

Abstract
Interindividual variability in susceptibility to HIV‐1 infection, its transmission, disease progression, and response to antiviral therapy has been attributed to host determinants and variability in multiple genes. Although most people exposed to the virus go on to develop full‐blown disease at variable intervals, a proportion of them, labeled as long‐term nonprogressors or exposed uninfected, possess ‘natural resistance’ to infection. A better understanding of genetic and immunologic basis of such a natural resistance to infection would bear important implications in designing therapeutic vaccine designs. The genetic variants that could influence susceptibility to HIV‐1 and limit AIDS vary in different populations and among individuals. Meta‐analyses of large cohort studies have identified numerous ‘AIDS restriction genes’ that regulate HIV cell entry (particularly chemokine coreceptors and their ligands), acquired and innate immunity (major histocompatibility complex, killer cell immunoglobulin‐like receptor, and cytokines), and others [tripartite interaction motif 5 α (TRIM5α) and apolipoprotein B mRNA‐editing enzyme, catalytic polypeptide‐like 3G] that influence outcome of HIV infection. Studies carried out in the Indian population with regard to genetic polymorphisms in chemokine receptors have shown that (i) the protective CCR5 Δ32 variant is rare, (ii) CCR5HHE carrying *59402A is associated with increased likelihood of infection and development of AIDS, and (iii) the Indian population generally has low CCL3L1 copy numbers (∼2.3). These data have implications in developing screening tests that could identify people at higher or lower risk of infection and rate of disease progression, predict vaccine responsiveness in clinical trials and understand the pathogenic mechanisms.
Keywords: AIDS, chemokine receptors, chemokines, genes, HIV‐1, susceptibility, virus entry
Background
The AIDS caused by the HIV‐1 has already claimed nearly 21 million lives. More than 33.2 million people are currently living with this infection worldwide (http://data.unaids.org/pub/EPISlides/2007/2007epiupdate.en.pdf; http://www.nacoonline.org/NACO). The situation is particularly alarming in regions like the Sub‐Saharan Africa where 22 million people are infected with HIV‐1, and the adult prevalence rate is ∼5%. Similarly, there are 4.2 million infected people living across South East Asia. According to the recent census published by the National AIDS control organization, there are ∼2.5 million HIV‐infected people residing in the Indian subcontinent with the adult prevalence rate of ∼0.36% (http://www.nacoonline.org/NACO). The menace of HIV‐1/AIDS has emerged as one of the biggest health hazards of today and a matter of global priority for researchers, clinicians, policy makers, social workers, and funding agencies alike. To this effect, a Joint United Nations Program on HIV/AIDS (UNAIDS) has been created by a constellation of important organizations like the UNHCR, UNICEF, WFP, UNDP, UNFPA, UNODC, ILO, UNESCO, WHO, and world bank. The program has committed its ‘millennium development goal 6’ as ‘to combat HIV/AIDS, malaria, and other diseases’ and targets ‘to have halted and begun to reverse the spread of HIV/AIDS’ by 2015.
According to the most widely recognized consensus, HIV‐1 appears to have evolved from the closely related simian immunodeficiency virus (SIV) in sub‐Saharan Africa and was transferred from nonhuman primates to humans through zoonosis during the mid‐1930s. After this initial transfer, the virus underwent mysterious changes and sudden mutations to evolve into HIV‐1. It began to spread among humans across the globe and ultimately became a true epidemic. On the other hand, HIV‐2 is relatively less transmittable and is largely confined to West Africa. It is believed that this strain may have originated from the Sooty Mangabey (Cercocebus atys), an Old World monkey residing predominantly in Guinea‐Bissau, Gabon, and Cameroon (http://en.wikipedia.org/wiki/AIDS_origin).
Since the discovery of the virus in the early 1980s, important milestones in understanding the pathology, structural biology, and clinical and immunological aspects of the infection have been achieved. The current focus is on defining the genetic and immunologic correlates of the host–pathogen interaction with a view to develop possible biomarkers for infection, transmission, and disease progression. Furthermore, despite impressive developments in antiretroviral therapy and control of the disease, a universal anti‐HIV vaccine still eludes the researchers. Table 1 summarizes some of the important milestones achieved since the first instance of AIDS was documented in 1981.
Table 1.
Important milestones of research in HIV/AIDS biology, diagnosis, and genetics
| General | Diagnostics | Viral biology and therapeutics | Genetics | ||||
|---|---|---|---|---|---|---|---|
| Year | Event | Year | Event | Year | Event | Year | Event |
| 1959 | First case of AIDS confirmed retrospectively in Congo | 1985 | First ELISA test kit for screening of abs against HIV | 1987 | AZT (Zidovudine) approved as the first anti‐HIV drug | 2004 | First meta‐analysis of AIDS restriction genes in multiple cohorts compiled (9) |
| 1981 | Pneumocystis carinii outbreak and Kaposi’s sarcoma reported as gay compromise syndrome or gay‐related immune deficiency | 1987 | Western blot blood test kit for confirmation of infection | 1995 | Triple combination therapy approved as more effective ART | 2007 | First whole‐genome scan identified susceptibility loci in MHC at 6p21 (10) |
| 1982 | Syndrome renamed as AIDS | 1996 | Introduction of Amplicor HIV‐1 monitor test for viral load | 1998 | First human trial of AIDS vaccine begins in the United States | 2008 | Whole‐genome association identified susceptibility loci on 8q24.3 (11) |
| 1983 | Luc Montagnier isolated lymphadenopathy‐associated virus – HIV‐1 | 2001 | Launch of TrueGene HIV‐1 genotyping kit and Open Gene DNA sequencing | 2007 | Crystal structure of gp120 in complex with neutralizing ab b12 | ||
| 1984 | Identification of HIV‐1 (HTLVIII) by Robert Gallo | 2002 | Versant HIV RNA3.0 assay (bDNA) for viral load estimation | 2008 | Novel host‐derived factors for HIV‐1 life cycle identified (8) | ||
| 1986 | Global AIDS strategy launched by WHO | 2002 | Development of OraQuick Rapid HIV‐1 ab test | 2008 | Nobel prize awarded to Luc Montagnier, Francoise Barre‐Sinoussi and Harald zur Hausen for their discoveries of HIV and HPV | ||
| 1996 | Launching of UNAIDS | 2004 | Introduction of OraSure rapid test for HIV‐2 | ||||
| 1999 | Source of origin of HIV traced to chimpanzee in West Central Africa | 2004 | Launching of Multispot HIV‐1/HIV‐2 rapid test | ||||
ab, antibody; ART, antiretroviral therapy; bDNA, branched DNA; ELISA, enzyme‐linked immunosorbent assay; MHC, major histocompatibility complex; UNAIDS, Joint United Nations Program on HIV/AIDS.
It took more than four decades to develop a vaccine for polio or for chicken pox; similarly, it may take still a few more years to develop a successful vaccine against HIV‐1. Lack of suitable animal model systems, inherent hypervariability of the virus, and its extraordinary ability to adapt and evolve in the host are some of the major hurdles responsible for the delay in developing vaccine against HIV (Table 2). The virus has devised its own mechanisms to escape immune recognition and surveillance through extensive glycosylation and surface masking. For example, the envelope glycoproteins gp120 and gp41 possess immunogenic epitopes, but most of these are not exposed on the virus surface and therefore not available for an effector immune response. Likewise, the gp120 CD4‐binding site is a recessed cavity in the virus and therefore inaccessible for the antibodies. Importantly, virus infection results in downregulation of human leukocyte antigen (HLA) class I expression and effectively perturbs antigen presentation. Likewise, antibodies to gp120 CD4‐binding site have been reported to hinder antigen presentation and suppress gp120‐specific CD4 T‐cell responses (1). The CD4 T cells decline in number, and their response is compromised in HIV‐1‐infected patients, although antigen stimulation can induce chemokine secretion and suppress HIV infection in vitro (2). HIV‐1‐infected T cells form a virological synapse with noninfected CD4+ T cells to efficiently transfer HIV‐1 virions from cell to cell. The viral gp120 participates in the formation of a virological synapse (3), plausibly leading to dysregulated T‐cell activation, which is in analogy with the immunological synapse that normally results in regulated T‐cell activation.
Table 2.
Major obstacles encountered for the development of a successful vaccine against HIV‐1
| Lack of suitable animal model systems |
| Hypervariability of the virus, particularly because of the extensive mutations in viral protein env and others |
| Extensive glycosylation and surface masking of viral proteins (e.g., gp120 and gp41) |
| Inaccessibility of antibody epitopes (e.g., CD4‐binding site is a recessed cavity) |
| Reduced antigen presentation (because of downregulation of human leukocyte antigen class I and development of antibodies against gp120 CD4‐binding site) |
| Decline in CD4 T‐cell numbers and dysfunction |
| Inability to induce broadly neutralizing antibodies against the virus |
| Rapid emergence of CTL escape mutants |
| Population‐specific variability in host genetic and immunologic response factors |
| Synergistic opportunistic infections and complications thereof |
Host genetic factors are important determinants that influence susceptibility to virus infection and subsequent progression to AIDS. The genetic contributions have largely been known through studies on naturally resistant individuals, for example the long‐term nonprogressors (LTNPs) or the exposed uninfected (EU) persons. A set of genes is known to govern virus entry and/or development of effective innate and adaptive immune responses against the virus. This article reviews important host genetic determinants that are associated with resistance/susceptibility to HIV‐1 infection and slow/fast disease progression and response to therapy.
Host genetics: individual variability in response to HIV/AIDS
Numerous studies have shown that an extensive interindividual variability exists in response to HIV infection. This includes susceptibility to virus, its transmission, and course of disease progression, with phenotypes ranging from undetectable, asymptomatic state to fatal AIDS 4, 5. Individuals are not equally susceptible to infection and have differences in their viral set points, rates of decline of CD4 T cells, levels of viremia, emergence of CTL escape mutants, and development of opportunistic infections resulting in varying incubation periods of the AIDS virus. While a majority of individuals infected with HIV‐1 progress to AIDS, if untreated (called as ‘progressors’), most of them can, however, be turned ‘aviremic’ by antiretroviral therapy (Figure 1). More importantly, ∼5%–10% of infected individuals do not develop AIDS for more than 7 years, remain naturally aviremic, and are referred to as LTNPs. This group of naturally resistant people represent ‘spontaneous controllers’ and include EUs partners of commercial sex workers as well as the discordant couples (6).
Figure 1.

Spontaneous controllers vs disease progressors following HIV‐1 infection. The former include exposed uninfected (EUs) like the discordant couples and commercial sex worker and long‐term nonprogressors (LTNPs). The latter compose of rapid and slow progressors as determined by their rate of disease progression. ART, antiretroviral therapy.
Spontaneous controllers
Spontaneous controllers possess a level of ‘natural resistance’ against HIV as they can combat infection after initial viral exposure and limit disease progression. Developing resistance against virus, acquisition is one of the currently defined goals of the first generation vaccines (7). Therefore, the underlying mechanisms that protect these people from HIV infection could hold the key to develop such an anticipated vaccine against the virus. The ‘elite’ or aviremic controllers, as they are often referred to, represent ∼1% of total infected individuals and maintain viremia at almost undetectable levels of <75 copies of viral RNA per milliliter. The ‘intermediate’ or ‘viremic’ controllers manage to maintain their viremia at <2000 RNA copies per milliliter, despite no treatment (Figure 1), and do not allow consecutive episodes of viral blips of >200 copies (http://www2.massgeneral.org/aids/hiv_elite_controllers.asp). The progressors, on the other hand, permit virus propagation to >10,000 RNA copies per milliliter, but once on antiretroviral therapy (ART), their viral loads can be suppressed to undetectable levels.
An international collaborative HIV controller consortium has recently been established with the purpose of recruiting elite controllers from different ethnic groups (7). This effort has already led to a collection of more than 300 elite controllers who are being checked through whole‐genome scans for their immune response to HIV and for the possible genetic mechanisms that may be responsible for spontaneous control of the virus (http://www2.massgeneral.org/aids/hiv_elite_controllers.asp and http://www.iasusa.org). The prime focus of the consortium was to understand host, viral, and immunologic parameters that are associated with effective containment of HIV infection.
Recent studies have shown specific immunologic and genetic features associated predominantly with elite controllers that could be correlated to their ability to resist virus. For example, such persons have an overrepresentation of protective HLA‐B*57 and B*27 alleles, high ratios of functional CD4:CD8 T cells, high generalized T activation levels (CD38+DR+), high proportions of polyfunctional IL2+γIFN+ CD4+ T helpers, CD8+ CTL responses, preferential targeting of gag and env, and high HIV‐specific antibody levels (7). However, not all elite controllers possess these resistance factors, indicating an involvement of additional hitherto undetected restriction factors. For example, Brass et al. (8) have reported that HIV uses at least 250 host‐derived HIV dependency factors for gaining entry into target cells and completing its life cycle. Hence, multiple genetic factors are expected to be involved in disease pathogenesis and progression following HIV infection. A study carried out by O’Brien and colleagues (9) using five different cohorts (Multicenter Hemophilia Cohort Study, AIDS Link to Intravenous Experience, Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study and San Francisco City Cohort) has shown the existence of a number of ‘AIDS restriction genes’ that could determine the genetic propensity of an individual or a population and help predict survival kinetics following HIV infection 4, 5, 10.
Multiple genes control HIV‐1/AIDS
Structural genomic approaches aimed at defining chromosomal localization of a host of human genetic variants that regulate susceptibility to HIV infection, rate of CD4 decline, viral load, disease progression, and response to therapy have shown interesting results. More than 10 such set of genes that regulate expression of chemokines, cytokines, HIV‐1 coreceptors, and immune response following HIV‐1 infection have been defined with their genetic loci located on variable chromosomes (Figure 2).
Figure 2.

Chromosomal localization of human genes involved in HIV/AIDS susceptibility, transmission, progression, and response to therapy. For the sake of convenience, individual genes have been grouped according to their function at the bottom of the figure. APOBEC3, apolipoprotein B mRNA‐editing enzyme, catalytic polypeptide‐like 3G; DC‐SIGN, dendritic cell‐specific intracellular adhesion molecule‐3‐grabbing nonintegrin; HLA, human leukocyte antigen; KIR, killer cell immunoglobulin‐like receptor; MIP‐1, macrophage inflammatory protein‐1; PARC, pulmonary and activation‐regulated chemokine; RANTES, regulated upon activation, normally T‐expressed, and presumably secreted; SDF‐1, stromal cell‐derived factor 1; TRIM5α, tripartite interaction motif 5 α.
Consortium approaches and their meta‐analyses have shown that only a fraction of the observed phenotypic differences can be explained by variability at such loci. Some of the genetic associations that have been established with HIV/AIDS conclusively involve (i) HLA polymorphic loci and their associated genes including HCP5, RNF39, and ZNRD1 on the short arm of chromosome 6, (ii) chemokine receptors CCR2 and CCR5 that act as HIV entry coreceptors and chemokine ligands like CCL3L1, CCL4L1, and CCL5, (iii) antiviral restriction factor tripartite interaction motif 5 α (TRIM5α) on chromosome 11p15, (iv) apolipoprotein B mRNA‐editing enzyme, catalytic polypeptide‐like 3G (APOBEC3) on chromosome 22q13, (v) dendritic cell‐specific intracellular adhesion molecule‐3‐grabbing nonintegrin (DC‐SIGN) on chromosome 19p13, (vi) KIR3DS1 on chromosome 19q13, (vii) interferon regulatory factor 1, and (viii) LY6 family of G [glycosylphosphatidylinositol (GPI)]‐anchored proteins and others. Of these, two genetic regions, namely HLA‐HCP5‐RNF39‐ZNRD1 region at 6p21 (10) and the rs2572886‐LY6 region at 8q24 (11), have been established through genome‐wide association studies on a variety of population groups.
Genes influencing viral entry
Chemokines and their receptors
Chemokines are low‐molecular‐weight potent chemoattractants produced by a variety of cell types that include T cells, macrophages, natural killer (NK) cells, B cells, fibroblasts, and mast cells. These are involved in cell trafficking and immunomodulation of inflammation and immune responses. In certain instances, their receptors serve as entry portals for pathogens to gain entry into target cells and establish infection. For example, the Duffy antigen receptor for chemokines (DARC) is used by Plasmodium vivax, while CCR5 and CXCR4 are preferentially opted by the HIV‐1.
Vast majority of primary HIV‐1 isolates are predominantly CCR5 tropic and gradually tend to become CXCR4 tropic during late infection. All non‐syncitium‐inducing (NSI) strains of HIV‐1 require chemokine receptor CCR5 to gain entry into target cells, while the syncitium‐inducing (SI) virions use CXCR4 to enter (Figure 3). The CCR5 expression is upregulated under the influence of IL‐2 and IL‐10, while IL‐4 induces CXCR4 expression. The chemokines CCL3 [macrophage inflammatory protein‐1 (MIP‐1)α], CCL4 (MIP‐1β), and CCL5 [regulated upon activation, normally T‐expressed, and presumably secreted (RANTES)] are natural ligands of CCR5 coreceptor that can block entry of NSI virions. Similarly, stromal cell‐derived factor 1 (SDF‐1) is a ligand for CXCR4 that is primarily used by SI viruses. Therefore, the availability of chemokine receptors expressed on cell surface and the levels of their ligands as well as cytokines are critical elements as they can enhance/suppress HIV entry. Functional genetic polymorphisms are known to occur in these proteins that affect their levels of expression and therefore might modulate their molecular interactions.
Figure 3.

Diagrammatic representation of binding of HIV to chemokine coreceptors CCR5 and CXCR4 expressed on the CD4 T cell. The level of expression of chemokine receptor CCR5 and its ligands MIP‐1α, MIP‐1β, and RANTES; and receptor CXCR4 and its ligand SDF‐1 may enhance or suppress risk of HIV infection. MIP‐1, macrophage inflammatory protein‐1; RANTES, regulated upon activation, normally T‐expressed, and presumably secreted.
Of the known 50 genes for chemokines, 16 corresponding to the CC chemokine family are clustered together on chromosome 17q12. The primary HIV coreceptor CCR5 and CCR2 are located on chromosome 3p21 and constitute the extended CCR5 haplogroups.
CCR2: A valine‐to‐isoleucine substitution at position 64 (V64I or G190A) in the first transmembrane region of CCR2 has been associated with delayed progression to AIDS by ∼2–4 years than homozygous individuals carrying the wild‐type allele (12). The protective allele ‘A’ occurs at a population frequency of 8%–10% among Caucasians, 15%–17% in Chinese, ∼12% in the North Indians (13), and 3%–17% in the South Indian populations (14). In an earlier study, Louisirirotchanakul et al. (15) reported that CCR264I homozygosity is associated with a reduced risk of acquiring infection among the HIV‐discordant couples in Thailand. However, in the Indian population, this polymorphism was not found to be protective against HIV‐1 (13). Because the genetic loci for CCR2 and CCR5 are in strong linkage disequilibrium, their combined analysis is greatly helpful to define CCR2–CCR5 extended haplotypes for disease association purposes.
CCR5: Multiple polymorphic variations have been described in the CCR5 gene (16). Of these, a natural knockout deletion of 32 bases, i.e. CCR5 Δ32, introduces a premature stop codon resulting in truncated protein product (17). In homozygous state, this deletion provides near complete protection in HIV‐infected individuals and up to 2–4 years delay in disease progression in the heterozygous state 17, 18. The CCR5 Δ32 is found at a frequency of 10%–16% in Northern Europe, and this frequency decreases in a southeast cline toward Mediterranean and gradually disappears in the African and Asian populations. It is extremely rare or almost absent in the Indian population (19).
Based on a unique constellation of additional multisite polymorphisms in CCR5 regulatory 5′ region, a number of CCR5 haplotypes have been identified 16, 20, as illustrated in Figure 4. These have been designated based on the nucleotide position in the 5′ UTR region and are referred to as haplogroups A to G. Of these, HHE has been associated with accelerated disease progression in Caucasians (16) and Thais (21) but not in African Americans. In the latter, it is the HHD haplotype that shows positive association with fast progression to AIDS (22). Similarly, HHC has been associated with fast disease progression in African Americans but with slow rate of progression in the Thai population. The HHG*2 and HHF*2 have been associated with slower progression among Caucasian and African American populations, respectively, plausibly because of the protective effects of Δ32 and CCR264I, respectively. In the Indian population, HHE has been implicated with susceptibility to infection and development of AIDS (23). Whether this haplotype also influences disease progression is not clear because long‐term follow‐up is desirable to reach such a conclusion.
Figure 4.

Organization of CCR2 and CCR5 haplotypes on chromosome 3p21 and their association with HIV disease progression. There are seven haplogroups encompassing single nucleotide polymorphisms corresponding to CCR2 V64I, CCR5 cis region, and the CCR5 deletion polymorphism. @Numbering system 1 is based on GenBank accession numbers AF031236 and AF031237 (15), whereas 2 is relative to the translational site and 3 is based on accession number U95626. #The promoter alleles (P1 to P10) are numbered according to Martin et al. (20).
CCL3–CCL4–CCL18 cluster: Genes coding for CCL3 (MIP‐1α), CCL4 (MIP‐1β), and CCL18 [pulmonary and activation‐regulated chemokine (PARC)] are clustered together within a 47‐kb region on chromosome 17q12. These are potent chemokines produced by macrophages, NK cells, fibroblasts, and T cells. Of these, CCL3 and CCL4 are natural ligands for the primary HIV coreceptor CCR5, and their genetic polymorphisms have been implicated in HIV‐1 acquisition and disease progression (24), although these associations are complicated because of strong linkage disequilibrium between them.
MIP‐1 is a monokine that is involved in the acute inflammatory state in the recruitment and activation of polymorphonuclear leukocytes. MIP‐1α (or the small inducible cytokine A3: SCYA3) and MIP‐1β (or the small inducible cytokine A4: SCYA4) are ∼60% identical at protein level, their genes are separated by 14 kb, and they are organized in a head‐to‐head fashion. The gene for CCL18, also referred to as the small inducible cytokine subfamily A member 18 (SCYA18) or the PARC, contains three exons and is 7.2 kb long. It is expressed at high levels in the lung, in alveolar macrophages, and in follicular dendritic cells of the germinal centers of lymph nodes. It is involved in the recruitment of Th2 cells and basophils in asthma allergic reactions and is upregulated by IL‐4.
CCL3L1: Human CC chemokine ligand 3 like 1 gene (CCL3L1) also referred to as the macrophage inflammatory protein‐1α (MIP‐1αP) or the small inducible cytokine A3‐like 1 (SCYA3L1) is a natural ligand of HIV coreceptor CCR5 and a potent HIV‐1 suppressive chemokine that can physically block the entry of HIV‐1. It is located on human chromosome 17q11.2 (Figure 5) and shares 96% amino acid homology with CCL3. Although the CCL3 gene exists as a single copy per haploid genome, the copy number of the CCL3L1 gene varies among different individuals and population groups.
Figure 5.

Comparative genomic analysis of CCL3L1 copy numbers in different populations compared with the chimpanzee. (A) Gene map showing localization of CCL3L1 and other chemokines on chromosome 17q12. (B) Copy number variations in CCL3L1 in the Japanese, Asian Indian, and African populations 25, 28.
The CCL3L1 and CCL4L1 genes harbor several single nucleotide polymorphisms (SNPs) and hotspots for duplication, resulting in distinct haplotypes and copy number variations, respectively, in different individuals (24). The copy numbers are the highest in Africans, followed by Asians, Amerindians, Central/South Asians, Middle East individuals and Europeans (25). It has been suggested that because CCL3L1 is a potent ligand of CCR5, variations in copy numbers and thus their expression might influence entry of the virus. Using real‐time quantitative polymerase chain reaction, Townson et al. (26) reported that CCL3L1 occurs as two copies per diploid genome in most Caucasians (49%), although 10%–20% individuals could have one, three, or four copies.
Copy number variations in CCL3L1 have been reported to be associated with susceptibility to HIV‐1 infection 25, 27. However, such variations do not seem to have a significant effect on disease progression (27). Furthermore, subjects with two or less copies of CCL3L1 have significantly higher risk of acquiring HIV infection. A recent study carried out in the Japanese population (27) has shown that the average copy number of CCL3L1 in the HIV‐1‐infected subjects is significantly lower than that in healthy controls (3.35 ± 0.24 vs 5.00 ± 0.22, P < 0.001). On the contrary, studies in the North Indian population showed that the copy number variation in CCL3L1 has no effect on acquisition (2.13 ± 0.15 in HIV‐infected individuals compared with 2.34 ± 0.17 in healthy controls) (28). It is worth mentioning that chimpanzees that are naturally resistant to HIV‐1 possess very high (∼9–10) CCL3L1 copy numbers 25, 29.
Based on the combination of genotypes for CCL3L1 copy numbers and CCR5 deletion mutation, genetic risk groups can be defined that could help assess AIDS risk to HIV infection 30, 31. Individuals with CCL3L1 high copy numbers and CCR5 deletion point toward low risk compared with those with CCL3L1 low copy numbers and CCR5 non‐deletion genotypes. Furthermore, these genotypes have been shown to influence CD4 recovery and immune reconstitution after highly active antiretroviral therapy (31). A recent study reported a correlation between higher CCL3L1 copy numbers and greater CD4+ and CD8+ T‐cell responses to HIV‐1 gag protein in the African population (32).
RANTES: The chemokine RANTES, also referred to as small inducible cytokine A5 (SCYA5), is not only a mediator of inflammation but also a significant regulator of differentiation in development. It is a natural ligand for CCR1, CCR3, and CCR5 and a potent inhibitor of infection by NSI CCR5 tropic HIV‐1. It has been reported that a transversion of −28C to G in the promoter of RANTES increases its transcription, and this polymorphism has been associated with delayed progression to AIDS in the Japanese population (33). Another SNP, In1.1T/C (168923T/C), is found in a regulatory element in the first intron of RANTES. The 1n1.1C is associated with downregulated expression and has been implicated in accelerated progression to AIDS in African Americans and European Americans (34). A recent study reported that the RANTES In1.1 T allele and haplotype II (ACT) may be a risk factor for HIV‐1 transmission, while RANTES In1.1 C allele may confer risk for disease progression among North Indians (35). These data need to be replicated in larger cohorts, also in other population groups.
SDF‐1: The SDF‐1 or the CXCL12 is a highly potent α chemokine that binds CXCR4 (NPY3R), a coreceptor for SI HIV‐1 strains. It induces intracellular actin polymerization in lymphocytes and plays an important role in basal extravazation of lymphocytes and monocytes. It is therefore involved directly in regulating immune surveillance rather than in inflammation. SDF1–CXCR4 interactions are essential for the homing and retention of hematopoietic progenitor cells in the bone marrow and have been shown to control the navigation of progenitor cells between the bone marrow and the blood. SDF1 is a ligand for CXCR4 and a potent entry inhibitor for SI viruses that generally emerge during late‐stage HIV infection.
The gene for SDF‐1 is ∼10 kb long and located on human chromosome 10q11.1. It exists in two isoforms: alpha and beta obtained as a consequence of alternative splicing. The alpha form is derived from exons 1–3, while the beta form contains additional sequence from exon 4. A common polymorphism, i.e. presence of A at the SNP G801A in the 3′ UTR of SDF‐1β, has been shown to have a recessive protective effect against HIV‐1 and associated with delayed onset of AIDS (36). The 801A allele is found at a frequency of 0.211 in Caucasians, 0.160 in Hispanics, 0.057 in African Americans, 0.257 in Asians, and 0.24 in case of North Indians 37, 38 and 0.117–0.35 in South Indians (13).
CXCR1–CXCR2: CXCR1 (IL‐8RA) and CXCR2 (IL‐8RB) are receptors for IL‐8, a proinflammatory cytokine involved in chemoattraction and activation of neutrophils. Their genes are colocalized in a region spanning ∼26 kb on chromosome 2q35 (Figure 6) and have ∼25% similarity in sequence to other neutrophil chemoattractants, fMet‐Leu‐Phe and C5a. Studies have indicated a dysregulated elevated IL‐8 production and reduced expression of IL‐8 receptors, CXCR1 and CXCR2, in HIV‐1‐infected patients. Reduced CXCR1 activity upon HIV‐1 infection because of cross‐receptor‐mediated internalization with the major coreceptors CCR5 and CXCR4 has also been reported (39). These observations suggest that CXCR1 and CXCR2 could affect AIDS‐related conditions.
Figure 6.
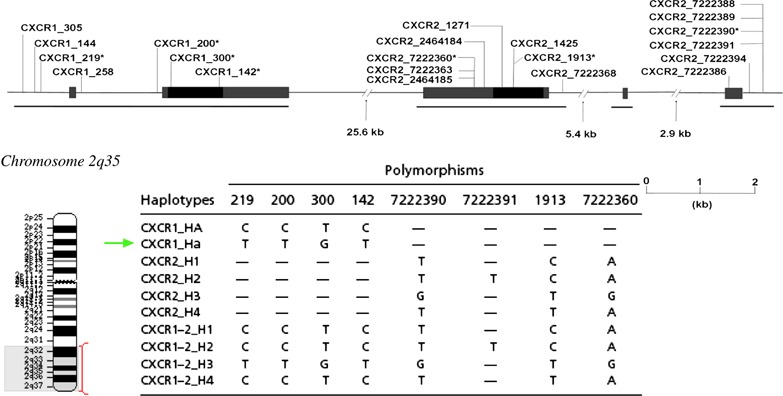
Gene map of CXCR1 and CXCR2 on chromosome 2q35 with polymorphic positions and CXCR1 haplotypes indicated. At least 10 CXCR1 haplotypes are known (40). Arrow indicates haplotype ‘Ha’ that has been associated with delayed progression in Caucasians.
The CXCR1 and CXCR2 genes consist of several polymorphisms and, depending on their linkage disequilibrium, have been classified into distinct haplotypes (40). Two SNPs T92G (CXCR1 −300) and C1003T (CXCR1 −142), in particular, result in nonsynonymous substitutions M31R in the N‐terminal extracellular domain and R335C in the C‐terminal intracellular domain, respectively. These two SNPs are an integral part of the CXCR1 haplotype ‘Ha’. A recent genetic study on the French GRIV cohort composing of 84 rapid and 253 slow progressors identified a strong association of CXCR1 haplotype Ha with protection against rapid progression to AIDS (40). It was suggested that the inhibitory effect of CXCR1 Ha could be mediated by suppressing CD4 and CXCR4 expression. This could be caused by either the direct involvement of palmitoylation of the cysteine introduced in the C‐terminus of CXCR1 or some other unknown CXCR1‐mediated signaling that regulate expression and intracellular trafficking of CD4 and CXCR4.
Duffy antigen receptor for chemokines
It has recently been shown that the DARC, alternatively called as Glycoprotein D, conventionally used in the Duffy‐based blood grouping system and a known receptor for P vivax, can aid HIV‐1 attachment to RBCs and its trans‐infection to target cells (41). The Duffy system is located on chromosome 1q21‐22. The −46T‐C SNP in particular, in the promoter of DARC, is widely prevalent in populations of African descent, and its homozygous −46CC genotype results in selective loss of DARC expression on RBCs. This genotype has been shown to be associated not only with a high risk of HIV‐1 infection but, on the contrary, also with slower progression in terms of death or development of dementia. It has been suggested that there is interplay between DARC and chemokines that ultimately determine the amount of free virus bound to RBCs for eventual trans‐infection.
Dendritic cell‐specific intracellular adhesion molecule‐3‐grabbing nonintegrin
DC‐SIGN (CD209) and its related protein DC‐SIGNR are C‐type lectins known to bind multiple pathogens such as HIV‐1, HIV‐2, Mycobacterium tuberculosis, Helicobacter pylori, Klebsiella pneumoniae, dengue virus, Ebola virus, hepatitis C virus, cytomegalovirus, measles, coronavirus, Leishmania pifanoi, Shistosoma mansoni, and Candida albicans. The DC‐SIGN and DC‐SIGNR show 77% homology at amino acid sequence level, act as cell adhesion and pathogen recognition receptors, and are involved in both innate and adaptive immunity. The two genes appear to have originated from a common ancestral gene by duplication and are therefore located in close proximity in head‐to‐head orientation on chromosome 19p13.
Polymorphisms in DC‐SIGN and DC‐SIGNR genes have recently been studied in Caucasian and African populations, and several novel SNPs were identified in 5′ and 3′ UTR regions (42). These investigators have reported several significant differences in the prevalence of different alleles and haplotypes in the two population groups. In general, the African population shows greater genetic diversity in DC‐SIGN than other populations. The DC‐SIGN promoter variant −336C affects an Sp1‐binding site and leads to decreased expression of DC‐SIGN. This particular allele has been associated with susceptibility to parenteral HIV‐1 infection in the European Americans (43). This association has not been reported in patients known to be at risk for mucosally acquired infection.
A total of eight DC‐SIGN haplotypes have been reported (Table 3). Of these, haplotype 4, which contains all the high‐risk SNPs, is associated with parenterally acquired infection (OR = 2.62, P = 0.018). On the other hand, the haplotype containing all the protective SNPs (−139T, −336T, −939T, −1180T, and −1466T) is quite rare (frequency ≤ 0.001), making it difficult to test its influence on the actual risk following HIV‐1 infection. Although this study did not find any significant association with HIV‐1 disease progression, further evidence would be required to suggest that DC‐SIGN variants that bind the envelope glycoprotein of both R5 and X4 viruses more efficiently might also lead to enhanced virus infection.
Table 3.
Major DC‐SIGN haplotypes identified in the promoter region and investigated in HIV infection and disease progression studies [adapted from Martin et al. (43)]
| Haplotype | Nucleotide at position | ||||||
|---|---|---|---|---|---|---|---|
| −139 | −336 | −939 | −1089 | −1180 | −1466 | −1530 | |
| 1 | C | T | T | C | T | C | A |
| 2 | T | T | C | C | A | T | A |
| 3 | C | T | T | C | T | T | A |
| 4 | C | C | C | C | A | C | A |
| 5 | T | T | C | A | A | T | A |
| 6 | C | C | C | C | A | C | C |
| 7 | T | T | C | C | A | C | A |
| 8 | C | C | C | C | T | C | A |
DC‐SIGN, Dendritic cell‐specific intracellular adhesion molecule‐3‐grabbing nonintegrin.
Genes involved in anti‐HIV immune response
In addition to the genes that act at the level of viral entry, there are others that are critically involved in HIV restriction and influence the ensuing immune response. These include, for example, the antiviral APOBEC3G gene family and the virus restriction factor TRIM5α that are described briefly in the following section.
Apolipoprotein B mRNA‐editing enzyme, catalytic polypeptide‐like 3G
APOBEC3G is a host antiviral factor. It is a cytidine deaminase that induces G‐to‐A hypermutation in newly synthesized retroviral DNA, resulting in instability of the nascent viral transcripts or lethal mutations (44). However, this APOBEC3G‐mediated innate immune mechanism is counteracted by the HIV‐1 vif, thus allowing the virus to escape through mutations and even develop drug resistance to lamivudine (3TC) (45). It has been shown that residues asp128 in APOBEC3G in humans and lys128 in African green monkeys are involved in binding with viral vif, resulting in its degradation. Furthermore, an interchange of the asp128 with lys128 in human APOBEC3G switches its sensitivity from HIV vif to SIV vif (46). APOBEC3G functions as a potent post‐entry restriction factor for HIV‐1 in naïve CD4 T cells but is not protective in activated CD4 T cells. In the latter, it is recruited into an enzymatically inactive high‐molecular‐mass ribonucleoprotein complex that depends on RNAse for its conversion to its low‐molecular‐mass and active form. This inactivated form of APOBEC3G following T‐cell activation has been correlated with permissivity for HIV infection (47). It has been suggested that creation of HIV vif resistant APOBEC3G mutations could provide an effective gene therapy approach against the virus (48).
The gene for APOBEC3G exists as a tandem array of seven APOBEC genes or pseudogenes on chromosome 22q12‐q13.2. It has been reported that the antiviral activity of the APOBEC3H has been lost in the majority of human populations excluding Africans through two polymorphisms, and this may have important consequences for susceptibility to retroviral infections (49).
Tripartite interaction motif 5 α
The antiviral factor TRIM5α protects humans and other primates against a broad range of retroviruses in a species‐specific manner. It targets the capsid molecules of the incoming retrovirus in the cytosol and disrupts its replication. Human TRIM5α can block N‐tropic Murine leukemia virus (MLV) and equine anemia virus, but is much less efficient in restricting HIV‐1 replication, although TRIM5α escape mutants have been identified late in infection. Two SNPs (H43Y and R136Q) have been reported to affect the antiviral activity of this restriction factor. The 43Y allele is believed to impair the E3 ligase activity of TRIM and favors progression to AIDS. The other polymorphism 136Q is associated with greater anti‐HIV activity and has earlier been reported to be more frequently observed in high‐risk seronegative African Americans as compared with HIV seropositive patients. Recently, van Manen et al. (50) have analyzed the effect of polymorphisms in TRIM5α on the clinical course of HIV‐1 infection in homosexual men enrolled in Amsterdam cohort studies (ACS). The study reported an accelerated disease progression in individuals homozygous for 43Y and established an in vitro protective effect of 136Q against the X4 viruses that infect naïve T cells. Furthermore, these investigators reported that the 136Q allele along with −2GG genotype in the 5′ UTR of TIM5α is associated with rapid progression to AIDS.
Conclusions
A preventive vaccine against HIV/AIDS still eludes researchers but would possibly involve both cellular and humoral immune responses against the virus. It is expected that a first generation vaccine against HIV would be able to reduce viremia to <2000 copies RNA per milliliter and/or prevent disease progression, thereby resulting in practically improved disease outcome and reduced transmission.
Genetic determinants of HIV infection and progression to AIDS can be grouped into two broad categories. First category involves those genes that govern viral entry and hence susceptibility. These include chemokine receptors and their ligands as well as others like DC‐SIGN, DARC, APOBEC3G, and TRIM5. The second category primarily influences the immune response following HIV infection and includes immune response genes in the MHC region, KIRs, and the genetic polymorphisms in cytokines.
Genetic studies aimed at identifying host factors associated with natural resistance against the retrovirus would be helpful in developing screening tests that could identify people who have a greater or lower predisposition to HIV infection and who could progress at fast/slow pace to AIDS. Furthermore, genetic testing could help in clinical diagnosis of drug hypersensitivity reactions prior to treatment and could possibly be translated in avoiding dug toxicities at an individual level. Because genetic variability is involved at several steps of the cascade of antiviral immune surveillance, these could help in predicting vaccine responsiveness in a population‐specific manner and help in our understanding of disease pathogenesis.
Nevertheless, several key issues have recently emerged through multidisciplinary approaches involving intensive international collaborations. It is clear that HIV‐1/AIDS pathogenesis involves complex counter‐interactive responses mediated by both viral and host‐derived factors. These are manifested differently in different individuals depending on host genomic and immunomic architecture. A number of host‐derived factors have been identified through case–control studies and multi‐cohort meta‐analyses. Whole‐genome scans, functional genomics, and other experimental approaches have provided new insights into our understanding of the host–pathogen interactions. Future studies focused primarily on the mechanisms of protection from the virus in naturally resistant people, especially the elite controllers could unravel the enigmatic approaches that could be used in favorable prognosis, vaccine design, therapy, and clinical practice.
Acknowledgments
The authors wish to thank the Department of Biotechnology and the Indian Council of Medical Research, Government of India, for their financial support. The training obtained by GK through the Fogarty’s AIDS International Training and Research Program fellowship program at the New York School of Medicine is gratefully acknowledged.
References
- 1. Visciano ML, Tuen M, Chen PD, Hioe CE. Antibodies to the CD4‐binding site of HIV‐1 gp120 suppress gp120‐specific CD4 T cell response while enhancing antibody response. Infect Agent Cancer 2008: 18: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kaur G, Tuen M, Virland D et al Antigen stimulation induces HIV envelope gp120‐specific CD4+ T cells to secrete CCR5 ligands and suppress HIV infection. Virology 2007: 369: 214–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vasiliver‐Shamis G, Tuen M, Wu T et al HIV‐1 envelope gp120 induces a stop signal and virological synapse formation in non‐infected CD4 T cells. J Virol 2008: 82: 9445–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Telenti A, Goldstein DB. Genomics meets HIV. Nat Rev Microbiol 2006: 4: 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Winkler C, An P, Brien SJO. Patterns of ethnic diversity among the genes that influence AIDS. Hum Mol Genet 2004: 13: R9–R19. [DOI] [PubMed] [Google Scholar]
- 6. Deeks SG, Walker BD. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity 2007: 27: 406–16. [DOI] [PubMed] [Google Scholar]
- 7. Baker BM, Block BL, Rothchild AC, Walker BD. Elite control of HIV infection: implications for vaccines and treatments. Expert Opin Biol Ther 2009: 9: 55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brass AL, Dykxhoorn DM, Benita Y et al Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008: 319: 921–6. [DOI] [PubMed] [Google Scholar]
- 9. O’Brien SJ, Nelson GW. Human genes that limit AIDS. Nat Genet 2004: 36: 565–74. [DOI] [PubMed] [Google Scholar]
- 10. Fellay J, Shianna KV, Ge D et al A whole‐genome association study of major determinants for host control of HIV‐1. Science 2007: 317: 944–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Loeuillet C, Deutsch S, Cluffi A et al In vitro whole‐genome analysis identifies a susceptibility locus for HIV‐1. PLoS Biol 2008: 6: e32. doi:10.1371/journal.pbio.0060032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smith MW, Dean M, Carrington M et al Contrasting genetic influence of CCR2 and CCR5 variants on HIV‐1 infection and disease progression. Hemophilia Growth and Development Study (HGDS), Multicenter AIDS Cohort Study (MACS), Multicenter Hemophilia Cohort Study (MHCS), San Francisco City Cohort (SFCC), ALIVE Study. Science 1997: 277: 959–65. [DOI] [PubMed] [Google Scholar]
- 13. Kaur G, Singh P, Kumar N et al Distribution of CCR2 polymorphism in HIV‐1 infected and healthy subjects in North India. Int J Immunogenet 2007: 34: 153–6. [DOI] [PubMed] [Google Scholar]
- 14. Ramana GV, Vasanthi A, Khaja M et al Distribution of HIV‐1 resistance‐conferring polymorphic alleles SDF‐1‐3’A, CCR2‐64I and CCR5‐Delta32 in diverse populations of Andhra Pradesh, South India. J Genet 2001: 80: 137–40. [DOI] [PubMed] [Google Scholar]
- 15. Louisirirotchanakul S, Liu H, Roongpisuthipong A et al Genetic analysis of HIV‐1 discordant couples in Thailand: association of CCR2 64I homozygosity with HIV‐1 negative status. J Acquir Immune Defic Syndr 2002: 29: 314–5. [DOI] [PubMed] [Google Scholar]
- 16. Gonzalez E, Bamshad M, Sato N et al Race‐specific HIV‐1 disease‐modifying effects associated with CCR5 haplotypes. Proc Natl Acad Sci U S A 1999: 96: 12004–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu R, Paxton WA, Choe S et al Homozygous defects in HIV‐1 coreceptor accounts for resistance of some multiply‐exposed individuals to HIV‐1 infection. Cell 1996: 86: 367–77. [DOI] [PubMed] [Google Scholar]
- 18. Dean M, Carrington M, Winkler C et al Genetic restriction of HIV‐1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science 1996: 273: 1856–62. [DOI] [PubMed] [Google Scholar]
- 19. Singh P, Kaur G, Sharma G, Mehra NK. Immunogenetic basis of HIV‐1 infection, transmission and disease progression. Vaccine 2008: 26: 2966–80. [DOI] [PubMed] [Google Scholar]
- 20. Martin MP, Dean M, Smith MW et al Genetic acceleration of AIDS progression by a promoter variant of CCR5. Science 1998: 282: 1907–11. [DOI] [PubMed] [Google Scholar]
- 21. Nguyen L, Li M, Chaowanachan T et al CCR5 promoter haplogroups associated with HIV‐1 discordant progression in Thai injection drug users. AIDS 2004: 18: 1327–33. [DOI] [PubMed] [Google Scholar]
- 22. Kostrikis LG, Neumann AU, Thomson B et al A polymorphism in the regulatory region of the CC chemokine receptor 5 gene influences perinatal transmission of HIV 1 to African American infants. J Virol 1999: 73: 10264–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaur G, Singh P, Rapthap CC et al Polymorphism in the CCR5 gene promoter and HIV‐1 infection in North Indians. Hum Immunol 2007: 68: 454–61. [DOI] [PubMed] [Google Scholar]
- 24. Modi WS, Lautenberger J, An P et al Genetic variation in the CCL18‐CCL3‐CCL4 chemokine gene cluster influences HIV type 1 transmission and AIDS disease progression. Am J Hum Genet 2006: 79: 120–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gonzalez E, Kulkarni H, Bolivar H et al The influence of CCL3L1 gene‐containing segmental duplications on HIV‐1/AIDS susceptibility. Science 2005: 307: 1434–40. [DOI] [PubMed] [Google Scholar]
- 26. Townson JR, Barcellos LF, Nibbs RJB. Gene copy number regulates the production of the human chemokine CCL3‐L1. J Immunol 2002: 32: 3016–26. [DOI] [PubMed] [Google Scholar]
- 27. Nakajima T, Ohtani H, Naruse T et al Copy number variations of CCL3L1 and long‐term prognosis of HIV‐1 infection in asymptomatic HIV‐infected Japanese with hemophilia. Immunogenetics 2007: 59: 793–8. [DOI] [PubMed] [Google Scholar]
- 28. Nakajima T, Kaur G, Mehra N, Kimura A. HIV‐1/AIDS susceptibility and copy number variation in CCL3L1, a gene encoding a natural ligand for HIV‐1 co‐receptor CCR5. Cytogenet Genome Res (in press). [DOI] [PubMed] [Google Scholar]
- 29. Shao W, Tang J, Song W et al CCL3L1 and CCL4L1: variable gene copy number in adolescents with and without HIV‐1 infection. Genes Immun 2007: 8: 224–31. [DOI] [PubMed] [Google Scholar]
- 30. Kulkarni H, Marconi VC, Agan BK et al Role of CCL3L1‐CCR5 genotypes in the epidemic spread of HIV‐1 and evaluation of vaccine efficacy. PLoS ONE 2008: 3: e3671. doi:10.1371/journal.pone.0003671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ahuja SK, Kulkarni H, Catano G et al CCL3L1‐CCR5 genotype influences durability of immune recovery during antiretroviral therapy of HIV‐1 infected individuals. Nat Med 2008: 14: 413–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shalekoff S, Meddows‐Taylor S, Schramm DB et al Host CCL3L1 gene copy number in relation to HIV‐1 specific CD4+ and CD8+ T cell responses and viral load in South African women. J Acquir Immune Defic Syndr 2008: 48: 245–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu H, Chao D, Nakayama EE et al Polymorphism in RANTES chemokine promoter affects HIV‐1 disease progression. Proc Natl Acad Sci U S A 1999: 96: 4581–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. An P, Nelson GW, Wang L et al Modulating influence on HIV/AIDS by interacting RANTES gene variants. Proc Natl Acad Sci U S A 2002: 99: 10002–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rathore A, Chatterjee A, Sivarama P, Yamamoto N, Singhal PK, Dhole TN. Association of RANTES ‐403 G/A, ‐28 C/G and In1.1 T/C polymorphism with HIV‐1 transmission and progression among North Indians. J Med Virol 2008: 80: 1133–41. [DOI] [PubMed] [Google Scholar]
- 36. Winkler C, Modi W, Smith MW et al Genetic restriction of AIDS pathogenesis by an SDF‐1 chemokine gene variant. Science 1998: 279: 389–93. [DOI] [PubMed] [Google Scholar]
- 37. Ramamoorti N, Kumarvelu J, Shanmugasundaram GK, Rani K, Banerjea AC. 2001 High frequency of G to A transition mutation in the stromal cell derived factor‐1 gene in India, a chemokine that blocks HIV‐1 (X4) infection: multiple proteins bind to 3’‐untranslated region of SDF‐1 RNA. Genes Immun 2001: 2: 408–10. [DOI] [PubMed] [Google Scholar]
- 38. Suresh P, Wanchu A, Sachdeva RK, Bhatnagar A. Gene polymorphisms in CCR5, CCR2, CX3CR1, SDF‐1 and RANTES in exposed but uninfected partners of HIV‐1 infected individuals in North India. J Clin Immunol 2006: 26: 476–84. [DOI] [PubMed] [Google Scholar]
- 39. Richardson RM, Tokunaga K, Marjoram R, Sata T, Snyderman R. Interleukin‐8‐mediated heterologous receptor internalization provides resistance to HIV‐1 infectivity. Role of signal strength and receptor desensitization. J Biol Chem 2003: 278: 15867–73. [DOI] [PubMed] [Google Scholar]
- 40. Vasilescu A, Terashima Y, Enomoto M et al A haplotype of the human CXCR1 gene protective against rapid disease progression in HIV‐1+ patients. Proc Natl Acad Sci U S A 2007: 104: 3354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. He W, Neil S, Kulkarni H et al Duffy antigen receptor for chemokines mediates trans‐infection of HIV‐1 from red blood cells to target cells and affects HIV‐AIDS susceptibility. Cell Host Microbe 2008: 4: 52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Boily‐Larouche G, Zijenah LS, Mbizvo M, Ward BJ, Roger M. DC‐SIGN and DC‐SIGNR genetic diversity among different ethnic populations: potential implications for pathogen recognition and disease susceptibility. Hum Immunol 2007: 68: 523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Martin MP, Lederman MM, Hutcheson HB et al Association of DC‐SIGN promoter polymorphism with increased risk for parenteral, but not mucosal, acquisition of human immunodeficiency virus type 1 infection. J Virol 2004: 78: 14053–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang J, Webb DM. Rapid evolution of primate antiviral enzyme APOBEC3G. Hum Mol Genet 2004: 13: 1785–91. [DOI] [PubMed] [Google Scholar]
- 45. Mulder LC, Harari A, Simon V. Cytidine deamination induced HIV‐1 drug resistance. Proc Natl Acad Sci U S A 2008: 105: 5501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schrofelbauer B, Chen D, Landau NR. A single amino acid of APOBEC3G controls its species‐specific interaction with virion infectivity factor (Vif). Proc Natl Acad Sci U S A 2004: 101: 3927–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chiu YL, Soros VB, Kreisberg JF, Stopak K, Yonemoto W, Greene WC. Cellular APOBEC3G restricts HIV‐1 infection in resting CD4+ T cells. Nature 2005: 435: 108–14. [DOI] [PubMed] [Google Scholar]
- 48. Xu H, Svarovskaia ES, Barr R et al A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV‐1 virion infectivity factor‐induced depletion. Proc Natl Acad Sci U S A 2004: 101: 5652–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. OhAinle M, Kerns JA, Li MM, Malik HS, Emerman M. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe 2008: 4: 249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Van Manen D, Rits MA, Beugeling C, Van Dort K, Schuitemaker H, Kootstra NA. The effect of Trim5 polymorphisms on the clinical course of HIV‐1 infection. PLoS Pathog 2008: 4: e18. [DOI] [PMC free article] [PubMed] [Google Scholar]