

Abstract
A highly effective medicine is urgently required to cure coronavirus disease 2019 (COVID-19). For the purpose, we developed a molecular docking based webserver, namely D3Targets-2019-nCoV, with two functions, one is for predicting drug targets for drugs or active compounds observed from clinic or in vitro/in vivo studies, the other is for identifying lead compounds against potential drug targets via docking. This server has its unique features, (1) the potential target proteins and their different conformations involving in the whole process from virus infection to replication and release were included as many as possible; (2) all the potential ligand-binding sites with volume larger than 200 Å3 on a protein structure were identified for docking; (3) correlation information among some conformations or binding sites was annotated; (4) it is easy to be updated, and is accessible freely to public (https://www.d3pharma.com/D3Targets-2019-nCoV/index.php). Currently, the webserver contains 42 proteins [20 severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) encoded proteins and 22 human proteins involved in virus infection, replication and release] with 69 different conformations/structures and 557 potential ligand-binding pockets in total. With 6 examples, we demonstrated that the webserver should be useful to medicinal chemists, pharmacologists and clinicians for efficiently discovering or developing effective drugs against the SARS-CoV-2 to cure COVID-19.
KEY WORDS: COVID-19, SARS-CoV-2, Target prediction, Multi-conformation, Multi-site, Docking, D3Targets-2019-nCoV
Graphical abstract
D3Targets-2019-nCoV is a webserver built for the purpose to find effective medicines against the SARS-CoV-2 to cure COVID-19, with two functions, one is for predicting target proteins for drugs or active compounds, and the other is for identifying lead compounds against potential drug targets via docking.

1. Introduction
Severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2)1, 2, 3 has caused more than 2800 deaths as of 29 February 2020 worldwide. Drug researchers and clinicians are working intensively hard to discover and test drugs against the coronavirus disease 2019 (COVID-19). For example, intravenous treatment of remdesivir has been reported to be helpful to improve the clinical condition for the first confirmed patient of SARS-CoV-2 infection in the United States, although the safety and efficacy are still needed to test in randomized controlled trials4. Nevertheless, there are many incoming or ongoing clinical studies in China, but no drug has been approved to be effective for COVID-19 so far.
In order to discover effective drugs against the virus, virtual screening studies have been performed based on several proteins, including 3C-like proteinase (Mpro)5, 6, 7, 8, 9, 10, 11, 12, angiotensin converting enzyme 2 (ACE2)13, papain-like proteinase (PLpro)14, and furin15. However, the clinic study did not discover desirable medicine. Therefore, more potential drug targets should be explored for virtual screening and drug design. As the whole process of viral disease involves not only the proteins encoded by the SARS-CoV-2 itself, but also the proteins of human being. Thus, both the viral protein and human protein should be included for drug discovery and development, and the virtual screening against more potential target proteins could reduce false negative and improve the success rate to quickly find cures for the disease. Moreover, multi-site docking is also indispensable for deep exploration of protein functions and drug discovery. On one hand, the functional sites of some proteins are unclear or not unique, which should be comprehensively considered. On the other hand, there may be allosteric sites on the protein, which may have distinct advantages for regulating protein function. In conclusion, multi-target and multi-site based virtual screening is a more expected and comprehensive approach for finding targeted therapeutic drugs, which can improve the accuracy and robustness of prediction as much as possible.
There are at least 126 planning or ongoing clinical studies against the SARS-CoV-2 as of 29 February 2020 according to the information from Chinese Clinical Trial Registry as shown in Supporting Information Table S1. For example, chloroquine, an anti-plasmodium drug, has been found partially effective in clinic. However, its exact target protein and function mechanism against the virus and COVID-19 are unknown. In addition, many groups are working on the cytopathic effect assay for discovering drugs or active compounds against the SARS-CoV-2. This kind of in vitro study may result in compounds with good activity yet unknown targets. Therefore, identifying potential drug targets will be of great importance to understand the underlying mechanism of how the drug works, and to provide information for further drug development. Thus, a platform is expected to provide reverse docking function for predicting target protein for mechanism unknown but active compounds and drugs.
To the best of our knowledge, there is no webserver available to perform virtual screening for discovering drugs based on the multi-target and multi-site strategy and to run reverse docking for predicting potential drug target. Here, we report a platform, namely D3Targets-2019-nCoV webserver, with two functions, one is for predicting drug targets for active compounds or drugs observed from clinic test or in vitro/in vivo study, and the other is for identifying lead compounds against a specific or multiple drug targets via protein structure based virtual screening. We hope this platform could help the medicinal chemists, pharmacologists and clinicians to efficiently discover and develop drugs against the SARS-CoV-2 to cure COVID-19.
2. Materials and methods
2.1. Potential target proteins included in the D3Targets-2019-nCoV database
In order to find potential target proteins as many as possible for discovering drugs to cure COVID-19, it is necessary to consider both the SARS-CoV-2 proteins and human proteins involved in the whole process of virus infection, replication and release. In total, there are 42 potential proteins included in the database as of 21 March 2020, among which 20 proteins are encoded by the virus itself, and 22 proteins are encoded by human genome (Table 1).
Table 1.
Information of the D3Targets-2019-nCoV database.
| No. | Target full name | Target abbreviation | Protein ID/UniProtKB | State/conformation | Marka | Number of pocketsb | Orthosteric pocketsc | PDB ID | Sequence similarity (%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Host translation inhibitor nsp 1 | Nsp 1 | QHD43415.1 | /d | V1 | 3 | / | 2HSX | 86.09 |
| 2 | Non-structural protein 2 | Nsp 2 | QHD43415.1 | / | V2 | 13 | / | / | / |
| 3 | Papain-like proteinase | PLP/PLpro | QHD43415.1 | Monomer | V3-1 | 6 | 1 | 3E9S | 82.86 |
| Dimer | V3-2 | 9 | 1 | 5Y3E | 82.80 | ||||
| 4 | ADP ribose phosphatase | ADRP | QHD43415.1 | Monomer | V4-1* | 1 | 1 | 6W02 | / |
| Dimer | V4-2* | 5 | 2, 3 | 6W02 | / | ||||
| 5 | Non-structural protein 4 | Nsp 4 | QHD43415.1 | / | V5 | 7 | / | / | / |
| 6 | 3C-like proteinase | 3CLpro/Mpro | QHD43415.1 | Monomer | V6-1 | 4 | 2 | 1Z1I | 96.01 |
| Monomer | V6-2* | 7 | 1 | 5R82 | / | ||||
| Dimer | V6-3 | 6 | 3, 4 | 2Z9J | 96.08 | ||||
| Dimer | V6-4* | 7 | 3, 4 | 6Y2G | / | ||||
| MD1# | V6-5 | 6 | 1 | 1Z1I | 96.01 | ||||
| MD2# | V6-6 | 6 | 2 | 1Z1I | 96.01 | ||||
| MD3# | V6-7 | 6 | 3 | 1Z1I | 96.01 | ||||
| MD4# | V6-8 | 7 | 3 | 1Z1I | 96.01 | ||||
| 7 | Non-structural protein 6 | Nsp 6 | QHD43415.1 | / | V7 | 7 | / | / | / |
| 8 | Non-structural protein 7 | Nsp 7 | QHD43415.1 | / | V8 | 1 | / | 1YSY | 98.80 |
| 9 | Non-structural protein 8 | Nsp 8 | QHD43415.1 | / | V9 | 1 | / | 2AHM | 97.42 |
| 10 | Non-structural protein 9 | Nsp 9 | QHD43415.1 | Monomer | V10-1 | 3 | / | 1UW7 | 97.35 |
| Monomer | V10-2* | 3 | / | 6W4B | / | ||||
| Dimer | V10-3 | 7 | / | 1QZ8 | 97.35 | ||||
| Dimer | V10-4* | 6 | / | 6W4B | / | ||||
| 11 | Non-structural protein 10 | Nsp 10 | QHD43415.1 | / | V11 | 3 | / | 5NFY | 98.47 |
| 12 | RNA-dependent RNA polymerase | RdRp | QHD43415.1 | / | V12-1 | 10 | / | 6NUR | 96.35 |
| +Mg2+ | V12-2 | 10 | / | 6NUR | 96.35 | ||||
| 13 | Helicase | / | QHD43415.1 | Monomer | V13-1 | 11 | / | 6JYT | 99.83 |
| Dimer | V13-2 | 21 | / | 6JYT | 99.83 | ||||
| 14 | Guanine-N7 methyltransferase | N7 Mtase | QHD43415.1 | / | V14 | 12 | 1 | 5NFY | 94.88 |
| 15 | Uridylate-specific endoribonuclease | NendoU | QHD43415.1 | Monomer | V15-1 | 7 | / | 2H85 | 88.12 |
| Monomer | V15-2* | 8 | / | 6W01 | / | ||||
| Dimer | V15-3* | 15 | / | 6W01 | / | ||||
| 16 | 2ʹ-O-Methyltransferase | 2ʹ-O-Mtase | QHD43415.1 | / | V16 | 5 | 1 | 2XYR | 93.49 |
| 17 | ORF7a protein | / | QHD43421.1 | / | V17 | 1 | / | 1YO4 | 91.57 |
| 18 | Spike protein | S protein | QHD43416.1 | Closed | V18-1 | 25 | / | 5X58 | 76.48 |
| Closed | V18-2* | 30 | / | 6VXX | / | ||||
| Open | V18-3 | 27 | / | 5 × 5B | 76.48 | ||||
| Open | V18-4* | 42 | / | 6VYB | / | ||||
| Heptad repeat 1 | V18-5 | 12 | / | 5ZVM | 87.50 | ||||
| S2 subunit | V18-6* | 4 | 6LXT | / | |||||
| 19 | Envelope protein | E protein | QHD43418.1 | Monomer | V19-1 | 1 | / | 2MM4 | 91.38 |
| Pentamer | V19-2 | 2 | / | 5X29 | 91.38 | ||||
| 20 | Nucleocapsid phosphoprotein | N protein | QHD43423.2 | C terminal | V20-1 | 1 | / | 2GIB | 96.04 |
| N terminal monomer | V20-2 | 4 | / | 2OFZ | 92.06 | ||||
| N terminal monomer | V20-3* | 3 | / | 6VYO | / | ||||
| N terminal-tetramer | V20-4* | 16 | / | 6VYO | / | ||||
| 21 | Angiotensin converting enzyme 2 | ACE2 | P59594 | / | H1* | 6 | / | 1R42 | / |
| 22 | Cathepsin L | CTSL | P07711 | / | H2* | 5 | 1 | 2XU3 | / |
| 23 | Transmembrane protease serine 2 | TMPRSS2 | O15393 | / | H3 | 10 | / | / | / |
| 24 | C type lectin domain family four member M | CLEC4M | Q9H2X3 | / | H4* | 2 | / | 1XAR | / |
| 25 | AP2-associated protein kinase 1 | AAK1 | Q2M2I8 | / | H5* | 12 | 1, 6 | 5TE0 | / |
| 26 | Cyclophilin A | CypA | P62937 | / | H6* | 3 | 1 | 4N1M | / |
| 27 | Disintegrin and metalloproteinase domain-containing protein 17 | ADAM17 | P78536 | / | H7-1 | 5 | 1 | 2I47 | 99.21 |
| +Zn2+ | H7-2 | 5 | 1 | 2I47 | 99.21 | ||||
| 28 | Furin | / | P09958 | / | H8* | 8 | 1 | 5JXG | / |
| 29 | Tyrosine-protein kinase ABL2 | ABL2 | P42684 | / | H9* | 3 | 1 | 3HMI | / |
| 30 | Eukaryotic initiation factor 4A-I | eIF4A | P60842 | / | H10* | 8 | 2 | 5ZC9 | / |
| 31 | Dihydroorotate dehydrogenase | DHODH | Q02127 | / | H11* | 6 | 1 | 3U2O | / |
| 32 | Glycogen synthase kinase-3 beta | GSK3β | P49841 | / | H12* | 13 | 1 | 1J1B | / |
| 33 | Heterogeneous nuclear ribonucleoprotein A1 | HNRNPA1 | P09651 | / | H13* | 4 | / | 1U1Q | / |
| 34 | Calnexin | / | P27823 | / | H14-1 | 7 | / | 1JHN | 95.73 |
| +Ca2+ | H14-2 | 7 | / | 1JHN | 95.73 | ||||
| 35 | Mitogen-activated protein kinase 8 | JNK1 | P45983 | / | H15 | 12 | 1 | 2G01 | 99.44 |
| 36 | Mitogen-activated protein kinase 9 | JNK2 | P45984 | / | H16 | 6 | 1 | 3NPC | 98.31 |
| 37 | Mitogen-activated protein kinase 10 | JNK3 | P53779 | / | H17* | 7 | 1 | 1JNK | / |
| 38 | RAC-alpha serine/threonine-protein kinase | AKT1 | P31749 | / | H18* | 8 | 1 | 3O96 | / |
| 39 | RAC-beta serine/threonine-protein kinase | AKT2 | P31751 | / | H19* | 12 | 1 | 3D0E | / |
| 40 | RAC-gamma serine/threonine-protein kinase | AKT3 | Q9Y243 | / | H20* | 5 | / | 2X18 | / |
| 41 | Caveolin-2 | CAV2 | P51636 | / | H21 | 2 | / | / | / |
| 42 | cGMP-specific 3ʹ,5ʹ-cyclic phosphodiesterase | PDE5 | O76074 | / | H22* | 10 | 1 | 2H44 | / |
*Crystal structures of SARS-CoV-2 or human proteins downloaded from PDB.
#Conformations clustered from molecular dynamics trajectories.
V refers to the SARS-CoV-2 protein, and H refers to the human protein.
The number of predicted binding pockets by D3Pockets on the protein with the PPV greater than 200 Å3.
The orthosteric ligand binding sites assigned based on its sequence similarity (>80%) to that of its homologous proteins.
Not available.
2.2. Protein structure and 3D models
We performed literature survey and found 13 crystal structures of 6 proteins encoded by SARS-CoV-2 as well as 16 human proteins that may be the potential target proteins to cure COVID-19 with three-dimensional (3D) structures available from Protein Data Bank (PDB)16. Therefore, their 3D structures were downloaded directly from PDB (Table 1).
Due to the missing residues of the viral crystal structures in comparison with the complete amino acid sequences, the 3D structures of total 19 viral proteins except ADP ribose phosphatase were reconstructed in two different ways, viz., homology modeling and de novo prediction. Among them, 16 proteins were modelled with SWISS-MODEL server (https://swissmodel.expasy.org/; the Center for Molecular Life Sciences, Switzerland)17 based on 22 homology structures in PDB, which are 2HSX, 3E9S, 5Y3E, 1Z1I, 2Z9J, 1YSY, 2AHM, 1UW7, 1QZ8, 5NFY, 6NUR, 6JYT, 2H85, 2XYR, 1YO4, 5X58, 5X5B, 5ZVM, 2MM4, 5X29, 2GIB, and 2OFZ (Table 1). Other three SARS-CoV-2 proteins have sequence similarity lower than 30% with the structures in PDB, therefore, we modelled their structures by Robetta (http://robetta.bakerlab.org/; Baker lab, USA)18 for de novo prediction, which are non-structural protein 2, non-structural protein 4, and non-structural protein 6 (Table 1). In addition, magnesium ions (Mg2+) are essential for the activity of the RNA-dependent RNA polymerase (RdRp). Therefore, we modelled two Mg2+ from the superimposition of the HCV RdRp (PDB ID: 1NB6)19 palm domain to SARS-CoV-2 RdRp palm domain.
As for the rest 6 human proteins without available crystal structures, 4 of them were modelled by SWISS-MODEL, and 2 were modelled by Robetta because of the poor sequence similarity (<30%), which are transmembrane protease serine 2 (TMPRSS2) and caveolin-2. The biological functions of each protein are summarized in Supporting Information Table S2.
In future we will keep our database updating regularly when new experimental and simulated structures of the potential target proteins related to COVID-19 are available. The update information could be found in the “updated” section in the D3Targets-2019-nCoV webserver.
2.3. Detection of potential binding pockets
Since most of the drug binding pockets of SARS-CoV-2 proteins are unknown and very difficult to be obtained from experiments in a short time, a new method we reported recently, namely D3Pockets (https://www.d3pharma.com/D3Pocket/index.php)20, was applied to systematically predict all the potential binding pockets for each protein. Due to the irregular shape of the predicted binding pockets, we used cuboid pseudo-pocket volume (PPV) to characterize the size of protein pockets by Eq. (1). Once submitting a hydrogenated homology-modeled protein to the D3pockets webserver, the coordinates of predicted binding pockets in the protein could be predicted, among which pockets with the PPV greater than 200 Å3 were selected for molecular docking in this study.
| (1) |
where and are the maximum and minimum values of the X coordinate in the pocket file, which was downloaded from the D3pockets server, respectively; and are the maximum and minimum values of the Y coordinate in the pocket file; and are the maximum and minimum values of the Z coordinate in the pocket file.
It is critical to choose appropriate docking box parameters for accurate prediction in molecular docking. Based on the coordinate file of predicted binding pockets by D3pockets, the center of the docking box is obtained by Eq. (2), and the size of the docking box is obtained by Eq. (3) which extends 10 Å in each dimension of the cubic box.
| (2) |
| (3) |
where x, y and z are the 3D coordinate centers of the docking box. And a, b and c are the widths of the docking box.
2.4. Molecular dynamics simulation on SARS-CoV-2 Mpro
For exploring druggable conformations as many as possible for a potential drug target, molecular dynamics (MD) simulations were performed, with SARS-CoV-2 Mpro as an example in this study, to sample more protein conformations by using our newly developed MD method, namely velocity-scaling optimized replica exchange molecular dynamics (vsREMD)21. The details of the vsREMD method have been described in our previous study21. Briefly, in the vsREMD, a set of replicas are simulated in explicit solvent environment at different temperatures, but exchange between neighboring replicas solely utilizes the sum of intra-protein interaction (Ppp) and protein–solvent interaction as the criterion by Eq. (4):
| (4) |
where β is the inverse of temperature 1/kBT. To obtain the correct ensemble after exchange moves between replica 1 and replica 2, the vsREMD uses Eqs. (5), (6) below to rescale uniformly the velocities of all particles.
| (5) |
| (6) |
where and are the velocities and kinetic energies of replica 1 before exchange, respectively; is the intra-solvent interaction; is the velocity of replica 1 after exchange. The same meaning for replica 2.
The initial structure of the SARS-CoV-2 Mpro was obtained from homology modelling with 1Z1I as template. AMBER99SB∗-ILDNP22 force field was used to model the protein. The simulation system was solvated in a cubic box of TIP3P water molecules with a 10.0 Å buffer along each dimension. To remove bad contacts formed during the system preparation, the simulation system was minimized using steepest descent algorithm. Then the system was heated to 300 from 0 K in 2 ns with a harmonic restraint (10 kcal/mol·Å−2) for the solute. The bonds connecting hydrogen atoms were constrained by the LINCS algorithm (an algorithm reseting bond's length; 1997)23 and the time step was set to 2.0 fs. The long-range electrostatic interactions were treated by Particle mesh Ewald (PME, long-range electrostatic algorithms; 1993)24 with the non-bonded cutoff of 12 Å. The vsREMD was run at 24 different temperatures from 300 to 450 K (300.0, 305.3, 310.8, 316.3, 321.9, 327.6, 333.5, 339.4, 345.4, 351.6, 357.8, 364.2, 370.7, 377.3, 384.0, 390.8, 397.8, 404.8, 412.0, 419.4, 426.8, 434.4, 442.1 and 450.0). Exchanges were attempted every 1000 steps. For each replica, the overall simulation time lasted for 50 ns.
After the vsREMD simulation, the representative conforamtions obtained from the trajectory were clustered and the correltation among different clusters and potential binding pockets were analyzed with D3Pockets20.
2.5. Molecular docking
Hydrogens were added to the protein structures by pdb2pqr25. The format of protein structures was converted to pdbqt by using the script prepare_receptor4.py in MGLTools (version 1.5.6)26. The pockets generated by D3Pockets are used to generate box and grid for docking. For the small molecule submitted for docking, the script prepare_ligand4.py in MGLTools (version 1.5.6) is used to convert its format of either mol2 or sdf to the format of pdbqt26. All the docking process was performed with smina (https://sourceforge.net/projects/smina/; open-source software, version Smina Sep 25 2019)27, which is a fork of AutoDock Vina28 with improved docking performance. The detailed formulas and parameters for the scoring function were introduced in Supporting Information Table S3. The random seed was explicitly set to 0. The exhaustiveness of the global search was set to 8 (exhaustiveness), and 1 binding mode (num_modes) was generated and reported for each small molecule against each docking pocket.
For the docking against RdRp, the interaction between Mg2+ and oxygen could not be handled by the default scoring function. Hence, a custom scoring function (0.3 atom_type_gaussian (t1 = Magnesium, t2 = OxygenXSAcceptor, o = 0, _w = 3, _c = 8)) is applied (Table S3).
2.6. Multi-protein and multi-pocket docking platform and webserver
All protein conformations and binding pockets were collated and used as the backend database of the D3Targets-2019-nCoV server. In the meantime, the molecular docking function was also embedded in the server. By submitting the compound file in format of mol2 or sdf via the website, the users can easily run a molecular docking job and download the result files including docking scores and the coordinates of the docked protein–ligand complexes from the D3Targets-2019-nCoV server. The workflow of D3Targets-2019-nCoV server is illustrated in Fig. 1.
Figure 1.

The workflow of the D3Targets-2019-nCoV server for predicting drug targets and for multi-target and multi-site based virtual screening.
3. Results and discussion
3.1. Overview of target proteins in the D3Targets-2019-nCoV database
The information of the 42 proteins and 69 confromations collected in the D3Targets-2019-nCoV database were summarized in Table 1, including the protein name, state and conformation, number of pockets, orthosteric pockets, sequence similarity, PDB IDs of the downloaded structures or of the templates used for homology modeling, etc. For those proteins without PDB ID information, their structuers were modeled with Robetta. The pocket number on each conformation was also summarized in Table 1.
3.2. D3Targets-2019-nCoV server
The D3Targets-2019-nCoV server was developed based on PHP, and hosted on a Linux server. The average running time for a docking job of a small molecule against all the 42 proteins and 557 binding sites is about an hour, but could be much longer if the molecule has large number of rotatable bonds. Upon a user uploading a molecular file, the D3Targets-2019-nCoV server will create a new job number and put it in queue immediately. The job status including “Computing”, “Waiting” and “Finished” will be shown on the result page. When the job is completed, the user can browse the result page to obtain docking scores for selected target proteins and can also download the docking results and the coordinate files related to ligand–protein interaction.
3.3. Input and output
The graphical interface of the D3Targets-2019-nCoV webserver is shown in Fig. 2. The webserver supports small molecule files in several formats, such as sdf, mol2, mol, smiles, and pdb formats, which will be converted to mol2 format ultimately by Open Babel (version 2.4.0)29 during the processing. Although 3D structures can be generated from the 2D coordinates and optimized under MMFF94 force field by RDKit (open-source cheminformatics software, version 2019.09.3, GitHub, Inc.), the chiral properties might be wrong in the conversion process. Therefore, we strongly recommend the users submitting 3D chemical structure in the format of mol2 or sdf, especially for the compounds with chiral centers.
Figure 2.

Graphical interface for input (A) and output (B) of D3Targets-2019-nCoV.
Registration is encouraged to make the result only visible to the user, which is free of charge. After registration, the user can login to upload a small molecule. If a user is interested in one or some specific proteins, the target list is customized to be selected manually by the user, while the default is to run docking against all the proteins and sites. The output is presented in ascending order of ligand–protein docking score. The docking results could be downloaded from the webserver as an archive file.
3.4. Potential allosteric binding pockets predicted with SARS-CoV-2 Mpro as an example
D3Targets-2019-nCoV server has collected some and will continue to collect potential allosteric binding pockets to further improve its performance. As SARS-CoV-2 Mpro is a promising target6, we selected it as an example to demonstrate how we predict potential allosteric binding pockets. Based on the vsREMD simulation trajectories at 300 K, we applied D3Pockets to explore the dynamic properties of potential pockets in SARS-CoV-2 Mpro. As shown in Fig. 3, there are grid points colored from blue to red that compose a pocket. The redder the grid points are, the more stable the sub-pocket regions throughout the MD trajectory. Therefore, for SARS-CoV-2 Mpro, four relatively stable pockets are observed (Fig. 3). Pocket 1, where the intrinsic ligand binds to, has more stable points than Pockets 2, 3, and 4. Pocket 2, which is far from Pocket 1, has a positive volume correlation (0.60) with Pocket 1 (substrate binding site), suggesting that when Pocket 1 gets bigger, Pocket 2 gets bigger as well (Fig. 4A). Similarly, Pocket 3 also has a positive volume correlation (0.69) with Pocket 1 (Fig. 4B). There is no correlation between Pockets 1 and 4. It is well known that a pocket with strong correlation with the substrate binding pocket could be used as an allosteric site for drug discovery and development30, 31, 32. Therefore, Pockets 2 and 3 are two potential allosteric binding pockets, which were included in the D3Targets-2019-nCoV webserver. The cutoff of correlation coefficient to identify the allosteric binding pockets was set to 0.5 by default with the program D3Pockets20.
Figure 3.

Pocket stability of SARS-CoV-2 Mpro. The protein and the binding pockets were prepared with PyMOL (The PyMOL molecular graphics system, version 2.4.2019, Schrodinger LLC.).
Figure 4.

Pocket correlation of SARS-CoV-2 Mpro.
3.5. Case study
RdRp was recognized as a challenging protein for molecular docking, thus, we selected remdesivir as an example to test the reliability of D3Targets-2019-nCoV server. Remdesivir is a RdRp inhibitor and has been reported to be effective in inhibiting severe acute respiratory syndrome-related coronavirus (SARS-CoV) and SARS-CoV-2 in vitro33,34. It should be noted that remdesivir is a prodrug, and the active form is the transformed nucleoside triphosphate (NTP, Fig. 5)35. Therefore, NTP was submitted to D3Targets-2019-nCoV for the target prediction.
Figure 5.
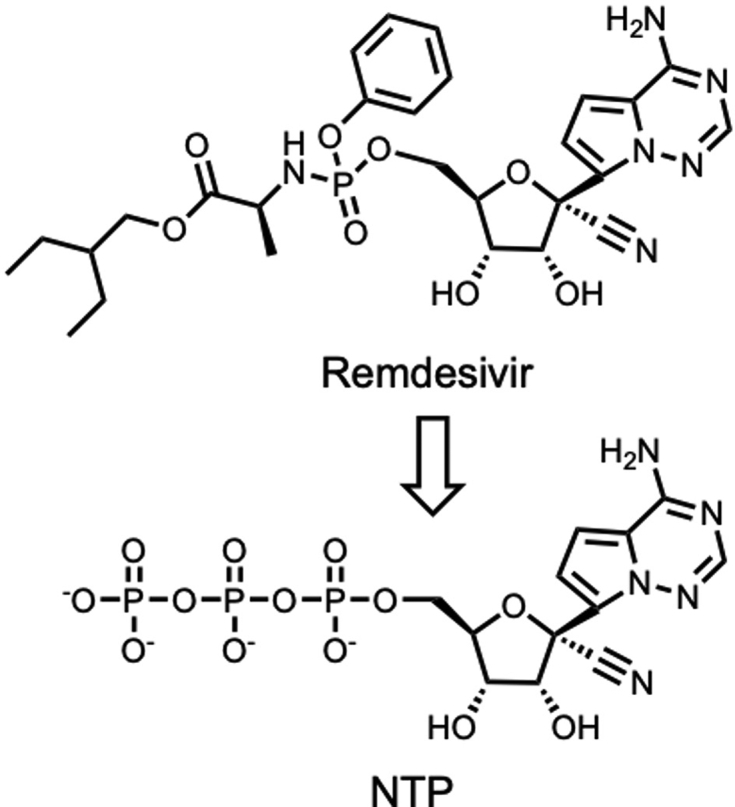
Remdesivir is converted to its pharmacologically active NTP in human cells.
Table 2 presented the top 10 target proteins with high docking scores predicted by the server for NTP. The target protein of remdesivir, RdRp, has a score of −11.37 kcal/mol in Pocket 1, which ranks the 3rd among all protein pockets (42 proteins, 69 conformations and 557 binding pockets). However, it should be noted that docking score alone does not perform well in many cases according to our experience. We recommend the users to check the top 10 docking results carefully to identify potential target protein.
Table 2.
Top 10 proteins and their docking scores for NTP.
| No. | Protein | State/Conformation | Pocket code | Docking score |
|---|---|---|---|---|
| 1 | Eukaryotic initiation factor 4A-I | / | 2 | −11.88 |
| 2 | Dihydroorotate dehydrogenase | / | 1 | −11.54 |
| 3 | RNA-dependent RNA polymerase | +Mg2+ | 1 | −11.37 |
| 4 | cGMP-specific 3ʹ,5ʹ-cyclic phosphodiesterase | / | 1 | −10.35 |
| 5 | Spike protein | Open | 8 | −10.28 |
| 6 | ADP ribose phosphatase | Dimer | 2 | −10.15 |
| 7 | Mitogen-activated protein kinase 10 | / | 1 | −9.98 |
| 8 | Uridylate-specific endoribonuclease | Monomer | 1 | −9.84 |
| 9 | Guanine-N7 methyltransferase | / | 1 | −9.67 |
| 10 | RAC-alpha serine/threonine-protein kinase | / | 1 | −9.40 |
One superiority to use the docking method to predict the target is that the docked binding mode of the compound is useful for the mutagenesis validation and is also useful to ligand optimization. According to the binding mode, the residues, e.g., TRP-800, ASP-761, and CYS-622 of NTP were found to form hydrogen bonds with the RdRp (Fig. 6), and there is a strong electrostatic attraction between the phosphate group of NTP and two Mg2+. Thus, the residues and the cations are essential for the strong binding of NTP to RdRp. Comparing with the newly reported RdRp–remdesivir complex structure36, 4 of the 6 key interaction residues, viz., D618, D623, D760 and D761, are included in our predicted key residues (Fig. 6). Therefore, the docking results by D3Targets-2019-nCoV webserver should be acceptable.
Figure 6.

Binding mode of NTP to the RdRp from the docking simulation.
To further test the reliability of D3Targets-2019-nCoV server, we performed literature survery and found 5 more active compounds with experimental informaiton of target protein and bioactivity. Therefore, we performed target predition for the 5 active compounds to test whether the predicted targets are ranked in top 10 among the 42 potential target proteins. The predicted results were summarized in Table 3, in which all the experimentally reported targets were predicted to be among top 5, demonstrating that the results from D3Targets-2019-nCoV should be acceptable at least to the systems we tested.
Table 3.
Case studies of potential antiviral agents against the SARS-CoV-2.
|
Antiviral agent |
Target | Molecular structure | Antiviral activity (μmol/L) | Score | Rank | Ref. |
|---|---|---|---|---|---|---|
| Remdesivir-active form | RdRp | 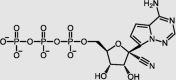 |
EC50 = 0.77 | −11.37 | 3/42 | 34 |
| Favipiravir-active form | RdRp | 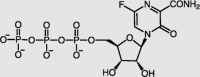 |
EC50 = 61.88 | −10.74 | 2/42 | 34,37,38 |
| Ribavirin-active form | RdRp | 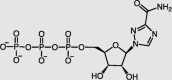 |
EC50 = 109.50 | −10.39 | 2/42 | 34,38,39 |
| Penciclovir-active form | RdRp | 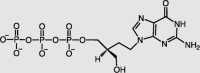 |
EC50 = 95.96 | −8.82 | 5/42 | 34,40 |
| N3 compound | 3CLpro | 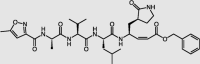 |
EC50 = 16.77 | −9.45 | 4/42 | 41 |
| Teriflunomide | DHODH | 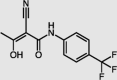 |
IC50 = 0.31 | −9.26 | 2/42 | 42 |
3.6. Discussion
We have presented the user-friendly D3Targets-2019-nCoV webserver for predicting drug targets and for multi-target and multi-site based virtual screening against COVID-19. Compared with existing reverse docking platforms43,44, the D3Targets-2019-nCoV webserver is specifically focused on COVID-19, including not only the experimental structures and homology models, but also the conformations with potential allosteric binding sites predicted by enhanced sampling method (vsREMD)21 and D3Pockets20, respectively. The abundant conformations and binding pockets make the webserver much possible to successfully predict target proteins and to discover hit compounds. It is also important to notice that other reverse docking platforms should be tried as the true targets of an active compound may not be included in D3Targets-2019-nCoV due to the unknown pathogenic mechanisms of COVID-19. Because the protein conformation obtained by homology modelling may not be the most stable conformation, and some docking models still need further optimization, for example, the positions of two Mg2+ on the RdRp, the workflow of D3Targets-2019-nCoV needs to be further improved.
In order to keep the D3Targets-2019-nCoV server fresh and active, we will continue to update the webserver in the future. To make better use of the docking platform, some applications are listed as follows: (1) target prediction of multi-target drugs; (2) target prediction of old drugs and novel active compounds; (3) virtual screening of drugs for specified targets; (4) prediction of binding modes of drugs to known targets, and (5) users can download protein models for free from the webserver for molecular dynamics simulation, molecular docking or structure–activity relationship studies, etc. In particular, predicting target based only on the highest docking score may not be reliable. Therefore, we strongly recommend that the users take into account the docked binding mode and docking scores together with protein function. The D3Targets-2019-nCoV webserver will keep regularly update when the new structures of potential target proteins related to COVID-19 are available and meet one of the 4 criteria, viz., natural mutant, RMSDs of the overall structure or ligand binding pockets or any key residue in the pockets ≥2 Å to any structures in the webserver.
4. Conclusions
SARS-CoV-2 has caused more than 2,800 deaths as of 29 February 2020 worldwide. Although there is no effective drug approved, many clinical trials are incoming or ongoing in China. Moreover, many groups are working on the cytopathic effect assay for discovering active compound. In many cases, the target protein of the discovered active compounds might be unknown. Therefore, identifying potential drug targets will be of great importance. Here, we developed a webserver for predicting potential target protein for active compounds and for virtual screening against multi-target and multi-pockets. The webserver database currently has 42 proteins (20 viral proteins and 22 human proteins related to virus infection, replication and release) with 69 conformations collected by means of downloading directly from PDB, homology and de novo modeling, and the MD simulation. With the program D3Pockets, 557 potential ligand binding pockets were successfully predicted and used for molecular docking by the server. Each submitted compound will be docked to all the binding pockets by smina, and the docking results will be presented in ascending order of docking score. Tests with 6 active compounds/drugs with experimental reported target proteins and bioactivity data demonstrated the potential usefulness of the D3Targets-2019-nCoV server. The webser is accessible via internet free of charge at https://www.d3pharma.com/D3Targets-2019-nCoV/index.php.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (2017YFB0202601 and 2016YFA0502301).
Author contributions
Weiliang Zhu and Zhijian Xu conceived and designed the study. Yulong Shi and Xinben Zhang wrote the script code for the webserver. Yulong Shi, Kaijie Mu, Cheng Peng, Zhengdan Zhu, Xiaoyu Wang and Yanqing Yang collected the data, performed homology modelling and molecular dynamics simulation, and developed docking models. Yulong Shi and Cheng Peng performed the data analysis. Weiliang Zhu, Zhijian Xu, Yulong Shi and Cheng Peng wrote the paper.
Conflicts of interest
The authors have no conflicts of interest to declare.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2020.04.006.
Contributor Information
Zhijian Xu, Email: zjxu@simm.ac.cn.
Weiliang Zhu, Email: wlzhu@simm.ac.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holshue M.L., DeBolt C., Lindquist S., Lofy K.H., Wiesman J., Bruce H. First case of 2019 novel coronavirus in the United States. N Engl J Med. 2020;382:929–936. doi: 10.1056/NEJMoa2001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu X., Wang X.J. Potential inhibitors for 2019-nCoV coronavirus M protease from clinically approved medicines. J Genet Genomics. 2020;47:119–121. doi: 10.1016/j.jgg.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu Z., Peng C., Shi Y., Zhu Z., Mu K., Wang X. Nelfinavir was predicted to be a potential inhibitor of 2019-nCoV main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. bioRxiv. 2020 doi: 10.1101/2020.01.27.921627. Available from: [DOI] [Google Scholar]
- 7.Beck B.R., Shin B., Choi Y., Park S., Kang K. Predicting commercially available antiviral drugs that may act on the novel coronavirus (2019-nCoV), Wuhan, China through a drug–target interaction deep learning model. bioRxiv. 2020 doi: 10.1101/2020.01.31.929547. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y., Zhang J., Wang N., Li H., Shi Y., Guo G. Therapeutic drugs targeting 2019-nCoV main protease by high-throughput screening. bioRxiv. 2020 doi: 10.1101/2020.01.28.922922. Available from: [DOI] [Google Scholar]
- 9.Anh-Tien T., Francesco G., Michael H., Fuqiang B., Artem C. Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 billion compounds. Mol Inf. 2020;39:2000028. doi: 10.1002/minf.202000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alessandro C. Virtual screening of an FDA approved drugs database on two COVID-19 coronavirus proteins. ChemRxiv. 2020 doi: 10.26434/chemrxiv.11847381.v1. Available from: [DOI] [Google Scholar]
- 11.Yu Wai C., Chin-Pang Y., Kwok-Yin W. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CLpro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. ChemRxiv. 2020 doi: 10.26434/chemrxiv.11831103.v2. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen D.D., Gao K., Chen J., Wang R., Wei G.W. Potentially highly potent drugs for 2019-nCoV. bioRxiv. 2020 doi: 10.1101/2020.02.05.936013. Available from: [DOI] [Google Scholar]
- 13.Cui Q., Huang C., Ji X., Zhang W., Zhang F., Wang L. Possible inhibitors of ACE2, the receptor of 2019-nCoV. Preprints. 2020 doi: 10.20944/preprints202002.0047.v1. Available from: [DOI] [Google Scholar]
- 14.Rimanshee A., Amit D., Vishal P., Mukesh K. Potential inhibitors against papain-like protease of novel coronavirus (SARS-CoV-2) from FDA approved drugs. ChemRxiv. 2020 doi: 10.26434/chemrxiv.11860011.v2. Available from: [DOI] [Google Scholar]
- 15.Li H., Wu C., Yang Y., Liu Y., Zhang P., Wang Y. Furin, a potential therapeutic target for COVID-19. ChinaXiv. 2020 doi: 10.12074/202002.00062. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burley S.K., Berman H.M., Bhikadiya C., Bi C., Chen L., Di Costanzo L. RCSB Protein Data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2018;47:D464–D474. doi: 10.1093/nar/gky1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim D.E., Chivian D., Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004;32:W526–W531. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Farrell D., Trowbridge R., Rowlands D., Jager J. Substrate complexes of hepatitis C virus RNA polymerase (HC-J4): structural evidence for nucleotide import and de-novo initiation. J Mol Biol. 2003;326:1025–1035. doi: 10.1016/s0022-2836(02)01439-0. [DOI] [PubMed] [Google Scholar]
- 20.Chen Z., Zhang X., Peng C., Wang J., Xu Z., Chen K. D3Pockets: a method and web server for systematic analysis of protein pocket dynamics. J Chem Inf Model. 2019;59:3353–3358. doi: 10.1021/acs.jcim.9b00332. [DOI] [PubMed] [Google Scholar]
- 21.Wang J., Peng C., Yu Y., Chen Z., Xu Z., Cai T. Exploring conformational change of adenylate kinase by replica exchange molecular dynamic simulation. Biophys J. 2020;118:1009–1018. doi: 10.1016/j.bpj.2020.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aliev A.E., Kulke M., Khaneja H.S., Chudasama V., Sheppard T.D., Lanigan R.M. Motional timescale predictions by molecular dynamics simulations: case study using proline and hydroxyproline sidechain dynamics. Proteins. 2014;82:195–215. doi: 10.1002/prot.24350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J Comput Chem. 1997;18:1463–1472. [Google Scholar]
- 24.Darden T., York D., Pedersen L. Particle mesh Ewald: an N⋅log (N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089. [Google Scholar]
- 25.Dolinsky T.J., Nielsen J.E., McCammon J.A., Baker N.A. PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koes D.R., Baumgartner M.P., Camacho C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J Chem Inf Model. 2013;53:1893–1904. doi: 10.1021/ci300604z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open Babel: an open chemical toolbox. J Cheminf. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suel G.M., Lockless S.W., Wall M.A., Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat Struct Biol. 2003;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 31.Gerek Z.N., Ozkan S.B. Change in allosteric network affects binding affinities of PDZ domains: analysis through perturbation response scanning. PLoS Comput Biol. 2011;7 doi: 10.1371/journal.pcbi.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma X., Meng H., Lai L. Motions of allosteric and orthosteric ligand-binding sites in proteins are highly correlated. J Chem Inf Model. 2016;56:1725–1733. doi: 10.1021/acs.jcim.6b00039. [DOI] [PubMed] [Google Scholar]
- 33.Agostini M.L., Andres E.L., Sims A.C., Graham R.L., Sheahan T.P., Lu X. Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio. 2018;9 doi: 10.1128/mBio.00221-18. e00221-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Warren T.K., Jordan R., Lo M.K., Ray A.S., Mackman R.L., Soloveva V. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature. 2016;531:381–385. doi: 10.1038/nature17180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao Y., Yan L., Huang Y., Liu F., Zhao Y., Cao L. Structure of RNA-dependent RNA polymerase from 2019-nCoV, a major antiviral drug target. bioRxiv. 2020 doi: 10.1101/2020.03.16.993386. Available from: [DOI] [Google Scholar]
- 37.Furuta Y., Komeno T., Nakamura T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93:449–463. doi: 10.2183/pjab.93.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tchesnokov E.P., Feng J.Y., Porter D.P., Gotte M. Mechanism of inhibition of Ebola virus RNA-dependent RNA polymerase by remdesivir. Viruses. 2019:11. doi: 10.3390/v11040326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo H., Sun S., Yang Z., Tang X., Wang Y. Strategies for ribavirin prodrugs and delivery systems for reducing the side-effect hemolysis and enhancing their therapeutic effect. J Contr Release. 2015;209:27–36. doi: 10.1016/j.jconrel.2015.04.016. [DOI] [PubMed] [Google Scholar]
- 40.Earnshaw D.L., Bacon T.H., Darlison S.J., Edmonds K., Perkins R.M., Vere Hodge R.A. Mode of antiviral action of penciclovir in MRC-5 cells infected with herpes simplex virus type 1 (HSV-1), HSV-2, and varicella-zoster virus. Antimicrob Agents Chemother. 1992;36:2747–2757. doi: 10.1128/aac.36.12.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. bioRxiv. 2020 doi: 10.1101/2020.02.26.964882. Available from: [DOI] [Google Scholar]
- 42.Xiong R., Zhang L., Li S., Sun Y., Ding M., Wang Y. Novel and potent inhibitors targeting DHODH, a rate-limiting enzyme in de novo pyrimidine biosynthesis, are broad-spectrum antiviral against RNA viruses including newly emerged coronavirus SARS-CoV-2. bioRxiv. 2020 doi: 10.1101/2020.03.11.983056. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H., Gao Z., Kang L., Zhang H., Yang K., Yu K. TarFisDock: a web server for identifying drug targets with docking approach. Nucleic Acids Res. 2006;34:W219–W224. doi: 10.1093/nar/gkl114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kharkar P.S., Warrier S., Gaud R.S. Reverse docking: a powerful tool for drug repositioning and drug rescue. Future Med Chem. 2014;6:333–342. doi: 10.4155/fmc.13.207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.