

Abstract
The serine/threonine kinase AKT, also known as protein kinase B (PKB), is the major substrate to phosphoinositide 3-kinase (PI3K) and consists of three paralogs: AKT1 (PKBα), AKT2 (PKBβ) and AKT3 (PKBγ). The PI3K/AKT pathway is normally activated by binding of ligands to membrane-bound receptor tyrosine kinases (RTKs) as well as downstream to G-protein coupled receptors and integrin-linked kinase. Through multiple downstream substrates, activated AKT controls a wide variety of cellular functions including cell proliferation, survival, metabolism, and angiogenesis in both normal and malignant cells. In human cancers, the PI3K/AKT pathway is most frequently hyperactivated due to mutations and/or overexpression of upstream components. Aberrant expression of RTKs, gain of function mutations in PIK3CA, RAS, PDPK1, and AKT itself, as well as loss of function mutation in AKT phosphatases are genetic lesions that confer hyperactivation of AKT. Activated AKT stimulates DNA repair, e.g. double strand break repair after radiotherapy. Likewise, AKT attenuates chemotherapy-induced apoptosis. These observations suggest that a crucial link exists between AKT and DNA damage. Thus, AKT could be a major predictive marker of conventional cancer therapy, molecularly targeted therapy, and immunotherapy for solid tumors. In this review, we summarize the current understanding by which activated AKT mediates resistance to cancer treatment modalities, i.e. radiotherapy, chemotherapy, and RTK targeted therapy. Next, the effect of AKT on response of tumor cells to RTK targeted strategies will be discussed. Finally, we will provide a brief summary on the clinical trials of AKT inhibitors in combination with radiochemotherapy, RTK targeted therapy, and immunotherapy.
Keywords: AKT/PKB, receptor tyrosine kinases, radiochemotherapy, DNA repair, Solid Tumors, molecular targeting
1. Structure, activation and function of AKT
The AKT/protein kinase B (PKB) genes encode serine/threonine kinases of 57 kilodaltons (kDa). There are three AKT paralogs, AKT1/PKBα, AKT2/PKBβ, and AKT3/PKBγ, that are the products of distinct genes localized on chromosomes 14, 19 and 1, respectively. The AKT1, AKT2, and AKT3 genes belong to a class of genes known as oncogenes that when altered via mutation, copy number, protein, or RNA, have the potential to cause normal cells to become cancerous. AKT paralogs consist of an N-terminal pleckstrin homology (PH) domain, a central kinase domain, and a C-terminal regulatory domain. The 100 amino acid PH domain of AKT interacts with membrane lipid products such as phosphatidylinositol 4,5‑ bisphosphate (PIP2) and phosphatidylinositol (3,4,5)‑ trisphosphate (PIP3) for recruitment to the cell membrane where activation occurs by phosphorylation at threonine (T) and serine (S) residues. The PH domain of AKT is separated by 39 amino acids from kinase domain [1]. The AKT kinase domain contains threonine residues for phosphorylation, e.g. T308 on AKT1, T309 on AKT2, and T305 on AKT3. The C-terminal domain contains the serine phosphorylation site of AKT, which is necessary for full AKT activity [2]. Serine phosphorylation residues on AKT1, AKT2, and AKT3 are S473, S474, and S472, respectively. As tested in non-small cell lung cancer cells (NSCLC), phosphorylation of AKT at threonine but not serine residues correlates with AKT kinase activity [3].
AKT is activated by a variety of signaling cascades, but binding of natural ligands to receptor tyrosine kinases (RTK) [4–7], G-protein coupled receptors (GPCR) [8–11], and integrin-linked kinase (ILK) [12–15] are the major mechanisms by which AKT is recruited to the cell membrane and activated by phosphorylation at serine and threonine residues. GPCRs can also indirectly activate the PI3K/AKT pathway by transactivating RTK and integrin [16]. So far, RTKs are the largest family of membrane-bound receptors that have been identified, with 20 families and 58 transmembrane receptors [4]. Ligand binding to the receptors results in activating phosphoinositide 3-kinase (PI3K), which contains regulatory subunits for binding to the receptors and catalytic subunits for the generation of PIP3 by phosphorylation of PIP2 [17]. PIP3 recruits AKT to the cell membrane followed by conformational changes in its structure and phosphorylation at T308 by the PI3K-dependent kinase-1 (PDK-1) [18]. For full activation, AKT needs to be phosphorylated at both threonine and serine residues [2, 19, 20]. In contrast to the phosphorylation of threonine residue by PDK1, so far no unique kinase has been discovered to be responsible for AKT phosphorylation at serine residues. Rather, a variety of kinases have been demonstrated to phosphorylate AKT at serine residues, downstream to PI3K. Among them are the PI3K-related kinases, DNA-PKcs [21, 22], ATM [2], ILK [23, 24], and the rapamycin insensitive companion of mammalian target of rapamycin (RICTOR) [25].
There are conflicting results for the role of DNA-PKcs on AKT phosphorylation. The initial studies for the role of DNA-PKcs were performed by employing the DNA-PKcs deficient glioblastoma cell line MO59J and DNA-PKcs proficient MO59K cells [22]. According to the results from our group and others, these cell lines have several other differences, e.g. ATM expression that impact activation of AKT. We previously showed that S473 phosphorylation of AKT induced by ionizing radiation (IR) is impaired in MO59J cells [26]. However, neither IR-induced AKT phosphorylation nor epidermal growth factor receptor (EGFR) ligand-induced phosphorylation of AKT at S473 were affected in DNA-PKcs knockout HCT116 cells [26]. Since HCT116 cells are the only available isogenic cells for DNA-PKcs, it is difficult to conclude if DNA-PKcs is a functional AKT-S473 kinase. Thus, according to the existing reports on the role of DNA-PKcs on AKT phosphorylation and conflicting reports on this issue, the best statement in this regard would be a cell line- and context-dependent function of DNA-PKcs on serine phosphorylation. To date, studies on phosphorylation of AKT have been centered on T308 and S473 on AKT1. Currently, it is unclear which pathways involved in AKT1 phosphorylation are applicable for phosphorylating AKT2 and AKT3, a question that remains to be answered. AKT also presents a PH domain-dependent, growth factor-independent activation step, which is marked by constitutive phosphorylation of T50 and potentially S124 [27]. This phosphorylation is most likely involved in stabilization of the AKT protein [28]. AKT is also known to be phosphorylated at the T474 residue resulting in approximately 50% of AKT activity in response to insulin growth factor-1 (IGF-1) [29] and epidermal growth factor (EGF) [30]. In addition to phosphorylation of AKT, other post-translational modifications such as ubiquitination, acetylation, glycosylation, oxidation, and SUMOylation might be important for additional functions of AKT paralogs. Different AKT post-translational modifications and their consequences for the AKT activity has been reviewed by Risso et al. [31].
Following activation of AKT through upstream receptors, cells use protein phosphatases as negative regulators to dephosphorylate AKT and turn off the AKT functions. Phosphatases and tensin homolog deleted on chromosome 10 (PTEN) is one of the serine-threonine phosphatases that deactivates AKT by dephosphorylating PIP3 [32]. PIP3 has also been described to be the target of Inositol polyphosphate 4-phosphatase B (INPP4B). Thus, PTEN and INPP4B, by reversing PIP3 to PIP2, interferes with accumulation of AKT and PDK1 to the cell membrane and, likewise, leads to releasing inactive AKT from the membrane to the cytoplasm. AKT signaling is also terminated after direct targeting by protein phosphatase 2A (PP2A) and PH domain leucine-rich repeat protein phosphatase (PHLPP) at T308 and S473, respectively [33]. PHLPP consists of two isoforms, PHLPP1 and PHLPP2, which selectively terminate AKT-signaling pathways through the inactivation of different AKT paralogs, i.e. modulating the phosphorylation of HDM2 and GSK-3 alpha through dephosphorylating AKT2, and modulating the phosphorylation of p27 through dephosphorylating AKT3 [34]. The specific impact of phosphatases on cell proliferation and survival has been comprehensively reviewed by Narla et al. [35]. In addition to phosphatases, phosphorylation of AKT is also regulated by AKT interacting proteins such as CTMP, Trb3 and Keratin K10, which negatively regulate AKT activation [28]. AKT paralogs regulate a wide variety of cellular responses, e.g. cell growth, proliferation, survival, metabolism, and angiogenesis. Details on the cellular functions of AKT have been reviewed previously [36, 37].
2. AKT/PKB and its role in human cancers
2.1. Activation status of AKT in tumor cells from different origins
Various studies have reported that the overexpression of phosphorylated AKT (pAKT) is a key defect in many types of solid tumors. pAKT is overexpressed due to an aberrant activation of upstream of growth factor receptors via numerous mechanisms. Furthermore, AKT is also an important signaling molecule with over 100 downstream target substrates that are involved in cell survival and proliferation. Shao et al. [38] recently showed that there was a close correlation between AKT copy number variations and its gene expression in human cancer cell lines and patient samples.
Breast cancer
It has been reported that pAKT overexpression is a negative prognostic factor for breast cancer in terms of both overall survival (OS) and disease-free survival (DFS) [39]. In 2018, Luo et al. [40] demonstrated that AKT was activated in 42.4% of breast invasive ductal carcinoma (BIDC) by immunohistochemical analysis (IHC). There was significantly higher expression of pAKT in BIDC compared with the noncancerous control breast tissues (P = 0.001). They also found that high expression of insulin receptor substrate 1 was significantly associated with positive expression of pAKT. Park et al. [41] reported that pAKT expression was confined to the invasive tumor components, with no staining in normal breast epithelial cells by IHC. In primary breast cancer tissues, 37 cases (29.1%) had a high nuclear pAKT score and 46 cases (36.2%) exhibited a high cytoplasmic pAKT score. These researchers also found that pAKT expression was significantly correlated with HER2 overexpression (p<0.001). Another study with 252 primary human breast carcinoma specimens investigated the incidence of AKT activation and the relationships between AKT activation and other tumor markers by IHC staining [42]. They observed that 33.3% of primary breast cancers were positive for pAKT expression. In addition, they demonstrated that AKT activation was significantly associated with resistance to endocrine therapy in 36 metastatic breast cancers. Collectively, these data revealed that pAKT might be a useful predictor of resistance to endocrine therapy, and inhibition of AKT may increase the efficacy of HER2 or endocrine therapy in breast cancer.
Lung cancer
The activation of AKT and its downstream effectors have been investigated in non-small cell lung cancer (NSCLC) extensively. Either AKT-S473 or -T308 were phosphorylated in most NSCLC specimens, but phosphorylation was detected rarely in surrounding normal lung tissues. For example, Tsurutani et al. [43] showed that AKT activation was specific for NSCLC tumors versus surrounding tissue (73.4% vs 0%; P<0.05). They also observed that AKT activation was more frequent in adenocarcinomas than in squamous cell carcinomas of the lung (78.1% vs 68.5%; P=0.04) as well as being associated with shorter OS for all stages of disease (log-rank P=0.04). In 2009, Yoshizawa et al. [44] reported that pAKT was overexpressed in 78% of NSCLC. Once again, pAKT was detected more frequently in adenocarcinomas (43%) than squamous cell carcinomas (36%), but this observation did not reach statistical significance. The 5-year survival rate was significantly lower in patients with pAKT positive tumors. Jin et al. [45] examined 99 NSCLC patients for the relationship between the level of pAKT expression and the risk of brain metastasis. The data showed that patients with high level expression of pAKT had higher cumulative probabilities of developing brain metastases. Investigations by Dobashi and colleagues [46] utilized 135 lung carcinomas to examine the incidence of pAKT dysregulation and its correlation with EGFR alterations by IHC. They found that 35% of cases exhibited increased copy number of the AKT gene. In addition, nuclear accumulation of pAKT was more frequent in tumors with EGFR mutations. Based on these findings, they concluded that a combination of AKT-targeted therapies with conventional treatments might be required for improved therapy of NSCLC.
Head and Neck Squamous Cell Carcinoma
In 2007, Molinolo et al. [47] established a Head and Neck Squamous Cell Carcinoma (HNSCC) tissue microarray using 1300 cases. They found that both AKT phosphorylated forms, pAKT-S473 and pAKT-T308, were present in the cytoplasm. In addition, some nuclear pAKT-S473 expression was detected. Cluster analysis using 327 HNSCC samples and 9 normal oral tissues showed that EGFR activation was not closely related with the activation of AKT and its downstream target mTOR. They also demonstrated that there was a significant correlation between pAKT-T308, pAKT-S473, and pS6. The PI3K/AKT signaling pathway was also aberrantly activated in HNSCC. Mutations of PIK3CA was one of the most common alterations in HNSCC tumors. Mundi et al. [48] recently summarized the frequency of mutation in the PI3K/AKT pathway genes such as PIK3CA, PTEN, PIK3R1, PIK3CG, and AKT1 in different type of cancers. Another study utilized 116 patients who were diagnosed with advanced oropharyngeal SCC [49]. Molecular analysis showed 25% of patients had HPV16-related tumors whereas 75% patients had non-HPV16-related tumors. Further analysis revealed that pAKT-S473 was highly expressed in non-HPV16-related tumors (53%) compared to HPV-related tumors (18%). This result showed a significant correlation (p<0.02). Activation of AKT has also been correlated with poor prognosis in HNSCC. Islam et al. [50] performed IHC and selected HNSCC biopsies that were positive for vascular endothelial growth factor A (VEGFA). IHC analysis revealed that patients whose tumors expressed both VEGFA and pAKT-S473 had a poor prognosis. These results suggested further research is necessary to determine the most effective approaches for suppression of PI3K/AKT signaling in HNSCCs.
Pancreatic cancer
KRAS is one of the major stimulators of PI3K/AKT pathways and is mutated in about 90% of pancreatic cancers. Consequently, the PI3K/AKT signaling is one of the most commonly deregulated signaling pathways in pancreatic cancer [51]. Mao et al. [52] utilized a pancreatic tissue microarray with 91 pancreatic cancer cases and 51 normal pancreatic tissues to determine the expression of AKT by IHC. The results showed 59% of tumor tissues exhibited AKT positive staining, while only 27% of normal pancreatic tissues had AKT positive staining. They also evaluated inhibition of the PI3K/AKT pathway with LY294002 using the BxPC-3 xenograft model. Both the tumor volume and the weight were inhibited by this PI3K inhibitor. In 2017, Massihnia et al. [53] investigated pAKT expression levels in a pancreatic ductal adenocarcinoma (PDAC) tissue microarray with a cohort of radically resected tumors (n=100). They found a significant correlation between high pAKT protein expression and both shorter OS and progression-free survival (PFS). In 2018, Sinkala et al. [54] performed survival, clustering, and integrative pathway and network analysis with 185 pancreatic ductal adenocarcinoma (PDAC) patients’ datasets from TCGA. These datasets comprised clinical information of cellular transcription data, protein expression data, genomic mutations, and copy number alterations. They found that alterations in specific PI3K-AKT pathway genes were more apparent in one of the subtypes called quasi-mesenchymal PDAC. They also demonstrated that AKT was altered in 27% of all PDAC tumors.
Glioblastoma multiform
Glioblastoma multiform (GBM) exhibits very complex pathogenesis that involves mutations as well as alterations of various cell signaling pathways including PI3K/AKT. The occurrence of GBM is frequently associated with molecular changes in EGFR and the PI3K/AKT signaling pathway. Mizoguchi et al. [55] performed IHC to explore the status of EGFR, AKT and other markers in a series of 55 GBMs and 27 anaplastic astrocytomas. AKT activation correlated significantly with positive EGFR IHC staining (p=0.001). In 2013, Wuchty et al. [56] combined matched genomic alteration and gene expression data from GBM patients. They used a nonlinear machine learning approach to examine associations between genomic copy number alterations and gene expression of signaling pathways. These results suggested that combination therapy with EGFR inhibitors and drugs targeting the PI3K/AKT/PTEN pathway would be a potentially useful approach for GBM treatment. To determine if AKT signaling was altered in GBM cell lines, Gallia et al. [57] used 16 established GBM cell lines to examine phoisphorylation levels of AKT. They found moderate to very strong phosphorylation levels of AKT in most of GBM cell lines. One of the PI3K/AKT inhibitors, A-443654, resulted in reduced cell proliferation of GBM cells. These results suggested that PI3K/AKT pathway is important in GBM pathogenesis.
2.2. Radiochemotherapy-induced AKT activation and its impact on treatment outcome
RTKs are the major membrane bound receptors that stimulate AKT in cells treated with the appropriate ligands. About 65% of these receptors are known to be dysregulated in different human cancers [4], which leads to stimulating AKT activation. Among these receptors, erbB/EGFR, platelet-derived growth factor receptor (PDFGR), vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR) are those families with the most frequent mutations and dysregulations. The other components of the PI3K/AKT pathway are also frequently mutated in human cancers and lead to enhanced phosphorylation of AKT. These include gain of function mutations in PIK3CA encoding the p110α protein in the catalytic subunit of PI3K, RAS isoforms (KRAS, HRAS and NRAS), as well as loss of function mutations in AKT phosphatases PTEN and PHLPP. Constitutive activation of AKT is achieved by the mutation in AKT paralogs as well. For example, the E17K mutation in AKT1 gene causes permanent AKT1 kinase activity and phosphorylation at T308 and S473, in association with its membrane localization. This results in constitutive activation of AKT substrates independent of upstream membrane-bound receptors, as reported for tumors from different origin, e.g. lung, breast, bladder, endometrial and colorectal [58–61]. The E17K mutation has also been reported in AKT3 gene that results in full AKT3 activity by phosphorylation at S472 and T305 [62].
Besides upregulation and hyperactivation of AKT in tumor cells as well as activation of AKT induced by ligands, conventional cancer treatment modalities, i.e. radiotherapy and chemotherapy, induce activation of AKT. Exposure to IR in the range of clinically relevant doses induces a ligand independent immediate phosphorylation of AKT that occurs within 0 to 60 minutes after irradiation [63–72]. It is also known that IR stimulates autocrine release of EGFR ligand, i.e. transforming growth factor α (TGFα) that stimulates EGFR and, consequently, EGFR-dependent activation of mitogen-activated protein kinase (MAPK)/extracellular regulated kinase (ERK1/2) within 2 to 24 hours [73, 74]. According to this report, the delayed activation of EGFR can result in a second phase of activation of AKT, in a ligand-dependent manner, as shown for example at 6 hours after irradiation in NSCLC cell line A549 [75]. IR induces phosphorylation of AKT through PI3K [64, 69, 76], and the level of phosphorylation is radiation-dose independent [75]. Almost all of the investigation related to IR and phosphorylation of AKT have been performed using antibodies against phospho-AKT1 S473 and T308. Thus, further investigation will be necessary to study phosphorylation status of AKT2 and AKT3 after irradiation.
Chemotherapy agents also induce phosphorylation of AKT. Cisplatin is an effective DNA-damaging antitumor agent for treating variety of human cancers that is known to induce AKT phosphorylation in tumor cells from different entities [72, 77, 78]. Activation of AKT by cisplatin leads to cisplatin-resistance, as downregulation of AKT and targeting PI3K reverses platinum resistance of human ovarian cancer cells [79] and triple negative breast cancer (TNBC) cells [80], respectively. Likewise, it was shown that the specific AKT paralogs AKT2 and AKT3 might be involved in chemoresistance to cisplatin in uterine cancers [81]. In a different study, Girouard et al. demonstrated that AKT1 and AKT2 but not AKT3 are the molecular mechanisms that govern chemoresistance of endometrial carcinomas [82]. AKT inhibitor MK2206 that is in clinical trials was shown to improve cisplatin’s effect in gastric cancer cells [83]. Together, it seems that the activation of the AKT pathway is one of the major mechanisms involved in intrinsic and acquired resistance to chemotherapy agents. Due to the well-described function of AKT in cell survival, protein synthesis, and proliferation, AKT activity can evade the cytotoxic effect of chemotherapeutic agents, leading to chemoresistance. This is mainly due to protecting cells from drug-induced apoptosis [77] through the apoptosis intrinsic pathway, e.g. by inactivating proapoptotic proteins BAD and caspase 9 and stimulating anti-apoptotic proteins MCl-1, or through upregulation of survivin as an inhibitor of apoptosis. Details on inhibition of apoptosis by AKT activity after chemotherapy have been reviewed by Huang and Hung [84].
Following radiotherapy, cells use a complex network of signal transduction known as DNA damage response (DDR) to protect themselves against the effects of IR. As an initial step in DDR, cells are transiently arrested in different cell cycles, i.e. G1/S and G2/M checkpoints. Following cell-cycle arrest, cells employ different repair mechanisms such as nucleotide excision repair, base excision repair, mismatch repair, and double strand breaks (DSBs) repair pathways to fix DNA damage. DSBs are the major cause of IR-induced cell death in radiotherapy, and clinical data exists indicating prognostic value of pAKT1-S473 for radiotherapy response such as in HNSCC and cervical cancer [85, 86]. IR-induced DSBs are repaired by either homologous recombination (HR) during G2/M phase or non-homologous end-joining (NHEJ) through the entire cell cycle [87]. Details of DSB repair pathways have been comprehensively discussed and reviewed previously by other investigators [88–90]. Activated AKT stimulates repair of radiation-induced DSBs [6, 7, 91, 92]. In this context, DNA-PKcs is the major component in NHEJ repair of DSB that directly interacts with the C-terminal domain of AKT1 [93, 94]. In this complex, activation of AKT1 phosphorylates DNA-PKcs and stimulates function of DNA-PKcs in DSB repair. As a consequence to the complex formation of AKT with DNA-PKcs, it is expected that hyperactivation of AKT, e.g. due to deregulation of RTKs, PI3K, AKT, RAS, and PTEN, leads to efficient DSB repair and radiotherapy resistance as reported before from different laboratories [68, 94–97]. Among different RTK members, the role of EGFR in DSB repair has been well investigated [98]. EGFR, through phosphorylating ATM at tyrosine (Y) site Y370, induces Chk2 activity [99], which leads to G1 cell cycle arrest, a prerequisite for NHEJ and HR, which are also stimulated by EGFR. EGFR activates DNA-PKcs [98, 100], which is involved in NHEJ and forms a complex with BRCA1 to facilitate HR [101]. Additionally, EGFR tyrosine kinase activity stimulates PCNA phosphorylation at Y211, which is necessary for the stabilization of the chromatin-bound PCNA protein and its function [102]. Y72 phosphorylation of histone 4 (H4), which accelerates DNA synthesis and repair, is also partially dependent on EGFR [103]. IGF1R is the second well-studied RTK that was shown to be expressed in the nucleus and mediates radioresistance through stimulating DSB repair [104, 105]. A very recent publication by Chalmers’ group showed that VEGFR also activates DNA repair via AKT and DNA-PKcs in 3D conditions [106]. Further studies support the link between the PI3K/AKT activity and radiation resistance in different tumor entities both in preclinical in vitro and in vivo studies [64, 67, 70, 96, 97, 107–110]. AKT1 is involved in Rad51 protein expression as well [111]. AKT1 knockdown leads to reduced Rad51 foci formation after irradiation and enhanced frequency of residual BRCA1 foci as an indication for deficient HR [111]. Regarding the role of AKT in HR, similar data was reported in PTEN-mutated PI3K-hyperactivated cells [112], so activation of AKT seems to be necessary for Rad51 protein expression. This issue needs to be further investigated in more detail. In addition to the well-described function of AKT, especially the AKT1 paralog, in repair of DSBs through NHEJ and HR, a recent report by Villafañez and colleagues showed that AKT inhibition impairs translesion DNA synthesis activation and replication fork processes. This finding was remarkable when the combination of UV irradiation and AKT inhibition led to robust synthetic lethal induction in HR-deficient cells [113], indicating two parallel mechanisms stimulating post-irradiation cell survival. In terms of applicability of this finding to cancer therapy, it can be suggested that combining AKT inhibitors with replication stress inducing chemotherapy agents such as cisplatin may improve treatment outcome. In support of this conclusion, Nagel et al. showed that inhibition of the replication stress response machinery, such as by using Chk1 and ATM inhibitors, acts synergistically in combination with cisplatin chemotherapy. [114].
To function as a DNA repair stimulus, AKT needs to be expressed in the nucleus immediately after irradiation. So far, there is no solid data supporting nuclear translocation of AKT after irradiation or stimulation with growth factor receptor ligands. In a previous report, we could show that exposure to IR or stimulation with EGF induces phosphorylation of AKT1 at S473 in the cytoplasmic and nuclear fractions of NSCLC A549 cells. However, careful analysis of AKT1 expression revealed that neither IR nor EGF induce nuclear translocation of AKT [26]. Radiation-induced phosphorylation of nuclear AKT depends on HER2 [26, 66] as the major heterodimerization partner for other erbB members, EGFR, HER3, and HER4. However, IR does induce nuclear translocation of HER2 in NSCLC cells [26]. Thus, it can be concluded that nuclear AKT is directly phosphorylated in the nucleus independent of the cytoplasmic fraction. In line with this conclusion, Nneguen et al. [115] reported that treatment with nerve growth factor induces strong phosphorylation of nuclear AKT at S473, which, to us, makes it clear that the level of phosphorylation of AKT in the nuclear fraction does not correlate to the level of translocated AKT. Phosphorylation of AKT1 at T308 depends on PI3K and PDK1. Both of the kinases are found in the nucleus [116, 117]. Additionally, the kinases involved in AKT1 phosphorylation at S473, i.e. DNA-PKcs [21, 22] and ATM [2] are mainly localized in the nucleus. Thus, as outlined in Fig. 1, in principle cell nuclei contain all necessary components to initiate phosphorylation of AKT in the nucleus. In this regard, Rubio et al. [118] showed the presence of a nuclear-associated signaling cascade involving PI3K and PDK that presumably influences activation of nuclear AKT, independent of the cytoplasmic fraction. The idea of direct phosphorylation of AKT in the nucleus is also supported by the fact that in order for AKT to stimulate the NHEJ repair pathway, it needs to be translocated to the nucleus immediately after irradiation. This observation is not supported by literature, and our unpublished data also indicates that stimulation of AKT by neither IR nor by receptor ligands can induce nuclear accumulation of AKT within 24 hours after stimulation.
Fig. 1.
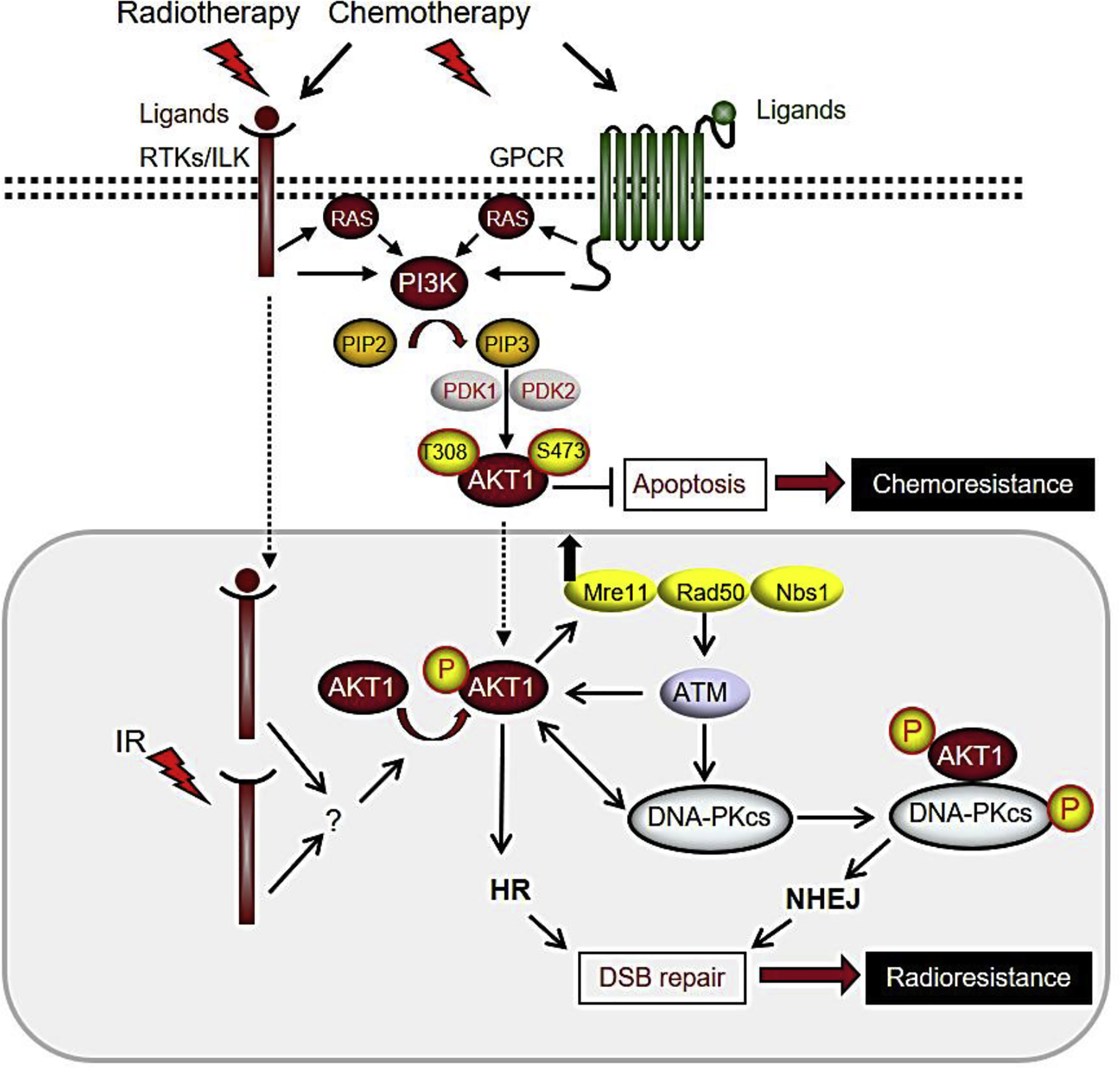
Activation of AKT mediates therapy resistance in solid tumors. Ionizing radiation, chemotherapy agents, and ligand-induced activation of membrane-bound receptors induce activation of AKT. AKT activity stimulates DNA repair leading to inhibition of apoptosis (induced by chemotherapy) and preventing mitotic catastrophe (induced by irradiation). IR-induced AKT activation in nuclear fraction is independent of AKT phosphorylation in cytoplasmic fraction. Please see the text for more details on the depicted signaling pathways.
2.3. Reactivation of AKT after RTK targeted therapies
In the last few decades, molecular targeting of RTKs have been applied as treatment for various solid tumors. There are two major avenues of RTK targeted therapies – monoclonal antibodies (mAbs) and small-molecule inhibitors. Specific mAbs bind to unique epitopes on the RTK whereas small molecule inhibitors block the kinase domain and prevent the phosphorylation of the RTKs.
Despite great progress in cancer treatment resulting from RTK molecular targeting therapies, acquired resistance occurs frequently due to the various types of feedback mechanisms including reactivation of AKT. Yoon et al. [12] inhibited the mTOR pathway in melanoma cells and found that pAKT-S473 was upregulated after 48 hours. Jacobsen et al. [119] showed that AKT pathway activation is a convergent feature in EGFR-mutant PC9 NSCLC cells with acquired resistance to EGFR tyrosine kinase inhibitors (TKIs, such as gefitinib and erlotinib). They found that combining an EGFR TKI with an AKT inhibitor induced significant growth inhibition in vitro and in vivo. They also examined clinical samples and found that pAKT was increased in the majority of EGFR-mutant patients after progression on EGFR TKIs. Furthermore, the high levels of pAKT in patients prior to EGFR-TKI treatment correlates with significantly worse PFS and OS after first-line EGFR-TKI treatment. In 2013, Lee and colleagues [120] investigated possible mechanisms responsible for acquired resistance to HER2-tageted therapy in gastric cancer. They applied Ingenuity Pathway Analysis to explore the extent to which activation of signaling pathways contributes to resistance to EGFR/HER2-targeted therapy. They found that lapatinib (dual EGFR and HER2 TKI)-resistant HER2-positive gastric cancer cells upregulated phosphorylation of EGFR/HER2, and MET appeared to be closely related to activation of PI3K/AKT and ERK1/2. Stuhlmiller et al. [121] sought to understand the bypass mechanisms toward HER2 inhibition using lapatinib-induced kinome adaptation in a panel of HER2-positive breast cancer cell lines. They found that activation of AKT was inhibited at 4 hours by lapatinib but became reactivated over 72 hours in SKBR-3 and BT474 luminal HER2-positive breast cancer cells. Our laboratory established a series of cetuximab-resistant cells in vitro following long-term exposure to cetuximab in NSCLC cells [122]. We found that total protein levels and activation of EGFR were upregulated in cetuximab-resistant cells. Furthermore, AKT has increased activity in cetuximab-resistant cells. In addition, the heightened activation of AKT substrates including c-jun, GsK3β, eIF4e, rps6, IKKα, IRs-1 and Raf1 was observed in cetuximab-resistant cells compared to cetuximab-sensitive parental control cells [123]. Gilles et al. [124] reported that acquired erlotinib-resistant HNSCC cells had reduced EGFR/pEGFR expression levels and increased AKT/pAKT expression levels. They also found increased AXL/pAXL expression levels by phospho-RTK array analysis in erlotinib-resistant HNSCC cells. Using glioblastoma cells, Martinho et al. [125] assessed AXL inhibition in two glioblastoma cell lines with distinct AXL levels using sunitinib or cediranib (pan-RTK inhibitors). They found that AXL inhibition by shRNA induced a cellular rewiring of several growth signaling pathways through activation of EGFR and AKT pathway in these glioblastoma cells. These data suggested that deregulation of one signaling pathway can sometimes alleviate or bypass the “oncogene addiction” in another pathway.
3. Targeting AKT/PKB
3.1. Targeting AKT/PKB in combination with radiochemotherapy
AKT is frequently hyperactivated in tumors from different entities. As discussed above, activated AKT stimulates DSB repair and blocks apoptosis after radiochemotherapy. Thus, targeting AKT will hamper DSB repair and enhance DNA damage-mediated mitotic catastrophe and apoptosis [126, 127]. Consequently, molecular targeting of AKT may be a promising strategy to treat cancers. This approach can be effective especially in those tumors with resistance to RTK antagonists or tumors with mutations in the components of PI3K/AKT pathway such as in genes encoding PI3K, AKT phosphatases, or AKT itself. Targeting AKT is achieved using either ATP competitive inhibitors that target the ATP binding pocket or allosteric inhibitors that function by blocking the kinase activity of AKT and preventing phosphorylation and activation of AKT by PDK1 and PDK2 [128].
Several AKT inhibitors such as AZD5363, GDC-0068 (ipatasertib), and MK-2206 have been tested in early phases of clinical trials as monotherapies or in combination therapies. A partial response to the catalytic AKT inhibitor AZD5363 that inhibits all AKT paralogs [129] was observed in patients with metastasized breast and ovarian cancer with tumors containing the AKT1-E17K mutation following monotherapy in phase I clinical trials [130]. In the same study, the modest monotherapy activity of AZD5363 was reported in the MGH-U3 bladder cancer model with an activating mutation in FGFR3 [130]. From this study, it was concluded that hyperactivation of AKT, e.g. due to the E17K mutation, may predict clinical response to AZD5363. This conclusion was further supported by the effect of AZD5363 in Japanese patients with advanced solid tumors [131]. Likewise, in a phase II trial, effectiveness of AZD5363 was shown in PIK3CA-mutated breast and gynecologic cancers [132]. Importance of hyperactivation of AKT as predictive marker for response to AKT inhibitors is confirmed in patients with solid tumors treated with AKT inhibitor GDC-0068. The marked effect, tested by metabolic PET response after treatment with GDC-0068, was observed in those patients with hyperactivation of the PI3K/AKT pathway due to decreased PTEN expression, PIK3CA mutations, and AKT1 mutation [133]. Since the majority of the AKT inhibitors were applied as monotherapy in patients without selection for mutations in the AKT pathway, often a limited anti-tumor activity by the AKT inhibitors was reported. Therefore, the investigations on AKT inhibitors have focused on the combination of the inhibitors with standard cancer therapy approaches. MK-2206 is an allosteric AKT inhibitor that has been investigated in several early phase I clinical trials to test toxicity and the anti-tumor activity in combination with the traditional therapeutic approaches (Table 1). In some studies, AKT inhibitors were applied with other molecular targeting approaches as well. KRAS mutation leads to the activation of the MAPK and PI3K/AKT pathways. In this regard, it has been shown that combination of MK-2206 with MEK inhibition may be a therapeutic strategy in KRAS-mutated solid tumors [134]. There are also studies that reported no advantage of the combination of MK-2206 with other targeting approaches. In this context, in a phase II trial of the combination of MK-2206 with the aromatase inhibitor anastrozole, MK-2206 did not add to the efficacy of anastrozole alone in PIK3CA-mutant ER+ breast cancer [135]. In this study, a lack of effect of MK-2206 on AKT substrate PRAS40 was shown [135]. This might be due to the compensatory activation of AKT substrates through alternative parallel pathways. Overlapping toxicities in combination therapies is often a limiting factor to achieve clinical activity. This issue has been also addressed in a phase II trial of the combination of AKT inhibitor MK-2206 with MEK inhibitor selumetinib that led to no objective responses in CRC patients stratified for KRAS mutation [136].
Table 1:
Clinical trials of the AKT antagonists in combination with other treatment approaches in patients with solid tumors.
| Drug | Combination | Tumor type | Outcome | Reference |
|---|---|---|---|---|
| AZD5363, KI | CT / phase 1 | mCRPC | RP2D: 320 mg BID | [152] |
| Ipatasertib, KI | HAT-abiraterone / phase 2 | mCRPC | Superior antitumor activity to HT | [153] |
| CT / phase II | mTNBC | Longer PFS | [154] | |
| MK-2206, AI | MEK inhibitor / phase 1 | KRAS-mutated solid tumors | Durable response | [134] |
| CT / phase 1 | advanced solid tumors | Well-tolerated, antitumor activity | [155] | |
| CT / phase 1 | breast cancer | Antitumor activity | [156] | |
| Anastrozole (aromatase inhibitor) / phase 1 | ER+ breast cancer | Well-tolerated | [157] | |
| Anastrozole (aromatase inhibitor) / phase 2 | PIKCA-Mutant ER+/HER2-Breast Cancer | No advantage of the combination | [135] | |
| Selumetinib (MEK inhibitor) | CRC | No objective response | [136] | |
| Perifosine | RT / phase 1 | NSCLC, prostate, oesophageal, colon and bladder cancer | Recommended phase II, 150 mg/day, started one week prior to RT | [141] |
| mTOR inhbitior / phase 1 | recurrent pediatric solid tumors. | Well-tolerated | [144] | |
| CT / phase 2 | mCRC | Promising clinical activity | [142] | |
| Nelfinavir | RT / Phase 1 | rectal cancer | Well tolerated and good tumor regression | [148] |
| CRT / Phase 1 | rectal cancer | Nelfinavir 750 mg recommended phase II | [149] | |
| CRT / Phase 1 | NSCLC | Acceptable toxicity and promising activity | [150] | |
| CRT / Phase 1 | pancreatic cancer | Acceptable toxicity and promising activity | [151] | |
| CRT / Phase 2 | pancreatic cancer | Acceptable toxicity and promising activity | [147] |
CT: chemotherapy CRT: chemoradiotherapy; RT: radiotherapy; HT: hormone therapy; NSCLC: Non-small cell lung cancer; RP2D: recommended phase II dose; mCRPC: metastatic castration resistant prostate cancer; mTNBC: metastatic triple-negative breast cancer; PFS: progression-free survival; KI: kinase inhibitor; AI: allosteric inhibitor; CRC: colorectal cancer; mCRC: metastatic colorectal cancer
Besides these well-described AKT kinase and allosteric inhibitors, there are compounds that are known to block AKT activity. Perifosine is one of those compounds in that its partial activity is AKT-dependent by targeting the PH domain of AKT, thereby preventing AKT accumulation to the plasma membrane [137]. Perifosine as a monotherapy was well tolerated in neuroblastoma in phase I studies [138, 139] and with efficacy demonstrated in ovarian cancer patients with PIK3CA mutations and endometrial cancer patients with PIK3CA wild-type [140]. Absence of PTEN expression was supposed to be predictive of clinical efficacy of perifosine in monotherapy [140]. So far, the results obtained from combinational studies with radiotherapy in a phase I trial and with chemotherapy in a phase II trial are promising (Table 1) [141, 142]. Similar to the AKT kinase or allosteric inhibitors, these class of compounds have also been investigated in combination with other molecular targeting approaches. Basal AKT activity as well as reactivation of AKT after targeting mammalian target of rapamycin (mTOR) is one of the mechanisms involved in limited efficacy of mTOR targeting strategies. Thus, as suggested by Holler et al. [143], co-targeting of AKT and mTOR might be an efficient approach to improve effect of DNA damaging cancer treatment strategies. To this aim, the combination of perifosine with mTOR inhibitor temsirolimus in a phase I clinical trial for recurrent pediatric solid tumors was shown to be safe and feasible [144]. A recent phase II trial by Kaley et al. [145] demonstrated that perifosine is tolerable but ineffective as monotherapy for glioblastoma multiform (GBM). Based on preclinical data, these authors suggested the combination of perifosine with other therapeutic approaches [145]. Nelfinavir as a human immunodeficiency virus protease inhibitor has been described to be an effective compound to block AKT dependent survival pathway and induce radiosensitization [146]. Acceptable toxicity and promising activity of nelfinavir in the combination with radiochemotherapy of NSCLC, rectal cancers, and pancreatic cancers has been reported [147–151] (Table 1).
Altogether, the so far obtained results from preclinical investigations as well as early-phase clinical trials support the rationale for targeting AKT only in combination with other cancer treatment approaches.
3.2. Targeting AKT in combination with RTK targeted therapies or immunotherapy
It has been reported that inhibition of the PI3K/AKT pathway with RTK inhibitors may be an effective strategy for cancer therapy in the setting of acquired resistance [158]. In a pre-clinical study with cetuximab-resistant cells, the combination of cetuximab with the allosteric AKT inhibitor MK-2206 resulted in further decreases in proliferation than either drug alone [123]. Stuhlmiller et al. [121] showed that the combination treatment with JQ1, an inhibitor of BET family bromodomains (major epigenetic regulators), plus lapatinib was required to substantially suppress transcription of many adaptive response RTKs implicated in resistance including HER2, HER3, DDR1, FGFR2, IGF1R, and MET. They also showed that this drug combination prevented reactivation of AKT/p70-S6K signaling. Using 3 different pharmacological inhibitors of growth factor receptors, Yoon et al. [12] found that inhibition of IGF1R/IR, but not EGFR or PDGFR, blocked AKT re-phosphorylation that is mediated by mTOR inhibition. Song et al. [159] explored the drug resistance mechanisms of the KRAS or BRAF mutant colorectal cancer (CRC) cells. They found that inhibition of AKT increased the phosphorylation of RTKs in KRAS/BRAF mutant CRC cells. The combination of AKT inhibitor (MK2206) and RTK inhibitors (Lapatinib, OSI-906 or jnj38877605) showed a synergistic effect in KRAS/BRAF mutant CRC cells. Feng et al. [160] found increased FGFR signaling in advanced prostate cancer. Their studies demonstrate additive effects on cell proliferation by AZD4547 (FGF receptor kinase inhibitor) and AZD5363 (AKT kinase inhibitor) in prostate cancer cell lines in vitro and in vivo. Another in vivo study that was conducted by Crafter et al. [161] demonstrated that the effects of the combination of an AKT inhibitor (AZD5363) and a novel EGFR/HER2/HER3 signaling inhibitor (AZD8931) in the HCC1954 breast cancer xenograft model. HCC1954 cells are a HER2-amplified, PIK3CA mutant cell line that is resistant to therapy with the anti-HER2 therapeutic antibody trastuzumab. Monotherapy with AZD5363 or AZD8931 inhibited tumor growth by 42% and 39% respectively, compared with vehicle controls. The combination of AZD5363 and AZD8931 was well tolerated and caused pronounced tumor regression that was sustained for the duration of the dosing period.
We identified 17 clinical trials focused on the treatment of solid malignancies evaluating PI3K/AKT inhibitors in combination with RTK inhibitors or immunotherapy (Table 2, https://clinicaltrials.gov). Cancer immunotherapy, specifically, the development of anti-programmed cell death 1 (PD-1) and anti-programmed cell death 1 ligand 1 (PD-L1) antibodies as immune-checkpoint inhibitors, has recently become a viable option for the treatment of many advanced cancer types. In 2019, Larkin et al. [162] reported that 5-year OS was 52% for advanced melanoma patients who received anti-CTLA-4 and anti-PD-1 combination therapy. This was a landmark study because a decade ago only 5% these patients were still alive after five years. Some of the targeted therapies actually inhibit not only tumor cells, but also directly modulate immune cells [163] [164] [165] [166]. Recently, Kashikara et al. [164] showed that expression of TAM (Tyro3, AXL, MerTK) family receptors in MCF10A normal breast epithelial cells induces chemoresistance, proliferation, and AKT activation. Furthermore, they examined the effect of treating TNBC cells with a TAM inhibitor (BMS-777607) plus an anti-PD-1 mAb [163]. Combinatorial BMS-777607 and anti-PD-1 mAb treatment showed enhanced antitumor effects compared with monotherapy (P<0.0001), as well as reduced lung metastatic nodules in vivo (P<0.01). Using lung cancer models, Lastwika et al. [167] revealed a strong association between PD-L1 expression and activation of the AKT/mTOR pathway in lung cancer. They showed that inhibitors of PI3K (LY294002), AKT (TCN-P), or mTOR (rapamycin) decreased PD-L1 expression in NSCLC cell lines. In addition, rapamycin decreased PD-L1 expression levels in lung tumor in vivo models. Despite the impressive developments of immunotherapy in the treatment of patients with metastatic cancer, the majority of patients still relapsed, demonstrating that like RTK targeted therapies, acquired resistance to immunotherapies frequently occur [168] [169] [170]. A further understanding of the effect of targeted therapy on tumor cells and immune cells may provide valuable insights to help design new strategies for combination therapies.
Table 2.
Clinical trials: PI3K/AKT inhibitors in combination with RTK inhibitors or immunotherapy
| Clinical trial number | Study Title | Drug | Cancer | Phase |
|---|---|---|---|---|
| NCT01705340 | Akt Inhibitor MK2206, lapatinib ditosylate, and trastuzumab in treating patients with Locally advanced or metastatic HER2-positive breast, gastric, or gastroesophageal cancer that cannot be removed by surgery | MK-2206 in combination with trastuzumab & lapatinib | Adenocarcinoma of the gastroesophageal junction, HER2-positive breast cancer, male breast cancer, recurrent breast cancer, recurrent esophageal cancer, recurrent gastric cancer, stage IIIC breast cancer, stage IIIC esophageal cancer, stage IIIC gastric cancer, stage IV breast cancer, stage IV esophageal cancer, stage IV gastric cancer | Phase 1 |
| NCT01235897 | MK-2206, paclitaxel and trastuzumab in treating patients with HER2-overexpressing solid tumor malignancies | MK2206+ weekly paclitaxel with or without trastuzumab | Advanced solid tumors | Phase 1 |
| NCT01971515 | First-in-human dose escalation trial in subjects with advanced malignancies | MSC236331 8A, a dual p70S6K/Akt inhibitor plus trastuzumab or tamoxifen | Solid tumor | Phase 1 |
| NCT01281163 | Lapatinib ditosylate and Akt inhibitor MK2206 in treating women with metastatic breast cancer | MK-2206 + lapatinib | Patients with HER2 positive metastatic breast cancer | Phase 1 |
| NCT01245205 | Akt inhibitor MK2206 in combination with lapatinib ditosylate in patients with advanced or metastatic solid tumors or breast cancer | MK-2206 + lapatinib | Refractory solid tumors followed by dose-expansion, advanced HER2+ breast cancer | Phase 1 |
| NCT01147211 | Dose defining study for MK-2206 combined with gefitinib in NSCLC | MK-2206 + gefitinib | NSCLC population enriched with EGFR mutation | Phase 1 |
| NCT02705859 | Phase Ib/II trial of copanlisib in combination with trastuzumab in HER2-positive breast cancer. (Panther Study) | Copanlisib + trastuzumab | Pretreated recurrent or metastatic HER2-positive breast cancer | Phase 1 |
| NCT02646748 | Pembrolizumab combined with itacitinib (INCB039110) and/or pembrolizumab combined with INCB050465in advanced solid tumors | Pembrolizumab + INCB combinations | Advanced solid tumors | Phase 1 |
| NCT01589861 | Safety and efficacy of BKM120 and lapatinib in HER2+/PI3K-activated, trastuzumab-resistant advanced breast cancer | Oral BKM120 + lapatinib | HER2+/PI3K-activated, trastuzumab-resistant locally advanced, recurrent and metastatic breast cancer. | Phase 1, Phase 2 |
| NCT03772561 | Phase I study of AZD5363 + Olaparib + durvalumab in patients with advanced or metastatic solid tumor malignancies | AZD5363 + Olaparib + Durvaluma b | Solid tumor, adult | Phase 1, Phase 2 |
| NCT03742102 | A study of novel anti-cancer agents in patients with metastatic triple negative breast cancer. | durvalumab + paclitaxel, durvalumab + paclitaxel + capivasertib durvalumab + paclitaxel + danvatirsen durvalumab + paclitaxel + oleclumab | Triple negative breast cancer | Phase 1, Phase 2 |
| NCT01816984 | PI3K inhibitor BKM120 and cetuximab in treating patients with recurrent or metastatic head and neck cancer | Pan-class I PI3K inhibitor NVP-BKM120 + cetuximab | Patients with recurrent/metastatic head and neck cancer | Phase 1, Phase 2 |
| NCT02822482 | Copanlisib in association with cetuximab in patients with recurrent and/or metastatic HNSCC harboring a PI3KCA mutation/amplification and/or a PTEN loss | Copanlisib, a selective PI3K inhibitor + cetuximab | Patients with recurrent and/or metastatic HNSCC harboring a PI3KCA mutation/amplification and/or a PTEN loss. | Phase 1, Phase 2 |
| NCT01870726 | Safety and efficacy of INC280 and buparlisib (BKM120) in patients with recurrent glioblastoma | INC280 (c-MET Inhibitor) + buparlisib (BKM120) | Patients with recurrent glioblastoma | Phase 1, Phase 2 |
| NCT01487265 | Trial of erlotinib and BKM120 in patients with advanced NSCLC previously sensitive to erlotinib | Erlotinib & BKM120 | Patients with advanced NSCLC previously sensitive to erlotinib | Phase 2 |
| NCT01294306 | MK2206 and erlotinib hydrochloride in treating patients with advanced NSCLC who have progressed after previous response to erlotinib hydrochloride therapy | MK-2206 + erlotinib | Patients with advanced NSCLC who have progressed after previous response (including stable disease) with erlotinib therapy | Phase 2 |
| NCT01816594 | NeoPHOEBE: neoadjuvant trastuzumab + BKM120 in combination with weekly paclitaxel in HER2-positive primary breast cancer | BKM120 + trastuzumab & paclitaxel | HER2-positive primary breast cancer prior to definitive surgery (neo-adjuvant setting) | Phase 2 |
4. Perspective and Conclusions
In this review, we discussed the role of AKT in human cancer, mechanisms of resistance to therapy, and ongoing strategies that target AKT in combination with other treatments to improve clinical outcomes. So far, tremendous effort has been devoted to targeting upstream stimulators of PI3K/AKT pathway such as RTKs in combination with DNA damage-inducing therapies, e.g. radiotherapy and chemotherapy. Despite some promising long-term responses, many of the tumors that initially respond to treatment eventually manifest resistance. The PI3K/AKT pathway is a central signaling hub that is crucial for tumor cell survival. Successful inhibition of PI3K/AKT in tumors is likely to provide a significant clinical benefit for cancer patients. Therefore, approaches that simultaneously target AKT in combination with radiochemotherapy or other molecularly targeted strategies including immunotherapy may provide a more effective approach to improve cancer treatment outcomes for the future.
Acknowledgements
We would like to thank Dr. Roger Wiseman at the Department of Pathology and Laboratory Medicine, University of Wisconsin for his critical reading of the manuscript.
Funding
Research reported in this publication was supported, in part, by the Wisconsin Head & Neck Cancer SPORE Grant (PMH and DLW, NIH P50 DE026787), MT’s research is supported, in part, by the German Research Council, Deutsche Forschungsgemeinschaft (DFG TO 685/2-1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Barnett SF, Defeo-Jones D, Fu S, Hancock PJ, Haskell KM, Jones RE, Kahana JA, Kral AM, Leander K, Lee LL, Malinowski J, McAvoy EM, Nahas DD, Robinson RG, Huber HE, Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors, Biochem J, 385 (2005) 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Viniegra JG, Martinez N, Modirassari P, Hernandez Losa J, Parada Cobo C, Sanchez-Arevalo Lobo VJ, Aceves Luquero CI, Alvarez-Vallina L, Ramon y Cajal S, Rojas JM, Sanchez-Prieto R, Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM, J Biol Chem, 280 (2005) 4029–4036. [DOI] [PubMed] [Google Scholar]
- [3].Vincent EE, Elder DJ, Thomas EC, Phillips L, Morgan C, Pawade J, Sohail M, May MT, Hetzel MR, Tavare JM, Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer, Br J Cancer, 104 (2011) 1755–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Blume-Jensen P, Hunter T, Oncogenic kinase signalling, Nature, 411 (2001) 355–365. [DOI] [PubMed] [Google Scholar]
- [5].Regad T, Targeting RTK Signaling Pathways in Cancer, Cancers (Basel), 7 (2015) 1758–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Toulany M, Targeting DNA Double-Strand Break Repair Pathways to Improve Radiotherapy Response, Genes (Basel), 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Toulany M, Rodemann HP, Phosphatidylinositol 3-kinase/Akt signaling as a key mediator of tumor cell responsiveness to radiation, Semin Cancer Biol, 35 (2015) 180–190. [DOI] [PubMed] [Google Scholar]
- [8].Rozengurt E, Mitogenic signaling pathways induced by G protein-coupled receptors, J Cell Physiol, 213 (2007) 589–602. [DOI] [PubMed] [Google Scholar]
- [9].Hossain MS, Mineno K, Katafuchi T, Neuronal Orphan G-Protein Coupled Receptor Proteins Mediate Plasmalogens-Induced Activation of ERK and Akt Signaling, PLoS One, 11 (2016) e0150846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ohtsuka H, Iguchi T, Hayashi M, Kaneda M, Iida K, Shimonaka M, Hara T, Arai M, Koike Y, Yamamoto N, Kasahara K, SDF-1alpha/CXCR4 Signaling in Lipid Rafts Induces Platelet Aggregation via PI3 Kinase-Dependent Akt Phosphorylation, PLoS One, 12 (2017) e0169609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Werner K, Neumann D, Seifert R, High constitutive Akt2 activity in U937 promonocytes: effective reduction of Akt2 phosphorylation by the histamine H2-receptor and the beta2-adrenergic receptor, Naunyn Schmiedebergs Arch Pharmacol, 389 (2016) 87–101. [DOI] [PubMed] [Google Scholar]
- [12].Yoon SO, Shin S, Karreth FA, Buel GR, Jedrychowski MP, Plas DR, Dedhar S, Gygi SP, Roux PP, Dephoure N, Blenis J, Focal Adhesion- and IGF1R-Dependent Survival and Migratory Pathways Mediate Tumor Resistance to mTORC1/2 Inhibition, Mol Cell, 67 (2017) 512–527 e514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Guidetti GF, Canobbio I, Torti M, PI3K/Akt in platelet integrin signaling and implications in thrombosis, Adv Biol Regul, 59 (2015) 36–52. [DOI] [PubMed] [Google Scholar]
- [14].Muller EJ, Williamson L, Kolly C, Suter MM, Outside-in signaling through integrins and cadherins: a central mechanism to control epidermal growth and differentiation?, J Invest Dermatol, 128 (2008) 501–516. [DOI] [PubMed] [Google Scholar]
- [15].Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S, Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase, Proc Natl Acad Sci U S A, 95 (1998) 11211–11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Overland AC, Insel PA, Heterotrimeric G proteins directly regulate MMP14/membrane type-1 matrix metalloprotease: a novel mechanism for GPCR-EGFR transactivation, J Biol Chem, 290 (2015) 9941–9947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu P, Cheng H, Roberts TM, Zhao JJ, Targeting the phosphoinositide 3-kinase pathway in cancer, Nat Rev Drug Discov, 8 (2009) 627–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P, Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha, Curr Biol, 7 (1997) 261–269. [DOI] [PubMed] [Google Scholar]
- [19].Kearney AL, Cooke KC, Norris DM, Zadoorian A, Krycer JR, Fazakerley DJ, Burchfield JG, James DE, Serine 474 phosphorylation is essential for maximal Akt2 kinase activity in adipocytes, J Biol Chem, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA, Mechanism of activation of protein kinase B by insulin and IGF-1, EMBO J, 15 (1996) 6541–6551. [PMC free article] [PubMed] [Google Scholar]
- [21].Bozulic L, Surucu B, Hynx D, Hemmings BA, PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival, Mol Cell, 30 (2008) 203–213. [DOI] [PubMed] [Google Scholar]
- [22].Feng J, Park J, Cron P, Hess D, Hemmings BA, Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase, J Biol Chem, 279 (2004) 41189–41196. [DOI] [PubMed] [Google Scholar]
- [23].Persad S, Attwell S, Gray V, Delcommenne M, Troussard A, Sanghera J, Dedhar S, Inhibition of integrin-linked kinase (ILK) suppresses activation of protein kinase B/Akt and induces cell cycle arrest and apoptosis of PTEN-mutant prostate cancer cells, Proc Natl Acad Sci U S A, 97 (2000) 3207–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Persad S, Attwell S, Gray V, Mawji N, Deng JT, Leung D, Yan J, Sanghera J, Walsh MP, Dedhar S, Regulation of protein kinase B/Akt-serine 473 phosphorylation by integrin-linked kinase: critical roles for kinase activity and amino acids arginine 211 and serine 343, J Biol Chem, 276 (2001) 27462–27469. [DOI] [PubMed] [Google Scholar]
- [25].Sarbassov DD, Guertin DA, Ali SM, Sabatini DM, Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex, Science, 307 (2005) 1098–1101. [DOI] [PubMed] [Google Scholar]
- [26].Toulany M, Schickfluss TA, Fattah KR, Lee KJ, Chen BP, Fehrenbacher B, Schaller M, Chen DJ, Rodemann HP, Function of erbB receptors and DNA-PKcs on phosphorylation of cytoplasmic and nuclear Akt at S473 induced by erbB1 ligand and ionizing radiation, Radiother Oncol, 101 (2011) 140–146. [DOI] [PubMed] [Google Scholar]
- [27].Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, McCormick F, Feng J, Tsichlis P, Akt activation by growth factors is a multiple-step process: the role of the PH domain, Oncogene, 17 (1998) 313–325. [DOI] [PubMed] [Google Scholar]
- [28].Hanada M, Feng J, Hemmings BA, Structure, regulation and function of PKB/AKT--a major therapeutic target, Biochim Biophys Acta, 1697 (2004) 3–16. [DOI] [PubMed] [Google Scholar]
- [29].Conus NM, Hannan KM, Cristiano BE, Hemmings BA, Pearson RB, Direct identification of tyrosine 474 as a regulatory phosphorylation site for the Akt protein kinase, J Biol Chem, 277 (2002) 38021–38028. [DOI] [PubMed] [Google Scholar]
- [30].Chen R, Kim O, Yang J, Sato K, Eisenmann KM, McCarthy J, Chen H, Qiu Y, Regulation of Akt/PKB activation by tyrosine phosphorylation, J Biol Chem, 276 (2001) 31858–31862. [DOI] [PubMed] [Google Scholar]
- [31].Risso G, Blaustein M, Pozzi B, Mammi P, Srebrow A, Akt/PKB: one kinase, many modifications, Biochem J, 468 (2015) 203–214. [DOI] [PubMed] [Google Scholar]
- [32].Maehama T, Dixon JE, The tumor suppressor PTEN /MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate, J Biol Chem, 273 (1998) 13375–13378. [DOI] [PubMed] [Google Scholar]
- [33].Gao T, Furnari F, Newton AC, PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth, Mol Cell, 18 (2005) 13–24. [DOI] [PubMed] [Google Scholar]
- [34].Brognard J, Sierecki E, Gao T, Newton AC, PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms, Mol Cell, 25 (2007) 917–931. [DOI] [PubMed] [Google Scholar]
- [35].Narla G, Sangodkar J, Ryder CB, The impact of phosphatases on proliferative and survival signaling in cancer, Cell Mol Life Sci, 75 (2018) 2695–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Manning BD, Toker A, AKT/PKB Signaling: Navigating the Network, Cell, 169 (2017) 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Manning BD, Cantley LC, AKT/PKB signaling: navigating downstream, Cell, 129 (2007) 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Shao X, Lv N, Liao J, Long J, Xue R, Ai N, Xu D, Fan X, Copy number variation is highly correlated with differential gene expression: a pan-cancer study, BMC Med Genet, 20 (2019) 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yang ZY, Di MY, Yuan JQ, Shen WX, Zheng DY, Chen JZ, Mao C, Tang JL, The prognostic value of phosphorylated Akt in breast cancer: a systematic review, Sci Rep, 5 (2015) 7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Luo J, Feng J, Wen Q, Qoyawayma C, Wang W, Chen L, Lu J, Zhan Y, Xu L, Zang H, Fan S, Chu S, Elevated expression of IRS-1 associates with phosphorylated Akt expression and predicts poor prognosis of breast invasive ductal carcinoma, Hum Pathol, 79 (2018) 9–17. [DOI] [PubMed] [Google Scholar]
- [41].Park SS, Kim SW, Activated Akt signaling pathway in invasive ductal carcinoma of the breast: correlation with HER2 overexpression, Oncol Rep, 18 (2007) 139–143. [PubMed] [Google Scholar]
- [42].Tokunaga E, Kimura Y, Mashino K, Oki E, Kataoka A, Ohno S, Morita M, Kakeji Y, Baba H, Maehara Y, Activation of PI3K/Akt signaling and hormone resistance in breast cancer, Breast Cancer, 13 (2006) 137–144. [DOI] [PubMed] [Google Scholar]
- [43].Tsurutani J, Fukuoka J, Tsurutani H, Shih JH, Hewitt SM, Travis WD, Jen J, Dennis PA, Evaluation of two phosphorylation sites improves the prognostic significance of Akt activation in non-small-cell lung cancer tumors, J Clin Oncol, 24 (2006) 306–314. [DOI] [PubMed] [Google Scholar]
- [44].Yoshizawa A, Fukuoka J, Shimizu S, Shilo K, Franks TJ, Hewitt SM, Fujii T, Cordon-Cardo C, Jen J, Travis WD, Overexpression of phospho-eIF4E is associated with survival through AKT pathway in non-small cell lung cancer, Clin Cancer Res, 16 (2010) 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jin Y, Yuan Y, Yi M, Han H, Liu B, Li Q, Phosphorylated-Akt overexpression is associated with a higher risk of brain metastasis in patients with non-small cell lung cancer, Biochem Biophys Rep, 18 (2019) 100625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dobashi Y, Kimura M, Matsubara H, Endo S, Inazawa J, Ooi A, Molecular alterations in AKT and its protein activation in human lung carcinomas, Hum Pathol, 43 (2012) 2229–2240. [DOI] [PubMed] [Google Scholar]
- [47].Molinolo AA, Hewitt SM, Amornphimoltham P, Keelawat S, Rangdaeng S, Meneses Garcia A, Raimondi AR, Jufe R, Itoiz M, Gao Y, Saranath D, Kaleebi GS, Yoo GH, Leak L, Myers EM, Shintani S, Wong D, Massey HD, Yeudall WA, Lonardo F, Ensley J, Gutkind JS, Dissecting the Akt/mammalian target of rapamycin signaling network: emerging results from the head and neck cancer tissue array initiative, Clin Cancer Res, 13 (2007) 4964–4973. [DOI] [PubMed] [Google Scholar]
- [48].Mundi PS, Sachdev J, McCourt C, Kalinsky K, AKT in cancer: new molecular insights and advances in drug development, Br J Clin Pharmacol, 82 (2016) 943–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Horn D, Freudlsperger C, Holzinger D, Kunzmann K, Plinkert P, Dyckhoff G, Hoffmann J, Freier K, Hess J, Upregulation of pAKT(Ser473) expression in progression of HPV-positive oropharyngeal squamous cell carcinoma, Head Neck, 39 (2017) 2397–2405. [DOI] [PubMed] [Google Scholar]
- [50].Islam MR, Ellis IR, Macluskey M, Cochrane L, Jones SJ, Activation of Akt at T308 and S473 in alcohol, tobacco and HPV-induced HNSCC: is there evidence to support a prognostic or diagnostic role?, Exp Hematol Oncol, 3 (2014) 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Murthy D, Attri KS, Singh PK, Phosphoinositide 3-Kinase Signaling Pathway in Pancreatic Ductal Adenocarcinoma Progression, Pathogenesis, and Therapeutics, Front Physiol, 9 (2018) 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Mao Y, Xi L, Li Q, Cai Z, Lai Y, Zhang X, Yu C, Regulation of cell apoptosis and proliferation in pancreatic cancer through PI3K/Akt pathway via Polo-like kinase 1, Oncol Rep, 36 (2016) 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Massihnia D, Avan A, Funel N, Maftouh M, van Krieken A, Granchi C, Raktoe R, Boggi U, Aicher B, Minutolo F, Russo A, Leon LG, Peters GJ, Giovannetti E, Phospho-Akt overexpression is prognostic and can be used to tailor the synergistic interaction of Akt inhibitors with gemcitabine in pancreatic cancer, J Hematol Oncol, 10 (2017) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sinkala M, Mulder N, Martin DP, Integrative landscape of dysregulated signaling pathways of clinically distinct pancreatic cancer subtypes, Oncotarget, 9 (2018) 29123–29139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Mizoguchi M, Betensky RA, Batchelor TT, Bernay DC, Louis DN, Nutt CL, Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: correlation with EGFR status, tumor grade, and survival, J Neuropathol Exp Neurol, 65 (2006) 1181–1188. [DOI] [PubMed] [Google Scholar]
- [56].Wuchty S, Vazquez A, Bozdag S, Bauer PO, Genome-wide associations of signaling pathways in glioblastoma multiforme, BMC Med Genomics, 6 (2013) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Gallia GL, Tyler BM, Hann CL, Siu IM, Giranda VL, Vescovi AL, Brem H, Riggins GJ, Inhibition of Akt inhibits growth of glioblastoma and glioblastoma stem-like cells, Mol Cancer Ther, 8 (2009) 386–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Cohen Y, Shalmon B, Korach J, Barshack I, Fridman E, Rechavi G, AKT1 pleckstrin homology domain E17K activating mutation in endometrial carcinoma, Gynecol Oncol, 116 (2010) 88–91. [DOI] [PubMed] [Google Scholar]
- [59].Askham JM, Platt F, Chambers PA, Snowden H, Taylor CF, Knowles MA, AKT1 mutations in bladder cancer: identification of a novel oncogenic mutation that can co-operate with E17K, Oncogene, 29 (2010) 150–155. [DOI] [PubMed] [Google Scholar]
- [60].Malanga D, Scrima M, De Marco C, Fabiani F, De Rosa N, De Gisi S, Malara N, Savino R, Rocco G, Chiappetta G, Franco R, Tirino V, Pirozzi G, Viglietto G, Activating E17K mutation in the gene encoding the protein kinase AKT1 in a subset of squamous cell carcinoma of the lung, Cell Cycle, 7 (2008) 665–669. [DOI] [PubMed] [Google Scholar]
- [61].Bleeker FE, Felicioni L, Buttitta F, Lamba S, Cardone L, Rodolfo M, Scarpa A, Leenstra S, Frattini M, Barbareschi M, Grammastro MD, Sciarrotta MG, Zanon C, Marchetti A, Bardelli A, AKT1(E17K) in human solid tumours, Oncogene, 27 (2008) 5648–5650. [DOI] [PubMed] [Google Scholar]
- [62].Masure S, Haefner B, Wesselink JJ, Hoefnagel E, Mortier E, Verhasselt P, Tuytelaars A, Gordon R, Richardson A, Molecular cloning, expression and characterization of the human serine/threonine kinase Akt-3, Eur J Biochem, 265 (1999) 353–360. [DOI] [PubMed] [Google Scholar]
- [63].Contessa JN, Hampton J, Lammering G, Mikkelsen RB, Dent P, Valerie K, Schmidt-Ullrich RK, Ionizing radiation activates Erb-B receptor dependent Akt and p70 S6 kinase signaling in carcinoma cells, Oncogene, 21 (2002) 4032–4041. [DOI] [PubMed] [Google Scholar]
- [64].Toulany M, Dittmann K, Baumann M, Rodemann HP, Radiosensitization of Ras-mutated human tumor cells in vitro by the specific EGF receptor antagonist BIBX1382BS, Radiother Oncol, 74 (2005) 117–129. [DOI] [PubMed] [Google Scholar]
- [65].Valerie K, Yacoub A, Hagan MP, Curiel DT, Fisher PB, Grant S, Dent P, Radiation-induced cell signaling: inside-out and outside-in, Mol Cancer Ther, 6 (2007) 789–801. [DOI] [PubMed] [Google Scholar]
- [66].Toulany M, Minjgee M, Kehlbach R, Chen J, Baumann M, Rodemann HP, ErbB2 expression through heterodimerization with erbB1 is necessary for ionizing radiation- but not EGF-induced activation of Akt survival pathway, Radiother Oncol, 97 (2010) 338–345. [DOI] [PubMed] [Google Scholar]
- [67].Li HF, Kim JS, Waldman T, Radiation-induced Akt activation modulates radioresistance in human glioblastoma cells, Radiat Oncol, 4 (2009) 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Toulany M, Kehlbach R, Florczak U, Sak A, Wang S, Chen J, Lobrich M, Rodemann HP, Targeting of AKT1 enhances radiation toxicity of human tumor cells by inhibiting DNA-PKcs-dependent DNA double-strand break repair, Mol Cancer Ther, 7 (2008) 1772–1781. [DOI] [PubMed] [Google Scholar]
- [69].Gupta AK, Bakanauskas VJ, Cerniglia GJ, Cheng Y, Bernhard EJ, Muschel RJ, McKenna WG, The Ras radiation resistance pathway, Cancer Res, 61 (2001) 4278–4282. [PubMed] [Google Scholar]
- [70].Zhang T, Cui GB, Zhang J, Zhang F, Zhou YA, Jiang T, Li XF, Inhibition of PI3 kinases enhances the sensitivity of non-small cell lung cancer cells to ionizing radiation, Oncology reports, 24 (2010) 1683–1689. [DOI] [PubMed] [Google Scholar]
- [71].Toulany M, Minjgee M, Kehlbach R, Chen J, Baumann M, Rodemann HP, ErbB2 expression through heterodimerization with erbB1 is necessary for ionizing radiation- but not EGF-induced activation of Akt survival pathway, Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology, (2010). [DOI] [PubMed] [Google Scholar]
- [72].Winograd-Katz SE, Levitzki A, Cisplatin induces PKB/Akt activation and p38(MAPK) phosphorylation of the EGF receptor, Oncogene, 25 (2006) 7381–7390. [DOI] [PubMed] [Google Scholar]
- [73].Dent P, Reardon DB, Park JS, Bowers G, Logsdon C, Valerie K, Schmidt-Ullrich R, Radiation-induced release of transforming growth factor alpha activates the epidermal growth factor receptor and mitogen-activated protein kinase pathway in carcinoma cells, leading to increased proliferation and protection from radiation-induced cell death, Mol Biol Cell, 10 (1999) 2493–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hagan M, Wang L, Hanley JR, Park JS, Dent P, Ionizing radiation-induced mitogen-activated protein (MAP) kinase activation in DU145 prostate carcinoma cells: MAP kinase inhibition enhances radiation-induced cell killing and G2/M-phase arrest, Radiat Res, 153 (2000) 371–383. [DOI] [PubMed] [Google Scholar]
- [75].Qu YY, Hu SL, Xu XY, Wang RZ, Yu HY, Xu JY, Chen L, Dong GL, Nimotuzumab enhances the radiosensitivity of cancer cells in vitro by inhibiting radiation-induced DNA damage repair, PLoS One, 8 (2013) e70727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kim IA, Bae SS, Fernandes A, Wu J, Muschel RJ, McKenna WG, Birnbaum MJ, Bernhard EJ, Selective inhibition of Ras, phosphoinositide 3 kinase, and Akt isoforms increases the radiosensitivity of human carcinoma cell lines, Cancer Res, 65 (2005) 7902–7910. [DOI] [PubMed] [Google Scholar]
- [77].Belyanskaya LL, Hopkins-Donaldson S, Kurtz S, Simoes-Wust AP, Yousefi S, Simon HU, Stahel R, Zangemeister-Wittke U, Cisplatin activates Akt in small cell lung cancer cells and attenuates apoptosis by survivin upregulation, Int J Cancer, 117 (2005) 755–763. [DOI] [PubMed] [Google Scholar]
- [78].Peng DJ, Wang J, Zhou JY, Wu GS, Role of the Akt/mTOR survival pathway in cisplatin resistance in ovarian cancer cells, Biochem Biophys Res Commun, 394 (2010) 600–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Hahne JC, Honig A, Meyer SR, Gambaryan S, Walter U, Wischhusen J, Haussler SF, Segerer SE, Fujita N, Dietl J, Engel JB, Downregulation of AKT reverses platinum resistance of human ovarian cancers in vitro, Oncol Rep, 28 (2012) 2023–2028. [DOI] [PubMed] [Google Scholar]
- [80].Gohr K, Hamacher A, Engelke LH, Kassack MU, Inhibition of PI3K/Akt/mTOR overcomes cisplatin resistance in the triple negative breast cancer cell line HCC38, BMC Cancer, 17 (2017) 711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Gagnon V, Mathieu I, Sexton E, Leblanc K, Asselin E, AKT involvement in cisplatin chemoresistance of human uterine cancer cells, Gynecol Oncol, 94 (2004) 785–795. [DOI] [PubMed] [Google Scholar]
- [82].Girouard J, Lafleur MJ, Parent S, Leblanc V, Asselin E, Involvement of Akt isoforms in chemoresistance of endometrial carcinoma cells, Gynecol Oncol, 128 (2013) 335–343. [DOI] [PubMed] [Google Scholar]
- [83].Tao K, Yin Y, Shen Q, Chen Y, Li R, Chang W, Bai J, Liu W, Shi L, Zhang P , Akt inhibitor MK-2206 enhances the effect of cisplatin in gastric cancer cells, Biomed Rep, 4 (2016) 365–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Huang WC, Hung MC, Induction of Akt activity by chemotherapy confers acquired resistance, J Formos Med Assoc, 108 (2009) 180–194. [DOI] [PubMed] [Google Scholar]
- [85].Gupta AK, McKenna WG, Weber CN, Feldman MD, Goldsmith JD, Mick R, Machtay M, Rosenthal DI, Bakanauskas VJ, Cerniglia GJ, Bernhard EJ, Weber RS, Muschel RJ, Local recurrence in head and neck cancer: relationship to radiation resistance and signal transduction, Clin Cancer Res, 8 (2002) 885–892. [PubMed] [Google Scholar]
- [86].Kim TJ, Lee JW, Song SY, Choi JJ, Choi CH, Kim BG, Lee JH, Bae DS, Increased expression of pAKT is associated with radiation resistance in cervical cancer, Br J Cancer, 94 (2006) 1678–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Hoeijmakers JH, Genome maintenance mechanisms for preventing cancer, Nature, 411 (2001) 366–374. [DOI] [PubMed] [Google Scholar]
- [88].Iliakis G, Backup pathways of NHEJ in cells of higher eukaryotes: cell cycle dependence, Radiother Oncol, 92 (2009) 310–315. [DOI] [PubMed] [Google Scholar]
- [89].Mladenov E, Magin S, Soni A, Iliakis G, DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy, Front Oncol, 3 (2013) 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Jeggo PA, Geuting V, Lobrich M, The role of homologous recombination in radiation-induced double-strand break repair, Radiother Oncol, 101 (2011) 7–12. [DOI] [PubMed] [Google Scholar]
- [91].Szymonowicz K, Oeck S, Malewicz NM, Jendrossek V, New Insights into Protein Kinase B/Akt Signaling: Role of Localized Akt Activation and Compartment-Specific Target Proteins for the Cellular Radiation Response, Cancers (Basel), 10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Toulany M, DNA Repair Pathways as a Potential Target for Radiosensitization In: Anscher M Va-lerie Keds. Strategies to Enhance the Therapeutic Ratio of Radiation as a Cancer Treatment, Springer; , (2016) 253–287. [Google Scholar]
- [93].Park J, Feng J, Li Y, Hammarsten O, Brazil DP, Hemmings BA, DNA-dependent protein kinase-mediated phosphorylation of protein kinase B requires a specific recognition sequence in the C-terminal hydrophobic motif, J Biol Chem, 284 (2009) 6169–6174. [DOI] [PubMed] [Google Scholar]
- [94].Toulany M, Lee KJ, Fattah KR, Lin YF, Fehrenbacher B, Schaller M, Chen BP, Chen DJ, Rodemann HP, Akt promotes post-irradiation survival of human tumor cells through initiation, progression, and termination of DNA-PKcs-dependent DNA double-strand break repair, Mol Cancer Res, 10 (2012) 945–957. [DOI] [PubMed] [Google Scholar]
- [95].Kao GD, Jiang Z, Fernandes AM, Gupta AK, Maity A, Inhibition of phosphatidylinositol-3-OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation, J Biol Chem, 282 (2007) 21206–21212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Minjgee M, Toulany M, Kehlbach R, Giehl K, Rodemann HP, K-RAS(V12) induces autocrine production of EGFR ligands and mediates radioresistance through EGFR-dependent Akt signaling and activation of DNA-PKcs, Int J Radiat Oncol Biol Phys, 81 (2011) 1506–1514. [DOI] [PubMed] [Google Scholar]
- [97].Oeck S, Al-Refae K, Riffkin H, Wiel G, Handrick R, Klein D, Iliakis G, Jendrossek V, Activating Akt1 mutations alter DNA double strand break repair and radiosensitivity, Sci Rep, 7 (2017) 42700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Toulany M, Kasten-Pisula U, Brammer I, Wang S, Chen J, Dittmann K, Baumann M, Dikomey E, Rodemann HP, Blockage of epidermal growth factor receptor-phosphatidylinositol 3-kinase-AKT signaling increases radiosensitivity of K-RAS mutated human tumor cells in vitro by affecting DNA repair, Clin Cancer Res, 12 (2006) 4119–4126. [DOI] [PubMed] [Google Scholar]
- [99].Lee HJ, Lan L, Peng G, Chang WC, Hsu MC, Wang YN, Cheng CC, Wei L, Nakajima S, Chang SS, Liao HW, Chen CH, Lavin M, Ang KK, Lin SY, Hung MC, Tyrosine 370 phosphorylation of ATM positively regulates DNA damage response, Cell Res, 25 (2015) 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Dittmann K, Mayer C, Fehrenbacher B, Schaller M, Raju U, Milas L, Chen DJ, Kehlbach R, Rodemann HP, Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase, The Journal of biological chemistry, 280 (2005) 31182–31189. [DOI] [PubMed] [Google Scholar]
- [101].Nowsheen S, Cooper T, Stanley JA, Yang ES, Synthetic lethal interactions between EGFR and PARP inhibition in human triple negative breast cancer cells, PLoS One, 7 (2012) e46614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Wang SC, Nakajima Y, Yu YL, Xia W, Chen CT, Yang CC, McIntush EW, Li LY, Hawke DH, Kobayashi R, Hung MC, Tyrosine phosphorylation controls PCNA function through protein stability, Nat Cell Biol, 8 (2006) 1359–1368. [DOI] [PubMed] [Google Scholar]
- [103].Chou RH, Wang YN, Hsieh YH, Li LY, Xia W, Chang WC, Chang LC, Cheng CC, Lai CC, Hsu JL, Chang WJ, Chiang SY, Lee HJ, Liao HW, Chuang PH, Chen HY, Wang HL, Kuo SC, Chen CH, Yu YL, Hung MC, EGFR modulates DNA synthesis and repair through Tyr phosphorylation of histone H4, Dev Cell, 30 (2014) 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Waraky A, Lin Y, Warsito D, Haglund F, Aleem E, Larsson O, Nuclear insulin-like growth factor 1 receptor phosphorylates proliferating cell nuclear antigen and rescues stalled replication forks after DNA damage, J Biol Chem, 292 (2017) 18227–18239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Codony-Servat J, Cuatrecasas M, Asensio E, Montironi C, Martinez-Cardus A, Marin-Aguilera M, Horndler C, Martinez-Balibrea E, Rubini M, Jares P, Reig O, Victoria I, Gaba L, Martin-Richard M, Alonso V, Escudero P, Fernandez-Martos C, Feliu J, Mendez JC, Mendez M, Gallego J, Salud A, Rojo F, Castells A, Prat A, Rosell R, Garcia-Albeniz X, Camps J, Maurel J, Nuclear IGF-1R predicts chemotherapy and targeted therapy resistance in metastatic colorectal cancer, Br J Cancer, 117 (2017) 1777–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Gomez-Roman N, Chong MY, Chahal SK, Caragher SP, Jackson MR, Stevenson KH, Dongre SA, Chalmers AJ, Radiation responses of 2D and 3D glioblastoma cells: a novel, 3D-specific radioprotective role of VEGF/Akt signaling through functional activation of NHEJ, Mol Cancer Ther, (2019). [DOI] [PubMed] [Google Scholar]
- [107].Bussink J, van der Kogel AJ, Kaanders JH, Activation of the PI3-K/AKT pathway and implications for radioresistance mechanisms in head and neck cancer, The lancet oncology, 9 (2008) 288–296. [DOI] [PubMed] [Google Scholar]
- [108].Toulany M, Dittmann K, Kruger M, Baumann M, Rodemann HP, Radioresistance of K-Ras mutated human tumor cells is mediated through EGFR-dependent activation of PI3K-AKT pathway, Radiother Oncol, 76 (2005) 143–150. [DOI] [PubMed] [Google Scholar]
- [109].Zhan M, Han ZC, Phosphatidylinositide 3-kinase/AKT in radiation responses, Histol Histopathol, 19 (2004) 915–923. [DOI] [PubMed] [Google Scholar]
- [110].Liu Y, Cui B, Qiao Y, Zhang Y, Tian Y, Jiang J, Ma D, Kong B, Phosphoinositide-3-kinase inhibition enhances radiosensitization of cervical cancer in vivo, International journal of gynecological cancer : official journal of the International Gynecological Cancer Society, 21 (2011) 100–105. [DOI] [PubMed] [Google Scholar]
- [111].Mueck K, Rebholz S, Harati MD, Rodemann HP, Toulany M, Akt1 Stimulates Homologous Recombination Repair of DNA Double-Strand Breaks in a Rad51-Dependent Manner, Int J Mol Sci, 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Philip CA, Laskov I, Beauchamp MC, Marques M, Amin O, Bitharas J, Kessous R, Kogan L, Baloch T, Gotlieb WH, Yasmeen A, Inhibition of PI3K-AKT-mTOR pathway sensitizes endometrial cancer cell lines to PARP inhibitors, BMC Cancer, 17 (2017) 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Villafanez F, Garcia IA, Carbajosa S, Pansa MF, Mansilla S, Llorens MC, Angiolini V, Guantay L, Jacobs H, Madauss KP, Gloger I, Gottifredi V, Bocco JL, Soria G, AKT inhibition impairs PCNA ubiquitylation and triggers synthetic lethality in homologous recombination-deficient cells submitted to replication stress, Oncogene, 38 (2019) 4310–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Nagel R, Avelar AT, Aben N, Proost N, van de Ven M, van der Vliet J, Cozijnsen M, de Vries H, Wessels LFA, Berns A, Inhibition of the Replication Stress Response Is a Synthetic Vulnerability in SCLC That Acts Synergistically in Combination with Cisplatin, Mol Cancer Ther, 18 (2019) 762–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Xuan Nguyen TL, Choi JW, Lee SB, Ye K, Woo SD, Lee KH, Ahn JY, Akt phosphorylation is essential for nuclear translocation and retention in NGF-stimulated PC12 cells, Biochem Biophys Res Commun, 349 (2006) 789–798. [DOI] [PubMed] [Google Scholar]
- [116].Lim MA, Kikani CK, Wick MJ, Dong LQ, Nuclear translocation of 3’-phosphoinositide-dependent protein kinase 1 (PDK-1): a potential regulatory mechanism for PDK-1 function, Proc Natl Acad Sci U S A, 100 (2003) 14006–14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Metjian A, Roll RL, Ma AD, Abrams CS, Agonists cause nuclear translocation of phosphatidylinositol 3-kinase gamma. A Gbetagamma-dependent pathway that requires the p110gamma amino terminus, J Biol Chem, 274 (1999) 27943–27947. [DOI] [PubMed] [Google Scholar]
- [118].Rubio M, Avitabile D, Fischer K, Emmanuel G, Gude N, Miyamoto S, Mishra S, Schaefer EM, Brown JH, Sussman MA, Cardioprotective stimuli mediate phosphoinositide 3-kinase and phosphoinositide dependent kinase 1 nuclear accumulation in cardiomyocytes, J Mol Cell Cardiol, 47 (2009) 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Jacobsen K, Bertran-Alamillo J, Molina MA, Teixido C, Karachaliou N, Pedersen MH, Castellvi J, Garzon M, Codony-Servat C, Codony-Servat J, Gimenez-Capitan A, Drozdowskyj A, Viteri S, Larsen MR, Lassen U, Felip E, Bivona TG, Ditzel HJ, Rosell R, Convergent Akt activation drives acquired EGFR inhibitor resistance in lung cancer, Nat Commun, 8 (2017) 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Lee YY, Kim HP, Kang MJ, Cho BK, Han SW, Kim TY, Yi EC, Phosphoproteomic analysis identifies activated MET-axis PI3K/AKT and MAPK/ERK in lapatinib-resistant cancer cell line, Exp Mol Med, 45 (2013) e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Stuhlmiller TJ, Miller SM, Zawistowski JS, Nakamura K, Beltran AS, Duncan JS, Angus SP, Collins KA, Granger DA, Reuther RA, Graves LM, Gomez SM, Kuan PF, Parker JS, Chen X, Sciaky N, Carey LA, Earp HS, Jin J, Johnson GL, Inhibition of Lapatinib-Induced Kinome Reprogramming in ERBB2-Positive Breast Cancer by Targeting BET Family Bromodomains, Cell Rep, 11 (2015) 390–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, Gondi V, Hsu KT, Harari PM, Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members, Oncogene, 27 (2008) 3944–3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Iida M, Brand TM, Campbell DA, Starr MM, Luthar N, Traynor AM, Wheeler DL, Targeting AKT with the allosteric AKT inhibitor MK-2206 in non-small cell lung cancer cells with acquired resistance to cetuximab, Cancer Biol Ther, 14 (2013) 481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Giles KM, Kalinowski FC, Candy PA, Epis MR, Zhang PM, Redfern AD, Stuart LM, Goodall GJ, Leedman PJ, Axl mediates acquired resistance of head and neck cancer cells to the epidermal growth factor receptor inhibitor erlotinib, Mol Cancer Ther, 12 (2013) 2541–2558. [DOI] [PubMed] [Google Scholar]
- [125].Martinho O, Zucca LE, Reis RM, AXL as a modulator of sunitinib response in glioblastoma cell lines, Exp Cell Res, 332 (2015) 1–10. [DOI] [PubMed] [Google Scholar]
- [126].Hemstrom TH, Sandstrom M, Zhivotovsky B, Inhibitors of the PI3-kinase/Akt pathway induce mitotic catastrophe in non-small cell lung cancer cells, Int J Cancer, 119 (2006) 1028–1038. [DOI] [PubMed] [Google Scholar]
- [127].Hou H, Zhang Y, Huang Y, Yi Q, Lv L, Zhang T, Chen D, Hao Q, Shi Q, Inhibitors of phosphatidylinositol 3’-kinases promote mitotic cell death in HeLa cells, PLoS One, 7 (2012) e35665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Cherrin C, Haskell K, Howell B, Jones R, Leander K, Robinson R, Watkins A, Bilodeau M, Hoffman J, Sanderson P, Hartman G, Mahan E, Prueksaritanont T, Jiang G, She QB, Rosen N, Sepp-Lorenzino L, Defeo-Jones D, Huber HE, An allosteric Akt inhibitor effectively blocks Akt signaling and tumor growth with only transient effects on glucose and insulin levels in vivo, Cancer Biol Ther, 9 (2010) 493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Davies BR, Greenwood H, Dudley P, Crafter C, Yu DH, Zhang J, Li J, Gao B, Ji Q, Maynard J, Ricketts SA, Cross D, Cosulich S, Chresta CC, Page K, Yates J, Lane C, Watson R, Luke R, Ogilvie D, Pass M, Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background, Mol Cancer Ther, 11 (2012) 873–887. [DOI] [PubMed] [Google Scholar]
- [130].Davies BR, Guan N, Logie A, Crafter C, Hanson L, Jacobs V, James N, Dudley P, Jacques K, Ladd B, D’Cruz CM, Zinda M, Lindemann J, Kodaira M, Tamura K, Jenkins EL, Tumors with AKT1E17K Mutations Are Rational Targets for Single Agent or Combination Therapy with AKT Inhibitors, Mol Cancer Ther, 14 (2015) 2441–2451. [DOI] [PubMed] [Google Scholar]
- [131].Tamura K, Hashimoto J, Tanabe Y, Kodaira M, Yonemori K, Seto T, Hirai F, Arita S, Toyokawa G, Chen L, Yamamoto H, Kawata T, Lindemann J, Esaki T, Safety and tolerability of AZD5363 in Japanese patients with advanced solid tumors, Cancer Chemother Pharmacol, 77 (2016) 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Banerji U, Dean EJ, Perez-Fidalgo JA, Batist G, Bedard PL, You B, Westin SN, Kabos P, Garrett MD, Tall M, Ambrose H, Barrett JC, Carr TH, Cheung SYA, Corcoran C, Cullberg M, Davies BR, de Bruin EC, Elvin P, Foxley A, Lawrence P, Lindemann JPO, Maudsley R, Pass M, Rowlands V, Rugman P, Schiavon G, Yates J, Schellens JHM, A Phase I Open-Label Study to Identify a Dosing Regimen of the Pan-AKT Inhibitor AZD5363 for Evaluation in Solid Tumors and in PIK3CA-Mutated Breast and Gynecologic Cancers, Clin Cancer Res, 24 (2018) 2050–2059. [DOI] [PubMed] [Google Scholar]
- [133].Saura C, Roda D, Rosello S, Oliveira M, Macarulla T, Perez-Fidalgo JA, Morales-Barrera R, Sanchis-Garcia JM, Musib L, Budha N, Zhu J, Nannini M, Chan WY, Sanabria Bohorquez SM, Meng RD, Lin K, Yan Y, Patel P, Baselga J, Tabernero J, Cervantes A, A First-in-Human Phase I Study of the ATP-Competitive AKT Inhibitor Ipatasertib Demonstrates Robust and Safe Targeting of AKT in Patients with Solid Tumors, Cancer Discov, 7 (2017) 102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Tolcher AW, Khan K, Ong M, Banerji U, Papadimitrakopoulou V, Gandara DR, Patnaik A, Baird RD, Olmos D, Garrett CR, Skolnik JM, Rubin EH, Smith PD, Huang P, Learoyd M, Shannon KA, Morosky A, Tetteh E, Jou YM, Papadopoulos KP, Moreno V, Kaiser B, Yap TA, Yan L, de Bono JS, Antitumor activity in RAS-driven tumors by blocking AKT and MEK, Clin Cancer Res, 21 (2015) 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Ma CX, Suman V, Goetz MP, Northfelt D, Burkard ME, Ademuyiwa F, Naughton M, Margenthaler J, Aft R, Gray R, Tevaarwerk A, Wilke L, Haddad T, Moynihan T, Loprinzi C, Hieken T, Barnell EK, Skidmore ZL, Feng YY, Krysiak K, Hoog J, Guo Z, Nehring L, Wisinski KB, Mardis E, Hagemann IS, Vij K, Sanati S, Al-Kateb H, Griffith OL, Griffith M, Doyle L, Erlichman C, Ellis MJ, A Phase II Trial of Neoadjuvant MK-2206, an AKT Inhibitor, with Anastrozole in Clinical Stage II or III PIK3CA-Mutant ER-Positive and HER2-Negative Breast Cancer, Clin Cancer Res, 23 (2017) 6823–6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Do K, Speranza G, Bishop R, Khin S, Rubinstein L, Kinders RJ, Datiles M, Eugeni M, Lam MH, Doyle LA, Doroshow JH, Kummar S, Biomarker-driven phase 2 study of MK-2206 and selumetinib (AZD6244, ARRY-142886) in patients with colorectal cancer, Invest New Drugs, 33 (2015) 720–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Gills JJ, Dennis PA, Perifosine: update on a novel Akt inhibitor, Curr Oncol Rep, 11 (2009) 102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Matsumoto K, Shichino H, Kawamoto H, Kosaka Y, Chin M, Kato K, Mugishima H, Phase I study of perifosine monotherapy in patients with recurrent or refractory neuroblastoma, Pediatr Blood Cancer, 64 (2017). [DOI] [PubMed] [Google Scholar]
- [139].Kushner BH, Cheung NV, Modak S, Becher OJ, Basu EM, Roberts SS, Kramer K, Dunkel IJ, A phase I/Ib trial targeting the Pi3k/Akt pathway using perifosine: Long-term progression-free survival of patients with resistant neuroblastoma, Int J Cancer, 140 (2017) 480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Hasegawa K, Kagabu M, Mizuno M, Oda K, Aoki D, Mabuchi S, Kamiura S, Yamaguchi S, Aoki Y, Saito T, Yunokawa M, Takehara K, Okamoto A, Ochiai K, Kimura T, Phase II basket trial of perifosine monotherapy for recurrent gynecologic cancer with or without PIK3CA mutations, Invest New Drugs, 35 (2017) 800–812. [DOI] [PubMed] [Google Scholar]
- [141].Vink SR, Schellens JH, Beijnen JH, Sindermann H, Engel J, Dubbelman R, Moppi G, Hillebrand MJ, Bartelink H, Verheij M, Phase I and pharmacokinetic study of combined treatment with perifosine and radiation in patients with advanced solid tumours, Radiother Oncol, 80 (2006) 207–213. [DOI] [PubMed] [Google Scholar]
- [142].Bendell JC, Nemunaitis J, Vukelja SJ, Hagenstad C, Campos LT, Hermann RC, Sportelli P, Gardner L, Richards DA, Randomized placebo-controlled phase II trial of perifosine plus capecitabine as second- or third-line therapy in patients with metastatic colorectal cancer, J Clin Oncol, 29 (2011) 4394–4400. [DOI] [PubMed] [Google Scholar]
- [143].Holler M, Grottke A, Mueck K, Manes J, Jucker M, Rodemann HP, Toulany M, Dual Targeting of Akt and mTORC1 Impairs Repair of DNA Double-Strand Breaks and Increases Radiation Sensitivity of Human Tumor Cells, PLoS One, 11 (2016) e0154745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Becher OJ, Gilheeney SW, Khakoo Y, Lyden DC, Haque S, De Braganca KC, Kolesar JM, Huse JT, Modak S, Wexler LH, Kramer K, Spasojevic I, Dunkel IJ, A phase I study of perifosine with temsirolimus for recurrent pediatric solid tumors, Pediatr Blood Cancer, 64 (2017). [DOI] [PubMed] [Google Scholar]
- [145].Kaley TJ, Panageas KS, Mellinghoff IK, Nolan C, Gavrilovic IT, DeAngelis LM, Abrey LE, Holland EC, Lassman AB, Phase II trial of an AKT inhibitor (perifosine) for recurrent glioblastoma, J Neurooncol, 144 (2019) 403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Gupta AK, Cerniglia GJ, Mick R, McKenna WG, Muschel RJ, HIV protease inhibitors block Akt signaling and radiosensitize tumor cells both in vitro and in vivo, Cancer Res, 65 (2005) 8256–8265. [DOI] [PubMed] [Google Scholar]
- [147].Wilson JM, Fokas E, Dutton SJ, Patel N, Hawkins MA, Eccles C, Chu KY, Durrant L, Abraham AG, Partridge M, Woodward M, O’Neill E, Maughan T, McKenna WG, Mukherjee S, Brunner TB, ARCII: A phase II trial of the HIV protease inhibitor Nelfinavir in combination with chemoradiation for locally advanced inoperable pancreatic cancer, Radiother Oncol, 119 (2016) 306–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Hill EJ, Roberts C, Franklin JM, Enescu M, West N, MacGregor TP, Chu KY, Boyle L, Blesing C, Wang LM, Mukherjee S, Anderson EM, Brown G, Dutton S, Love SB, Schnabel JA, Quirke P, Muschel R, McKenna WG, Partridge M, Sharma RA, Clinical Trial of Oral Nelfinavir before and during Radiation Therapy for Advanced Rectal Cancer, Clin Cancer Res, 22 (2016) 1922–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Buijsen J, Lammering G, Jansen RL, Beets GL, Wals J, Sosef M, Den Boer MO, Leijtens J, Riedl RG, Theys J, Lambin P, Phase I trial of the combination of the Akt inhibitor nelfinavir and chemoradiation for locally advanced rectal cancer, Radiother Oncol, 107 (2013) 184–188. [DOI] [PubMed] [Google Scholar]
- [150].Rengan R, Mick R, Pryma D, Rosen MA, Lin LL, Maity AM, Evans TL, Stevenson JP, Langer CJ, Kucharczuk J, Friedberg J, Prendergast S, Sharkoski T, Hahn SM, A phase I trial of the HIV protease inhibitor nelfinavir with concurrent chemoradiotherapy for unresectable stage IIIA/IIIB non-small cell lung cancer: a report of toxicities and clinical response, J Thorac Oncol, 7 (2012) 709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Brunner TB, Geiger M, Grabenbauer GG, Lang-Welzenbach M, Mantoni TS, Cavallaro A, Sauer R, Hohenberger W, McKenna WG, Phase I trial of the human immunodeficiency virus protease inhibitor nelfinavir and chemoradiation for locally advanced pancreatic cancer, J Clin Oncol, 26 (2008) 2699–2706. [DOI] [PubMed] [Google Scholar]
- [152].Crabb SJ, Birtle AJ, Martin K, Downs N, Ratcliffe I, Maishman T, Ellis M, Griffiths G, Thompson S, Ksiazek L, Khoo V, Jones RJ, ProCAID: a phase I clinical trial to combine the AKT inhibitor AZD5363 with docetaxel and prednisolone chemotherapy for metastatic castration resistant prostate cancer, Invest New Drugs, 35 (2017) 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].de Bono JS, De Giorgi U, Rodrigues DN, Massard C, Bracarda S, Font A, Arranz Arija JA, Shih KC, Radavoi GD, Xu N, Chan WY, Ma H, Gendreau S, Riisnaes R, Patel PH, Maslyar DJ, Jinga V, Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss, Clin Cancer Res, 25 (2019) 928–936. [DOI] [PubMed] [Google Scholar]
- [154].Kim SB, Dent R, Im SA, Espie M, Blau S, Tan AR, Isakoff SJ, Oliveira M, Saura C, Wongchenko MJ, Kapp AV, Chan WY, Singel SM, Maslyar DJ, Baselga J, investigators L, Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial, Lancet Oncol, 18 (2017) 1360–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Molife LR, Yan L, Vitfell-Rasmussen J, Zernhelt AM, Sullivan DM, Cassier PA, Chen E, Biondo A, Tetteh E, Siu LL, Patnaik A, Papadopoulos KP, de Bono JS, Tolcher AW, Minton S, Phase 1 trial of the oral AKT inhibitor MK-2206 plus carboplatin/paclitaxel, docetaxel, or erlotinib in patients with advanced solid tumors, J Hematol Oncol, 7 (2014) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Gonzalez-Angulo AM, Krop I, Akcakanat A, Chen H, Liu S, Li Y, Culotta KS, Tarco E, Piha-Paul S, Moulder-Thompson S, Velez-Bravo V, Sahin AA, Doyle LA, Do KA, Winer EP, Mills GB, Kurzrock R, Meric-Bernstam F, SU2C phase Ib study of paclitaxel and MK-2206 in advanced solid tumors and metastatic breast cancer, J Natl Cancer Inst, 107 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Ma CX, Sanchez C, Gao F, Crowder R, Naughton M, Pluard T, Creekmore A, Guo Z, Hoog J, Lockhart AC, Doyle A, Erlichman C, Ellis MJ, A Phase I Study of the AKT Inhibitor MK-2206 in Combination with Hormonal Therapy in Postmenopausal Women with Estrogen Receptor-Positive Metastatic Breast Cancer, Clin Cancer Res, 22 (2016) 2650–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Hervieu A, Kermorgant S, The Role of PI3K in Met Driven Cancer: A Recap, Front Mol Biosci, 5 (2018) 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Song Q, Sun X, Guo H, Yu Q, Concomitant inhibition of receptor tyrosine kinases and downstream AKT synergistically inhibited growth of KRAS/BRAF mutant colorectal cancer cells, Oncotarget, 8 (2017) 5003–5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Feng S, Shao L, Castro P, Coleman I, Nelson PS, Smith PD, Davies BR, Ittmann M, Combination treatment of prostate cancer with FGF receptor and AKT kinase inhibitors, Oncotarget, 8 (2017) 6179–6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Crafter C, Vincent JP, Tang E, Dudley P, James NH, Klinowska T, Davies BR, Combining AZD8931, a novel EGFR/HER2/HER3 signalling inhibitor, with AZD5363 limits AKT inhibitor induced feedback and enhances antitumour efficacy in HER2-amplified breast cancer models, Int J Oncol, 47 (2015) 446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, Cowey CL, Schadendorf D, Wagstaff J, Dummer R, Ferrucci PF, Smylie M, Hogg D, Hill A, Marquez-Rodas I, Haanen J, Guidoboni M, Maio M, Schoffski P, Carlino MS, Lebbe C, McArthur G, Ascierto PA, Daniels GA, Long GV, Bastholt L, Rizzo JI, Balogh A, Moshyk A, Hodi FS, Wolchok JD, Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma, N Engl J Med, 381 (2019) 1535–1546. [DOI] [PubMed] [Google Scholar]
- [163].Kasikara C, Davra V, Calianese D, Geng K, Spires TE, Quigley M, Wichroski M, Sriram G, Suarez-Lopez L, Yaffe MB, Kotenko SV, De Lorenzo MS, Birge RB, Pan-TAM Tyrosine Kinase Inhibitor BMS-777607 Enhances Anti-PD-1 mAb Efficacy in a Murine Model of Triple-Negative Breast Cancer, Cancer Res, 79 (2019) 2669–2683. [DOI] [PubMed] [Google Scholar]
- [164].Kasikara C, Kumar S, Kimani S, Tsou WI, Geng K, Davra V, Sriram G, Devoe C, Nguyen KN, Antes A, Krantz A, Rymarczyk G, Wilczynski A, Empig C, Freimark B, Gray M, Schlunegger K, Hutchins J, Kotenko SV, Birge RB, Phosphatidylserine Sensing by TAM Receptors Regulates AKT-Dependent Chemoresistance and PD-L1 Expression, Mol Cancer Res, 15 (2017) 753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Kim JE, Kim Y, Li G, Kim ST, Kim K, Park SH, Park JO, Park YS, Lim HY, Lee H, Sohn TS, Kim KM, Kang WK, Lee J, MerTK inhibition by RXDX-106 in MerTK activated gastric cancer cell lines, Oncotarget, 8 (2017) 105727–105734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Yokoyama Y, Lew ED, Seelige R, Tindall EA, Walsh C, Fagan PC, Lee JY, Nevarez R, Oh J, Tucker KD, Chen M, Diliberto A, Vaaler H, Smith KM, Albert A, Li G, Bui JD, Immuno-oncological Efficacy of RXDX-106, a Novel TAM (TYRO3, AXL, MER) Family Small-Molecule Kinase Inhibitor, Cancer Res, 79 (2019) 1996–2008. [DOI] [PubMed] [Google Scholar]
- [167].Lastwika KJ, Wilson W 3rd, Li QK, Norris J, Xu H, Ghazarian SR, Kitagawa H, Kawabata S, Taube JM, Yao S, Liu LN, Gills JJ, Dennis PA, Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer, Cancer Res, 76 (2016) 227–238. [DOI] [PubMed] [Google Scholar]
- [168].Restifo NP, Smyth MJ, Snyder A, Acquired resistance to immunotherapy and future challenges, Nat Rev Cancer, 16 (2016) 121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].O’Donnell JS, Massi D, Teng MWL, Mandala M, PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux, Semin Cancer Biol, 48 (2018) 91–103. [DOI] [PubMed] [Google Scholar]
- [170].Fares CM, Van Allen EM, Drake CG, Allison JP, Hu-Lieskovan S, Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients?, Am Soc Clin Oncol Educ Book, 39 (2019) 147–164. [DOI] [PubMed] [Google Scholar]