

Abstract
Myoepithelial tumors (MET) represent a clinicopathologically heterogeneous group of tumors, ranging from benign to highly aggressive lesions. Although MET arising in soft tissue, bone or viscera share morphologic and immunophenotypic overlap with their salivary gland and cutaneous counterparts, there is still controversy regarding their genetic relationship. Half of MET of soft tissue and bone harbor EWSR1 or FUS related fusions, while MET arising in the salivary gland and skin often show PLAG1 and HMGA2 gene rearrangements. Regardless of the site of origin, the gold standard in diagnosing a MET relies on demonstrating its ‘myoepithelial immunophenotype’ of positivity for EMA/CK and S100 protein or GFAP. However, the morphologic spectrum of MET in soft tissue and bone is quite broad and the above immunoprofile is non-specific, being shared by other pathogenetically unrelated neoplasms. Moreover, rare MET lack a diagnostic immunoprofile but show instead the characteristic gene fusions. In this study we analyzed a large cohort of 66 genetically confirmed MET spanning various clinical presentations, to better define their morphologic spectrum and establish relevant pathologic-molecular correlations. Genetic analysis was carried out by FISH for EWSR1/FUS rearrangements and potential partners, and/or by targeted RNA sequencing. 82% showed EWSR1 rearrangement, while 18% had FUS abnormalities. EWSR1-POU5F1 occurred with predilection in malignant MET in children and young adults and these tumors had nested epithelioid morphology and clear cytoplasm. In contrast, EWSR1/FUS-PBX1/3 fusions were associated with benign and sclerotic spindle cell morphology. Tumors with EWSR1-KLF17 showed chordoma-like morphology. Our results demonstrate striking morphologic-molecular correlations in MET of bone, soft tissue and viscera, which might have implications in their clinical behavior.
Keywords: myoepithelial tumors, EWSR1, FUS, POU5F1, PBX1, PBX3
1. INTRODUCTION
The spectrum of myoepithelial tumors (MET) represents a family of lesions with variable terminology, which based on anatomic location and the presence of ductal structures are designated as pleomorphic adenoma in the salivary gland and benign mixed tumor in the skin or soft tissue1,2. Tumors lacking ducts or an overt biphasic epithelial-myoepithelial phenotype are often designated as myoepithelioma or myoepithelial tumor at various sites, being composed of pure populations of epithelioid or spindle cells embedded in variable stromal components. Moreover, the MET arising in soft tissue and bone lack a corresponding cell of origin, compared to the lesions from salivary gland or skin, which appear to relate to the normal myoepithelial cells surrounding tubulo-acinar glandular structures3. Furthermore, significant differences exist between assessing risk of malignancy of MET at various anatomic sites; salivary gland malignant MET are defined based on the extent of capsular invasion4,5, while malignancy in soft tissue MET relates to nuclear pleomorphism and mitotic activity1. To complicate things further, the emerging genetic signatures of MET from different sites have suggested a dichotomy of molecular events, with PLAG1 and HMGA2 gene abnormalities being prevalent in salivary gland and skin6,7, while in bone, soft tissue and other viscera recurrent EWSR1 and FUS related gene fusions are detected in half of MET8. Despite this conflicting evidence toward a unified family of MET, the current diagnostic criteria have been rather homogeneous across different clinical presentations and molecular phenotypes, relying on demonstrating the so-called ‘myoepithelial immunophenotype’. This immunoprofile consists of co-expression of cytokeratins (CK) or epithelial membrane antigen (EMA) in combination with S100 protein, calponin or glial fibrillary acidic protein (GFAP). If in the salivary gland location these diagnostic criteria appear reliable, in soft tissue and bone MET the above immunoprofile is far from specific, encompassing other look-alike mesenchymal neoplasms2,9. Furthermore, the pathogenetic relationship between the fusion-positive and fusion-negative cohorts of MET sharing similar histology and immunoprofile has not been yet elucidated. It is possible that some of the molecularly negative cases might represent alternative diagnoses, which may be inappropriately lumped together based on histologic or immunohistochemical grounds.
In this study we evaluate the clinicopathologic features of a large cohort of MET selected based on their positive gene fusion signature, spanning a broad spectrum of morphologies and clinical presentations. We further sought to investigate whether fusion type correlates with histologic features, myoepithelial marker expression, and histologic grade.
2. MATERIAL AND METHODS
We retrieved 66 molecularly confirmed MET from the consultation files of the authors (CRA, CDF, BD), 30 of which were included in earlier publications8,10,11. The molecular results were obtained by FISH in 50 cases. In 13 cases the molecular diagnosis was established by one of the following platforms of targeted sequencing, including 9 cases using targeted RNA sequencing, 2 cases by Archer and 2 cases being tested by Foundation One. In 3 cases the diagnosis of a MET was confirmed by conventional karyotyping results. When material was available, FISH was also performed to confirm the RNA sequencing or karyotype results.
Clinical data, including age, gender, and anatomic site, as well as IHC results were retrieved from pathology reports. Hematoxylin and eosin-stained slides from resection specimens were re-reviewed by two of us (AS, CRA). For each tumor, special emphasis was put on the cytomorphology of myoepithelial cells (epithelioid, plasmacytoid, spindle, small cell), histologic growth pattern (solid sheets, nests, reticular pattern, fascicular growth) and presence of stromal changes (myxoid, myxohyaline, sclerotic)3,12. Criteria for malignancy included moderate to severe nuclear atypia (nuclear enlargement and hyperchromasia), easily discerned prominent nucleoli, and mitotic activity, typically defined as ≥5 MF/10 HPFs1,12. In most cases immunohistochemical stains for CK, EMA, S100 and GFAP were available for review, however, more than half of the cases had a much wider spectrum of immunostains performed, including markers for smooth muscle differentiation such as calponin, SMA, etc. As the 4 immunostains mentioned above are the most sensitive markers defining myoepithelial phenotype in soft tissue and bone anatomic sites, and most cases in the current series had these results available, we only included these markers. Although most cases fulfilled the previous pre-requisite criteria of a positive ‘myoepithelial immunoprofile’, the few cases that fell short of this requirement were not excluded, as our main inclusion criteria was the confirmed molecular signature. However, cutaneous myoepithelial tumors with EWSR1-PBX3 fusions diagnostic of syncytial myoepithelioma and benign cutaneous myoepithelial tumors/ benign mixed tumors with PLAG1/HMGA2 gene rearrangements were not included in this investigation.
2.1. Fluorescence In Situ Hybridization.
FISH was used on FFPE sections for the examination of EWSR1, FUS, POU5F1, PBX1, PBX3, ZNF444, KLF15 or KLF17 gene rearrangements8,10. Tumors were first tested for EWSR1 gene rearrangements, and if negative were subsequently tested for FUS gene abnormalities. MET positive for EWSR1 and FUS rearrangements were further tested to interrogate a potential fusion partner, including: POU5F1, PBX1, PBX3, ZNF444, KLF15 and KLF17. If no partner was detected, further FISH testing was performed in most cases to exclude alternative diagnoses, including abnormalities in FLI1, ERG, NR4A3, ATF1, CREB1, and CREM genes. Custom probes made by bacterial artificial chromosomes (BAC) clones flanking the genes of interest according to UCSC genome browser (http://genome.ucsc.edu) and obtained from BACPAC sources of Children’s Hospital of Oakland Research Institute (Oakland, CA; http://bacpac.chori.org). DNA from each BAC was isolated according to the manufacturer’s instructions. The BAC clones were labeled with fluorochromes (fluorescent-labeled dUTPs, Enzo Life Sciences, New York, NY) by nick translation and validated on normal metaphase chromosomes. The 4 μm-thick FFPE slides were deparaffinized, pretreated, and hybridized with denatured probes. After overnight incubation, the slides were washed, stained with 4’,6-diamidino-2-phenylindole, mounted with an antifade solution, and then examined on a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany) controlled by Isis 5 software (Metasystems).
2.2. Targeted RNA sequencing.
In 8 cases analyzed by targeted RNA sequencing, RNA was extracted from FFPE tissue using Amsbio’s ExpressArt FFPE Clear RNA Ready kit (Amsbio LLC, Cambridge, MA). The fragment length was assessed with an RNA 6000 chip on an Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA). RNA-seq libraries were prepared using 20 to 100 ng total RNA with the TruSight RNA Fusion Panel (Illumina, San Diego, CA). Targeted RNA sequencing was performed on an Illumina MiSeq platform. Reads were independently aligned with STAR (version 2.3) against the human reference genome (hg19) and analyzed by STAR-Fusion.
2.3. Anchored Multiplex RNA Sequencing (Archer Dx).
The detailed procedure for the two cases studied by Anchored Multiplex RNA sequencing assay has been previously described13. In short, unidirectional gene-specific primers were designed to target specific exons in 62 genes known to be involved in oncogenic fusions in solid tumors. In brief, RNA was extracted from formalin-fixed paraffin embedded (FFPE) specimens, followed by cDNA synthesis and library preparation. Anchored Multiplex polymerase chain reaction amplicons were sequenced on Illumina Miseq, and the data was analyzed using the Archer software.
3. RESULTS
The current series included 66 MET which were positive for gene rearrangements or gene fusions typically seen in MET. There were 38 females and 28 males, with a wide age range at diagnosis (1–64 years; median age 27 years). About one-third of patients (n=21) were younger than the age of 18, while nine patients were older than 50 years of age. Cases had a wide anatomic distribution, with 48 tumors arising in diverse soft tissue locations, of which 30 were extremity-based, 7 occurred in the trunk, and 5 located in the head and neck. Twelve tumors presented as primary bone tumors, of which 6 originated in long bones, 2 in pelvic bones, 2 in the mandible and one each in the vertebral body and navicular bone. Six tumors were located in visceral organs, including 4 in the lung, and one each in the kidney and urinary bladder.
By FISH, targeted RNA sequencing, or cytogenetics, EWSR1 rearrangement was found in the large majority of MET, occurring in 54 (82%) cases, while FUS rearrangement was detected in 12 (18%) cases. The EWSR1 fusion partners included POU5F1 (n=15, 28%), PBX3 (n=10, 19%), PBX1 (n=6, 11%), ZNF444 (n=3, 6%), KLF15 (n=2, 4%), and KLF17 (n=1, 2%). In 17 (31%) cases no EWSR1 partner was identified. Among the 12 MET cases with FUS gene rearrangements, only two partners were identified, including the most common KLF17 (n=8, 67%) gene and less frequently POU5F1 (n=2, 17%) (Fig 1). No fusion partner was detected in 2 MET with FUS rearrangements.
Figure 1.

Distribution of gene fusions in the present cohort of 66 MET of soft tissue, bone, and visceral organs.
Microscopically, the MET lesions included in this study showed a heterogenous morphologic spectrum, often composed of epithelioid to ovoid cells embedded in a variable amount of myxoid or collagen-rich stroma (Fig 2). At low power, the tumors often displayed a multinodular growth pattern, with mostly well-defined, non-encapsulated borders. Rare cases showed a more infiltrative growth pattern within subcutis or muscle. The degree of cellularity varied significantly between cases and between different areas within one lesion, which inversely correlated with the amount of stromal component. Thus, some cases resembled primitive undifferentiated round cell tumors, while others were deceptively bland, hypocellular and markedly fibrotic. Although most tumors had a predominant epithelioid phenotype, arranged in cords, nests and sheets, at least focal areas of cells with ovoid or short spindle cells with palely eosinophilic cytoplasm organized in vague fascicles were also noted. At higher power, the epithelioid cells showed moderate amounts of pale to densely eosinophilic cytoplasm and round, often eccentric uniform nuclei, with fine chromatin and only a mild to moderate degree of pleomorphism. Infrequent patterns included foci of more basaloid epithelioid cells with scant cytoplasm (15%) and plasmacytoid (10%) morphology. A consistent feature seen in most MET was the presence of a prominent but variable extracellular matrix, alternating from densely sclerotic, hyalinized, fibromyxoid, myxochondroid or purely myxoid. None of the lesions displayed ducts or glandular structures. Notably, several of these histologic patterns were associated with certain gene fusions; these phenotype-genotype correlations are summarized below. In this series, the number of malignant MET (n=30) was only slightly smaller than the MET with benign histology (n=36), undoubtedly reflecting referral bias of diagnostically challenging malignant examples. Typical features of malignancy were moderate to severe cytonuclear atypia and prominent nucleoli, whereas mitoses were often ≥ 5 / 10 HPFs. Necrosis was only rarely observed, almost exclusively in tumors with undifferentiated morphology. The majority of tumors (n=51; 81%) expressed S100 or GFAP in combination with EMA or cytokeratin. Six (10%) MET expressed either only epithelial markers or only S100, whereas 6 MET lacked all four myoepithelial markers. In 3 cases the IHC data were not available.
Figure 2. Morphologic spectrum of MET with POU5F1 gene rearrangements.
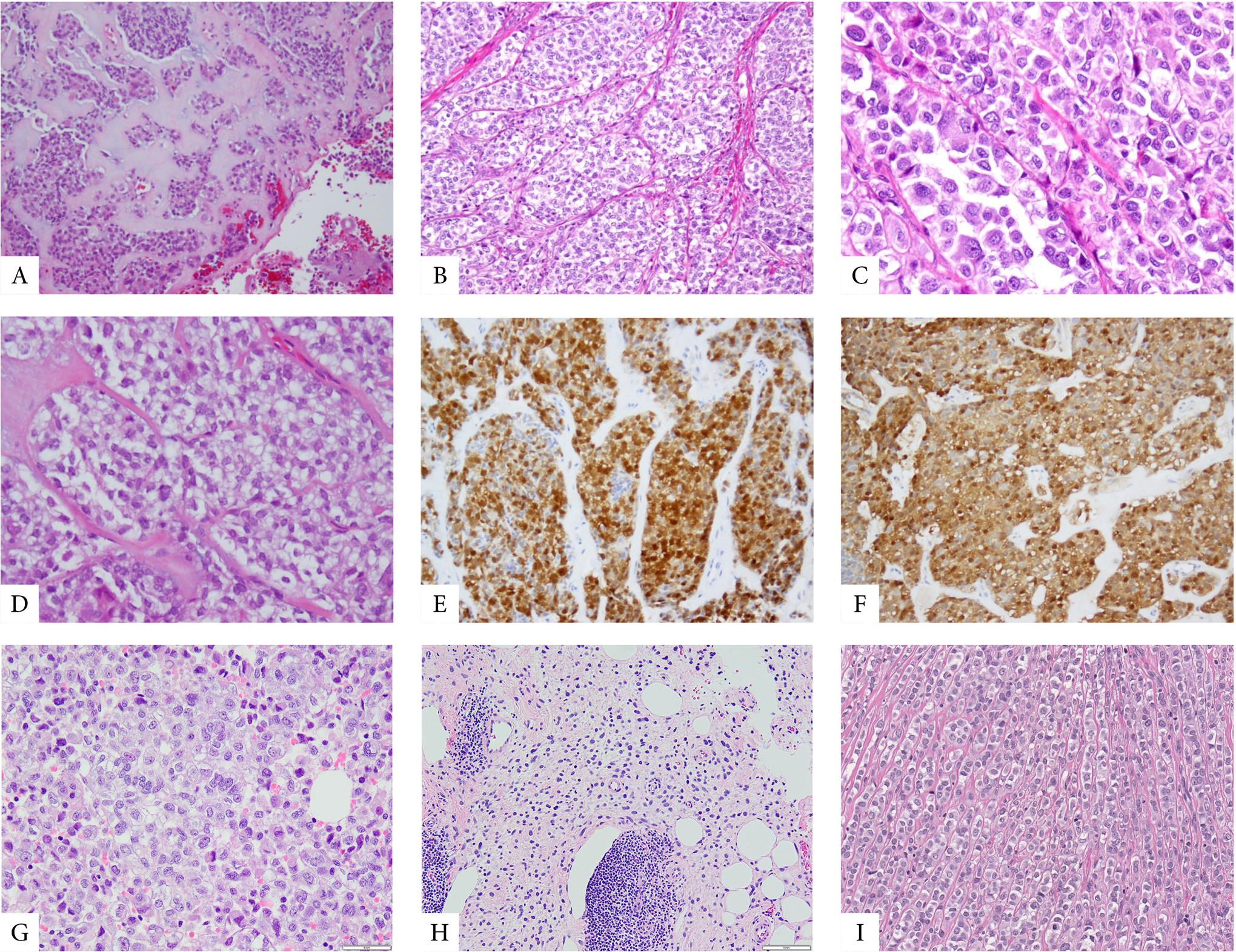
A-H. MET with EWSR1-POU5F1 showing at low power a lobulated growth within a prominent myxochondroid stroma (A), often a well-defined nested pattern (B-D), being composed of epithelioid cells with overtly malignant features and lightly eosinophilic cytoplasm (C) or clear cytoplasm (D). These classic examples often show diffuse positivity for cytokeratin (E) and S100 protein (F). Rare examples showed unusual features such as more solid growth of epithelioid cells with scant clear cell cytoplasm (G) or an infiltrative growth pattern within subcutaneous fat, with ill-defined cell borders (H). Rare FUS-POU5F1 positive cases showed epithelioid morphology with light eosinophilic to clear cytoplasm organized in nests or linear arrangements (I).
3.1. EWSR1-POU5F1 fusions are associated with malignant MET with nested epithelioid growth with clear cell morphology
There were 15 MET with EWSR1-POU5F1 fusions and 2 harboring FUS-POU5F1 fusions (Supplem Table 1). These 17 tumors occurred in 10 females and 7 male patients, with an age range of 3–49 years (median age 26 years). Tumors with EWSR1-POU5F1 had the following anatomic distribution: 11 were located in soft tissue (all in the extremities), 2 in the bone (mandible and pelvic bone), and in one case each located in the kidney and vulva. Notably, 11/15 (73%) MET with EWSR1-POU5F1 fusions had overtly malignant features, which occurred preferentially in children and young adults, 9 patients being younger than the age of 30. Morphologically, these 11 malignant MET with EWSR1-POU5F1 fusions consisted of nests and sheets of epithelioid cells, often with clear cell cytoplasm, showing moderate pleomorphism, prominent nucleoli, and increased mitotic activity, but no necrosis (Fig 2). By IHC, the malignant MET consistently expressed S100 with either EMA or CK, except for one case showing only cytokeratin expression. Four EWSR1-POU5F1 positive MET were benign, displaying classic histology, with epithelioid cells arranged in a reticular pattern within a myxoid stroma, alternating with a spindle cell component admixed with a variable fibromyxoid stroma. Three of the 4 benign EWSR1-POU5F1-positive MET lacked markers of myoepithelial differentiation (S100, EMA and CK). The two FUS-POU5F1-positive MET cases (arising in the trunk of a 34-year-old female and in the hand of a 31-year-old male, respectively) displayed benign classic histology, with epithelioid and ovoid spindle cells in a collagen-rich stroma.
3.2. MET with EWSR1-PBX1/3 fusions are often benign and associated with bland spindle cell and sclerotic morphology
There were 10 MET with EWSR1-PBX3 and 6 with EWSR1-PBX1 fusions. These 16 patients had an age range at diagnosis of 2–75 years (median 29 years). Eight tumors arose in extremity soft tissue locations, 7 presented as primary bone tumors and one occurred in the lung. In fact, EWSR1-PBX1/3 fusions were the most common gene fusions in primary skeletal MET, occurring in 7 out of 12 (58%) cases. Most tumors with EWSR1-PBX1/3 (13/16, 81%) consisted of benign appearing spindle cells arranged in fascicles, often embedded in hyaline sclerotic stroma (Fig 3). Only 3/16 (19%) of MET cases with EWSR1-PBX1/3 showed malignant cytologic features. By IHC, 13 tumors showed positivity for EMA and S100 (Fig 3), but lacked cytokeratin expression. Two cases showed only S100 protein positivity, while one case, occurring in the bone, was negative for all markers possibly related to decalcification.
Figure 3. Pathologic features of MET harboring EWSR1-PBX1/3 fusions.
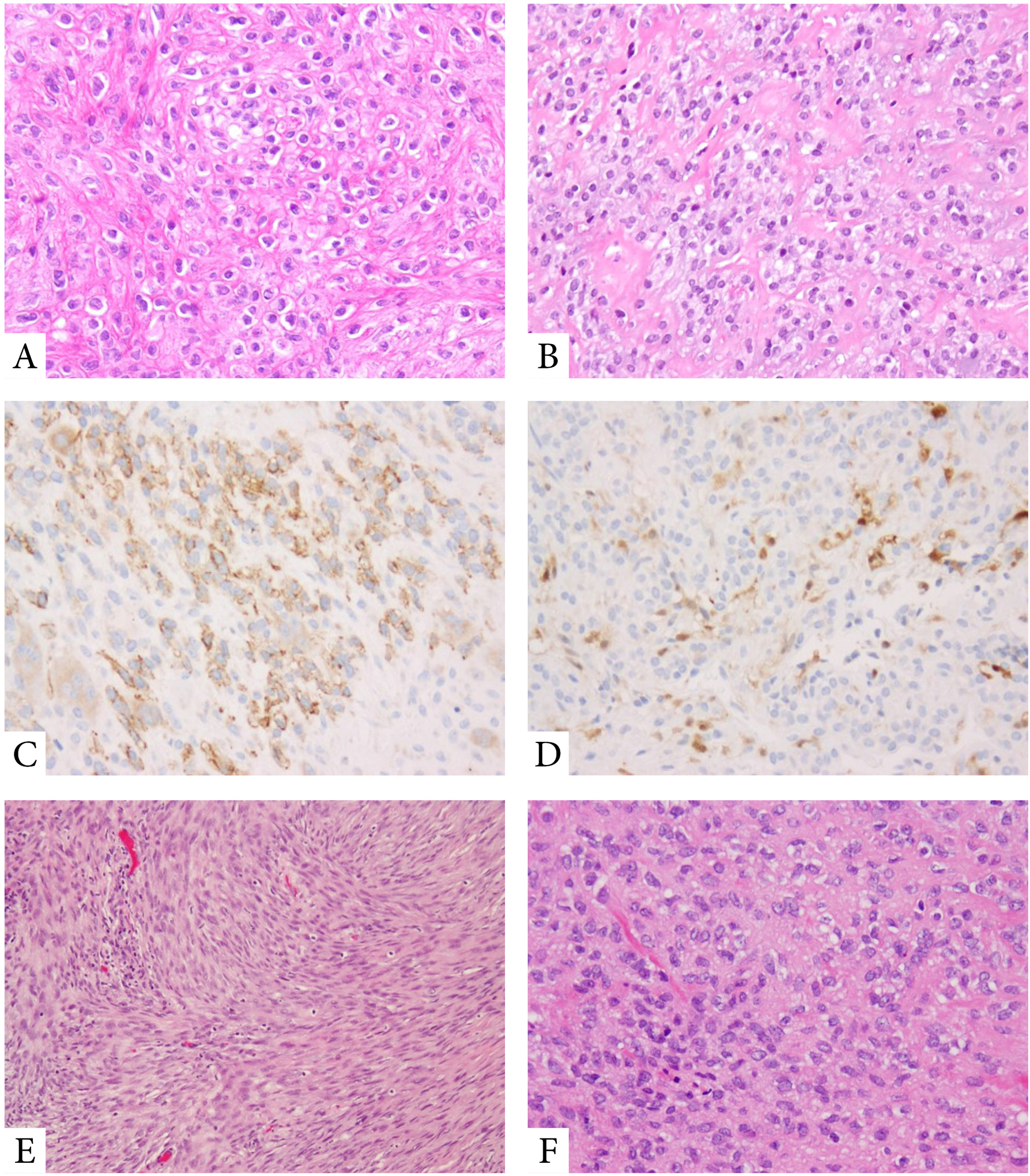
Tumors with EWSR1-PBX1 fusions often show a bland epithelioid to ovoid phenotype, with scant clear cytoplasm, embedded in a delicate fibrous collagenous stroma (A,B). Tumors are frequently positive for EMA (C) and S100 protein (D). MET with EWSR1-PBX3 fusions display a more ovoid to spindle cell appearance, with benign histologic features (E,F).
3.3. FUS-KLF17 fusions are associated with chordoma-like morphology (so-called parachordoma)
Eight MET harbored FUS-KLF17 fusions. Patients had an age range at diagnosis of 8–58 years (median 33 years). Five tumors were located in soft tissue, two in the lung or pleura, one in the periosteum of bone, and one intradurally involving the lumbar spine. Microscopically, the predominant pattern seen in 6/8 tumors consisted of radiating cords of epithelioid cells with eosinophilic or clear cytoplasm. The tumors were often embedded in ample myxoid or myxohyaline stroma, reminiscent of chordoma pattern (so-called parachordoma)14 (Fig 4). Three of the cases (37%) were associated with malignant histologic features. By IHC, all tumors were consistently positive for cytokeratins (or EMA) and S100.
Figure 4. Morphologic appearances of rare genetic subtypes of MET.
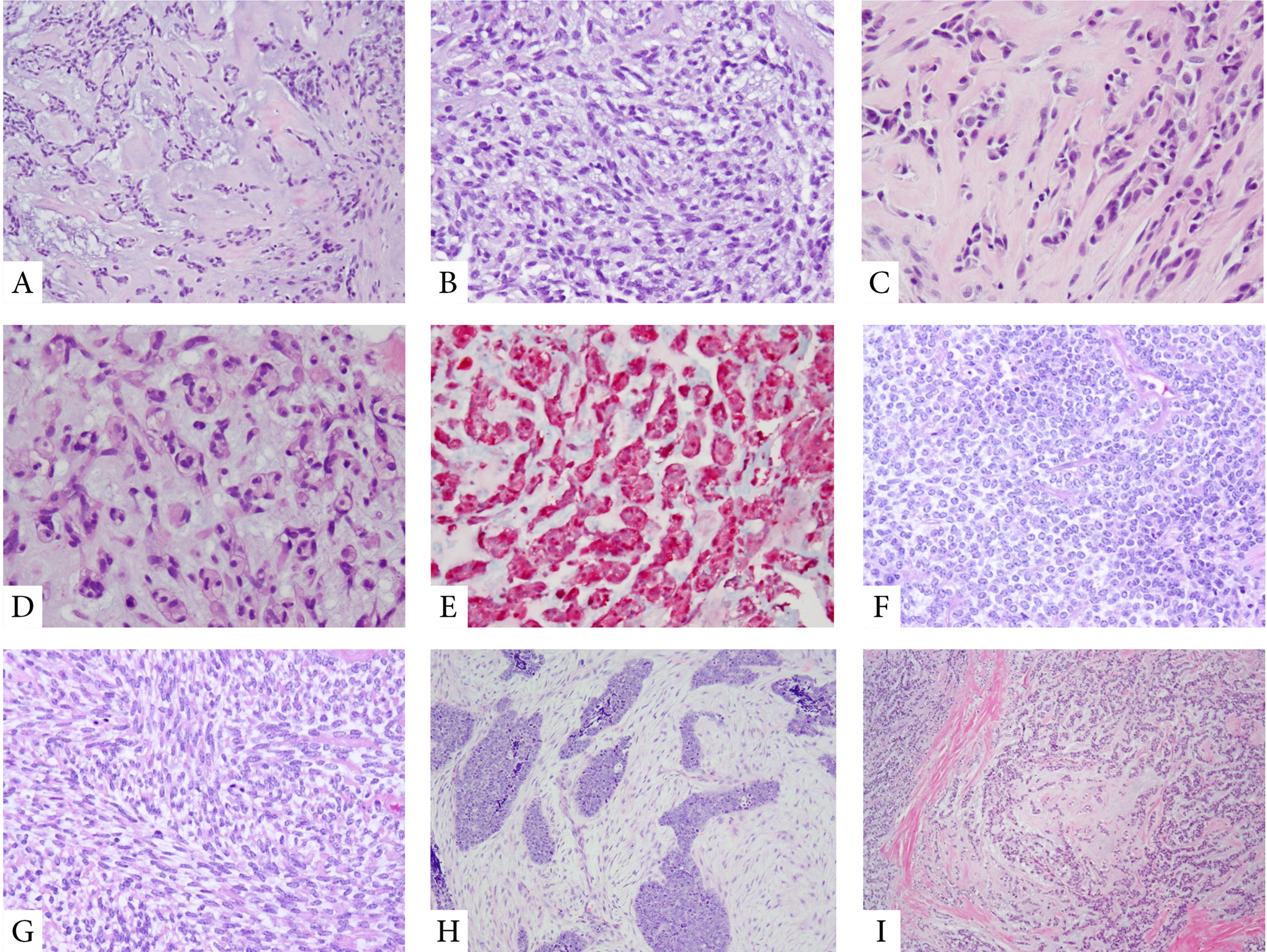
FUS-KLF17-positive MET show often a diffusely myxoid or fibromyxoid stromal component with ovoid to epithelioid cells arranged in cords and a reticular pattern, reminiscent of chordoma phenotype (parachordoma) (A-D). These tumors are often S100 protein positive (E). MET with EWSR1-ZNF444 are often malignant and may display an undifferentiated round to spindle cell phenotype (F,G), with variable stromal component, ranging from very scant to prominent and desmoplastic (H). A rare tumor with EWSR1-KLF15 fusions showing cords of epithelioid cells embedded in a variably myxoid or fibromyxoid stroma (I).
3.4. MET with rare gene fusions (EWSR1-ZNF444, EWSR1-KLF15, and EWSR1-KLF17)
Three MET harbored EWSR1-ZNF444 fusions. One of the cases which was previously reported8, occurred in a 64 year-old female as a 13 cm malignant pulmonary MET with classic morphologic features and coexpression of cytokeratin and S100. The patient subsequently developed multiple local and distant recurrences over a 10 year-follow-up period. Two additional new cases with this fusion occurred as a temporal head mass in a 2 year-old female and as a thigh mass in a 44-year-old female. Both cases had overtly malignant features, with an undifferentiated small blue round cell phenotype, and revealed a non-specific immunophenotype (negative for S100, EMA, and cytokeratin). Notably, the malignant MET in the two-year-old resembled a desmoplastic small blue round cell tumor, consisting of small nests of undifferentiated small blue round cells embedded in a loosely arranged spindled stromal cells (Fig 4). The tumor from the 44 year-old from the thigh was initially diagnosed as a Ewing sarcoma with EWSR1 gene rearrangement, until the Archer results showed a EWSR1-ZNF444 fusion.
Two MET had EWSR1-KLF15 fusions. The first case concerned a visceral MET arising in the urinary bladder of a 12-year-old female, which showed highly variable but classic histology and immunoreactivity for S100, EMA, and cytokeratin. The second case was a malignant soft tissue MET arising in the thigh of a 7-year-old male. This tumor had an uncommon biphasic appearance, being composed of reticular cords of epithelioid cells and nests of malignant undifferentiated round cells (Fig 4). By IHC tumor was positive for S100 and cytokeratin, while negative for EMA.
The single MET with an EWSR1-KLF17 fusion previously reported represented a benign MET, occurring in the foot of a 20-year-old male11. Morphologically, it was composed of radiating cords and clusters of epithelioid cells arranged in myxoid and hyalinized stroma. The tumor cells expressed cytokeratin, EMA, and S100.
4. DISCUSSION
The molecular abnormalities of bone and soft tissue MET have been recently elucidated as harboring EWSR1/FUS-related gene fusions in at least half of the cases, involving EWSR1 and FUS with various partner genes encoding for transcription factors, including POU5F1, PBX1, PBX3, ZNF444, KLF15 and KLF178,10,11,15. Presumably other novel gene fusions not yet defined might be responsible for the other half of MET lacking EWSR1/FUS gene abnormalities. In contrast, MET arising in the skin and salivary gland frequently show distinct PLAG1 and HMGA2 related gene fusions6,7. Controversy still remains regarding the pathogenetic link between fusion-positive versus fusion-negative MET, as well as the relationship between MET characterized by either EWSR1/FUS or PLAG1/HMGA2 type fusions. To avert these challenges, the current study focused on molecularly confirmed MET arising in soft, tissue, bone and viscera, which were characterized in the majority but not all cases by a classic morphologic appearance and displayed a ‘myoepithelial’ immunophenotype.
In keeping with prior published data, the current study cohort showed a predilection for young adults (median age 27 years) and extremity soft tissue locations (72%), whereas skeletal MET (18%) had a highly variable anatomic distribution in long bones, pelvic bones, and mandible. Although our results reveal a wide morphologic spectrum of architectural and cytologic heterogeneity, common phenotypes emerged, including reticular or trabecular growth patterns with prominent myxoid stroma, or areas of more nested or solid growth and hyalinized stroma1,8,12. Tumor cells ranged from epithelioid, ovoid to short spindled, typically containing uniform nuclei and eosinophilic to clear cytoplasm. Another common histotype was that of a deceptively bland spindle cell neoplasm associated with a prominent fibrotic stromal component. A small subset of malignant myoepithelial tumors displayed undifferentiated round cell features showing histologic overlap with small blue round cell tumors. By IHC, the majority (81%) of fusion-positive MET expressed cytokeratins (CK) and/or EMA in combination with S100 protein or GFAP, in keeping with the well-established myoepithelial immunoprofile. Among the 12 cases (20%) that did not meet the IHC criteria (either displaying positivity for only one marker or negativity for all), there were 4 tumors with classic, predominant epithelioid morphology and EWSR1/FUS-POU5F1 fusions, 3 tumors with classic predominant spindle cell morphology and EWSR1-PBX1/3 fusions, and 5 tumors with undifferentiated round cell morphology, of which 1 EWSR1-ZNF444, 2 EWSR1-POU5F1, and 1 each showed EWSR1 or FUS rearrangement with no partner identified.
Conversely, some of the tumors that were initially diagnosed as MET based on morphologic findings, immunophenotype and/or EWSR1/FUS gene rearrangements, were reclassified as other mesenchymal neoplasms based on the subsequent NGS or FISH results of variant EWSR1 gene fusions, involving other gene partners such as CREM, ATF1, FLI1, etc. Our results further emphasize the significant challenges in diagnosing myoepithelial tumors, especially at the malignant/high grade end of the spectrum, without a comprehensive molecular analysis. A particular pitfall is the overlap with the increasing family of round cell sarcomas, in particular with Ewing sarcoma-like tumors, which now encompass variant morphologies and immunoprofiles16. In fact, three tumors occurring in children were initially misinterpreted as Ewing sarcoma at the outside institution based on an EWSR1 gene rearrangement positive result and treated with Ewing sarcoma regimens.
Although some morphologic-genotypic correlations were observed in our initial molecular study8, our current larger investigation was able to draw more robust associations. First, MET with EWSR1-POU5F1 fusions represents the most common molecular subset (28%), being prevalent in children or young adults, presenting in the deep soft tissues of the extremities. Morphologically, tumors often displayed nested epithelioid morphology with clear cytoplasm, and thin fibrous septa. The majority of MET with EWSR1-POU5F1 fusions (11/15; 73%) showed microscopic features in keeping with malignant behavior. Second, most MET with EWSR1-PBX1/3 (12/15, 80%) consisted of benign appearing spindle cells organized in fascicles or embedded in hyaline sclerotic stroma. This subset of pure spindle cell MET expressed S100 and EMA, but typically lacked cytokeratin expression. PBX1/3 fusions were most prevalent among skeletal MET, found in more than half of bone tumors. Third, chordoma-like morphology (parachordoma) was observed in 6/8 MET with FUS-KLF17 fusions, of which 3 had a benign appearance and 3 had features of malignancy.
In addition, there were three MET with EWSR1-ZNF444 fusions. Notably, all 3 tumors had malignant microscopic features. Two cases represented soft tissue tumors with undifferentiated round cell morphology that lacked myoepithelial markers; these presented in the flank of a 44-year-old woman and the temple of a 2-year-old girl. The third case was a visceral lung MET in a 64-year-old woman, which showed malignant epithelioid histology, expression of CK and S100, and an aggressive clinical behavior with multiple metastatic implants over a 10-year period, as illustrated in a previous publication8.
Other rare fusion genes were EWSR1-KLF15 and EWSR1-KLF17. EWSR1-KLF15 was detected in two MET, both occurring in children, aged 7 and 12-years. The first case concerned a thigh mass with an unusual biphasic malignant phenotype consisting of cords of epithelioid cells and nests of undifferentiated round cells. The second case was a benign visceral MET of the urinary bladder with classic morphology. Both EWSR1-KLF15 fused MET expressed myoepithelial markers. Three other malignant MET with EWSR1-KLF15 fusions and myoepithelial marker expression have been described in the literature. Strikingly, like our first case, these 3 MET had variable biphasic malignant morphology with solid sheets of malignant epithelioid cells and areas with undifferentiated and small blue round cell morphology. One case was a parotid tumor in a 20-year-old woman that had metastasized to the lung17, whereas the other two cases were large renal childhood tumors (in girls aged 4 and 6 years) that also developed lung metastases18. These data indicate that MET with EWSR1-KLF15 mainly occur in children and are strongly associated with undifferentiated round cell morphology and clinically malignant behavior. The single MET with EWSR1-KLF17 was described in an earlier publication by our group11. This tumor presented in the foot of a 74-year old male and showed benign but classic histology.
Several soft tissue and bone tumors with overlapping morphologic features, including trabecular cords of epithelioid cells in myxoid, myxohyaline or sclerotic stroma, and co-expression of S100 and/or CK/EMA, enter the differential diagnosis of MET. One of the closest mimics is extraskeletal myxoid chondrosarcoma (EMC), which shows bland ovoid to epithelioid cells interconnected in trabecular and cribriform networks in an ample myxoid stroma. EMC does not have a specific immunoprofile, but expression of S100 and EMA is found in 20–40% of cases, whereas it usually lacks cytokeratin expression19. EMC diagnosis relies on the identification of its characteristic EWSR1-NR4A3 or TAF15-NR4A3 fusion genes20. Another group of look-alike tumors are chordoma and chordoma periphericum, which represent true notochordal related neoplasms. Due to striking overlapping morphologic features and immunoprofile (coexpression of CK, EMA and S100), the distinction between chordomas and soft tissue MET, previously designated as parachordomas, has been problematic in the past. Chordomas typically occur in the axial skeleton, while rare extra-axial tumors have been reported at any site21. It was later recognized that only the true notochordal lesions are positive for the transcription factor brachyury, while MET (parachordomas) are not22. Ossifying fibromyxoid tumor (OFMT) is another tumor closely resembling the morphology and immunoprofile of MET. OFMTs are typically composed of cords and trabeculae of uniform ovoid cells with bland nuclei set in variable collagenous and myxoid stroma. OFMTs often show a thick fibrous capsule and a peripheral shell of ossification. By IHC, OFMT may show expression of S100 (60–70%) or desmin (50%), whereas CK and EMA expression is less common23. The large majority of OFMT show recurrent PHF1 gene rearrangements which can confirm the diagnosis in challenging cases24,25. Epithelioid MPNST (EMPNST) is another rare soft tissue tumor with lobular architecture, cords of epithelioid cells with eosinophilic cytoplasm, within a collagenous stroma. EMPNST typically exhibits diffuse immunostaining for S100 and SOX10, and variably positivity for CK, while showing loss of SMARCB1 expression in the majority of cases26. However, SOX10 expression and loss of SMARCB1 do not exclude MET. Some of the deceptively bland and fibrotic MET may mimic either sclerosing epithelioid fibrosarcoma (SEF) or a low-grade fibromyxoid sarcoma (LGFMS). A further pitfall can occur if a positive EWSR1 or FUS gene rearrangement is documented. However, MUC4 is usually negative in MET, while it is diffusely and strongly expressed in most SEF (75%) and all LGFMS27. As our results highlight, 5 (8%) MET cases showed an undifferentiated round cell morphology, closely resembling a round cell sarcoma, including Ewing family of tumors or occasionally desmoplastic round cell tumor. Of interest, the first reported case of an EWSR1-POU5F1 positive tumor, occurring in the pelvis in a 39-year old woman, showed an undifferentiated round cell phenotype28, mimicking a lesion in the Ewing sarcoma family. The challenge also stems from the overlapping EWSR1/FUS gene abnormalities detected in both tumor categories. In these cases, establishing their fusion gene partners by additional molecular techniques that complement FISH break-apart assays for EWSR1 and FUS is recommended for a more definitive subclassification.
In conclusion, the histopathologic classification of MET presenting in soft tissue, bone and visceral organs remains challenging, as these tumors show remarkable heterogeneity in morphology, rather non-specific IHC marker expression, and high variability in clinical presentation and behavior. In this expanded series of 66 MET, several genotype-phenotype correlations are emerging, which provide diagnostic utility in daily practice and may serve as future roadmap for potential therapeutic target discovery. Of particular importance is the recognition that rare MET subsets with undifferentiated (small blue) round cell morphology may harbor uncommon fusion genes, e.g. EWSR1-ZNF444, or EWSR1-KLF15. Our results further show a good but imperfect concordance between the so-called ‘myoepithelial immunoprofile’, used currently as the diagnostic mainstay, and the presence of fusion gene alterations. In fact 12 (20%) cases lacked this immunoprofile, including seven with classic morphologic features and either characteristic EWSR1/FUS-POU5F1 or PBX1/3 related fusions. In contrast, the remaining 5 cases had an atypical phenotype, composed of undifferentiated round cell morphology, and being associated with variable gene fusions, including EWSR1-POU5F1 and EWSR1-ZNF444. Further studies are needed to establish the relationship of these undifferentiated tumors harboring so-called ‘myoepithelial gene fusions’ with other round cell sarcomas in the family of Ewing sarcoma-like spectrum.
Supplementary Material
Disclosures:
Supported in part by: P50 CA 140146-01 (CRA), P50 CA217694 (CRA), P30 CA008748, Cycle for Survival (CRA), Kristin Ann Carr Foundation (CRA)
Footnotes
Data Availability Statement: The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflict of interest: none
REFERENCES
- 1.Hornick JL, Fletcher CD. Myoepithelial tumors of soft tissue: a clinicopathologic and immunohistochemical study of 101 cases with evaluation of prognostic parameters. Am J Surg Pathol. 2003;27:1183–1196. [DOI] [PubMed] [Google Scholar]
- 2.Song W, Flucke U, Suurmeijer AJH. Myoepithelial Tumors of Bone. Surg Pathol Clin. 2017;10:657–674. [DOI] [PubMed] [Google Scholar]
- 3.Shah AA, Mulla AF, Mayank M. Pathophysiology of myoepithelial cells in salivary glands. J Oral Maxillofac Pathol. 2016;20:480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katabi N, Gomez D, Klimstra DS, et al. Prognostic factors of recurrence in salivary carcinoma ex pleomorphic adenoma, with emphasis on the carcinoma histologic subtype: a clinicopathologic study of 43 cases. Hum Pathol. 2010;41:927–934. [DOI] [PubMed] [Google Scholar]
- 5.Kong M, Drill EN, Morris L, et al. Prognostic factors in myoepithelial carcinoma of salivary glands: a clinicopathologic study of 48 cases. Am J Surg Pathol. 2015;39:931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antonescu CR, Zhang L, Shao SY, et al. Frequent PLAG1 gene rearrangements in skin and soft tissue myoepithelioma with ductal differentiation. Genes Chromosomes Cancer. 2013;52:675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katabi N, Ghossein R, Ho A, et al. Consistent PLAG1 and HMGA2 abnormalities distinguish carcinoma ex-pleomorphic adenoma from its de novo counterparts. Hum Pathol. 2015;46:26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antonescu CR, Zhang L, Chang NE, et al. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. A molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer. 2010;49:1114–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thway K, Fisher C. Myoepithelial tumor of soft tissue: histology and genetics of an evolving entity. Adv Anat Pathol. 2014;21:411–419. [DOI] [PubMed] [Google Scholar]
- 10.Agaram NP, Chen HW, Zhang L, et al. EWSR1-PBX3: a novel gene fusion in myoepithelial tumors. Genes Chromosomes Cancer. 2015;54:63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang SC, Chen HW, Zhang L, et al. Novel FUS-KLF17 and EWSR1-KLF17 fusions in myoepithelial tumors. Genes Chromosomes Cancer. 2015;54:267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gleason BC, Fletcher CD. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol. 2007;31:1813–1824. [DOI] [PubMed] [Google Scholar]
- 13.Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20:1479–1484. [DOI] [PubMed] [Google Scholar]
- 14.Fisher C Parachordoma exists--but what is it? Adv Anat Pathol. 2000;7:141–148. [DOI] [PubMed] [Google Scholar]
- 15.Flucke U, Mentzel T, Verdijk MA, et al. EWSR1-ATF1 chimeric transcript in a myoepithelial tumor of soft tissue: a case report. Hum Pathol. 2012;43:764–768. [DOI] [PubMed] [Google Scholar]
- 16.Diaz-Perez JA, Nielsen GP, Antonescu C, et al. EWSR1/FUS-NFATc2 rearranged round cell sarcoma: clinicopathological series of 4 cases and literature review. Hum Pathol. 2019;90:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cajaiba MM, Jennings LJ, Rohan SM, et al. Expanding the Spectrum of Renal Tumors in Children: Primary Renal Myoepithelial Carcinomas With a Novel EWSR1-KLF15 Fusion. Am J Surg Pathol. 2016;40:386–394. [DOI] [PubMed] [Google Scholar]
- 18.Stevens TM, Qarmali M, Morlote D, et al. Malignant Ewing-Like Neoplasm With an EWSR1-KLF15 Fusion: At the Crossroads of a Myoepithelial Carcinoma and a Ewing-Like Sarcoma. A Case Report With Treatment Options. Int J Surg Pathol. 2018;26:440–447. [DOI] [PubMed] [Google Scholar]
- 19.Flucke U, Tops BB, Verdijk MA, et al. NR4A3 rearrangement reliably distinguishes between the clinicopathologically overlapping entities myoepithelial carcinoma of soft tissue and cellular extraskeletal myxoid chondrosarcoma. Virchows Arch. 2012;460:621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agaram NP, Zhang L, Sung YS, et al. Extraskeletal myxoid chondrosarcoma with non-EWSR1-NR4A3 variant fusions correlate with rhabdoid phenotype and high-grade morphology. Hum Pathol. 2014;45:1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Righi A, Sbaraglia M, Gambarotti M, et al. Extra-axial chordoma: a clinicopathologic analysis of six cases. Virchows Arch. 2018;472:1015–1020. [DOI] [PubMed] [Google Scholar]
- 22.Tirabosco R, Mangham DC, Rosenberg AE, et al. Brachyury expression in extra-axial skeletal and soft tissue chordomas: a marker that distinguishes chordoma from mixed tumor/myoepithelioma/parachordoma in soft tissue. Am J Surg Pathol. 2008;32:572–580. [DOI] [PubMed] [Google Scholar]
- 23.Graham RP, Dry S, Li X, et al. Ossifying fibromyxoid tumor of soft parts: a clinicopathologic, proteomic, and genomic study. Am J Surg Pathol. 2011;35:1615–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suurmeijer AJH, Song W, Sung YS, et al. Novel recurrent PHF1-TFE3 fusions in ossifying fibromyxoid tumors. Genes Chromosomes Cancer. 2019;58:643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antonescu CR, Sung YS, Chen CL, et al. Novel ZC3H7B-BCOR, MEAF6-PHF1, and EPC1-PHF1 fusions in ossifying fibromyxoid tumors--molecular characterization shows genetic overlap with endometrial stromal sarcoma. Genes Chromosomes Cancer. 2014;53:183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaefer IM, Dong F, Garcia EP, et al. Recurrent SMARCB1 Inactivation in Epithelioid Malignant Peripheral Nerve Sheath Tumors. Am J Surg Pathol. 2019;43:835–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doyle LA, Wang WL, Dal Cin P, et al. MUC4 is a sensitive and extremely useful marker for sclerosing epithelioid fibrosarcoma: association with FUS gene rearrangement. Am J Surg Pathol. 2012;36:1444–1451. [DOI] [PubMed] [Google Scholar]
- 28.Yamaguchi S, Yamazaki Y, Ishikawa Y, et al. EWSR1 is fused to POU5F1 in a bone tumor with translocation t(6;22)(p21;q12). Genes Chromosomes Cancer. 2005;43:217–222. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.