenveloped viruses and additional manufacturing steps reliably contribute to inactivation and/or removal of viruses, the second step might not be required.10 In the United States, the manufacture of plasma‐derived products must include at least one effective step for removal and/or inactivation of viruses.14 Further, careful validation is required for each inactivation or removal step. Various methods of virus inactivation and removal used in concentrates available in many countries are presented in Table 1.15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32
Table 1.
Virus inactivation and/or removal methods for selected plasma‐derived clotting factor products1, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32
| Brand name (manufacturer) | Concentrated coagulation factor(s) | Virus inactivation and removal methods | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Precipitation | Chromatography | Pasteurization | S/D | Dry heat | Lyophilization | Vapor heat | Virus filtration | ||
| Alphanate SD (Grifols) | FVIII, VWF | x | xa | x | x | x | |||
| Biostate (CSL Ltd.) | FVIII, VWF | x | xb | x | x | x | |||
| Humate‐P/Haemate‐P (CSL Behring) | FVIII, VWF | x | x | x | |||||
| Koate DVI (Talecris Biotherapeutics) | FVIII, VWF | x | x | x | |||||
| Wilate (Octapharma) | FVIII, VWF | xc | x | x | x | ||||
| Hemofil M (Baxter) | FVIII | xc | x | x | |||||
| Monoclate‐P (CSL Behring) | FVIII | xa | x | x | |||||
| AlphaNine SD (Grifols) | F IX | xc | x | x | x | ||||
| Bebulin VH (Baxter) | F IX | xc | x | x | x | ||||
| Mononine (CSL Behring) | F IX | xa | x | x | |||||
| Profilnine SD (Grifols) | F IX | xc | x | x | |||||
| Fibrogammin P/Corifact (CSL Behring) | FXIII | x | xc | x | x | x | |||
| Beriplex P/N/Kcentra (CSL Behring) | FII, FVII, F IX, FX | x | xc | x | x | x | |||
| Octaplex (Octapharma) | FII, FVII, F IX, FX | xc | x | x | x | ||||
| Haemocomplettan P (CSL Behring) | Fibrinogen | x | x | x | |||||
Immunoaffinity chromatography.
Size‐exclusion chromatography.
Ion‐exchange chromatography.
To estimate the potential of specific steps for reducing prion infectivity, the EMA recommends evaluation studies for prions.7, 9 Established processes for the production of clotting factor concentrates have been shown to reduce prion infectivity.7, 33, 34
Biologic products, including recombinant clotting factor products, are required to undergo complementary pathogen reduction steps analogous to those of plasma‐derived clotting factor concentrates.35, 36 Validation requirements are also similar for recombinant proteins.35, 36 The safety focus in plasma‐derived products has received greater attention because of the history of virus transmissions before 1990. However, the potential for virus contamination is an issue for all biologic products.
Plasma Screening
Adherence to strict donor selection criteria and screening of donations effectively reduces the virus load to be inactivated and/or removed from the plasma pool for fractionation. Currently, no screening tests are able to detect prion infectivity in donors,37, 38, 39 although an experimental blood test for prion infection was recently developed.40 As a precaution, the EMA, the US Food and Drug Administration (FDA), and WHO recommend deferral of donors at risk of developing Creutzfeldt‐Jakob disease (CJD) and its variant (vCJD) using a series of questions to exclude donations at risk from pooling and further processing.7, 8, 9
Although screening for viral markers by serology and virus nucleic acid by nucleic acid testing (NAT) ensures that nearly all plasma units entering production are free of HBV, HCV, and HIV, inactivation and removal steps are necessary to reduce any viruses that may enter the plasma pool during a “window period” before markers can be detected.6 In 1999, it was estimated that this window for serologic testing could result in the identification of up to 1 in 10 plasma pools containing an HIV‐infected unit and up to one in five containing an HBV‐ or HCV‐infected unit.2 For a particular NAT assay and pool configuration, NAT has been shown to decrease the diagnostic window from 22 to 11 days for HIV‐1, 59 to 34 days for HBV, and 82 to 23 days for HCV, thereby decreasing the amount of virus entering the plasma pool for fractionation.6 This reduces the residual risk by decreasing both the number of potentially infected units in the pool and the concentration of virus. However, screening does not sufficiently ensure the safety of products, as tests have limits of detection and are target specific.13 In addition, the size of the minipool for NAT impacts its effectiveness.
Industry Standards
The plasma industry developed two sets of standards through the Plasma Protein Therapeutics Association.41 The International Quality Plasma Program focuses on donor and center management. The second standard, known as Quality Standards of Excellence, Assurance, and Leadership (QSEAL), addresses manufacturing.41 Part of QSEAL is the inventory hold standard, which was introduced to address the “window period” described previously; this standard allows removal of previously donated units from donors who subsequently test positive for a virus (i.e., removal of window donations). This has been highly effective in reducing the risk of donations from infected donors who are nonreactive in serology (and NAT) at the time of donation from entering a manufacturing pool.42 The inventory hold is not a quarantine, as it would not interdict an infectious unit in the NAT window period if the donor did not return. The effectiveness of the hold is related to the large number of donors who donate frequently. All source plasma donations used for manufacturing are equivalent to donations for transfusion in terms of pathogen safety. In addition to manufacturing standards, QSEAL includes qualified donor and virus marker standards identical to those in International Quality Plasma Program.41
Validation Studies
Validation of production steps is necessary to estimate pathogen inactivation and/or removal capacity and provides confidence in the safety of a plasma‐derived clotting factor product.6, 43 The virus reduction capacity of a manufacturing process is tested in a dedicated laboratory using a validated scaled‐down version of the manufacturing process by employing process intermediates spiked with high titers of an appropriate virus and then determining the virus reduction factor achieved.6 Viruses selected for validation should closely resemble those that pose the greatest threat of contamination, such as hepatitis viruses and HIV. For some viruses, no practical cell culture system is available; thus, specific model viruses are used. The selected viruses should include different genome, size, and envelope characteristics to represent the wide range of the physicochemical properties of viruses. This allows manufacturers to fully test the virus reduction capacity of the manufacturing process and to provide indirect evidence that it reduces viruses in general.13, 14, 43 Reduction factors greater than 4 log are generally considered effective.43, 44 Some virus reduction factors achieved by individual inactivation and/or removal steps are presented in Table 2.17, 19, 32, 45, 46, 47, 48
Table 2.
Virus reduction factors (log) for relevant viruses reported for individual process stepsa
| Process step evaluated | Virus reduction factors (log) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Enveloped viruses | Non‐enveloped viruses | |||||||||
| HIV, | HCV model, | HBV model | HAV and model | B19V and model | ||||||
| HIV‐1 | BVDV | BHV | PRV | HAV | POL | B19V | PPV | CPV | MMV | |
| Lyophilization17, 45 | 1.2 | 1.7 | 1.3 | – | 2.1 | 3.4 | – | <1.0 | – | |
| Precipitation | ||||||||||
| Cryoprecipitation32 | – | – | – | 1.6 | 1.5 | – | – | 1.5 | – | |
| 3.5% PEG precipitation17 | <1.0 | <1.0 | <1.0 | – | – | 3.3 | – | 1.2 | – | |
| Al(OH)3 adsorption/glycine precipitation/NaCl precipitation32 | 3.6 | 2.4 | – | 3.7 | 2.4 | – | – | 3.4 | – | |
| S/D17, 46 | ≥11.1 | ≥4.5 | ≥8.0 | ≥8.5 | NA | NA | NA | NA | NA | |
| Dry heat17, 45, 46 | 5.2b | ≥4.9c | 2.1c | 4.9d | ≥5.8c | ≥2.5c | 4.1d | 4.1c | – | |
| Pasteurizatione, 32 | ≥6.4 | ≥8.9 | – | 4.6 | 4.2 | – | ≥3.9 | 1.1 | – | |
| Chromatography17 | ≥2.0 | <1.0 | 7.6 | – | – | <1.0 | – | <1.0 | – | |
| Virus filtration19, 47 | >6.4f | 6.6** | – | >6.0f | 6.8** | – | – | – | ≤1.0f | |
| Vapor heat19, 48 | >6.8 | >7.1 | – | >7.4 | >4.5 | – | ≥3.5g | – | ≤1.0 | |
Dashes indicate that no data are available.
100°C for 30 min.
80°C for 72 hr.
100°C for 120 min.
In aqueous solution at 60°C for 10 hr.
35‐nm filtration.
35‐ to15‐nm sequential filtration.
60°C for 500 min, followed by 80°C for 60 min.
BHV = bovine herpes virus (a nonspecific model virus for HBV); CPV = canine parvovirus (a model virus for B19V); MMV = mice minute virus (a model virus for B19V); NA = not applicable; PEG = polyethylene glycol; POL = poliovirus Sabin type 2.
Removal of prions from plasma‐derived products is evaluated similarly to virus validation studies, based on a valid scaled‐down version of the manufacturing process.33, 49, 50 For prion detection, the quantitative animal bioassay is considered most appropriate, although the in vitro methods of Western blot assay, enzyme‐linked immunosorbent assay, and conformation‐dependent immunoassay are also used for the detection of misfolded prion protein.
Pathogen Inactivation Methods
Pasteurization
Pasteurization by heating the protein in aqueous stabilized solution at 60°C for 10 to 11 hours inactivates both lipid membrane–enveloped and a range of nonenveloped viruses (Table 3).13 Coagulation factors are heat sensitive; therefore, stabilizers (usually sugars, amino acids, or acetate) are added to preserve protein integrity and are removed after pathogen inactivation. Homogeneity of temperature throughout pasteurization must be validated by temperature mapping techniques. Studies on inactivation of HIV, HAV, and B19V by pasteurization in a FVIII/VWF concentrate have demonstrated virus reduction factors of at least 6.4, 4.2, and at least 3.9 log, respectively.6, 32 Table 4 is an example of virus reduction factors observed in an actual product. Additional studies have demonstrated that pasteurization inactivates a wide range of enveloped and nonenveloped viruses, including bovine viral diarrhea virus (BVDV; a specific model virus for HCV), pseudorabies virus (PRV; a nonspecific model virus for HBV), herpes simplex virus‐1, West Nile virus (WNV), and poliovirus.6
Table 3.
Common methods for virus inactivation*
| Treatment | Treatment conditions | Advantages | Limitations | Relevant properties to be recorded |
|---|---|---|---|---|
| Pasteurization |
|
|
|
|
| Terminal dry heat |
or
|
|
|
|
| Vapor heat |
|
|
|
|
| S/D |
|
|
|
|
* Adapted from the World Health Organization.13
TNBP = tri(n‐butyl)phosphate.
Table 4.
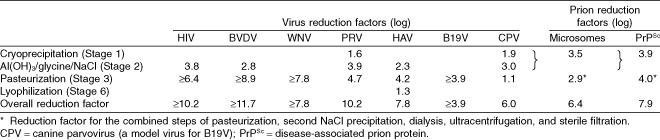
S/D treatment
Lipid membranes of enveloped viruses are disrupted by S/D mixtures, thereby preventing binding to and infection of cells by these viruses.51 Organic solvents (e.g., tri(n‐butyl)phosphate), combined with a nonionic detergent (polyethylene glycol octylphenyl ether [Triton X‐100] or polysorbate 80), are typically used for treatment; S/D compounds are removed by chromatography or oil extraction.51 Product solutions are filtered to remove viruses entrapped in particles and thus protected from exposure to S/D.13 Homogeneous mixing and maintenance of a consistent temperature throughout incubation must be validated. Plasma Protein Therapeutics Association member companies have compiled data on effective virus inactivation by S/D treatment for FVIII, F IX, intravenous immunoglobulin, and intramuscular immunoglobulin.52 Virus inactivation was evaluated in 308 studies reflecting production conditions, as well as variables significantly beyond process specification; the results demonstrated that product class, process temperature, protein concentration, and pH were not substantially involved in virus inactivation. In contrast, the concentration of S/D was critical when it was significantly below that specified for the process.52 For a FVIII and VWF concentrate, S/D treatment studies demonstrated reduction factors of more than 7.5 log for HIV, Sindbis virus (SINV), and PRV;46 for a F IX concentrate, reduction factors were greater than 4.5 log for HIV, SINV, vesicular stomatitis virus, BVDV, and PRV.47
An important limitation of S/D treatment is specificity for lipid‐enveloped viruses;4, 53 there have been reports of patients with hemophilia with HAV or B19V infections after receiving S/D‐treated FVIII concentrates.3, 5, 54, 55, 56 Therefore, a second virus reduction step is required in the manufacture of such plasma‐derived products to close this gap in virus safety.
Dry heat treatment of lyophilized products
Most plasma‐derived concentrates are lyophilized and, especially those treated with S/D, subsequently treated with dry heat to inactivate nonenveloped viruses that resist S/D treatment. Lyophilization confers a certain degree of virus inactivation;2, 57, 58, 59 the moisture content of lyophilized products undergoing dry heat treatment should be kept low (typically <2%), as residual moisture may affect product stability,45 although higher levels may enhance inactivation of some viruses.60
Dry heat treatment of lyophilized products has demonstrated favorable results for inactivation of relevant or model viruses of HAV, HBV, HCV, and HIV.45, 46, 61, 62 Dry heating of a lyophilized FVIII product at 80°C for 72 hours inactivated a wide range of viruses,60 and dry heating of FVIII or F IX concentrates reduced the risk of HCV transmission.61 An additional study demonstrated that terminal dry heating of a lyophilized FVIII concentrate at 100°C for 30 minutes inactivated HAV and HIV to below detectable levels within 10 minutes, while retaining approximately 95% of FVIII activity,45 whereas heating a lyophilized FVIII and VWF concentrate at 100°C for 120 minutes reduced a wide array of viruses (HIV, SINV, PRV, reovirus type‐3, HAV, and B19V) by more than 4 log.46 Although B19V was reduced by dry heat in validation studies, the reduction factor may not be sufficient for complete inactivation of the virus load in the final product; asymptomatic B19V infection was detected in a patient who received FVIII concentrate treated at 80°C for 72 hours.63
Vapor heat
One drawback to lyophilization is that in addition to stabilizing coagulation factors, the removal of water can also stabilize potential viruses in the product.62 By adding water vapor to lyophilized products before heating, higher levels of virus inactivation can be achieved at equivalent temperatures.13 Vapor heating of lyophilized products targets enveloped and nonenveloped viruses and has demonstrated inactivation of hepatitis viruses and HIV.19 Lyophilization and vapor heat at 60°C inactivated approximately 6 log of HAV in spiked FVIII concentrates within 8 to 10 hours and similarly reduced HAV titers in F IX concentrates within 3 hours.64 Among 20 patients with hemophilia who received vapor‐heated F IX infusions, none developed markers for infection with HCV or HIV during 6 to 15 months of follow‐up, suggesting that vapor heat–treated concentrates may be associated with a low risk of viral infection.65 However, vapor heat at 60°C may be insufficient to inactivate HBV completely, depending on the virus load in the starting material (batch related) and the amount of product given to patients not vaccinated against HBV; there are reports of four hemophilia patients not vaccinated against HBV who became infected with HBV after receiving vapor‐heated FVIII concentrate.66
Methods of Pathogen Removal
Partition processes: fractionation and chromatography
In addition to virus inactivation, viruses can also be physically removed from plasma‐derived products by fractionation (precipitation or chromatography) and filtration (Table 5). Before the 1980s, plasma fractionation was mainly considered a step in protein purification; no dedicated virus reduction step (except for pasteurization of human albumin) was implemented during the manufacturing process. Later virus validation studies showed that fractionation reliably contributes to the overall virus reduction capacity for plasma‐derived proteins.47, 68, 69 Although ethanol precipitation is the most widely used plasma fractionation method worldwide,13 it is not used for the production of coagulation factor concentrates, as this treatment denatures the desired proteins.
Table 5.
Selected common methods for virus removal*
| Treatment | Treatment conditions | Advantages | Limitations | Relevant properties |
|---|---|---|---|---|
| Chromatography |
|
|
|
|
| Virus filtration |
|
|
|
|
* Adapted from the World Health Organization.13
After initial cryoprecipitation, many clotting factor concentrates are purified chromatographically. In a study by Roberts and colleagues,69 purification of F IX concentrate using metal chelate affinity chromatography reduced poliovirus and SINV by reduction factors of 4.0 and 6.5 log, respectively. Some products are further purified using affinity chromatography mediated by an antibody against the protein of interest.44 Affinity chromatography has been used to purify FVIII concentrates, resulting in reduction factors of 8.3 log for encephalomyocarditis virus,68 4.3 log for SINV,68 4.2 log for porcine parvovirus (PPV; a model virus for B19V),21 and 5.3 log for HAV.21 Purification of a FVIII concentrate using monoclonal antibody (MoAb) affinity chromatography contributed to overall virus reduction capabilities of 5 to 6 log for HIV, SINV, vesicular stomatitis virus, and PRV before pasteurization.23 For a F IX concentrate, MoAb affinity chromatography alone resulted in reduction factors of at least 5.8 log for enveloped viruses HIV, BVDV, PRV, and WNV and reduction factors of 3.3 and 6.6 log for nonenveloped viruses HAV and parvovirus, respectively.24 An important consideration exists in the reuse of chromatography columns;13 viruses adhere to resins and cannot be completely washed out. Therefore, equipment and material must be sanitized with validated chemical and/or physical treatments to inactivate and remove viruses to ensure no cross‐contamination of subsequent batches of product.13
Virus filtration
Viruses may be removed from clotting factor concentrates via filtration employing retentive filters with pores smaller than the virus diameter (virus filtration, also called nanofiltration).67 Filtration may not be feasible for products containing proteins of a comparable or larger size to the viruses or pore size of the filter; thus, filtration is mainly limited to smaller‐molecular‐weight coagulation factors (e.g., F IX or FVIII). However, a VWF concentrate filtered through 35‐nm pore membranes resulted in reduction factors of at least 5.0 log of enveloped viruses HIV, BVDV, and PRV, although filtration removed large VWF multimers as well.70 Filtration of a FVIII concentrate using 35‐ and 15‐nm sequential filtration resulted in removal of enveloped (HIV, BVDV, and PRV) and nonenveloped (HAV and PPV) viruses.71 Virus filtration alone resulted in virus reduction factors of at least 3.6 log, while combination of S/D, chromatography, and virus filtration yielded reduction factors of at least 5.1 log for all tested viruses. Virus filtration of a FXIII concentrate using a 20‐nm filter resulted in a reduction of enveloped (HIV, BVDV, PRV) and nonenveloped (HAV) viruses by at least 5.5 log and a reduction of a nonenveloped parvovirus by 3.4 log.72 Table 6 is an example of virus reduction achieved through the addition of a filtration step. In a safety study on F IX concentrate, a final virus filtration step was added following fractionation, chromatography, and S/D treatment.47 The 35‐ to 15‐nm sequential filtration step resulted in a reduction factor of at least 6.8 log for HAV and at least 6.6 log for BVDV.47 Filtration of another F IX concentrate using two filters (20‐nm mean pore size) in series very effectively removed enveloped and nonenveloped viruses.24
Table 6.
Virus reduction factors for a pasteurized FXIII concentrate utilizing ion‐exchange chromatography and virus filtration72
| Stages | Virus reduction factor (log) | ||||||
|---|---|---|---|---|---|---|---|
| Enveloped viruses | Nonenveloped viruses | ||||||
| HIV | BVDV | WNV | PRV | HAV | CPV | B19V | |
| Adsorption to Al(OH)3/Vitacel and defibrination | 6.9 | ||||||
| Ion‐exchange chromatography | 5.0 | 3.3 | ≥8.0 | 3.4 | 3.7 | ||
| Pasteurization (heat treatment at 60°C for 10 hr) | ≥7.7 | ≥8.1 | ≥7.4 | 4.3 | 1.0 | ≥4.0 | |
| 20N/20N virus filtration | ≥6.1 | ≥5.0 | ≥7.4 | ≥6.4 | ≥5.6 | 6.1 | |
| Overall virus reduction factor | ≥18.8 | ≥16.4 | ≥14.8 | ≥21.3 | ≥13.3 | 10.8 | NA |
CPV = canine parvovirus (a model virus for B19V); NA = not applicable; PRV = pseudorabies virus (a nonspecific model virus for HBV).
Risk Assessment Regarding Virus Transmission
An appropriate margin of safety with respect to blood‐borne viruses is achieved when the overall virus reduction factor clearly exceeds the amount of virus potentially entering the manufacturing pool. In Europe, quantitative risk assessments are required.10 An estimate of virus particle content of the finished product is determined based on the worst case scenario of virus concentration in the starting material and the amount of plasma required to produce one vial, divided by the virus reduction factor demonstrated in validation studies. For this calculation, the following variables must be considered:10
-
Virus load in the plasma volume needed for production of one vial of product
Volume of manufacturing pool
Potential virus load in a donation from an infected donor remaining below the limit of detection of a NAT assay
Volume of donation
-
Number of viremic donations entering a manufacturing pool taking into account
Epidemiology in donor population
Inventory hold and look‐back procedure
Virus doubling time, if more than one donation from an infected donor could enter the manufacturing pool
Product yield
Virus reduction factors demonstrated in validation studies
A high margin of safety is documented when fewer than one virus particle per million doses is expected; this is based on the sterility assurance level with respect to bacteria, molds, and yeasts by pharmacopoeias worldwide. As viruses cannot replicate in a cell‐free medium such as the dissolved final product, application of the sterility assurance level for viruses is conservative.
Prion Risk Reduction
Transmissible spongiform encephalopathies are a group of prion‐associated, fatal neurodegenerative diseases. Three possible cases of prion transmission resulting in vCJD and one probable case of asymptomatic infection have been reported in patients in the United Kingdom who received non–leukoreduced red blood cells from asymptomatic infected individuals.73 To date, no recipients of pooled plasma‐derived products have developed vCJD;39 however, there is one report of asymptomatic prion detection at autopsy in a hemophilia patient,74 probably due to prion‐contaminated FVIII concentrate.74 As prion screening tests are currently unavailable, donor selection and manufacturing processes for prion removal are important.39 As requested by the WHO, EMA, and FDA, restrictions on plasma donation have been imposed in many countries.6, 7, 8, 9, 33 Plasma‐derived proteins are leukoreduced by plasma preparation, decreasing the risk for prion transmission. Additionally, manufacturing steps for plasma fractionation and virus removal, including precipitation, chromatography, and filtration (Table 4), have also proven efficacy in evaluation studies.6, 34, 75 Inactivation of prions in plasma‐derived clotting factor concentrates is not possible, as the needed techniques denature coagulation factors.33 Appropriate cleaning and sanitization methods for material and equipment used in the production of plasma‐derived products inactivate and remove prion proteins; thus, batch‐to‐batch segregation can be achieved and batch‐to‐batch contamination excluded.76, 77, 78, 79
Emerging Threats
Emerging microbial pathogens or parasites are less of a threat because of sterile filtration, which, in combination with other steps before preparation of the finished product, removes these pathogens. However, “new emerging viruses” must be assessed very carefully with regard to potential impact on the safety of the plasma pool for fractionation and the final products. New emerging viruses may be novel, zoonotic viruses that are encountered as humans enter new geographic areas with previously undiscovered animal viruses that cross the species barrier to enter the human disease chain (e.g., HIV, yellow fever virus, severe acute respiratory syndrome coronavirus, Menangle virus, Hendra virus, Nipah virus, hantaviruses, monkeypox virus). Hepatitis E virus has been detected in different regions of the world and is considered a zoonosis; in particular, Genotype 3, mainly detected in domestic and wild pigs, has been found in humans in developed countries.80, 81 Human parvovirus 4 (PARV4) and related parvoviruses have been identified in cows, pigs, primates, and humans.82, 83, 84, 85, 86 Furthermore, a “new” virus can emerge and/or reemerge in new geographic regions; for example, WNV was known in Africa and the Middle East but emerged in North America in 1999.87 Improved diagnostic methods have resulted in detection of previously unknown viruses (which should not be considered as emerging), such as B19V in 1975,88 HCV in 1989,89 human herpesvirus Type 8 in 1996,90 and TT virus in 1997.91 To meet this challenge, the epidemiology of emerging pathogens in the donor population and the potential infectious virus load in a donation during asymptomatic incubation must be addressed. Diligent surveillance of available information may result in a (temporal) deferral of donors based on geographic risk.
PARV4 can reach a high load in an infected person,92 but the prevalence of the virus is low in many regions.93, 94 Contamination of plasma pools for fractionation with PARV4 has been reported,95 resulting in final products with PARV4 DNA. However, PARV4 DNA in current products is undetectable96 or lower than in products manufactured between 1970 and 1980,97, 98 possibly because of thorough screening of donors and testing of donations for pathogens that may be associated with PARV4. Some manufacturing processes for coagulation factor concentrates remove and/or inactivate low levels of PARV4.98 Dedicated virus reduction steps in the manufacture of coagulation factor concentrates also reduced arthropod‐transmitted viruses such as WNV,99 other flaviviruses (dengue virus),100 and togaviruses (Chikungunya virus);101 reduction of these viruses during manufacture of clotting factor concentrates was also demonstrated using model viruses for validation. These data demonstrate that the measures currently taken are also relevant for emerging viruses.
Discussion and Conclusions
The combination of inactivation and removal methods, in addition to donation screening, has essentially eliminated transmission of viruses of principal concern, namely HAV, HBV, HCV, HIV, and B19V. As shown in validation studies, a strategy involving different steps for inactivation and removal is optimal for targeting viruses with different physicochemical properties. For instance, although S/D is highly effective against enveloped viruses, it is ineffective against nonenveloped viruses (e.g., HAV and B19V); thus, a second step, often dry heating of the lyophilized product, is implemented. Procedures for purification and concentration of the desired protein are usually less effective in virus removal than dedicated reduction steps, such as inactivation by heat and S/D treatment or removal by virus filtration. As diagnostic techniques advance, emerging pathogens may be identified,92 and their genomes may be detected in the final product (e.g., PARV4), depending on the manufacturing process.96, 98 The methods employed make it extremely likely that any new pathogen would be inactivated or removed to a high degree, but diligence must be maintained.
Immune globulin and other plasma products, such as α1‐proteinase inhibitor and C1 esterase inhibitor, are subject to similar pathogen reduction measures. In addition, low pH can be utilized for immune globulin products to inactivate certain viruses. Thus, the same high safety record has been observed with these products in recent years. Albumin has always been pasteurized to prevent pathogen transmission.
A product similar to fresh‐frozen plasma (FFP), but pooled and treated with S/D, has been available in Europe and was recently licensed in the United States (Octaplas).102 Enveloped viruses are effectively inactivated by S/D treatment, and chromatographic methods reduce prion transmission potential. This product relies on testing of the donations for protection against nonenveloped virus, specifically HAV, B19V, and more recently, hepatitis E virus. Improved virus safety compared with FFP is based solely on the reductions observed with S/D methods for blood products.103
Over the past 20 years, fears of potential infection and the introduction of recombinant factor products have fueled a shift toward recombinant products as the first choice for treatment of patients with hemophilia. However, opinions on the role of recombinant and plasma‐derived concentrates for treatment vary. The United Kingdom Hemophilia Center Directors Organization recommends recombinant products for all patients with hemophilia, whereas other European countries recommend them as the first choice for children only. In contrast, in Germany and Austria, there are no clear recommendations on use of recombinant factor concentrates in patients with hemophilia. Furthermore, patients with other factor deficiencies for which no recombinant product is currently available (e.g., von Willebrand disease) must use plasma‐derived concentrates.
Physicians currently have fewer safety concerns using plasma‐derived products than in the past; in fact, 45% of FVIII consumption and approximately 65% of F IX consumption in Germany are plasma‐derived. For patients with hemophilia A and inhibitors against FVIII, plasma‐derived FVIII products containing VWF are widely used as second‐line treatment for immune tolerance induction. Due to the improved and comprehensive pathogen reduction steps, physicians and patients should have confidence in the safety of plasma‐derived products for the treatment of bleeding disorders.
Conflict of Interest
RK has acted as a consultant and a paid speaker for CSL Behring, Bayer, Baxter, Pfizer, Novo Nordisk, and Sobi. AG and TLS are employees of CSL Behring.
Acknowledgments
Medical writing support was provided by Amanda Sheldon, PhD, at Complete Publication Solutions, LLC; this support was funded by CSL Behring.
Funded by CSL Behring.
References
- 1. Kcentra™ (Prothombin Complex Concentrate [Human]). Marburg, Germany: CSL Behring GmbH; 2013.
- 2. Tabor E. The epidemiology of virus transmission by plasma derivatives: clinical studies verifying the lack of transmission of hepatitis B and C viruses and HIV type 1. Transfusion 1999;39:1160‐1168. [DOI] [PubMed] [Google Scholar]
- 3. Mannucci PM, Gdovin S, Gringeri A, et al. Transmission of hepatitis A to patients with hemophilia by factor VIII concentrates treated with organic solvent and detergent to inactivate viruses. The Italian Collaborative Group. Ann Intern Med 1994;120:1‐7. [DOI] [PubMed] [Google Scholar]
- 4. Mannucci PM. Clinical evaluation of viral safety of coagulation factor VIII and IX concentrates. Vox Sang 1993;64:197‐203. [DOI] [PubMed] [Google Scholar]
- 5. Azzi A, Ciappi S, Zakvrzewska K, et al. Human parvovirus B19 infection in hemophiliacs first infused with two high‐purity, virally attenuated factor VIII concentrates. Am J Hematol 1992;39:228‐230. [DOI] [PubMed] [Google Scholar]
- 6. Gröner A. Pathogen safety of plasma‐derived products—Haemate‐P/Humate‐P. Haemophilia 2008;14(s5):54‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. European Medicines Agency . CHMP position statement on Creutzfeldt‐Jakob disease and plasma‐derived and urine‐derived medicinal products. London, UK: EMA; 2010.
- 8. US Food and Drug Administration . Guidance for industry: revised preventive measures to reduce the possible risk of transmission of Creutzfeldt‐Jakob disease (CJD) and variant Creutzfeldt‐Jakob disease (vCJD) by blood and blood products. 2002. [cited 2013 Jan 25]. Available from: http://www.fda.gov/cber/guidelines.html
- 9. World Health Organization . WHO guidelines on transmissible spongiform encephalopathies in relation to biological and pharmaceutical products. Geneva, Switzerland: WHO; 2003.
- 10. European Medicines Agency . Guideline on plasma‐derived medicinal products. London, UK: EMA; 2011.
- 11. Farrugia A, Penrod J, Bult JM. Payment, compensation and replacement—the ethics and motivation of blood and plasma donation. Vox Sang 2010;99:202‐211. [DOI] [PubMed] [Google Scholar]
- 12. Simon TL. Monetary compensation for plasma donors: a record of safety. Transfusion 1998;38:883‐886. [DOI] [PubMed] [Google Scholar]
- 13. World Health Organization . Annex 4. Guidelines on viral inactivation and removal procedures intended to assure the viral safety of human blood plasma products. WHO Technical Report, Series No. 924. Geneva, Switzerland; 2004:150‐224.
- 14. US Food and Drug Administration . Guide to inspections of viral clearance process for plasma derivatives. 2009. [cited 2013 Jan 25]. Available from: http://www.fda.gov/ICECI/Inspections/InspectionGuides/ucm074866.htm
- 15. Josephson CD, Abshire TC. Clinical uses of plasma and plasma fractions: plasma‐derived products for hemophilias A and B, and for von Willebrand disease. Best Pract Res Clin Haematol 2006;19:35‐49. [DOI] [PubMed] [Google Scholar]
- 16. Mazurier C. Composition, quality control, and labeling of plasma‐derived products for the treatment of von Willebrand disease. Semin Thromb Hemost 2006;32:529‐536. [DOI] [PubMed] [Google Scholar]
- 17. Alphanate® (Antihemophilic Factor/von Willebrand Factor Complex [Human]). Los Angeles, CA: Grifols Biologicals, Inc.; 2007.
- 18. AlphaNine® SD (Coagulation Factor XI [Human]). Los Angeles, CA: Grifols Biologicals, Inc.; 2011.
- 19. Bebulin (Factor IX Complex). Westlake Village, CA: Baxter Healthcare Corporation; 2011.
- 20. Biostate® (Coagulation Factor VIII/von Willebrand Factor Complex [Human]). Broadmeadows, Australia: CSL Limited; 2010.
- 21. Hemofil M Antihemophilic Factor (Human). Glendale, CA: Baxter Healthcare Corporation; 2002.
- 22. Koate® ‐DVI Antihemophilic Factor (Human). Research Triangle Park, NC: Talecris Biotherapeutics, Inc.; 2006.
- 23. Monoclate‐P® (Antihemophilic Factor [Human]). Kankakee, IL: CSL Behring; 2010.
- 24. Mononine® (Coagulation Factor IX [Human]). Kankakee, IL: CSL Behring; 2011.
- 25. Profilnine® SD (Factor IX Complex). Los Angeles, CA: Grifols Biologicals Inc.; 2011.
- 26. Wilate® (von Willebrand Factor/Coagulation Factor VIII Complex [Human]). Hoboken, NJ: Octapharma USA Inc.; 2009.
- 27. Corifact® (Factor XIII Concentrate [Human]). Kankakee, IL: CSL Behring LLC; 2011.
- 28. Franchini M, Lippi G. Prothrombin complex concentrates: an update. Blood Transfus 2010;8:149‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gröner A. Reply. Pereira A. Cryoprecipitate versus commercial fibrinogen concentrate in patients who occasionally require a therapeutic supply of fibrinogen: risk comparison in the case of an emerging transfusion‐transmitted infection. Haematologica 2007;92:846‐9. Haematologica 2008;93:e24‐26; author reply e27. [DOI] [PubMed] [Google Scholar]
- 30. Beriplex® P/N (Human Prothrombin Complex). Ottawa, ON: CSL Behring Canada, Inc.; 2010.
- 31. Octaplex® (Human Prothrombin Complex). Vienna, Austria: Octapharma Pharmazeutika Produktionsges m.b.H.; 2010.
- 32. Humate‐P® (Antihemophilic Factor/von Willebrand Factor Complex [Human]). Kankakee, IL: CSL Behring; 2012.
- 33. Burdick MD, Pifat DY, Petteway SR Jr, et al. Clearance of prions during plasma protein manufacture. Transfus Med Rev 2006;20:57‐62. [DOI] [PubMed] [Google Scholar]
- 34. Cai K, Gröner A, Dichtelmüller HO, et al. Prion removal capacity of plasma protein manufacturing processes: a data collection from PPTA member companies. Transfusion 2013;53:1894‐1905. [DOI] [PubMed] [Google Scholar]
- 35. International Conference on Harmonization . Guidance for industry: Q5A viral safety evaluation of biotechnology products derived from cell lines of human or animal origin. Rockville, MD: US FDA; 1997. [PubMed]
- 36. European Medicines Agency . Note for guidance on quality of biotechnological products: viral safety evaluation of biotechnology products derived from cell lines of human or animal origin. London, UK: EMA; 1997.
- 37. Working Group “Overall Blood Supply Strategy with Regard to Variant Creutzfeldt‐Jakob Disease (vCJD).” Report of the Working Group “Overall Blood Supply Strategy with Regard to Variant Creutzfeldt‐Jakob Disease (vCJD)”: statement on the development and implementation of test systems suitable for the screening of blood donors for vCJD—dated September 17, 2008. Transfus Med Hemother 2009;36:79‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ironside JW. Variant Creutzfeldt‐Jakob disease: an update. Folia Neuropathol 2012;50:50‐56. [PubMed] [Google Scholar]
- 39. Turner ML, Ludlam CA. An update on the assessment and management of the risk of transmission of variant Creutzfeldt‐Jakob disease by blood and plasma products. Br J Haematol 2009;144:14‐23. [DOI] [PubMed] [Google Scholar]
- 40. Edgeworth JA, Farmer M, Sicilia A, et al. Detection of prion infection in variant Creutzfeldt‐Jakob disease: a blood‐based assay. Lancet 2011;377(9764):487‐493. [DOI] [PubMed] [Google Scholar]
- 41. Plasma Protein Therapeutics Association . [cited 2013 Jan 25]. Available from: http://www.pptaglobal.org/
- 42. Whitaker BI, Schreiber GB, Simon TL. The residual risk of a window period (WP) unit entering a pool of source plasma. Transfusion 1998;38(Suppl):81S. [Google Scholar]
- 43. European Medicines Agency . Note for guidance on virus validation studies: the design, contribution and interpretation of studies validating the inactivation and removal of viruses. London, UK: EMA; 1996.
- 44. Johnston A, Adcock W. The use of chromatography to manufacture purer and safer plasma products. Biotechnol Genet Eng Rev 2000;17:37‐70. [DOI] [PubMed] [Google Scholar]
- 45. Kim IS, Choi YW, Kang Y, et al. Dry‐heat treatment process for enhancing viral safety of an antihemophilic factor VIII concentrate prepared from human plasma. J Microbiol Biotechnol 2008;18:997‐1003. [PubMed] [Google Scholar]
- 46. Stadler M, Gruber G, Kannicht C, et al. Characterisation of a novel high‐purity, double virus inactivated von Willebrand Factor and Factor VIII concentrate (Wilate). Biologicals 2006;34:281‐288. [DOI] [PubMed] [Google Scholar]
- 47. Johnston A, Macgregor A, Borovec S, et al. Inactivation and clearance of viruses during the manufacture of high purity factor IX. Biologicals 2000;28:129‐136. [DOI] [PubMed] [Google Scholar]
- 48. Berting A, Modrof J, Unger U, et al. Inactivation of parvovirus B19 during STIM‐4 vapor heat treatment of three coagulation factor concentrates. Transfusion 2008;48:1220‐1226. [DOI] [PubMed] [Google Scholar]
- 49. European Medicines Agency . Guideline on the investigation of manufacturing processes for plasma‐derived medicinal products with regard to vCJD risk. London, UK: EMA; 2004.
- 50. Vey M, Baron H, Weimer T, et al. Purity of spiking agent affects partitioning of prions in plasma protein purification. Biologicals 2002;30:187‐196. [DOI] [PubMed] [Google Scholar]
- 51. Solheim BG. Pathogen reduction of blood components. Transfus Apher Sci 2008;39:75‐82. [DOI] [PubMed] [Google Scholar]
- 52. Dichtelmüller HO, Biesert L, Fabbrizzi F, et al. Robustness of solvent/detergent treatment of plasma derivatives: a data collection from Plasma Protein Therapeutics Association member companies. Transfusion 2009;49:1931‐1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chandra S, Groener A, Feldman F. Effectiveness of alternative treatments for reducing potential viral contaminants from plasma‐derived products. Thromb Res 2002;105:391‐400. [DOI] [PubMed] [Google Scholar]
- 54. Mannucci PM, Santagostino E, Di Bona E, et al. The outbreak of hepatitis A in Italian patients with hemophilia: facts and fancies. Vox Sang 1994;67(Suppl 1):31‐35. [PubMed] [Google Scholar]
- 55. Chudy M, Budek I, Keller‐Stanislawski B, et al. A new cluster of hepatitis A infection in hemophiliacs traced to a contaminated plasma pool. J Med Virol 1999;57:91‐99. [DOI] [PubMed] [Google Scholar]
- 56. Jee YM, Go U, Cheon D, et al. Detection of hepatitis A virus from clotting factors implicated as a source of HAV infection among haemophilia patients in Korea. Epidemiol Infect 2006;134:87‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Saez A, Bosh N, Boadas N, et al. Pharmacokinetics and acute tolerance of a double virus inactivated plasma derived factor VIII concentrate. Haemophilia 1999;5:260‐265. [DOI] [PubMed] [Google Scholar]
- 58. Smales CM, Pepper DS, James DC. Protein modification during anti‐viral heat‐treatment bioprocessing of factor VIII concentrates, factor IX concentrates, and model proteins in the presence of sucrose. Biotechnol Bioeng 2002;77:37‐48. [DOI] [PubMed] [Google Scholar]
- 59. Unger U, Poelsler G, Modrof J, et al. Virus inactivation during the freeze‐drying processes as used for the manufacture of plasma‐derived medicinal products. Transfusion 2009;49:1924‐1930. [DOI] [PubMed] [Google Scholar]
- 60. Roberts PL, Dunkerley C, McAuley A, et al. Effect of manufacturing process parameters on virus inactivation by dry heat treatment at 80 degrees C in factor VIII. Vox Sang 2007;92:56‐63. [DOI] [PubMed] [Google Scholar]
- 61. Effect of dry‐heating of coagulation factor concentrates at 80 degrees C for 72 hours on transmission of non‐A, non‐B hepatitis. Study Group of the UK Haemophilia Centre Directors on Surveillance of Virus Transmission by Concentrates. Lancet 1988;2(8615):814‐816. [PubMed] [Google Scholar]
- 62. Arrighi S, Rossi R, Borri MG, et al. “In vitro” and in animal model studies on a double virus‐inactivated factor VIII concentrate. Thromb Haemost 1995;74:868‐873. [PubMed] [Google Scholar]
- 63. Blümel J, Schmidt I, Effenberger W, et al. Parvovirus B19 transmission by heat‐treated clotting factor concentrates. Transfusion 2002;42:1473‐1481. [DOI] [PubMed] [Google Scholar]
- 64. Barrett PN, Meyer H, Wachtel I, et al. Inactivation of hepatitis A virus in plasma products by vapor heating. Transfusion 1997;37:215‐220. [DOI] [PubMed] [Google Scholar]
- 65. Shapiro A, Abe T, Aledort LM, et al. Low risk of viral infection after administration of vapor‐heated factor VII concentrate or factor IX complex in first‐time recipients of blood components. International Factor Safety Study Group. Transfusion 1995;35:204‐208. [DOI] [PubMed] [Google Scholar]
- 66. Mannucci PM, Zanetti AR, Colombo M. Prospective study of hepatitis after factor VIII concentrate exposed to hot vapour. Br J Haematol 1988;68:427‐430. [DOI] [PubMed] [Google Scholar]
- 67. Burnouf T, Radosevich M. Nanofiltration of plasma‐derived biopharmaceutical products. Haemophilia 2003;9:24‐37. [DOI] [PubMed] [Google Scholar]
- 68. Lawrence JE. Affinity chromatography to remove viruses during preparation of plasma derivatives. Dev Biol Stand 1993;81:191‐197. [PubMed] [Google Scholar]
- 69. Roberts PL, Walker CP, Feldman PA. Removal and inactivation of enveloped and non‐enveloped viruses during the purification of a high‐purity factor IX by metal chelate affinity chromatography. Vox Sang 1994;67(Suppl):69‐71. [PubMed] [Google Scholar]
- 70. Mazurier C, Poulle M, Samor B, et al. In vitro study of a triple‐secured von Willebrand factor concentrate. Vox Sang 2004;86:100‐104. [DOI] [PubMed] [Google Scholar]
- 71. Chtourou S, Porte P, Nogre M, et al. A solvent/detergent‐treated and 15‐nm filtered factor VIII: a new safety standard for plasma‐derived coagulation factor concentrates. Vox Sang 2007;92:327‐337. [DOI] [PubMed] [Google Scholar]
- 72. Gröner A, Nowak T, Popp B, et al. High margin of pathogen safety of a plasma‐derived FXIII concentrate. Haemophilia 2012;18(Suppl 3):31. [Google Scholar]
- 73. Ironside JW. Variant Creutzfeldt‐Jakob disease. Haemophilia 2010;16(Suppl 5):175‐180. [DOI] [PubMed] [Google Scholar]
- 74. Peden A, McCardle L, Head MW, et al. Variant CJD infection in the spleen of a neurologically asymptomatic UK adult patient with haemophilia. Haemophilia 2010;16:296‐304. [DOI] [PubMed] [Google Scholar]
- 75. Ludlam CA, Turner ML. Managing the risk of transmission of variant Creutzfeldt Jakob disease by blood products. Br J Haematol 2006;132:13‐24. [DOI] [PubMed] [Google Scholar]
- 76. Flechsig E, Hegyi I, Enari M, et al. Transmission of scrapie by steel‐surface‐bound prions. Mol Med 2001;7:679‐684. [PMC free article] [PubMed] [Google Scholar]
- 77. Käsermann F, Kempf C. Sodium hydroxide renders the prion protein PrPSc sensitive to proteinase K. J Gen Virol 2003;84(Pt 11):3173‐3176. [DOI] [PubMed] [Google Scholar]
- 78. Bauman PA, Lawrence LA, Biesert L, et al. Critical factors influencing prion inactivation by sodium hydroxide. Vox Sang 2006;91:34‐40. [DOI] [PubMed] [Google Scholar]
- 79. Gröner A, Boose JA. Evaluation of a cleaning procedure for its capacity to remove prions from equipment used in the production of plasma derivatives. Blood 2002;100(s):139b. [Google Scholar]
- 80. Okamoto H. Genetic variability and evolution of hepatitis E virus. Virus Res 2007;127:216‐228. [DOI] [PubMed] [Google Scholar]
- 81. Juhl D, Baylis SA, Blümel J, et al. Seroprevalence and incidence of hepatitis E virus infection in German blood donors. Transfusion 2014;54:49‐56. [DOI] [PubMed] [Google Scholar]
- 82. Adlhoch C, Kaiser M, Ellerbrok H, et al. High prevalence of porcine Hokovirus in German wild boar populations. Virol J 2010;25:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Adlhoch C, Kaiser M, Loewa A, et al. Diversity of parvovirus 4‐like viruses in humans, chimpanzees, and monkeys in hunter‐prey relationships. Emerg Infect Dis 2012;18:859‐862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Jones MS, Kapoor A, Lukashov VV, et al. New DNA viruses identified in patients with acute viral infection syndrome. J Virol 2005;79:8230‐8236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lau SK, Woo PC, Tse H, et al. Identification of novel porcine and bovine parvoviruses closely related to human parvovirus 4. J Gen Virol 2008;89(Pt 8):1840‐1848. [DOI] [PubMed] [Google Scholar]
- 86. Sharp CP, LeBreton M, Kantola K, et al. Widespread infection with homologues of human parvoviruses B19, PARV4, and human bocavirus of chimpanzees and gorillas in the wild. J Virol 2010;84(19):10289‐10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Asnis DS, Conetta R, Teixeira AA, et al. The West Nile virus outbreak of 1999 in New York: the Flushing Hospital experience. Clin Infect Dis 2000;30:413‐418. [DOI] [PubMed] [Google Scholar]
- 88. Cossart YE, Field AM, Cant B, et al. Parvovirus‐like particles in human sera. Lancet 1975;1(7898):72‐73. [DOI] [PubMed] [Google Scholar]
- 89. Choo QL, Kuo G, Weiner AJ, et al. Isolation of a cDNA clone derived from a blood‐borne non‐A, non‐B viral hepatitis genome. Science 1989;244(4902):359‐362. [DOI] [PubMed] [Google Scholar]
- 90. Moore PS, Gao SJ, Dominguez G, et al. Primary characterization of a herpesvirus agent associated with Kaposi's sarcomae. J Virol 1996;70:549‐558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Nishizawa T, Okamoto H, Konishi K, et al. A novel DNA virus (TTV) associated with elevated transaminase levels in posttransfusion hepatitis of unknown etiology. Biochem Biophys Res Commun 1997;241:92‐97. [DOI] [PubMed] [Google Scholar]
- 92. Sharp CP, Lail A, Donfield S, et al. Virologic and clinical features of primary infection with human parvovirus 4 in subjects with hemophilia: frequent transmission by virally inactivated clotting factor concentrates. Transfusion 2012;52:1482‐1489. [DOI] [PubMed] [Google Scholar]
- 93. Eis‐Hübinger AM, Drexler JF, Reber U, et al. Absence of detection of novel human parvoviruses in German plasma donations. Transfusion 2010;50:266‐267. [DOI] [PubMed] [Google Scholar]
- 94. Vallerini D, Barozzi P, Quadrelli C, et al. Parvoviruses in blood donors and transplant patients, Italy. Emerg Infect Dis 2008;14:185‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Fryer JF, Kapoor A, Minor PD, et al. Novel parvovirus and related variant in human plasma. Emerg Infect Dis 2006;12:151‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Modrow S, Wenzel JJ, Schimanski S, et al. Prevalence of nucleic acid sequences specific for human parvoviruses, hepatitis A and hepatitis E viruses in coagulation factor concentrates. Vox Sang 2011;100:351‐358. [DOI] [PubMed] [Google Scholar]
- 97. Fryer JF, Hubbard AR, Baylis SA. Human parvovirus PARV4 in clotting factor VIII concentrates. Vox Sang 2007;93:341‐347. [DOI] [PubMed] [Google Scholar]
- 98. Schneider B, Fryer JF, Oldenburg J, et al. Frequency of contamination of coagulation factor concentrates with novel human parvovirus PARV4. Haemophilia 2008;14:978‐986. [DOI] [PubMed] [Google Scholar]
- 99. Kreil TR, Berting A, Kistner O, et al. West Nile virus and the safety of plasma derivatives: verification of high safety margins, and the validity of predictions based on model virus data. Transfusion 2003;43:1023‐1028. [DOI] [PubMed] [Google Scholar]
- 100. Xie YW, Chan PK, Szeto CK, et al. Clearance of dengue virus in the plasma‐derived therapeutic proteins. Transfusion 2008;48:1342‐1347. [DOI] [PubMed] [Google Scholar]
- 101. Leydold SM, Farcet MR, Kindermann J, et al. Chikungunya virus and the safety of plasma products. Transfusion 2012;52:2122‐2130. [DOI] [PubMed] [Google Scholar]
- 102. Octaplas® (Pooled Plasma [Human]). Vienna, Austria: Octapharma Pharmazeutika Produktionsges m.b.H.; 2013.
- 103. Horowitz B, Prince AM, Horowitz MS, et al. Viral safety of solvent‐detergent treated blood products. Dev Biol Stand 1993;81:147‐161. [PubMed] [Google Scholar]