

Abstract
All single‐stranded ‘positive‐sense’ RNA viruses that infect mammalian, insect or plant cells rearrange internal cellular membranes to provide an environment facilitating virus replication. A striking feature of these unique membrane structures is the induction of 70–100 nm vesicles (either free within the cytoplasm, associated with other induced vesicles or bound within a surrounding membrane) harbouring the viral replication complex (RC). Although similar in appearance, the cellular composition of these vesicles appears to vary for different viruses, implying different organelle origins for the intracellular sites of viral RNA replication. Genetic analysis has revealed that induction of these membrane structures can be attributed to a particular viral gene product, usually a non‐structural protein. This review will highlight our current knowledge of the formation and composition of virus RCs and describe some of the similarities and differences in RNA‐membrane interactions observed between the virus families Flaviviridae and Picornaviridae.
Keywords: cellular ultrastructure, membrane rearrangements, virus‐host interactions, virus RNA replication
Membrane wrapping is an important principle that many viruses use during their life cycles. As a virologist, I have learnt to appreciate that many years of evolution have ‘educated’ viruses in the ways of the cell. We have learnt many new aspects of cell biology from existing virus models, and I am sure that many more insights are yet to come. In this context, various laboratories have toiled long and hard to understand the cellular principles associated with virus replication. All RNA viruses, whether they infect mammalian, insect or plant cells, induce membrane structures. Replication of positive‐sense RNA viruses is intimately linked to unique membrane structures that ultimately wrap around the active replication complexes (RCs), providing a membrane‐bounded microenvironment in which RNA synthesis can occur. In many cases, these consist of small vesicles, approximately 70–100 nm in diameter, that accumulate in the perinuclear region of infected cells 1, 2, 3, 4, 5, 6, 7). These vesicles or spherules appear to be common to a number of virus families including alphaviruses (e.g. Semliki Forest virus), rubiviruses (e.g. Rubella virus), alphavirus‐like superfamily (e.g. Brome mosaic virus), flaviviruses (e.g. Kunjin virus), tombusviruses (e.g. Carnation Italian ringspot virus) and alphanodaviruses (e.g. Flock House virus) 4, 5, 6, 8, 9, 10, 11, 12, 13, 14, 15, 16). In each case, the spherules/vesicles themselves present as invaginations of a cellular membrane, of differing origins. Thus, the lumen is enclosed or bounded by a membrane but still has access to cytoplasmic constituents via an open neck. The presence of readily visible threads within these spherules/vesicles has lead to the belief that they contain the viral RNA. This idea has been strengthened by immunolabelling the spherules/vesicles with antibodies to double‐stranded RNA (5, 6, 10, 17) or exogenously added bromouridine (4, 14, 18), in situ hybridization (19, 20) or treatment of infected cells with RNAse leading to the digestion and thus absence of the threads after embedding and microscopy (21). Hepatitis C virus (HCV), a member of the Hepacivirus genera within the Flaviviridae family of viruses, appears to induce more heterogenously sized vesicles within the perinuclear region (1, 22). These vesicles were also shown to contain the viral RNA by in situ hybridization and in vivo labelling with bromouridine triphosphate (22). In contrast to the above morphology, coronaviruses and arteriviruses induce double‐membrane vesicles (DMV) of approximately 80–100 nm in diameter (7, 23, 24). In situ experiments with BrUTP and conjugated riboprobes have established that these DMVs house the replicating RNA (7, 23, 24). Picornaviruses on the other hand still induce dramatic cytoplasmic vesiculation of cellular membranes to wrap the active replicating viral RNA. However, these membranes are much more heterogenous in size and shape (3, 25, 26, 27). It is quite clear though that in each virus family the RC becomes surrounded by cellular membranes. More than likely, this wrapping of the RC ensures protection from host‐response proteins recognizing the viral RNA, i.e. protein kinase R, but in addition also provides a stable and confined surface area for the polymerase and RC to assemble and function. As most of these structures appear morphologically similar, common modes of biogenesis must exist that are ‘shared’ by the virus families. It appears that almost each genera has its own way of exploiting host‐cell machinery. This is especially true for the picornaviruses.
As there have been two recent reviews on RCs of other virus families (28, 29), I will concentrate on two similar virus families – the Flaviviridae and the Picornaviridae that differ in their mechanisms of RNA replication. Both families contain members of great health concerns for humans and domesticated animals.
The flavivirus replication complex and associated membranes
The Flaviviridae family consists of three genera: Flavivirus, Pestivirus and Hepacivirus (30). As the majority of the studies investigating the RC have been analysed using the flavivirus Kunjin virus (KUNV) as a model, this review will only discuss the Flavivirus genus. The Flavivirus genus comprises over 60 species, by far the largest member of the Flaviviridae family. These include a range of pathogens, such as dengue virus (DENV), West Nile virus (WNV), Yellow fever virus (YFV) and Tick‐borne encephalitis virus (TBEV) that are important to both man and animals (30) (World Health Organization, Fact Sheet no. 117, 2002). Superficially, the flaviviruses appear to be relatively simple in structure and replication. They have a membrane and a genome consisting of an 11‐kb single positive‐sense RNA molecule that encodes one long open reading frame with no overlapping gene sequences or subgenomic RNA species. The translated proteins are post‐ and co‐translationally cleaved by both host and viral proteases to yield the mature proteins (Figure 1) (30). The genes encoding the three structural proteins core (C), premembrane (prM) and envelope (E) are the first to be translated and are the only proteins found in secreted virions. The remaining seven non‐structural (NS) proteins appear to play roles primarily in replication of the viral RNA (31); however, recent evidence suggests that some also contribute to virion assembly and/or maturation (32, 33).
Figure 1.
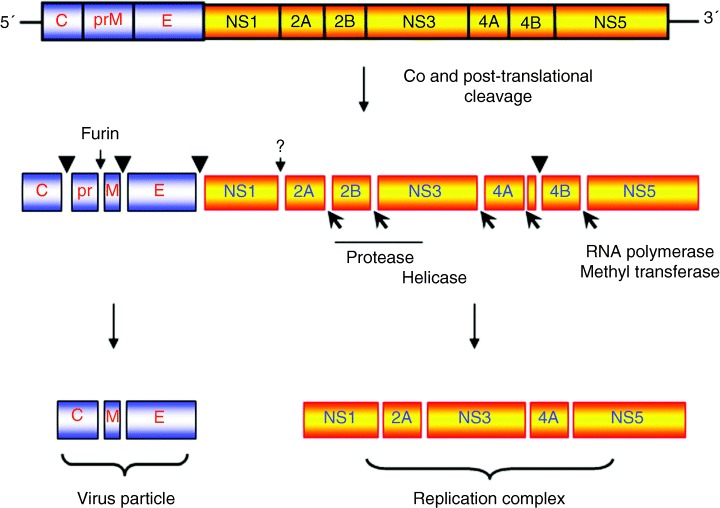
Schematic representation of the Flavivirus genome organization and polyprotein‐processing events associated with replication. Cleavage sites depicted with a ▾ represent cleavage by host‐signal peptidase within the lumen of the endoplasmic reticulum. Sites represented with a  depict cleavage by the viral‐encoded protease NS3 with cofactor NS2B. The cleavage between NS1 and NS2A is currently not well understood but is performed by a host‐cell protease in the lumen of the endoplasmic recticulum.
depict cleavage by the viral‐encoded protease NS3 with cofactor NS2B. The cleavage between NS1 and NS2A is currently not well understood but is performed by a host‐cell protease in the lumen of the endoplasmic recticulum.
Flavivirus infection of cultured cells is associated with a reasonably long latent period of infection, approximately 12 h (34). Subsequently, replication increases exponentially which is associated with the induction and proliferation of unique and characteristic cytoplasmic membrane structures (35). These membranes have been described as convoluted membranes (CM), paracrystalline arrays (PC) and small vesicular structures (SMS; after chemical fixation) or vesicle packets [VPs; after cryofixation (36); Figure 2]. Our studies with WNV strain KUNV have provided a comprehensive and detailed analysis of the composition and function of the individual membranes during KUNV replication (31, 37).
Figure 2.
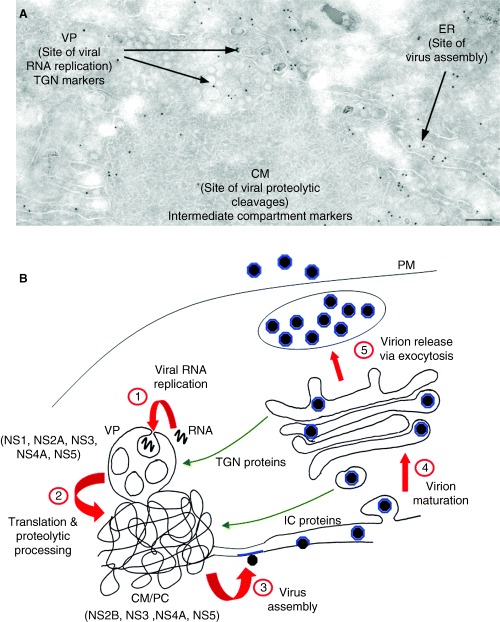
Ultrastructural observations of Kunjin virus‐infected Vero cells at 24 h post‐infection. Panel A reveals immunogold labelling of a thawed cryosection prepared from Kunjin virus‐infected cells with antibodies raised against protein disulphide isomerase (PDI) and 10 nm protein‐A gold. Note the labelling of the lumen of the rER continuous with the convoluted membranes (CM) but no labelling within this structure. Adjacent vesicle packets (VPs) are also devoid of the PDI antibodies. Magnification bar represents 200 nm. Schematic representation of the flavivirus life cycle is depicted in PanelB.Step 1 proposes the incorporation of cytoplasmic viral RNA into the VP for replication by the replication complex. (2) The transcribed viral RNA is then transported to the CM/PC for translation and proteolytic cleavage via both host‐ and viral‐encoded proteases. (3) Subsequently, the RNA is encapsidated by the core protein, and virion assembly proceeds via budding into the ER lumen. (4) Virion maturation occurs via transit through the Golgi apparatus before release through the plasma membrane (PM) via exocytosis (5). Redistribution of trans‐Golgi network (TGN) and intermediate compartment (IC) host proteins to the VP and CM/paracrystalline arrays (PC), respectively, are highlighted.
The first indication that these induced membranes were associated with RNA replication came from the seminal experiments by Chu and Westaway (38). Membrane fractions were isolated from cytoplasmic extracts separated by sucrose density centrifugation and analysed for protein composition and viral RNA species [either after direct separation or for RNA‐dependent RNA polymerase (RdRp) activity in the isolated fractions]. Electron microscopy (EM) of the RdRp‐active fractions revealed a striking correlation of KUNV NS proteins implicated in viral RNA synthesis with the KUNV‐induced membranes in infected cells. Immunolabelling of thawed cryosections prepared from DENV‐infected cells revealed a significant accumulation of NS1 within distinct morphological membrane structures, that we termed VP (6, 36). Double‐labelling with antibodies to dsRNA revealed co‐localization of both antibodies only within the VP, strongly suggesting the VPs were the intracellular sites of DENV RNA replication (6). In situ hybridization of resin‐embedded sections from DENV‐infected cells supported the proposal that the VPs are the same structure as the SMS (19). We could show that the NS proteins NS1, NS2A, NS3, NS4A, NS5 and dsRNA were all localized to VP (17, 39), whereas NS2B, NS3, NS4A and NS5 localized to CM/PC (17, 39), and NS4B localized to proliferated endoplasmic recticulum (ER) and the nucleus (40). Based on the known protease activity of NS3 that works in concert with its cofactor NS2B, we proposed the CM/PC to be sites of protein translation and proteolytic cleavage of the polyprotein by the KUNV protease NS2B‐NS3 (17). Whereas localization of dsRNA and KUNV proteins associated with RNA replication (17) indicated that the VPs constitute the sites of viral RNA synthesis. The role of VP in viral RNA synthesis was additionally confirmed by in situ labelling of nascent RNA with bromouridine (18).
The immunolocalization data, together with molecular and biochemical results 41, 42, 43, 44), led us to propose the following model of the events during flavivirus RNA replication (31). Our assumption is that on completion of translation, NS5 binds the 3′ end of the (+) RNA molecule. NS2A and NS3 then also bind to the RNA and/or NS5. This preforming RC is then directed to the cytoplasmic face of a membrane through interaction with two additional proteins, NS4A and NS2A. Importantly, this interaction only occurs in the presence of viral RNA (39). At this point, we predicted that the RC‐bound viral RNA becomes wrapped in the VP (step 1 in Figure 2B) and is contained within this invagination where efficient replication of the viral RNA continues, hidden from the host‐surveillance proteins. This is analogous to models proposed for alphaviruses and alphavirus‐like superfamily (8, 14), whereby access of the cytoplasmic viral RNA to the active viral RC housed within the indented vesicular pits is through an open neck. It is presumed that other cellular factors and nucleotides required for efficient RNA replication can also pass through the vesicle neck. Interestingly, although NS1 translocates into the ER lumen, it is able to interact with NS4A presumably via regions of NS4A that transverse the ER membrane. However, genetic studies have suggested that this interaction is mediated by residues in NS4A on the cytoplasmic face of the membrane (45). How this interaction occurs is not fully understood. As cycloheximide treatment does not affect replication, it is likely that the RC is used for multiple rounds of replication/membrane recruitment of the RC facilitated by its confined location within the VP (18). Following replication within the VP, the RNA is exported to the CM/PC for translation and proteolytic processing (step 2 in Figure 2B). It is known that translation and replication are coupled and that replication of a nascent RNA molecule is required for packaging (46). Following translation, the RNA molecule is then transported to the rER where assembly of the nucleocapsid and immature virion occurs (47) (Step 3 in figure 2B).
An important focus for virologists is the cellular origins of membranes and proteins involved in viral RNA replication. For flaviviruses, this process appears bewilderingly complex. As described above, there are three distinct membrane structures associated with the flavivirus life cycle – the rER, CM/PC and VP – all of which are to some extent related to membranes derived from the early secretory pathway (Figure 2). Interestingly, each of these membrane structures contains a distinct set of viral proteins with apparently different functions (see above). Furthermore, each of the membrane structures appears to contain a different subset of host proteins (48). Significantly, the trans‐Golgi marker β‐1,4‐galactosyltransferase (GalT) localized to the VPs in KUNV‐infected Vero cells (48); GalT was especially concentrated in membranes of the vesicles within the packet and not the bounding membrane itself. Other trans‐Golgi network (TGN) markers such as p230 and TGN46 also localize within the VP, whereas p200 and γ‐adaptin were not observed (Jason Mackenzie, unpublished observations). Notably, although much of the GalT, TGN46 and p230 redistributed to the VP, a subset of these proteins remained associated with the Golgi apparatus, which appeared morphologically unperturbed and had the normal distribution of the Golgi cis‐ and medial‐cisternae protein Giantin (48). An intact Golgi apparatus is a strict requirement for flavivirus replication, because the fully assembled virions exit the cell through the Golgi apparatus (47), and furin cleavage of prM (which occurs at the trans side of the Golgi) to M is essential for virus infectivity (49). In contrast to TGN proteins, ERGIC‐53, a recognized marker for the intermediate compartment (IC) (50), did not localize to the VP but accumulated within the CM/PC. Protein disulphide isomerase (PDI; an rER resident protein) was observed to label rER continuous with the CM/PC but was not significantly found within the CM/PC (48). Thus, it would appear that a subset of TGN/Golgi protein associates intimately with the ER/IC‐derived CM/PC and that the CM/PC are physically continuous with the rER.
Our data offer insights into the long‐standing controversy about the organization of the IC. In one, still popular view, the IC is considered as a compartment distinct from the ER and the Golgi 51, 52, 53). Others propose that the IC is continuous with the ER and the cis Golgi (52, 53). Our data, in flavivirus‐infected cells, show clearly that the rER and IC appear as continuous structures: the rER (identified with anti‐PDI antibodies) and the CM/PC (defined as the IC by its labelling with anti‐ERGIC‐53 antibodies). Our observations strongly support a transport model, whereby proteins are transported from the rER to the IC via a continuous membrane rather than via vesicular steps.
Brefeldin A (BFA) is a fungal metabolite and a potent inhibitor of anterograde membrane transport due to its effects on the activity of ADP ribosylation factor (ARF). Among other activities, the action of BFA prevents coatomer proteins binding to membranes (54), culminating in the disassembly of the Golgi apparatus and redistribution of Golgi proteins to the ER. Our studies with BFA‐treated, KUNV‐infected cells have clearly indicated that anterograde membrane and/or protein transport are required for biogenesis of the KUNV membrane structures, but only at an early step in the replication cycle (48). Membrane induction was not inhibited once the membranes themselves were formed, after the latent period (48). Additionally, GalT localized within the VP was also resistant to dispersion; this is in contrast to the redistribution of GalT observed in uninfected cells treated under similar conditions (48).
It is not clear at present which of the flaviviral proteins are responsible for membrane induction. However, after examination of cell lines bearing KUNV replicons of variable replication efficiencies, the ‘induced’ membranes were only found in cell lines that harboured efficiently replicating replicons (55). This suggested that one or more of the NS proteins is required for membrane induction and indicated a direct correlation between the extents of viral protein synthesis/accumulation and membrane proliferation/alteration. Analysis of the cellular origins of these membranes in replicon‐transduced cells was in total agreement with that observed during virus infection, except for the notable difference in the distribution of dsRNA and GalT (55). The subcellular distribution of the replicon dsRNA appeared as small foci scattered throughout the cytoplasm by immunofluorescence (IF) that presented as isolated foci on the cytoplasmic face of the ER when observed by cryo‐immunoelectron microscopy with anti‐dsRNA antibodies (55). This labelling pattern is strikingly similar to the pattern obtained in cell lines harbouring HCV replicons, using in situ hybridization or in vivo labelling of nascent viral RNA with BrUTP (22). Recent evidence has indicated that the HCV NS4B proteins has the capacity to induce membrane rearrangements resembling those observed in cell lines replicating a HCV replicon (1, 56). However, our recent results argue that at least the KUNV polyprotein NS2B‐NS3‐NS4A by itself contains the required attributes to induce cytoplasmic membrane structures analogous to those observed during flavivirus infection (Jason Mackenzie, unpublished observations).
Replication complex of the picornaviridae
Investigation of the RCs of picornaviruses has been pioneered by the elegant and comprehensive studies of Kurt Bienz and Denise Egger (Basel, Switzerland). Their systematic approach has been an inspiration for other investigators (including myself) who have studied the RCs of their own viruses.
Like the Flaviviridae, the Picornaviridae genomic RNA is translated as a polyprotein, albeit initiated by an internal ribosomal entry site and post‐ and co‐translationally cleaved by host‐ and viral‐encoded proteases (Figure 3). The mature proteins assemble on the 3′ end of the (+)‐strand RNA to initiate (–)‐strand synthesis. It is proposed that this occurs on the cytoplasmic face of membranes facilitated by the interaction of the hydrophobic viral proteins with host membranes. Poliovirus (PV) proteins 2B, 2C and 3A are all tightly associated with ER membranes, and 2C binds specifically to the 3′ end of the minus‐strand RNA, and 3Dpol specifically associates with the input positive‐sense RNA to generate minus‐strand RNA. Specific protein–protein interactions within the RC are difficult to dissect, as all the PV proteins, both structural and NS, reside within the RC (Figure 4). Furthermore, the presence of different polyprotein species makes it difficult to specifically identify individual proteins by immunolabelling. An antibody raised against one protein could also recognize the polyprotein it is derived from. The early stages of replication likely occur on rER membranes until sufficient viral protein accumulates and cellular membranes are rearranged.
Figure 3.
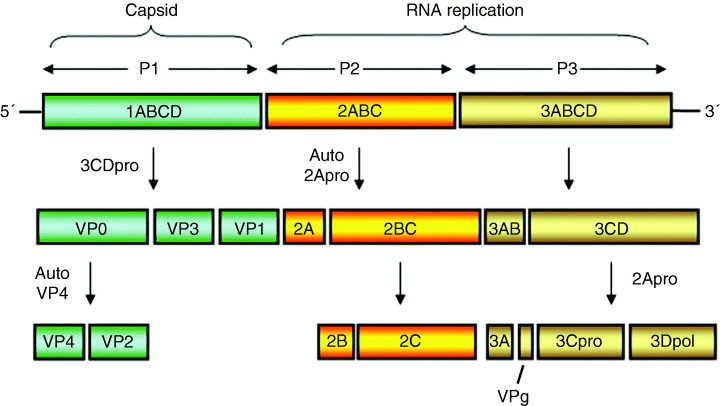
Schematic representation of the genomic organization of the Picornavirus genome and polyprotein species generated via virus‐specific proteolysis. Many of these polyprotein species have specific roles during replication of picornaviruses.
Figure 4.
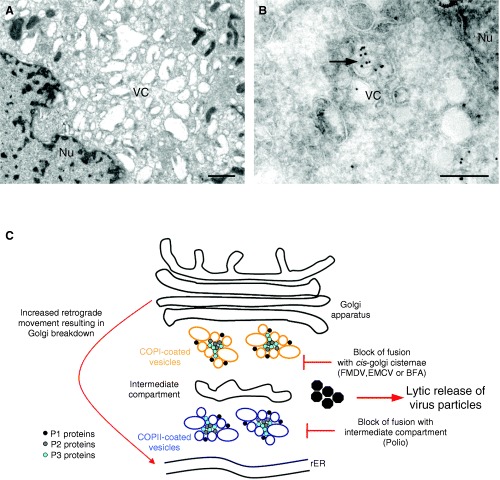
Ultrastructural analysis of Parechovirus‐infected BSC‐1 cells at 6 h post‐infection. In (A), the resin‐embedded section reveals vesicle clusters (VCs) accumulating in the perinuclear region. The vesicles range in size from approximately 100–800 nm. B) immunogold labelling of cryosections from parechovirus‐infected cells reveals the vesicle clusters heavily decorated with antibodies to dsRNA and 10 nm protein‐A gold.C)Schematic representation of membrane rearrangements observed during picornavirus infection. Anterograde transport carriers are restricted at different stages of the secretory pathway depending on the different viruses investigated. This restriction in transport of protein and membrane to the Golgi apparatus leads to the eventual disintegration of the Golgi due to the redistribution of Golgi proteins and membrane to the ER via retrograde movement. Nu, nucleus. Magnification bars represent 1 µm and 200 nm in Panels A and B, respectively.
The initial connection between ‘induced’ vesicles and RNA replication was revealed by high‐resolution autoradiography (2) and subsequently extended using detailed in situ hybridization and immunogold labelling of prepared embedded sections from PV‐infected cells (57, 58); similar techniques were used on membranes isolated from sucrose density gradient fractions containing active RNA polymerase activity 58, 59, 60). These experiments showed not only that the viral RNA and proteins involved in replication were associated with the proliferating membranes, but also that these membranes supported active replication (61). More intriguing was the appearance of the isolated membranes which were observed as vesicle rosettes (60). An interesting property of these rosettes was that they readily dissociate at low temperature or under low ionic conditions into tubulo vesicular structures and can reassemble into a functional complex on increased incubation at 30 °C (61).
Infection of mammalian cells with members of the Picornaviridae family results in a dramatic vesiculation and disintegration of internal membrane structures (3, 25, 62, 63, 64). The vesicles within these clusters range in size from 70 to 500 nm in diameter and appear to accumulate within the perinuclear area (Figure 4). Evidence for a requirement for continuous lipid synthesis to facilitate viral RNA replication has emerged from two independent observations. Firstly, experiments using the phospholipid synthesis inhibitor cerulenin showed that replication and infectivity of members of the Picornaviridae was greatly reduced in the presence of the drug 65, 66, 67). Subsequent studies indicated that many other RNA viruses are equally sensitive to the action of cerulenin suggesting a broad spectrum activity against viruses requiring lipid moieties (68, 69). Secondly, PV‐induced vesicles, and thus the RC formed from defective viruses, cannot support the replication of superinfecting wild‐type PV (26, 70). This suggests that each membranous RC must be formed in cis from nascent translated viral proteins recruiting cellular membranes. This inability to ‘recycle’ RCs may explain some of the difficulties in trans‐complementing defective PV genomes (71).
One model in the PV field speculates that the virally induced membranes in the peri‐nuclear area of the cell reflect the inhibition of the host secretory pathway leading to an accumulation of anterograde transport carriers. Such a process is potentially blocked by BFA, as this drug stops anterograde transport by inhibiting the ARF‐exchange factor resulting in dissociation of the COP1 coat. However, experiments using BFA have revealed that different viruses within the Picornaviridae family have different susceptibilities to BFA (3, 72, 73) and may use different host membrane components. Poliovirus and Echovirus 11 are extremely sensitive to BFA treatment (3, 73), whereas foot‐and‐mouth disease virus (FMDV) and encephalomyocarditis virus (EMCV) are resistant to BFA effects (3, 27), and Parechovirus displays some partial sensitivity (3). For the susceptible viruses, the effects of BFA can be rationalized by results indicating a role for host coat proteins in the formation of picornavirus RCs. Poliovirus appears to use COPII proteins early in the formation of the RC to induce the viral vesicles (74). However, late in infection, most cellular proteins are also associated with the PV vesicles (75). Based on these observations, it was proposed that the PV vesicle induction using COPII components occurred in a manner similar to the formation of transport vesicles in uninfected cells (74). Like anterograde transport vesicles, the PV vesicles were also observed to exclude ER‐resident marker proteins (e.g. p63) (74, 76, 77). Nevertheless, the apparent COPII‐dependent formation of the PV vesicles does not explain why PV replication is sensitive to BFA. The dynamics and activity of COPII proteins is generally assumed to be unaffected by this drug (78). Rather, BFA sensitivity and some in vitro experiments strongly implicate ARF directly in the virus replication process (79, 80). Recent data has implied that the PV proteins 3A or 3CD can induce ARF translocation to membranes facilitating active PV replication (79). It is interesting to note that the PV 2C protein is a GTPase (81, 82), and a single point mutation within 2C was observed to render PV resistant to BFA (83). Perhaps, the PV 2C protein may stabilize coatomer proteins on the vesicles within the clusters. In contrast to PV, Echovirus 11 infections lead to an active redistribution of COPI to the viral replication sites, and infection is also extremely sensitive to BFA (3). Therefore, a direct role of COPI proteins can be assumed in the replication of Echovirus 11. Interestingly, Parechovirus infection dispersed COPI throughout the cytoplasm (3), similar to the action of BFA itself, with some COPI also observed within the RC. Additionally, we and others have observed GalT to localize to the RC (Jason Mackenzie and E. Gazina, unpublished observations) (62) suggesting a possible Golgi origin of these vesicles. In contrast, EMCV and FMDV appear to require neither COPI or COPII and are both resistant to the effects of BFA (3, 27). In this context, it is surprising that very little research has sought to investigate the role of the IC during picornavirus replication, especially considering that the IC represents a crossroads within the secretory pathway, where a switching of COPII to COPI proteins on transport carriers is thought to occur (84). Experiments with FMDV showed a slight dispersion of ERGIC‐53 from the perinuclear region to scattered foci within the cytoplasm (27). However, our preliminary data suggest some excellent co‐localization of Echovirus 11 and EMCV RC with anti‐ERGIC‐53 antibodies by IF (Figure 5). The distribution of ERGIC‐53 appears relatively unaffected during these infections with coincident labelling observed both in the perinuclear region and in isolated cytoplasmic foci (Figure 5).
Figure 5.
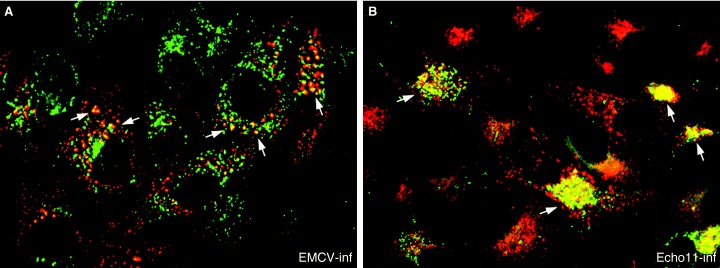
Immunofluorescene analysis of EMCV‐infected (A) and Echovirus 11‐infected (B) BSC‐1 cells at 6 h post‐infection. Co‐localization of anti‐dsRNA (labelled with Alexa Fluor 488; green) and anti‐ERGIC53 (labelled with Alexa Fluor 568; red) antibodies is observed within discrete cytoplasmic foci representing the picornavirus replication complex (highlighted with arrowheads). Dual localization is recognized as a yellow hue in both panels.
The results summarized above strongly implicate a dynamic association of ARF, COPI and COPII components with picornavirus replication, possibly regulated and/or stabilized by the viral proteins 2C, 3A and 3CD. The effect of BFA may relate to the activity of protein 2C, to release COPII and/or bind and stabilize COPI to the vesicle membranes. This stabilizing of COPI and ARF to cellular membranes may explain why large vesicles are observed during picornavirus infection, as the COPI‐bound vesicles would retain the capacity to fuse to other membranes bearing the appropriate tether. Whether or not the COP proteins are directly required for RNA replication and/or virus assembly is not currently known, but siRNA knock‐down experiments may provide a better understanding in the future. It has been speculated that perhaps this dismantling of the secretory pathway is an active measure to prevent cell‐surface expression of immunoregulatory molecules such as MHC I 85, 86, 87). It is important to note that picornaviruses do not require an intact secretory pathway for virion maturation, as the virions have no membranes and consist of assembled capsid subunits that are released via a lytic process (rather than through exocytosis; Figure 4C). Additionally, the capsid proteins required for virus assembly are also intimately associated with the vesicle clusters (61, 88) (Figure 4C), exemplifying the organized membranous microenvironment facilitating virus replication.
Another model proposed by the Kirkegaard laboratory (Stanford, USA) suggests a role for autophagy in the generation of the PV vesicle clusters (75, 77). Autophagy is a membrane‐dependent engulfment of cytoplasmic constituent destined for degradation typified by the presence of DMV structures (89). Using high‐pressure freezing and EM, the PV vesicles were presented as DMVs resembling autophagic vesicles (75). Earlier studies investigating Mouse Hepatitis virus (MHV) and Equine Arterivirus RNA replication revealed the presence of DMVs harbouring the viral RC (23, 24, 90). The studies by van der Meer et al. (24) identified the presence of the endosomal markers lamp‐1 and endocytosed BSA‐gold in the MHV RC; however, the role of autophagy was not investigated. Recent studies on MHV have suggested that the process of autophagy may contribute to the formation and stability of MHV RCs, as MHV replication was impaired in an autophagy knock‐out cell line (91). Immunogold labelling indicated the presence of the autophagosomal protein LC392, 93, 94) as well as cellular markers from different organelles within the PV RC (75). Additionally, when MHV‐ and PV‐infected cells were incubated in the presence of a chemical inhibitor of autophagy, 3‐methyladenine, there was decreased viral replication (91, 94). Consistent with this biogenesis model is the fact that although dramatic breakdown of the Golgi apparatus occurs during PV infection, an intact Golgi apparatus or trans‐Golgi network is not essential for the formation of autophagic vacuoles (95). Yet in contrast, BFA does not seem to prevent the formation of autophagic vacuoles but rather tends to increase the volume fraction of autophagic vacuoles (95). These contradictory results could suggest that more than one mechanism underlies PV vesicle formation, perhaps implying a combined use of anterograde membrane transport and autophagy. It can be envisaged that on prevention and accumulation of anterogade transport vesicles, the Golgi apparatus breaks down due to the retrograde movement of Golgi enzymes and proteins to the ER (as observed during treatment of uninfected cells with BFA). This accumulation of proteins within the ER may trigger host‐cell responses to signal proteolysis of the accumulated proteins within the ER, possibly initiating autophagy. With such a rapid life cycle, the window of opportunity for researchers to dissect the steps of picornavirus replication is limited.
To gain additional insights into the process of membrane induction in the absence of viral replication, viral proteins have been expressed individually. Such studies have been successful in identifying viral factors involved in membrane proliferation for a number of picornaviruses. In each case, expression of proteins from the P2 and P3 region of the genome leads to membrane alterations analogous to those observed during wild‐type infection. Further experiments indicate a role for the polyprotein precursor 2BC and 2C itself in inducing vesicle clusters96, 97, 98). Expression of each of the individual proteins alone has highlighted the membrane association and destabilizing effects of proteins 2B, 2C and 3A (99, 100). Both 2B and 3A are efficient immobilizers of membrane and protein trafficking through the secretory pathway, with 2B displaying some specificity for the Golgi apparatus (101). By contrast, 3A appears to restrict protein export from the ER (102) perhaps inducing the dispersion of coat proteins from newly forming transport vesicles. However, recent observations have indicated that only the FMDV protein 2BC can inhibit protein trafficking to the cell plasma membrane, and FMDV proteins 2B, 2C and 3A do not convey these properties (100). On the other hand, protein 2C does not block protein transport, yet induces vesicle clusters analogous to those observed in the cytoplasm during wild‐type infection (98, 103). The PV protein 3D – the virus encoded RdRp – has been the focus of many structural analyses. The goal of these studies has been to identify peptide‐binding pockets that might be targets for chemical inhibitors. Nevertheless, some interesting observations were made after EM analysis of the purified protein. The higher order structure of purified 3D was observed to be an ordered sheet or lattice (104). These arrays also had the capacity to form tubules. It was thus proposed that 3D is recruited to the membrane via interaction with 3AB and that this physical interaction between the 3D molecules induces assembly of the observed lattices forming a uniform surface area that can subsequently be utilized for RNA replication (104).
Concluding Remarks
In reviewing our current knowledge of the replication of two well‐studied families of viruses, it is clear that we still have a long way to go. Like all viruses, the subversion of cellular machinery and pathways is crucial for virus propagation and survival. The specific interaction and recruitment of host factors and distinct membrane domains to virus replication sites imply different strategies for the manipulation of host components. Dissection of the steps taken during biogenesis of the viral RC has highlighted some basic cell biology principles, especially relating to membrane traffic from the ER to IC and Golgi apparatus and possible subdomains within the trans‐Golgi. Questions still remain as to why different picornaviruses use distinct host components in producing morphologically similar vesicle structures. Also, what role does the apparent dynamic exchange of ARF and COP proteins play in the virus lifecycle? Is it merely a mechanism to prevent surface expression and secretion of immune regulatory molecules or is it directly involved in the assembly process? In contrast, flaviviruses redistribute trans‐Golgi proteins to their sites of viral RNA replication. What roles do host glycosylation and sorting proteins play in viral RNA replication and why do positive‐sense RNA viruses wrap their replicative machinery? Is it simply to ensure greater efficiency of replication by concentrating proteins and molecules within a desired membranous environment? Is it to provide a greater membrane surface area for RC assembly and increased cellular machinery for translation and subsequent post‐translational modifications? Or are the viruses hiding potential cell activators (i.e. dsRNA) from host surveillance molecules like double‐stranded RNA‐dependent protein kinase (PKR)? In any case, viruses have provided us with models to understand and dissect many cellular trafficking pathways and will no doubt continue to do so. I hope that eventually we can ‘wrap up’ the events associated with the replication of these families of pathogenic positive‐sense RNA viruses.
Acknowledgments
I thank Gareth Griffiths and Jacomine Krijnse‐Locker for critical review of the manuscript and making it a much better manuscript than the original. I thank Elena Gazina for the Parechovirus‐infected cells and for the ERGIC‐53 immunofluorescence pictures. I also want to acknowledge support of our research by the National Health and Medical Research Council of Australia.
References
- 1. Egger D, Wolk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol 2002;76: 5974–5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bienz K, Egger D, Pasamontes L. Association of polioviral proteins of the P2 genomic region with the viral replication complex and virus‐induced membrane synthesis as visualized by electron microscopic immunocytochemistry and autoradiography. Virology 1987;160: 220–226. [DOI] [PubMed] [Google Scholar]
- 3. Gazina EV, Mackenzie JM, Gorrell RJ, Anderson DA. Differential requirements for COPI coats in formation of replication complexes among three genera of Picornaviridae. J Virol 2002;76: 11113–11122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kujala P, Ikaheimonen A, Ehsani N, Vihinen H, Auvinen P, Kaariainen L. Biogenesis of the Semliki Forest virus RNA replication complex. J Virol 2001;75: 3873–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Magliano D, Marshall JA, Bowden DS, Vardaxis N, Meanger J, Lee JY. Rubella virus replication complexes are virus‐modified lysosomes. Virology 1998;240: 57–63. [DOI] [PubMed] [Google Scholar]
- 6. Mackenzie JM, Jones MK, Young PR. Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication. Virology 1996;220: 232–240. [DOI] [PubMed] [Google Scholar]
- 7. Pedersen KW, van der Meer Y, Roos N, Snijder EJ. Open reading frame 1a‐encoded subunits of the arterivirus replicase induce endoplasmic reticulum‐derived double‐membrane vesicles which carry the viral replication complex. J Virol 1999;73: 2016–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Froshauer S, Kartenbeck J, Helenius A. Alphavirus RNA replicase is located on the cytoplasmic surface of endosomes and lysosomes. J Cell Biol 1988;107 (6 Pt 1):2075–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kujala P, Ahola T, Ehsani N, Auvinen P, Vihinen H, Kaariainen L. Intracellular distribution of rubella virus nonstructural protein P150. J Virol 1999;73: 7805–7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee JY, Marshall JA, Bowden DS. Characterization of rubella virus replication complexes using antibodies to double‐stranded RNA. Virology 1994;200: 307–312. [DOI] [PubMed] [Google Scholar]
- 11. Lee JY, Marshall JA, Bowden DS. Replication complexes associated with the morphogenesis of rubella virus. Arch Virol 1992;122: 95–106. [DOI] [PubMed] [Google Scholar]
- 12. Ng ML. Ultrastructural studies of Kunjin virus‐infected Aedes albopictus cells. J Gen Virol 1987;68: 577–582. [DOI] [PubMed] [Google Scholar]
- 13. Hatta T, Bullivant S, Matthews RE. Fine structure of vesicles induced in chloroplasts of Chinese cabbage leaves by infection with turnip yellow mosaic virus. J Gen Virol 1973;20: 37–50. [DOI] [PubMed] [Google Scholar]
- 14. Schwartz M, Chen J, Janda M, Sullivan M, den Boon J, Ahlquist P. A positive‐strand RNA virus replication complex parallels form and function of retrovirus capsids. Mol Cell 2002;9: 505–514. [DOI] [PubMed] [Google Scholar]
- 15. Russo M, Di Franco A, Martelli GP. Cytopathology in the identification and classification of tombusviruses. Intervirology 1987;28: 134–143. [DOI] [PubMed] [Google Scholar]
- 16. Miller DJ, Schwartz MD, Ahlquist P. Flock house virus RNA replicates on outer mitochondrial membranes in Drosophila cells. J Virol 2001;75: 11664–11676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Westaway EG, Mackenzie JM, Kenney MT, Jones MK, Khromykh AA. Ultrastructure of Kunjin virus‐infected cells: colocalization of NS1 and NS3 with double‐stranded RNA, and of NS2B with NS3, in virus‐ induced membrane structures. J Virol 1997;71: 6650–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Westaway EG, Khromykh AA, Mackenzie JM. Nascent flavivirus RNA colocalized in situ with double‐stranded RNA in stable replication complexes. Virology 1999;258: 108–117. [DOI] [PubMed] [Google Scholar]
- 19. Grief C, Galler R, Cortes LM, Barth OM. Intracellular localisation of dengue‐2 RNA in mosquito cell culture using electron microscopic in situ hybridisation. Arch Virol 1997;142: 2347–2357. [DOI] [PubMed] [Google Scholar]
- 20. Restrepo‐Hartwig M, Ahlquist P. Brome mosaic virus RNA replication proteins 1a and 2a colocalize and 1a independently localizes on the yeast endoplasmic reticulum. J Virol 1999;73: 10303–10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Di Franco A, Russo M, Martelli GP. Ultrastructural and origin of cytoplasmic multivesicular bodies induced by Carnation Italian Ringspot virus. J Gen Virol 1984;65: 1233–1237. [Google Scholar]
- 22. Gosert R, Egger D, Lohmann V, Bartenschlager R, Blum HE, Bienz K, Moradpour D. Identification of the hepatitis C virus RNA replication complex in Huh‐7 cells harboring subgenomic replicons. J Virol 2003;77: 5487–5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gosert R, Kanjanahaluethai A, Egger D, Bienz K, Baker SC. RNA replication of mouse hepatitis virus takes place at double‐membrane vesicles. J Virol 2002;76: 3697–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van der Meer Y, Snijder EJ, Dobbe JC, Schleich S, Denison MR, Spaan WJ, Locker JK. Localization of mouse hepatitis virus nonstructural proteins and RNA synthesis indicates a role for late endosomes in viral replication. J Virol 1999;73: 7641–7657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bienz K, Egger D, Rasser Y, Bossart W. Intracellular distribution of poliovirus proteins and the induction of virus‐specific cytoplasmic structures. Virology 1983;131: 39–48. [DOI] [PubMed] [Google Scholar]
- 26. Egger D, Bienz K. Recombination of poliovirus RNA proceeds in mixed replication complexes originating from distinct replication start sites. J Virol 2002;76: 10960–10971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Knox C, Moffat K, Ali S, Ryan M, Wileman T. Foot‐and‐mouth disease virus replication sites form next to the nucleus and close to the Golgi apparatus, but exclude marker proteins associated with host membrane compartments. J Gen Virol 2005;86: 687–696. [DOI] [PubMed] [Google Scholar]
- 28. Salonen A, Ahola T, Kaariainen L. Viral RNA replication in association with cellular membranes. Curr Top Microbiol Immunol 2005;285: 139–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Novoa RR, Calderita G, Arranz R, Fontana J, Granzow H, Risco C. Virus factories: associations of cell organelles for viral replication and morphogenesis. Biol Cell 2005;97: 147–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lindenbach BD, Rice CM. Flaviviridae: the viruses and their replication, 4th edn In:Fields BN, Knipe DN, Howley PM, editors. Fields Virology. New York: Lippincott‐Raven; 2001, pp.991–1041. [Google Scholar]
- 31. Westaway EG, Mackenzie JM, Khromykh AA. Replication and gene function in Kunjin virus. Curr Top Microbiol Immunol 2002;267: 323–351. [DOI] [PubMed] [Google Scholar]
- 32. Liu WJ, Sedlak PL, Kondratieva N, Khromykh AA. Complementation analysis of the flavivirus Kunjin NS3 and NS5 proteins defines the minimal regions essential for formation of a replication complex and shows a requirement of NS3 in cis for virus assembly. J Virol 2002;76: 10766–10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kummerer BM, Rice CM. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J Virol 2002;76: 4773–4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Westaway EG. Replication of flaviviruses In:Schlesinger RW, editor. Togaviruses. New York: Academic Press; 1980, pp.531–581. [Google Scholar]
- 35. Murphy FA. Morphology and morphogenesis In:Monath TP, editor. St. Louis Encephalitis. Washington DC: American Public Health Association Inc, 1980, pp.65–103. [Google Scholar]
- 36. Mackenzie JM, Jones MK, Young PR. Improved membrane preservation of flavivirus‐infected cells with cryosectioning. J Virol Methods 1996;56: 67–75. [DOI] [PubMed] [Google Scholar]
- 37. Westaway EG, Mackenzie JM, Khromykh AA. Kunjin RNA replication and applications of Kunjin replicons. Adv Virus Res 2003;59: 99–140. [DOI] [PubMed] [Google Scholar]
- 38. Chu PW, Westaway EG. Molecular and ultrastructural analysis of heavy membrane fractions associated with the replication of Kunjin virus RNA. Arch Virol 1992;125: 177–191. [DOI] [PubMed] [Google Scholar]
- 39. Mackenzie JM, Khromykh AA, Jones MK, Westaway EG. Subcellular localization and some biochemical properties of the flavivirus Kunjin nonstructural proteins NS2A and NS4A. Virology 1998;245: 203–215. [DOI] [PubMed] [Google Scholar]
- 40. Westaway EG, Khromykh AA, Kenney MT, Mackenzie JM, Jones MK. Proteins C and NS4B of the flavivirus Kunjin translocate independently into the nucleus. Virology 1997;234: 31–41. [DOI] [PubMed] [Google Scholar]
- 41. Khromykh AA, Westaway EG. Completion of Kunjin virus RNA sequence and recovery of an infectious RNA transcribed from stably cloned full‐length cDNA. J Virol 1994;68: 4580–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Khromykh AA, Westaway EG. Subgenomic replicons of the flavivirus Kunjin: construction and applications. J Virol 1997;71: 1497–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rice CM, Grakoui A, Galler R, Chambers TJ. Transcription of infectious yellow fever RNA from full‐length cDNA templates produced by in vitro ligation. New Biol 1989;1: 285–296. [PubMed] [Google Scholar]
- 44. Lindenbach BD, Rice CM. trans‐Complementation of yellow fever virus NS1 reveals a role in early RNA replication. J Virol 1997;71: 9608–9617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lindenbach BD, Rice CM. Genetic interaction of flavivirus nonstructural proteins NS1 and NS4A as a determinant of replicase function. J Virol 1999;73: 4611–4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Khromykh AA, Varnavski AN, Sedlak PL, Westaway EG. Coupling between replication and packaging of flavivirus RNA: evidence derived from the use of DNA‐based full‐length cDNA clones of Kunjin virus. J Virol 2001;75: 4633–4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mackenzie JM, Westaway EG. Assembly and maturation of the flavivirus Kunjin virus appear to occur in the rough endoplasmic reticulum and along the secretory pathway, respectively. J Virol 2001;75: 10787–10799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mackenzie JM, Jones MK, Westaway EG. Markers for trans‐Golgi membranes and the intermediate compartment localize to induced membranes with distinct replication functions in flavivirus‐infected cells. J Virol 1999;73: 9555–9567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stadler K, Allison SL, Schalich J, Heinz FX. Proteolytic activation of tick‐borne encephalitis virus by furin. J Virol 1997;71: 8475–8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schweizer A, Fransen JA, Bachi T, Ginsel L, Hauri HP. Identification, by a monoclonal antibody, of a 53‐kD protein associated with a tubulo‐vesicular compartment at the cis‐side of the Golgi apparatus. J Cell Biol 1988;107: 1643–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pelham HR. Sorting and retrieval between the endoplasmic reticulum and Golgi apparatus. Curr Opin Cell Biol 1995;7: 530–535. [DOI] [PubMed] [Google Scholar]
- 52. Mironov AA, Mironov AA Jr, Beznoussenko GV, Trucco A, Lupetti P, Smith JD, Geerts WJ, Koster AJ, Burger KN, Martone ME, Deerinck TJ, Ellisman MH, Luini A. ER‐to‐Golgi carriers arise through direct en bloc protrusion and multistage maturation of specialized ER exit domains. Dev Cell 2003;5: 583–594. [DOI] [PubMed] [Google Scholar]
- 53. Krijnse‐Locker J, Ericsson M, Rottier PJ, Griffiths G. Characterization of the budding compartment of mouse hepatitis virus: evidence that transport from the RER to the Golgi complex requires only one vesicular transport step. J Cell Biol 1994;124: 55–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Donaldson JG, Klausner RD. ARF: a key regulatory switch in membrane traffic and organelle structure. Curr Opin Cell Biol 1994;6: 527–532. [DOI] [PubMed] [Google Scholar]
- 55. Mackenzie JM, Khromykh AA, Westaway EG. Stable expression of noncytopathic Kunjin replicons simulates both ultrastructural and biochemical characteristics observed during replication of Kunjin virus. Virology 2001;279: 161–172. [DOI] [PubMed] [Google Scholar]
- 56. Konan KV, Giddings TH Jr, Ikeda M, Li K, Lemon SM, Kirkegaard K. Nonstructural protein precursor NS4A/B from hepatitis C virus alters function and ultrastructure of host secretory apparatus. J Virol 2003;77: 7843–7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Troxler M, Pasamontes L, Egger D, Bienz K. In situ hybridization for light and electron microscopy. a comparison of methods for the localization of viral RNA using biotinylated DNA and RNA probes. J Virol Methods 1990;30: 1–14. [DOI] [PubMed] [Google Scholar]
- 58. Bienz K, Egger D. Immunocytochemistry and in situ hybridization in the electron microscope: combined application in the study of virus‐infected cells. Histochem Cell Biol 1995;103: 325–338. [DOI] [PubMed] [Google Scholar]
- 59. Bienz K, Egger D, Pfister T, Troxler M. Structural and functional characterization of the poliovirus replication complex. J Virol 1992;66: 2740–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bienz K, Egger D, Troxler M, Pasamontes L. Structural organization of poliovirus RNA replication is mediated by viral proteins of the P2 genomic region. J Virol 1990;64: 1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Egger D, Pasamontes L, Bolten R, Boyko V, Bienz K. Reversible dissociation of the poliovirus replication complex: functions and interactions of its components in viral RNA synthesis. J Virol 1996;70: 8675–8683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Krogerus C, Egger D, Samuilova O, Hyypia T, Bienz K. Replication complex of human parechovirus 1. J Virol 2003;77: 8512–8523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gosert R, Egger D, Bienz K. A cytopathic and a cell culture adapted hepatitis A virus strain differ in cell killing but not in intracellular membrane rearrangements. Virology 2000;266: 157–169. [DOI] [PubMed] [Google Scholar]
- 64. Monaghan P, Cook H, Jackson T, Ryan M, Wileman T. The ultrastructure of the developing replication site in foot‐and‐mouth disease virus‐infected BHK‐38 cells. J Gen Virol 2004;85 (Pt 4):933–946. [DOI] [PubMed] [Google Scholar]
- 65. Molla A, Paul AV, Wimmer E. Effects of temperature and lipophilic agents on poliovirus formation and RNA synthesis in a cell‐free system. J Virol 1993;67: 5932–5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Guinea R, Carrasco L. Phospholipid biosynthesis and poliovirus genome replication, two coupled phenomena. Embo J 1990;9: 2011–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Carette JE, Stuiver M, Van Lent J, Wellink J, Van Kammen A. Cowpea mosaic virus infection induces a massive proliferation of endoplasmic reticulum but not Golgi membranes and is dependent on de novo membrane synthesis. J Virol 2000;74: 6556–6563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Perez L, Guinea R, Carrasco L. Synthesis of Semliki Forest virus RNA requires continuous lipid synthesis. Virology 1991;183: 74–82. [DOI] [PubMed] [Google Scholar]
- 69. Perez L, Carrasco L. Cerulenin, an inhibitor of lipid synthesis, blocks vesicular stomatitis virus RNA replication. FEBS Lett 1991;280: 129–133. [DOI] [PubMed] [Google Scholar]
- 70. Egger D, Teterina N, Ehrenfeld E, Bienz K. Formation of the poliovirus replication complex requires coupled viral translation, vesicle production, and viral RNA synthesis. J Virol 2000;74: 6570–6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Teterina NL, Zhou WD, Cho MW, Ehrenfeld E. Inefficient complementation activity of poliovirus 2C and 3D proteins for rescue of lethal mutations. J Virol 1995;69: 4245–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. O'Donnell VK, Pacheco JM, Henry TM, Mason PW. Subcellular distribution of the foot‐and‐mouth disease virus 3A protein in cells infected with viruses encoding wild‐type and bovine‐attenuated forms of 3A. Virology 2001;287: 151–162. [DOI] [PubMed] [Google Scholar]
- 73. Maynell LA, Kirkegaard K, Klymkowsky MW. Inhibition of poliovirus RNA synthesis by brefeldin A. J Virol 1992;66: 1985–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rust RC, Landmann L, Gosert R, Tang BL, Hong W, Hauri HP, Egger D, Bienz K. Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J Virol 2001;75: 9808–9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Schlegel A, Giddings TH Jr, Ladinsky MS, Kirkegaard K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J Virol 1996;70: 6576–6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schweizer A, Rohrer J, Slot JW, Geuze HJ, Kornfeld S. Reassessment of the subcellular localization of p63. J Cell Sci 1995;108: 2477–2485. [DOI] [PubMed] [Google Scholar]
- 77. Suhy DA, Giddings TH Jr, Kirkegaard K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy‐like origin for virus‐induced vesicles. J Virol 2000;74: 8953–8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ward TH, Polishchuk RS, Caplan S, Hirschberg K, Lippincott‐Schwartz J. Maintenance of Golgi structure and function depends on the integrity of ER export. J Cell Biol 2001;155: 557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Belov GA, Fogg MH, Ehrenfeld E. Poliovirus proteins induce membrane association of GTPase ADP‐ribosylation factor. J Virol 2005;79: 7207–7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cuconati A, Molla A, Wimmer E. Brefeldin A inhibits cell‐free, de novo synthesis of poliovirus. J Virol 1998;72: 6456–6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rodriguez PL, Carrasco L. Poliovirus protein 2C has ATPase and GTPase activities. J Biol Chem 1993;268: 8105–8110. [PubMed] [Google Scholar]
- 82. Rodriguez PL, Carrasco L. Poliovirus protein 2C contains two regions involved in RNA binding activity. J Biol Chem 1995;270: 10105–10112. [DOI] [PubMed] [Google Scholar]
- 83. Crotty S, Saleh MC, Gitlin L, Beske O, Andino R. The poliovirus replication machinery can escape inhibition by an antiviral drug that targets a host cell protein. J Virol 2004;78: 3378–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hauri HP, Kappeler F, Andersson H, Appenzeller C. ERGIC‐53 and traffic in the secretory pathway. J Cell Sci 2000;113: 587–596. [DOI] [PubMed] [Google Scholar]
- 85. Deitz SB, Dodd DA, Cooper S, Parham P, Kirkegaard K. MHC I‐dependent antigen presentation is inhibited by poliovirus protein 3A. Proc Natl Acad Sci USA 2000;97: 13790–13795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Neznanov N, Kondratova A, Chumakov KM, Angres B, Zhumabayeva B, Agol VI, Gudkov AV. Poliovirus protein 3A inhibits tumor necrosis factor (TNF) ‐induced apoptosis by eliminating the TNF receptor from the cell surface. J Virol 2001;75: 10409–10420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Neznanov N, Chumakov KP, Ullrich A, Agol VI, Gudkov AV. Unstable receptors disappear from cell surface during poliovirus infection. Med Sci Monit 2002;8: BR391–BR396. [PubMed] [Google Scholar]
- 88. Pfister T, Pasamontes L, Troxler M, Egger D, Bienz K. Immunocytochemical localization of capsid‐related particles in subcellular fractions of poliovirus‐infected cells. Virology 1992;188: 676–684. [DOI] [PubMed] [Google Scholar]
- 89. Levine B, Klionsky DJ. Development by self‐digestion: molecular mechanisms biological functions autophagy. Dev Cell 2004;6: 463–477. [DOI] [PubMed] [Google Scholar]
- 90. van der Meer Y, van Tol H, Locker JK, Snijder EJ. ORF1a‐encoded replicase subunits are involved in the membrane association of the arterivirus replication complex. J Virol 1998;72: 6689–6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem 2004;279: 10136–10141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kirkegaard K, Taylor MP, Jackson WT. Cellular autophagy: surrender, avoidance and subversion by microorganisms. Nat Rev Microbiol 2004;2: 301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sodeik B, Schramm B, Suomalainen M, Locker JK. Meeting report: EMBO workshop ‘Cell Biology of Virus Infection’, September 25–29, 2004, EMBL, Heidelberg, Germany. Traffic 2005;6: 351–356. [DOI] [PubMed] [Google Scholar]
- 94. Jackson WT, Giddings THJ, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol 2005;3: e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Purhonen P, Pursiainen K, Reunanen H. Effects of brefeldin A on autophagy in cultured rat fibroblasts. Eur J Cell Biol 1997;74: 63–67. [PubMed] [Google Scholar]
- 96. Aldabe R, Carrasco L. Induction of membrane proliferation by poliovirus proteins 2C and 2BC. Biochem Biophys Res Commun 1995;206: 64–76. [DOI] [PubMed] [Google Scholar]
- 97. Cho MW, Teterina N, Egger D, Bienz K, Ehrenfeld E. Membrane rearrangement and vesicle induction by recombinant poliovirus 2C and 2BC in human cells. Virology 1994;202: 129–145. [DOI] [PubMed] [Google Scholar]
- 98. Teterina NL, Bienz K, Egger D, Gorbalenya AE, Ehrenfeld E. Induction of intracellular membrane rearrangements by HAV proteins 2C and 2BC. Virology 1997;237: 66–77. [DOI] [PubMed] [Google Scholar]
- 99. Aldabe R, Barco A, Carrasco L. Membrane permeabilization by poliovirus proteins 2B and 2BC. J Biol Chem 1996;271: 23134–23137. [DOI] [PubMed] [Google Scholar]
- 100. Moffat K, Howell G, Knox C, Belsham GJ, Monaghan P, Ryan MD, Wileman T. Effects of foot‐and‐mouth disease virus nonstructural proteins on the structure and function of the early secretory pathway: 2BC but not 3A blocks endoplasmic reticulum‐to‐golgi transport. J Virol 2005;79: 4382–4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sandoval I, Carrasco L. Poliovirus infection and expression of the poliovirus protein 2B provoke the disassembly of the Golgi complex, the organelle target for the antipoliovirus drug Ro‐090179. J Virol 1997;71: 4679–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Doedens JR, Kirkegaard K. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. Embo J 1995;14: 894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Teterina NL, Gorbalenya AE, Egger D, Bienz K, Ehrenfeld E. Poliovirus 2C protein determinants of membrane binding and rearrangements in mammalian cells. J Virol 1997;71: 8962–8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Lyle JM, Bullitt E, Bienz K, Kirkegaard K. Visualization and functional analysis of RNA‐dependent RNA polymerase lattices. Science 2002;296: 2218–2222. [DOI] [PubMed] [Google Scholar]