

Abstract
BACKGROUND
Red blood cell (RBC) transfusion risks could be reduced if a robust technology for pathogen inactivation of RBC (PI‐RBCs) were to be approved.
MATERIALS AND METHODS
Estimates of per‐unit and per‐patient aggregate infectious risks for conventional RBCs were calculated; the latter used patient diagnosis as a determinant of estimated lifetime exposure to RBC units. Existing in vitro data for the two technologies under development for producing PI‐RBCs and the status of current clinical trials are reviewed.
RESULTS
Minimum and maximum per‐unit risk were calculated as 0.0003% (1 in 323,000) and 0.12% (1 in 831), respectively. The minimum estimate is for known lower‐risk pathogens while the maximal estimate also includes an emerging infectious agent (EIA) and endemic area Babesia risk. Minimum and maximum per‐patient lifetime risks by diagnosis grouping were estimated as 1.5 and 3.3%, respectively, for stem cell transplantation (which includes additional risk for cytomegalovirus transmission); 1.2 and 3.7%, respectively, for myelodysplastic syndrome; and 0.2 and 44%, respectively, for hemoglobinopathy.
DISCUSSION
There is potential for PI technologies to reduce infectious RBC risk and to provide additional benefits (e.g., prevention of transfusion‐associated graft‐versus‐host disease and possible reduction of alloimmunization) due to white blood cell inactivation. PI‐RBCs should be viewed in the context of having a fully PI‐treated blood supply, enabling a blood safety paradigm shift from reactive to proactive. Providing insurance against new EIAs. Further, when approved, the use of PI for all components may catalyze operational changes in blood donor screening, laboratory testing, and component manufacturing.
ABBREVIATIONS
- EIA(s)
emerging infectious agent(s)
- GSH
glutathione
- HSCT
hematopoietic stem cell transplantation
- LR
leukoreduced
- MDS
myelodysplastic syndrome
- PI
pathogen inactivation
- SCD
sickle cell disease
- TA‐GVHD
transfusion‐associated graft‐versus‐host disease
- TT‐CMV
transfusion‐transmitted cytomegalovirus
- TTB
transfusion‐transmitted babesiosis
- WB
whole blood
Although transfusion safety has increased greatly, risks are still associated with red blood cell (RBC) transfusion. These could be reduced if a robust technology was approved for pathogen inactivation of RBCs (PI‐RBCs) by applying the PI technology either to the RBC component or to the parent whole blood (WB) unit. In a recent risk/benefit publication on PI platelets (PLTs) prepared using the Intercept system,1 we emphasized that such an analysis should calculate benefits and risks on a per‐patient rather than a per‐unit basis. However, because this is a more difficult task for RBCs, we used a combined approach of calculating a risk per RBC unit and, when possible, a per‐patient risk. Since the availability of PI‐RBCs would result in the potential to transfuse a full complement of PI blood components (RBC, PLTs, plasma), we also discuss operational benefits that could be achieved under a full PI scenario.
CATEGORIZING RBC RECIPIENTS AND ESTIMATING NUMBER OF TRANSFUSED RBC UNITS
The 2011 HHS National Blood Collection and Utilization Survey estimated a mean per‐patient RBC dose of 2.75 units annually,2 and a 5‐year retrospective study in a regional hospital system reported a mean of 2.9 (±2.7) RBC units per transfused inpatient.3 However, there is substantial interpatient variability in RBC units transfused due to clinical diagnoses of the patient, the indication for transfusion, long established physician practice patterns, and the presence or absence of patient blood management programs.
Figure 1 provides a theoretical schema for understanding a recipient's risk of acquiring a transfusion‐transmitted infection, which is dependent on two factors: the number of units transfused (e.g., a higher risk with more units) and whether transfusion occurs when an undetected emerging infectious agent (EIA) is in the blood supply. This latter time‐related risk is higher for recipients whose transfusion exposure spans a longer time interval.1, 4 Factors relevant to clinical outcome of a transfusion‐transmitted infection include the expected length of recipient survival due to underlying disease and the increased susceptibility of different patient populations (based on their degree of immunosuppression) to adverse clinical outcomes secondary to infectious disease transmission.5 Thus, a logical way to categorize RBC recipients is both by number of units transfused and by the time interval over which transfusions occur. In Table 1, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 which forms the basis of a per‐patient risk analysis for selected patient groups, we synthesized existing RBC usage and transfusion practice data for illustrative diagnoses into a five‐tiered classification scheme based on acute (single transfusion episode), intermittent (multiple episodes), or chronic (often lifetime) RBC transfusion therapy.
Figure 1.
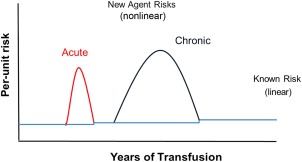
Quantitating transfusion risk over time. In the absence of additional interventions, known per‐unit infectious risks are consistent over time. These risks change when an EIA enters the blood supply. The figure indicates two types of EIAs: an acute agent and a chronic agent.* A past example of an acute EIA is West Nile virus and a past example of a chronic EIA is HIV. In contrast to known agents, EIA risks will vary over time. The intervals between an acute or chronic EIA entering the blood supply and the application of a successful intervention for that agent have been estimated as 1.5 and 5 years, respectively. After recognition of the EIA and development of a screening test, the risk from that agent will be decreased but a small residual risk will remain, thereby slightly increasing the overall per‐unit risk above the previous level. This is indicated (though not to scale) by the stepwise increase in the horizontal line. *An acute agent is present only transiently (usually days to weeks) until the donor resolves the viremia or parasitemia. In contrast, the donor retains the chronic agent in their blood for many years (perhaps an entire lifetime) while remaining asymptomatic and capable of blood donation.
Table 1.
Patients receiving RBC transfusions get exposed to different numbers of RBC units with different time frames of exposurea
| RBC transfusion category | Diagnosis or procedure | Number of transfusion episodes | Total RBC unit exposureb (time) | Immune suppressed | Use of irradiated blood |
|---|---|---|---|---|---|
| Acute | Cardiac surgery6, 7 | Single | 3c | No | No |
| Acute | Trauma8 | Single | 5c | Suppressed cell immunity | No |
| Intermittent | ICU9 | Variable | 3.5c | No | No |
| Intermittent | Cardiovascular disease10 | Variable | 3c | No | No |
| Sustained over limited time frame | HSCT11, 12 | Multiple | 10‐20 (3‐6 months) | Yes | Yes |
| Chronic but time‐limited | MDS13 | Multiple |
13/year (3 years) |
Immunosuppressed in many cases | Nod |
| Chronic, lifelong |
SCD14 Thalassemia17 |
Multiple | 24c/year (30 years15, 16) 15/year (50 years18, 19) |
Asplenic No |
Nod |
These data are taken from representative publications for each RBC transfusion category and may not be fully reflective of all practice patterns. Depending on how the data were presented in the cited publication(s), they are expressed as a median, mean, or range thereof.
The data include only the patients who received transfusions.
Median.
Not routinely; may be irradiated if hospital‐wide policies for hematology‐oncology patients or for pediatric patients require.
RISK REDUCTION BY PI
Infectious risks
Exclusively or predominantly from RBC transfusion
Babesiosis is a malaria‐like illness transmitted by infected ticks and by transfusion. In healthy persons, infection is generally asymptomatic or mild and transient. However, clinically severe and even fatal disease has occurred in at‐risk ill populations, especially patients who are immunosuppressed.20, 21
Babesia spp. are intraerythrocytic protozoan parasites. B. microti is the primary agent of babesiosis in the United States and is highly endemic in the Northeast (Connecticut, Massachusetts, New Jersey, New York, Rhode Island) and the Upper Midwest (Minnesota and Wisconsin), areas that include 16% of the US population.22 Recently, geographic expansion has been reported to neighboring states.22, 23 In the absence of surveillance in all 50 states, additional geographic areas where Babesia transmissions occur may go unrecognized.
A comprehensive CDC review reported 162 transfusion‐transmitted babesiosis (TTB) cases (28 fatalities) in the United States between 1979 and 2009.20 Since cases were compiled by passive surveillance, this is very likely an underestimate. B. microti was the agent in 159 cases while three involved B. duncani. All but four cases were from RBC units with transmission occurring throughout the storage period. Almost 80% were reported from 1999 to 2009; whether this represents increased transfusion transmission or lack of recognition of past cases or both is not known. Although nearly 90% of cases were in the seven highly endemic states, cases also occurred in nonendemic states.20, 22 These were attributed to shipment of RBC units from an endemic to a nonendemic area, a donor from an endemic area donating while visiting a nonendemic area, or a nonendemic area donor having acquired the infection while visiting an endemic area.
There are no FDA‐licensed blood donor screening assays. Recently, B. microti donor screening using serologic (automated immunofluorescence or enzyme‐linked immunosorbent assay) and nucleic acid test (NAT) assays under an investigational new drug procedure has been ongoing on a portion of the inventory in several states.24, 25, 26, 27 Seroprevalence in these states ranged between 0.4 and 1.2% (17% being polymerase chain reaction [PCR] positive) and 1 in 10,000 donors showed PCR positivity without antibody.27 Although PCR positivity increases the risk of transfusion transmission, PCR‐negative but seropositive units may also transmit; furthermore low‐level parasitemia or infectivity may be intermittent over a period of months to a year.27, 28, 29 Whereas there have been no TTB cases from Babesia‐negative RBC units, the TTB rate in these regions from unscreened units has ranged from 1 in 20,000 to 1 in 31,000 over the same time interval.22
These data suggest that a dual serology and NAT approach is needed to maximize risk reduction.26, 27 Given that B. microti is a mostly intracellular organism, this would require PCR testing of cellular material, which is logistically more challenging than the plasma NAT performed for other pathogens. Further, since Babesia infection is regional, a blood center's impetus to screen donations upon FDA licensure is likely to vary. In nonendemic areas, reluctance may be due to concerns about adding unnecessary cost for little safety gain and the loss of donors due to false‐positive test results.30 Finally, there is no legal or ethical precedent or model for regional screening of the US blood supply. These issues could be made moot by the use of PI‐RBCs.
Transfusion‐transmitted malaria is rare in the United States with an annual incidence of less than one case.31 The major mechanism to reduce risk is the use of multiple predonation questions, including travel to a malaria‐endemic area. Due to poor specificity for detecting malarial infections, large numbers of individuals who are not infected with malaria are deferred, thereby impacting blood availability.32 Furthermore, errors in eliciting travel history lead to a large number of biologic deviation reports to FDA, which has a negative impact on blood operations and staff productivity. The introduction of a robust PI‐RBC method that can inactivate all Plasmodium species in all their intra‐ and extracellular forms may allow elimination of the nonspecific and complex donor questioning used in the United States and eliminate malarial antibody testing that is used in many countries to shorten the deferral period.
Agents transmitted by RBCs or other components
Sepsis resulting from RBC bacterial contamination is rare but does occur. In France, this occurred at a rate of 1 per 2.6 million transfused RBC units from 2000 to 2008;33 the rate in Germany was 1 in 1.9 million over a similar time frame.34, 35 In France, all seven septic cases were caused by Gram‐negative bacteria, but in only a single case was the isolate (Yersinia enterocolitica) one that is commonly considered to be psychrophilic.33 In Germany, multiple species of Gram‐positive and Gram‐negative organisms were reported.34, 35 In the United States, no RBC‐mediated fatalities due to bacterial sepsis have been reported to the FDA in the past 5 years.36
Analogous to PLTs, it is likely that RBC bacterial contamination occurs more frequently than clinically detected sepsis.37 As demonstrated in Table 2, 7% to 15% of RBC cocomponents associated with bacterially contaminated WB‐derived PLTs contain bacteria; these data justify the policy of quarantining and either discarding or culturing RBC units associated with culture‐positive PLT pools. PI‐RBCs may prevent RBC‐mediated Gram‐negative sepsis as well as any potential deleterious effects from transfused Gram‐positive bacteria.33, 38, 39, 40, 41
Table 2.
Bacterial contamination rates for WB‐derived PC and associated results or disposition of RBC cocomponents
| Period | Country (PC pool size) | Number of pools tested | Bacterial incidencea/106 | PLT pools results | RBC cocomponent results or disposition |
|---|---|---|---|---|---|
| 2000‐200833 | France (4/5) | 320,000 | 25 | 6 Gram‐positive/2 Gram‐negative (1 death) | NA |
| 2008‐201138 | United States (2‐6) | 70,867 | 99 | 7 (+) pools by POC test |
1/7 (15%) RBCs (+) (CoNS); Culture‐negative RBCs transfused |
| 2003‐201039 | Wales (4) | 37,594 | 771 | 29 (+) pools (116 units) |
7/105 (7%) RBCs; 7 Gram‐positive |
| 2005‐201040 | Canada (4 [BC]; 5 [PRP]) | 228,142 (BC) (51,151 [PRP]) | 127 176 | 29 BC and 9 PRP culture (+) | NA |
| 200841 | United States (5) | 20,275 | 965 | 20 culture (+) | 130 RBC units retrieved and discardedb |
This refers to bacterial contamination and is not a measure of clinical sepsis.
The 130 RBC units were not cultured.
BC = buffy coat; NA = no data reported; PRP = platelet rich plasma.
Transfusion‐transmitted cytomegalovirus (TT‐CMV) can be a serious medical complication in specific immunosuppressed populations such as CMV‐seronegative hematopoietic stem cell transplantation (HSCT) recipients.42, 43 Strategies to reduce TT‐CMV include the use of leukoreduced (LR) cellular products or CMV serology testing or both.42, 43 Despite these strategies, there is consensus that TT‐CMV residual risk persists. As shown in Fig. 2, several recent transfusion transmission and/or donor‐based PCR studies indicate that per‐unit risk is approximately 0.1%.42, 43, 44, 45, 46, 47, 48 A recent editorial indicated that, in the absence of PI, complex testing algorithms would be needed to reduce this residual CMV risk and would result in substantial loss of transfusable RBC units.42
Figure 2.
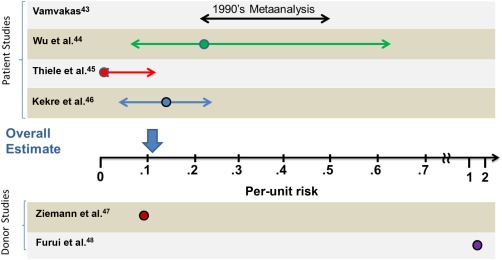
CMV risk: historical data and recent studies. The graph depicts the per‐unit risk as quantified in the different publications. The circles indicate the mean values. Patient studies are grouped above and donor studies are depicted under the x‐axis. The length of the arrows corresponds to the 95% confidence intervals, when reported, or high and low estimates. The overall estimate is depicted with the vertical arrow above the x‐axis and takes into account that only the approximately 50% of patients who are CMV seronegative are at risk for acquiring TT‐CMV.
Anaplasma phagocytophilum, the agent of human granulocytic anaplasmosis, is an intracellular, Gram‐negative bacterium with neutrophil tropism.49, 50 Nine transfusion‐transmitted cases have been reported; in seven cases, the implicated blood product was an RBC.49, 51 Transmission has occurred with both LR and non‐LR RBC units; in one case the unit had been stored for 30 days. Eight of the cases were reported since 2007, indicating an incidence of about one case annually.49, 51 Similar to B. microti, these observations illustrate how increased scrutiny may uncover a pathogen prevalence and level of risk that had previously escaped the attention of public health and transfusion medicine specialists. Currently, there is no blood donor screening for this agent.
EIAs
Table 3 contains risk estimates for a theoretical EIA entering the US blood supply as well as recent data for EIAs detected in specific non‐US and US locations.4, 52 ‐ 56The recent chikungunya epidemic in the Caribbean, including the US territory of Puerto Rico with some autochthonous cases in Florida,57 validate the model and indicate that acute agents can rapidly materialize and not be limited to traditional geographical boundaries. Currently for dengue and chikungunya, risk is posed by donors with recent travel to “epidemic” regions, as illustrated by the very high estimated peak incidence of chikungunya in La Reunion (1500 per 105 donations)53, 58 and Thailand (38.2‐52.3 per 105 donations).52, 59 This has prompted travel‐related donor deferrals in some European and Asian countries, with similar policies under consideration elsewhere.60 However, such deferrals have inherent nonspecificity and may require periodic revision to account for new epidemics. Extrapolating from data showing the effectiveness of PLT and plasma PI61, 62 against these agents, a reasonable assumption is that PI‐RBC technology will also be effective. If so, application of PI to all components could obviate the need for EIA‐related travel deferral.
Table 3.
Calculated and actual prevalence of EIAs
| Prevalence in blood donations (%) | ||
|---|---|---|
| Chronic agent | Acute agent | |
| Model EIA4 (range) | 0.045 (0.01‐0.08) | 0.025 (0.007‐0.075) |
| CHIKV | 0.038‐0.052 (Thailand) 52 0.4 (Reunion)53, 54 | |
| DENV | 0.07 mean;55 0.45 max | |
| HEV | 0.01 (US);a 0.035 (UK)56 | |
S.L. Stramer, personal communication, 2015.
Overall RBC infectious risks without PI
Per‐unit risks for individual pathogens are summarized in Tables 4 and 5,1, 27, 33, 46, 50, 51, 63, 64, 65 which also provide an aggregate per‐unit risk, expressed as a minimum (0.00031%, based on lower risk pathogens) and a maximum (0.12031%, representing composite risk due to Babesia in an endemic area, an acute EIA, and lower risk pathogens). These risks are increased by 0.1% for HSCT patients due to their susceptibility to CMV transmission. Table 6 combines aggregate per‐unit risk estimates from Table 5 with the number of transfused units from Table 1 to calculate a minimum and maximum per‐recipient risk for different patient categories.66 At the minimum risk levels, it is estimated that 1 in 67 HSCT recipients will acquire an infection during their period of intensive posttransplant transfusion support and that approximately 1 in 450 (0.22%) patients with hemoglobinopathies will do so over their entire course of transfusion therapy (approx. 30‐50 years). At the maximum levels, infectious risk increases to 43% to 45% (1 in 2) for hemoglobinopathy patients. Risk is also high for patients with myelodysplastic syndrome (MDS; 1 in 27) and HSCT recipients (1 in 30) and is not insignificant (1 in 400 to 1 in 150) for other categories of patients who receive fewer units.
Table 4.
Per unit risk in transfused RBC under current donor testing protocols in the United States
| Pathogen | Risk | Method of estimation |
|---|---|---|
| Higher‐risk pathogens | ||
| B. microti 27 |
0.076% (1 in 1316) |
Antibody and PCR data in endemic areasaunder IND screeningb |
| CMV1, 46 |
0.1% (1 in 1000)a |
Detection of infection in transfused recipients and PCR data in donors |
| EIA | ||
| Acute‐type agent4 |
0.025% (1 in 4000) |
Mathematical modelingc |
| Chronic‐type agent4 |
0.045% (1 in 2222) |
Mathematical modelingc |
| Lower‐risk pathogens | ||
| Plasmodia—all species | Rare | Clinical case reporting (<1 TT case per year in United States) |
| Bacteria33 |
0.00005% (1 in 2 million) Clinical Sepsis |
Based on French and German Data No documented clinical cases in the United States in past 5 years; May be more common for subclinical cases |
| A. phagocytophilum 50, 51 | Rare |
Clinical case reporting (<1 TT case per year in United States); May be more common for subclinical cases |
| HIV63 |
0.00007% (1 in 1.5 million) |
Mathematical modelingd |
| HCV63 |
0.00009% (1 in 1.1 million) |
Mathematical modelingd |
| HBV64 |
0.0001% (1 in 1 million) |
Mathematical modelingd |
| WNV65 | Rare | Clinical case reporting (<1 TT case per year in United States) |
Rare in nonendemic areas.
Assumes that all PCR‐positive donations, regardless of antibody status, would be infectious.
Using data from previously detected EIAs.
Using NAT donor screening data and a window period model.
IND = investigational new drug.
Table 5.
Aggregate single‐unit risks in transfused RBC under current donor testing protocols in the United States
| Aggregate risk category | Risk elementsa | Risk |
|---|---|---|
| Minimum |
• HIV + HCV+ HBV • Bacteria • Babesia‐nonendemic area |
0.00031% (1 in 322,600) |
| Minimum + CMVb |
• HIV + HCV+ HBV • Bacteria • CMV risk for immunocompromised patients • Babesia‐nonendemic area |
0.10031% (1 in 996) |
| Maximum |
• HIV + HCV+ HBV • Bacteria • Babesia‐endemic area • New chronic EIA |
0.12031% (1 in 831) |
| Maximum CMVb |
• HIV + HCV+ HBV • Bacteria • CMV risk for immunocompromised patients • Babesia‐endemic area • New chronic EIA |
0.22031% (1 in 454) |
This column contains the components that are then summed together to provide the total risk (shown in the right‐hand column), for each aggregate risk category. The numbers for each risk element are taken from Table 4.
(HSCT patients).
Table 6.
Aggregate lifetime patient risks due to RBC transfusion for different patient categories under current testing algorithms in the United States
| Aggregate risk per patient (%) | |||
|---|---|---|---|
| Diagnosis | RBC unit exposure | Minimuma 1 | Maximumb 2 |
| Cardiac surgery | 3 | 0.0009 (1/107,000) | 0.36 (1/277) |
| Trauma | 5 | 0.0016 (1/65,000) | 0.60 (1/167) |
| ICU | 3.5 | 0.0011 (1/91,000) | 0.42 (1/238) |
| Cardiovascular disease | 3 | 0.0009 (1/107,000) | 0.36 (1/277) |
| HSCT | 15 | 1.49 (1/67) | 3.25 (1/31) |
| MDS | 39 | 0.012 (1/8,000) | 3.76 (1/27) |
| SCD | 720 | 0.22 (1/450) | 43.17 (1/2) |
| Thalassemia | 750 | 0.23 (1/430) | 45.13 (1/2) |
The method of calculating risk when large numbers of units are transfused as described by Kleinman et al.66
Lifetime risks, except for cardiovascular disease and ICU patient groups. In the latter groups, risk is for a single hospitalization or ICU stay. Lifetime risk would increase for patients transfused on multiple occasions.1 Minimum per‐unit risk is 0.00031% for all patient groups except for HSCT patients, where minimum risk is 0.10031% based on potential sequelae from TT‐CMV infection.2 Maximum per‐unit risk is 0.12031% for the first four patient groups and 0.22031% for HSCT patients. For patients with MDS, SCD, and thalassemia, risk is 0.12031% for a 1.5‐year period (when a new acute EIA is in the blood supply) and 0.07631% (due to Babesia) when transfused during other time intervals.
Noninfectious risks
Transfusion‐associated graft‐versus‐host disease
Transfusion‐associated graft‐versus‐host disease (TA‐GVHD), an almost uniformly fatal condition, is prevented by completely inactivating T lymphocytes in RBCs or LR‐RBCs.67 This is currently accomplished by gamma irradiation, which although highly effective in preventing GVHD, has multiple limitations.68 Rare TA‐GVHD cases are still reported likely due to substandard treatment or failure to apply the procedure uniformly to all cellular units for patients at risk, due either to inappropriate institutional criteria or to incorrect patient diagnosis.68, 69, 70, 71, 72, 73, 74
Gamma irradiation is known to damage RBC membranes causing acute and delayed hemolysis and to damage the Na‐K pump resulting in potassium leakage from the RBCs.75 Consequently, storage of irradiated RBCs is limited to 28 days postirradiation.76
In the United States in 2011, an estimated 13.4% of transfused RBCs were irradiated.2 It is likely that the criteria for irradiation varied among institutions. Similarly, the length of time that an irradiated RBC unit is stored before being transfused may also vary. In the scenario of batch mode irradiation with subsequent storage of units, there is the theoretical concern that these RBC units may function less optimally than nonirradiated RBCs and therefore should not be given to patients not at risk of TA‐GVHD. The alternative scenario of irradiating units just before product issue poses logistic challenges and is only possible if the institution has its own blood irradiator. Finally, facilities with irradiators have been subject to increasing regulatory scrutiny due to bioterrorism concerns, making the continued use of this equipment less desirable and more expensive.76
PI‐RBCs and PI‐WB procedures have been found effective in inactivating white blood cells (WBCs) and T cells and when applied routinely, could replace the use of gamma irradiation, and solve logistic challenges.77, 78 Data for one of these technologies indicate that PI‐RBCs show lower hemolysis, lack of effect on the Na‐K pump, and lower extracellular potassium and protein levels, resulting in better in vitro function than gamma‐irradiated RBCs.79
RBC alloimmunization
Since PI is known to prevent donor WBCs from exerting their immunologic effects, PI could theoretically affect the immune system's presentation of RBC antigens and thereby influence RBC alloimmunization rates; therefore, the impact of LR on RBC alloimmunization may help predict whether PI treatment of RBC would have a similar effect.80, 81, 82
The development of RBC alloantibodies has well‐known potential deleterious consequences. The rate of RBC alloimmunization in transfused hospitalized patients (excluding patients with hemoglobinopathies) has been measured at 1.8% and 4% in two large studies.83, 84 The rate increases with the number of RBC units transfused; however, the majority of antibodies are formed early in the course of transfusion therapy. Specific primary diagnoses are associated with higher rates: 18% to 47% in sickle cell disease (SCD) patients in the absence of phenotypic matching,85 5% to 30% in thalassemia,17, 86, 87, 88, 89 15% in MDS,90 and 9% in patients with malignant hematology diagnoses.91
A prospective small randomized control trial in 404 cardiac surgery patients examined the effect of universal LR on RBC alloimmunization.92 Although the rate was lower in the LR group than in the non‐LR group (3.4% vs. 7.1%), the difference was not significant. A smaller single‐hospital study using a retrospective noncontemporaneous study design demonstrated a decreased alloimmunization rate in recipients of LR versus non‐LR RBCs based on comparing data from two 1‐year intervals separated by 14 years.93 This same study showed that LR resulted in a decreased alloimmunization rate in acute myeloid leukemia patients whereas a different study showed no effect of LR in MDS patients.90 Other smaller studies in thalassemia patients have suggested an association of LR with a decreased RBC alloimmunization rate;94 however, these studies have had small sample sizes and have potential confounding factors. In summary, the available data do not allow for a firm conclusion. Unlike LR (which still leaves a small number of viable WBCs in the blood product),80, 81 PI renders WBCs nonviable and stops protein production and antigen expression, thus establishing a theoretical basis for why PI might reduce alloimmunization even if LR does not.95, 96
Patients with leukemia usually receive both RBC and PLT transfusions. Current data suggest that PI treatment of PLTs may reduce the rate of HLA alloimmunization in this patient group.96 It is possible that PI‐RBCs may show the same benefit. If so, the application of PI to both components may protect against HLA alloimmunization and may improve the odds of finding a compatible HSCT donor for patients with leukemia as well as for patients with hemoglobinopathies who may become future HSCT candidates.97, 98 These possibilities need to be examined in the clinic.
It is important for PI clinical trial protocols to include an assessment of RBC alloimmunization rates. Since the overall RBC alloimmunization rate is low in the general transfused population,83 it is unlikely that pivotal clinical trials will be powered to adequately evaluate this phenomenon and data from routine use will be required.
APPROACHES FOR PRODUCING A PI‐RBC PRODUCT
There are two conceptual approaches to obtaining PI‐RBCs (Fig. 3). WB can be separated into components and then PI can be applied to the RBCs, or PI treatment can be applied to the WB unit. The PI‐WB unit can be transfused as WB or, alternatively, could subsequently undergo further processing to produce components;99 the latter approach has the logistic advantage of producing multiple PI components from a single PI application. However, if the WB unit is stored before processing, this approach may require compromises since component storage requirements are conflicting and will be difficult to satisfy simultaneously (e.g., RBCs and WB are stored refrigerated, PLTs at RT, and plasma frozen).
Figure 3.
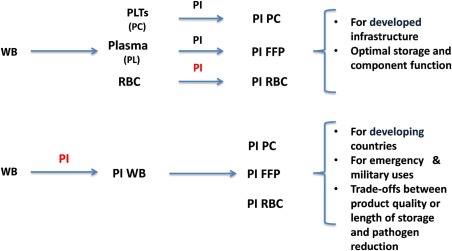
Conceptual approaches for PI of blood products.
In developed countries, targeted blood component therapy for specific indications has been standard practice for many decades. Specialized storage containers have been tailored to each component and additive solutions developed to optimize quality and extend shelf life.100 Furthermore, individual‐component PI technology is compatible with apheresis component collection, which has become an important part of the blood supply chain.101 In contrast, in countries with little infrastructure or in acute trauma situations (particularly in military conflicts), the need for WB transfusion is greater and suggests the value of the application of PI to WB units.102, 103
Two methods are in commercial development for supplying PI‐RBC products: WB photochemical inactivation using riboflavin and ultraviolet (UV) light (Mirasol System)104 and RBC chemical inactivation using S‐303 and glutathione (GSH; Intercept System).105 In addition, use of the S‐303 and GSH system to treat WB is being pursued for the developing world.106 The basic characteristics of the two systems are summarized in Fig. 4. The riboflavin and UV WB system utilizes the same riboflavin dose and illuminator as for plasma and PLT PI, but uses a much higher UV dose, corresponding to a significantly longer illumination time (Fig. 4).104
Figure 4.
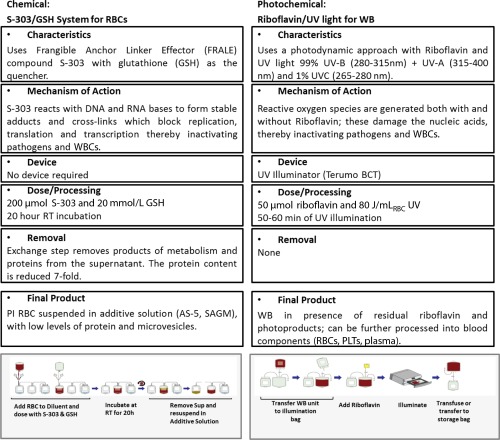
The S‐303 and GSH system, now in its second generation, utilizes a chemical system featuring a fast‐acting compound (S‐303) that reacts with nucleic acid bases to form stable adducts and cross‐links with a mode of action similar to the Intercept systems for PLTs and plasma, but without the use of an illuminator.105 To minimize nonspecific reactions with molecules in the extracellular domain, GSH is included in the process. Because of its size, GSH does not penetrate cell or viral membranes, so when added to the RBC unit, it remains exclusively in the extracellular space. This allows quenching of extracellular reactions without a significant impact on pathogen and WBC inactivation. The modifications present in the second‐generation system were implemented to reduce the formation of immunogenic adducts on the surface of PI‐RBCs.107, 108, 109 The second‐generation system uses the same dose of the active ingredient S‐303, a buffered version of the quencher GSH at 10‐fold higher concentration for improved quenching, and includes an exchange step after the overnight incubation that allows the effective removal of proteins and electrolytes from the RBC supernatant.109, 110
DATA FOR PI‐RBC AND PI‐WB SYSTEMS
Licensure of PI‐RBCs or PI‐WB requires in vitro studies of RBC quality during storage, in vitro inactivation studies for representative transfusion‐transmitted pathogens, in vivo recovery and survival studies of transfused RBCs in healthy volunteers to validate the maximum allowable length of product storage, and clinical trials of safety and efficacy in relevant patient populations. After licensure, postmarketing hemovigilance studies will allow the further characterization of PI‐RBC safety and efficacy.
RBC quality during storage has been summarized in the literature.104, 105, 110, 111 PI studies are ongoing by both PI manufacturers; available data are summarized in Table 7.77, 78, 99, 104, 105, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121 These inactivation data still need to be validated by full‐unit studies with multiple replicates, and hence care should be taken in their interpretation; nevertheless, a few points emerge. For the S‐303 and GSH system, the extent of PI has remained comparable between the first‐ and second‐generation approaches. For the riboflavin and UV system, the limited data indicate that inactivation of some pathogens is lower in the WB system than in the PLT and plasma systems, despite the higher dose of UV light used (80 J/mLRBC vs. 6.2 J/mL PLASMA). This is consistent with the lower efficiency for UV light delivery in the presence of hemoglobin (Hb)‐containing RBCs.99
Table 7.
Compilation of published PI data in RBCs for S‐303 and GSH and in WB by riboflavin and UV
| Mean log reduction | |||
|---|---|---|---|
| Pathogen | S‐303 and GSH | Riboflavin and UV (@ 80 J/mLRBC) | |
| Viruses | |||
| HIV ‐ cell free | >6.5112, a | ||
| HIV‐ cell associated | >5.9105 | 4.5113 | |
| BVDV (surrogate for HCV) | >4.8105 | ||
| DHBV (surrogate for HBV) | >5.1121 | 6.3112, a | |
| CMV model viruses | |||
| HSV | >6.0112, a | ||
| IBR | 1.599 | ||
| VSV | 5.7112 | 4.5104 | |
| Bluetongue | ≥6.0112 | >5105 | 199 |
| Adeno Type 5 | >7.4105 | ||
| WNV | >6.0a, 119 | ||
| SARS | >6.5120, a | ||
| CPV | 3.899 | ||
| HAV | 1.599 | ||
| Parasites | |||
| B. microti | >5.5112 | >4.9119, a | >4.73114 |
| Plasmodium falciparum | >6.8119, a | >6.4118 | |
| T. cruzi | >5.4112, a | >5.3119, a | >3.5116 |
| Leishmania donovani | 2.3117 | ||
| Bacteria | |||
| Y. enterocolitica | ≥ 6.8105 | 7.4112, a | 299, b |
| Serratia marcescens | 5.1105 | 4.1112, a | |
| Serratia liquifaciens | 299, b | ||
| Pseudomonas aeruginosa | 4.5112, a | ||
| Escherichia coli | ≥6.7105 | 7.4112, a | |
| Staphylococcus aureus | 5.1105 | >5.1112, a | |
| Staphylococcus epidermidis | >6.9112, a | ||
| Listeria monocytogenes | >7.1112, a | ||
| WBCs | >577, a | 4.778 | |
Inactivation achieved with first‐generation system (0.2/2 mmol/L GSH).
These data are from low‐titer experiments and inactivation of higher bacterial titers was not evaluated.
CPV = canine parvovirus; DHBV = duck hepatitis virus; HSV = herpes simplex virus; IBR = infectious bovine rhinotracheitis virus; SARS = severe acute respiratory syndrome; VSV = vesicular stomatitis virus.
Each technology has undergone recovery and survival studies independently performed by the same investigator (Table 8).122, 123 The S‐303 and GSH system was tested in 27 healthy volunteers in two centers using a crossover design.122 At 35 days of storage, the 24‐hour recovery of autologous treated RBCs compared favorably with control RBCs (88% vs. 90%, p = 0.31), meeting FDA requirements. The survival of S‐303 RBC (T50) was lower than that of control RBCs, but within the normal range (32.7 days vs. 39.5 days; p = 0.0001; normal, 28‐35 days).122 In a different study,123 RBCs stored for 42 days were manufactured from riboflavin and UV‐treated autologous WB units prepared using variable UV doses (22, 33, and 44 J/mLRBC). Only five of 11 subjects met the FDA requirement of more than 75% recovery at 24 hours; mean RBC survival was 24 ± 9 days. There was a trend toward lower recovery and lower survival for higher illumination doses. These data, along with recently published in vitro data at Storage Days 21 and 42, indicate that a storage time shorter than 42 days will be required for an 80 J/mLRBC dose.111
Table 8.
Clinical experience
| Study | Number | Description | Endpoints | Results |
|---|---|---|---|---|
| A. With the S‐303 and GSH PI‐RBC system | ||||
| First generation | ||||
| US Phase III chronic study | 50 | Transfusion‐dependent SCD patients; two‐arm double‐blinded crossover design | Blood utilization | Terminated |
| US Phase III acute study124 | 148 | CV surgery patients | Composite endpoint of MI, renal failure, and mortality124 | Met primary endpoint, early termination |
| Second generation | ||||
| US Phase II study 122 | 27 | Healthy volunteers; crossover design | 24‐hour recovery: 88.0 ± 8.5 days (T) vs. 90.1 ± 6.9 (C) | Met primary endpoint, completed |
| EU Phase III acute study125 | 50 | CV surgery patients; two‐arm design |
Primary efficacy: mean Hb content per RBC component Primary safety: adverse events over 90 days (related and unrelated to study RBC components) compared between the treatment groups |
Met primary endpoint with similar AE profile between arms126 |
| EU Phase III chronic study127 | 70 | Transfusion‐dependent thalassemia Major patients; crossover design |
Primary efficacy: Hb consumption (g Hb/kg body weight/day). Primary safety: incidence of a treatment‐emergent antibody with confirmed specificity to S‐303 RBCs over 12 months |
In progress |
| B. With the riboflavin and UV PI‐WB system | ||||
| US Phase II Study–IMPROVE123, 128 |
12 (4/4/3) |
Feasibility trial to evaluate recovery and survival in RBCs obtained from WB units treated with the Mirasol system. Three study arms each using a different UV dose (22, 33, and 44 J/mLRBC) |
Primary: 24‐hr posttransfusion RBC recovery Secondary: RBC survival; SAE |
Terminated Recovery ≥ 75% 22‐J dose: 1 of 4 33‐J dose: 1 of 4 44‐J dose: 1 of 3 Survival ≥ 28 days 22‐J dose: 2 of 4 33‐J dose: 2 of 4 44‐J dose: 0 of 4 |
| US Phase II study–IMPROVE II1 29 | 29 | To evaluate, as per FDA criteria, the 24‐hr posttransfusion RBC recovery in healthy adult subjects of LR‐RBCs, derived from Mirasol‐treated fresh WB units, and stored refrigerated for 21 days. |
Primary: 24‐hr posttransfusion RBC recovery Secondary: RBC survival, AUC; SAE; neoantigenicity |
Completed Data not yet reported |
| Ghana Phase‐III Study–AIMS1 30 | 250 | Treatment of WB with the Mirasol system: prevention of Malaria caused by transfusion |
Primary: TT malaria Secondary: TT bacterial infections |
Completed |
AUC = area under curve; CV = cardiovascular; MI = myocardial infarction; SAE = severe adverse event.
Table 8122‐131 also reports other Phase II and III studies, primarily using information from the http://ClinicalTrials.gov website.124, 125, 126, 127, 128, 129, 130, 131 For the first‐generation S‐303 and GSH system, a Phase III trial in SCD patients was terminated early when two patients developed apparent RBC antibodies, but no sequela.108
The companion Phase III first‐generation PI‐RBC study in cardiovascular surgery patients with acute anemia, conducted simultaneously with the SCD study, met its primary noninferiority composite endpoint despite its early termination due to the SCD study findings.124 After S‐303 reformulation, two Phase III clinical studies with the second‐generation S‐303 and GSH RBC system are in progress in Europe, targeting the indications of acute and chronic anemia in cardiovascular patients and patients with thalassemia, respectively.125, 127
For riboflavin and UV, an additional Phase II study (presumably using an 80 J/mLRBC dose) has finished recruiting.129 A Phase III study on the prevention of TT‐malaria among recipients of PI‐WB is under way in Africa.130
POTENTIAL ADVERSE EFFECTS OR RISKS OF PI‐RBCs
Potential risks of transfusing PI‐RBCs include toxicology‐based adverse side effects, increased RBC alloimmunization, and reduced clinical benefit to patients (i.e., efficacy). Extensive toxicology data for both PI‐RBC systems are available in the literature.132, 133 Such data have been reviewed by regulatory agencies and found robust enough to authorize Phase II and III clinical trial work. With regard to the primary chemical agents, S‐303 completely decomposes during the 20‐hour treatment process and, in addition, the chemically inert reaction by‐products are significantly reduced through the exchange step. Riboflavin and its photodegradation products have a toxicology profile of “generally regarded as safe.” Nevertheless, potential long‐term toxicology risks can only be definitively assessed by collecting routine use data.
As mentioned, two SCD patients in a first‐generation S‐303 and GSH Phase III trial developed antibodies; these were found to be directed against adducts formed on the RBC surface during S‐303 treatment. Further characterization of the antibodies showed they were low titer (2‐8), were inhibited by acridine compounds (thereby pinpointing specificity for the anchor part of the S‐303 molecule), and did not cause phagocytosis of S‐303–treated RBCs in an in vitro model of RBC clearance.134 In a rabbit mismatch transfusion model,135 first‐generation S‐303 RBCs circulated normally in naive animals and did not cause the formation of antibodies. However the S‐303 RBCs showed accelerated clearance when the animals were immunized with KLH‐acridine compounds. These observations led to the technology modifications incorporated in the second‐generation system. When second‐generation S‐303 RBCs were transfused to the same preimmunized animals, they exhibited normal circulation. Finally, sera from the two antibody‐positive patients were negative when cross‐matched against second‐generation S‐303 RBCs.
Despite these encouraging observations, concerns still exist regarding alloantibody formation due to RBC alterations caused by second‐generation S‐303 treatment. Thus, a primary aim of the second‐generation S‐303 thalassemia clinical trial is to monitor RBC immunogenicity. However, given the small size of any clinical trial, negative results will not be sufficient to resolve this issue. This will require an ongoing hemovigilance program to monitor routinely used product (as has been done for PI‐PLTs and PI‐plasma)136, 137, 138, 139 to achieve the numbers required to assess this potential transfusion complication. Similar clinical trial endpoints and hemovigilance monitoring will be required to determine if the riboflavin system affects RBC immunogenicity.
If an increased frequency of alloantibody formation were to be found, this should be viewed in the context of the known high alloimmunization rate in chronic RBC recipients;80 ,82 that is, it may be that the benefits of PI‐RBCs will exceed a small risk conferred by the development of additional antibodies, especially if these antibodies do not cause hemolysis and/or result in an increased difficulty in finding compatible RBC units.
The potential for decreased efficacy of PI‐RBCs has not yet been fully assessed and will require Phase III clinical trial data as well as routine use data. For S‐303 RBCs, in vitro RBC quality assessment and in vivo volunteer studies indicate that at 35 days of storage, these cells would be expected to function as well as non–PI‐RBCs when transfused. With regard to the riboflavin WB system, a full set of similar data is not yet available but preliminary results suggest that the shelf life of this product may be limited to shorter than 35 days.
DISCUSSION
RBC transfusion carries multiple risks, each of which (excepting some infections in highly endemic areas) is relatively small. Because of this, it appears unlikely that a laboratory screening intervention to minimize any one of these risks will be implemented. The chief deterrent is cost, but other factors such as lack of concern by clinicians (due to underrecognition of cases, underappreciation of the potential for severe outcomes, and the availability of treatment) and blood center concerns of unnecessary donor deferrals due to false‐positive test results may also influence inaction. PI offers a solution to this dilemma in that multiple risks can be obviated by a single intervention. Since PI will come at additional cost, an economic analysis leading to a decision to implement PI may also need to factor in protection against an EIA that could result in a large number of transfusion transmissions and severe recipient outcomes.4, 5 In this worst‐case scenario, failure to have implemented PI may create lack of trust in the entire blood system.
The potential specific benefits of PI‐RBC technology include:
Reducing risk to essentially zero for most current transfusion‐transmitted pathogens (HIV, HCV, HBV, HTLV, WNV, syphilis, CMV, Babesia).
Substantially decreasing or completely eliminating risk for pathogens with the potential for a very high genomic titer (e.g., dengue).61, 140 Depending upon the robustness of the PI technology and the infectious dose of a particular pathogen, full inactivation of such pathogens could be achieved for all units or a substantial proportion of cases depending on the pathogen load.
A greater margin of protection against TA‐GVHD than irradiation due to robust WBC inactivation.67, 69, 70, 71, 72, 73, 74, 141
The possible reduction of WBC alloimmunization would provide HSCT candidate recipients with a higher likelihood of successful HLA matching to potential HSCT donors.
Operational benefits in blood manufacturing and inventory management. Dual RBC inventories for both CMV and irradiation status could be eliminated and recall of RBC units associated with bacterially contaminated PLTs could be discontinued.
Eliminating a blind spot of the current testing paradigm, which requires that a pathogen be detectable by NAT in a plasma sample or that the donor has developed a robust serologic response to an intracellular organism (e.g., Babesia); PI would obviate the need to implement a more logistically complex cellular‐based PCR platform.
Eliminating donor screening questions including travel to malaria‐endemic areas, a history of babesiosis or Chagas disease, and travel to WNV‐endemic areas (asked ex‐US). Since malaria travel deferrals are very common, their elimination could have a significant impact on the number of eligible donors.
With regard to cost containment, PI implementation for all components (RBCs/PLTs/plasma) should allow for the discontinuation of some donor screening tests and/or the modification of existing screening protocols.142 Syphilis testing would no longer be needed as transfusion transmission risk is exceedingly low, serologic testing has limited ability to detect early infection, and PI methods have high efficacy in killing the organism in each component type (shown for PLTs and plasma and still to be verified for PI‐RBCs).143, 144, 145 CMV antibody, Trypanosoma cruzi antibody, and HBsAg testing (assuming that HBV NAT is implemented) could also be eliminated.
A robust PI technology that can inactivate 5 to 8 infectious log of most pathogens would allow for modification of NAT protocols. There would be no need to perform individual NAT for HIV, HCV, HBV, or WNV since minipool testing would be adequate to detect any units with high viral load that otherwise might theoretically escape the full effects of PI. Minipools could be made larger as is currently done in source plasma testing.146 Consideration could also be given to removing some serologic assays that might be considered redundant; these include anti‐HBc, anti‐HCV, and even anti‐HIV (although the latter might trigger public concern). Of course, modification of blood donor screening protocols would require regulatory authority approval and it would probably take some years of routine use with systematic hemovigilance efforts to accumulate the data required for such changes to be made.
A fully PI‐treated blood supply would shape the response to threats from new EIAs, in that there would be less pressure to develop screening assays.142 For years, this has been the case for fractionated plasma derivatives that routinely undergo PI. For example, recipients of these products were protected from WNV transmission at a time when transmission to recipients of blood components occurred.146 Also, as has been seen with regard to agents posing a transfusion transmission risk that does not reach a crisis level and do not have compelling business cases due to factors such as geographic or seasonal variations in incidence/prevalence (e.g., Babesia and dengue), new blood donor screening assay development cannot be relied upon to protect the blood supply.30 The approach of PI‐treated components may ultimately be less complex and less expensive than continued assay development.
Two PI‐RBC and WB systems are in different levels of development and each may have its role with riboflavin and UV best suited for developing countries and S‐303 and GSH for developed countries. Further studies with the actual system(s) used are still needed to demonstrate inactivation of CMV, spirochetes, selected EIAs, multiple bacterial species, and WBCs—the latter for replacement of gamma irradiation. The assessment of alloimmunization with PI‐treated RBCs should be investigated with the realization that routine use is the only way to achieve the numbers required to assess this transfusion complication and other potential severe adverse events. Finally, PI‐RBCs may still have its limitations since some pathogens (Parvovirus B19, HAV, HEV) are at least partially resistant to inactivation.
In summary, PI‐RBCs should be viewed in the context of having a fully PI‐treated blood supply, thereby shifting the blood safety paradigm from reactive to proactive142 and as providing insurance against known and unknown pathogens that may enter the blood supply or are currently un(der)recognized.4
CONFLICT OF INTEREST
SK is a paid consultant to Cerus Corporation; AS is employed by Cerus Corporation.
REFERENCES
- 1. Kleinman S, Reed W, Stassinopoulos A. A patient‐oriented risk‐benefit analysis of pathogen‐inactivated blood components: application to apheresis platelets in the United States. Transfusion 2013;53:1603‐18. [DOI] [PubMed] [Google Scholar]
- 2. Whitaker B, Hinkins S. The 2011 national blood collection and utilization survey report [Internet]. Washington (DC); US Department of Health and Human Services; 2013 [cited 2014 Dec]. Available from: http://www.hhs.gov/ash/bloodsafety/2011-nbcus.pdf
- 3. Roubinian NH, Escobar GJ, Liu V, et al. Trends in red blood cell transfusion and 30‐day mortality among hospitalized patients. Transfusion 2014;54:2678‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kleinman S, Cameron C, Custer B, et al. Modeling the risk of an emerging pathogen entering the Canadian blood supply. Transfusion 2010;50:2592‐606. [DOI] [PubMed] [Google Scholar]
- 5. Custer B, Agapova M, Martinez R. The cost‐effectiveness of pathogen reduction technology as assessed using a multiple risk model. Transfusion 2010;50:2461‐73. [DOI] [PubMed] [Google Scholar]
- 6. Horvath KA, Acker MA, Chang H, et al. Blood transfusion and infection after cardiac surgery. Ann Thorac Surg 2013;95:2194‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jin R, Zelinka ES, McDonald J, et al. Effect of hospital culture on blood transfusion in cardiac procedures. Ann Thorac Surg 2013;95:1269‐74. [DOI] [PubMed] [Google Scholar]
- 8. Holcomb JB, del Junco DJ, Fox EE, et al. The prospective, observational, multicenter, major trauma transfusion (PROMMTT) study: comparative effectiveness of a time‐varying treatment with competing risks. JAMA Surg 2013;148:127‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aubron C, Syres G, Nichol A, et al. A pilot feasibility trial of allocation of freshest available red blood cells versus standard care in critically ill patients. Transfusion 2012;52:1196‐202. [DOI] [PubMed] [Google Scholar]
- 10. Heddle NM, Eikelboom J, Liu Y, et al. Exploratory studies on the age of transfused blood and in‐hospital mortality in patients with cardiovascular diagnoses. Transfusion 2015;55:364‐72. [DOI] [PubMed] [Google Scholar]
- 11. Xenocostas A, Yee A, Wong CJ, et al. RBC transfusion requirements after allogeneic marrow transplantation: impact of the before‐transplant Hb level on transfusion and early survival. Transfusion 2003;43:373‐82. [DOI] [PubMed] [Google Scholar]
- 12. Solh M, Brunstein C, Morgan S, et al. Platelet and red blood cell utilization and transfusion independence in umbilical cord blood and allogeneic peripheral blood hematopoietic cell transplants. Biol Blood Marrow Transplant 2011;17:710‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ramsey SD, McCune JS, Blough DK, et al. Patterns of blood product use among patients with myelodysplastic syndrome. Vox Sang 2012;102:331‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roberts DO, Covert B, Lindsey T, et al. Directed blood donor program decreases donor exposure for children with sickle cell disease requiring chronic transfusion. Immunohematology 2012;28:7‐12. [PubMed] [Google Scholar]
- 15. Meny GM. Transfusion protocols for patients with sickle cell disease: working toward consensus? Immunohematology 2012;28:1‐2. [PubMed] [Google Scholar]
- 16. Platt OS, Brambilla D, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death in sickle cell disease. N Engl J Med 1994;330:1639–44. [DOI] [PubMed] [Google Scholar]
- 17. el‐Danasoury AS, Eissa DG, Abdo RM, et al. Red blood cell alloimmunization in transfusion‐dependent Egyptian patients with thalassemia in a limited donor exposure program. Transfusion 2012;52:43‐7. [DOI] [PubMed] [Google Scholar]
- 18. Borgna‐Pignatti C, Rugolotto S, De Stefano P, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica 2004;89:1187‐93. [PubMed] [Google Scholar]
- 19. Modell B, Khan M, Darlison M. Survival in beta‐thalassaemia major in the UK: data from the UK Thalassaemia Register. Lancet 2000;355:2051‐2. [DOI] [PubMed] [Google Scholar]
- 20. Herwaldt BL, Linden JV, Bosserman E, et al. Transfusion‐associated babesiosis in the United States: a description of cases. Ann Intern Med 2011;155:509‐19. [DOI] [PubMed] [Google Scholar]
- 21. Vannier E, Krause PJ. Human babesiosis. N Engl J Med 2012;366:2397‐407. [DOI] [PubMed] [Google Scholar]
- 22. Sher G, Markowitz M. Babesiosis [Internet]. AABB Association Bulletin #14‐05. Bethesda (MD): AABB; 2014 [cited 2015 Jun]. Available from: http://www.aabb.org/programs/publications/bulletins/Documents/ab14-05.pdf
- 23. Centers for Disease Control and Prevention (CDC). Babesiosis surveillance ‐ 18 States, 2011. MMWR Morb Mortal Wkly Rep 2012;61:505‐9. [PubMed] [Google Scholar]
- 24. Levin AE, Williamson PC, Erwin JL, et al. Determination of Babesia microti seroprevalence in blood donor populations using an investigational enzyme immunoassay. Transfusion 2014;54:2237‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Young C, Chawla A, Berardi V, et al. Preventing transfusion‐transmitted babesiosis: preliminary experience of the first laboratory‐based blood donor screening program. Transfusion 2012;52:1523‐9. [DOI] [PubMed] [Google Scholar]
- 26. Moritz ED, Winton CS, Johnson ST, et al. Investigational screening for Babesia microti in a large repository of blood donor samples from nonendemic and endemic areas of the United States. Transfusion 2014;54:2226‐36. [DOI] [PubMed] [Google Scholar]
- 27. Moritz E, Winton C, Townsend R, et al. Extended Babesia microti investigational screening studies: implications for transfusion‐transmitted Babesiosis (TTB). Transfusion 2014;54 Suppl 2B:26A‐7A. 23521109 [Google Scholar]
- 28. Leiby DA, Johnson ST, Won KY, et al. A longitudinal study of Babesia microti infection in seropositive blood donors. Transfusion 2014;54:2217‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Johnson ST, Cable RG, Leiby DA. Lookback investigations of Babesia microti‐seropositive blood donors: seven‐year experience in a Babesia‐endemic area. Transfusion 2012;52:1509‐16. [DOI] [PubMed] [Google Scholar]
- 30. Katz LM, Sayers M. Screening for babesiosis: where is the policy? Transfusion 2014;54:2154‐6. [DOI] [PubMed] [Google Scholar]
- 31. Mungai M, Tegtmeier G, Chamberland M, et al. Transfusion‐transmitted malaria in the United States from 1963 through 1999. N Engl J Med 2001;344:1973‐8. [DOI] [PubMed] [Google Scholar]
- 32. Spencer B, Steele W, Custer B, et al. Risk for malaria in United States donors deferred for travel to malaria‐endemic areas. Transfusion 2009;49:2335‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lafeuillade B, Eb F, Ounnoughene N, et al. Residual risk and retrospective analysis of transfusion‐transmitted bacterial infection reported by the French National Hemovigilance Network from 2000 to 2008. Transfusion 2015;55:636‐46. [DOI] [PubMed] [Google Scholar]
- 34. Keller‐Stanislawski B, Lohmann A, Günay S, et al. The German Haemovigilance System‐‐reports of serious adverse transfusion reactions between 1997 and 2007. Transfus Med 2009;19:340‐9. [DOI] [PubMed] [Google Scholar]
- 35. Funk MB, Lohmann A, Guenay S, et al. Transfusion‐Transmitted Bacterial Infections ‐ Haemovigilance Data of German Blood Establishments (1997‐2010). Transfus Med Hemother 2011;38:266‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fatalities reported to FDA following blood collection and transfusion: annual summary for fiscal year 2013 [Internet]. Silver Spring (MD): U.S. Food and Drug Administration; 2013 [cited 2015 June]. Available from: http://www.fda.gov/BiologicsBloodVaccines/SafetyAvailability/ReportaProblem/TransfusionDonationFatalities/ucm391574.htm
- 37. Jacobs MR, Good CE, Lazarus HM, et al. Relationship between bacterial load, species virulence, and transfusion reaction with transfusion of bacterially contaminated platelets. Clin Infect Dis 2008;46:1214‐20. [DOI] [PubMed] [Google Scholar]
- 38. Harm SK, Delaney M, Charapata M, et al. Routine use of a rapid test to detect bacteria at the time of issue for nonleukoreduced, whole blood‐derived platelets. Transfusion 2013;53:843‐50. [DOI] [PubMed] [Google Scholar]
- 39. Pearce S, Rowe GP, Field SP. Screening of platelets for bacterial contamination at the Welsh Blood Service. Transfus Med 2011;21:25‐32. [DOI] [PubMed] [Google Scholar]
- 40. Jenkins C, Ramírez‐Arcos S, Goldman M, et al. Bacterial contamination in platelets: incremental improvements drive down but do not eliminate risk. Transfusion 2011;51:2555‐65. [DOI] [PubMed] [Google Scholar]
- 41. Benjamin RJ, Kline L, Dy BA, et al. Bacterial contamination of whole‐blood‐derived platelets: the introduction of sample diversion and prestorage pooling with culture testing in the American Red Cross. Transfusion 2008;48:2348‐55. [DOI] [PubMed] [Google Scholar]
- 42. Roback JD, Josephson CD. New insights for preventing transfusion‐transmitted cytomegalovirus and other white blood cell‐associated viral infections. Transfusion 2013;53:2112‐6. [DOI] [PubMed] [Google Scholar]
- 43. Vamvakas EC. Is white blood cell reduction equivalent to antibody screening in preventing transmission of cytomegalovirus by transfusion? A review of the literature and meta‐analysis. Transfus Med Rev 2005;19:181‐99. [DOI] [PubMed] [Google Scholar]
- 44. Wu Y, Zou S, Cable R, et al. Direct assessment of cytomegalovirus transfusion‐transmitted risks after universal leukoreduction. Transfusion 2010;50:776‐86. [DOI] [PubMed] [Google Scholar]
- 45. Thiele T, Kruger W, Zimmermann K, et al. Transmission of cytomegalovirus (CMV) infection by leukoreduced blood products not tested for CMV antibodies: a single‐center prospective study in high‐risk patients undergoing allogeneic hematopoietic stem cell transplantation (CME). Transfusion 2011;51:2620‐6. [DOI] [PubMed] [Google Scholar]
- 46. Kekre N, Tokessy M, Mallick R, et al. Is cytomegalovirus testing of blood products still needed for hematopoietic stem cell transplant recipients in the era of universal leukoreduction? Biol Blood Marrow Transplant 2013;19:1719‐24. [DOI] [PubMed] [Google Scholar]
- 47. Ziemann M, Juhl D, Görg S, et al. The impact of donor cytomegalovirus DNA on transfusion strategies for at‐risk patients. Transfusion 2013;53:2183‐9. [DOI] [PubMed] [Google Scholar]
- 48. Furui Y, Satake M, Hoshi Y, et al. Cytomegalovirus (CMV) seroprevalence in Japanese blood donors and high detection frequency of CMV DNA in elderly donors. Transfusion 2013;53:2190‐7. [DOI] [PubMed] [Google Scholar]
- 49. Townsend RL, Moritz ED, Fialkow LB, et al. Probable transfusion‐transmission of Anaplasma phagocytophilum by leukoreduced platelets. Transfusion 2014;54:2828‐32. [DOI] [PubMed] [Google Scholar]
- 50. Shields K, Cumming M, Rios J, et al. Transfusion‐associated Anaplasma phagocytophilum infection in a pregnant patient with thalassemia trait: a case report. Transfusion 2014; [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 51.AABB fact sheet for Anaplasma phagocytophylum [Internet]. Bethesda (MD): AABB; 2009 [cited 2015 Jun]. Available from: http://www.aabb.org/tm/eid/Pages/eidpostpub.aspx
- 52. Appassakij H, Promwong C, Rujirojindakul P, et al. The risk of blood transfusion‐associated Chikungunya fever during the 2009 epidemic in Songkhla Province, Thailand. Transfusion 2014;54:1945-52. [DOI] [PubMed] [Google Scholar]
- 53. Brouard C, Bernillon P, Quatresous I, et al. Estimated risk of Chikungunya viremic blood donation during an epidemic on Reunion Island in the Indian Ocean, 2005 to 2007. Transfusion 2008;48:1333-41. [DOI] [PubMed] [Google Scholar]
- 54. Petersen LR, Epstein JS. Chikungunya virus: new risk to transfusion safety in the Americas. Transfusion 2014;54:1911-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Petersen LR, Tomashek KM, Biggerstaff BJ. Estimated prevalence of dengue viremia in Puerto Rican blood donations, 1995 through 2010. Transfusion 2012;52:1647‐51. [DOI] [PubMed] [Google Scholar]
- 56. Hewitt PE, Ijaz S, Brailsford SR, et al. Hepatitis E virus in blood components: a prevalence and transmission study in southeast England. Lancet 2014;384:1766-73. [DOI] [PubMed] [Google Scholar]
- 57.Chikungunya in the Americas; 11 Autochthonous cases in Florida Chikungunya in the Americas [Internet]. Atlanta (GA): Centers for Disease Control and Prevention; last updated 2015 Jun 22 [cited 2015 Aug 6]. Available from: http://www.cdc.gov/chikungunya/pdfs/2014Table1-final.pdf
- 58. Rasongles P, Angelini‐Tibert MF, Simon P, et al. Transfusion of platelet components prepared with photochemical pathogen inactivation treatment during a Chikungunya virus epidemic in Ile de La Reunion. Transfusion 2009;49:1083‐91. [DOI] [PubMed] [Google Scholar]
- 59. Appassakij H, Khuntikij P, Kemapunmanus M, et al. Viremic profiles in asymptomatic and symptomatic chikungunya fever: a blood transfusion threat? Transfusion 2013;53:2567‐74. [DOI] [PubMed] [Google Scholar]
- 60. Lieshout‐Krikke RW, Oei W, Habets K, et al. Travel behavior and deferral of Dutch blood donors: consequences for donor availability. Transfusion 2015;55:79‐85. [DOI] [PubMed] [Google Scholar]
- 61. Musso D, Richard V, Broult J, et al. Inactivation of dengue virus in plasma with amotosalen and ultraviolet A illumination. Transfusion 2014;54:2924‐30. [DOI] [PubMed] [Google Scholar]
- 62. Tsetsarkin KA, Sampson‐Johannes A, Sawyer L, et al. Photochemical inactivation of chikungunya virus in human apheresis platelet components by amotosalen and UVA light. Am J Trop Med Hyg 2013;88:1163‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zou S, Stramer SL, Dodd RY. Donor testing and risk: current prevalence, incidence, and residual risk of transfusion‐transmissible agents in US allogeneic donations. Transfus Med Rev 2012;26:119‐28. [DOI] [PubMed] [Google Scholar]
- 64. Stramer SL, Notari EP, Krysztof DE, et al. Hepatitis B virus testing by minipool nucleic acid testing: does it improve blood safety? Transfusion 2013;53:2449‐58. [DOI] [PubMed] [Google Scholar]
- 65. Kelly S. Fatal West Nile Virus infection after probable transfusion‐associated transmission‐‐Colorado, 2012. MMWR Morb Mortal Wkly Rep 2013;62:622‐4. [PMC free article] [PubMed] [Google Scholar]
- 66. Kleinman S, Busch MP, Korelitz JJ, et al. The incidence/window period model and its use to assess the risk of transfusion‐transmitted human immunodeficiency virus and hepatitis C virus infection. Transfus Med Rev 1997;11:155‐72. [DOI] [PubMed] [Google Scholar]
- 67. Luban NL, Drothler D, Moroff G, et al. Irradiation of platelet components: inhibition of lymphocyte proliferation assessed by limiting‐dilution analysis. Transfusion 2000;40:348‐52. [DOI] [PubMed] [Google Scholar]
- 68. Pelszynski MM, Moroff G, Luban NL, et al. Effect of gamma irradiation of red blood cell units on T‐cell inactivation as assessed by limiting dilution analysis: implications for preventing transfusion‐associated graft‐versus‐host disease. Blood 1994;83:1683‐9. [PubMed] [Google Scholar]
- 69. Triulzi D, Duquesnoy R, Nichols L, et al. Fatal transfusion‐associated graft‐versus‐host disease in an immunocompetent recipient of a volunteer unit of red cells. Transfusion 2006;46:885‐8. [DOI] [PubMed] [Google Scholar]
- 70. O'Brien KL, Pereira SE, Wagner J, et al. Transfusion‐associated graft‐versus‐host disease in a liver transplant recipient: an unusual presentation and review of the literature. Transfusion 2013;53:174‐80. [DOI] [PubMed] [Google Scholar]
- 71. Rososhansky S, Badonnel MC, Hiestand LL, et al. Transfusion‐associated graft‐versus‐host disease in an immunocompetent patient following cardiac surgery. Vox Sang 1999;76:59‐63. [DOI] [PubMed] [Google Scholar]
- 72. Gorman TE, Julius CJ, Barth RF, et al. Transfusion‐associated graft‐vs‐host disease. A fatal case caused by blood from an unrelated HLA homozygous donor. Am J Clin Pathol 2000;113:732‐7. [DOI] [PubMed] [Google Scholar]
- 73. Gilstad C, Roschewski M, Wells J, et al. Fatal transfusion‐associated graft‐versus‐host disease with concomitant immune hemolysis in a group A combat trauma patient resuscitated with group O fresh whole blood. Transfusion 2012;52:930‐5. [DOI] [PubMed] [Google Scholar]
- 74. Kopolovic I, Ostro J, Tsubota H, et al. A systematic review of transfusion‐associated graft versus host disease (TA‐GVHD). Blood 2015;126:406‐14. [DOI] [PubMed] [Google Scholar]
- 75. Moroff G, Holme S, AuBuchon JP, et al. Viability and in vitro properties of AS‐1 red cells after gamma irradiation. Transfusion 1999;39:128‐34. [DOI] [PubMed] [Google Scholar]
- 76. Mintz PD. Cesium cessation? An advantage of pathogen reduction treatments. Transfusion 2011;51:1369‐76. [DOI] [PubMed] [Google Scholar]
- 77. Hanson D, Propst M, Dupuis K, et al. High titer leukocyte inactivation in INTERCEPT red blood cells (RBC). Transfus Clin Biol 2001;8:1‐292. [Google Scholar]
- 78. Fast LD, Nevola M, Tavares J, et al. Treatment of whole blood with riboflavin plus ultraviolet light, an alternative to gamma irradiation in the prevention of transfusion‐associated graft‐versus‐host disease? Transfusion 2013;53:373‐81. [DOI] [PubMed] [Google Scholar]
- 79. Knutson F. Quality of inactivated red cells compared to gamma irradiated red cells. Vox Sang 2013;105 Suppl 1:151‐2. [Google Scholar]
- 80. Matteocci A, Pierelli L. Red blood cell alloimmunization in sickle cell disease and in thalassaemia: current status, future perspectives and potential role of molecular typing. Vox Sang 2014;106:197‐208. [DOI] [PubMed] [Google Scholar]
- 81. Yazdanbakhsh K, Ware RE, Noizat‐Pirenne F. Red blood cell alloimmunization in sickle cell disease: pathophysiology, risk factors, and transfusion management. Blood 2012;120:528‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hendrickson JE, Tormey CA, Shaz BH. Red blood cell alloimmunization mitigation strategies. Transfus Med Rev 2014;28:137‐44. [DOI] [PubMed] [Google Scholar]
- 83. Higgins JM, Sloan SR. Stochastic modeling of human RBC alloimmunization: evidence for a distinct population of immunologic responders. Blood 2008;112:2546‐53. [DOI] [PubMed] [Google Scholar]
- 84. Zalpuri S, Zwaginga JJ, le Cessie S, et al. Red‐blood‐cell alloimmunization and number of red‐blood‐cell transfusions. Vox Sang 2012;102:144‐9. [DOI] [PubMed] [Google Scholar]
- 85. Lasalle‐Williams M, Nuss R, Le T, et al. Extended red blood cell antigen matching for transfusions in sickle cell disease: a review of a 14‐year experience from a single center (CME). Transfusion 2011;51:1732‐9. [DOI] [PubMed] [Google Scholar]
- 86. Sirchia G, Zanella A, Parravicini A, et al. Red cell alloantibodies in thalassemia major. Results of an Italian cooperative study. Transfusion 1985;25:110‐2. [DOI] [PubMed] [Google Scholar]
- 87. Spanos T, Karageorga M, Ladis V, et al. Red cell alloantibodies in patients with thalassemia. Vox Sang 1990;58:50‐5. [DOI] [PubMed] [Google Scholar]
- 88. Cheng CK, Lee CK, Lin CK. Clinically significant red blood cell antibodies in chronically transfused patients: a survey of Chinese thalassemia major patients and literature review. Transfusion 2012;52:2220‐4. [DOI] [PubMed] [Google Scholar]
- 89. Ameen R, Al‐Shemmari S, Al‐Humood S, et al. RBC alloimmunization and autoimmunization among transfusion‐dependent Arab thalassemia patients. Transfusion 2003;43:1604‐10. [DOI] [PubMed] [Google Scholar]
- 90. Sanz C, Nomdedeu M, Belkaid M, et al. Red blood cell alloimmunization in transfused patients with myelodysplastic syndrome or chronic myelomonocytic leukemia. Transfusion 2013;53:710‐5. [DOI] [PubMed] [Google Scholar]
- 91. Schonewille H, Haak HL, van Zijl AM. Alloimmunization after blood transfusion in patients with hematologic and oncologic diseases. Transfusion 1999;39:763‐71. [DOI] [PubMed] [Google Scholar]
- 92. van de Watering L, Hermans J, Witvliet M, et al. HLA and RBC immunization after filtered and buffy coat‐depleted blood transfusion in cardiac surgery: a randomized controlled trial. Transfusion 2003;43:765‐71. [DOI] [PubMed] [Google Scholar]
- 93. Blumberg N, Heal JM, Gettings KF. WBC reduction of RBC transfusions is associated with a decreased incidence of RBC alloimmunization. Transfusion 2003;43:945‐52. [DOI] [PubMed] [Google Scholar]
- 94. Singer ST, Wu V, Mignacca R, et al. Alloimmunization and erythrocyte autoimmunization in transfusion‐dependent thalassemia patients of predominantly Asian descent. Blood 2000;96:3369‐73. [PubMed] [Google Scholar]
- 95. Fiebig E, Hirschkorn DF, Maino VC, et al. Assessment of donor T‐cell function in cellular blood components by the CD69 induction assay: effects of storage, gamma radiation, and photochemical treatment. Transfusion 2000;40:761‐70. [DOI] [PubMed] [Google Scholar]
- 96. Osselaer J, Vandendaele M, Guldenpfennig M, et al. Influence on HLA alloimmunisation frequency in hematology patients supported with INTERCEPT pathogen inactivated (PI) platelet components (PC) [abstract]. Transfusion 2011;51 Suppl 3:122A. [Google Scholar]
- 97. Zimring JC, Hair GA, Deshpande SS, et al. Immunization to minor histocompatibility antigens on transfused RBCs through crosspriming into recipient MHC class I pathways. Blood 2006;107:187‐9. [DOI] [PubMed] [Google Scholar]
- 98. Horan JT, Liesveld JL, Fenton P, et al. Hematopoietic stem cell transplantation for multiply transfused patients with sickle cell disease and thalassemia after low‐dose total body irradiation, fludarabine, and rabbit anti‐thymocyte globulin. Bone Marrow Transplant 2005;35:171‐7. [DOI] [PubMed] [Google Scholar]
- 99. Goodrich RP, Doane S, Reddy HL. Design and development of a method for the reduction of infectious pathogen load and inactivation of white blood cells in whole blood products. Biologicals 2010;38:20‐30. [DOI] [PubMed] [Google Scholar]
- 100. Prowse CV, de Korte D, Hess JR, et al. Commercially available blood storage containers. Vox Sang 2014;106:1‐13. [DOI] [PubMed] [Google Scholar]
- 101. Prowse CV. Component pathogen inactivation: a critical review. Vox Sang 2013;104:183‐99. [DOI] [PubMed] [Google Scholar]
- 102. Hassall O, Maitland K, Pole L, et al. Bacterial contamination of pediatric whole blood transfusions in a Kenyan hospital. Transfusion 2009;49:2594‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Gonzalez EA, Moore FA, Holcomb JB, et al. Fresh frozen plasma should be given earlier to patients requiring massive transfusion. J Trauma 2007;62:112‐9. [DOI] [PubMed] [Google Scholar]
- 104. Marschner S, Goodrich R. Pathogen reduction technology treatment of platelets, plasma and whole blood using riboflavin and UV light. Transfus Med Hemother 2011;38:8‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Henschler R, Seifried E, Mufti N. Development of the S‐303 pathogen inactivation technology for red blood cell concentrates. Transfus Med Hemother 2011;38:33‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Mufti NA, Erickson AC, North AK, et al. Treatment of whole blood (WB) and red blood cells (RBC) with S‐303 inactivates pathogens and retains in vitro quality of stored RBC. Biologicals 2010;38:14‐9. [DOI] [PubMed] [Google Scholar]
- 107. Stassinopoulos A, inventor ; Cerus Corp., assignee. Quenching methods for red blood cell inactivation process. US patent 7,655,392. 2010 Feb 2.
- 108. Conlan MG, Stassinopoulos A, Garratty G, et al. Antibody formation to S‐303‐treated RBCS in the setting of chronic RBC transfusion. Blood 2004;104 Suppl 10:112A. [Google Scholar]
- 109. Mufti N, Erickson A, North A, inventors ; Cerus Corp., assignee. Quenching methods for red blood cell pathogen inactivation. United States patent US 8,900,805. 2014 Dec 2.
- 110. Winter KM, Johnson L, Kwok M, et al. Red blood cell in vitro quality and function is maintained after S‐303 pathogen inactivation treatment. Transfusion 2014;54:1798‐807. [DOI] [PubMed] [Google Scholar]
- 111. Schubert P, Culibrk B, Karwal S, et al. Whole blood treated with riboflavin and ultraviolet light: quality assessment of all blood components produced by the buffy coat method. Transfusion 2014; [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 112. Schlenke P. Pathogen inactivation technologies for cellular blood components: an update. Transfus Med Hemother 2014;41:309‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Keil S, Rapaport R, Doane S, et al. Viral reduction of intracellular HIV using the Mirasol system for whole blood. Vox Sang 2013;103(Suppl 1):144. [Google Scholar]
- 114. Tonnetti L, Thorp AM, Reddy HL, et al. Riboflavin and ultraviolet light reduce the infectivity of Babesia microti in whole blood. Transfusion 2013;53:860‐7. [DOI] [PubMed] [Google Scholar]
- 115. El Chaar M, Atwal S, Freimanis GL, et al. Inactivation of Plasmodium falciparum in whole blood by riboflavin plus irradiation. Transfusion 2013;53:3174‐83. [DOI] [PubMed] [Google Scholar]
- 116. Tonnetti L, Thorp AM, Reddy HL, et al. Evaluating pathogen reduction of Trypanosoma cruzi with riboflavin and ultraviolet light for whole blood. Transfusion 2012;52:409‐16. [DOI] [PubMed] [Google Scholar]
- 117. Tonnetti L, Thorp AM, Reddy HL, et al. Reduction of Leishmania donovani infectivity in whole blood using riboflavin and ultraviolet light. Transfusion 2014;55:326‐9. [DOI] [PubMed] [Google Scholar]
- 118. Olver C, Martinez C, Keil S, et al. Treatment of P. falciparum contaminated whole blood units using the mirasol system for whole blood. Vox Sang 2014;107:5(suppl):131. [Google Scholar]
- 119. Dupuis K, Bernard K, Jones S, et al. Helinx technology inactivates pathogens of emerging importance in red blood cell concentrates. Blood 2003;102:816a. [Google Scholar]
- 120. Dupuis K, Sampson‐Johannes A, Corash L. The causative agent of SARS in platelets and red blood cell concentrates is inactivated by Helinx technology. Blood 102 2003;11:816a. [Google Scholar]
- 121. Hanson D, Dupuis K, Arnold D, et al. Inactivation of important viral contaminants in all blood components [abstract]. Transfusion 2012;52:225A. [Google Scholar]
- 122. Cancelas JA, Dumont LJ, Rugg N, et al. Stored red blood cell viability is maintained after treatment with a second‐generation S‐303 pathogen inactivation process. Transfusion 2011;51:2367‐76. [DOI] [PubMed] [Google Scholar]
- 123. Cancelas JA, Rugg N, Fletcher D, et al. In vivo viability of stored red blood cells derived from riboflavin plus ultraviolet light‐treated whole blood. Transfusion 2011;51:1460‐8. [DOI] [PubMed] [Google Scholar]
- 124. Benjamin RJ, McCullough J, Mintz PD, et al. Therapeutic efficacy and safety of red blood cells treated with a chemical process (S‐303) for pathogen inactivation: a Phase III clinical trial in cardiac surgery patients. Transfusion 2005;45:1739‐49. [DOI] [PubMed] [Google Scholar]
- 125.Study to assess S303 RBCs and evaluate safety and efficacy in patients requiring transfusion support of acute anemia [Internet]. http://ClinicalTrials.gov identifier: NCT01716923. Bethesda (MD): U.S. National Institutes of Health; 2012 [cited 2015 Jun]. Available from: http://clinicaltrials.gov/ct2/show/NCT01716923
- 126. Brixner V, Kiessling A, Madlener K, et al. Clinical safety and efficacy of red blood cell components treated with the second generation S‐303 pathogen and leukocyte inactivation system ‐ a randomized controlled double‐blind phase 3 study in patients requiring transfusion support of acute anemia. Vox Sang 2015;109(Suppl. 1):28. [Google Scholar]
- 127.Study to evaluate efficacy and safety of S303 treated red blood cells (RBCs) in subjects with thalassemia major requiring chronic RBC transfusion [Internet]. http://ClinicalTrials.gov identifier: NCT01740531. Bethesda (MD): U.S. National Institutes of Health; 2012 [cited 2015 Jun]. Available from: http://clinicaltrials.gov/ct2/show/NCT01740531
- 128.Feasibility study of Mirasol‐treated whole blood red cell recovery and survival (IMPROVE) [Internet]. http://ClinicalTrials.gov identifier: NCT00742001. Bethesda (MD): U.S. National Institutes of Health; 2008 [cited 2015 Jun]. Available from: http://clinicaltrials.gov/ct2/show/NCT00742001
- 129.Inactivation of whole blood with Mirasol (IMPROVEII) [Internet]. http://ClinicalTrials.gov identifier: NCT01907906. Bethesda (MD): U.S. National Institutes of Health; 2013 [cited 2015 Jun]. Available from: http://clinicaltrials.gov/ct2/show/NCT01907906?term=NCT01907906rank=1
- 130.Clinical and biological efficacy of Mirasol‐treated fresh whole blood for the prevention of transfusion‐transmitted malaria (AIMS) [Internet]. http://ClinicalTrials.gov identifier: NCT02118428. Bethesda (MD): U.S. National Institutes of Health; 2014 [cited 2015 Jun]. Available from: http://clinicaltrials.gov/ct2/show/NCT02118428?term=NCT02118428rank=1
- 131. Owusu‐Ofori S. Riboflavin and UV illumination treatment of whole blood units (WBU) in a Ghanaian blood center [abstract]. Transfusion 2013;53:202A‐3A. 22574682 [Google Scholar]
- 132. North A, Ciaravino V, Mufti N, et al. Preclinical pharmacokinetic and toxicology assessment of red blood cells prepared with S‐303 pathogen inactivation treatment. Transfusion 2011;51:2208‐18. [DOI] [PubMed] [Google Scholar]
- 133. Reddy HL, Doane SK, Keil SD, et al. Development of a riboflavin and ultraviolet light‐based device to treat whole blood. Transfusion 2013;53 Suppl 1:131S‐6S. [DOI] [PubMed] [Google Scholar]
- 134. Sacks DA, Nance SJ, Garratty G, et al. Monocyte monolayer assay as a predictor of severity of hemolytic disease of the fetus and newborn. Am J Perinatol 1993;10:428‐31. [DOI] [PubMed] [Google Scholar]
- 135. Ness PM, Shirey RS, Weinstein MH, et al. An animal model for delayed hemolytic transfusion reactions. Transfus Med Rev 2001;15:305‐17. [DOI] [PubMed] [Google Scholar]
- 136. Osselaer JC, Cazenave JP, Lambermont M, et al. An active haemovigilance programme characterizing the safety profile of 7437 platelet transfusions prepared with amotosalen photochemical treatment. Vox Sang 2008;94:315‐23. [DOI] [PubMed] [Google Scholar]
- 137. Osselaer JC, Messe N, Hervig T, et al. A prospective observational cohort safety study of 5106 platelet transfusions with components prepared with photochemical pathogen inactivation treatment. Transfusion 2008;48:1061‐71. [DOI] [PubMed] [Google Scholar]
- 138. Cazenave JP, Waller C, Kientz D, et al. An active hemovigilance program characterizing the safety profile of 7483 transfusions with plasma components prepared with amotosalen and UVA photochemical treatment. Transfusion 2010;50:1210‐9. [DOI] [PubMed] [Google Scholar]
- 139. Knutson F, Osselaer J, Pierelli L, et al. Prospective, active haemovigilance study with combined cohort analysis of 19 175 transfusions of platelet components prepared with amotosalen‐UVA photochemical treatment. Vox Sang 2015;109:343-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Stramer SL, Linnen JM, Carrick JM, et al. Dengue viremia in blood donors identified by RNA and detection of dengue transfusion transmission during the 2007 dengue outbreak in Puerto Rico. Transfusion 2012;52:1657‐66. [DOI] [PubMed] [Google Scholar]
- 141. Corash L, Lin L. Novel processes for inactivation of leukocytes to prevent transfusion‐associated graft‐versus‐host disease. Bone Marrow Transplant 2004;33:1‐7. [DOI] [PubMed] [Google Scholar]
- 142. Alter HJ. Pathogen reduction: a precautionary principle paradigm. Transfus Med Rev 2008;22:97‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Orton SL, Liu H, Dodd RY, et al. Prevalence of circulating Treponema pallidum DNA and RNA in blood donors with confirmed‐positive syphilis tests. Transfusion 2002;42:94‐9. [DOI] [PubMed] [Google Scholar]
- 144. Cable RG. Evaluation of syphilis testing of blood donors. Transfus Med Rev 1996;10:296‐302. [DOI] [PubMed] [Google Scholar]
- 145. Lin L, Dikeman R, Molini B, et al. Photochemical treatment of platelet concentrates with amotosalen and long‐wavelength ultraviolet light inactivates a broad spectrum of pathogenic bacteria. Transfusion 2004;44 1496‐504. [DOI] [PubMed] [Google Scholar]
- 146. Leydold SM, Farcet MR, Kindermann J, et al. Chikungunya virus and the safety of plasma products. Transfusion 2012;52:2122‐30. [DOI] [PubMed] [Google Scholar]