

Summary
A porcine epidemic diarrhoea outbreak first occurred in southern China at the end of 2010 and afterwards the disease spread throughout the country. Spike gene is divergent and important for understanding the genetic relations of porcine epidemic diarrhoea virus field strains, the epidemiological status of the virus and vaccine development. In this study, S1 regions of spike gene of 1,235 selected strains collected from 2012 to 2017 in China were clustered along with 25 references of spike sequences mainly from China. The phylogenetic analysis demonstrates that these sequences of S1 regions were genetically more diverse with time. In all strains, G1a, G1b, G2a and G2b clusters accounted for 1.9%, 9.6%, 32.2% and 56.3%, respectively, namely G2a and G2b were the two most prevalent clusters in China. Furthermore, we made a more detailed classification for G2 group based on phylogenetic tree, in which G2a was divided into two subgroups, and G2b was separated into four subgroups.
Keywords: G2 group, phylogenetic analysis, porcine epidemic diarrhoea virus, spike gene
1. INTRODUCTION
Porcine epidemic diarrhoea (PED) is a highly contagious, intestinal infectious disease in pigs, characterized by severe diarrhoea, vomiting and dehydration. Pigs at all ages are susceptible to the disease. However, mortality is high in young pigs, especially in piglets younger than one‐week old. Since its first discovery in the late 1970s (Chasey & Cartwright, 1978; Pensaert & de Bouck, 1978), PED has continued to cause severe economic impact in swine industry worldwide. Starting from the end of 2010, a PED outbreak occurred in several pig‐producing provinces in southern China due to new porcine epidemic diarrhoea virus (PEDV) variants. Subsequently, the disease spread throughout the country and caused enormous economic losses to the pork industry (Chen et al., 2011, 2012, 2013; Li, Li, et al., 2012; Li, Zhu, et al., 2012; Yang et al., 2013).
PEDV, a causative agent of PED, is an enveloped single‐stranded positive‐sense RNA virus belonging to the order Nidovirales, family Coronaviridae and genus Alphacoronavirus. At present, four PEDV variants belonging to G1a, G1b, G2a and G2b groups were currently characterized (Lee, 2015; Sun et al., 2015). The PEDV genome consists of seven open reading frames (ORFs) as following: ORF1a, ORF1b, spike (S), ORF3, envelope (E), membrane (M) and nucleoprotein (N) (Kocherhans, Bridgen, Ackermann, & Tobler, 2001). The S protein is a glycosylated protein involved with viral pathogenesis and further divided into S1 and S2 domains. The S gene is divergent and important for understanding of the genetic relations of PEDV field strains, the epidemiological status of the virus and vaccine development (Park, Kim, Song, Moon, & Park, 2011; Stott et al., 2017; Sun et al., 2012).
In order to control and prevent PEDV infection, it is necessary for us to further investigate the prevalence of PEDV and the molecular characteristics of the S gene of Chinese PEDV field strains during 2012–2017. In this study, we first detected PEDV in samples collected from 400 farms, then analysed sequence characteristics of S gene in field strains and performed phylogenetic analysis of these field strains. These results would provide us the information on the evolutionary dynamics of PEDV in China.
2. RESULTS
2.1. Sample collection and detection
From January 2012 to April 2017, we collected 1,542 porcine faecal samples from pigs potentially affected by PEDV from 400 farms in China. These farms distributed in 15 provinces, including Guangdong, Jiangsu, Jiangxi, Hubei, Anhui, Liaoning, Hunan, Guangxi, Sichuan, Guizhou, Shaanxi, Yunnan, Chongqing, Hebei and Shanxi (Figure 1).
Figure 1.
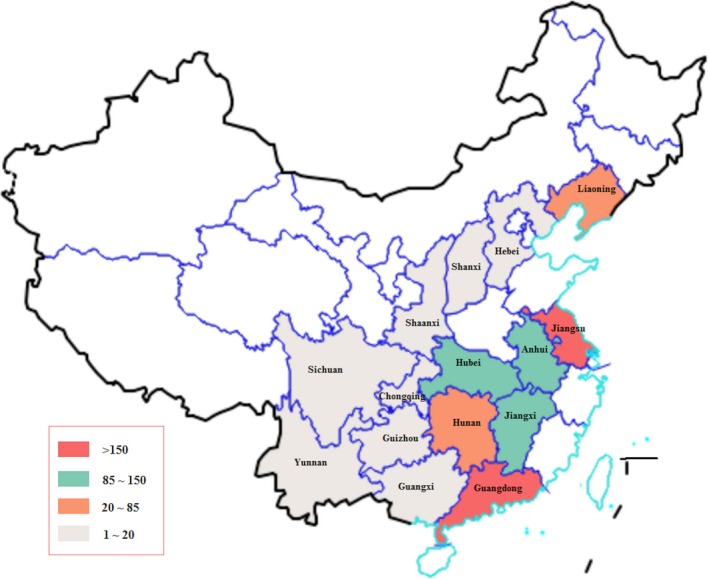
Geographic distribution of collected PEDV samples. The samples were collected from 15 provinces in China. The colours correspond to the number of collected positive samples [Colour figure can be viewed at http://wileyonlinelibrary.com]
Among them, 1,235 samples (80.1%) were positive for PEDV after RT‐PCR detection. The number of positive samples collected from 2012 to 2017 was 42, 172, 164, 271, 411 and 175, respectively (Figure 2a). We found that there were more positive samples collected from October to March than other periods, and this time range increased from 2012 to 2017. As shown in Figure 2b, most of the positive samples were collected from Guangdong (570), Jiangsu (195), Jiangxi (111), Hubei (103) and Anhui (95).
Figure 2.
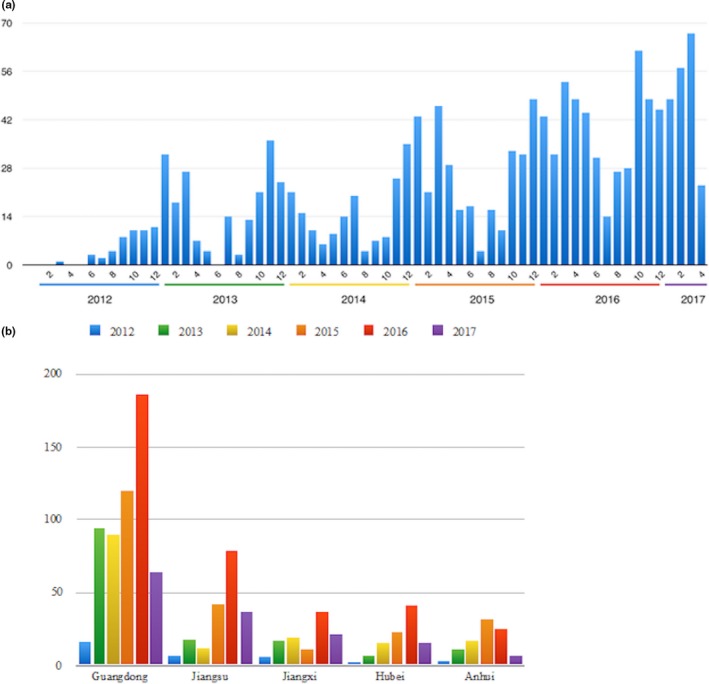
Summary of isolated PEDV strains. (a) The distribution of collected samples in time from 2012 to 2017. (b) The number of strains collected in five provinces each year from 2012 to 2017 [Colour figure can be viewed at http://wileyonlinelibrary.com]
2.2. S1 region sequencing
S1 region of spike gene was sequenced for all 1,235 positive samples with Sanger method. The length of these partial spike gene sequences ranged from 1,142 to 1,802. The nucleotide identity compared to CV777 strain ranged from 75% to 95.49%, indicating that S1 region was very diverse and had a high mutation rate. The median nucleotide identities compared to CV777 strain for strains in each year were 88.94%, 88.94%, 88.89%, 88.45%, 88.40% and 88.30%, respectively, indicating that S1 regions of spike sequences were genetically more diverse with time (Figure 3a).
Figure 3.
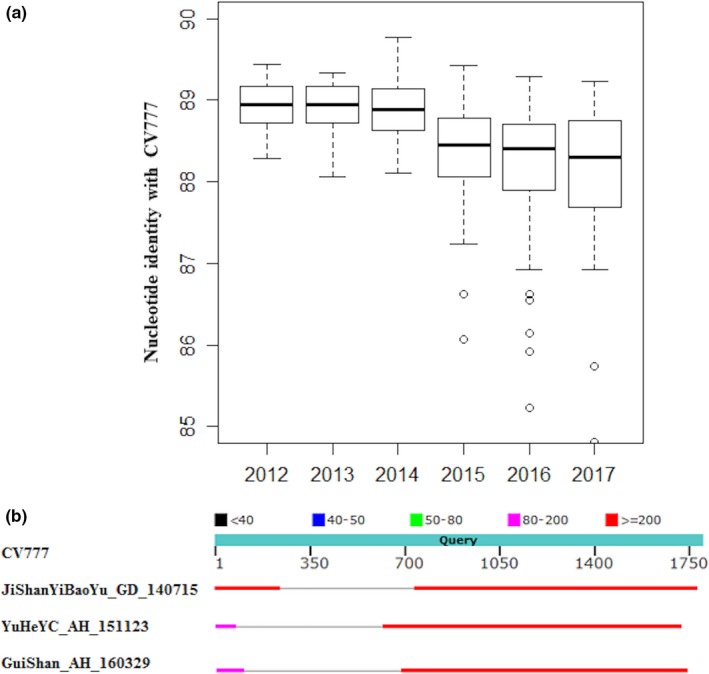
(a) Box plot of nucleotide identities compared to CV777 for strains from 2012 to 2017. All strains were separated into six groups according to their collection time, and their nucleotide identities with CV777 strain were calculated. (b) The comparison of sequences for S indel strains and CV777 strain. Three S indel strains were aligned with CV777 strain. Grey line indicated a deletion in S indel strains compared with CV777 strain [Colour figure can be viewed at http://wileyonlinelibrary.com]
We also identified three indel strains from these samples, namely JiShanYiBaoYu_GD_140715, YuHeYC_AH_151123 and GuiShan_AH_160329. These three indel strains were isolated from Guangdong (JiShanYiBaoYu_GD_140715) at 2014 and Anhui (YuHeYC_AH_151123, GuiShan_AH_160329) at 2015 and 2016, respectively. The deletion events occurred in different positions for all three indel strains. The locations of indels compared to CV777 strain were at 75 ‐ 615 (541 nt), 100 ‐ 681 (582 nt) and 243 ‐ 733 (491 nt), respectively (Figure 3b).
2.3. Phylogenetic analysis
All positive samples were grouped based on partial spike gene sequences using clades previously reported (Lee, 2015) in which PEDV evolved into two separated clades including G1 and G2. Each clade was further divided into two subclades including subclades G1a, G1b, G2a and G2b. In our data, G1a, G1b, G2a and G2b accounted for 1.9% (24 strains), 9.6% (118 strains), 32.2% (398 strains) and 56.3% (695 strains), respectively (Figure 4). The nucleotide identities of sequences in each group compared to CV777 were 88.1% ~ 95.49% for G1a, 92.52% ~ 95.39% for G1b, 75% ~ 89.25% for G2a and 79.39% ~ 89.44% for G2b. Obviously, G2a and G2b are the two most prevalent in China.
Figure 4.
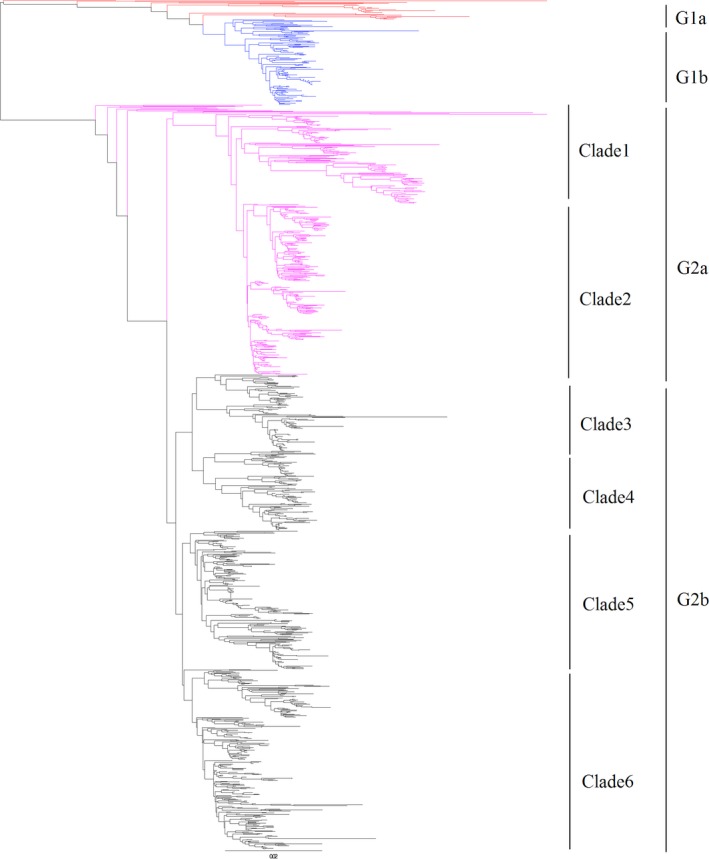
The phylogenetic tree based on partial spike gene of Chinese PEDV sequences along with reference sequences. PEDV is evolved into two genogroups including G1 and G2. G1 and G2 are each further divided into two subgroups including G1a, G1b, G2a and G2b. Novel clades for G2a and G2b were defined as clades 1 to 6 [Colour figure can be viewed at http://wileyonlinelibrary.com]
We also made a more detailed classification for G2 group, in which G2a was divided into two subgroups, and G2b was separated into four subgroups (Figure 4). There were 146 and 252 strains in clade 1 and clade 2 for G2a, while there were 113, 118, 204 and 260 strains in clade 3, clade 4, clade 5 and clade 6 for G2b.
2.4. Origin of transmission
From Figure 5, we can see that G2a and G2b were widely distributed in China compared to G1a and G1b. In the detailed classification of G2 group, strains of clades 2, 5 and 6 were more widely distributed in China than those of clades 1, 3 and 4. In our samples, G1b and G2b were first detected at 2012 while G1a and G2a were first detected at 2014 and 2013, respectively (Table 1). In the detailed subgroups, clades 5 and 6 were first detected at 2012, which belonged to typical variant strains, and clades 1 and 3 were first detected at 2013, and clades 2 and 4 were first detected at 2014.
Figure 5.
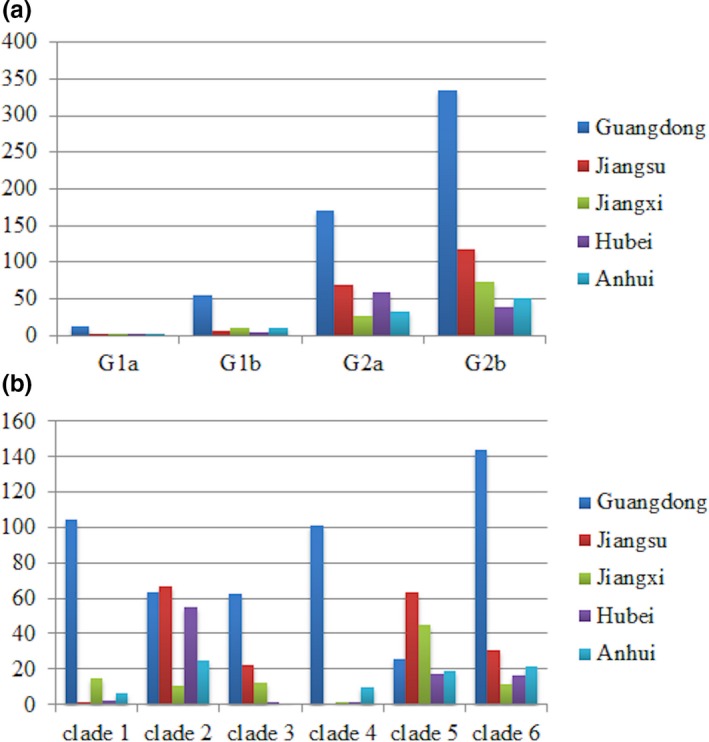
The distribution of PEDV strains in five provinces. A. Classical genogroups. B. Novel detailed subgroups for G2a and G2b [Colour figure can be viewed at http://wileyonlinelibrary.com]
Table 1.
Summary of strains in each group collected at different years
| 2012 | 2013 | 2014 | 2015 | 2016 | 2017 | Total | ||
|---|---|---|---|---|---|---|---|---|
| G1a | 0 | 0 | 2 | 8 | 12 | 2 | 24 | |
| G1b | 3 | 29 | 28 | 22 | 21 | 15 | 118 | |
| G2a | ||||||||
| Clade 1 | 0 | 2 | 8 | 23 | 87 | 26 | 146 | |
| Clade 2 | 0 | 0 | 10 | 66 | 112 | 64 | 252 | |
| Total | 0 | 2 | 18 | 89 | 199 | 90 | 398 | |
| G2b | ||||||||
| Clade 3 | 0 | 25 | 17 | 10 | 49 | 12 | 113 | |
| Clade 4 | 0 | 0 | 19 | 33 | 45 | 21 | 118 | |
| Clade 5 | 11 | 24 | 26 | 49 | 65 | 29 | 204 | |
| Clade 6 | 28 | 92 | 55 | 59 | 20 | 6 | 260 | |
| Total | 39 | 141 | 117 | 151 | 179 | 68 | 695 | |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Next, we tried to analyse the origin of clades 1, 2, 3 and 4 according to the collection time. Two strains of G2a (clade 1), DaChuanDing_GD_130401 and TianTang_GD_130530, were collected at April and May 2013 in Guangdong. There were 25 strains of clade 3 detected at 2013, and 88% of them were collected in Guangdong. The 10 strains of clade 2 at 2014 were collected in Hubei (three strains), Anhui (four strains) and Jiangsu (two strains). The strains of clade 4 at 2014 were all detected in Guangdong. These results indicated that geographically Guangdong might be the origin of clades 1, 3 and 4, and Hubei, Anhui and Jiangsu might be the origins of clade 2.
3. DISCUSSION
PEDV S gene plays an important role in the molecular epidemiology and in the genetic variation of PEDV field strains. In order to control and prevent PEDV infection, it is necessary for us to further investigate the prevalence of PEDV and the molecular characteristics of the S genes of Chinese PEDV field strains during 2012–2017. In this study, the genetic epidemiology of 1,235 PEDV samples collected between 2012 and 2017 in China was investigated.
These samples were collected from 400 farms in 15 provinces, which was one of the most widespread surveys of PEDV in China. Based on the phylogenetic analysis of a large scale of sequences of partial spike gene, several important findings involving the genetic evolution of PEDV in China were unveiled. We found that G2a and G2b are the two most prevalent in China, and they have evolved into six different subclades. And we got some clues of origins of subclades by analysing the time and region of collected PEDV samples in each subclade. Besides, we also got one S indel strain in Guangdong and two S indel strains in Anhui.
Through comparing S1 sequences of all strains with CV777, we found PEDV showed a significant decrease in nucleotide identity with CV777 with time, which meant more diversity generated due to a high mutation rate of coronavirus. To some extent, this also indicated a change in prevalence of PEDV. And further phylogenetic analysis confirmed our assumption. Using the typical classification method (Lee, 2015), we divided all strains into four groups, in which G2a and G2b are the two most prevalent in China. Traditionally, there was more than one group of strains prevalent in one region, which made PEDV prevention more difficult and complicated. For example, all four groups of PEDV strains existed in Guangdong, Jiangsu and Jiangxi.
Benefit from the large amount of S1 sequences and widespread sample collections, we could make a detailed classification of PEDV groups and try to analyse the origins. We divided G2 group into six clades, in which G2a contained two clades and G2b contained four clades. Clades 1 and 4 were mainly prevalent in Guangdong, while clades 2, 3, 5 and 6 were prevalent in a wider area. According to the first appearance time in our samples, we separated them into three stages: clades 5 and 6 were in early stage, clades 1 and 3 were in middle stage, and clades 2 and 4 were in late stage. In other word, clades 1, 2, 3 and 4 might contain new mutant strains in G2 group. We concluded that Guangdong might be the origin of clades 1, 3 and 4, and Hubei, Anhui and Jiangsu might be the origins of clade 2.
Taken together, these results provided us a clear landscape of genetic epidemiology of PEDV circulating in China in 2012–2017 and would help in the areas of accurate PEDV prevention.
4. MATERIALS AND METHODS
4.1. Source of specimens and PEDV isolation
Between January 2012 and April 2017, porcine faecal samples (n = 1542) were collected from pigs affected by PEDV from 400 farms in 15 provinces (Guangdong, Jiangsu, Jiangxi, Hubei, Anhui, Liaoning, Hunan, Guangxi, Sichuan, Guizhou, Shaanxi, Yunnan, Chongqing, Hebei and Shanxi). Samples were suspended in PBS and clarified by centrifugation. The supernatant was filtered through 0.22‐μm filters and stored at −80°C until use.
4.2. Cloning, plasmid purification and sequence determination
Sequencing was performed as previously reported (Temeeyasen et al., 2014). Briefly, total RNA was extracted from the supernatant using the NucleoSpin® RNA Virus kit in accordance with the manufacturer's instructions. cDNA was synthesized from the extracted RNA using M‐MuLV Reverse Transcriptase. To amplify the S1 domain of S genes, PCR amplification was performed using previously reported primers (Lee, Park, Kim, & Lee, 2010) and using Platinum® Tag DNA Polymerase High Fidelity.
4.3. Phylogenetic analysis
The nucleotide sequences were aligned using the multiple sequence alignment software Clustal Omega 1.2.1 (Sievers et al., 2011). A phylogenetic analysis was performed based on the nucleotide sequences of partial spike genes of 1,235 PEDV strains collected from 2012 to 2017 along with 25 other PEDV spike sequences available in GenBank from the previous study (Lee, 2015) with ClustalX2 (Larkin et al., 2007). FigTree 1.4.3 was used to view the phylogenetic tree and classify sequences to groups. The genogroup described in another study (Lee, 2015) was used to classify and compare with the sequences in this study.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
ACKNOWLEDGEMENTS
This work was supported by the National Key Research and Development Program (grant no. 2016YFD0500101).
Wen Z, Li J, Zhang Y, et al. Genetic epidemiology of porcine epidemic diarrhoea virus circulating in China in 2012–2017 based on spike gene. Transbound Emerg Dis. 2018;65:883–889. 10.1111/tbed.12825
REFERENCES
- Chasey, D. , & Cartwright, S. (1978). Virus‐like particles associated with porcine epidemic diarrhoea. Research in Veterinary Science, 25, 255–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Liu, X. , Shi, D. , Shi, H. , Zhang, X. , & Feng, L. (2012). Complete genome sequence of a porcine epidemic diarrhea virus variant. Journal of Virology, 86, 3408 10.1128/JVI.07150-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Liu, X. , Shi, D. , Shi, H. , Zhang, X. , Li, C. , … Feng, L. (2013). Detection and molecular diversity of spike gene of porcine epidemic diarrhea virus in China. Viruses, 5, 2601–2613. 10.3390/v5102601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Wang, C. , Shi, H. , Qiu, H. , Liu, S. , Shi, D. , … Feng, L. (2011). Complete genome sequence of a Chinese virulent porcine epidemic diarrhea virus strain. Journal of Virology, 85, 11538–11539. 10.1128/JVI.06024-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocherhans, R. , Bridgen, A. , Ackermann, M. , & Tobler, K. (2001). Completion of the porcine epidemic diarrhoea coronavirus (PEDV) genome sequence. Virus Genes, 23, 137–144. 10.1023/A:1011831902219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin, M. A. , Blackshields, G. , Brown, N. P. , Chenna, R. , McGettigan, P. A. , McWilliam, H. , … Higgins, D. G. (2007). Clustal W and Clustal X version 2.0. Bioinformatics, 23, 2947–2948. 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- Lee, C. (2015). Porcine epidemic diarrhea virus: An emerging and re‐emerging epizootic swine virus. Virology Journal, 12, 193 10.1186/s12985-015-0421-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, D. K. , Park, C. K. , Kim, S. H. , & Lee, C. (2010). Heterogeneity in spike protein genes of porcine epidemic diarrhea viruses isolated in Korea. Virus Research, 149, 175–182. 10.1016/j.virusres.2010.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , Li, H. , Liu, Y. , Pan, Y. , Deng, F. , Song, Y. , … He, Q. (2012). New variants of porcine epidemic diarrhea virus, China, 2011. Emerging Infectious Diseases, 18, 1350–1353. 10.3201/eid1803.120002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z. L. , Zhu, L. , Ma, J. Y. , Zhou, Q. F. , Song, Y. H. , Sun, B. L. , … Bee, Y. Z. (2012). Molecular characterization and phylogenetic analysis of porcine epidemic diarrhea virus (PEDV) strains in south China. Virus Genes, 45, 181–185. 10.1007/s11262-012-0735-8 [DOI] [PubMed] [Google Scholar]
- Park, S. J. , Kim, H. K. , Song, D. S. , Moon, H. J. , & Park, B. K. (2011). Molecular characterization and phylogenetic analysis of porcine epidemic diarrhea virus (PEDV) field isolates in Korea. Archives of Virology, 156, 577–585. 10.1007/s00705-010-0892-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pensaert, M. B. , & de Bouck, P. (1978). A new coronavirus‐like particle associated with diarrhea in swine. Archives of Virology, 58, 243–247. 10.1007/BF01317606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers, F. , Wilm, A. , Dineen, D. G. , Gibson, T. J. , Karplus, K. , Li, W. , … Higgins, D. G. (2011). Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology, 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stott, C. J. , Temeeyasen, G. , Tripipat, T. , Kaewprommal, P. , Tantituvanont, A. , Piriyapongsa, J. , & Nilubol, D. (2017). Evolutionary and epidemiological analyses based on spike genes of porcine epidemic diarrhea virus circulating in Thailand in 2008‐2015. Infection, Genetics and Evolution, 50, 70–76. 10.1016/j.meegid.2017.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, R. Q. , Cai, R. J. , Chen, Y. Q. , Liang, P. S. , Chen, D. K. , & Song, C. X. (2012). Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerging Infectious Diseases, 18, 161–163. 10.3201/eid1801.111259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, M. , Ma, J. , Wang, Y. , Wang, M. , Song, W. , Zhang, W. , … Yao, H. (2015). Genomic and epidemiological characteristics provide new insights into the phylogeographical and spatiotemporal spread of porcine epidemic diarrhea virus in Asia. Journal of Clinical Microbiology, 53, 1484–1492. 10.1128/JCM.02898-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temeeyasen, G. , Srijangwad, A. , Tripipat, T. , Tipsombatboon, P. , Piriyapongsa, J. , Phoolcharoen, W. , … Nilubol, D. (2014). Genetic diversity of ORF3 and spike genes of porcine epidemic diarrhea virus in Thailand. Infection, Genetics and Evolution, 21, 205–213. 10.1016/j.meegid.2013.11.001 [DOI] [PubMed] [Google Scholar]
- Yang, X. , Huo, J. Y. , Chen, L. , Zheng, F. M. , Chang, H. T. , Zhao, J. , … Wang, C. Q. (2013). Genetic variation analysis of reemerging porcine epidemic diarrhea virus prevailing in central China from 2010 to 2011. Virus Genes, 46, 337–344. 10.1007/s11262-012-0867-x [DOI] [PMC free article] [PubMed] [Google Scholar]