

Abstract
The hydrophobic molecules of the metabolome – also named the lipidome – constitute a major part of the entire metabolome. Novel technologies show the existence of a staggering number of individual lipid species, the biological functions of which are, with the exception of only a few lipid species, unknown. Much can be learned from pathogens that have evolved to take advantage of the complexity of the lipidome to escape the immune system of the host organism and to allow their survival and replication. Different types of pathogens target different lipids as shown in interaction maps, allowing visualization of differences between different types of pathogens. Bacterial and viral pathogens target predominantly structural and signaling lipids to alter the cellular phenotype of the host cell. Fungal and parasitic pathogens have complex lipidomes themselves and target predominantly the release of polyunsaturated fatty acids from the host cell lipidome, resulting in the generation of eicosanoids by either the host cell or the pathogen. Thus, whereas viruses and bacteria induce predominantly alterations in lipid metabolites at the host cell level, eukaryotic pathogens focus on interference with lipid metabolites affecting systemic inflammatory reactions that are part of the immune system. A better understanding of the interplay between host–pathogen interactions will not only help elucidate the fundamental role of lipid species in cellular physiology, but will also aid in the generation of novel therapeutic drugs.
Keywords: bacteria, fungi, host–pathogen interactions, lipidome, lipids, metabolome, parasites, viruses
The lipidome constitutes a major part of the entire cellular metabolome with functions in cell structure, membrane dynamics and organization, energy supply and storage, and cellular signaling. Pathogens have adapted in different ways to take advantage of the complexity of the host cell lipidome in order to escape the immune system and to allow their survival and replication. Here we review the lipid targeting by various types of pathogens and provide interaction maps, allowing visualization of differences between viruses, bacteria, fungi and parasites.
The metabolome encompasses all small molecules that are present in a biological system (Figure 1). Unlike genes and proteins, the functions of which are subject to epigenetic regulation and post‐translational modifications, respectively, metabolites serve as direct signatures of biochemical activity and tightly correlate with phenotype 1. These properties make the cellular metabolome an attractive target for pathogens in order to introduce phenotypic perturbations that allow their survival and replication.
Figure 1.
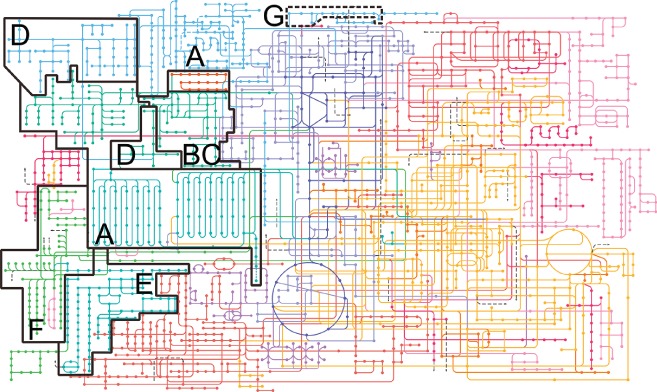
Contribution of lipid metabolic pathways to the KEGG map of metabolism. The metabolic map was constructed based on the KEGG (Kyoto Encyclopedia of Genes and Genomes) database (http://www.kegg.jp/) 245. The graphical presentation is based on the Genome‐Linked Application for Metabolic Maps (http://glamm.lbl.gov) 246 with a minor modification that allows visualization of elongation and desaturation of palmitic‐ to stearic‐ and oleic acid, respectively. Lipid classification into eight main categories (A–H) is according to the 2005 convention on lipid nomenclature 19: A, fatty acids; B, glycerolipids; C, glycerophospholipids; D, sphingolipids; E, sterols; F, prenol lipids; G, saccharolipids. Polyketides (lipid category H) are not commonly found in mammalian hosts and are not depicted. Saccharolipids (lipid category G) are shown as a dotted line and not discussed in this review as they are not constituents of the mammalian lipidome. In this graphical pathway representation, cholesterol esters, lyso‐phospholipids and bis(monoacylglycero)phosphate (BMP) species are lacking.
With recent developments in instrumentation (mass spectroscopy), bioinformatics and software, our knowledge on the identification and quantification of metabolites, as well as on the metabolic pathways and metabolite fluxes is rapidly expanding. Metabolites are loosely defined as just about any small molecule with a molecular weight −2000 Da that is metabolized by an organism 2. The METLIN metabolite database (http://metlin.scripps.edu/) is one of the most comprehensive freely accessible databases and currently contains more than 240 000 (possible) entries and approximately 60 000 different structures 3, 4. The human metabolome database (http://www.hmdb.ca/) contains over 40 000 metabolite entries consisting of detected and expected metabolites 2. The metabolites in both databases include water‐soluble and water‐insoluble or lipid metabolites. The analysis of lipid metabolites by mass spectrometry poses additional requirements and constraints to the experiments due to their inherent hydrophobic nature. Lipids must first be extracted using a procedure that involves phase separation into a hydrophobic and a hydrophilic phase. In addition, mass spectrometry analyses require that the hydrophobic lipids must be charged prior to MS analysis. This only became routine with the advent of electrospray‐mass spectrometry in the late 1980s 5, 6. Hence, metabolomic analyses often refer to the water‐soluble metabolome whereas lipidomic analyses refer (by definition) to water‐insoluble analyses. Despite these additional experimental requirements and constraints, the lipidomic field is now rapidly expanding, providing a glimpse of its contribution to the metabolome 6, 7, 8, 9, 10, 11, 12, 13, 14, 15. The LIPID MAPS Structure Database (http://www.lipidmaps.org/), the largest public lipid‐only database, contains over 37 500 unique structures of biologically relevant lipids 16, 17. Current estimates suggest that the entire lipidome may encompass approximately 180 000 different lipid species 18. Thus, of all the molecules contained in the metabolome, the lipids constitute the largest subset, including tens of thousands of distinct lipid molecular species existing in cells and tissues 12.
Hydrophobic Metabolome
Each dot of the KEGG‐based metabolic map (Figure 1) represents a single metabolite with its own unique chemical structure that is connected by line(s) indicating the metabolic pathway(s) each metabolite is involved in. The metabolic pathways involving lipid metabolites are outlined on the metabolic map. Recently, lipids have been divided into eight categories containing distinct classes and subclasses based on their chemical structure 19, 20 and the presence of these categories within the metabolome is indicated by individual panels. In this overview of the metabolome, the contribution of lipids may appear under‐represented relative to their abundance with the metabolome. This is mainly due to the fact that in this pathway representation, generic lipid classes are shown without sufficient fatty acid (side) chain information (relevant for all panels) and without carbohydrate head group variations (relevant for panel G). With these variations in fatty acid and glycolipid head group composition, an estimated number of 180 000 lipids has been predicted to exist 18 and this number does not even include the vast number of possible (per)oxidized lipids, that can be generated spontaneously during radical formation (e.g. stress) or by enzymatic reactions 21.
The enormous structural diversity of lipids requires complex regulation at multiple spatial and temporal scales and mainly due to innovations in lipidomic techniques we are only beginning to understand the biological role of lipids in health and disease. We are long past the dogma that lipids have an essential role as component of the lipid bilayer of biomembranes and in energy storage utilizing cellular lipid droplets and plasma lipoproteins. Research over the last decades has identified an additional role of lipids in cellular signaling, a structural role in membrane microdomain organization and dynamics, and a regulatory role in membrane trafficking. The importance of lipids as part of the metabolome is also evident from the numerous lipid‐related pathologies including lipid storage diseases such as fatty liver, obesity, and atherosclerosis, neurodegenerative diseases such as Alzheimer's disease, cancer, inflammation, and infectious diseases.
Pathogens and Lipids
Pathogenic micro‐organisms have evolved many strategies to circumvent host defenses and exploit the host cellular machinery. Specific virulence factors disable or subvert vesicular trafficking pathways to and from the host cell surface, which promotes pathogen entry, replication or escape 22, 23, 24, 25. The direct link of the metabolome with the cellular phenotype as well as the abundance of hydrophobic metabolites within the metabolome (hydrophobic metabolome or lipidome) makes lipids an attractive target for pathogens to modulate host cell processes 26, 27, 28, 29, 30. In many instances, this results in altered intracellular trafficking of pathogens after entry into host cells, resulting in escape from the default pathway toward lysosomes (for reviews, see e.g. 22 31, 32, 33).
Microbial pathogens can be classified into viral, bacterial, fungal and parasitic microbes. Obviously, these different types of pathogens have very different types of interactions with host cells and this is reflected in their usage of host cell lipids. An overview of the targeting of host cell lipids by pathogens is shown in Figure 2. Of note, we have excluded references to lipid rafts (discussed elsewhere e.g. 34, 35, 36) as in most instances it is not clear whether this is due to cholesterol, sphingolipids or indirect effects because of destabilization of lipid raft structures. Comparison of host cell lipid usage by viral (Figure 2A), bacterial (Figure 2B), fungal (Figure 2C) and parasitic (Figure 2D) pathogens shows that they each have a remarkable distinct and different preference for individual lipid categories in order to modulate host cell responses.
Figure 2.
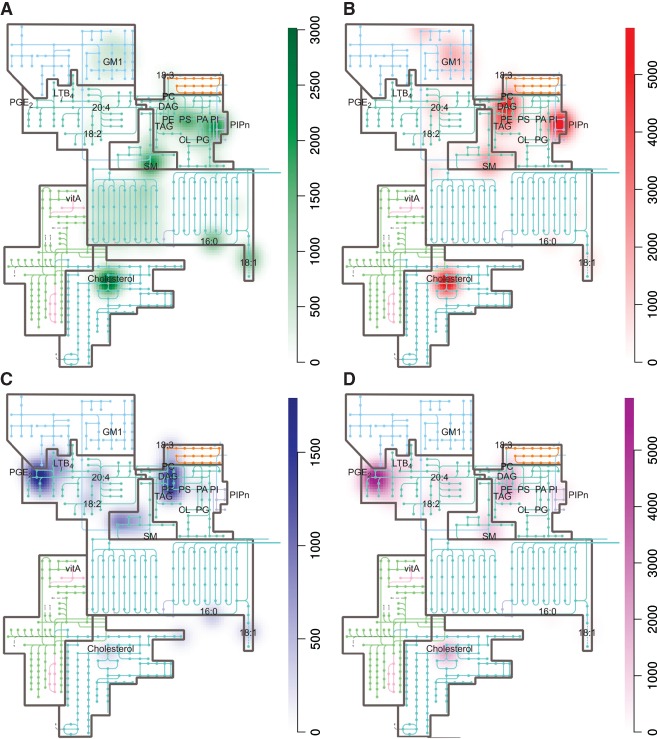
The involvement of lipids in host–pathogen interactions. Heat maps were generated for different types of pathogens (panels A–D) based on the weighted involvements of lipids in host–pathogen interactions (the list of pathogens and their lipid targets and weight factor is described in Table S1, Supporting Information). Heat maps show the frequency of involvements of specific host cell lipid (sub)classes (e.g. phosphatidylserine) and/or species (e.g. cholesterol) for viruses (A), bacteria (B), fungi (C), and parasites (D). Increased coloring indicates increased frequency. The heat maps were constructed with an algorithm using the R‐package for spatial statistics (spatstat) 247.
Viruses have no intrinsic lipid metabolism and by definition affect host cell lipids to modulate virus replication, host cell responses and/or to acquire lipids for their viral envelope. There is a strong focus on signaling lipids such as phosphoinositides 37, 38, 39, 40, 41, 42, 43, 44, 45 and phosphatidylserine (PS) 46, 47, 48, 49, 50 (Figure 2A). In addition, many viruses target cholesterol 51, 52, 53, 54, 55, 56, 57, 58, a structural membrane lipid specific for host cells that allows the virus to modulate the physical properties of intracellular membranes. During the genome replication stage, there are marked differences in the virus–host lipidome interaction even between viruses from the same family 40, 59, 60, whereas unrelated viruses might use similar strategies to usurp the host lipidome [e.g. the requirement for phosphatidylinositol‐4‐phosphate (PI(4)P) and cholesterol appears to be widespread] 37, 40, 45, 60, 61, 62, 63. Finally, viral activation of fatty acid synthase (FAS) for the production of new membrane compounds is frequently observed 59, 64, 65, 66, 67, 68, 69, 70.
For the other types of pathogens, bacteria, parasites and fungi, the situation is more complex. Prokaryotic or eukaryotic micro‐organisms not only target host cells to modulate host cell responses directly, but they can also produce lipids themselves, or acquire specific lipids from the host and/or convert these lipids, necessary for microbe growth or to influence host cell responses 26. This is highly relevant – but beyond the scope of this review – as it not only may provide important insights into the development of new therapeutic strategies, but also because the outcome of these lipid interactions may either lead to commensalism or to host damage/disease 26.
The interaction map of bacteria with host cell lipids (Figure 2B) shows that bacteria preferentially target phosphoinositides 71, 72, 73, 74, 75, 76, 77, 78, cholesterol 79, 80, 81, 82, 83, sphingomyelin (SM) 79, 84, 85, 86 and neutral lipids 87, 88, 89, 90. Most bacteria are not capable of synthesizing these specific lipid classes. For example, the major phospholipids in Escherichia coli are phosphatidylethanolamine (PE, 70%), phosphatidylglycerol (20%) and cardiolipin (5%) 91. It is tempting to speculate that by targeting these host‐cell specific lipids, bacteria are not in danger of affecting their own metabolism. Indeed, bacteria hardly target host cell PS, a minor lipid in bacteria, but needed for the synthesis of PE, whereas viruses do (compare Figure 2A and B).
The interaction map of fungi with host cell lipids is very different from that of viruses and bacteria and these pathogens appear to focus on structural phospholipids (PC and PE) 92, 93, sphingolipids 94, 95, 96 and eicosanoids 97, 98, 99 (Figure 2C). The targeting of neutral lipids is possibly related to the generation of precursors for phospholipid biosynthesis or eicosanoids to induce inflammatory responses.
Parasites are in many aspects not comparable to other pathogens. In general, in accordance with their parasitic way of life, parasites have discarded pathways of de novo lipid synthesis but have selectively retained several biosynthetic pathways that modify host lipids 100, 101. Although lipids such as fatty acids, phospholipids and sterols are obtained from their host or are synthesized from building blocks obtained from the host, less abundant lipids that are more difficult to acquire (e.g. specific unsaturated fatty acids, eicosanoids, ecdysteroids and quinones) are synthesized by the parasite, often by modification of more abundant substrates 102, 103, 104. Comparison of the four different panels shows that viruses, bacteria and parasites target glycolipids whereas fungi do not. Many viruses and bacteria use glyco(sphingo)lipids as receptors, but whether these lipids are indeed crucial for infection has been addressed in only a few cases. For more details about carbohydrates as virus (co)receptors, we refer to other reviews 85, 105, 106.
Viruses
Viruses constitute a highly diverse group of pathogens that are mainly classified by phenotypic characteristics, such as host organisms, the diseases they cause, their structure (e.g. presence or absence of a lipid bilayer, the ‘envelope’), and the nucleic acid type of the viral genome, which is either single‐ or double‐stranded DNA or RNA. Single‐stranded genomes are then designated as positive‐strand (+RNA), i.e. containing directly translatable information like mRNA, or negative‐strand (−RNA). Viruses are obligate intracellular pathogens and require host cells in order to replicate. A viral replication cycle basically comprises the following steps: receptor binding (attachment to the host cell), cell entry (often via endocytosis), uncoating (release of the viral genome into the cell), genome replication, virion assembly (packaging of new genomes into virus particles), and virus release (by budding or cell lysis). Viruses can interact with the host lipidome in two ways, namely by exploiting pre‐existing molecules or by actively altering host lipid metabolism. The role of lipids in (i) vital entry, (ii) genome replication, (iii) budding and (iv) innate immune response will be briefly addressed.
Viral entry
During entry, viruses usually specifically exploit one of the cellular endocytic routes and the lipids functioning in these pathways, but do not actively modulate the lipidome yet. The pathways and lipids exploited by viruses span the entire cellular repertoire. Despite this large degree of variation, cholesterol (category E) appears to be important for the entry of many different viruses (see e.g. 22 107). The most straightforward explanation is the fundamental importance of cholesterol for the organization of membranes and the functioning of endocytic pathways.
Viral genome replication
To support the replication of their genome, viruses can actively modulate the host lipidome. Most DNA viruses (e.g. herpes viruses) replicate their genomes inside the nucleus, but some [e.g. vaccinia virus (VACV)] assemble structures in the cytoplasm to complete their replication cycles (reviewed in 108 109). All +RNA viruses [e.g. poliovirus (PV), hepatitis C virus (HCV), dengue virus (DENV), West‐Nile virus (WNV) and SARS‐ and MERS‐coronavirus] reorganize host cell membranes into membranous structures in the cytoplasm that serve as a scaffold for viral RNA synthesis/replication (referred to as ‘viral factories’, ‘membranous web’ or ‘replication organelles’). While all +RNA viruses generate replication organelles, the morphology of these organelles varies greatly among virus families. Each virus family hijacks membranes from a different cellular organelle (e.g. Golgi, ER, mitochondria, endosomes or lysosomes), exploits a distinct set of host factors, and modulates specific lipid metabolic pathways to build replication organelles with a unique protein and lipid composition (reviewed in 110 111).
Fatty acids (FAs, category A) have been implicated in the replication of many different viruses. VACV was reported to rely on the synthesis and mitochondrial import of FAs and on β‐oxidation for ATP production 69. Many +RNA viruses use FAs to synthesize lipids to build their replication organelles. PV was reported to enhance uptake and prevent routing of FAs to lipid droplets 64, whereas DENV recruited FAS to its replication organelles and increased the activity of the enzyme to ensure high amounts of local FA synthesis 66. Also other viruses, e.g. WNV, require FAS activity, as their replication is inhibited by FAS inhibitors 59. Furthermore, the cellular energy regulator AMPK, which is an inhibitor of FAS, restricted infection of a number of unrelated viruses including the ‐RNA virus Rift Valley Fever Virus 68 and HCV. At least HCV actively counteracts this restrictive mechanism by inactivating AMPK 112. Finally, not only the amount of FAs may be important, but also chain length and saturation. PV specifically enhances the uptake of long chain FAs 64 and the replication of HCV and the integrity of its membranous web depend on the enzyme stearoyl‐CoA desaturase 1, which converts the saturated FA stearate into the mono‐unsaturated FA oleate 113.
Recently, PI(4)P (category C) and the PI(4)P‐synthesizing enzymes PI4KIIIα and PI4KIIIβ have been shown to play a pivotal role in +RNA virus replication. HCV utilizes predominantly PI4KIIIα to generate large pools of PI(4)P in its membranous web that are essential for viral RNA replication 38, 39, 41, 42, 43, 114, 115. PI4KIIIα activity was shown to be important for the integrity of the membranous web and the PI(4)P levels affect the phosphorylation status of one of the viral proteins (NS5A), thereby influencing the process of viral RNA replication 40, 44. Enteroviruses (e.g. PV, Coxsackievirus B3 and most probably also the other enteroviruses) on the other hand rely on PI4KIIIβ for a PI(4)P‐rich environment in their replication organelles 37, 116, 117. It has been suggested that the PI(4)P‐rich environment attracts the viral polymerase to the replication organelles to replicate the viral RNA 37. A new role of PI(4)P in infection was recently uncovered for both HCV and enteroviruses, being the recruitment of oxysterol‐binding protein (OSBP) to the replication organelles 60, 63 (see below). Also the replication organelles of the ‐RNA virus Junin virus were reported to contain PI(4)P, but the significance of this finding is still unclear 118. Recent research into the importance of phospholipids for virus replication has focussed on PI(4)P, but there is emerging evidence that also other phospholipids play a role. In DENV‐infected mosquito cells, PE (primarily lysoPE16:0) and PA are upregulated, but the role of these lipids in replication remains to be established 65. Most strikingly, influenza A virus (IAV), a ‐RNA virus that replicates in the nucleus, upregulates the synthesis of ether‐phosphatidylcholine (ether‐PC) species in peroxisomes, which appears to be important for replication, although the role of the ether‐PCs is unknown 119.
For virus genome replication, the importance of sphingolipids (category D) has only been covered by a few studies. It was noted that two Ceramide species (Cer18:1/16:0 and Cer18:0/16:0) are specifically enriched on replication organelles in DENV‐infected mosquito cells, but the role of these lipids remains to be shown 65. During HCV infection, the SM synthases SMS1 and SMS2 are upregulated and inhibition of SMS activity impairs replication. A number of specific SM and Cer species, in particular SM18:1/16:0 and SM18:1/24:0, are enriched on the membranous web, where they interact with and increase the activity of NS5B, the viral RNA‐dependent RNA‐polymerase 120.
As mentioned above, HCV and enteroviruses were reported to use PI(4)P to recruit OSBP to replication organelles. OSBP is an important regulator of cellular lipid homeostasis that shuttles cholesterol from ER to Golgi 121. It is suggested that in infected cells OSBP is important for the accumulation of cholesterol (category E) on the replication organelles 60, 63. To accommodate the requirements for cholesterol, HCV upregulates the synthesis of cholesterol (and FAs) via SREBP activation 122, whereas enteroviruses, which shut down host protein synthesis, acquire cholesterol by increasing uptake of the lipid and possibly by rerouting cholesterol from lipid droplets 61, 62. The roles of cholesterol in virus replication have only begun to be unravelled. For HCV, OSBP and cholesterol appear to be important for the integrity of the membranous web 60. Enteroviruses were proposed to require cholesterol for optimal proteolytic processing of viral proteins by a mechanism that has yet to be elucidated 61, although a role in replication organelle organization similar to HCV remains possible. Other viruses also require cholesterol or modulate its synthesis for efficient replication (e.g. DENV, Norwalk virus; 51 58), although the role of cholesterol in the replication of these viruses is still under investigation. Interestingly, hepatitis B and C viruses, which are in distinct virus families, accumulate the cholesterol biosynthetic intermediates 7‐dehydrocholesterol and desmosterol respectively 123, 124. The importance of these lipids for infection is not known, but their function may extend beyond that of mere biosynthetic intermediates.
Viral budding
During budding, many – but not all – enveloped viruses acquire specific lipids, leading to an envelope with a lipid composition different from the donor membrane (reviewed in e.g. 125). These lipids are needed for efficient budding and release, optimal stability of the viral particle and entry. Viruses may obtain envelopes with a specific lipid composition by modulating host lipid metabolism and/or by budding from membrane microdomains (rafts) with a specific lipid composition. For example, the human immunodeficiency virus (HIV) obtains a raft‐like envelope rich in sphingolipids (category D) presumably by budding from lipid rafts modulated by the viral protein Nef 126, 127. Many other viruses (e.g. IAV and WNV) also have envelopes enriched in SM and ceramide species 119, 128. Vaccinia virus has a PS‐rich envelope (category C), which makes it resemble apoptotic bodies and provides an entry route via PS‐dependent macropinocytosis 46. PS‐dependent entry may be a widespread mechanism used by many different viruses, such as DENV virus and the −RNA ebola virus (EBOV) (reviewed in 107).
Innate antiviral response
Finally, cells can alter their lipidome to mediate antiviral defense mechanisms. When cells sense a viral infection, they produce and secrete interferons as danger signals. In response to interferons, a collection of interferon‐stimulated genes (ISGs) is upregulated to combat the viral infection at several levels. Cholesterol‐25‐hydroxylase, which synthesizes the cholesterol derivative 25‐hydroxycholesterol (25HC) (category E), was found to be such an ISG 129 and 25HC levels were highly elevated upon infection 130. 25HC was suggested to alter the properties of host and virus membranes (e.g. IAV, HIV and EBOV) and reduce their permissiveness for fusion 129. Additionally, 25HC inhibits the synthesis of a wide variety of lipids and thus may restrict viruses that require lipid synthesis. Another ISG, IFITM3, was reported to disrupt OSBP function and alter cellular cholesterol homeostasis, which inhibited infection of at least two different enveloped viruses 131. However, viruses in turn have developed mechanisms to neutralize the innate defense mechanism. For example, WNV was reported to redistribute cellular cholesterol not only to enhance replication, but also to disrupt the cholesterol‐ and raft‐dependent functioning of interferon signaling thereby reducing the innate immune response 132.
Pathogenic Bacteria
Intracellular bacterial pathogens induce a variety of metabolic changes in the affected host cells to promote their own survival and replication 133, 134, 135, 136. By secreting virulence factors termed effectors, bacteria trigger uptake (invasion), intracellular replication and interfere with inflammation processes 23, 137, 138. Modulation of lipid metabolism by virulent bacteria is a common approach in their strategy to invade and propagate in the host cell 28. Thus, lipids are not only used as a nutrient source but also to influence the host cell metabolism, physiology and membrane trafficking, enabling pathogen survival and replication. While the targeting of plasma membrane lipid rafts and the modulation of phosphoinositides seems characteristic for influencing their invasion, targeting of host cell cholesterol (esters), triacylglycerols and phosphoinositides facilitates the intracellular survival of a number of pathogens. FAs are the predominant target for interference with inflammation. The role of lipids in (i) bacterial uptake, (ii) replication and (iii) inflammation will be briefly addressed.
Bacterial invasion
Pathogenic bacteria evolved different strategies to invade their host. For example, Streptococcus, Salmonella and Coxiella induce their uptake in nonphagocytic cells by triggering receptor‐mediated endocytosis 139, by actin rearrangements 140, 141 or by targeting lipid raft microdomains at the plasma membrane 142. Interference with the mechanism of uptake was shown to decrease the virulence of these pathogens suggesting that the entry mechanism is tightly linked to the intracellular survival of the bacteria. Cholesterol is considered a crucial factor in the uptake of host cells by Salmonella 143, 144, 145, Helicobacter 146, Vibrio cholera 147, 148, Shigella flexneri 79 and Listeria 149.
Another target for intracellular bacteria to affect their invasion and subsequent diversion of host membrane trafficking pathways is the manipulation of phosphoinositides 71, 74, 150, 151. All different phosphoinositide species are targeted during the bacterial‐host interplay. Pathogens such as Salmonella and Mycobacteria e.g. directly interfere with the PI(3)P levels of the plasma membrane during entry by secreting effector molecules. The phosphatase SopB secreted by Salmonella increases the PI(3)P levels at the site of engulfment to recruit the host Rab 5 and PI(3)P kinase Vps34 to the newly formed Salmonella‐containing vacuole 152, 153. By contrast, Mycobacteria decreases PI(3)P levels to interfere with the intracellular maturation 154. Similar seemingly contrasting phenomena are observed for other phosphoinositides. For the benefit of Shigella, PI(4,5)P2 levels are decreased in an bacterial effector driven way to produce PI(4)P and PI(5)P 155, but successful Yersinia invasion requires transient production of PI(4,5)P2 at the site of bacterial entry 73. It will require a better understanding of the entire (hydrophobic) metabolome to understand how opposing regulation of phosphoinositide metabolites can both favor pathogen survival.
Bacterial replication
Once inside the host cell, bacterial pathogens use different strategies to subvert the host‐cell trafficking pathways in order to resist intracellular killing 156. Interfering with the host cell lipid metabolism is a powerful mechanism used by a number of intracellular bacteria to create a replicative niche and to inhibit autophagosomal induction, vacuole acidification and apoptosis from inside the pathogenic vacuole 28.
Many bacteria interfere with phosphoinositide metabolism not only to promote host cell entry but also to facilitate survival 74, 150. During invasion, targeting of host cell phosphoinositide metabolism is aimed at interference with the signal transduction pathways and cytoskeletal architecture. During intracellular survival, the role of phosphoinositides in the host cell membrane dynamics is targeted 157. Phosphoinositides contribute to organelle identity by recruitment of specific effector proteins 75, 158. To subvert vesicular trafficking of infected host cells, Legionella was shown to secret effector proteins that are anchored to the Legionella‐containing vacuole in a PI(4)P‐dependent manner 159. Bacteria that reside in a bacterial containing vacuole for successful replication like Salmonella 160, 161, 162, Shigella 163 and Mycobacteria 164, continuously manipulate phosphoinositide metabolism from within the vacuole by secreting phosphatases (SopB, IpgD and SapM, respectively).
Many intracellular pathogens also interfere with cholesterol metabolism by acquisition of host cell cholesterol. Accumulation of cholesterol on the vacuole of Coxiella 165, 166, Salmonella 81, 145, 167, Chlamydia 80 and Mycobacteria 82 was shown to be essential for intracellular survival. The Salmonella‐containing vacuole for example contains up to 30% of the cellular cholesterol pool 81 by redirecting exocytic vesicles from the Golgi complex rich in cholesterol and sphingolipids 168. A similar strategy is used by Chlamydia 33. Although the function of this recruitment is not clear, a role in membrane‐trafficking pathways has been suggested 169. A role as a nutritional source can also not be excluded, similar to Mycobacterial cholesterol acquisition 170. Coxiella, a pathogen that persists for 4–5 days in the infected host cell, causes an increase of cellular cholesterol levels by up to 70% 166. It is not clear whether this is a pathogen driven process or a response by the host cell to maintain cholesterol homeostasis. Nevertheless, it is an evident example of pathogen‐induced (de)regulation of host lipid metabolism.
Several intracellular bacteria also interfere with neutral lipid metabolism, either directly (e.g. secretion of lipases) or indirectly by interference with lipid droplet homeostasis. Besides their role in energy (TAG) and cholesterol ester storage, lipid droplets are involved in host cell lipid transport and metabolism, membrane trafficking, intracellular signaling, and production of inflammatory mediators 171, 172. Although interference with lipid droplet biogenesis and turnover is a strategy used by many bacteria, the exact role of this interference is not well understood in most cases. This is illustrated by Salmonella, which actively interferes with host cell lipid droplets by secreting SSeJ and SseL effector molecules, however with opposing effects. SseJ mediates the esterification of cholesterol in cell membranes which results in enhanced lipid droplet biogenesis 173. SseL prevents the accumulation of lipid droplets via its deubiquitinase activity 87. Other bacteria like Mycobacteria and Chlamydia induce a massive increase of (TAG) containing lipid droplets in the infected macrophages and in both cases lipid droplet accumulation during infection was shown to be crucial for bacterial survival 174. Mycobacteria utilize lipid droplets as a nutritional source 88 and hijack lipid droplets as part of their strategy in acquiring host cell iron 175. Chlamydia targets lipid droplets to interfere with inflammation by replacing the lipid droplet core protein ADRP with bacterial derived effector molecules 89, 90.
Bacterial inflammation
Inflammation is the body's immediate response in attempt to prevent the spread and infection of microbial pathogens 176, 177. Lipid mediators, such as prostaglandins and leukotrienes (lipid category A) are key players in inducing inflammation 178. They are synthesized from phospholipid‐derived polyunsaturated FAs (PUFAs) like arachidonic acid (ω6) and ω3 fatty acids 177. Both Gram‐negative and Gram‐positive bacteria are able to induce their synthesis via triggering signal transduction pathways that enhance phospholipase activities and/or cyclooxygenase COX‐2 expression levels in target cells 179. Salmonella alters host cell signaling in the intestinal epithelial cells by delivering the effector protein SpiC to the cytosol of infected macrophages. This action promotes an immunosuppressive phenotype which impairs bacterial killing 180. Escherichia coli, Vibrio cholera, Mycobacteria, Streptococcus and Pseudomonas use similar mechanisms to induce the COX‐2 expression and promote prostaglandin production for their own benefits 179. Bacterial pathogens also interfere with the biosynthesis of oxidized FAs that act as anti‐inflammatory molecules. Pseudomonas aeruginosa secretes a lipo‐oxygenase that converts host arachidonic acid for local 15‐HETE production 181. In this way, the host‐immune defense can be subverted by generating local ‘stop signals’ 182.
Fungi
Of the 1.5 million species of fungi, there are only around 300 known to cause pathology in humans 183, 184. Pathogenic species such as the relatively common Trichophyton (causing athlete's foot), Crytococcus neoformans (causing meningoencephalitis) and Aspergillus spp. (causing infections of ear, eye and nails) are often opportunistic and systemic infections normally only occur in immunocompromised individuals 185. Thus, fungi do not typically enter the host cell but affect host lipid metabolism by extracellular secretion of factors. Indeed, fungi are rich sources for molecules that interact with the lipid metabolism of mammals. Particularly, well studied are molecules that interfere with various steps in the synthesis of sphingolipids because of their applicability in fundamental research and cancer therapy 186, 187. Sphingolipids are key molecules in protein trafficking, lipid homeostasis and cell differentiation and their balanced occurrence is essential. Serine palmitoyltransferase (SPT) catalyzes the first, committing‐ and rate limiting step in sphingolipid de novo synthesis. Sphingofungins, lipoxamycin and myriocin are fungal structural analogs of the SPT intermediate‐ or end products with potent and highly selective inhibitory activities 188, 189. Also subsequent steps in the synthesis of the sphingolipid backbone are efficiently inhibited by fungal metabolites: ceramide synthase is sensitive to Fumonisins (isolated from Fusarium verticillioides), AAL‐toxin (Alternaria alternata) and Australifungins (nonpathogenic Sporormiella australis) 190, 191, 192. It is interesting to note that all these fungi‐derived molecules interfere with early steps in sphingolipid synthesis, i.e. prior to the synthesis of sphingomyelin in host cells (Figure 2C). This is probably related to the fact that after the synthesis of (dihydro‐)ceramide, the fungal‐ and mammalian anabolic metabolism of phosphosphingolipids diverge. Whereas mammals synthesize SM, fungi use inositol as an headgroup, leading to inositol phosphorylceramide (IPC), which plays a crucial role in pathogenesis of Cryptococcus neoformans 193, 194. Therefore, these inhibitors are also potent antifungals and it has been suggested that these inhibitors are used to gain advantage over fungi competing for the same resources 185.
Most pathogenic fungi also secrete phospholipases to facilitate adhesion, cell entry, and lysis 92. Phospholipases are hydrolytical enzymes that degrade membrane phospholipids, leading to membrane dysfunction or even disruption of the cell. In this case, targeting of the most abundant host cell membrane phospholipids, PC and PE, is effective in interference with membrane integrity (Figure 2C). The action of phospholipases also results in the generation of bioactive signaling molecules. In the case of phospholipase C action, diacylglycerol is generated which leads to activation of protein kinase C and tilts the survival/death balance toward survival and proliferation. In the case of phospholipase A or B, FAs are released that can be consumed by fungi and/or oxidized to generate potent signaling molecules with a profound impact on the immune system. These oxidation products are derived from PUFAs in the host cell. Arachidonic acid is used as a precursor in the generation of eicosanoids, a family of oxygenated C20 fatty acids that include prostaglandins, leukotrienes and thromboxanes 195 (Figure 2D). Enzymatic oxidation of arachidonic acid is achieved by the actions of either lipo‐oxygenases, cyclooxigenases or CYP enzymes, but also nonenzymatic reactions (lipid peroxidation) can lead to the formation of bioactive arachidonic acid metabolites known as isoprostanes 196. Similarly, other PUFAs such as eicosapentaenoic acid (20:5) or docosahexaenoic acid (22:6) can serve as precursors for bioactive oxidation products in mammalian hosts. Analysis of the genomes of numerous fungi suggests the ubiquitous presence of enzymes forming oxidized FAs 197. This makes oxidized FAs ideal candidates for host–pathogen interaction and there are indeed numerous reports of fungal derived oxidized FAs interacting with the immune system of the host 198. For instance, the major pathogens Cryptococcus neoformans, Candida albicans and Aspergillus fumigatus, all synthesize and secrete anti‐inflammatory prostaglandins PGE2 en PGD2 97, 98. Linked to decreased pulmonary function are PGF2a, TXB2, 6‐keto PGF1a and isoprostanes, which are released by the respiratory pathogen Aspergillus fumigatus 97, 99, 199. It has been suggested that the immunosuppressive molecules secreted by fungi are necessary to establish a sustainable population of (nonpathogenic) yeast in e.g. the bowel and that fungal dysbiosis leads to disease 200.
As with the fungal‐derived molecules that interfere with sphingolipid metabolism in both host cells and in fungi, oxidized FAs can also act both on host cells and on fungi. Fungi use oxidized FAs for regulation of fungal cell growth and differentiation, but these processes are still poorly understood. For instance, PGE2 and TXB2 induce germ tube formation in C. albicans and there is strong evidence that other oxidized FAs are involved in the development of the sexual stage in this and other yeasts 201. Also the formation of biofilms, conglomerates of fungi protected by a mucuous layer, is directed by prostaglandins 202. Thus, fungal derived oxidized FAs modulate the host's immune system, while the same molecules influence normal fungal form and function. Here we see a typical example of signaling by oxidized FAs that is well conserved across biological kingdoms and that is optimally exploited as inter‐kingdom signaling molecules 198. Whereas virulence factors from bacteria, viruses and parasites have evolved to optimally exploit the resources of the host, it seems that fungal effectors establish a stable commensalism based on complex bi‐directional fungi–host interactions mediated by e.g. oxidized FAs, which may run out‐of‐control in the case of pathogenic fungi.
Parasites
Two completely different types of parasites exist: unicellular parasitic protozoa and parasitic helminths (worms). For the purpose of this review, parasitic protozoa can be divided in (i) protozoa that live inside cells of their host (e.g. erythrocytes or macrophages) and (ii) those that live outside the cells of the host, e.g. parasitizing the digestive and urogenital tract or the bloodstream of the host (see e.g. Table 1). Interactions with the lipid metabolism of the host can be strikingly different in these different niches. Parasitic helminths, however, for obvious reasons of magnitude never live inside cells of the host, but are always extracellular. However, with respect to metabolic interactions with their host a clear distinction can be made between helminths that live in, e.g. the digestive tract of the host, compared with helminths that live truly inside the body of their host (in, e.g. the lymphatic system or bloodstream). These differences in niche between the various types of parasites, protozoa as well as helminths, result in differences in the ways parasites interact with the lipid metabolism of their hosts.
Table 1.
Protozoa versus helminths: Different niches of unicellular and multicellular parasites
| Parasite type | Intra‐ or extra ‐cellular | Example (parasite group) | Example (species) | Intracellular locationa | Extracellular locationa |
|---|---|---|---|---|---|
| Protozoa | Intracellular | Apicomplexa | Plasmodium spp. | Erythrocytes | |
| Toxoplasma gondii | Nucleated cells | ||||
| Trypanosomatidae | Leishmania spp. | Macrophages | |||
| Trypanosoma cruzi | (Heart) Muscle cells | ||||
| Microsporida | Enterocytozoon spp. | Mucosal cells | |||
| Extracellular | Diplomonadida | Giardia lamblia | Digestive tract | ||
| Amoeba | Entamoeba histolytica | Digestive tract | |||
| Parabasalia | Trichomonas vaginalis | Urogenital tract | |||
| Trypanosomatidae | Trypanosoma brucei | Bloodstream | |||
| Helminths | Intracellular | None | None | – | |
| Extracellular | Nematodes (roundworms) | Ascaris lumbricoides | Digestive tract | ||
| Wuchereria bancrofti | Lymphatics / bloodstream | ||||
| Cestodes (tapeworms) | Taenia saginata | Digestive tractb | |||
| Echinococcus granulosus | Digestive tractc | ||||
| Trematodes (flukes) | Fasciola hepatica | Bile duct | |||
| Schistosoma mansoni | Bloodstream | ||||
In final host.
+ cysts in muscle tissue in mammalian host (cattle).
+ cysts in liver and lungs of intermediate mammalian hosts.
In general, most parasites affect the host FA metabolism (lipid category A) by catabolism of host lipids. Subsequently, the parasites often take up the lipid degradation products, including FAs, from their host 100, 203, 204, 205, 206, 207. Some parasites even lost the ability to synthesize FAs themselves de novo, such as the blood‐dwelling helminths Schistosoma spp. 208, 209, and thus depend entirely on FA uptake from the host. After uptake by the parasite these FAs are often remodeled, for instance, by chain elongation or by alteration of the desaturation of the FA moiety 104, 203, 210. Many parasites take up PUFAs, such as arachidonic acid, to produce eicosanoids that subsequently affect the host immune reaction 211. For instance, prostaglandin PGE2 is synthesized by many parasites including parasitic protozoa (e.g. Entamoeba histolytica, Toxoplasma gondii and Trypanosoma spp.), as well as by nematode, cestodes and trematode helminth species 212. Prostaglandins produced by the parasite affect the host in many processes including vascular tone, hemostasis, chemotaxis, activation and skewing of various types of immune cells 212, 213.
Similarly, parasites do not take up intact TAGs from the host and they tend to use phospholipases (affecting lipids in category C) to be able to take up free FAs and lyso compounds as building blocks for their own complex lipid biosynthesis. TAG metabolism (lipid category B) and trafficking has been shown to be essential for parasite development and proliferation in Plasmodium‐infected erythrocytes 214, 215. Accumulation of TAG in lipid droplets in P. falciparum‐infected erythrocytes was shown to be strikingly pronounced during intraerythrocytic proliferation from trophozoite to the schizont stage, whereas TAG degradation became active from schizont to the segmented schizont stage. Presumably, TAG is not essential as a source of energy for the parasite, because the capacity for FA oxidation is very limited. Possibly TAG provides a source of FA groups for the glycerophospholipid biosynthesis that is required for membrane biosynthesis for merozoite release 215. Glycerophospholipids (lipid category C) of the host cell play a role in the invasion process of various intracellular protozoan parasites, as surface associated phospholipase enzymes of Toxoplasma, Cryptosporidium, Entamoeba, Plasmodium and Trypanosoma cruzi are supposed to be involved in their invasion of the host cells by creating pores in the host cell membrane, or by altering the host membrane fluidity 207. Phospholipase activity has also been shown to play a role in differentiation and intracellular development of these protozoan pathogens.
A biphasic generation of ceramide (lipid category D) is triggered in macrophages by the protozoan parasite Leishmania 216, 217. First, attachment of the parasite to the macrophage membrane activates acid sphingomyelinase, which catalyzes the formation of ceramide from SM. Inhibition of acid sphingomyelinase resulted in reduced uptake and infection with the parasite, which shows that ceramides are important for entry into the host cell. Subsequently, de novo synthesis generates ceramide that will probably reduce the cellular cholesterol level and displace the cholesterol from the membrane, leading to enhanced membrane fluidity, disruption of rafts, and impaired antigen‐presentation to the T cells. SM is likely to be an important lipid in Toxoplasma, as the SM levels in Toxoplasma infected cells are increased. Toxoplasma salvages sphingolipids from the host Golgi through the rerouting of selected Rab vesicles to the parasitophorous vacuole 218.
Parasites do not synthesize sterols (lipid category E) themselves and instead sterols are usually taken up from the host 204. In mammalian cells infected with Toxoplasma gondii, the uptake of low‐density lipoproteins (LDL) is upregulated and its cargo is subsequently diverted such that LDL‐derived cholesterol is not first transferred to the plasma membrane of the host cell but directly to the parasitophorous vacuole 219, 220, 221. In addition, in Toxoplasma infected cells cholesterol synthesis is suggested to be uncoupled from LDL uptake, such that the normally negative feedback on HMG‐CoA reductase, the enzyme that controls the biosynthetic flux to cholesterol, is dysregulated 204, 222.
Cholesterol from the host is not only required for parasite replication, it is also important in host cell invasion by Toxoplasma, because cholesterol depletion in the host cell plasma membrane blocks parasite internalization 223. Although cholesterol‐enriched parasite apical organelles termed rhoptries discharge lipids during cell entry and contribute to the parasitophorous vacuolar membrane (PVM) formation, rhoptry cholesterol appeared not to be essential for this process. However, host plasma membrane cholesterol was shown to be incorporated into the forming PVM during invasion, through a caveolae‐independent mechanism, which suggests that host cholesterol controls entry of an intracellular pathogen 27, 223. Similar results were obtained for entry of the parasitic protozoan Leishmania donovani into macrophages and Trypanosoma cruzi into HeLa cells, as cholesterol depletion from the plasma membrane inhibited entry of the parasite 224, 225, 226.
Relevance to Membrane Trafficking
Research on the involvement of lipids in host–pathogen interactions has significantly contributed to our molecular understanding of intracellular membrane trafficking and dynamics in eukaryotic cells. A few examples will be given for each type of pathogen.
Notable contributions to the membrane trafficking field come from lipid related research on protein toxins internalization, yielding insight in retrograde transport from the plasma membrane to the endoplasmic reticulum. Shiga and cholera toxin are internalized into the host cell in a cholesterol‐sphingolipid dependent manner 79. Retrograde transport was known to operate between PM and TGN, but these studies showed that it continued through Golgi cisternae to the ER 227, 228, 229. The targeting and interconversion of various phosphoinositides by pathogen kinases and phosphatases contributed to our understanding of the importance of phosphoinositide signatures to subcellular organelle identity 71, 230.
In the 1980s, fumonisins were isolated from the fungus Fusarium moniliforme 231. The usability in biochemical research of these analogs of intermediates in sphingolipid biosynthesis was quickly recognized and fumonisins (together with other fungal sphingolipid analogs such as myriocin discussed above) have been widely used in the elucidation of the role of sphingolipids in the trafficking of proteins and lipids 232, 233.
Viruses have evolved to utilize a wide variety of cellular molecules and pathways. The finding by Helenius and colleagues that Semliki Forest virus (SFV), an enveloped virus, fused in a prelysosomal compartment 234 was critical to the discovery and characterization of the organelles that we now know as ‘endosomes’ 235. The dependence of SFV on luminal acidification and cholesterol 236 suggested two important properties of endosomes. Since those pioneering studies, viruses have been indispensable tools for the elucidation of endocytic pathways and many other aspects of membrane and lipid biology 22, 237.
During the obligate intracellular liver stage, Plasmodium berghei parasites are surrounded by vesicles from the host late endocytic pathway allowing transport of material toward the parasite interior 238. The Plasmodium parasitophorous vacuole (PV) membrane displays long tubular extensions that pervade the host cytoplasm, which increases the surface of exchange between the host cell and the PV. Although Plasmodium is unable to synthesize sterols, it does contain cholesterol that must be diverted from host cell compartments and properly delivered to the PV. Interestingly, the PV membrane forms tight associations with the host endoplasmic reticulum (ER) shortly after invasion and during schizogony 239. The gathering of host ER to the PV membrane offers an attractive mechanism to situate the host lipid biosynthetic machinery in close proximity to the PV and may be reminiscent of the currently intensely investigated organellar contact sites that allow rapid transport and/or exchange of lipids.
These examples from the various types of pathogens exemplify the relevance of lipid targeting by pathogens to affect host cell membrane trafficking. Due to the lack of experimental resolution, most of these studies address the effect of lipid classes. In only a very few cases, the role of a specific lipid species in membrane dynamics could be identified. Notable examples are the role of the ganglioside GM1 as the receptor of cholera toxin (although in this case the importance of the fatty acid composition is not known)240, and a highly specific interaction of a single sphingomyelin species, SM 18, with the transmembrane domain the COPI machinery protein p24 241. Lipidomic and/or metabolomic studies on host–pathogen interactions bear great potential to contribute to advancement of intracellular (lipid) trafficking pathways in eukaryotic cells. First of all, systematic lipidomic analysis of lipids allows the simultaneous screening of virtually all lipids in one assay. Second, using mass spectrometry, the analysis can be performed with much higher sensitivity, allowing the identification of minor (signaling) components. Third, the new generation of mass spectrometers has a much higher resolution, allowing precise annotation of molecular species.
Conclusions and Outlook
The hydrophobic metabolome encompasses an unprecedented number of lipid species and deserve more attention as being a significant – if not largest – part of the entire metabolome. Even current estimates are probably conservative as there are almost unlimited possibilities with spontaneous and enzymatically catalyzed lipid oxidation products. It is becoming increasingly evident that even these oxidized lipids can have important cellular functions by interacting with lipid‐binding proteins 242. In addition, there are numerous pathogen‐specific lipids such as lipopolysaccharides containing a lipid A moiety in Gram‐negative bacteria 243 and unique lyso‐PS species 244 that are not considered in this review but that further increase the complexity of the entire lipidome. Yet, the function of the individual lipid species is largely unknown and only in a few cases we start seeing a glimpse of their biological function.
During evolution, microbial pathogens have exploited the potential of the hydrophobic metabolome and now take full advantage of the complexity of the lipidome to influence the host cell phenotype 28. The different types of microbial pathogens do so in a very different way. Simple (unicellular and prokaryotic) microbial pathogens such as bacteria target host cell lipids that are generally not synthesized by bacteria. These include structural membrane lipids such as cholesterol and phosphatidylcholine as well as signaling lipids such as phosphoinositides and sphingomyelin. In addition, they target neutral lipids for fatty acid production for a variety of functions. In broad outlines, viruses and bacteria target the same group of lipids (compare Figure 2A and B) with one notable exception: whereas several viruses target PS in the host cells, this is only sporadically reported for bacteria. The complex eukaryotic microbial pathogens, fungi and parasites, interact with the host cell lipidome in a very different way as compared to bacteria and viruses (compare Figure 2C and D with Figure 2A and B). The most notable differences between ‘simple’ and ‘complex’ microbial pathogens are (i) the lack of cholesterol and signaling lipid (phosphoinositides and sphingomyelin) targeting by fungi and parasites; and (ii) the abundant targeting of the eicosanoid pathways in the host cells by fungi and parasites, often dealing with systemic inflammations in the host organism.
All pathogens target the PC, PE, diacylglycerol and TAG area of the metabolic map (Figure 2). These lipids are located in close proximity for obvious reasons, i.e. having DAG as a shared precursor. The hydrolysis of these lipids can, however, yield very different fatty acids. TAG species generally contain saturated and mono/di unsaturated fatty acids with an average chain length of 16–18 carbon atoms. In contrast, PC and PE will contain mostly long chain (C20–C24) PUFAs at the sn‐2 position. Hence, it is expected that complex pathogens predominantly target the phospholipids in this metabolic area to generate arachidonic acid, a central precursor for eicosanoid synthesis. Bacteria and viruses also target this metabolic area, but for other reasons, e.g. the generation of fatty acids needed for membrane formation.
Here we have presented one of the first visualizations of the lipid‐based interaction of pathogens with host cells. Based on available data in the literature, we have categorized approximately 500 interactions using the cellular metabolome as a template. At this relatively low resolution, hotspots for the involvement of host–pathogen interactions can already be identified. Novel lipidomic technologies are rapidly being created and implemented such as Imaging mass spectrometry and single cell metabolomics/lipidomics. High‐resolution mass spectrometry will allow precise annotation of lipid species and combined with automated high‐throughput analysis and novel bioinformatics tools, we will soon witness the generation of enormous amounts of high density data allowing spatiotemporal resolution of the involvement of lipids in host–pathogen interactions. Therefore, we expect the resolution of the interaction maps to increase dramatically in the near future, allowing the identification of novel hotspots for the involvement of lipids in host–pathogen interactions with potential applications in fundamental and applied research.
GLOSSARY
- 25HC
25‐hydroxycholesterol
- AMPK
AMP‐activated protein kinase
- DENV
dengue virus
- EBOV
Ebola virus
- FAS
fatty acid synthase
- FAs
fatty acids
- HCV
hepatitis C virus
- HIV
human immunodeficiency virus
- IAV
influenza A virus
- ISG
interferon‐stimulated gene
- OSBP
oxysterol‐binding protein
- PA
phosphatidic acid
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PI(4)P
phosphatidylinositol‐4‐phosphate
- PS
phosphatidylserine
- PUFA
polyunsaturated fatty acid
- PV
poliovirus
- PVM
parasitophorous vacuolar membrane
- SFV
Semliki Forest virus
- SM
sphingomyelin
- SPT
serine palmitoyltransferase
- TAG
triacylglycerol
- VACV
vaccinia virus
- WNV
West‐Nile virus
Supporting information
Table S1: List of lipid‐based interactions between host cells and pathogens that was used for the generation of Figure 2. A weighting of less than one was assigned if one specific interaction can be located at different nodes in the metabolic pathway. For example, phospholipase A2 activity of pathogens increases host levels of unesterified polyunsaturated fatty acids (18:2, 18:3, 20:4), commonly found at the sn‐2 position of phospholipase A2 substrates. In this case, the three fatty acids will be assigned a weighting factor of 0.33. Similarly, distinct interactions of a pathogen that target the same lipid in the host will result in weighting factors larger than 1. For example, Listeria has been described to affect a lipase and a phospholipase C, both resulting in increased levels of DAG.
Acknowledgments
We apologize to our colleagues whose work could not be cited due to space restrictions. Research in the authors' laboratories is supported by grants from the Netherlands Organization for Scientific Research (NWO): VENI‐722.012.066 (JRPS), VENI‐863.12.005 (HMvdS), ALW‐820.02.018 and VICI‐91812628 (FJMvK); by the BE‐Basic R&D Program (http://www.be-basic.org), which was granted an Economic Structure Enhancement Fund (FES) subsidy from the Dutch Ministry of Economic Affairs, Agriculture and Innovation (EL&I) (JBH and JFB); and by an NIH CENTER U19AI109725 grant to JBH.
References
- 1. Patti GJ, Yanes O, Siuzdak G. Innovation: metabolomics – the apogee of the omics trilogy. Nat Rev Mol Cell Biol 2012;13:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, Djoumbou Y, Mandal R, Aziat F, Dong E, Bouatra S, Sinelnikov I, Arndt D, Xia J, Liu P, et al. HMDB 3.0 – The Human Metabolome Database in 2013. Nucleic Acids Res 2013;41:D801–D807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smith CA, O'Maille G, Want EJ, Qin C, Trauger SA, Brandon TR, Custodio DE, Abagyan R, Siuzdak G. METLIN: a metabolite mass spectral database. Ther Drug Monit 2005;27:747–751. [DOI] [PubMed] [Google Scholar]
- 4. Tautenhahn R, Cho K, Uritboonthai W, Zhu Z, Patti GJ, Siuzdak G. An accelerated workflow for untargeted metabolomics using the METLIN database. Nat Biotechnol 2012;30:826–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989;246:64–71. [DOI] [PubMed] [Google Scholar]
- 6. Griffiths WJ, Wang Y. Mass spectrometry: from proteomics to metabolomics and lipidomics. Chem Soc Rev 2009;38:1882–1896. [DOI] [PubMed] [Google Scholar]
- 7. Wenk MR. The emerging field of lipidomics. Nat Rev Drug Discov 2005;4:594–610. [DOI] [PubMed] [Google Scholar]
- 8. Van Meer G. Cellular lipidomics. EMBO J 2005;24:3159–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dennis EA. Lipidomics joins the omics evolution. Proc Natl Acad Sci USA 2009;106:2089–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wenk MR. Lipidomics: new tools and applications. Cell 2010;143:888–895. [DOI] [PubMed] [Google Scholar]
- 11. Shevchenko A, Simons K. Lipidomics: coming to grips with lipid diversity. Nat Rev Mol Cell Biol 2010;11:593–598. [DOI] [PubMed] [Google Scholar]
- 12. Harkewicz R, Dennis EA. Applications of mass spectrometry to lipids and membranes. Annu Rev Biochem 2011;80:301–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brouwers JF. Liquid chromatographic–mass spectrometric analysis of phospholipids: chromatography, ionization and quantification. Biochim Biophys Acta 2011;1811:763–775. [DOI] [PubMed] [Google Scholar]
- 14. Han X, Yang K, Gross RW. Multi‐dimensional mass spectrometry‐based shotgun lipidomics and novel strategies for lipidomic analyses. Mass Spectrom Rev 2012;31:134–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brügger B. Lipidomics: analysis of the lipid composition of cells and subcellular organelles by electrospray ionization mass spectrometry. Annu Rev Biochem 2014;83:79–98. [DOI] [PubMed] [Google Scholar]
- 16. Fahy E, Sud M, Cotter D, Subramaniam S. LIPID MAPS online tools for lipid research. Nucleic Acids Res 2007;35:W606–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sud M, Fahy E, Cotter D, Brown A, Dennis EA, Glass CK, Merrill AH, Murphy RC, Raetz CRH, Russell DW, Subramaniam S. LMSD: LIPID MAPS structure database. Nucleic Acids Res 2007;35:D527–D532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yetukuri L, Ekroos K, Vidal‐Puig A, Oresic M. Informatics and computational strategies for the study of lipids. Mol Biosyst 2008;4:121–127. [DOI] [PubMed] [Google Scholar]
- 19. Fahy E, Subramaniam S, Brown HA, Glass CK, Merrill AH, Murphy RC, Raetz CRH, Russell DW, Seyama Y, Shaw W, Shimizu T, Spener F, van Meer G, VanNieuwenhze MS, White SH, et al. A comprehensive classification system for lipids. J Lipid Res 2005;46:839–861. [DOI] [PubMed] [Google Scholar]
- 20. Fahy E, Subramaniam S, Murphy RC, Nishijima M, Raetz CRH, Shimizu T, Spener F, van Meer G, Wakelam MJO, Dennis EA. Update of the LIPID MAPS comprehensive classification system for lipids. J Lipid Res 2009;50(Suppl):S9–S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Higdon A, Diers AR, Oh JY, Landar A, Darley‐Usmar VM. Cell signalling by reactive lipid species: new concepts and molecular mechanisms. Biochem J 2012;442:453–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mercer J, Schelhaas M, Helenius A. Virus entry by endocytosis. Annu Rev Biochem 2010;79:803–833. [DOI] [PubMed] [Google Scholar]
- 23. Ham H, Sreelatha A, Orth K. Manipulation of host membranes by bacterial effectors. Nat Rev Microbiol 2011;9:635–646. [DOI] [PubMed] [Google Scholar]
- 24. Hilbi H, Haas A. Secretive bacterial pathogens and the secretory pathway. Traffic 2012;13:1187–1197. [DOI] [PubMed] [Google Scholar]
- 25. Guichard A, Nizet V, Bier E. RAB11‐mediated trafficking in host‐pathogen interactions. Nat Rev Microbiol 2014;12:624–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heung LJ, Luberto C, Del Poeta M. Role of sphingolipids in microbial pathogenesis. Infect Immun 2006;74:28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wenk MR. Lipidomics of host–pathogen interactions. FEBS Lett 2006;580:5541–5551. [DOI] [PubMed] [Google Scholar]
- 28. Van der Meer‐Janssen YPM, van Galen J, Batenburg JJ, Helms JB. Lipids in host–pathogen interactions: pathogens exploit the complexity of the host cell lipidome. Prog Lipid Res 2010;49:1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chukkapalli V, Heaton NS, Randall G. Lipids at the interface of virus–host interactions. Curr Opin Microbiol 2012;15:512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vromman F, Subtil A. Exploitation of host lipids by bacteria. Curr Opin Microbiol 2014;17:38–45. [DOI] [PubMed] [Google Scholar]
- 31. Sviridov D, Bukrinsky M. Interaction of pathogens with host cholesterol metabolism. Curr Opin Lipidol 2014;25:333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haneburger I, Hilbi H. Phosphoinositide lipids and the Legionella pathogen vacuole. Curr Top Microbiol Immunol 2013;376:155–173. [DOI] [PubMed] [Google Scholar]
- 33. Elwell CA, Engel JN. Lipid acquisition by intracellular Chlamydiae. Cell Microbiol 2012;14:1010–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Van der Goot FG, Harder T. Raft membrane domains: from a liquid‐ordered membrane phase to a site of pathogen attack. Semin Immunol 2001;13:89–97. [DOI] [PubMed] [Google Scholar]
- 35. Mañes S, del Real G, Martínez‐A C. Pathogens: raft hijackers. Nat Rev Immunol 2003;3:557–568. [DOI] [PubMed] [Google Scholar]
- 36. Simons K, Gerl MJ. Revitalizing membrane rafts: new tools and insights. Nat Rev Mol Cell Biol 2010;11:688–699. [DOI] [PubMed] [Google Scholar]
- 37. Hsu N‐Y, Ilnytska O, Belov G, Santiana M, Chen Y‐H, Takvorian PM, Pau C, van der Schaar H, Kaushik‐Basu N, Balla T, Cameron CE, Ehrenfeld E, van Kuppeveld FJM, Altan‐Bonnet N. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010;141:799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Berger KL, Cooper JD, Heaton NS, Yoon R, Oakland TE, Jordan TX, Mateu G, Grakoui A, Randall G. Roles for endocytic trafficking and phosphatidylinositol 4‐kinase III alpha in hepatitis C virus replication. Proc Natl Acad Sci USA 2009;106:7577–7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tai AW, Benita Y, Peng LF, Kim SS, Sakamoto N, Xavier RJ, Chung RT. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 2009;5:298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Reiss S, Rebhan I, Backes P, Romero‐Brey I, Erfle H, Matula P, Kaderali L, Poenisch M, Blankenburg H, Hiet M‐S, Longerich T, Diehl S, Ramirez F, Balla T, Rohr K, et al. Recruitment and activation of a lipid kinase by Hepatitis C Virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 2011;9:32–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vaillancourt FH, Pilote L, Cartier M, Lippens J, Liuzzi M, Bethell RC, Cordingley MG, Kukolj G. Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology 2009;387:5–10. [DOI] [PubMed] [Google Scholar]
- 42. Trotard M, Lepere‐Douard C, Regeard M, Piquet‐Pellorce C, Lavillette D, Cosset FL, Gripon P, Le Seyec J. Kinases required in hepatitis C virus entry and replication highlighted by small interference RNA screening. FASEB J 2009;23:3780–3789. [DOI] [PubMed] [Google Scholar]
- 43. Borawski J, Troke P, Puyang X, Gibaja V, Zhao S, Mickanin C, Leighton‐Davies J, Wilson CJ, Myer V, Cornellataracido I, Baryza J, Tallarico J, Joberty G, Bantscheff M, Schirle M, et al. Class III phosphatidylinositol 4‐kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J Virol 2009;83:10058–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reiss S, Harak C, Romero‐Brey I, Radujkovic D, Klein R, Ruggieri A, Rebhan I, Bartenschlager R, Lohmann V. The lipid kinase phosphatidylinositol‐4 kinase III alpha regulates the phosphorylation status of Hepatitis C virus NS5A. PLoS Pathog 2013;9:e1003359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sasaki J, Ishikawa K, Arita M, Taniguchi K. ACBD3‐mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J 2012;31:754–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mercer J, Helenius A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 2008;320:531–535. [DOI] [PubMed] [Google Scholar]
- 47. Vanlandschoot P, Leroux‐Roels G. Viral apoptotic mimicry: an immune evasion strategy developed by the hepatitis B virus? Trends Immunol 2003;24:144–147. [DOI] [PubMed] [Google Scholar]
- 48. Callahan MK, Popernack PM, Tsutsui S, Truong L, Schlegel RA, Henderson AJ. Phosphatidylserine on HIV envelope is a cofactor for infection of monocytic cells. J Immunol 2003;170:4840–4845. [DOI] [PubMed] [Google Scholar]
- 49. Shimojima M, Takada A, Ebihara H, Neumann G, Fujioka K, Irimura T, Jones S, Feldmann H, Kawaoka Y. Tyro3 family‐mediated cell entry of Ebola and Marburg viruses. J Virol 2006;80:10109–10116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Coil DA, Miller AD. Enhancement of enveloped virus entry by phosphatidylserine. J Virol 2005;79:11496–11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rothwell C, Lebreton A, Young Ng C, Lim JY, Liu W, Vasudevan S, Labow M, Gu F, Gaither LA. Cholesterol biosynthesis modulation regulates dengue viral replication. Virology 2009;389:8–19. [DOI] [PubMed] [Google Scholar]
- 52. Scheiffele P, Rietveld A, Wilk T, Simons K. Influenza viruses select ordered lipid domains during budding from the plasma membrane. J Biol Chem 1999;274:2038–2044. [DOI] [PubMed] [Google Scholar]
- 53. Aizaki H, Lee K‐J, Sung VM‐H, Ishiko H, Lai MMC. Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology 2004;324:450–461. [DOI] [PubMed] [Google Scholar]
- 54. Amako Y, Sarkeshik A, Hotta H, Yates J, Siddiqui A. Role of oxysterol binding protein in hepatitis C virus infection. J Virol 2009;83:9237–9246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Paul D, Hoppe S, Saher G, Krijnse‐Locker J, Bartenschlager R. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J Virol 2013;87:10612–10627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liao Z, Cimakasky LM, Hampton R, Nguyen DH, Hildreth JE. Lipid rafts and HIV pathogenesis: host membrane cholesterol is required for infection by HIV type 1. AIDS Res Hum Retroviruses 2001;17:1009–1019. [DOI] [PubMed] [Google Scholar]
- 57. Zheng Y‐H, Plemenitas A, Fielding CJ, Peterlin BM. Nef increases the synthesis of and transports cholesterol to lipid rafts and HIV‐1 progeny virions. Proc Natl Acad Sci USA 2003;100:8460–8465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chang KO. Role of cholesterol pathways in norovirus replication. J Virol 2009;83:8587–8595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Martín‐Acebes MA, Blázquez A‐B, Jiménez de Oya N, Escribano‐Romero E, Saiz J‐C. West Nile Virus replication requires fatty acid synthesis but is independent on phosphatidylinositol‐4‐phosphate lipids. PLoS One 2011;6:e24970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang H, Perry JW, Lauring AS, Neddermann P, De Francesco R, Tai AW. Oxysterol‐binding protein is a phosphatidylinositol 4‐kinase effector required for HCV replication membrane integrity and cholesterol trafficking. Gastroenterology 2014;146:1373–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ilnytska O, Santiana M, Hsu N‐Y, Du W‐L, Chen Y‐H, Viktorova EG, Belov G, Brinker A, Storch J, Moore C, Dixon JL, Altan‐Bonnet N. Enteroviruses harness the cellular endocytic machinery to remodel the host cell cholesterol landscape for effective viral replication. Cell Host Microbe 2013;14:281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Roulin PS, Lötzerich M, Torta F, Tanner LB, van Kuppeveld FJM, Wenk MR, Greber UF. A phosphatidylinositol 4‐phosphate and cholesterol counter‐current model for the formation of rhinovirus replication compartments at ER‐Golgi interface. Cell Host Microbe 2014;16:677–690. [DOI] [PubMed] [Google Scholar]
- 63. Arita M. Phosphatidylinositol‐4 kinase III beta and oxysterol‐binding protein accumulate unesterified cholesterol on poliovirus‐induced membrane structure. Microbiol Immunol 2014;58:239–256. [DOI] [PubMed] [Google Scholar]
- 64. Nchoutmboube JA, Viktorova EG, Scott AJ, Ford LA, Pei Z, Watkins PA, Ernst RK, Belov GA. Increased long chain acyl‐Coa synthetase activity and fatty acid import is linked to membrane synthesis for development of picornavirus replication organelles. PLoS Pathog 2013;9:e1003401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Perera R, Riley C, Isaac G, Hopf‐Jannasch AS, Moore RJ, Weitz KW, Pasa‐Tolic L, Metz TO, Adamec J, Kuhn RJ. Dengue virus infection perturbs lipid homeostasis in infected mosquito cells. PLoS Pathog 2012;8:e1002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Heaton NS, Perera R, Berger KL, Khadka S, Lacount DJ, Kuhn RJ, Randall G. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc Natl Acad Sci USA 2010;107:17345–17350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kapadia SB, Chisari FV. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc Natl Acad Sci USA 2005;102:2561–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Moser TS, Schieffer D, Cherry S. AMP‐activated kinase restricts Rift Valley fever virus infection by inhibiting fatty acid synthesis. PLoS Pathog 2012;8:e1002661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Greseth MD, Traktman P. De novo fatty acid biosynthesis contributes significantly to establishment of a bioenergetically favorable environment for vaccinia virus infection. PLoS Pathog 2014;10:e1004021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yang W, Hood BL, Chadwick SL, Liu S, Watkins SC, Luo G, Conrads TP, Wang T. Fatty acid synthase is up‐regulated during hepatitis C virus infection and regulates hepatitis C virus entry and production. Hepatology 2008;48:1396–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pizarro‐Cerdá J, Cossart P. Subversion of phosphoinositide metabolism by intracellular bacterial pathogens. Nat Cell Biol 2004;6:1026–1033. [DOI] [PubMed] [Google Scholar]
- 72. Drecktrah D, Knodler LA, Steele‐Mortimer O. Modulation and utilization of host cell phosphoinositides by Salmonella spp. Infect Immun 2004;72:4331–4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wong K‐W, Isberg RR. Arf6 and phosphoinositol‐4‐phosphate‐5‐kinase activities permit bypass of the Rac1 requirement for beta1 integrin‐mediated bacterial uptake. J Exp Med 2003;198:603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hilbi H. Modulation of phosphoinositide metabolism by pathogenic bacteria. Cell Microbiol 2006;8:1697–1706. [DOI] [PubMed] [Google Scholar]
- 75. Krauss M, Haucke V. Phosphoinositide‐metabolizing enzymes at the interface between membrane traffic and cell signalling. EMBO Rep 2007;8:241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bakowski MA, Braun V, Lam GY, Yeung T, Heo WD, Meyer T, Finlay BB, Grinstein S, Brumell JH. The phosphoinositide phosphatase SopB manipulates membrane surface charge and trafficking of the Salmonella‐containing vacuole. Cell Host Microbe 2010;7:453–462. [DOI] [PubMed] [Google Scholar]
- 77. Harding CR, Mattheis C, Mousnier A, Oates CV, Hartland EL, Frankel G, Schroeder GN. LtpD is a novel Legionella pneumophila effector that binds phosphatidylinositol 3‐phosphate and inositol monophosphatase IMPA1. Infect Immun 2013;81:4261–4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pizarro‐Cerdá J, Kühbacher A, Cossart P. Phosphoinositides and host–pathogen interactions. Biochim Biophys Acta 2014. (In press). [DOI] [PubMed] [Google Scholar]
- 79. Lafont F, Tran Van Nhieu G, Hanada K, Sansonetti P, van der Goot FG. Initial steps of Shigella infection depend on the cholesterol/sphingolipid raft‐mediated CD44‐IpaB interaction. EMBO J 2002;21:4449–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Carabeo RA, Mead DJ, Hackstadt T. Golgi‐dependent transport of cholesterol to the Chlamydia trachomatis inclusion. Proc Natl Acad Sci USA 2003;100:6771–6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Catron DM, Sylvester MD, Lange Y, Kadekoppala M, Jones BD, Monack DM, Falkow S, Haldar K. The Salmonella‐containing vacuole is a major site of intracellular cholesterol accumulation and recruits the GPI‐anchored protein CD55. Cell Microbiol 2002;4:315–328. [DOI] [PubMed] [Google Scholar]
- 82. De Chastellier C, Thilo L. Cholesterol depletion in Mycobacterium avium‐infected macrophages overcomes the block in phagosome maturation and leads to the reversible sequestration of viable mycobacteria in phagolysosome‐derived autophagic vacuoles. Cell Microbiol 2006;8:242–256. [DOI] [PubMed] [Google Scholar]
- 83. Gilk SD, Cockrell DC, Luterbach C, Hansen B, Knodler LA, Ibarra JA, Steele‐Mortimer O, Heinzen RA. Bacterial colonization of host cells in the absence of cholesterol. PLoS Pathog 2013;9:e1003107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gupta VR, Patel HK, Kostolansky SS, Ballivian RA, Eichberg J, Blanke SR. Sphingomyelin functions as a novel receptor for Helicobacter pylori VacA. PLoS Pathog 2008;4:e1000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hanada K. Sphingolipids in infectious diseases. Jpn J Infect Dis 2005;58:131–148. [PubMed] [Google Scholar]
- 86. Moore ER, Fischer ER, Mead DJ, Hackstadt T. The chlamydial inclusion preferentially intercepts basolaterally directed sphingomyelin‐containing exocytic vacuoles. Traffic 2008;9:2130–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Arena ET, Auweter SD, Antunes LCM, Vogl AW, Han J, Guttman JA, Croxen MA, Menendez A, Covey SD, Borchers CH, Finlay BB. The deubiquitinase activity of the Salmonella pathogenicity island 2 effector, SseL, prevents accumulation of cellular lipid droplets. Infect Immun 2011;79:4392–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Daniel J, Maamar H, Deb C, Sirakova TD, Kolattukudy PE. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy‐like phenotype in lipid‐loaded macrophages. PLoS Pathog 2011;7:e1002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kumar Y, Cocchiaro J, Valdivia RH. The obligate intracellular pathogen Chlamydia trachomatis targets host lipid droplets. Curr Biol 2006;16:1646–1651. [DOI] [PubMed] [Google Scholar]
- 90. Cocchiaro JL, Kumar Y, Fischer ER, Hackstadt T, Valdivia RH. Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc Natl Acad Sci USA 2008;105:9379–9384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Dowhan W, Mileykovskaya E, Bogdanov M. Diversity and versatility of lipid|–protein interactions revealed by molecular genetic approaches. Biochim Biophys Acta 2004;1666:19–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ghannoum MA. Potential role of phospholipases in virulence and fungal pathogenesis. Clin Microbiol Rev 2000;13:122–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Cox GM, McDade HC, Chen SC, Tucker SC, Gottfredsson M, Wright LC, Sorrell TC, Leidich SD, Casadevall A, Ghannoum MA, Perfect JR. Extracellular phospholipase activity is a virulence factor for Cryptococcus neoformans. Mol Microbiol 2001;39:166–175. [DOI] [PubMed] [Google Scholar]
- 94. Rhome R, Del Poeta M. Sphingolipid signaling in fungal pathogens. Adv Exp Med Biol 2010;688:232–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rheeder JP, Marasas WFO, Vismer HF. Production of fumonisin analogs by Fusarium species. Appl Environ Microbiol 2002;68:2101–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Merrill AH, Wang E, Gilchrist DG, Riley RT. Fumonisins and other inhibitors of de novo sphingolipid biosynthesis. Adv Lipid Res 1993;26:215–234. [PubMed] [Google Scholar]
- 97. Kupfahl C, Tsikas D, Niemann J, Geginat G, Hof H. Production of prostaglandins, isoprostanes and thromboxane by Aspergillus fumigatus: identification by gas chromatography‐tandem mass spectrometry and quantification by enzyme immunoassay. Mol Immunol 2012;49:621–627. [DOI] [PubMed] [Google Scholar]
- 98. Noverr MC, Phare SM, Toews GB, Coffey MJ, Huffnagle GB. Pathogenic yeasts Cryptococcus neoformans and Candida albicans produce immunomodulatory prostaglandins. Infect Immun 2001;69:2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tsitsigiannis DI, Bok J‐W, Andes D, Nielsen KF, Frisvad JC, Keller NP. Aspergillus cyclooxygenase‐like enzymes are associated with prostaglandin production and virulence. Infect Immun 2005;73:4548–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Vial HJ, Eldin P, Tielens AGM, van Hellemond JJ. Phospholipids in parasitic protozoa. Mol Biochem Parasitol 2003;126:143–154. [DOI] [PubMed] [Google Scholar]
- 101. Van Hellemond JJ, Retra K, Brouwers JFHM, van Balkom BWM, Yazdanbakhsh M, Shoemaker CB, Tielens AGM. Functions of the tegument of schistosomes: clues from the proteome and lipidome. Int J Parasitol 2006;36:691–699. [DOI] [PubMed] [Google Scholar]
- 102. Coppens I. Targeting lipid biosynthesis and salvage in apicomplexan parasites for improved chemotherapies. Nat Rev Microbiol 2013;11:823–835. [DOI] [PubMed] [Google Scholar]
- 103. Ramakrishnan S, Serricchio M, Striepen B, Bütikofer P. Lipid synthesis in protozoan parasites: a comparison between kinetoplastids and apicomplexans. Prog Lipid Res 2013;52:488–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Brouwers JF, Versluis C, van Golde LMG, Tielens AGM. 5‐Octadecenoic acid: evidence for a novel type of fatty acid modification in schistosomes. Biochem J 1998;334:315–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Bomsel M, Alfsen A. Entry of viruses through the epithelial barrier: pathogenic trickery. Nat Rev Mol Cell Biol 2003;4:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Smith AE, Helenius A. How viruses enter animal cells. Science 2004;304:237–242. [DOI] [PubMed] [Google Scholar]
- 107. Mazzon M, Mercer J. Lipid interactions during virus entry and infection. Cell Microbiol 2014;16:1493–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Novoa RR, Calderita G, Arranz R, Fontana J, Granzow H, Risco C. Virus factories: associations of cell organelles for viral replication and morphogenesis. Biol Cell 2005;97:147–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Miller S, Krijnse‐Locker J. Modification of intracellular membrane structures for virus replication. Nat Rev Microbiol 2008;6:363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Romero‐Brey I, Bartenschlager R. Membranous replication factories induced by plus‐strand RNA viruses. Viruses 2014;6:2826–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Den Boon JA, Diaz A, Ahlquist P. Cytoplasmic viral replication complexes. Cell Host Microbe 2010;8:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Mankouri J, Tedbury PR, Gretton S, Hughes ME, Griffin SD, Dallas ML, Green KA, Hardie DG, Peers C, Harris M. Enhanced hepatitis C virus genome replication and lipid accumulation mediated by inhibition of AMP‐activated protein kinase. Proc Natl Acad Sci USA 2010;107:11549–11554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lyn RK, Singaravelu R, Kargman S, O'Hara S, Chan H, Oballa R, Huang Z, Jones DM, Ridsdale A, Russell RS, Partridge AW, Pezacki JP. Stearoyl‐CoA desaturase inhibition blocks formation of hepatitis C virus‐induced specialized membranes. Sci Rep 2014;4:4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Tai AW, Salloum S. The role of the phosphatidylinositol 4‐kinase PI4KA in hepatitis C virus‐induced host membrane rearrangement. PLoS One 2011;6:e26300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Zhang L, Hong Z, Lin W, Shao R‐X, Goto K, Hsu VW, Chung RT. ARF1 and GBF1 generate a PI4P‐enriched environment supportive of Hepatitis C virus replication. PLoS One 2012;7:e32135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Van der Schaar HM, Leyssen P, Thibaut HJ, de Palma A, van der Linden L, Lanke KH, Lacroix C, Verbeken E, Conrath K, Macleod AM, Mitchell DR, Palmer NJ, van de Poel H, Andrews M, Neyts J, et al. A novel, broad‐spectrum inhibitor of enterovirus replication that targets host cell factor phosphatidylinositol 4‐kinase IIIbeta. Antimicrob Agents Chemother 2013;57:4971–4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Spickler C, Lippens J, Laberge MK, Desmeules S, Bellavance E, Garneau M, Guo T, Hucke O, Leyssen P, Neyts J, Vaillancourt FH, Decor A, O'Meara J, Franti M, Gauthier A. Phosphatidylinositol 4‐kinase III beta is essential for replication of human rhinovirus and its inhibition causes a lethal phenotype in vivo. Antimicrob Agents Chemother 2013;57:3358–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Baird NL, York J, Nunberg JH. Arenavirus infection induces discrete cytosolic structures for RNA replication. J Virol 2012;86:11301–11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Tanner LB, Chng C, Guan XL, Lei Z, Rozen SG, Wenk MR. Lipidomics identifies a requirement for peroxisomal function during influenza virus replication. J Lipid Res 2014;55:1357–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Hirata Y, Ikeda K, Sudoh M, Tokunaga Y, Suzuki A, Weng L, Ohta M, Tobita Y, Okano K, Ozeki K, Kawasaki K, Tsukuda T, Katsume A, Aoki Y, Umehara T, et al. Self‐enhancement of hepatitis C virus replication by promotion of specific sphingolipid biosynthesis. PLoS Pathog 2012;8:e1002860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Mesmin B, Bigay J, Moser von Filseck J, Lacas‐Gervais S, Drin G, Antonny B. A four‐step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER‐Golgi tether OSBP. Cell 2013;155:830–843. [DOI] [PubMed] [Google Scholar]
- 122. Waris G, Felmlee DJ, Negro F, Siddiqui A. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J Virol 2007;81:8122–8130. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 123. Rodgers MA, Saghatelian A, Yang PL. Identification of an overabundant cholesterol precursor in hepatitis B virus replicating cells by untargeted lipid metabolite profiling. J Am Chem Soc 2009;131:5030–5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Rodgers MA, Villareal VA, Schaefer EA, Peng LF, Corey KE, Chung RT, Yang PL. Lipid metabolite profiling identifies desmosterol metabolism as a new antiviral target for Hepatitis C Virus. J Am Chem Soc 2012;134:6896–6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Lorizate M, Kräusslich H‐G. Role of lipids in virus replication. Cold Spring Harb Perspect Biol 2011;3:a004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Brügger B, Glass B, Haberkant P, Leibrecht I, Wieland FT, Kräusslich HG. The HIV lipidome: a raft with an unusual composition. Proc Natl Acad Sci USA 2006;103:2641–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Brügger B, Krautkramer E, Tibroni N, Munte CE, Rauch S, Leibrecht I, Glass B, Breuer S, Geyer M, Krausslich HG, Kalbitzer HR, Wieland FT, Fackler OT. Human immunodeficiency virus type 1 Nef protein modulates the lipid composition of virions and host cell membrane microdomains. Retrovirology 2007;4:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Martin‐Acebes MA, Merino‐Ramos T, Blazquez AB, Casas J, Escribano‐Romero E, Sobrino F, Saiz JC. The composition of West Nile virus lipid envelope unveils a role of sphingolipid metabolism on flavivirus biogenesis. J Virol 2014;88:12041–12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Liu SY, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, Pernet O, Guo H, Nusbaum R, Zack JA, Freiberg AN, Su L, Lee B, Cheng G. Interferon‐inducible cholesterol‐25‐hydroxylase broadly inhibits viral entry by production of 25‐hydroxycholesterol. Immunity 2013;38:92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, Lacaze P, Watterson S, Griffiths SJ, Spann NJ, Meljon A, Talbot S, Krishnan K, Covey DF, Wenk MR, et al. The transcription factor STAT‐1 couples macrophage synthesis of 25‐hydroxycholesterol to the interferon antiviral response. Immunity 2013;38:106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Amini‐Bavil‐Olyaee S, Choi YJ, Lee JH, Shi M, Huang IC, Farzan M, Jung JU. The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe 2013;13:452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Mackenzie JM, Khromykh AA, Parton RG. Cholesterol manipulation by West Nile Virus perturbs the cellular immune response. Cell Host Microbe 2007;2:229–239. [DOI] [PubMed] [Google Scholar]
- 133. Stein M‐P, Müller MP, Wandinger‐Ness A. Bacterial pathogens commandeer Rab GTPases to establish intracellular niches. Traffic 2012;13:1565–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ehrt S, Rhee K. Mycobacterium tuberculosis metabolism and host interaction: mysteries and paradoxes. Curr Top Microbiol Immunol 2013;374:163–188. [DOI] [PubMed] [Google Scholar]
- 135. Beisel WR. Metabolic response to infection. Annu Rev Med 1975;26:9–20. [DOI] [PubMed] [Google Scholar]
- 136. Gillmaier N, Götz A, Schulz A, Eisenreich W, Goebel W. Metabolic responses of primary and transformed cells to intracellular Listeria monocytogenes. PLoS One 2012;7:e52378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Cui J, Shao F. Biochemistry and cell signaling taught by bacterial effectors. Trends Biochem Sci 2011;36:532–540. [DOI] [PubMed] [Google Scholar]
- 138. Waterman SR, Holden DW. Functions and effectors of the Salmonella pathogenicity island 2 type III secretion system. Cell Microbiol 2003;5:501–511. [DOI] [PubMed] [Google Scholar]
- 139. Bonazzi M, Cossart P. Bacterial entry into cells: a role for the endocytic machinery. FEBS Lett 2006;580:2962–2967. [DOI] [PubMed] [Google Scholar]
- 140. Humphries AC, Way M. The non‐canonical roles of clathrin and actin in pathogen internalization, egress and spread. Nat Rev Microbiol 2013;11:551–560. [DOI] [PubMed] [Google Scholar]
- 141. Patel JC, Galán JE. Manipulation of the host actin cytoskeleton by Salmonella – all in the name of entry. Curr Opin Microbiol 2005;8:10–15. [DOI] [PubMed] [Google Scholar]
- 142. Rosenberger CM, Brumell JH, Finlay BB. Microbial pathogenesis: lipid rafts as pathogen portals. Curr Biol 2000;10:R823–R825. [DOI] [PubMed] [Google Scholar]
- 143. LaRock DL, Brzovic PS, Levin I, Blanc M‐P, Miller SI. A Salmonella typhimurium‐translocated glycerophospholipid:cholesterol acyltransferase promotes virulence by binding to the RhoA protein switch regions. J Biol Chem 2012;287:29654–29663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Misselwitz B, Dilling S, Vonaesch P, Sacher R, Snijder B, Schlumberger M, Rout S, Stark M, von Mering C, Pelkmans L, Hardt W‐D. RNAi screen of Salmonella invasion shows role of COPI in membrane targeting of cholesterol and Cdc42. Mol Syst Biol 2011;7:474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Huang F‐C. The critical role of membrane cholesterol in salmonella‐induced autophagy in intestinal epithelial cells. Int J Mol Sci 2014;15:12558–12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Wunder C, Churin Y, Winau F, Warnecke D, Vieth M, Lindner B, Zähringer U, Mollenkopf H‐J, Heinz E, Meyer TF. Cholesterol glucosylation promotes immune evasion by Helicobacter pylori. Nat Med 2006;12:1030–1038. [DOI] [PubMed] [Google Scholar]
- 147. Harris JR, Bhakdi S, Meissner U, Scheffler D, Bittman R, Li G, Zitzer A, Palmer M. Interaction of the Vibrio cholerae cytolysin (VCC) with cholesterol, some cholesterol esters, and cholesterol derivatives: a TEM study. J Struct Biol 2002;139:122–135. [DOI] [PubMed] [Google Scholar]
- 148. Zitzer A, Bittman R, Verbicky CA, Erukulla RK, Bhakdi S, Weis S, Valeva A, Palmer M. Coupling of cholesterol and cone‐shaped lipids in bilayers augments membrane permeabilization by the cholesterol‐specific toxins streptolysin O and Vibrio cholerae cytolysin. J Biol Chem 2001;276:14628–14633. [DOI] [PubMed] [Google Scholar]
- 149. Seveau S, Bierne H, Giroux S, Prévost M‐C, Cossart P. Role of lipid rafts in E‐cadherin‐ and HGF‐R/Met‐mediated entry of Listeria monocytogenes into host cells. J Cell Biol 2004;166:743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Weber SS, Ragaz C, Hilbi H. Pathogen trafficking pathways and host phosphoinositide metabolism. Mol Microbiol 2009;71:1341–1352. [DOI] [PubMed] [Google Scholar]
- 151. Behnia R, Munro S. Organelle identity and the signposts for membrane traffic. Nature 2005;438:597–604. [DOI] [PubMed] [Google Scholar]
- 152. Mallo GV, Espina M, Smith AC, Terebiznik MR, Alemán A, Finlay BB, Rameh LE, Grinstein S, Brumell JH. SopB promotes phosphatidylinositol 3‐phosphate formation on Salmonella vacuoles by recruiting Rab5 and Vps34. J Cell Biol 2008;182:741–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Roppenser B, Grinstein S, Brumell JH. Modulation of host phosphoinositide metabolism during Salmonella invasion by the type III secreted effector SopB. Methods Cell Biol 2012;108:173–186. [DOI] [PubMed] [Google Scholar]
- 154. Cambier CJ, Takaki KK, Larson RP, Hernandez RE, Tobin DM, Urdahl KB, Cosma CL, Ramakrishnan L. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature 2014;505:218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Niebuhr K, Giuriato S, Pedron T, Philpott DJ, Gaits F, Sable J, Sheetz MP, Parsot C, Sansonetti PJ, Payrastre B. Conversion of PtdIns(4,5)P(2) into PtdIns(5)P by the S.flexneri effector IpgD reorganizes host cell morphology. EMBO J 2002;21:5069–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Alonso A, García‐del Portillo F. Hijacking of eukaryotic functions by intracellular bacterial pathogens. Int Microbiol 2004;7:181–191. [PubMed] [Google Scholar]
- 157. Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature 2006;443:651–657. [DOI] [PubMed] [Google Scholar]
- 158. De Matteis MA, Di Campli A, Godi A. The role of the phosphoinositides at the Golgi complex. Biochim Biophys Acta 2005;1744:396–405. [DOI] [PubMed] [Google Scholar]
- 159. Weber SS, Ragaz C, Reus K, Nyfeler Y, Hilbi H. Legionella pneumophila exploits PI(4)P to anchor secreted effector proteins to the replicative vacuole. PLoS Pathog 2006;2:e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Dukes JD, Lee H, Hagen R, Reaves BJ, Layton AN, Galyov EE, Whitley P. The secreted Salmonella dublin phosphoinositide phosphatase, SopB, localizes to PtdIns(3)P‐containing endosomes and perturbs normal endosome to lysosome trafficking. Biochem J 2006;395:239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Terebiznik MR, Vieira OV, Marcus SL, Slade A, Yip CM, Trimble WS, Meyer T, Finlay BB, Grinstein S. Elimination of host cell PtdIns(4,5)P(2) by bacterial SigD promotes membrane fission during invasion by Salmonella. Nat Cell Biol 2002;4:766–773. [DOI] [PubMed] [Google Scholar]
- 162. Hernandez LD, Hueffer K, Wenk MR, Galán JE. Salmonella modulates vesicular traffic by altering phosphoinositide metabolism. Science 2004;304:1805–1807. [DOI] [PubMed] [Google Scholar]
- 163. Pendaries C, Tronchère H, Arbibe L, Mounier J, Gozani O, Cantley L, Fry MJ, Gaits‐Iacovoni F, Sansonetti PJ, Payrastre B. PtdIns5P activates the host cell PI3‐kinase/Akt pathway during Shigella flexneri infection. EMBO J 2006;25:1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Vergne I, Chua J, Lee H‐H, Lucas M, Belisle J, Deretic V. Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc Natl Acad Sci USA 2005;102:4033–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Howe D, Heinzen RA. Coxiella burnetii inhabits a cholesterol‐rich vacuole and influences cellular cholesterol metabolism. Cell Microbiol 2006;8:496–507. [DOI] [PubMed] [Google Scholar]
- 166. Gilk SD. Role of lipids in Coxiella burnetii infection. Adv Exp Med Biol 2012;984:199–213. [DOI] [PubMed] [Google Scholar]
- 167. Garner MJ, Hayward RD, Koronakis V. The Salmonella pathogenicity island 1 secretion system directs cellular cholesterol redistribution during mammalian cell entry and intracellular trafficking. Cell Microbiol 2002;4:153–165. [DOI] [PubMed] [Google Scholar]
- 168. Kuhle V, Abrahams GL, Hensel M. Intracellular Salmonella enterica redirect exocytic transport processes in a Salmonella pathogenicity island 2‐dependent manner. Traffic 2006;7:716–730. [DOI] [PubMed] [Google Scholar]
- 169. Haas A. The phagosome: compartment with a license to kill. Traffic 2007;8:311–330. [DOI] [PubMed] [Google Scholar]
- 170. Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci USA 2008;105:4376–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Ploegh HL. A lipid‐based model for the creation of an escape hatch from the endoplasmic reticulum. Nature 2007;448:435–438. [DOI] [PubMed] [Google Scholar]
- 172. Goodman JM. The gregarious lipid droplet. J Biol Chem 2008;283:28005–28009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Nawabi P, Catron DM, Haldar K. Esterification of cholesterol by a type III secretion effector during intracellular Salmonella infection. Mol Microbiol 2008;68:173–185. [DOI] [PubMed] [Google Scholar]
- 174. Stehr M, Elamin AA, Singh M. Cytosolic lipid inclusions formed during infection by viral and bacterial pathogens. Microbes Infect 2012;14:1227–1237. [DOI] [PubMed] [Google Scholar]
- 175. Luo M, Fadeev EA, Groves JT. Mycobactin‐mediated iron acquisition within macrophages. Nat Chem Biol 2005;1:149–153. [DOI] [PubMed] [Google Scholar]
- 176. Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta 1843;2014:2563–2582. [DOI] [PubMed] [Google Scholar]
- 177. Barbosa T, Rescigno M. Host–bacteria interactions in the intestine: homeostasis to chronic inflammation. Wiley Interdiscip Rev Syst Biol Med 2010;2:80–97. [DOI] [PubMed] [Google Scholar]
- 178. Tam VC. Lipidomic profiling of bioactive lipids by mass spectrometry during microbial infections. Semin Immunol 2013;25:240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Agard M, Asakrah S, Morici LA. PGE(2) suppression of innate immunity during mucosal bacterial infection. Front Cell Infect Microbiol 2013;3:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Uchiya K, Groisman EA, Nikai T. Involvement of Salmonella pathogenicity island 2 in the up‐regulation of interleukin‐10 expression in macrophages: role of protein kinase A signal pathway. Infect Immun 2004;72:1964–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Vance RE, Hong S, Gronert K, Serhan CN, Mekalanos JJ. The opportunistic pathogen Pseudomonas aeruginosa carries a secretable arachidonate 15‐lipoxygenase. Proc Natl Acad Sci USA 2004;101:2135–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti‐inflammatory and pro‐resolution lipid mediators. Nat Rev Immunol 2008;8:349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Hawksworth DL. The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycol Res 2001;105:1422–1432. [Google Scholar]
- 184. Taylor LH, Latham SM, Woolhouse ME. Risk factors for human disease emergence. Philos Trans R Soc Lond B Biol Sci 2001;356:983–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Underhill DM, Iliev ID. The mycobiota: interactions between commensal fungi and the host immune system. Nat Rev Immunol 2014;14:405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Plano D, Amin S, Sharma AK. Importance of sphingosine kinase (SphK) as a target in developing cancer therapeutics and recent developments in the synthesis of novel SphK inhibitors. J Med Chem 2014;57:5509–5524. [DOI] [PubMed] [Google Scholar]
- 187. Merrill AH. De novo sphingolipid biosynthesis: a necessary, but dangerous, pathway. J Biol Chem 2002;277:25843–25846. [DOI] [PubMed] [Google Scholar]
- 188. Delgado A, Casas J, Llebaria A, Abad JL, Fabrias G. Inhibitors of sphingolipid metabolism enzymes. Biochim Biophys Acta 2006;1758:1957–1977. [DOI] [PubMed] [Google Scholar]
- 189. Hanada K. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim Biophys Acta 2003;1632:16–30. [DOI] [PubMed] [Google Scholar]
- 190. Desai K, Sullards MC, Allegood J, Wang E, Schmelz EM, Hartl M, Humpf H‐U, Liotta DC, Peng Q, Merrill AH. Fumonisins and fumonisin analogs as inhibitors of ceramide synthase and inducers of apoptosis. Biochim Biophys Acta 2002;1585:188–192. [DOI] [PubMed] [Google Scholar]
- 191. Abbas HK, Paul RN, Riley RT, Tanaka T, Shier WT. Ultrastructural effects of AAL‐toxin TA from the fungus Alternaria alternata on black nightshade (Solanum nigrum L.) leaf discs and correlation with biochemical measures of toxicity. Toxicon 1998;36:1821–1832. [DOI] [PubMed] [Google Scholar]
- 192. Mandala SM, Harris GH. Isolation and characterization of novel inhibitors of sphingolipid synthesis: australifungin, viridiofungins, rustmicin, and khafrefungin. Methods Enzymol 2000;311:335–348. [DOI] [PubMed] [Google Scholar]
- 193. Heidler SA, Radding JA. Inositol phosphoryl transferases from human pathogenic fungi. Biochim Biophys Acta 2000;1500:147–152. [DOI] [PubMed] [Google Scholar]
- 194. Rittershaus PC, Kechichian TB, Allegood JC, Merrill AH, Hennig M, Luberto C, Del Poeta M. Glucosylceramide synthase is an essential regulator of pathogenicity of Cryptococcus neoformans. J Clin Invest 2006;116:1651–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Buczynski MW, Dumlao DS, Dennis EA. Thematic review series: proteomics – an integrated omics analysis of eicosanoid biology. J Lipid Res 2009;50:1015–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Galano J‐M, Mas E, Barden A, Mori TA, Signorini C, De Felice C, Barrett A, Opere C, Pinot E, Schwedhelm E, Benndorf R, Roy J, Le Guennec J‐Y, Oger C, Durand T. Isoprostanes and neuroprostanes: total synthesis, biological activity and biomarkers of oxidative stress in humans. Prostaglandins Other Lipid Mediat 2013;107:95–102. [DOI] [PubMed] [Google Scholar]
- 197. Andreou A, Brodhun F, Feussner I. Biosynthesis of oxylipins in non‐mammals. Prog Lipid Res 2009;48:148–170. [DOI] [PubMed] [Google Scholar]
- 198. Pohl CH, Kock JLF. Oxidized fatty acids as inter‐kingdom signaling molecules. Molecules 2014;19:1273–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Scher JU, Pillinger MH. The anti‐inflammatory effects of prostaglandins. J Investig Med 2009;57:703–708. [DOI] [PubMed] [Google Scholar]
- 200. Ott SJ, Kühbacher T, Musfeldt M, Rosenstiel P, Hellmig S, Rehman A, Drews O, Weichert W, Timmis KN, Schreiber S. Fungi and inflammatory bowel diseases: alterations of composition and diversity. Scand J Gastroenterol 2008;43:831–841. [DOI] [PubMed] [Google Scholar]
- 201. Kalo‐Klein A, Witkin SS. Prostaglandin E2 enhances and gamma interferon inhibits germ tube formation in Candida albicans. Infect Immun 1990;58:260–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Alem MAS, Douglas LJ. Prostaglandin production during growth of Candida albicans biofilms. J Med Microbiol 2005;54:1001–1005. [DOI] [PubMed] [Google Scholar]
- 203. Brouwers JF, Smeenk IM, van Golde LMG, Tielens AGM. The incorporation, modification and turnover of fatty acids in adult Schistosoma mansoni. Mol Biochem Parasitol 1997;88:175–185. [DOI] [PubMed] [Google Scholar]
- 204. Coppens I. Contribution of host lipids to Toxoplasma pathogenesis. Cell Microbiol 2006;8:1–9. [DOI] [PubMed] [Google Scholar]
- 205. Van Hellemond JJ, Tielens AGM. Adaptations in the lipid metabolism of the protozoan parasite Trypanosoma brucei. FEBS Lett 2006;580:5552–5558. [DOI] [PubMed] [Google Scholar]
- 206. Mazumdar J, Striepen B. Make it or take it: fatty acid metabolism of apicomplexan parasites. Eukaryot Cell 2007;6:1727–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Rub A, Arish M, Husain SA, Ahmed N, Akhter Y. Host–lipidome as a potential target of protozoan parasites. Microbes Infect 2013;15:649–660. [DOI] [PubMed] [Google Scholar]
- 208. Smith TM, Brooks TJ. Lipid fractions in adult Schistosoma mansoni. Parasitology 1969;59:293–298. [DOI] [PubMed] [Google Scholar]
- 209. Meyer F, Meyer H, Bueding E. Lipid metabolism in the parasitic and free‐living flatworms, Schistosoma mansoni and Dugesia dorotocephala. Biochim Biophys Acta 1970;210:257–266. [DOI] [PubMed] [Google Scholar]
- 210. Brouwers JF, Gadella BM, van Golde LMG, Tielens AGM. Quantitative analysis of phosphatidylcholine molecular species using HPLC and light scattering detection. J Lipid Res 1998;39:344–353. [PubMed] [Google Scholar]
- 211. Kubata BK, Duszenko M, Martin KS, Urade Y. Molecular basis for prostaglandin production in hosts and parasites. Trends Parasitol 2007;23:325–331. [DOI] [PubMed] [Google Scholar]
- 212. Belley A, Chadee K. Eicosanoid production by parasites: from pathogenesis to immunomodulation? Parasitol Today 1995;11:327–334. [DOI] [PubMed] [Google Scholar]
- 213. Da'dara A, Skelly PJ. Manipulation of vascular function by blood flukes? Blood Rev 2011;25:175–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Palacpac NMQ, Hiramine Y, Seto S, Hiramatsu R, Horii T, Mitamura T. Evidence that Plasmodium falciparum diacylglycerol acyltransferase is essential for intraerythrocytic proliferation. Biochem Biophys Res Commun 2004;321:1062–1068. [DOI] [PubMed] [Google Scholar]
- 215. Palacpac NMQ, Hiramine Y, Mi‐ichi F, Torii M, Kita K, Hiramatsu R, Horii T, Mitamura T. Developmental‐stage‐specific triacylglycerol biosynthesis, degradation and trafficking as lipid bodies in Plasmodium falciparum‐infected erythrocytes. J Cell Sci 2004;117:1469–1480. [DOI] [PubMed] [Google Scholar]
- 216. Majumder S, Dey R, Bhattacharjee S, Rub A, Gupta G, Bhattacharyya Majumdar S, Saha B, Majumdar S. Leishmania‐induced biphasic ceramide generation in macrophages is crucial for uptake and survival of the parasite. J Infect Dis 2012;205:1607–1616. [DOI] [PubMed] [Google Scholar]
- 217. Zhang K, Hsu F‐F, Scott DA, Docampo R, Turk J, Beverley SM. Leishmania salvage and remodelling of host sphingolipids in amastigote survival and acidocalcisome biogenesis. Mol Microbiol 2005;55:1566–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Romano JD, Sonda S, Bergbower E, Smith ME, Coppens I. Toxoplasma gondii salvages sphingolipids from the host Golgi through the rerouting of selected Rab vesicles to the parasitophorous vacuole. Mol Biol Cell 2013;24:1974–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Coppens I, Sinai AP, Joiner KA. Toxoplasma gondii exploits host low‐density lipoprotein receptor‐mediated endocytosis for cholesterol acquisition. J Cell Biol 2000;149:167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Sehgal A, Bettiol S, Pypaert M, Wenk MR, Kaasch A, Blader IJ, Joiner KA, Coppens I. Peculiarities of host cholesterol transport to the unique intracellular vacuole containing Toxoplasma. Traffic 2005;6:1125–1141. [DOI] [PubMed] [Google Scholar]
- 221. Sleat DE, Wiseman JA, El‐Banna M, Price SM, Verot L, Shen MM, Tint GS, Vanier MT, Walkley SU, Lobel P. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc Natl Acad Sci USA 2004;101:5886–5891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Blader IJ, Manger ID, Boothroyd JC. Microarray analysis reveals previously unknown changes in Toxoplasma gondii‐infected human cells. J Biol Chem 2001;276:24223–24231. [DOI] [PubMed] [Google Scholar]
- 223. Coppens I, Joiner KA. Host but not parasite cholesterol controls Toxoplasma cell entry by modulating organelle discharge. Mol Biol Cell 2003;14:3804–3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Pucadyil TJ, Tewary P, Madhubala R, Chattopadhyay A. Cholesterol is required for Leishmania donovani infection: implications in leishmaniasis. Mol Biochem Parasitol 2004;133:145–152. [DOI] [PubMed] [Google Scholar]
- 225. Tewary P, Veena K, Pucadyil TJ, Chattopadhyay A, Madhubala R. The sterol‐binding antibiotic nystatin inhibits entry of non‐opsonized Leishmania donovani into macrophages. Biochem Biophys Res Commun 2006;339:661–666. [DOI] [PubMed] [Google Scholar]
- 226. Fernandes MC, Cortez M, Geraldo Yoneyama KA, Straus AH, Yoshida N, Mortara RA. Novel strategy in Trypanosoma cruzi cell invasion: implication of cholesterol and host cell microdomains. Int J Parasitol 2007;37:1431–1441. [DOI] [PubMed] [Google Scholar]
- 227. Sandvig K, Skotland T, van Deurs B, Klokk TI. Retrograde transport of protein toxins through the Golgi apparatus. Histochem Cell Biol 2013;140:317–326. [DOI] [PubMed] [Google Scholar]
- 228. Sandvig K, van Deurs B. Delivery into cells: lessons learned from plant and bacterial toxins. Gene Ther 2005;12:865–872. [DOI] [PubMed] [Google Scholar]
- 229. Sandvig K, Bergan J, Kavaliauskiene S, Skotland T. Lipid requirements for entry of protein toxins into cells. Prog Lipid Res 2014;54:1–13. [DOI] [PubMed] [Google Scholar]
- 230. De Matteis MA, Godi A. PI‐loting membrane traffic. Nat Cell Biol 2004;6:487–492. [DOI] [PubMed] [Google Scholar]
- 231. Gelderblom WC, Jaskiewicz K, Marasas WF, Thiel PG, Horak RM, Vleggaar R, Kriek NP. Fumonisins‐‐novel mycotoxins with cancer‐promoting activity produced by Fusarium moniliforme. Appl Environ Microbiol 1988;54:1806–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Breslow DK, Collins SR, Bodenmiller B, Aebersold R, Simons K, Shevchenko A, Ejsing CS, Weissman JS. Orm family proteins mediate sphingolipid homeostasis. Nature 2010;463:1048–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Spiegel S, Merrill AH. Sphingolipid metabolism and cell growth regulation. FASEB J 1996;10:1388–1397. [DOI] [PubMed] [Google Scholar]
- 234. Marsh M, Bolzau E, Helenius A. Penetration of Semliki Forest virus from acidic prelysosomal vacuoles. Cell 1983;32:931–940. [DOI] [PubMed] [Google Scholar]
- 235. Helenius A, Mellman I, Wall D, Hubbard A. Endosomes. Trends Biochem Sci 1983;8:245–250. [Google Scholar]
- 236. Helenius A, Kartenbeck J, Simons K, Fries E. On the entry of Semliki forest virus into BHK‐21 cells. J Cell Biol 1980;84:404–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Pelkmans L, Helenius A. Insider information: what viruses tell us about endocytosis. Curr Opin Cell Biol 2003;15:414–422. [DOI] [PubMed] [Google Scholar]
- 238. Lopes da Silva M, Thieleke‐Matos C, Cabrita‐Santos L, Ramalho JS, Wavre‐Shapton ST, Futter CE, Barral DC, Seabra MC. The host endocytic pathway is essential for Plasmodium berghei late liver stage development. Traffic 2012;13:1351–1363. [DOI] [PubMed] [Google Scholar]
- 239. Bano N, Romano JD, Jayabalasingham B, Coppens I. Cellular interactions of Plasmodium liver stage with its host mammalian cell. Int J Parasitol 2007;37:1329–1341. [DOI] [PubMed] [Google Scholar]
- 240. King CA, Van Heyningen WE. Deactivation of cholera toxin by a sialidase‐resistant monosialosylganglioside. J Infect Dis 1973;127:639–647. [DOI] [PubMed] [Google Scholar]
- 241. Contreras F‐X, Ernst AM, Haberkant P, Björkholm P, Lindahl E, Gönen B, Tischer C, Elofsson A, von Heijne G, Thiele C, Pepperkok R, Wieland F, Brügger B. Molecular recognition of a single sphingolipid species by a protein's transmembrane domain. Nature 2012;481:525–529. [DOI] [PubMed] [Google Scholar]
- 242. Olkkonen VM, Li S. Oxysterol‐binding proteins: sterol and phosphoinositide sensors coordinating transport, signaling and metabolism. Prog Lipid Res 2013;52:529–538. [DOI] [PubMed] [Google Scholar]
- 243. Raetz CRH, Reynolds CM, Trent MS, Bishop RE. Lipid A modification systems in gram‐negative bacteria. Annu Rev Biochem 2007;76:295–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Van der Kleij D, Latz E, Brouwers JFHM, Kruize YCM, Schmitz M, Kurt‐Jones EA, Espevik T, de Jong EC, Kapsenberg ML, Golenbock DT, Tielens AGM, Yazdanbakhsh M. A novel host‐parasite lipid cross‐talk. Schistosomal lyso‐phosphatidylserine activates toll‐like receptor 2 and affects immune polarization. J Biol Chem 2002;277:48122–48129. [DOI] [PubMed] [Google Scholar]
- 245. Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res 2004;32:D277–D280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Bates JT, Chivian D, Arkin AP. GLAMM: genome‐linked application for metabolic maps. Nucleic Acids Res 2011;39:W400–W405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Baddely A, Turner R. spatstat: an R package for analyzing spatial point patterns. J Stat Softw 2005;12:1–42. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: List of lipid‐based interactions between host cells and pathogens that was used for the generation of Figure 2. A weighting of less than one was assigned if one specific interaction can be located at different nodes in the metabolic pathway. For example, phospholipase A2 activity of pathogens increases host levels of unesterified polyunsaturated fatty acids (18:2, 18:3, 20:4), commonly found at the sn‐2 position of phospholipase A2 substrates. In this case, the three fatty acids will be assigned a weighting factor of 0.33. Similarly, distinct interactions of a pathogen that target the same lipid in the host will result in weighting factors larger than 1. For example, Listeria has been described to affect a lipase and a phospholipase C, both resulting in increased levels of DAG.