

Summary
Porcine epidemic diarrhoea virus (PEDV) is the aetiologic agent of porcine epidemic diarrhoea (PED), a highly contagious enteric disease that is threatening the swine industry globally. Since PED was first reported in Southern Vietnam in 2009, the disease has spread throughout the country and caused substantial economic losses. To identify PEDVs responsible for the recent outbreaks, the full‐length spike (S) gene of 25 field PEDV strains collected from seven northern provinces of Vietnam was sequenced and analysed. The sequence analysis revealed that the S genes of Vietnamese PEDVs were heterogeneous and classified into four genotypes, namely North America and Asian non‐S INDEL, Asian non‐S INDEL, new S INDEL and classical S INDEL. This study reported the pre‐existence of US‐like PEDV strains in Vietnam. Thirteen Vietnamese variants had a truncated S protein that was 261 amino acids shorter than the normal protein. We also detected one novel variant with an 8‐amino acid insertion located in the receptor‐binding region for porcine aminopeptidase N. Compared to the commercial vaccine strains, the emerging Vietnamese strains were genetically distant and had various amino acid differences in epitope regions and N‐glycosylation sites in the S protein. The development of novel vaccines based on the emerging Vietnamese strains may be contributive to the control of the current PED outbreaks.
Keywords: genetic heterogeneity, porcine epidemic diarrhoea virus, S INDEL, spike gene, US‐like strains
1. INTRODUCTION
Porcine epidemic diarrhoea virus (PEDV) is the causative agent of porcine epidemic diarrhoea (PED), a devastating disease characterized by acute enteritis and severe watery diarrhoea followed by dehydration leading to death, and the virus is associated with high mortality rates in piglets (Song & Park, 2012). PEDV belongs to the family Coronaviridae in the order Nidovirales. This virus has a single‐stranded positive‐sense RNA genome of approximately 28 kb with a 5′ cap and a 3′ polyadenylated tail (Kocherhans, Bridgen, Ackermann, & Tobler, 2001; Pensaert & de Bouck, 1978). The PEDV genome is composed of the 5′ untranslated region and at least seven open reading frames (ORFs) that encode four structural proteins, namely spike (S), envelope (E), membrane (M) and nucleocapsid (N), and three major non‐structural proteins (1a, 1ab and ORF3) (Song & Park, 2012). Of the structural proteins, the S glycoprotein is the most diverse protein (Chen et al., 2013), and it plays a vital role in the induction of neutralizing antibodies and regulation of interactions with cellular receptors to mediate virus entry (Bosch, van der Zee, de Haan, & Rottier, 2003). It is also associated with growth adaptation in vitro and attenuation of virulence in vivo (Sato et al., 2011). The S protein has been utilized as an effective component for studying genetic relatedness among PEDV strains, examining their epidemiological status and facilitating vaccine development (Chiou et al., 2015; Lee, Park, Kim, & Lee, 2010; Puranaveja et al., 2009; Temeeyasen et al., 2014).
Porcine epidemic diarrhoea was first recognized in England in 1971 and later reported in several European countries (Pensaert & de Bouck, 1978; Wood, 1977). In Asia, PED has been reported since the 1980s, and it has become an economic concern for the swine industry in Japan (Takahashi, Okada, & Ohshima, 1983), China (Sun, Wang, Wei, Chen, & Feng, 2016), South Korea (Kweon et al., 1993), Thailand (Puranaveja et al., 2009) and Taiwan (Lin et al., 2014). Before 2010, the prevalence of PEDV infection was comparatively low with mostly isolated outbreaks (Song & Park, 2012). Since late 2010, new PEDV strains with increased pathogenicity compared to the classical strains emerged in China, and they are regarded as the first pandemic strains (Sun et al., 2016). In April 2013, highly pathogenic PEDV (US prototype, North American type) was first detected in the US, and it rapidly spread to 31 states as well as Mexico and Canada (Oka et al., 2014). Later, the second type of PEDV in the US with lower virulence in the field, designated S INDEL PEDV, was detected in samples collected beginning in June 2013 (Vlasova et al., 2014). Subsequently, US‐like strains have been reported in South Korea, Japan, Taiwan and European countries such as Ukraine, Belgium, Germany, France, Slovenia and Netherlands (Lin, Saif, Marthaler, & Wang, 2016). In Vietnam, PED outbreaks were firstly confirmed by reverse transcription polymerase chain reaction (RT‐PCR) in the southern provinces in early 2009 (Duy, Toan, Puranaveja, & Thanawongnuwech, 2011). For Northern Vietnam, PEDV has been detected in severe outbreaks since the beginning of 2010 using commercial immunochromatographic assay kits (Diep, Lan, Hoa, & Yamaguchi, 2014). A previous study of PED in Northern Vietnam conducted in 2012 and early 2013 (Diep et al., 2014) revealed that the disease primarily occurred in the cold season lasting from November to April. Pigs of all ages were affected, manifesting diarrhoea at a rate of 92.0%–100% in herds, and the mortality rate is as high as 93.8% (68.6% on average) in suckling pigs (Diep et al., 2014). A commercial PED live vaccine (strain KPEDV‐9) has been used in pig farms in Northern Vietnam since 2011. Another more common immunoprophylaxis applied in the field is the artificial infection of sows (i.e., the feedback method) during pregnancy using pooled faeces and intestine collected from infected piglets (Song, Moon, & Kang, 2015). However, the effectiveness of these preventive measurements in the field is questionable because PED still occurred and frequently recurred in pig farms in which vaccination or the feedback method was applied. The persistence and reoccurrence of PED have been becoming more common in infected farms. To date, PEDV continues to spread widely and cause severe economic losses for the national swine industry. To identify the PEDV strains responsible for the recent outbreaks occurred from 2012 to 2015 in Northern Vietnam, we sequenced and analysed the full‐length S gene of PEDVs obtained from 25 affected pig farms. This result will provide useful insights into the molecular epidemiology in Vietnam and serve as the basis for the development of effective vaccines for controlling the disease.
2. MATERIALS AND METHODS
2.1. Sample collection, RNA extraction and PEDV detection
Twenty‐five PEDV‐positive samples as confirmed by RT‐PCR collected from 25 herds located in seven different northern provinces of Vietnam (Fig. S1) between December 2012 and July 2015 were used in this study. The number of samples from each province was as follows: Hung Yen (n = 9), Ha Noi (n = 6), Hoa Binh (n = 3), Thai Binh (n = 3), Vinh Phuc (n = 2), Quang Ninh (n = 1) and Son La (n = 1). Small intestine and stool specimens were taken from sucking piglets, post‐weaning pigs and sows exhibiting acute watery diarrhoea at 25 pig farms. Intestine samples were collected from dead piglets, and the faecal samples were non‐invasively collected immediately after excretion. Therefore, no aggressive operation was conducted in pigs for sampling purposes. Samples were prepared in 20% suspensions through homogenization with Dulbecco's Modified Eagle's Medium with a low concentration of glucose. The suspensions were then vortexed and centrifuged at 2300 g for 10 min at 4°C. The clarified supernatants were stored at −80°C until use. Viral RNA was extracted from 300 μl of the samples and eluted in 30 μl of RNase‐free water using ReliaPrep™ RNA Cell Miniprep kits (Promega Corporation, Madison, WI, USA) in accordance with the manufacturer's instructions. PEDV was detected in the collected samples by RT‐PCR using a newly designed primer pair (pM‐F/pM‐R) for amplification of a partial M gene (Table S1). The expected size of amplified product was 463 bp. One‐tube RT‐PCR was performed using AccessQuick™ RT‐PCR System kits (Promega). Exactly 4 μl of RNA template was mixed with the reaction mixture, which contained 12.5 μl of AccessQuick™ Master Mix (2×), 0.5 μl of each specific primer (10 μM) and 0.5 μl of AMV reverse transcriptase (5 u/μl). Then, 7 μl of nuclease‐free water was added to reach the total reaction volume of 25 μl. RT‐PCR was performed using a PCR Thermal Cycler (Takara, Japan). Following a reverse transcription step of 45°C for 45 min and an incubation step of 94°C for 2 min, 35 cycles were performed as follows: 94°C for 30 s, 53°C for 30 s and 72°C for 30 s. The cycles were followed by a final 10‐min extension step at 72°C. The RT‐PCR products were analysed by 1.2% agarose gel electrophoresis and visualized by ultraviolet illumination after ethidium bromide staining.
2.2. Sequencing of the S gene
From extracted RNA, RT was first performed using random hexamer primers and oligo primers from Reverse Transcription System Kits (Promega). The full‐length S gene of PEDV was amplified using the primer pair FS‐F/FS‐R (Table S1) and KOD FX neo Kit (Toyobo Co., Japan). The PCR conditions were as follows: denaturation at 94°C for 2 min; 35 cycles of denaturation at 98°C for 10 s, annealing at 55°C for 30 s, and extension at 68°C for 2.5 min; and a final extension step at 68°C for 10 min. Then, the PCR products were used as templates for nested PCR to amplify five DNA fragments spanning the entire S gene using five published primer pairs (CS1–CS5) (Diep, Norimine, Sueyoshi, Lan, & Yamaguchi, 2017). The amplified PCR products were purified using a FastGene Gel/PCR Extraction Kit (NIPPON Genetics Co., Ltd, Japan) according to the manufacturer's protocol. All sequencing reactions were performed in duplicate, and sequences were determined in both directions using BigDye® Terminator v3.1 Cycle Sequencing Kits (Applied Biosystems, CA, USA). The products were analysed using ABI PRISM 3130xl Genetic Analyzers (Applied Biosystems).
2.3. Sequence analysis
Nucleotide and deduced amino acid sequences were assembled and aligned using Geneious version 9.1.6 software (http://www.geneious.com). The percentage sequence divergences at the nucleotide and amino acid levels were further calculated using the same software application. The obtained nucleotide sequences were deposited in GenBank under the accession numbers KX982553–KX982577 as shown in Table 1. Unrooted phylogenetic trees were constructed using molecular evolutionary genetics analysis (MEGA) software version 6.06 (Tamura, Stecher, Peterson, Filipski, & Kumar, 2013) with maximum likelihood method and bootstrap tests of 1,000 replicates. The best‐fit nucleotide substitution models for analysis were assessed. Phylogenetic trees based on the nucleotide sequences of the full‐length S gene were generated using the Tamura–Nei substitution model with a discrete gamma distribution (TN93+G). Prediction of N‐glycosylation sites was performed using services available at http://www.cbs.dtu.dk/services/NetNGlyc. To identify high‐specificity N‐glycosylation sites, any potential crossing 0.5 and Jury agreement (9/9) or potential greater 0.75 for asparagines that occurred within the Asn‐Xaa‐Ser/Thr triplet was used. The potential phosphorylation sites were determined using the NetPhos 3.1 server (http://www.cbs.dtu.dk/services/NetPhos/) and NetPhosBac 1.0 server (http://www.cbs.dtu.dk/services/NetPhosBac-1.0). Prediction of O‐glycosylation sites was performed using the NetOGlyc 3.1 Server (http://www.cbs.dtu.dk/services/NetOGlyc-3.1/).
Table 1.
Information of 25 Vietnamese PEDV isolates analysed in this study
| No | Isolates | Farm | Sample origin | Geographical origin | Collection time | Length of the S gene (nt) | Acce No |
|---|---|---|---|---|---|---|---|
| 1 | VN101/HY/2012 | Ata | Faeces | HY | 22‐Dec‐12 | 4,161 | KX982562 |
| 2 | VN01/HY/2013 | HT1 | Small intestine | HY | 20‐Jan‐13 | 4,161 | KX982553 |
| 3 | VN02/HY/2013 | HT2 | Small intestine | HY | 22‐Jan‐13 | 4,158 | KX982554 |
| 4 | VN06/HY/2013 | CT1 | Faeces | HY | 23‐Jan‐13 | 4,161 | KX982555 |
| 5 | VN12/HY/2013 | Tam1 | Faeces | HY | 31‐Jan‐13 | 4,161 | KX982557 |
| 6 | VN11/HY/2013 | CT2 | Faeces | HN | 1‐Feb‐13 | 4,161 | KX982556 |
| 7 | VN15/HY/2013 | Tam2 | Faeces | HY | 1‐Feb‐13 | 4,161 | KX982558 |
| 8 | VN19/HY/2013 | Tam3 | Faeces | HY | 5‐Feb‐13 | 4,161 | KX982559 |
| 9 | VN112/HN/2013 | CTd | Faeces | HN | 5‐Feb‐13 | 4,161 | KX982563 |
| 10 | VN44/QN/2013 | LaT | Faeces | QN | 20‐Feb‐13 | 4,161 | KX982560 |
| 11 | VN97/HN/2013 | Tdu1 | Faeces | HN | 18‐Apr‐13 | 4,161 | KX982561 |
| 12 | VN232/HB/2013 | Ama1 | Small intestine | HB | 10‐Oct‐13 | 4,143 | KX982564 |
| 13 | VN262/HB/2014 | Jaf1 | Faeces | HB | 12‐Feb‐14 | 4,152 | KX982565 |
| 14 | VN288/SL/2014 | Car1 | Small intestine | SL | 14‐Mar‐14 | 4,161 | KX982567 |
| 15 | VN292/HN/2014 | Car2 | Faeces | HN | 24‐Mar‐14 | 4,152 | KX982568 |
| 16 | VN297/HB/2014 | Jaf2 | Faeces | HB | 31‐Mar‐14 | 4,161 | KX982569 |
| 17 | VN344/HN/2014 | Tdu2 | Small intestine | HN | 13‐May‐14 | 4,161 | KX982570 |
| 18 | VN367/VP/2014 | Ama2 | Small intestine | VP | 11‐Jul‐14 | 4,152 | KX982571 |
| 19 | VN385/TB/2014 | Ama3 | Small intestine | TB | 24‐Jul‐14 | 4,161 | KX982572 |
| 20 | VN‐K2/HY/2015 | Han | Faeces | HY | 26‐Jan‐15 | 4,152 | KX982573 |
| 21 | VN‐K23/HN/2015 | Pro | Small intestine | HN | 6‐Apr‐15 | 4,161 | KX982574 |
| 22 | VN‐TH15/HY/2015 | Thu | Small intestine | HY | 7‐Apr‐15 | 4,182 | KX982576 |
| 23 | VN‐Jafa/HB/2015 | JaHB | Small intestine | TB | 18‐Jun‐15 | 4,161 | KX982577 |
| 24 | VN‐K28/TB/2015 | Ama4 | Small intestine | TB | 3‐Jul‐15 | 4,161 | KX982575 |
| 25 | VN270/VP/2014 | Yhu | Faeces | VP | 22‐Feb‐14 | 4,161 | KX982566 |
HY, Hung Yen; HN, Ha Noi; QN, Quang Ninh; HB, Hoa Binh; SL, Son La; VP, Vinh Phuc; TB, Thai Binh.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
3. RESULTS
3.1. Phylogenetic analysis of the S gene
To investigate the diversity and relatedness of PEDV strains circulating in Northern Vietnam, the full‐length S gene of Vietnamese PEDVs derived from 25 field samples collected in seven Northern provinces of Vietnam were sequenced and analysed. The phylogenetic tree of the identified S genes was constructed together with other classical and recent strains isolated in Europe, Asia and North America. Phylogenetic analysis based on the nucleotide sequences of the S gene revealed two major clusters that were designated genogroup 1 (G1) and genogroup 2 (G2). G2 was further divided into G2‐1 and G2‐2, and G1 was divided into G1‐1, G1‐2 and G1‐3 as shown in Figure 1. G1‐1 includes the prototype European strain CV777, other global historic S INDEL strains isolated between 1970 and 2010 (Lin et al., 2016), and vaccine strains that were developed in Asian countries such as KPEDV‐9 and attenuated DR13 from South Korea, P5‐V from Japan and attenuated CV777 from China. One Vietnamese strain (VN232/HB2013) in this study was clustered into this subgroup, and it was closely related to the previously reported Vietnamese strain HUA‐PED67 (2013) and Chinese strains CH7 (2011), JS‐2004‐2 (2004) and LJB03 (2006). G1‐2 consisted of two classical strains isolated in Japan, namely NK and KH. G1‐3 was composed of new emerging S INDEL strains. Notably, a Vietnamese strain in our study, VN262/HB/2014, was included in this subgroup. G2 primarily consisted of PEDV strains identified since 2010 in Asia, America and Europe. Twenty‐three Vietnamese field PEDV strains in this study were categorized into two subgroups of G2. Majority of Vietnamese strains in our study belonged to G2‐2, which comprised 17 of 25 current Vietnamese strains along with high virulent non‐S INDEL strains identified in North America and Asia. Excluding VN97/HN/2013 and VN344/HN/2014 in this subgroup, which were closely clustered with Chinese strains, 15 other Vietnamese strains were grouped closest to North American non‐S INDEL strains identified in US, Mexico, Canada, Japan, South Korea and Taiwan (Chiou et al., 2015; Suzuki et al., 2015; Vlasova et al., 2014). Notably, three Vietnamese strains, namely VN01/HY/2013, VN12/HY/2013 and VN385/TB/2014, were most closely related to three Japanese strains (AOM‐3/JPN/2014, 14JM126, 14JM226) and two US strains (USA/Minnesota90/2013, NPL‐PEDv/2013), and they formed a monophyletic branch. The remaining 12 Vietnamese strains were grouped in a segregated branch, and they were most closely related to the Japanese strain AOM‐2/JPN/2014 and US strain USA/Minnesota/271/2014 (US). Six PEDV Vietnamese strains in our study were included in subgroup G2‐1. Four of these strains (VN292/HN/2014, VN367/VP/2014, VN‐K2/HY/2015 and VN‐TH15/HY/2015) formed a monophyletic branch, and they were closely related to the previously reported Vietnamese strains HUA‐PED45 and HUA‐PED47 (Kim, Lim et al., 2015). The other Vietnamese strain in this subgroup, namely VN02/HY/2013, was closely grouped with reference strains recently isolated in Japan (13JM‐291), Thailand (BRC2) and Vietnam (VN/VAP1113_1/2013/VungTau, VN/KCHY_310113/2013/KhoaiChau and VN/JFP1013_1/2013/VinhAn).
Figure 1.
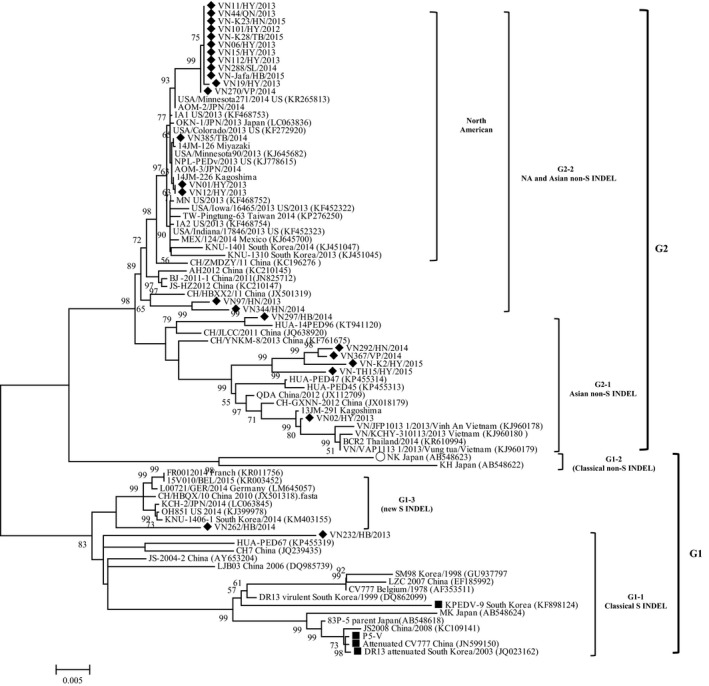
Phylogenetic tree based on the nucleotide sequences of the entire S genes of 25 Vietnamese PEDV strains isolated in this study and other reference strains identified worldwide. The tree was constructed by the maximum likelihood method using the Tamura–Nei substitution model with a discrete gamma distribution in the MEGA v.6.05 program. Numbers at nodes represent the percentages of 1,000 bootstrap replicates (values <50 are not shown). The scale bar indicates the number of nucleotide substitutions per site. The Vietnamese PEDV strains evaluated in this study are marked by solid diamond symbols, and vaccine strains are marked by solid square symbols
3.2. Sequence homology analysis
Pairwise alignment of the 25 field strains in our study revealed that their S genes were not homologous. The S genes of these Vietnamese field strains had 93.51%–100% nucleotide homologies with each other, as well as homologies of 92.13–95.16, 92.1–95.45, 92.01–95.40 and 92.08%–95.42% with the vaccine strains KPEDV‐9, P5‐V, attenuated CV777 and attenuated DR13, respectively (Table S2, S3). The deduced amino acid sequences of the S protein of these Vietnamese strains had 92.77%–100% identity with each other, as well as identities of 91.48–95.66, 91.05–95.52, 90.83–95.30 and 91.05%–95.44% identity with KPEDV‐9, P5‐V, attenuated CV777 and attenuated DR13, respectively. The S gene of VN232/HB/2013 shared 93.73%–96.79% nucleotide identity with those of other strains in G1‐1. The S gene of VN262/HB/2014 had 98.22%–98.58% nucleotide identity with those of other strains in G1‐3. VN262 had the highest nucleotide identity (98.58%) with S INDEL strains OH851 from USA and KCH‐2/JPN/2014 from Japan. The S genes of six Vietnamese field strains in G2‐1 had homologies of 96.71%–99.69% with each other and 96.52%–99.88% with reference strains in G2‐1. Notably, VN02/HY/2013 had the highest nucleotide and amino acid identities (99.88% and 99.86%, respectively) with the Japanese strain 13JM‐291. Vietnamese strains in subgroup G2‐2 in our study had 98.22–100 and 97.69%–100% nucleotide and amino acid identities with each other, respectively and 97.98–99.98 and 97.62%–99.93% nucleotide and amino acid identities with other reference strains in this subgroup, respectively. Compared with other reference PEDV strains, the Vietnamese field PEDVs in G2‐2 had the highest nucleotide (amino acid) identity with emerging North American (NA) strains. VN01/HY/2013, VN12/HY/2013 and VN385/TB/2014 had the highest nucleotide sequence identity with USA/Colorado/2013, USA/Minnesota90/2013, NPL‐PEDv/2013 (US), AOM‐3/JPN/2014 (Japan) and 14JM‐226 (Japan), ranging from 99.93% to 99.98% (99.86%–99.93%). Notably, of the 12 Vietnamese strains that formed a segregated branch in G2‐2, 10 strains, namely VN06/HY/2013, VN11/HY/2013, VN15/HY/2013, VN44/QN/2013, VN101/HY/2012, VN112/HY/2013, VN288/SL/2014, VN‐K23/HN/2015, VN‐K28/TB/2015 and VN‐Jafa/HB/2015, had the same sequence of the S gene, sharing 99.93% nucleotide (99.86% amino acid) identity with VN19/HY/2013 and 99.95% nucleotide (100.00% amino acid) identity with VN270/VP/2014. In comparison with the other reference strains, these 12 Vietnamese variants had the highest nucleotide identities (99.57%–99.64%) with USA/Colorado/2013, USA/Minnesota271/2014 and AOM‐2/JPN/2014. Among the Vietnamese strains in our study, VN232/2013 had the highest nucleotide (amino acid) identities with the vaccine strains, ranging from 94.51% to 94.85% (94.07%–94.43%).
3.3. Genetic analysis of the S gene
The sequence data revealed that the S genes of the Vietnamese field strains are 4,143–4,182 nucleotides in length (Table 1). The length of the S gene of Vietnamese PEDVs was more diverse than that of PEDVs identified in China (4,146–4,170 nucleotides) (Chen et al., 2013), Korea (4,161 nucleotides) (Kim, Lee et al., 2015), Japan and USA (4,152–4,161 nucleotides). Compared to the S gene (4,152 nucleotides) of prototype strain CV777, the S genes of most (18/25) Vietnamese strains were 9 nucleotides longer, consisting of 4,161 nucleotides, and the S gene of one field strain was 9 nucleotide shorter (4,143 nucleotides). The S genes of four field strains consisted of 4,152 nucleotides, that of one field strain was 6 nucleotides longer (4,158 nucleotides) and that of one field strain was 30 nucleotides longer (4,182 nucleotides). These changes were due to the presence of four deletions (three 3‐nucleotide deletions at positions 327–329, 401–403 and 3578–3580 and a 6‐nucleotide deletion at positions 464–469) and six insertions (three 3‐nucleotide insertions between positions 163 and 164, 172 and 173 and 401 and 402; a 9‐nucleotide insertion between positions 172 and 173; a 12‐nucleotide insertion between positions 170 and 171; and a 24‐nucleotide insertion between positions 1137 and 1138) accumulated in the S genes compared to that in CV777.
Although the S genes of 12 Vietnamese strains (VN06/HY/2013, VN11/HY/2013, VN15/HY/2013, VN44/QN/2013, VN101/HY/2012, VN112/HY/2013, VN288/SL/2014, VN‐K23/HN/2015, VN‐K28/TB/2015, VN‐Jafa/HB/2015 VN19/HY/2013 and VN270/VP/2014) in our study were 4,161 nucleotides in length, they encoded S proteins of 1,125 amino acids, which is 261 amino acids shorter than expected (1,386 amino acids). This consequence was due to one nucleotide substitution (C→T) at position 3376 (Fig. S3), leading to the formation of a stop codon (TAA) at positions 3,376–3,378 in the S gene. Likewise, the S gene of the field strain VN262/HB/2014 was 4,152 nucleotides in length but encoded a protein of 1,122 amino acids due to a similar nucleotide substitution at position 3367 of the S gene. The other Vietnamese PEDV strains in our study had S proteins of 1,380–1,393 amino acids. Compared to the reference strains and other Vietnamese strains, VN‐TH15/HY/2015 had a unique 8‐amino acid insertion (ITGSSTTP) between positions 380 and 381 of the S protein (Fig. S2). Correspondingly, at the nucleotide level, VN‐TH15/HY/2015 had a 24‐nucleotide insertion (CCATCACAGGCAGCAGCACGACTC) between positions 1137 (C) and 1138 (A) in the S gene compared to that in CV777.
Five Vietnamese strains in G2‐1 (except for VN297/HB/2014) have a 1‐amino acid deletion at position 1194 (Fig. S2). In comparison with the S protein of CV777, all Vietnamese strains in G2‐1 and G2‐2 examined in this study had insertions (a 4‐amino acid insertion between positions 55 and 56 and a 1‐amino acid insertion between positions 134 and 135) and a 2‐amino acid deletion at positions 158–159. These insertions and deletions were identical to those of previously reported strains isolated after 2010, in which the mutations were found in the hypervariable domain in the N‐terminus of the S protein (Kim, Lim et al., 2015; Temeeyasen et al., 2014; Vui, Tung, Inui, Slater, & Nilubol, 2014). In comparison with the sequence of the initial prototype strains in North America (high virulent NA prototype strains), the Vietnamese field strains VN262/HB/2014 and VN232/2013 possessed three deletions (a 1‐nucleotide deletion at position 166, an 11‐nucleotide deletion at positions 172–182 and a 3‐nucleotide deletion at positions 414–416) and a 6‐nucleotide insertion between positions 474 and 475 in the S gene. These insertions and deletion in the S gene are typical for the S INDEL type of PEDV (Vlasova et al., 2014; Lin et al., 2016). Of note, two classical Japanese strains in G1‐2 (NK and KH) do not possess these typical INDELs in the S gene. Therefore, these Japanese strains could be classified into classical non‐S INDEL type.
At least four epitope regions exhibiting neutralizing activities against PEDV have been identified in the S glycoprotein (Chang et al., 2002; Cruz, Kim, & Shin, 2008; Sun et al., 2008). These four regions include the COE domain (positions 499–638), SS2 (positions 748–755), SS6 (positions 764–771) and 2C10 (positions 1368–1374). Our amino acid analysis revealed that, in comparison with the vaccine strain KPEDV‐9, P5‐V, attenuated CV777 and attenuated DR13, all Vietnamese strains have amino acid substitutions at 19 positions in the COE domain (Figure 2). Particularly, compared to the vaccine strains, all Vietnamese strains (except VN262/HB/2014) have an amino acid substitution at position 549 (R→S or T→S) that produced an additional serine phosphorylation site as predicted using the NetPhos 3.1 and NetPhosBac 1.0 servers (Figure 2). The amino acid substitution at position 496 (I→T) found in five Vietnamese strains of G2‐1 and strains VN232/HB/2013 generated an additional threonine phosphorylation site as predicted using the NetPhos 3.1 Server. In comparison with the four vaccine strains, all Vietnamese PEDV strains had no change in epitopes SS2 and 2C10. For epitope SS6, compared to the vaccine strain KPEDV‐9, all Vietnamese strains had no changes. Compared to the vaccine strains P5‐V, attenuated CV777 and attenuated DR13, all Vietnamese PEDV strains had two different amino acids in epitope SS6 at positions 764 (L→S) and 766 (D→S). Notably, the amino acid substitution at position 766 resulted in the formation of a serine phosphorylation site in the S proteins of all Vietnamese strains as predicted using the NetPhos 3.1 service.
Figure 2.
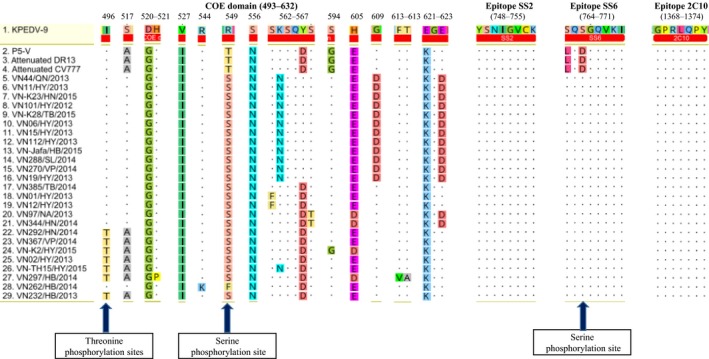
The amino acid difference in epitope regions of the S protein between Vietnamese isolates and four vaccine strains. The dots represent amino acids that are identical to those in strain KPEDV‐9. The four epitope regions in the S protein include the COE domain, SS2, SS6 and 2C10
Regarding high‐specificity N‐glycosylation, Vietnamese PEDV strains in our study have different patterns from each other and the vaccine strains (Table 2). The Vietnamese strains exhibited six to nine sites over the S protein, whereas the vaccine strain KPEDV‐9 exhibited seven sites. Three vaccine strains, namely P5‐V, attenuated CV777 and attenuated DR13, had the same pattern of high‐specificity N‐glycosylation, which occurred at eight sites. Compared to KPEDV‐9, all Vietnamese strains (except VN262/HB/2014 and VN232/HB/2013) lost one high‐specificity site at position 131 (NNTL/NKTN) but gained two additional sites at positions 62 (NSTW) and 118 (NATA /NTSA). Compared with P5‐V, attenuated CV777 and attenuated DR13, most Vietnamese strains lost three high‐specificity sites at positions 131 (NNTL/NKTN), 514 (NITV) and 566 (NVTN/NVTS) but gained three other sites at positions 62 (NSTW), 326 (NDTS) and 1271 (NRTG). Compared to all other strains, VN297/HB/2014 had a distinct high‐specificity N‐glycosylation site at position 873 (NISS) over the S protein.
Table 2.
The differences in the highly‐specific N‐glycosylation sites of the spike protein among the porcine epidemic diarrhoea virus of the emerging Vietnamese and vaccine strains
| No | Strain | Position | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 62 (NSTW) | 118 (NATA /NTSA) | 131 (NNTL/NKTN) | 218 (NVTS) | 326 (NDTS) | 353 (NSSN/NSSD) | 514 (NITV) | 566 (NVTN/NVTS) | 791 (NISI) | 873 (NISS) | 1259 (NKTL) | 1271 (NRTG) | ||
| 1 | VN06/HY/2013 | N | N | □ | N | N | N | □ | □ | N | □ | * | * |
| 2 | VN11/HY/2013 | N | N | □ | N | N | N | □ | □ | N | □ | * | * |
| 3 | VN15/HY/2013 | N | N | □ | N | N | N | □ | □ | N | □ | * | * |
| 4 | VN44/QN/2013 | N | N | □ | N | N | N | □ | □ | N | □ | * | * |
| 5 | VN101/HY/2012 | N | N | □ | N | N | N | □ | □ | N | □ | * | * |
| 6 | VN112/HN/2013 | N | N | □ | N | N | N | □ | □ | N | □ | * | * |
| 7 | VN‐K23/HN/2015 | N | N | □ | N | N | N | □ | □ | N | □ | * | * |
| 8 | VN‐K28/TB/2015 | N | N | □ | N | N | N | □ | □ | N | □ | * | * |
| 9 | VN‐Jafa/HB/2015 | N | N | □ | N | N | N | □ | □ | N | □ | * | * |
| 10 | VN288/SL/2014 | N | N | □ | N | N | N | □ | □ | N | □ | * | * |
| 11 | VN19/HY/2013 | N | N | □ | N | N | N | □ | □ | N | □ | * | * |
| 12 | VN270/VP/2014 | N | N | □ | N | N | N | □ | □ | N | □ | * | * |
| 13 | VN385/TB/2014 | N | N | □ | N | N | N | □ | □ | N | □ | N | N |
| 14 | VN01/HY/2013 | N | N | □ | N | N | N | □ | □ | N | □ | N | N |
| 15 | VN12/HY/2013 | N | N | □ | N | N | N | □ | □ | N | □ | N | N |
| 16 | VN97/HN/2013 | N | N | □ | N | N | N | □ | □ | N | □ | N | N |
| 17 | VN344/HN/2014 | N | N | □ | N | N | N | □ | □ | N | □ | N | N |
| 18 | VN297/HB/2014 | N | N | □ | N | N | N | □ | □ | N | N | N | N |
| 19 | VN292/HN/2014 | □ | N | □ | N | N | N | N | □ | N | □ | N | N |
| 20 | VN367/VP/2014 | □ | N | □ | N | N | N | N | □ | □ | □ | N | |
| 21 | VN‐K2/HY/2015 | □ | □ | □ | N | N | N | N | □ | N | □ | □ | N |
| 22 | VN‐TH15/HY/2015 | N | N | □ | N | N | N | N | □ | N | □ | □ | N |
| 23 | VN02/HY/2013 | N | N | □ | N | □ | N | N | □ | N | □ | N | N |
| 24 | VN262/HB/2014 | □ | □ | N | N | N | N | □ | N | N | □ | * | * |
| 25 | VN232/HB/2013 | □ | □ | N | N | N | N | N | □ | N | □ | N | N |
| 26 | KPEDV‐9 | □ | □ | N | N | N | N | □ | □ | N | □ | N | N |
| 27 | P5‐V | □ | N | N | N | □ | N | N | N | N | □ | N | □ |
| 28 | Attenuated CV777 | □ | N | N | N | □ | N | N | N | N | □ | N | □ |
| 29 | Attenuated DR13 | □ | N | N | N | □ | N | N | N | N | □ | N | □ |
The numbers identifying N‐glycosylation sites correspond to aa position within the consensus S protein sequence of the Vietnamese and Vaccine strains. N: N‐glycosylation is predicted to be formed at that site; □: No N‐glycosylation is predicted to be formed at the site; *: Prediction of N‐glycosylation was not applied at that site of the truncated S protein.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
4. DISCUSSION
Since 2010, PEDV has been reported to be responsible for severe outbreaks in Northern Vietnam. Subsequently, the virus has spread rapidly and become more complex. PED persistence and recurrent epidemics are often observed in PEDV‐infected herds. Outbreaks are also reported to occur throughout the year opposed to only in the cold season as previously reported. At present, the disease continues to widely circulate, becoming a major focus of farm owners. To better understand the prevailing PEDVs, full‐length S genes of PEDVs collected from different farms in Northern Vietnam were sequenced and analysed. The sequencing results revealed variations in length and various insertions and deletions in the S gene of Vietnamese strains. Sequence homology analysis of the S gene illustrated that the Vietnamese PEDVs were genetically diverse, displaying divergence in sequence identity with each other. Moreover, all Vietnamese strains were classified into four types corresponding to four subgroups. Taken together, these results indicated the heterogeneity among prevailing PEDVs in the northern provinces of Vietnam. They also suggested the need for multiple PEDV vaccine strains for preventing PED in Vietnam because the availability of vaccine strains that are genetically related to circulating viruses is critical (Song et al., 2015).
Prototype PEDV strains (NA non‐S INDEL type) were first identified in Iowa in the US at the end of April 2013 (Stevenson et al., 2013). Jarvis et al., (2016) reported that the evolution of US strains is estimated to have begun around July 2009 to August 2012. Although NA non‐S INDEL strains were proposed to have evolved from different emerging Chinese strains via recombination (Tian et al., 2014; Vlasova et al., 2014), no NA non‐S INDEL strains were isolated in China (Lin et al., 2016). The timing and mechanism of the appearance and global spread of the NA PEDV type remain unclear. In this study, we identified NA PEDV strains from PED outbreaks in South‐East Asia. Interestingly, some Vietnamese strains belonging to the NA type appeared earlier in Northern Vietnam than the earliest reported US strains. For example, VN101/HY/2012 was collected in December 2012. Although VN01/HY/2013 and VN12/HY/2013 were collected in January 2013, the strains were derived from two outbreaks that begin in December 2012. Therefore, the earliest PED outbreaks caused by NA strains in Northern Vietnam occurred as early as December 2012, approximately 4 months earlier than the first appearance of PEDV in the US. Additionally, sequence comparison and phylogenetic analysis indicated that the Vietnamese strains of the NA type were most similar to the emerging US strains. Whether the US‐like PEDVs in Vietnam evolved independently via recombination from the domestic PEDV strains or originated directly from China remains unanswered. These possibilities are both highly feasible. The possible reasons are first, the NA Vietnamese PEDV strains appeared closely with the estimated occurrence of NA emergence (2009–2012) as reported by Jarvis et al. and last, the existence of Chinese‐like emerging PEDV strains in Vietnam could mean potentially parent strains for recombination that generated the US‐like PEDVs. On the other hand, Northern Vietnam shares a long border with China, and the export of live pigs (finished stage) from Vietnam to China has been extremely active with weak oversight. Therefore, the transmission of new PEDV strains between the two countries is highly possible. Further study is needed to clarify the origin and evolution of the NA strain‐like PEDVs in Northern Vietnam.
In this study, the appearance of an early stop codon in the S gene was observed in 13 Vietnamese PEDVs. This resulted in the generation of a truncated S protein that was 261 amino acids shorter than expected. These 13 variants were collected from different pigs with diarrhoea. Therefore, it is suggested that these PEDV variants were infectious. Of note, among these variants, 10 of them collected during PED outbreaks occurring in six different provinces between 2012 and 2015 had the same S gene sequence. It suggested that the variants were likely transmitted from one source of origin. In our study, an earlier stop codon in the S gene was observed in both the NA PEDV type (12 variants) and S INDEL type (VN262/HB/2014). This suggested that the truncated S proteins in the Vietnamese variants resulted from recombination or mutation events having the same mechanism. Notably, the neutralizing epitope 2C10 was located in this deleted portion of the S protein. Thus, further study is needed to identify the differences in anti‐genicity and biological properties of the truncated S variants compared to the other PEDV strains.
In addition to the NA type, a Vietnamese strain (VN262/HB/2013) of the US‐like S INDEL type collected in October 2013 was reported for the first time in the present study. Phylogenetic and sequence analyses revealed that VN262/HB/2013 was clustered together with new S INDEL strains and shared the highest nucleotide sequence identity with the US strain OH851. Additionally, in the US and South Korea, where two US‐like strains (both NA non‐S INDEL and new S INDEL types) have been circulating since 2013, the outbreak‐causing PEDVs were believed to result from the introduction of multiple parental PEDV strains from abroad (Lee, 2015; Vlasova et al., 2014). Therefore, the presence of the new S INDEL type of PEDV in Northern Vietnam suggested a possibility that US‐like PEDV strains might have originated from Vietnam.
Two recent studies reported that five Vietnamese PEDV strains isolated in 2013 and early 2014 including VN/JFP1013_1/2013/Vinh An/Vietnam, VN/VAP1113_1/2013/Vung Tua/Vietnam, VN/KCHY‐310113/2013/KhoaiChau/HungYen/Vietnam (Vui et al., 2014), HUA‐PED45 and HUA‐PED47 2010 (Kim, Lim et al., 2015) were related to new Chinese variants that have emerged since 2010. The Vietnamese PEDV strain HUA‐14PED96 (accession number KT941120), which was identified in a Northern province, was reported to have a unique 72‐nucleotide deletion in the ORF1a gene (Choe et al., 2016). Consistent with the previous results, several Vietnamese PEDV strains in our study were genetically similar to the reported Vietnamese strains and re‐emerging Chinese strains. Four strains in our study, namely VN292/HN/2014, VN367/VP/2014, VN‐K2/HY/2015 and VN‐TH15/HY/2015, were phylogenetically similar to HUA‐PED45 and HUA‐PED47. Notably, the four Vietnamese strains collected from three adjacent provinces formed a well‐supported branch. These results indicated that these PEDV strains might have derived from a common ancestral virus, the progeny of which spread in this region. Interestingly, strain VN297/HB/2013 in our study was genetically most similar to HUA‐14PED96, and these two strains formed a monophyletic branch. This indicated that VN297/HB/2013 might also possess the same 72‐nucleotide deletion in ORF1a compared with HUA‐14PED96, although more extensive genome sequencing is required to clarify this finding. In this study, we identified one novel PEDV strain, VN‐TH15/HY/2015, with an 8‐amino acid insertion between positions 380 and 381 of the S protein, which are located in the receptor‐binding domain (amino acids 253–638) for porcine aminopeptidase N (Liu et al., 2015; Deng et al., 2016). It is revealed that the classical PEDV exhibited weaker sugar‐binding activity than the recently uncovered G2 isolates (Deng et al., 2016). Amino acid substitutions were also reported in the receptor‐binding region (Wang et al., 2016). Whether the 8‐amino acid insertion observed in VN‐TH15/HY/2015 influences the biological functions of PEDV requires further study.
Among the Vietnamese strains in G2‐1, homology and phylogenetic analyses revealed that strain VN02/HY/2013 collected in January 2013 was most closely related to the re‐emerging Japanese strain 13JM‐291 collected in late 2013 from an early outbreak in Kagoshima prefecture. Additionally, several US‐like strains (including NA and new S INDEL types) identified at the beginning of 2013 in Northern Vietnam were also most closely related to Japanese PEDV strains isolated in 2014. In addition, recent studies demonstrated that the re‐emerging PEDV outbreaks in Japan were caused by novel strains that were introduced from abroad (Suzuki et al., 2015; Van Diep et al., 2015). Therefore, the possibility that multiple parental PEDV strains introduced into Japan originated from Vietnam is quite notable. Considering this international dissemination route, we may recheck the quarantine process against PEDV and other epizootic agents.
In a previous study (Kim, Lim et al., 2015), five PEDV strains isolated in a central province (Quang Tri) of Vietnam were clustered into the S INDEL type of PEDV. Consistent with those data, in this study, we identified a strain (VN232/HB/2013) belonging to this type that was collected in a northern province (Hoa Binh) of Vietnam. The result indicated that this genotype of PEDV was distributed in both the northern and central regions of the country. These Vietnamese strains were most closely related to classical strains isolated in China (JS‐2004‐2, LJB03 and CH7) before 2010. Therefore, these Vietnamese strains were classified as the classical S INDEL type. The data also suggested that the classical S INDEL strains prevailing in Vietnam evolved from classical Chinese S INDEL strains.
Because of urgent demands, several PED vaccine strains produced in Asian countries such as P5‐V (Japan), attenuated CV777 (China) and attenuated DR13 (South Korea) were considered for import into Vietnam. Phylogenetic and sequence analyses revealed that the current PEDV strains in Vietnam were genetically distant from the aforementioned vaccine strains. Antigenic analysis illustrated that compared to the vaccine strains, the circulating Vietnamese PEDVs were conserved in epitopes SS2 and 2C10 but had various substitutions in the COE domain and epitope SS6. In particular, substitutions in three positions, namely 496, 549 and 766 in the epitope regions of the S protein, were predicted to result in the formation of new phosphorylation sites. Phosphorylation of viral proteins is a post‐translational modification which can have major impacts on viral infection, survival, replication and cytotoxicity in a host cell (Keating & Striker, 2012). Phosphorylation is considered to introduce a change in the charge of the protein which results in a conformational change and alters the activation status of the protein (Keck, Ataey, Amaya, Bailey, & Narayanan, 2015). Therefore, the result suggested that these mutations in the epitope regions may lead to changes in the antigenicity of prevailing Vietnamese PEDVs and consequently influence the efficacy of the vaccines.
Along with phosphorylation, other post‐translational modifications such as N‐glycosylation often strongly affect protein function. The changes in N‐glycosylation sites might influence the survival and transmission of the virus. They also play a major role in interactions with receptors and result in interference with virus recognition by the immune system of the host, therefore, influencing viral replication and infectivity (Meunier et al., 1999; Vigerust & Shepherd, 2007). A study of a lactate dehydrogenase‐elevating virus belonging to Coronaviridae reported that the acquisition or loss of N‐glycosylation sites in an envelope glycoprotein (VP‐3P) could result in changes in virulence and cellular tropism (Li et al., 2000). N‐glycosylation sites in the S gene of severe acute respiratory syndrome coronavirus were demonstrated to be crucial to virus infection (Han, Lohani, & Cho, 2007). In our study, variations in high‐specificity N‐glycosylation sites were found among the prevailing Vietnamese PEDVs as well as between the Vietnamese strains and vaccine strains. In particular, the high‐specificity sites 514 (NITV) and 566 (NVTN/NVTS) were promising markers of virus evolvement, as they are located in the COE region, meaning that small changes could possibly result in disturbances in the folding and conformation of the S protein. The substitution in N‐glycosylation sites also suggested the possibility of anti‐genic modifications of the S proteins in the emerging Vietnamese strains compared to those of the vaccine strains.
In conclusion, there are at least four genotypes of the PEDVs circulating in northern provinces of Vietnam. This study reported the prevalence of US‐like strains in South‐East Asia, and in particular, the NA type had already existed in Northern Vietnam before its first appearance in the US. Compare to the vaccine strains, Vietnamese PEDVs were genetically distant and had a variation in the epitope regions as well as N‐glycosylation sites in the S proteins. The development of novel vaccines based on the circulating Vietnamese strains may be necessary to protect pigs against the emerging PEDV strains. Our present study also provides useful insights into the molecular epidemiology of PED that could improve disease control in the future.
CONFLICT OF INTEREST
The authors declare no conflict of interests.
Supporting information
{kind=link}
ACKNOWLEDGEMENTS
The work was supported by grants from The Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan for global human resource and prevention of livestock epidemics by the Center for Animal Disease Control, University of Miyazaki (2014–2015).
Diep NV, Sueyoshi M, Izzati U, et al. Appearance of US‐like porcine epidemic diarrhoea virus (PEDV) strains before US outbreaks and genetic heterogeneity of PEDVs collected in Northern Vietnam during 2012–2015. Transbound Emerg Dis. 2018;65:e83–e93. 10.1111/tbed.12681
REFERENCES
- Bosch, B. J. , van der Zee, R. , de Haan, C. A. , & Rottier, P. J. (2003). The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. Journal of Virology, 77, 8801–8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, S. H. , Bae, J. L. , Kang, T. J. , Kim, J. , Chung, G. H. , Lim, C. W. , … Jang, Y. S. (2002). Identification of the epitope region capable of inducing neutralizing antibodies against the porcine epidemic diarrhea virus. Molecules and Cells, 14, 295–299. [PubMed] [Google Scholar]
- Chen, J. , Liu, X. , Shi, D. , Shi, H. , Zhang, X. , Li, C. , … Feng, L. (2013). Detection and molecular diversity of spike gene of porcine epidemic diarrhea virus in China. Viruses, 5, 2601–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou, H. Y. , Huang, Y. L. , Deng, M. C. , Chang, C. Y. , Jeng, C. R. , Tsai, P. S. , … Chang, H. W. (2015). Phylogenetic analysis of the spike (S) gene of the new variants of porcine epidemic diarrhoea virus in Taiwan. Transboundary and Emerging Diseases, 64, 157–166. [DOI] [PubMed] [Google Scholar]
- Choe, S. E. , Park, K. H. , Lim, S. I. , Le, V. P. , Hien, N. B. , Thach, P. N. , … An, D. J. (2016). Complete genome sequence of a porcine epidemic diarrhea virus strain from Vietnam, HUA‐14PED96, with a large genomic deletion. Genome Announcements, 4, e0002–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz, D. J. , Kim, C. J. , & Shin, H. J. (2008). The GPRLQPY motif located at the carboxy‐terminal of the spike protein induces antibodies that neutralize Porcine epidemic diarrhea virus. Virus Research, 132, 192–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, F. , Ye, G. , Liu, Q. , Navid, M. T. , Zhong, X. , Li, Y. , … Peng, G. (2016). Identification and comparison of receptor binding characteristics of the spike protein of two porcine epidemic diarrhea virus strains. Viruses, 8, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diep, N. V. , Lan, N. T. , Hoa, N. T. , & Yamaguchi, R. (2014). Epidemiological and pathologic characteristics of Porcine Epidemic Diarrhea outbreaks in Northern provinces of Vietnam (in Vietnamese). Vietnamese Journal of Veterinary Sciences and Techniques, 02, 43–56. [Google Scholar]
- Diep, N. V. , Norimine, J. , Sueyoshi, M. , Lan, N. T. , & Yamaguchi, R. (2017). Novel porcine epidemic diarrhea virus (PEDV) variants with large deletions in the spike (S) gene coexist with PEDV strains possessing an intact S gene in domestic pigs in Japan: A new disease situation. PLoS One, 12, e0170126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duy, D. T. , Toan, N. T. , Puranaveja, S. , & Thanawongnuwech, R. (2011). Genetic characterization of porcine epidemic diarrhea virus (PEDV) isolates from southern Vietnam during 2009‐2010 outbreaks. The Thai Journal of Veterinary Medicine, 41, 55–64. [Google Scholar]
- Han, D. P. , Lohani, M. , & Cho, M. W. (2007). Specific asparagine‐linked glycosylation sites are critical for DC‐SIGN‐ and L‐SIGN‐mediated severe acute respiratory syndrome coronavirus entry. Journal of Virology, 81, 12029–12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis, M. C. , Lam, H. C. , Zhang, Y. , Wang, L. , Hesse, R. A. , Hause, B. M. , … Marthaler, D. (2016). Genomic and evolutionary inferences between American and global strains of porcine epidemic diarrhea virus. Preventive Veterinary Medicine, 123, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating, J. A. , & Striker, R. (2012). Phosphorylation events during viral infections provide potential therapeutic targets. Reviews in Medical Virology, 22, 166–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck, F. , Ataey, P. , Amaya, M. , Bailey, C. , & Narayanan, A. (2015). Phosphorylation of single stranded RNA virus proteins and potential for novel therapeutic strategies. Viruses, 7, 5257–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. H. , Lee, J. M. , Jung, J. , Kim, I. J. , Hyun, B. H. , Kim, H. I. , … Lee, K. K. (2015). Genetic characterization of porcine epidemic diarrhea virus in Korea from 1998 to 2013. Archives of Virology, 160, 1055–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y. K. , Lim, S. I. , Lim, J. A. , Cho, I. S. , Park, E. H. , Le, V. P. , … An, D. J. (2015). A novel strain of porcine epidemic diarrhea virus in Vietnamese pigs. Archives of Virology, 160, 1573–1577. [DOI] [PubMed] [Google Scholar]
- Kocherhans, R. , Bridgen, A. , Ackermann, M. , & Tobler, K. (2001). Completion of the porcine epidemic diarrhoea coronavirus (PEDV) genome sequence. Virus Genes, 23, 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kweon, C. H. , Kwon, B. J. , Jung, T. S. , Kee, Y. J. , Hur, D. H. , Hwang, E. K. , … An, S. H. (1993). Isolation of porcine epidemic diarrhea virus (PEDV) in Korea. Korean Journal of Veterinary Research, 33, 249–254. [Google Scholar]
- Lee, C. (2015). Porcine epidemic diarrhea virus: An emerging and re‐emerging epizootic swine virus. Virology Journal, 12, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, D. K. , Park, C. K. , Kim, S. H. , & Lee, C. (2010). Heterogeneity in spike protein genes of porcine epidemic diarrhea viruses isolated in Korea. Virus Research, 149, 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, K. , Schuler, T. , Chen, Z. , Glass, G. E. , Childs, J. E. , & Plagemann, P. G. (2000). Isolation of lactate dehydrogenase‐elevating viruses from wild house mice and their biological and molecular characterization. Virus Research, 67, 153–162. [DOI] [PubMed] [Google Scholar]
- Lin, C. N. , Chung, W. B. , Chang, S. W. , Wen, C. C. , Liu, H. , Chien, C. H. , & Chiou, M. T. (2014). US‐like strain of porcine epidemic diarrhea virus outbreaks in Taiwan, 2013‐2014. Journal of Veterinary Medical Science, 76, 1297–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C. M. , Saif, L. J. , Marthaler, D. , & Wang, Q. (2016). Evolution, antigenicity and pathogenicity of global porcine epidemic diarrhea virus strains. Virus Research, 226, 20–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C. , Tang, J. , Ma, Y. , Liang, X. , Yang, Y. , Peng, G. , … Li, F. (2015). Receptor usage and cell entry of porcine epidemic diarrhea coronavirus. Journal of Virology, 89, 6121–6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier, J. C. , Fournillier, A. , Choukhi, A. , Cahour, A. , Cocquerel, L. , Dubuisson, J. , & Wychowski, C. (1999). Analysis of the glycosylation sites of hepatitis C virus (HCV) glycoprotein E1 and the influence of E1 glycans on the formation of the HCV glycoprotein complex. Journal of General Virology, 80(Pt 4), 887–896. [DOI] [PubMed] [Google Scholar]
- Oka, T. , Saif, L. J. , Marthaler, D. , Esseili, M. A. , Meulia, T. , Lin, C. M. , … Wang, Q. (2014). Cell culture isolation and sequence analysis of genetically diverse US porcine epidemic diarrhea virus strains including a novel strain with a large deletion in the spike gene. Veterinary Microbiology, 173, 258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pensaert, M. B. , & de Bouck, P. (1978). A new coronavirus‐like particle associated with diarrhea in swine. Archives of Virology, 58, 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puranaveja, S. , Poolperm, P. , Lertwatcharasarakul, P. , Kesdaengsakonwut, S. , Boonsoongnern, A. , Urairong, K. , … Thanawongnuwech, R. (2009). Chinese‐like strain of porcine epidemic diarrhea virus, Thailand. Emerging Infectious Diseases, 15, 1112–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, T. , Takeyama, N. , Katsumata, A. , Tuchiya, K. , Kodama, T. , & Kusanagi, K. (2011). Mutations in the spike gene of porcine epidemic diarrhea virus associated with growth adaptation in vitro and attenuation of virulence in vivo. Virus Genes, 43, 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, D. , Moon, H. , & Kang, B. (2015). Porcine epidemic diarrhea: A review of current epidemiology and available vaccines. Clinical and Experimental Vaccine Research, 4, 166–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, D. , & Park, B. (2012). Porcine epidemic diarrhoea virus: A comprehensive review of molecular epidemiology, diagnosis, and vaccines. Virus Genes, 44, 167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson, G. W. , Hoang, H. , Schwartz, K. J. , Burrough, E. R. , Sun, D. , Madson, D. , … Yoon, K. J. (2013). Emergence of Porcine epidemic diarrhea virus in the United States: Clinical signs, lesions, and viral genomic sequences. Journal of Veterinary Diagnostic Investigation, 25, 649–654. [DOI] [PubMed] [Google Scholar]
- Sun, D. , Feng, L. , Shi, H. , Chen, J. , Cui, X. , Chen, H. , … Tong, G. (2008). Identification of two novel B cell epitopes on porcine epidemic diarrhea virus spike protein. Veterinary Microbiology, 131, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, D. , Wang, X. , Wei, S. , Chen, J. , & Feng, L. (2016). Epidemiology and vaccine of porcine epidemic diarrhea virus in China: A mini‐review. Journal of Veterinary Medical Science, 78, 355–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, T. , Murakami, S. , Takahashi, O. , Kodera, A. , Masuda, T. , Itoh, S. , … Tsutsui, T. (2015). Molecular characterization of pig epidemic diarrhoea viruses isolated in Japan from 2013 to 2014. Infection, Genetics and Evolution, 36, 363–368. [DOI] [PubMed] [Google Scholar]
- Takahashi, K. , Okada, K. , & Ohshima, K. (1983). An outbreak of swine diarrhea of a new‐type associated with coronavirus‐like particles in Japan. Nihon Juigaku Zasshi, 45, 829–832. [DOI] [PubMed] [Google Scholar]
- Tamura, K. , Stecher, G. , Peterson, D. , Filipski, A. , & Kumar, S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution, 30, 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temeeyasen, G. , Srijangwad, A. , Tripipat, T. , Tipsombatboon, P. , Piriyapongsa, J. , Phoolcharoen, W. , … Nilubol, D. (2014). Genetic diversity of ORF3 and spike genes of porcine epidemic diarrhea virus in Thailand. Infection, Genetics and Evolution, 21, 205–213. [DOI] [PubMed] [Google Scholar]
- Tian, P. F. , Jin, Y. L. , Xing, G. , Qv, L. L. , Huang, Y. W. , & Zhou, J. Y. (2014). Evidence of recombinant strains of porcine epidemic diarrhea virus, United States, 2013. Emerging Infectious Diseases, 20, 1735–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Diep, N. , Norimine, J. , Sueyoshi, M. , Lan, N. T. , Hirai, T. , & Yamaguchi, R. (2015). US‐like isolates of porcine epidemic diarrhea virus from Japanese outbreaks between 2013 and 2014. Springerplus, 4, 756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigerust, D. J. , & Shepherd, V. L. (2007). Virus glycosylation: Role in virulence and immune interactions. Trends in Microbiology, 15, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlasova, A. N. , Marthaler, D. , Wang, Q. , Culhane, M. R. , Rossow, K. D. , Rovira, A. , … Saif, L. J. (2014). Distinct characteristics and complex evolution of PEDV strains, North America, May 2013‐February 2014. Emerging Infectious Diseases, 20, 1620–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vui, D. T. , Tung, N. , Inui, K. , Slater, S. , & Nilubol, D. (2014). Complete genome sequence of porcine epidemic diarrhea virus in Vietnam. Genome Announcements, 2, e00753–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, E. , Guo, D. , Li, C. , Wei, S. , Wang, Z. , Liu, Q. , … Sun, D. (2016). Molecular characterization of the ORF3 and S1 genes of porcine epidemic diarrhea virus non S‐INDEL strains in seven regions of China, 2015. PLoS One, 11, e0160561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood, E. N. (1977). An apparently new syndrome of porcine epidemic diarrhoea. Veterinary Record, 100, 243–244. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials