

The transport pathway of autophagy is characterized by double‐membrane vesicles called autophagosomes, which are formed through the concerted action of the conserved ATG. Until recently it was assumed that ATG proteins were exclusively involved in autophagy. A growing number of studies, however, have revealed that specific ATG proteins have roles beyond their involvement in autophagosome biogenesis. This article reviews this fast expanding new field of the autophagy research.
Keywords: apoptosis, ATG proteins, autophagy, degradation, immunity, infection, pathogens, subversion, unconventional
Abstract
Macroautophagy (hereafter referred to as autophagy) is an evolutionarily conserved intracellular catabolic transport route that generally allows the lysosomal degradation of cytoplasmic components, including bulk cytosol, protein aggregates, damaged or superfluous organelles and invading microbes. Target structures are sequestered by double‐membrane vesicles called autophagosomes, which are formed through the concerted action of the autophagy (ATG)‐related proteins. Until recently it was assumed that ATG proteins were exclusively involved in autophagy. A growing number of studies, however, have attributed functions to some of them that are distinct from their classical role in autophagosome biogenesis. Autophagy‐independent roles of the ATG proteins include the maintenance of cellular homeostasis and resistance to pathogens. For example, they assist and enhance the turnover of dead cells and microbes upon their phagocytic engulfment, and inhibit murine norovirus replication. Moreover, bone resorption by osteoclasts, innate immune regulation triggered by cytoplasmic DNA and the ER‐associated degradation regulation all have in common the requirement of a subset of ATG proteins. Microorganisms such as coronaviruses, Chlamydia trachomatis or Brucella abortus have even evolved ways to manipulate autophagy‐independent functions of ATG proteins in order to ensure the completion of their intracellular life cycle. Taken together these novel mechanisms add to the repertoire of functions and extend the number of cellular processes involving the ATG proteins.
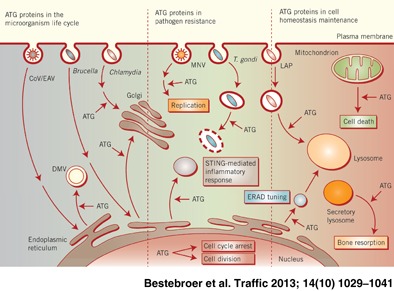
Autophagy, from the Greek ‘self‐eating’, participates in cellular and organismal homeostasis by disposing long‐lived proteins, protein aggregates and/or cellular regulators as well as obsolete or damaged organelles such as mitochondria 1, 2. Autophagy was initially found to be induced in response to starvation, but other cellular stresses, immunological signals and developmental cues, e.g. growth factor deprivation, endoplasmic reticulum (ER) and oxidative stress, are known to induce this pathway as well 3, 4. As a result autophagy is induced in numerous pathological situations and its impairment leads to various human diseases 5, 6.
Autophagosomes are generated at the phagophore assembly site (PAS), a specialized location where ATG proteins assemble to first mediate the formation of an initial cistern called phagophore and subsequently elongate it into an autophagosome (Figure 1A). During the expansion of the phagophore they selectively or non‐selectively sequester the cargo. The exact origin of the autophagosomal membranes remains unclear, but evidence points to multiple possible sources, including the ER, the outer mitochondrial membrane, the plasma membrane and endosomes 7. Complete autophagosomes then first fuse with endosomes to form amphisomes and finally with lysosomes to generate autolysosomes, where the autophagosomal inner membrane and cargo are degraded 8. The resulting metabolites are then transported into the cytoplasm where they can be used as either building blocks for the synthesis of new macromolecules or a source of energy.
Figure 1.
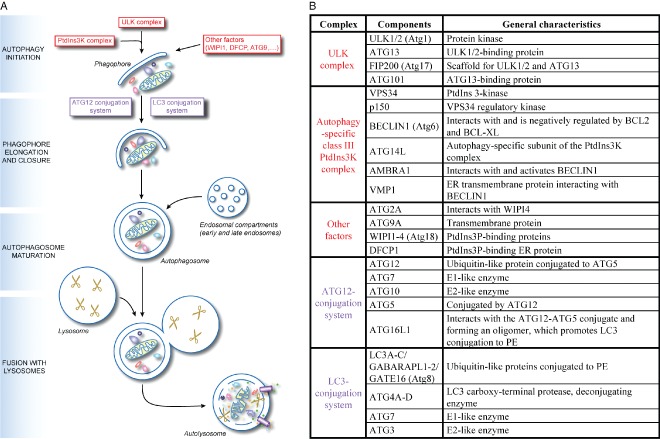
The mechanism of canonical autophagy and the conventional functions of the ATG proteins. A) The mechanism of autophagy. Upon induction, the early steps of autophagosome biogenesis entail the formation of the phagophore through the orchestrated action of the ULK complex, the PtdIns3K complex and other factors including ATG9, VMP1, AMBRA1, DFCP1 and the WIPI proteins. Subsequently the ATG12 and LC3 conjugation systems are key in mediating the expansion and nucleation of the phagophore into an autophagosome, an event that allows the sequestration of the cargo targeted for degradation. Once complete, autophagosomes first fuse with endosomal compartments to mature into amphisomes before fusing with lysosomes to form autolysosomes. Resident hydrolases (indicated with scissors) consume the autophagosomal cargo and internal vesicles into metabolites (amino acids, sugars, nucleotides and other basic molecules), which are subsequently transported into the cytoplasm by permeases and possibly other transporters (depicted as small violet pipes), where they are used as either a source of energy or as building blocks for the synthesis of new macromolecules. B) The key autophagy proteins and their organization in functional gene clusters. The ULK complex and the autophagy‐specific class III PtdIns3K complex, plus other factors, are involved in the initiation of autophagosome biogenesis (highlighted in red in panel A) whereas the ATG12‐ and LC3‐conjugation systems are mostly involved in the phagophore elongation (highlighted in violet in panel A). While the yeast homologues of the ATG2A, ATG9A and WIPI are interacting, there is no evidence this also holds true for their mammalian counterparts. Moreover, there are no indications that DFCP1 associate with these components. The yeast homologs of the mammalian ATGs proteins are indicated in between brackets. Adapted from 4.
The discovery of the ATG genes in yeast and their mammalian counterparts has allowed the molecular characterization of the autophagy process. In particular eighteen ATG proteins and five additional factors, grouped in five functional complexes, compose the core autophagy machinery in high eukaryotes 2 (Figure 1B). Autophagosome biogenesis is initiated by the UNC‐51‐like kinase (ULK) complex, composed of the ULK1/2 kinase, ATG13, FIP200 and ATG101. Mammalian target of rapamycin complex 1 (mTORC1) negatively regulates the ULK complex by phosphorylating ULK1/2 and ATG13 2. Inactivation of mTORC1 leads to the dephosphorylation, translocation and activation of the ULK complex from the cytosol to the PAS, where it recruits other elements of the ATG machinery such as the autophagy‐specific class III phosphatidylinositol (PtdIns) 3‐kinase (PtdIns3K) complex 2. This complex is formed by VPS34, p150, BECLIN1/ATG6 and ATG14L, and interacts with various factors such as AMBRA1 and VMP1, which regulate its localization and activity. The generation of phosphatiylinositol‐3‐phosphate (PtdIns3P) at the PAS triggers the recruitment of proteins binding this lipid, such as WIPI and DFCP1 proteins, which also mediate the formation of the phagophore 7. Another protein essential for the initial steps of autophagosome formation is the transmembrane protein ATG9. This protein is principally distributed to the trans‐Golgi network and late endosomes, and dynamically associates with the PAS 9, 10. While the precise function of ATG9 is still unknown, it has been proposed that it provides some of the initial membranes required to organize the PAS 11.
The elongation and closure of the phagophore are driven by two distinct but interconnected ubiquitin‐like conjugation systems. The first comprises the E1‐like ATG7 and the E2‐like ATG10 enzymes, which mediate the covalent conjugation of the ubiquitin‐like ATG12 to ATG5. The ATG12‐ATG5 conjugate subsequently binds to ATG16L1 to form the ATG12‐ATG5/ATG16L1 complex 2. This complex is essential for autophagosome biogenesis by targeting the second ubiquitin‐like conjugation system to the PAS and promoting its last step 2. The latter involves the conjugation of phosphatidylethanolamine (PE) to the yeast ATG8 homologs, which are represented by the microtubule‐associated protein light‐chain 3 (LC3) and γ‐aminobutyric acid receptor‐associated protein (GABARAP) subfamilies 12. After their synthesis, these ubiquitin‐like proteins are post‐translationally processed at the C‐terminus by the ATG4 protease to generate the so‐called non‐lipidated LC3‐I form. Upon autophagy induction, LC3‐I becomes conjugated to PE at its C‐terminus on both the inner and outer membrane of the growing phagophore through the action of ATG7 and the E2‐like enzyme ATG3. The lipidated form of LC3 is known as LC3‐II. The members of the ATG8/LC3 protein family appear to carry out several functions during autophagosome biogenesis. They have fusion properties 13, 14 that could be essential for the phagophore expansion and closure 15, 16. Moreover the pool of these proteins found in the interior of the autophagosomes plays a pivotal role in the recognition and selection of specific cargoes degraded by autophagy 17. Finally, LC3 is implicated in microtubule‐dependent transport of autophagosomes towards lysosomes 18.
Until recently, it was believed that ATG proteins were exclusively involved in autophagosome formation. A series of discoveries, however, has revealed that they are individually or as a functional group also involved in pathways distinct from autophagy 19. Here we will review these unconventional functions of the ATG proteins and highlight the different processes through which they are also crucial in maintaining cellular and organismal homeostasis.
LC3‐Associated Phagocytosis
Autophagy is induced upon phagocytic uptake of microbes and their subsequent recognition by toll‐like receptors (TLR). This pathway is not only responsible for the destruction of the microbe‐containing vacuoles but also of pathogens that have gained access to the cytoplasm by either entering the cells or escaping the phagolysosomal pathway 4, 20. ATG proteins also participate in microbe clearance through a mechanism distinct from classical autophagy. LC3‐associated phagocytosis (LAP) is a process by which components of the ATG machinery directly modify the phagosomal membrane to assist and enhance degradation of phagocytosed elements 21, 22 (Figure 2). Sanjuan and co‐workers 23 were the first to demonstrate that TLR signaling mediates the rapid recruitment of LC3 to phagosomes. They observed that macrophages fed with either zymosan (a TLR2 agonist), Escherichia coli or latex beads coated with TLR agonists such as Pam3CSK4 (a TLR1/2 agonist) or lipopolysaccharides (LPS; a TLR4 agonist) recruit LC3 to the phagosomal membrane. The LC3 recruitment concurred with enhanced degradation of the phagocytosed cargo by a more rapid and extensive acidification of the phagosomal compartment 23. Localized TLR signaling at this site was necessary for LAP because no association of LC3 with phagosomes was observed in tlr2 knockout macrophages. Recruitment of LC3 to phagosomes requires ATG5 and ATG7 as well as the PtdIns3K complex 23, with BECLIN1 associating prior to LC3. In contrast to canonical autophagy, LAP does not depend on ULK1 and it is not affected by rapamycin, chloroquine or starvation 23, 24. The absence of double‐membrane structures surrounding the engulfed material morphologically also distinguishes LC3‐positive phagosomes from autophagosomes 23.
Figure 2.
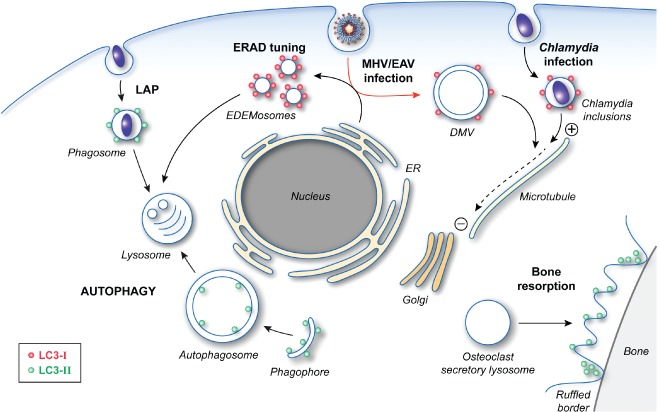
The cellular functions of LC3. After post‐translational processing by ATG4, LC3‐I is covalently linked to PE to generate LC3‐II through the action of two ubiquitin‐like conjugation systems. The formation of lipidated LC3 is essential for autophagosome formation by mediating the expansion of the phagophore. LC3‐II is also involved in LAP as well as the specialized secretion by osteoclasts, which is essential for the establishment of the ruffled borders for bone resorption. The role of LC3‐II in these two latter pathways is unclear, but its localization and function do not require an intact ATG machinery. Precursor LC3‐I is also carrying out cellular functions, which do not appear to involve most of the other ATG proteins. LC3‐I is recruited together with MAP1 proteins onto Chlamydia inclusions and allows the redistribution of these bacteria‐containing compartments in proximity of the Golgi through its interaction with the microtubules. LC3‐I is also associated with the surface of the EDEMosomes via its interaction with the protein cargo receptor Sel1L. The role of LC3‐I in the ERAD tuning pathway remains unknown, but it could act as an adaptor for either a protein vesicle coat or the cytoskeleton network. Viruses, such as MHV and EAV, hijack this transport route to generate their replicative DMVs, which are also decorated with LC3‐I (red arrow). LC3‐I is essential for both the ERAD tuning and the replication of these viruses. In this latter context, LC3‐I could mediate the microtubule‐dependent subcellular redistribution of DMVs.
LAP has been extensively studied in the context of Burkholderia pseudomallei infections. This intracellular bacterium escapes phagosomes upon endocytosis using a type III secretion system and replicates in the cytoplasm of the host cells. An initial study investigating the interaction between B. pseudomallei and autophagy suggested a possible role of autophagosomes in reducing intracellular bacterial survival in phagocytic and non‐phagocytic cells 25. Later, however, ultrastructural evidences have revealed that B. pseudomallei is subjected to LAP rather than to conventional autophagy and that the previously observed colocalization of LC3 with bacteria must result from the recruitment of LC3 onto phagosomal membranes. In particular, electron microscopy (EM) analyses showed that although numerous autophagosomes were found in the cytoplasm of infected cells, they did not contain bacteria 26. In contrast B. pseudomallei was either surrounded by a single membrane or was found free in the cytoplasm.
Most of B. pseudomallei break out of endosomes and avoid degradation by canonical autophagy through a still unknown mechanism. Bacteria that do not escape into the cytoplasm are mostly found in phagosomes positive for both LC3 and the lysosomal marker protein LAMP1. Consistently, B. pseudomallei type III secretion system mutants bopA and bipD, which are unable to escape phagosomes, display increased colocalization with LC3 and LAMP1, suggesting an enhanced routing towards the lysosome 26. Surprisingly, heat‐killed B. pseudomallei display diminished recruitment of LC3 to phagosomes, indicating that live bacteria and signaling routes other than TLR activation might be involved in LAP induction 26. Nevertheless, LAP and efficient killing of B. pseudomallei also require BECLIN1 and are enhanced by starvation and rapamycin treatments 27. Surprisingly, BECLIN1 knockdown does not affect LAP in rapamycin‐treated cells raising new questions about the possible existence of either different LC3 pools or a BECLIN1‐independent LAP [in analogy to BECLIN1‐independent autophagy 28] as well as alternative mechanisms for the PtdIns3P synthesis on phagosomal membranes. Importantly, LAP could be key in processing exogenous microbial antigens for MHC II presentation in specific cell types like dendritic cells 29.
LAP is not only employed as a cellular mechanism to combat pathogens. This type of modification of the endocytic compartments can also assist in the removal of apoptotic, necrotic or entotic cells. Dying cells have to be cleared from the extracellular milieu to avoid inflammatory and autoimmune reactions. Specific receptors on phagocytes recognize phosphatidylserine (PtdSer), which is normally absent on the surface of healthy cells but gets exposed after apoptosis or necrosis 30. Engagement of PtdSer and TIM4 is responsible for the engulfment of apoptotic or necrotic bodies and by a yet unidentified pathway, triggers the LAP of the dead cell bodies 24. Entosis is a novel form of non‐apoptotic cell death where an epithelial cell engulfs a living cell, which is then degraded in the lysosome 31. Similar to apoptotic and necrotic cell disposal, the limiting membranes of endosomes containing entotic cells rapidly recruit LC3 32. Both entosis and clearance of apoptotic and necrotic cells depend on ATG5 and ATG7 as well as on the presence of the components of the PtdIns3K complex, such as VPS34 and BECLIN1, on the limiting membrane of the compartment 24, 32. In contrast to classical autophagy, components of the ULK complex, including ULK1 or FIP200, are not required for LAP progression. Work in Caenorhabditis elegans on the removal of apoptotic corpses during embryonic development has also implicated ATG18/WIPI in LAP in addition to BECLIN1 33.
Interestingly, a recent study has reported a form of LAP that appears to be mainly non‐degradative as it is involved in the biogenesis of an organelle required to induce interferon‐α (IFN‐α) secretion 34. A feature of several autoimmune diseases is the phagocytosis of host DNA associated with antibodies by plasmacytoid dendritic cells. This leads to the activation of TLR9 that in turn induces the pathogenic secretion of type I interferons. TLR9 is an ER‐resident transmembrane protein, which has to relocate to the endolysosomal system in order to be proteolytically processed into the active form. The new data reveal that the DNA‐antibody complexes induce LAP, which results in the colocalization of LC3 and TLR9 in phagosomes 34. These events require ATG5, ATG7 and PtdIns3K activity but not the component of the ULK complex such as ULK1, ATG13 and FIP200 34. Collectively, these observations show that LAP plays a key role in the establishment of the so‐called TRL9‐positive interferon signaling compartments. The relevance of LAP in this process is elicited by the fact that this pathway is required for IFN‐α secretion in response to DNA‐antibody complexes.
Bone Resorption by Osteoclasts
LAP is not the only case in which cellular membranes are modified by LC3. This phenomenon has also been encountered in osteoclasts, specialized multinucleated cells of myeloid origin responsible for bone resorption. There is a fine balance between bone formation and resorption in order to maintain skeletal integrity (subject to growth and mechanical constraints), mineral homeostasis (calcium and phosphate levels) and the bone marrow environment 35. Osteoclasts hence play an essential role in maintaining the bone tissue homeostasis. These cells are adjacent to the bone and an actin ring seals their bone‐apposed membrane. The formation of the actin ring is regulated by microtubules and the small GTPase CDC42. The generated delimitated extracellular space is referred to as the resorptive organelle and the limiting membrane develops into a ruffled border increasing the surface through which ions, mainly protons and chloride, are pumped into the resorptive organelle in order to degrade the mineral matrix. In addition, lysosomal enzymes such as cathepsin K and collagenase digest the organic components of the bone upon delivery into the resorptive organelle through the fusion of osteoclast secretory lysosomes with the ruffled border 36.
Maturation of osteoclasts is associated with increased synthesis of LC3‐II, ATG4B and ATG5, which does not correlate with an enhanced autophagic flux 37. Upregulation of these proteins is probably due to the fact that formation of the ruffled border, fusion of secretory lysosomes with the ruffled border and bone resorption activity depends on the ATG12‐ATG5 conjugates, ATG7, ATG4B and LC3B 37, 38. In particular, LC3B localizes at the bone‐apposed membrane in association with the actin ring but is not present on secretory lysosomes, indicating an essential and direct role for LC3‐II in the process 38 (Figure 2). This observation has led to the hypothesis that LC3‐II‐decorated membranes promote fusion with secretory lysosomes in analogy with the role of LC3 in LAP. More recently, it has been shown that LC3 associates with microtubules in proximity of the actin ring and regulates CDC42 37. These data thus suggest that the pool of LC3‐II at the ruffled border could be a structural and/or a functional link between CDC42 microtubules, which are known to be essential for the initiation of the actin ring formation.
The ERAD Tuning, Coronaviruses and Chlamydia trachomatis
ER‐associated degradation (ERAD) is a cellular pathway responsible for the turnover of folding‐defective polypeptides generated in the ER 39. Upon recognition of their modified sugar chains by ERAD lectins, misfolded polypeptides are retrotranslocated through the dislocon complex into the cytosol and targeted for proteasomal degradation. The ER degradation‐enhancing α‐mannosidase‐like 1 (EDEM1) and osteosarcoma amplified 9 (OS‐9) are stress‐inducible positive regulators of the ERAD‐mediated protein disposal 40. Unlike the vast majority of ER resident chaperones, EDEM1 and OS‐9 are short‐lived and rapidly degraded in the endolysosomal system. The constitutive removal of these two regulators from the ER has been termed ERAD tuning 41. The current model is that it guarantees low ER intralumenal EDEM1 and OS‐9 concentrations in physiological conditions therefore avoiding premature turnover of nascent polypeptides and interference with the ongoing folding programs 41.
EDEM1 and OS‐9 specifically exit the ER in LC3‐I‐positive vesicles that have been named EDEMosomes 42, 43 (Figure 2). These carriers are distinct from autophagosomes as they are not labeled with the specific marker construct GFP‐LC3 and induction of autophagy by starvation decreases EDEM1 disposal possibly by reducing LC3 availability 42. Moreover deletion of ATG7 does not affect EDEM1 and OS‐9 turnover while LC3 does 44, 45. SEL1L is a type I membrane protein and is a component of the HRD1 dislocon. EDEM1 and OS‐9 selectively bind SEL1L in the lumen of the ER, while LC3‐I binds on the cytosolic side, probably assisting the formation of EDEMosomes and the incorporation of EDEM1 and OS‐9 in these vesicles 44. As a result SEL1L appears to act as an EDEMosome cargo receptor. The presence of misfolded proteins in the ER lumen outcompetes the binding of EDEM1 and OS‐9 to SEL1 and inhibits the formation of EDEMosomes. This stabilizes the SEL1‐HRD1 complex engaging the ERAD pathway while simultaneously increasing EDEM1 and OS‐9 intralumenal concentrations 44, 46, 47.
Coronaviruses (CoV) are enveloped viruses with a large single‐stranded, positive‐sense RNA as a genome and comprise the Severe Acute Respiratory Syndrome (SARS)‐CoV or the model virus Mouse Hepatitis Virus (MHV). They belong to the order of the Nidovirales and to the family of the Coronaviridae 48. After entry, they induce the formation of double membrane vesicles (DMVs) to which they target their replication and transcription complex 49. The exact source of DMVs membranes still remains elusive although several lines of evidence, including ultrastructural studies 50, support an ER origin. A possible role for autophagy in the generation of DMVs was proposed as the morphology of their double‐lipid bilayers is reminiscent of autophagosomes 51. However, conflicting results on the recruitment of LC3 onto DMVs membranes and the requirement of ATG5 for virus replication led to some confusion 52, 53, 54, 55. The issue was clarified by a series of experiments showing that MHV probably hijacks LC3‐I‐positive EDEMosome membranes 45 (Figure 2). Thus, similarly to EDEMosomes, MHV replication requires LC3 (in its non‐lipidated form, i.e. LC3‐I) but not an intact autophagy pathway. LC3‐I is non‐covalently associated to the MHV‐induced DMVs, which are distinct from the LC3‐II‐positive autophagosomes, and knockdown of LC3A and LC3B or SEL1L severely impairs MHV replication. Moreover, while the DMVs lack conventional ER protein markers, they contain EDEM1 and OS‐9. Together these lines of evidence have led to the hypothesis that CoV usurp the ERAD tuning pathway in order to form the DMVs essential for their replication 45. Whether the CoV exploit the totality or part of the ERAD tuning machinery and how these viruses generate DMVs from single membrane vesicles, e.g. the EDEMosomes, remains to be understood.
Identically to MHV, Equine Arteritis Virus (EAV) a member of the arteriviridae family that belongs to the order of the Nidovirales as well 56, also usurps the ERAD tuning pathway 57 (Figure 2). Thus the completion of EAV intracellular life cycle does not require ATG7 but depends on LC3‐I, which localizes to the DMVs induced by this virus 57. The striking analogy in the infection mechanisms of MHV and EAV raises the question whether the subversion of the ERAD tuning is a common strategy employed by other related RNA viruses that are part of different virus families.
Chlamydia trachomatis is an obligate intracellular bacterium that relies on its host for nutrient supply. It exhibits an alternate biphasic developmental cycle where infectious elementary bodies, which are metabolically inert, are responsible for the dissemination of the infection 58. Upon their uptake and internalization into the host cell, they generate membrane‐bound compartments termed inclusions and there they switch towards the metabolically active form that is no longer infectious 59. C. trachomatis‐containing inclusions escape the endocytic route before being acidified and travel to the perinuclear region near the Golgi, where their interaction with the exocytic pathway allows the bacterium to replicate 58. Eventually, C. trachomatis exits the host cell through lysis or extrusion.
Early findings suggested a potential interaction between C. trachomatis and autophagy, as the growth of this microbe in the inclusions was hindered after inhibition of autophagy by either addition of an excess of amino acids or 3‐methyladenine, a PtdIns3K inhibitor 60. The probable unspecific and pleiotropic effects of these treatments, which might be responsible for the C. trachomatis growth impairment, later discredited these results. Still, localization of endogenous LC3 at the periphery of the inclusions indicated the engagement of autophagy. Pachikara and colleagues 61 were the first to suggest that C. trachomatis had a more subtle interplay with the autophagy as neither the induction of this pathway by starvation or rapamycin, nor its inhibition upon ATG5 knockout had an effect on chlamydial life cycle. This notion was confirmed through the precise characterization of LC3 recruitment onto the inclusion membrane 62. Similarly to the ERAD tuning, only the LC3‐I form of endogenous LC3 decorated the inclusions whereas GFP‐LC3 failed to be recruited onto them (Figure 2). Moreover LC3 knockdown decreased bacterial progeny as well as inclusion size and infectivity. Interestingly LC3 and its interaction partners, the microtubule‐associated protein 1A and 1B (MAP1), colocalize with the bacterial inclusions situated in proximity of the Golgi at late time points during infection but not with the inclusions dispersed in the cytoplasm shortly after infection. Together with the observation that LC3 and MAP1 required microtubules to associate with the inclusion, these results suggest that the probable function of LC3 in C. trachomatis life cycle is mediating the relocalization of the inclusion in a microtubule‐dependent manner. It remains to be established whether C. trachomatis hijacks components of the ERAD tuning to usurp LC3.
Clearance of Brucella abortus Infection
B. abortus is another obligatory intracellular bacterium that can invade and replicate in several cell types. After phagocytic uptake and internalization, B. abortus resides in a modified compartment referred to as the Brucella‐containing vacuoles (BCVs). These vacuoles undergo limited fusion with endosomes and lysosomes thus avoiding degradation by the endolysosomal system 63. Acidification of the vacuole is necessary for the induction of B. abortus's VirB operon, which encodes for a type IV secretion system that injects bacterial effectors into the cytoplasm of the host cell and trigger BCVs translocation to the ER. There, B. abortus replicates in ER‐derived replicative BCVs (rBCVs), which are positive for ER marker proteins and are coated with ribosomes. Subsequently the rBCVs are converted into vacuoles with autophagic properties termed autophagic BCVs (aBCVs), which are essential for the completion of the intracellular life cycle of B. abortus and its cell‐to‐cell spread 63.
The aBCVs are negative for the ER marker protein calreticulin and positive for LAMP1 and RAB7, another protein that characterizes the late compartments of the endolysosomal system 63. They are acidic and enwrapped by double‐membrane crescents or multiple membranes. The aBCV formation depends on ATG proteins mediating the initiation of the autophagosome biogenesis (Figure 1B), such as ULK1, BECLIN1, ATG14L and PtdIns3P synthesis 63. In contrast depletion of ATG proteins essential for the expansion of the phagophore, like ATG5, ATG7, ATG16L1, ATG4B and LC3B (Figure 1B), or factors involved in autophagosome maturation, including UVRAG and RAB9, did not affect aBCV formation or B. abortus replication 63. Accordingly with these requirements for aBCV biogenesis, knockdown of BECLIN1 and ULK1 significantly reduced the establishment of secondary B. abortus infection foci while depletion of ATG5, ATG7 or LC3B did not affect the cell‐to‐cell spread of this bacterium 63.
Inhibition of Murine Norovirus Replication
Murine norovirus (MNV) is a non‐enveloped single‐stranded RNA virus used as a model to study human gastroenteritis caused by the norovirus family members 64. IFN‐γ has a central stage in integrating antiviral defenses, especially in the absence of type I interferons IFN‐α and IFN‐β, which can play a compensatory role 65. A recent study has revealed that the ATG12‐ATG5/ATG16L1 complex is necessary for the IFN‐γ‐mediated antiviral activity against MNV 66. In particular ATG5 is required for MNV control in IFN‐γ‐treated macrophages as this virus replicated normally in atg5 knockout cells, which display unaffected transcriptional changes and activation of the JAK‐STAT signaling upon IFN‐γ treatment. The restriction of MNV infection, however, does not involve autophagy because various treatments modulating autophagy (inhibition or activation) did not affect MNV replication, indicating that ATG5 has an autophagy‐independent key role in the antiviral activity triggered by IFN‐γ 66.
Expression of an ATG5 mutant form that cannot be conjugated with ATG12 or bind to ATG16L1 could not complement the lack of resistance against MNV infections in atg5‐deficient macrophages. In addition, atg16L1‐ and atg7‐deficient cells failed to control the virus upon IFN‐γ treatments. A dominant‐negative construct of ATG4B that blocks LC3 lipidation, however, did not have a negative impact on MNV infection restriction revealing that the LC3 conjugation system (which also involves ATG7, Figure 1B) does not participate to this activity. Collectively, these observations indicate the implication of the ATG12‐ATG5/ATG16L1 complex but not the entire ATG machinery in the IFN‐γ‐induced antiviral defense 66.
Interestingly ATG16L1 localizes to the replicative membranous structures induced by MNV and associates with the viral polymerase 66. In addition, ATG5 is responsible for the inhibition of the viral polymerase expression and the synthesis of positive and antisense viral genome as well as structural proteins. These autophagy‐independent functions of the ATG12‐ATG5/ATG16L1 complex could account for the inhibition of the formation of the viral replication complexes upon induction of IFN‐γ‐mediated responses in macrophages, but it could also inhibit the viral life cycle through additional not yet identified mechanisms.
Toxoplasma gondii Vacuole Disruption
ATG proteins are also involved in autophagy‐independent defense mechanisms against intracellular protozoan parasites, as observed during T. gondii infections. This parasite persists in non‐activated macrophages by inhibiting upon entry the fusion of the modified parasitophorous vacuole, in which it replicates, with the lysosome 67. Activated macrophages, however, can limit replication, kill and clear this pathogen through two independent mechanisms 68. The first involves the activation of autophagy by CD40‐CD40L signaling while the second mechanism entails the IFN‐γ‐induced disruption and stripping of the parasitophorous vacuole membrane, followed by the clearance of the damaged parasite and membranous debris. This pathway is modulated by the p47 GTPase IIGP1 and requires an autophagy‐independent function of ATG5 69. In particular, T. gondii‐infected atg5‐deficient macrophages exposed to IFN‐γ/LPS display intact parasitophorous vacuoles and they are consequently unable to clear the invading parasites 69. It appears that the lack of ATG5 hinders the rapid recruitment of IIGP1 onto the parasitophorous vacuole membrane. IIGP1 is instead found on intracellular vesicles, thus leading to the hypothesis of an impairment of this protein trafficking to the T. gondii vacuole in absence of ATG5. The involvement of autophagy in this process was excluded mostly through EM examinations, which failed to detect membrane structures resembling autophagosomes in proximity of the parasitophorous vacuole 69. While the cellular localization of LC3 in the atg5 knockout and control cells has not been investigated in this study, an earlier report revealed accumulations of LC3 onto disrupted parasitophorous vacuoles in the vicinity of intense IIGP1 signals in T. gondii‐infected, IFN‐γ‐stimulated astrocytes 70. The intriguing questions concerning the possible functions of ATG5 and LC3 in the immune response against T. gondii thus still remain unanswered.
Regulation of STING‐Induced Inflammatory Immune Response
Another example where a subset of ATG proteins has been shown to exert an autophagy‐independent function is during the stimulator of interferon genes (STING)‐mediated innate inflammatory immune response upon detection of cytoplasmic DNA. In this context, the ATG proteins intervene in a signal transduction pathway and execute a regulatory function rather than acting as immune effectors. Cytopalsmic double‐stranded oligonucleotides are recognized by TLRs or RIG‐I‐like receptors (RLRs), which subsequently trigger the innate inflammatory immune responses. On one hand, absent in melanoma 2 (AIM2) signaling through the inflammasome complex and activation of caspase 1 are responsible for the production of the pro‐inflammatory cytokines interleukin‐1β (IL‐1β) and IL‐18 71. On the other hand, STING activation leads to the phosphorylation of transcription factors IRF3 and STAT6, which subsequently translocate into the nucleus and induce the transcription of the IFN genes 72.
The precise mechanism of action of STING is largely unknown. Nevertheless, it has been shown that upon dsDNA recognition, this transmembrane protein translocates from the ER to the Golgi apparatus before associating and forming cytoplasmic foci with the TANK‐binding kinase 1 (TBK1), which regulates certain selective types of autophagy, and the autophagy cargo receptor p62 73, 74, 75. LC3 and ATG9a also localize to these punctate structures while ULK1, ATG5 and ATG14L do not. The independence of STING signaling from the two autophagy ubiquitin‐like systems is also underlined by the fact that atg7 and atg16l1 knockout cells display normal membrane trafficking of STING and IFN‐β secretion upon exposure to cytosolic dsDNA 73. In contrast the cytoplasmic TBK1‐positive STING puncta are largely increased in atg9a‐deficient cells in response to dsDNA stimulation as are IRF3 phosphorylation and IFN‐β, IL‐6 and CXCL10 production. This aberrant activation of the STING downstream signaling indicates that a subset of ATG proteins, independently from their implication in autophagosome formation, plays a role in the regulation of innate signaling upon detection of cytoplasmic DNA. The exact mechanism of such a modulation, however, still remains to be elucidated.
The Role of the ATG Proteins in Cell Division and Cell Cycle Arrest
As being part of the autophagy machinery, ATG7 is essential to overcome the post‐natal starvation period of newborn mice 76, 77. Recently, Lee et al. 78 have identified another function of ATG7 in response to metabolic stresses that is autophagy‐independent. Nutrient deprivation stimulates the exit from the cell division cycle, in particular from the S‐phase. ATG7 is required for this exit from the S‐phase but also to efficiently express the cyclin‐dependent kinase inhibitor (CDI) p21CDKN1A 78, which prevents cell cycle progression. This unconventional role of ATG7 is autophagy‐independent because it does not require the E1‐like conjugation activity of this protein. Moreover neither BECLIN1 knockdown or atg5 −/− knockout cells display abnormal exit from the S‐phase upon nutrient starvation. The role of ATG7 in the cell cycle arrest relies on its ability to bind to p53 in both the cytoplasm and the nucleus. The association between ATG7 and p53 is enhanced by metabolic stresses and together with p53, ATG7 binds to the promoter of p21CDKN1A gene inducing its expression (Figure 3D). Conversely, in the absence of ATG7, p53 induces the expression of pro‐apoptotic genes, which eventually trigger cell death pathways. atg7 −/− knockout cells also showed increased radical oxygen species (ROS) production and enhanced phosphorylation of p53 on Ser20, which are features indicating an activation of the DNA damage response pathway that eventually leads to a p53‐mediated cell death 79. Indeed genetic blocking of the DNA damage response mediated by the ROS‐CHK2‐p53 pathway was beneficial for the survival of the atg7 −/− mice. That is atg7 −/− chk2 −/− mice survived longer and displayed augmented p53‐dependent pro‐apoptotic gene expression compared to the atg7 −/− animals revealing that the ATG7‐mediated switch from p53‐dependent cell cycle arrest versus p53‐dependent cell death is probably an important etiological factor for the post‐natal lethality of mice as well 78, 80.
Figure 3.
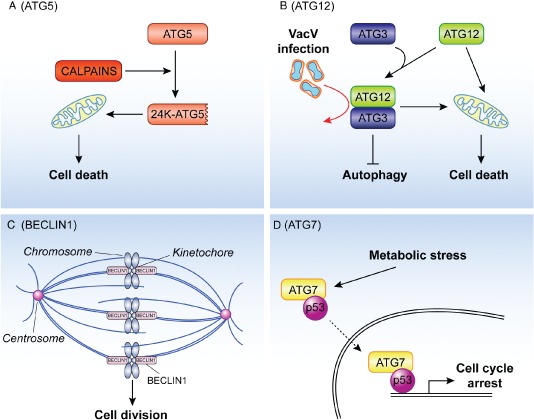
The unconventional roles of ATG proteins in cell death and cell division and proliferation. A) Apoptotic stimuli activate calpains and one of the substrates of these proteases is ATG5. The resulting 24 kDa truncated form of ATG5 (24K‐ATG5) associates with mitochondria and triggers cytochrome c release through a still unknown mechanism, thus participating to the initiation of the cell death programs. B) ATG12 participates in apoptosis at least in two distinct ways. First, it can become part of the ATG12‐ATG3 conjugate, which plays an important role in controlling mitochondria homeostasis and sensitizes cells to apoptosis triggered by the intrinsic mitochondrial pathway. The ATG12‐ATG3 conjugate is also stimulated upon vaccinia virus infection and it is required for the life cycle of this virus (red arrow). Second and independently from its capacity to be covalently linked to ATG5 or ATG3, ATG12 stimulates mitochondrial apoptosis through its binding and inhibition of the anti‐apoptotic function of the members of the BCL‐2 protein family. C) During mitosis BECLIN1 associates with ZWINT‐1 and mediates the interaction between the kinetochore and microtubules, a key event in cell division. Depletion of BECLIN1 causes a defect in the kinetochore protein assembling, misalignment of chromosomes and a general block in mitotic progression. D) Upon metabolic stress, ATG7 is found associated with p53 in the cytoplasm and nucleus. The nuclear complex positively regulates the expression of genes mediating the cell cycle arrest. It remains to be determined whether the nuclear ATG7‐p53 pool results from the translocation into the nucleus of the subpopulation of this complex formed in the cytoplasm (dashed arrow).
The autophagy‐independent function of ATG7 in controlling cell cycle arrest and preventing uncontrolled stimulation of cell death contributes to maintain cellular homeostasis, but it is not the only ATG protein participating in the regulation of cell division and proliferation. BECLIN1 was the first ATG protein that linked autophagy with tumor development as mono‐allelic loss of BECLIN1 can be observed in many tumors 81. Loss of BECLIN1 in tumor cells is accompanied with chromosomal instability and centrosomal abnormalities 82, 83. During mitosis BECLIN1 localizes to the kinetochore complex in a microtubule‐dependent manner and it binds ZWINT‐1 at this location (Figure 3C). ZWINT‐1 is a component of the KNL‐1/MIS12/NDC80 (KMN) complex, which mediates the interaction between the kinetochore and microtubules 84. Acting downstream of the KMN complex, BECLIN1 participates in the association of the kinetochore proteins and the subsequent anchoring of microtubules to the kinetochore (Figure 3C). As a result, depletion of BECLIN1 causes a defect in the kinetochore protein assembling, misalignment of chromosomes and a general block in mitotic progression. This novel mitosis‐specific function of BECLIN1 is independent of its function in autophagy or as a component of the PdtIns3K complex because downregulation of components of the PtdIns3K complex such as VPS34, VPS15 and UVRAG, or other ATG proteins including ATG5 or LC3 did not have negative effects on mitosis progression 82. Moreover the depletion of these proteins did not alter the cellular levels of the kinetochore proteins or BECLIN1 subcellular distribution during mitosis 82. Thus, in addition to its involvement in autophagy, one possibility could be that BECLIN1 controls tumor development and progression by also regulating chromosomal congressing during mitosis. This could, at least in part, explain why BECLIN1 is the mostly frequently ATG gene found altered in cancers and possibly why its knockout is embryonically lethal in mice while the ablation of most of the other ATG genes is not.
The Roles of Different ATG Proteins in Cell Death
ATG proteins can carry out unconventional functions upon post‐translational modifications that divert them into a different pathway than autophagy and ATG5 provides a first example. Yousefi and colleagues 85 have reported that a truncated form of this protein is involved in promoting apoptotic cell death. Apoptotic stimuli, like the CD95 ligand or anticancer drugs stimulate the activation of calpains, calcium‐dependent non‐lysosomal cysteine proteases, which also cleave full‐length ATG5 (33 kDa) at threonine 193 (Figure 3A). The resulting 24 kDa truncated form of ATG5 associates with mitochondria and triggers cytochrome c release and subsequent caspase 3 activation through a still unknown mechanism. In addition to its translocation onto mitochondria, truncated ATG5 binds to BCL‐XL in apoptotic cells, and thereby it possibly blocks its anti‐apoptotic activity. Importantly, the overexpression of truncated ATG5 does not stimulate autophagy, which suggests that the truncated and the full‐length ATG5 forms have separate biological roles. The involvement of autophagy in promoting cell death is a very controversial topic but recent reports support the notion that autophagy is in general not a cause for cell death 86, 87. Unconventional functions of ATG proteins, in contrast, may well promote and/or contribute to cell death. Thus, the function of the ATG5 can switch from being required for protective autophagy towards triggering apoptosis. Moreover the processing of ATG5 could also have inhibitory effect on autophagy by depleting the pool of this protein involved in this pathway thus further enhancing cell death. Similarly to ATG5, BECLIN 1 processing by caspase 3 also appears to generate a fragment with pro‐apoptotic properties 88, 89.
Radoshevich and colleagues 90 have recently identified different ATG12‐containing complexes with yet uncharacterized ATG12 partners. In this study, they revealed a novel conjugate of ATG12 in which this protein is fused with ATG3. ATG12 was generally known to be covalently linked to ATG5, which is essential for autophagosome biogenesis. The newly discovered ATG12‐ATG3 conjugate is generated through the autocatalytic activity of ATG3 that links ATG12 to the lysine 243 of ATG3 90. In contrast to the ATG12‐ATG5 conjugate, the ATG12‐ATG3 fusion also forms in the absence of ATG5 and is not required for mammalian autophagy. The ATG12‐ATG3 conjugate rather plays an important role in controlling mitochondria homeostasis 90 (Figure 3B). In its absence the tubular mitochondrial network is disrupted due to a reduced capacity of mitochondria to undergo homeotypic fusion. Moreover the ATG12‐ATG3 conjugate sensitizes cells to apoptosis triggered by the intrinsic mitochondrial pathway (Figure 3B).
The generation of the ATG12‐ATG3 conjugate is also stimulated upon vaccinia virus infection 91. Even in the absence of ATG5 and ATG7, vaccinia virus infection causes a massive LC3 lipidation. This event requires the ATG12‐ATG3 conjugate, the formation of which is stimulated by the virus and leads to a concomitant decrease of the cellular amounts of ATG12‐ATG5 91. The increased levels of LC3‐II, however, do not correlate with an induction of autophagy. In fact, vaccinia virus infection blocks this transport route. Thus the ATG12‐ATG3 conjugate functions in different cellular processes, which are distinct from autophagy and even counteract it. The stimulation of the formation of the ATG12‐ATG3 conjugate could therefore provide an additional mechanism for pathogens to inhibit antimicrobial activity of autophagy.
In addition to its autophagy‐independent function as part of the ATG12‐ATG3 conjugate, ATG12 also associates with members of the BCL‐2 protein family and stimulates mitochondrial apoptosis 92 (Figure 3B). The pro‐apoptotic function of ATG12 is likely mediated through its binding and inhibition of the antiapoptotic function of BCL‐2 and MCL‐1, also a member of the BCL‐2 protein family. The binding of ATG12 to the BCL‐2 protein family members is independent from its capacity to be covalently linked to ATG5 or ATG3 92. Thus, ATG proteins previously known to only play an essential role in the autophagic ubiquitin‐like conjugation systems are increasingly identified as important actors in cellular processes distinct from autophagy.
Conclusions and Perspectives
Most of the ATG genes in high eukaryotes have been identified through homology to yeast counterparts isolated with genetic screens aimed to uncover autophagy players. This has somehow biased the investigation of the cellular functions of the ATG proteins, but studies on their role particularly in immunity and infections have been fundamental in revealing that they are key components in other pathways as well. The emerging picture is that ATG proteins are part of these processes sometimes as the only component of the ATG machinery and sometimes together with their functional ATG gene cluster 19. Posttranslational modifications, such as lipidation, conjugation to other proteins and proteolytic processing, appear to be central in regulating and coordinating the different functions of each ATG gene products. A clear example of this multitask versatility is LC3 (Figure 2). Its lipidation and targeting to autophagosomal membranes require all ATG functional gene clusters 93. While the exact function of lipidated LC3 (LC3‐II) remains to be determined, its molecular characteristics are very likely exploited in LAP, establishment of the ruffled borders by osteoclasts (Figure 2), and possibly for trafficking though the Golgi apparatus 94, 95. Not surprisingly, the factors mediating the formation of LC3‐II, including the ATG12 conjugation system, are also involved in these processes. In contrast, the functions of LC3‐I in the ERAD tuning, the formation of coronaviral DMVs and subcellular redistribution of Chlamydia inclusions appear to implicate none of the other ATG components but rather the ability of this protein to interact with microtubules, at least in the context of the two infections 18, 62, 96. The fact that LC3‐I and LC3‐II have very divers molecular features and probably interact with different sets of partners is also underlined by the observation that tagging of LC3 at its N‐terminus is not interfering with the functions of LC3‐II but abolishes those of LC3‐I 12, 23, 38, 42, 45, 62.
An important consideration is that the ATG genes that are part of more than one pathway are potentially ideal molecular switches for the coordination of major cellular reprogrammings. The data on the unconventional functions of ATG5 underline the possibility of the existence of such scenario. Through its participation in autophagy, ATG5 has mainly a pro‐survival role 86, 87. Apoptotic cues, however, induce both the calpain‐dependent processing and ATG3conjugation of ATG5, and the resulting forms of this protein contribute to the cell signaling that leads to cell death 85, 90. In turn, these modifications probably cause a depletion of the ATG5 pool that can be engaged in autophagy thus leading to a downregulation of this pathway, which further enhances the apoptotic program.
The identification and the study of the unconventional functions of the ATG proteins is a new emerging field, which will very likely expand during the next years with scientists investigating the molecular roles of these proteins in so far unexplored tissues. Historically the atg5‐ and atg7‐knockout mice were the first the first generated atg gene knockout animals 76, 77, and as a result they were pivotal in revealing the unconventional functions of these two genes and the two ubiquitin‐like systems. Mouse models that lack most of the other ATG genes are now available, and they will also be a key tool to shed light into the physiological roles of the ATG proteins beyond autophagy. Nonetheless, a current certitude is that the role of autophagy cannot be exclusively assessed by depleting one single ATG protein or even a few of them that belong to the same functional group (Figure 1B). Thus, while most of the researchers are not yet familiar with the unconventional functions of the ATG genes; their discovery has already influenced our experimental approach to study autophagy.
Acknowledgments
The authors thank René Scriwanek for the assistance with the preparation of the figures. J. B. is supported by a ZonMW VENI grant (916.126.018). F. R. is supported by ECHO (700.59.003), ALW Open Program (821.02.017 and 822.02.014), DFG‐NWO cooperation (DN82‐303) and ZonMW VICI (016.130.606) grants.
References
- 1. Kraft C, Martens S. Mechanisms and regulation of autophagosome formation. Curr Opin Cell Biol 2012;24:496–501. [DOI] [PubMed] [Google Scholar]
- 2. Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010;22:124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010;40:280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011;469:323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013;368:651–662. [DOI] [PubMed] [Google Scholar]
- 6. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008;132:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mari M, Tooze SA, Reggiori F. The puzzling origin of the autophagosomal membrane. F1000 Biol Rep 2011;3:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Orsi A, Polson HE, Tooze SA. Membrane trafficking events that partake in autophagy. Curr Opin Cell Biol 2010;22:150–156. [DOI] [PubMed] [Google Scholar]
- 9. Young ARJ, Chan EYW, Hu XW, Köchl R, Crawshaw SG, High S, Hailey DW, Lippincott‐Schwartz J, Tooze SA. Starvation and ULK1‐dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci 2006;119:3888–3900. [DOI] [PubMed] [Google Scholar]
- 10. Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, Tooze SA. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell 2012;23:1860–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mari M, Griffith J, Rieter E, Krishnappa L, Klionsky DJ, Reggiori F. An Atg9‐containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol 2010;190:1005–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shpilka T, Mizushima N, Elazar Z. Ubiquitin‐like proteins and autophagy at a glance. J Cell Sci 2012;125:2343–2348. [DOI] [PubMed] [Google Scholar]
- 13. Weidberg H, Shpilka T, Shvets E, Abada A, Shimron F, Elazar Z. LC3 and GATE‐16N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev Cell 2011;20:444–454. [DOI] [PubMed] [Google Scholar]
- 14. Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin‐like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007;130:165–178. [DOI] [PubMed] [Google Scholar]
- 15. Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, Sawada N, Yamada A, Mizushima N, Uchiyama Y, Kominami E, Tanaka K, Komatsu M. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell 2008;19:4762–4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE‐16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 2010;29:1792–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin‐mediated recognition and beyond. Nat Cell Biol 2010;12:836–841. [DOI] [PubMed] [Google Scholar]
- 18. Monastyrska I, Rieter E, Klionsky DJ, Reggiori F. Multiple roles of the cytoskeleton in autophagy. Biol Rev Camb Philos Soc 2009;84:431–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Subramani S, Malhotra V. Non‐autophagic roles of autophagy‐related proteins. EMBO Rep 2013;14:143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Virgin HW, Levine B. Autophagy genes in immunity. Nat Immunol 2009;10:461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sanjuan M, Milasta S, Green D. Toll‐like receptor signaling in the lysosomal pathways. Immunol Rev 2009;227:203–220. [DOI] [PubMed] [Google Scholar]
- 22. Florey O, Overholtzer M. Autophagy proteins in macroendocytic engulfment. Trends Cell Biol 2012;22:374–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, Green DR. Toll‐like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007;450:1253–1257. [DOI] [PubMed] [Google Scholar]
- 24. Martinez J, Almendinger J, Oberst A, Ness R, Dillon C, Fitzgerald P, Hengartner M, Green D. Microtubule‐associated protein 1 light chain 3 alpha (LC3)‐associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci USA 2011;108:17396–17401. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25. Cullinane M, Gong L, Li X, Lazar‐Adler N, Tra T, Wolvetang E, Prescott M, Boyce J, Devenish R, Adler B. Stimulation of autophagy suppresses the intracellular survival of Burkholderia pseudomallei in mammalian cell lines. Autophagy 2008;4:744–753. [DOI] [PubMed] [Google Scholar]
- 26. Gong L, Cullinane M, Treerat P, Ramm G, Prescott M, Adler B, Boyce J, Devenish R. The Burkholderia pseudomallei type III secretion system and BopA are required for evasion of LC3‐associated phagocytosis. PLoS One 2011;6:e17852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li X, Prescott M, Adler B, Boyce J, Devenish R. Beclin 1 is required for starvation‐enhanced, but not rapamycin‐enhanced, LC3‐associated phagocytosis of Burkholderia pseudomallei in RAW 264.7 cells. Infec Immun 2012;81:271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Codogno P, Mehrpour M, Proikas‐Cezanne T. Canonical and non‐canonical autophagy: variations on a common theme of self‐eating? Nat Rev Mol Cell Biol 2012;13:7–12. [DOI] [PubMed] [Google Scholar]
- 29. Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, Mizushima N, Grinstein S, Iwasaki A. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 2010;32:227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ravichandran K. Find‐me and eat‐me signals in apoptotic cell clearance: progress and conundrums. J Exp Med 2010;207:1807–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Overholtzer M, Mailleux A, Mouneimne G, Normand G, Schnitt S, King R, Cibas E, Brugge J. A nonapoptotic cell death process, entosis, that occurs by cell‐in‐cell invasion. Cell 2007;131:966–979. [DOI] [PubMed] [Google Scholar]
- 32. Florey O, Kim S, Sandoval C, Haynes C, Overholtzer M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol 2011;13:1335–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li W, Zou W, Yang Y, Chai Y, Chen B, Cheng S, Tian D, Wang X, Vale RD, Ou G. Autophagy genes function sequentially to promote apoptotic cell corpse degradation in the engulfing cell. J Cell Biol 2012;197:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Henault J, Martinez J, Riggs JM, Tian J, Mehta P, Clarke L, Sasai M, Latz E, Brinkmann MM, Iwasaki A, Coyle AJ, Kolbeck R, Green DR, Sanjuan MA. Noncanonical autophagy is required for type I interferon secretion in response to DNA‐immune complexes. Immunity 2012;37:986–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boyce B, Rosenberg E, de Papp A, Duong LT. The osteoclast, bone remodelling and treatment of metabolic bone disease. Eur J Clin Invest 2012;42:1332–1341. [DOI] [PubMed] [Google Scholar]
- 36. Coxon FP, Taylor A. Vesicular trafficking in osteoclasts. Semin Cell Dev Biol 2008;19:424–433. [DOI] [PubMed] [Google Scholar]
- 37. Chung Y‐H, Yoon S‐Y, Choi B, Sohn D, Yoon K‐H, Kim W‐J, Kim D‐H, Chang E‐J. Microtubule‐associated protein light chain 3 regulates Cdc42‐dependent actin ring formation in osteoclast. Int J Biochem Cell Biol 2012;44:989–997. [DOI] [PubMed] [Google Scholar]
- 38. DeSelm CJ, Miller BC, Zou W, Beatty WL, van Meel E, Takahata Y, Klumperman J, Tooze SA, Teitelbaum SL, Virgin HW. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell 2011;21:966–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol 2005;7:766–772. [DOI] [PubMed] [Google Scholar]
- 40. Aebi M, Bernasconi R, Clerc S, Molinari M. N‐glycan structures: recognition and processing in the ER. Trends Biochem Sci 2010;35:74–82. [DOI] [PubMed] [Google Scholar]
- 41. Bernasconi R, Molinari M. ERAD and ERAD tuning: disposal of cargo and of ERAD regulators from the mammalian ER. Curr Opin Cell Biol 2011;23:176–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cali' T, Galli C, Olivari S, Molinari M. Segregation and rapid turnover of EDEM1 by an autophagy‐like mechanism modulates standard ERAD and folding activities. Biochem Biophys Res Commun 2008;371:405–410. [DOI] [PubMed] [Google Scholar]
- 43. Zuber C, Cormier JH, Guhl B, Santimaria R, Hebert DN, Roth J. EDEM1 reveals a quality control vesicular transport pathway out of the endoplasmic reticulum not involving the COPII exit sites. Proc Natl Acad Sci USA 2007;104:4407–4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bernasconi R, Galli C, Noack J, Bianchi S, de Haan CA, Reggiori F, Molinari M. Role of the SEL1L:LC3‐I complex as an ERAD tuning receptor in the mammalian ER. Mol Cell 2012;46:809–819. [DOI] [PubMed] [Google Scholar]
- 45. Reggiori F, Monastyrska I, Verheije MH, Cali T, Ulasli M, Bianchi S, Bernasconi R, de Haan CA, Molinari M. Coronaviruses hijack the LC3‐I‐positive EDEMosomes, ER‐derived vesicles exporting short‐lived ERAD regulators, for replication. Cell Host Microbe 2010;7:500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Iida Y, Fujimori T, Okawa K, Nagata K, Wada I, Hosokawa N. SEL1L protein critically determines the stability of the HRD1‐SEL1L endoplasmic reticulum‐associated degradation (ERAD) complex to optimize the degradation kinetics of ERAD substrates. J Biol Chem 2011;286:16929–16939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Christianson JC, Shaler TA, Tyler RE, Kopito RR. OS‐9 and GRP94 deliver mutant alpha1‐antitrypsin to the Hrd1‐SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol 2008;10:272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Masters P. The molecular biology of coronaviruses. Adv Virus Res 2006;66:193–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gosert R, Kanjanahaluethai A, Egger D, Bienz K, Baker S. RNA replication of mouse hepatitis virus takes place at double‐membrane vesicles. J Virol 2002;76:3697–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Knoops K, Kikkert M, Worm SH, Zevenhoven‐Dobbe JC, van der Meer Y, Koster AJ, Mommaas AM, Snijder EJ. SARS‐coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol 2008;6:e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. de Haan C, Reggiori F. Are nidoviruses hijacking the autophagy machinery? Autophagy 2008;4:276–279. [DOI] [PubMed] [Google Scholar]
- 52. Snijder E, van der Meer Y, Zevenhoven‐Dobbe J, Onderwater J, van der Meulen J, Koerten H, Mommaas A. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J Virol 2006;80:5927–5940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Prentice E, McAuliffe J, Lu X, Subbarao K, Denison M. Identification and characterization of severe acute respiratory syndrome coronavirus replicase proteins. J Virol 2004;78:9977–9986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhao Z, Thackray L, Miller B, Lynn T, Becker M, Ward E, Mizushima N, Denison M, Virgin H. Coronavirus replication does not require the autophagy gene ATG5. Autophagy 2007;3:581–585. [DOI] [PubMed] [Google Scholar]
- 55. Prentice E, Jerome W, Yoshimori T, Mizushima N, Denison M. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem 2004;279:10136–10141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. MacLachlan NJ, Balasuriya UB. Equine viral arteritis. Adv Exp Med Biol 2006;581:429–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Monastyrska I, Ulasli M, Rottier P, Guan J‐L, Reggiori F, de Haan C. An autophagy‐independent role for LC3 in equine arteritis virus replication. Autophagy 2012;9:164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Abdelrahman Y, Belland R. The chlamydial developmental cycle. FEMS Microbiol Rev 2005;29:949–959. [DOI] [PubMed] [Google Scholar]
- 59. Fields K, Hackstadt T. The chlamydial inclusion: escape from the endocytic pathway. Annu Rev Cell Dev Biol 2002;18:221–245. [DOI] [PubMed] [Google Scholar]
- 60. Al‐Younes H, Brinkmann V, Meyer T. Interaction of Chlamydia trachomatis serovar L2 with the host autophagic pathway. Infect Immun 2004;72:4751–4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pachikara N, Zhang H, Pan Z, Jin S, Fan H. Productive Chlamydia trachomatis lymphogranuloma venereum 434 infection in cells with augmented or inactivated autophagic activities. FEMS Microbiol Lett 2009;292:240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Al‐Younes HM, Al‐Zeer MA, Khalil H, Gussmann J, Karlas A, Machuy N, Brinkmann V, Braun PR, Meyer TF. Autophagy‐independent function of MAP‐LC3 during intracellular propagation of Chlamydia trachomatis . Autophagy 2011;7:814–828. [DOI] [PubMed] [Google Scholar]
- 63. Starr T, Child R, Wehrly T, Hansen B, Hwang S, Lopez‐Otin C, Virgin H, Celli J. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe 2012;11:33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wobus C, Thackray L, Virgin H. Murine norovirus: a model system to study norovirus biology and pathogenesis. J Virol 2006;80:5104–5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev 2001;14:778–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hwang S, Maloney N, Bruinsma M, Goel G, Duan E, Zhang L, Shrestha B, Diamond M, Dani A, Sosnovtsev S, Green K, Lopez‐Otin C, Xavier R, Thackray L, Virgin H. Nondegradative role of Atg5‐Atg12/ Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell Host Microbe 2012;11:397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sibley L. Toxoplasma gondii: perfecting an intracellular life style. Traffic 2003;4:581–586. [DOI] [PubMed] [Google Scholar]
- 68. Zhao Y, Wilson D, Matthews S, Yap G. Rapid elimination of Toxoplasma gondii by gamma interferon‐primed mouse macrophages is independent of CD40 signaling. Infec Immun 2007;75:4799–4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhao Z, Fux B, Goodwin M, Dunay I, Strong D, Miller B, Cadwell K, Delgado M, Ponpuak M, Green K, Schmidt R, Mizushima N, Deretic V, Sibley L, Virgin H. Autophagosome‐independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe 2008;4:458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Martens S, Parvanova I, Zerrahn J, Griffiths G, Schell G, Reichmann G, Howard J. Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47‐resistance GTPases. PLoS Pathog 2005;1:e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hornung V, Ablasser A, Charrel‐Dennis M, Bauernfeind F, Horvath G, Caffrey D, Latz E, Fitzgerald K. AIM2 recognizes cytosolic dsDNA and forms a caspase‐1‐activating inflammasome with ASC. Nature 2009;458:514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Burdette D, Vance R. STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol 2013;14:19–26. [DOI] [PubMed] [Google Scholar]
- 73. Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T, Yamamoto N, Kawai T, Ishii K, Takeuchi O, Yoshimori T, et al. Atg9a controls dsDNA‐driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci USA 2009;106:20842–20846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin‐induced cell death. J Cell Biol 2005;171:603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dotsch V, Bumann D, Dikic I. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011;333:228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 2004;432:1032–1036. [DOI] [PubMed] [Google Scholar]
- 77. Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation‐induced and constitutive autophagy in Atg7‐deficient mice. J Cell Biol 2005;169:425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJ, Motoyama N, Cao L, Finkel T. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 2012;336:225–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge SJ, Mak TW. DNA damage‐induced activation of p53 by the checkpoint kinase Chk2. Science 2000;287:1824–1827. [DOI] [PubMed] [Google Scholar]
- 80. Kageyama S, Komatsu M. Impaired G1‐arrest, autophagy, and apoptosis in Atg7‐knockout mice. Circ Res 2012;111:962–964. [DOI] [PubMed] [Google Scholar]
- 81. Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999;402:672–676. [DOI] [PubMed] [Google Scholar]
- 82. Fremont S, Gerard A, Galloux M, Janvier K, Karess RE, Berlioz‐Torrent C. Beclin‐1 is required for chromosome congression and proper outer kinetochore assembly. EMBO Rep 2013;14:364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 2007;21:1367–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Obuse C, Iwasaki O, Kiyomitsu T, Goshima G, Toyoda Y, Yanagida M. A conserved Mis12 centromere complex is linked to heterochromatic HP1 and outer kinetochore protein Zwint‐1. Nat Cell Biol 2004;6:1135–1141. [DOI] [PubMed] [Google Scholar]
- 85. Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU. Calpain‐mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 2006;8:1124–1132. [DOI] [PubMed] [Google Scholar]
- 86. Shen S, Kepp O, Kroemer G. The end of autophagic cell death? Autophagy 2012;8:1–3. [DOI] [PubMed] [Google Scholar]
- 87. Shen S, Kepp O, Michaud M, Martins I, Minoux H, Metivier D, Maiuri MC, Kroemer RT, Kroemer G. Association and dissociation of autophagy, apoptosis and necrosis by systematic chemical study. Oncogene 2011;30:4544–4556. [DOI] [PubMed] [Google Scholar]
- 88. Luo S, Rubinsztein DC. Apoptosis blocks Beclin 1‐dependent autophagosome synthesis: an effect rescued by Bcl‐xL. Cell Death Differ 2010;17:268–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wirawan E, Vande Walle L, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I, Roelandt R, De Rycke R, Verspurten J, Declercq W, Agostinis P, Vanden Berghe T, Lippens S, Vandenabeele P. Caspase‐mediated cleavage of Beclin‐1 inactivates Beclin‐1‐induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis 2010;1:e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Radoshevich L, Murrow L, Chen N, Fernandez E, Roy S, Fung C, Debnath J. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 2010;142:590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Moloughney JG, Monken CE, Tao H, Zhang H, Thomas JD, Lattime EC, Jin S. Vaccinia virus leads to ATG12‐ATG3 conjugation and deficiency in autophagosome formation. Autophagy 2011;7:1434–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Rubinstein AD, Eisenstein M, Ber Y, Bialik S, Kimchi A. The autophagy protein Atg12 associates with antiapoptotic Bcl‐2 family members to promote mitochondrial apoptosis. Mol Cell 2011;44:698–709. [DOI] [PubMed] [Google Scholar]
- 93. Suzuki K, Kubota Y, Sekito T, Ohsumi Y. Hierarchy of Atg proteins in pre‐autophagosomal structure organization. Genes Cells 2007;12:209–218. [DOI] [PubMed] [Google Scholar]
- 94. Sagiv Y, Legesse‐Miller A, Porat A, Elazar Z. GATE‐16, a membrane transport modulator, interacts with NSF and the Golgi v‐SNARE GOS‐28. EMBO J 2000;19:1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Legesse‐Miller A, Sagiv Y, Glozman R, Elazar Z. Aut7p, a soluble autophagic factor, participates in multiple membrane trafficking processes. J Biol Chem 2000;275:32966–32973. [DOI] [PubMed] [Google Scholar]
- 96. Hagemeijer MC, Verheije MH, Ulasli M, Shaltiel IA, de Vries LA, Reggiori F, Rottier PJ, de Haan CA. Dynamics of coronavirus replication‐transcription complexes. J Virol 2010;84:2134–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]