

Acute repiratory distress syndrome
Acute alveolar injury may be caused by a wide range of pulmonary insults that at their most severe result in what is termed the acute respiratory distress syndrome (ARDS). This was formerly known as the adult respiratory distress syndrome,1, 2, 3 a term introduced to emphasise its similarity to that seen in premature babies suffering from the effects of deficient pulmonary surfactant production, i.e. the infantile respiratory distress syndrome (see p. 43), but following the recognition that the causes of the adult respiratory distress syndrome may also operate in children, ‘adult’ has given way to ‘acute’. The causes of the syndrome differ markedly in infants and adults but whatever the cause, a common cycle of events is initiated (Fig. 4.1 ) so that the end-result is the same regardless of the cause. It is a common condition, which carries a high mortality, about 50% overall. Estimations of incidence show considerable international variability but one American study reported it to be 79 per 100 000 population.4
Figure 4.1.
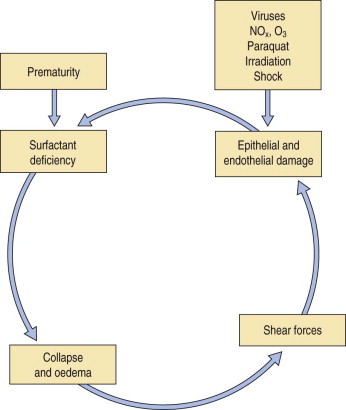
The cycle of events in the infantile and acute (formerly adult) respiratory distress syndromes. The infantile syndrome is initiated by the immature fetal lungs being unable to replenish spent surfactant whereas in the acute syndrome the cycle is initiated by a variety of causes that damage the delicate alveolar epithelium.
ARDS is a fulminant form of respiratory failure characterised by refractory hypoxaemia (P ao2/FIo2 < 200) and bilateral radiographic opacification in the absence of any evidence of an elevated left atrial pressure.5 These features indicate widespread alveolar collapse and exudation that cannot be attributed to left heart failure or other cause of pulmonary venous hypertension. There is generalised ground-glass opacification of the lungs, which is most pronounced in the dependent parts of the lungs. The opacification rapidly becomes increasingly dense until there is frank consolidation (Fig. 4.2 ).6 Air is then confined to the bronchi, which therefore appear black on plain radiographs where they stand out against the alveolar ‘white-out’ (so-called air bronchograms).7
Figure 4.2.
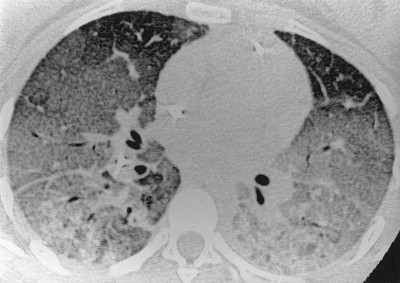
Acute respiratory distress syndrome. High-resolution computed tomography shows dense consolidation of both lungs, especially the dependent regions.
Functional studies confirm that little of the lung substance is ventilated. There is initially mild pulmonary hypertension but the pulmonary arterial constriction responsible for this is succeeded by vascular non-responsiveness so that the normal vasoconstrictor response to hypoxia is diminished. The consequent ventilation/perfusion mismatching aggravates the hypoxaemia. Other organs suffer both hypoxia and the effects of inflammatory mediators initiated by the pulmonary injury once these gain access to the general circulation. The end stages of ARDS are therefore frequently associated with multiple organ failure.8
Despite the variety of causes, most patients with ARDS show the same pattern of pathological changes, one that is termed diffuse alveolar damage (DAD). Much of this chapter is devoted to this pattern of injury but occasionally patients presenting with ARDS show other changes when subjected to biopsy or coming to autopsy. These are listed in Box 4.1 and are considered in other chapters but their chief pathological features are compared with those of DAD in Table 4.1 . Recognition of these other patterns may lead to a change in the patient's management. In that they include infection it is essential that some of the specimen goes to the microbiology department. Lung biopsy is also undertaken to assess the reversibility of the changes.9, 10 The process is potentially reversible in its exudative phase whereas widespread fibrosis with loss of the lung architecture is not.
Box 4.1. The pathological basis of the acute respiratory distress syndrome.
-
1
Diffuse alveolar damage (the typical pattern, considered in this chapter)
-
2Other causes (considered elsewhere):
-
•Pulmonary oedema due to high-altitude, rapid lung re-expansion or cerebral injury
-
•Unusual manifestations of bacterial pneumonia, e.g. streptococcal pneumonia, leptospiral pneumonia
-
•Acute fibrinous organising pneumonia
-
•Capillaritis/diffuse alveolar haemorrhage
-
•Acute eosinophilic pneumonia
-
•Unusual forms of malignant disease that affect the lungs diffusely, e.g. adenocarcinoma, intravascular lymphoma, ‘lymphangitis carcinomatosis’
-
•
Table 4.1.
Histological patterns associated with the acute respiratory distress syndrome
| Diffuse alveolar damage | Acute eosinophilic pneumonia | Acute fibrinous organising pneumonia | Capillaritis/diffuse alveolar haemorrhage | |
|---|---|---|---|---|
| Hyaline membranes | ++ | + | − | +/− |
| Eosinophils | − | + | − | − |
| Fibrin knots | +/− | +/− | ++ | +/− |
| Type II cell hyperplasia | ++ | +/− | +/− | − |
| Interstitial fibrosis | + | +/− | +/− | − |
| Capillaritis/haemorrhage | − | − | − | ++ |
Diffuse alveolar damage
DAD represents a non-specific pattern of acute alveolar injury caused by a variety of noxious agents.11, 12, 13, 14 It is the chief pathological basis of ARDS. The pathological changes of DAD can be divided into the overlapping phases of exudation, regeneration and repair.15
Exudative phase
To facilitate gas exchange the alveolar wall is highly specialised in structure. Unfortunately this specialisation renders it susceptible to injury by a wide variety of agents. The principal cells of the air–blood barrier, the type I alveolar epithelial cell and the capillary endothelial cell are exceptionally thin (see Fig. 1.27, p. 16) and this makes them particularly vulnerable to non-specific damage. Injury to these two cells underlies the development of DAD. At an early stage of alveolar injury the type I epithelial cells show cytoplasmic blebbing, which is soon followed by necrosis resulting in denudation of the basement membrane (see Fig. 7.2.5, p. 376).16, 17 Similar blebbing is seen in the alveolar capillary endothelium but denudation of the endothelial basement membrane is seldom observed, probably because of differences in the ways epithelial and endothelial cells regenerate (see below). The consequences of this damage include the escape of fibrin-rich exudates into the interstitial and air spaces, loss of the surface-active alveolar lining film and pulmonary collapse.
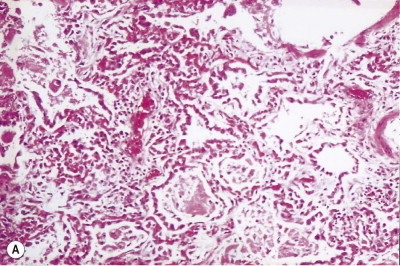
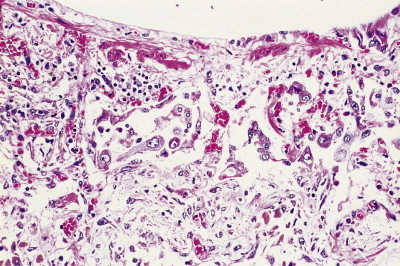
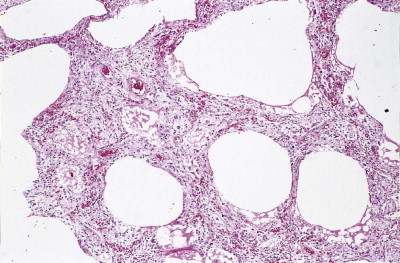
The exudative phase lasts about 1 week, during which the lungs are heavy, often weighing over 1 kg each, dark and airless. The changes are often patchy, with the dorsal and basal regions being most severely affected (Fig. 4.3 ).18, 19, 20 Slicing shows that they are wet, the cut surface exuding blood or heavily blood-stained watery fluid. Microscopically there is widespread collapse, intense congestion of the capillaries, interstitial oedema and distension of the lymphatics, a pattern that is sometimes known as congestive atelectasis (Fig. 4.4 ). Alternatively, there may be haemorrhagic oedema (Fig. 4.5 ). At the air–tissue interface, which in these collapsed lungs is at the respiratory bronchiole or alveolar duct level, respiratory movements compact a fibrin-rich exudate mixed with necrotic epithelial debris into a thin layer that covers an otherwise denuded epithelial basement membrane (Fig. 4.6 ), leading to the formation of hyaline membranes (Fig. 4.7 )11, 12, 13, 21, 22: these are identical to those that paediatric pathologists recognise as the hallmark of the infantile respiratory distress syndrome (compare Fig. 4.3 with Fig. 2.8, p. 44). Similar changes are also seen in acute eosinophilic pneumonia but here the hyaline membranes contain eosinophils, possibly in small focal collections that are easily overlooked (see p. 462).
Figure 4.3.
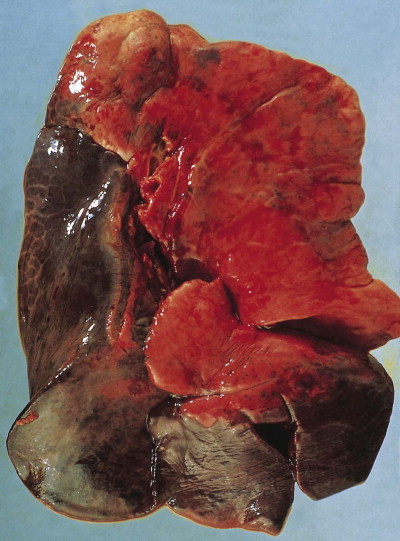
Shock lung. The lower lobe shows large irregular areas of collapse and congestion.
(Reproduced from Corrin (1980)18by permission of the editor of the Journal of Clinical Pathology.)
Figure 4.4.
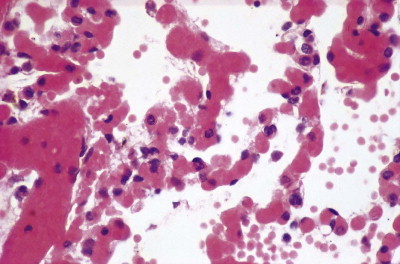
Congestive atelectasis in septic shock. There is severe capillary congestion and alveolar collapse.
Figure 4.5.
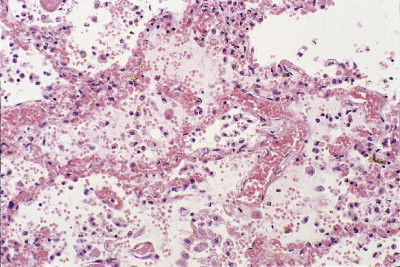
Haemorrhagic pulmonary oedema in septic shock. The alveolar capillaries are congested and the alveolar spaces are filled by oedema fluid in which there are free erythrocytes.
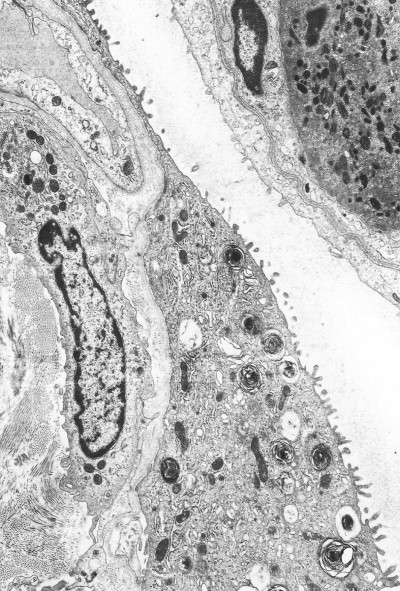
Figure 4.6.
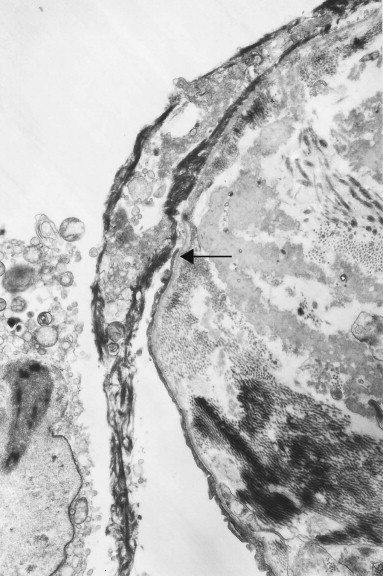
Diffuse alveolar damage. The alveolar epithelium terminates just above centre and from this point (arrow) upwards a mixture of electron-dense fibrin and cell debris (seen at the light microscopic level as a hyaline membrane) is closely applied to the denuded basement membrane. Part of a necrotic cell is seen within the alveolus. Transmission electron micrograph.
(Courtesy of Miss A Dewar, Brompton, UK.)
Figure 4.7.
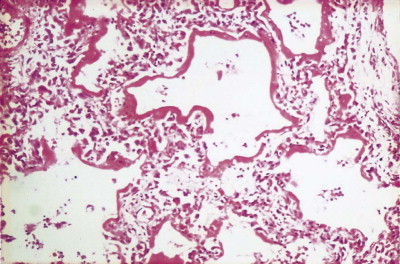
Diffuse alveolar damage, exudative phase. Hyaline membranes are a conspicuous feature. They often outline alveolar ducts. From a pregnant woman who suffered respiratory arrest due to angioneurotic oedema. She needed to be ventilated for 2 weeks before death and the oxygen concentration in the inspired air constantly increased to prevent hypoxaemia, reaching 70% for the last 8 days of her life.
(Courtesy of Dr D Melcher, Brighton, UK.)
The congested alveolar capillaries sometimes contain increased numbers of platelets or neutrophil leukocytes. This selective sequestration of formed blood elements in the microvasculature of the lungs is particularly noticeable in shock, and is considered in more detail under that heading below.
Regenerative phase
As with any exudative process, healing may be by resolution, which involves fibrinolysis and permits the lungs to return to normal, or repair, which involves fibrosis and leaves the lungs permanently scarred. Resolution and repair are both accompanied by epithelial and endothelial regeneration.
The regenerative (or proliferative) phase becomes prominent 1–2 weeks after the initial injury. It involves proliferation of both epithelial and connective tissue cells. The stem cell concerned in epithelial regeneration is the type II alveolar epithelial cell.23, 24 These cells first proliferate and then differentiate into type I cells, thereby re-epithelialising the denuded basement membranes. The dividing type II cells form a simple cuboidal epithelium (Fig. 4.8 ), or the alveoli may be lined by plump pleomorphic spindle cells that represent cell forms intermediate between types II and I epithelial cells (Figure 4.9, Figure 4.10 ). Eosinophilic cytoplasmic inclusions indicative of cell damage (see Fig. 7.1.25, p. 348) are often present,25, 26 and sometimes there is squamous metaplasia instead of orderly differentiation into type I cells, a change that the unwary pathologist may mistake for neoplasia.27
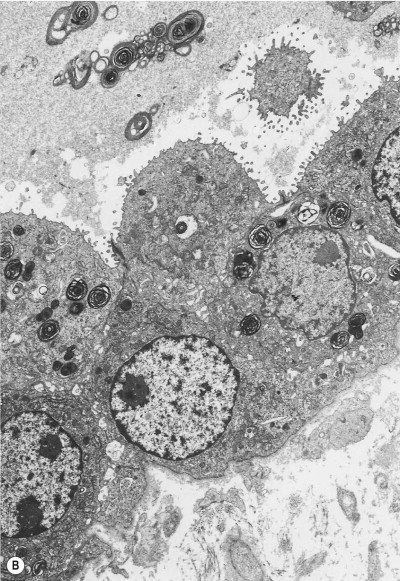
(B) Electron microscopy shows that such cells represent hyperplastic type II pneumocytes, which form a continuous row rather than being scattered singly in the corners of the alveoli. They are readily recognisable by their surface microvilli and the osmiophilic lamellar secretory vacuoles of alveolar surfactant.
(Courtesy of Mrs D Bowes, Midhurst, UK.)
Figure 4.8.
Diffuse alveolar damage, regenerative phase. (A) The alveoli have a simple cuboidal epithelial lining.
(Courtesy of Dr D Melcher, Brighton, UK.)
Figure 4.9.
Diffuse alveolar damage, regenerative phase. Regenerating alveolar epithelial cells are atypical, consisting of plump, elongated forms that represent type II pneumocytes differentiating into type I.
Figure 4.10.
Diffuse alveolar damage, regenerative phase. An alveolar epithelial cell having the squamous form of a type I cell but the microvilli and lamellar vacuoles of a type II cell. Such intermediate cells are indicative of epithelial regeneration.
The regenerating epithelium usually grows beneath the exudates lining the denuded basement membrane, casting the hyaline membranes off into the air space (Fig. 4.11 ), but it may grow over them so that they are incorporated into the interstitium (Figure 4.12, Figure 4.13 ),11, 12, 13 where their subsequent organisation contributes to the fibrosis.22 The regenerating epithelial cells may also bridge the mouths of collapsed alveoli so that these air spaces never re-expand and there is permanent shrinkage of the lung, a process termed atelectatic induration (Figure 4.14, Figure 4.15 ).28, 29, 30
Figure 4.11.
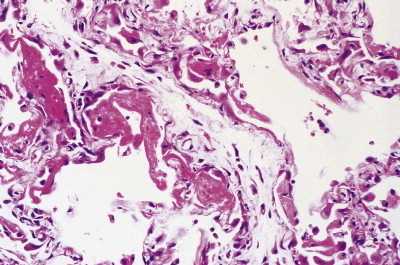
Diffuse alveolar damage, regenerative phase. The regenerating alveolar epithelium grows underneath the hyaline membranes, casting them off into the air space.
Figure 4.12.
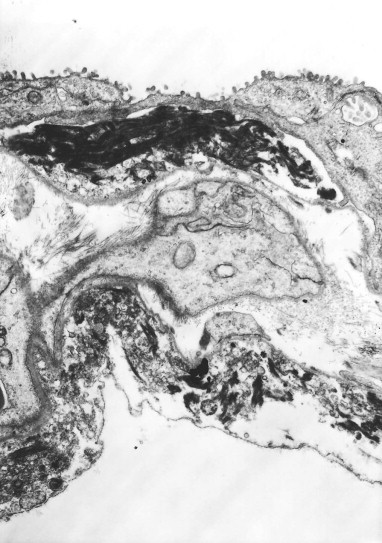
Diffuse alveolar damage, regenerative phase. The regenerating alveolar epithelium may also grow over the hyaline membranes to incorporate them into the alveolar wall. In this electron micrograph the hyaline membranes are largely represented by electron-dense fibrin which is closely applied to previously denuded alveolar epithelial basement membrane and are in turn covered by regenerating epithelium laying down a new basement membrane.
(Courtesy of Miss A Dewar, Brompton, UK.)
Figure 4.13.
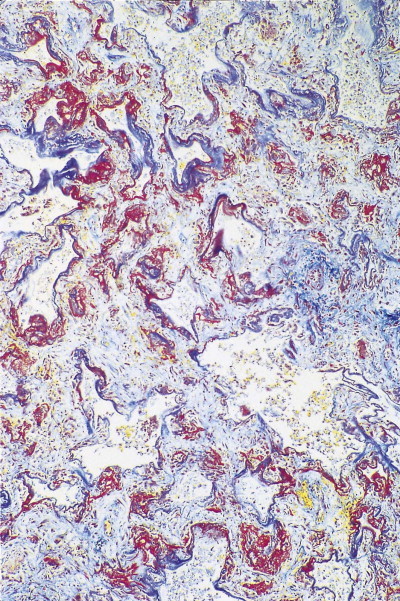
Diffuse alveolar damage, repair phase. Hyaline membranes, which are stained red, are in the process of being incorporated into the alveolar interstitium. Martius scarlet blue stain.
Figure 4.14.
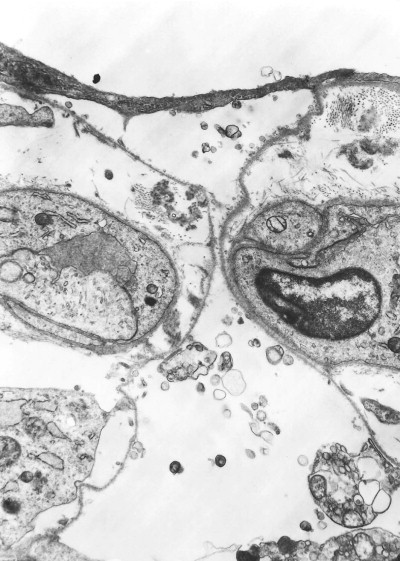
Atelectatic induration. Denuded alveolar epithelial basement membranes are closely apposed (centre). The regenerating alveolar epithelium (top) bridges the mouth of the alveolus, which consequently has little chance of ever re-expanding.
(Courtesy of Miss A Dewar, Brompton, UK.)
Figure 4.15.
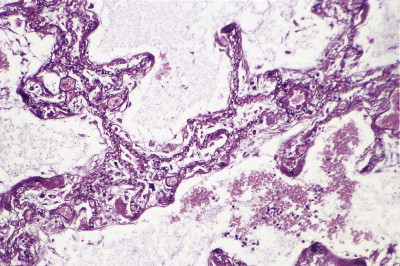
Atelectatic induration. Silver staining of the basement membranes shows that what appear to be thickened single alveolar walls represent the closely apposed walls of several collapsed alveoli.
In contrast to the type I epithelial cells, which have no regenerative powers and are replaced by differentiation of proliferating type II cells, endothelial cells are replaced by lateral spread of their own kind. An effete endothelial cell is first undermined by its healthy neighbours and only cast off when these have completely covered the basement membrane.31 Therefore, although segments of bare basement membrane have been described on the vascular side of the air–blood barrier,32, 33 they are not seen to the same extent as on the epithelial side. Nevertheless, thrombosis may complicate such endothelial damage21, 32 and subsequent organisation of such thrombi is probably responsible for some of the vascular remodelling that is seen in the repair phase of DAD. This remodelling consists of fibrocellular intimal thickening that narrows the lumen of small vessels throughout the lung and can be visualised as decreased background filling on postmortem arteriograms.34
Repair phase
If healing is by repair, interstitial connective tissue cells proliferate and, as in any scarring, myofibroblasts are involved at an early stage.35 Whilst myofibroblast contracture is beneficial when it promotes early closure of an open wound, in the lungs it largely results in harmful distortion of the bronchioloalveolar architecture and shrinkage of the lungs. Possible sources of the proliferating interstitial connective tissues include resting interstitial connective tissue cells, the bone marrow and the alveolar epithelium. Epithelial–mesenchymal transition is being increasingly recognised throughout the body and in the lung in conditions such as idiopathic pulmonary fibrosis, asthma and obliterative bronchiolitis.36, 37, 38
Whatever the source of the proliferating connective tissue cells, those with a fibroblastic phenotype lay down collagen, leading to the development of interstitial fibrosis.39, 40 Interactions between fibroblasts and the alveolar epithelium through gaps in the basement membrane have been described,41, 42, 43, 44, 45 suggesting that the regenerating epithelial cells play a further role in the underlying process of fibrosis. It is known that these cells synthesise fibrogenic cytokines such as tumour necrosis factor-α; the secretion of tumour necrosis factor-α into the underlying connective tissue would further promote interstitial fibrosis.46, 47
Fibroblasts also migrate into the alveolar exudates through defects in the epithelial basement membrane to lay down collagen within the hyaline membranes.48, 49 As epithelial cells grow over the newly formed connective tissue, a new basement membrane is formed, thereby incorporating the collagen into the interstitium,49 a process known as fibrosis by accretion. Involvement of the alveolar ducts results in these structures being lined by a ring of granulation tissue (Fig. 4.16 ). Less frequently, the organised exudates retain a predominantly intra-alveolar position, resulting in loose buds of granulation tissue similar to those seen in organising pneumonia due to other causes (see Box 6.2.1, p. 309). However, here they are more widespread and there is more prominent type II cell hyperplasia, interstitial fibroblastic proliferation and interstitial inflammation (Fig. 4.17 ). Nevertheless, such an organising pneumonia pattern is worth recognising as it carries a better prognosis than interstitial fibrosis.50 It is claimed that this intra-alveolar pattern of repair is particularly found when generalised sepsis is the cause of the initial damage whereas interstitial fibrosis is more characteristic of injury caused by cytotoxic drugs and the idiopathic cases.51
Figure 4.16.
‘Ring’ fibrosis, representing the organisation and incorporation of exudates lining the alveolar ducts into the walls of these airways.
Figure 4.17.
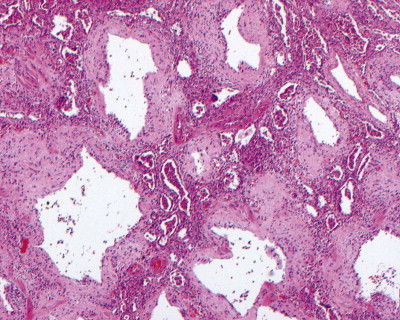
Diffuse alveolar damage, repair phase. (A) A lower lobe seen at autopsy shows diffuse consolidation, with occasional lobules showing more exudative appearances. (B) Microscopy shows diffuse intra-alveolar organisation with abundant interstitial inflammation.
An increase in lung collagen can be detected in patients with ARDS who survive longer than 14 days and this progressively increases with the duration of the disease.52 The identification of pulmonary fibrosis on transbronchial biopsy is closely related to mortality.53 Fibrosis can be well established by 2 weeks, at which time the lungs may be contracted and firm with a fine sponge-like pattern on their cut surfaces, representing bronchiolectasis and irregular microcystic distortion of the alveolar architecture (Figure 4.18, Figure 4.19 ).54, 55, 56, 57 The changes are similar to those seen in any fibrotic process but are reached remarkably quickly (see Fig. 4.19).22 However, at this early stage the fibrosis differs from that seen in chronic fibrosis in being more cellular and less collagenous: extensive fibroblast proliferation is evident. With small air cysts alternating with solid areas of fibrosis and foci of squamous metaplasia there is a resemblance to the bronchopulmonary dysplasia seen in the late stages of the infantile respiratory distress syndrome.55, 56 In survivors the granulation tissue undergoes progressive collagenisation and such patients may suffer from debilitating fibrotic lung disease. The pattern is then that of fibrotic non-specific interstitial pneumonia,58 making it important to determine whether there has been an episode of acute lung injury in such patients.
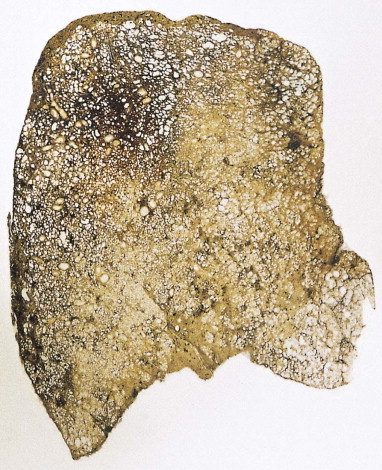
Figure 4.18.
Diffuse alveolar damage, repair phase. The repair process has resulted in interstitial fibrosis.
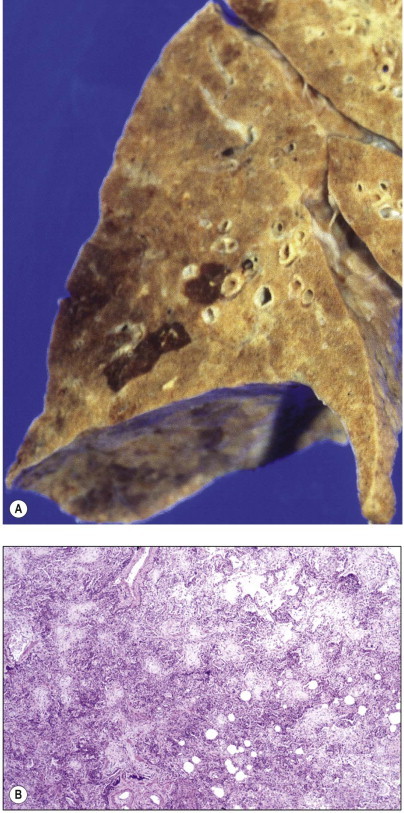
Figure 4.19.
Suicidal ingestion of kerosene resulting in advanced pulmonary fibrosis and honeycombing within 2 weeks of the initial injury.
Causes
The causes of DAD are quite diverse (Box 4.2 ). So too are the pathways by which the injurious agents reach the lungs. Some enter the lungs directly via the airways, e.g. oxygen in high concentrations, poisonous gases such as phosgene and metallic fumes such as those of mercury and cadmium. Other agents responsible for DAD penetrate the chest wall to damage the lungs (e.g. ionising radiation) and some reach the lungs via the blood stream, having been ingested or injected (e.g. paraquat and cytotoxic chemotherapeutic agents). The blood stream also conveys many of the endogenous factors that underlie the DAD of shock. In numerical terms, septic shock is the most important cause of DAD.59, 60
Box 4.2. Causes of diffuse alveolar damage.
Via the airways
Infection, especially viral
Inhaled smoke, fume and toxic gases, including oxygen in high concentrations
Aspiration of gastric contents
Through the chest wall
X-irradiation
Via the blood stream
- Various mediators generated in shock
- Reperfusion of ischaemic tissue
- Acute pancreatitis
- Cardiopulmonary bypass
- Transfusion of stored blood
- Fat embolism
- Paraquat ingestion
- Cytotoxic chemotherapeutic agents
Multiple causes may operate in one patient. For example, trauma may be combined with blood loss, fat embolism and sepsis, whilst therapeutic efforts to correct these may themselves be hazardous. The transfusion of stored blood is not without danger, whilst to prevent hypoxaemia, damaged lungs that require rest often have to be forcibly ventilated and subjected to injurious concentrations of oxygen, although it is known that this can only aggravate the injury to the lungs.22 The damaged lung also appears to be unduly susceptible to infection,61 partly because of impaired neutrophil migration into the air spaces.62 It is therefore common in clinical practice for the lungs to be subjected to several injurious agents and since these all contribute to a non-specific pattern of disease it may be difficult for the pathologist to distinguish the initiating factor from the effects of treatment. Consideration of events in the intensive care unit is essential in these circumstances. Many of the causes of DAD listed in Box 4.2 are dealt with elsewhere, leaving only a few to be considered here.
Shock
Shock is a state of prolonged hypotension, generally attributable to trauma, hypovolaemia, cardiac failure, sepsis or anaphylaxis.63, 64 The hypotension leads to inadequate tissue perfusion and, if this is not corrected, multiorgan failure is inevitable. At necropsy, the lungs are the organs most commonly affected.65, 66, 67
Severe pulmonary injury was well described in patients suffering from shock in the Second World War68 but it was not until the war in Vietnam that the importance of respiratory failure as a complication of shock was fully appreciated.69, 70 By this time there had been major improvements in medical care. Casualties could be transported rapidly by helicopter to well-equipped field hospitals where intensive care with mechanical ventilatory support was available. Despite this, injured patients often developed fatal respiratory insufficiency, typically after an interval of between 48 and 72 hours. A number of terms graphically described this syndrome – ‘shock lung’, ‘posttraumatic respiratory insufficiency’, ‘traumatic wet lung’ and ‘Da Nang lung’. Pathological examination showed congestive atelectasis or haemorrhagic oedema proceeding to fully developed DAD, as described above.
The pathogenesis of ‘shock lung’ is complex and requires special consideration. In hypovolaemic and cardiogenic shock, compensatory mechanisms such as peripheral vasoconstriction initially maintain cerebral oxygenation, but if the underlying cause is untreated, there follows a state of decompensation characterised by vascular unresponsiveness: vasodilatation develops, the blood pressure plummets and there is widespread hypoxic cell death. Anaphylactic and septic shock are characterised from the outset by such vasodilatation, which is caused by a variety of mediators that are released from inflammatory and other cells.71 The identification of the same mediators in experimental hypovolaemia72 suggests that the pathogenesis of shock may be similar regardless of the cause.
In numerical terms, septic shock is the most important cause of diffuse damage.59, 60 It represents the culmination of a clinicopathological continuum of infection-driven sepsis syndromes:
-
1
Sepsis – the systemic inflammatory response to infection characterised by normal heart rate, temperature, respiratory rate and white blood cell count
-
2
Severe sepsis – sepsis, as defined in 1, plus multiorgan dysfunction
-
3
Septic shock – severe sepsis, as defined in 2, plus refractory hypotension.
These terms have replaced ‘septicaemia’, which proved difficult to define and has now been abandoned. The multiorgan dysfunction is due to inflammatory cytokines released into the circulation from the site of infection, the common sites of which are, in decreasing frequency, lung, blood, intestine and peritoneum, urinary tract and surgical wounds. Gram-positive infections are slightly commoner than Gram-negative, which are slightly commoner than polymicrobial. The role of lipopolysaccharide derived from the cell walls of Gram-negative bacteria has been particularly well studied in the pathogenesis of septic shock.73, 74, 75, 76, 77 The many effects of this endotoxin include the release of tumour necrosis factor from monocytes, macrophages and polymorphonuclear leukocytes,74, 75, 76, 78, 79 the production of cytokines such as interleukin-1 and interferon-γ that act synergistically with tumour necrosis factor,80 the widespread induction of nitric oxide synthesis81, 82, 83 and the activation of both the coagulation and complement cascades.84 Exotoxins released by Gram-positive bacteria appear to act similarly. Some of the most fulminant forms of shock are seen with group A streptococci causing puerperal sepsis and necrotising fasciitis, meningococci, Staphylococcus aureus related to tampon retention and a meticillin-resistant strain that secretes an exotoxin known as Panton–Valentine leukocidin.85
Tumour necrosis factor has been demonstrated on the luminal surface of pulmonary endothelium in endotoxin-induced shock.86 It causes vascular smooth muscle to relax but this action is reduced if the endothelium is removed,87 indicating that tumour necrosis factor-induced vasodilatation is partially dependent upon the integrity of the endothelium. A factor that causes vascular dilatation has been detected as coming from the vascular endothelium and acting on the medial muscle coat of the vessel. At first termed endothelium-derived relaxing factor, this factor is now known to be nitric oxide, a remarkably simple chemical that has long been recognised to be poisonous.88 Fortunately, its half-life in the vessel wall is very short, timed in seconds rather than minutes. The enzyme responsible for its production (from l-arginine) is nitric oxide synthase, which is found in endothelium and can be induced in the vascular medial smooth muscle. Both the constitutive and inducible forms of the enzyme are activated by bacterial lipopolysaccharide and it therefore seems likely that in septic shock bacterial products act directly on the vessel wall resulting in the production of excess amounts of nitric oxide. Even momentarily increased levels of nitric oxide might be expected to cause arterial dilatation and hence capillary congestion. It would appear that in septic shock circulating bacterial products such as lipopolysaccharide cause vascular dilatation and possibly increased permeability by direct action on the blood vessels and indirectly through the induction of nitric oxide synthase and the release of the cytokines mentioned in the preceding paragraph. Pulmonary endothelial upregulation is indicated by widespread expression of intercellular adhesion molecule-1 (ICAM-1: CD54) in septic shock. Indeed, positive immunostaining for CD54 is proving to be a useful marker of shock.89
Nitric oxide is a powerful vasodilator but it also mediates many other processes throughout the body. In host defence nitric oxide plays a very different and more aggressive role. It enables macrophages to generate free oxygen radicals, the principal means by which these cells eliminate both bacteria and cancer cells, but which, if not inactivated, also damage healthy host cells.90 Macrophages release much more nitric oxide than endothelial cells but, as in the blood vessels, the amounts released are generally inactivated within seconds. However, overwhelming bacterial infections result in the release of very large amounts of nitric oxide and the overproduction of toxic oxygen radicals. Although the oxygen radicals are countered by the protective action of enzymes such as superoxide dismutase,91 their excessive release results in oxidation of lipids and protein sulphydryl groups, and DNA damage.92 Damaged cell membrane phospholipids release free arachidonic acid which in turn is degraded to produce leukotrienes, such as prostaglandins and thromboxane, that are capable of altering vessel calibre and permeability.
The direct vasodilatory action of nitric oxide and the toxic action of the free oxygen radicals that nitric oxide generates account for most of the pathological features of shock but there are other processes in the microvasculature of the lung that contribute to the pulmonary damage. The vascular engorgement characteristic of shock lung is occasionally accompanied by sequestration of neutrophil polymorphonuclear leukocytes in the pulmonary microvasculature (Figure 4.20, Figure 4.21 ).18, 18, 93, 94 Acting through the complement cascade,84, 95, 96 endotoxin activates these cells within the systemic circulation so that they lose their normal deformability and aggregate into microemboli, with the result that they cannot traverse the alveolar capillaries.73, 97, 98 Their arrest there is promoted by activated endothelial intercellular adhesion molecules.99, 100, 101, 102, 103 The unique position of the pulmonary capillaries in the circulation is probably responsible for the lungs being the organs most severely affected in shock.65, 66, 67 Trapped in the alveolar capillaries, activated neutrophils damage the alveolar wall by producing reactive oxygen radicals and releasing enzymes such as elastase, collagenase and cathepsins that are able to degrade protein constituents of the wall.104, 105, 106, 107, 108, 109, 110 Impairment of neutrophil migration into the air spaces has been referred to above.62
Figure 4.20.
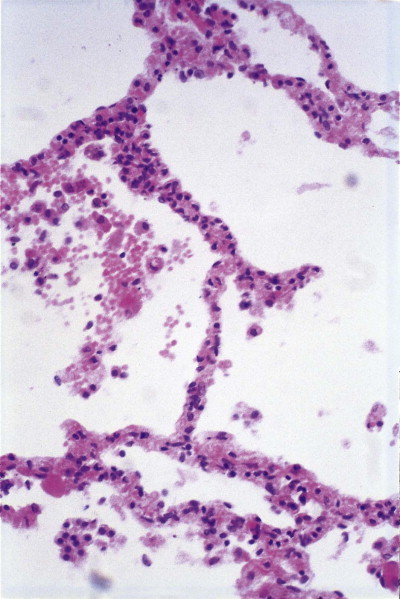
Shock lung. In shock, the alveolar walls are sometimes hypercellular due to the accumulation of neutrophil polymorphonuclear leukocytes.
Figure 4.21.
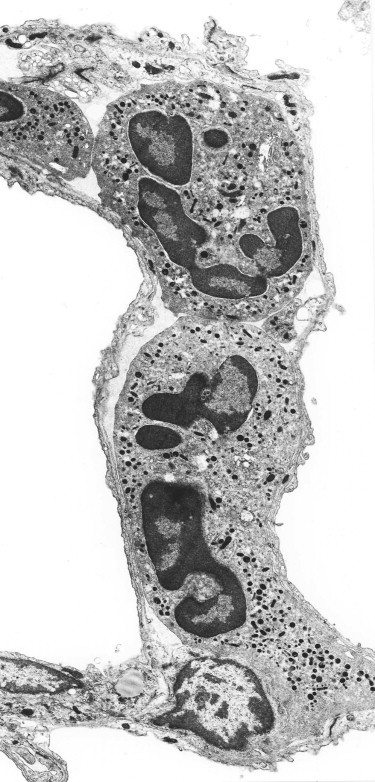
Shock lung. Electron microscopy shows that the neutrophil leukocytes are sequestered in the alveolar capillaries.
(Courtesy of Professor PK Jeffery, Brompton, UK.)
Patients suffering from ARDS frequently have haematological evidence of disseminated intravascular coagulation111 and it is then generally possible to demonstrate platelet and fibrin thrombi and increased numbers of megakaryocytes in their alveolar capillaries postmortem (Fig. 4.22 ).18, 112, 113, 114, 115, 116 It is uncertain whether intravascular coagulation initiates lung injury,73, 117, 118 but histamine released from platelets is likely to increase vascular permeability whilst fibrin degradation products, which are elevated in patients with the respiratory distress syndrome,119 are known to induce pulmonary oedema.120 Larger pulmonary thrombi, formed in situ or embolic, are common in patients with ARDS.34 Any infarction they cause increases the risk of interstitial emphysema and pneumothorax, forms of barotrauma to which all patients on ventilators are prone.
Figure 4.22.
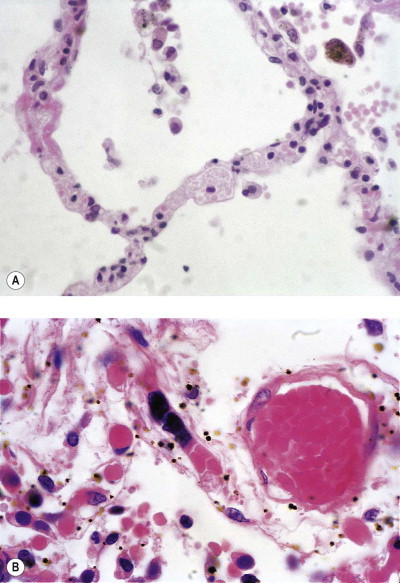
Shock lung. Shock is often accompanied by disseminated intravascular coagulation, consumptive coagulopathy and the release of megakaryocytes from the bone marrow. (A) Platelets fill the alveolar capillaries. (B) Megakaryocytes released from the bone marrow are arrested in the pulmonary capillary bed, from whence they release their platelets. Occasional megakaryocytes may be observed in normal lung but they are more noticeable in shock. They are seen as clumps of basophilic nuclear material within alveolar capillaries (centre). To the right a blood vessel contains a globular hyaline microthrombus.
Infection
In assessing the role of infection in causing DAD it is important to distinguish between primary infection of the lung and infection elsewhere in the body. Septic shock has been dealt with above and attention has been drawn to the increased risk of secondary lung infection when the lungs are already damaged.62 This section is confined to consideration of some primary pneumonias that can produce DAD.
DAD is characteristic of some viral infections, most recently the severe acute respiratory syndrome (SARS)-related coronavirus (see p. 163). Some patients with viral pneumonia succumb during the acute exudative phase and are found to have prominent hyaline membranes, whilst others suffer less severe damage, allowing regeneration and fibrosis. Indications that the disease is viral include the presence of specific inclusions, such as those seen in cytomegalovirus infection, or of syncytial giant cell formation, most typically seen in measles pneumonia.
The changes of DAD are not typically seen in bacterial pneumonia, but may occur in fulminating infection with organisms such as Streptococcus pyogenes. In pneumonic plague and anthrax pneumonia, the overwhelming alveolar damage leads to intensely haemorrhagic pulmonary oedema similar to that sometimes seen in shock. DAD may also accompany miliary tuberculosis and Pneumocystis pneumonia, particularly the non-reactive forms seen in the severely immunocompromised.
Aspiration of gastric contents
Pulmonary aspiration of gastric contents is a frequent event in unconscious or semiconscious patients. If the aspirated material is infected it is likely to cause pneumonia and lung abscess but if sterile and highly acid the consequences are liable to be even more dire. Mendelson reported 66 instances of patients aspirating stomach contents during obstetric anaesthesia. The aspiration of abundant solid material resulted in suffocation but Mendelson was more interested in the larger number of patients who aspirated liquids and developed pulmonary oedema. He suspected that the pH of the aspirate was important and confirmed this in experiments on rabbits.121 The aspiration of gastric acid is now known as Mendelson's syndrome. The mortality is high,122, 123 reaching 94% in some series.124
Ultrastructural studies on experimental animals in which fluids of differing pH and osmolarity have been instilled into the lungs show features of alveolar injury that are most severe when the fluid is strongly acid, but even distilled water or saline is able to produce minor damage. Disturbance of the osmotic gradient across the alveolar capillary membrane may therefore be an additional factor. Damage occurs to both epithelial and endothelial cells, which separate from their basement membranes.125 The most severe changes include necrosis and neutrophil exudation. Pulmonary haemorrhage is generally found and often shows a brown discoloration microscopically, due to the production of acid haematin (Fig. 4.23 ). The alveolar changes are accompanied by acute bronchitis and bronchiolitis with sloughing of the mucosa. The pathological changes after acid aspiration may be described as those of a severe chemical burn.
Figure 4.23.
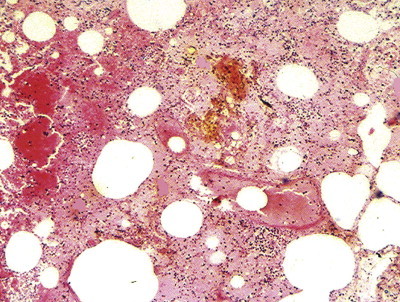
Aspiration of gastric acid (Mendelson's syndrome). There is haemorrhagic oedema and centrally the extravasated blood is discoloured due to the production of brown acid haematin.
Preventive measures include the administration of antacids to postoperative or obstetric patients, but this often results in colonisation of the stomach by Gram-negative bacteria and a bacterial rather than a chemical pneumonia if there is aspiration.126
Irradiation
Radiation may be environmental, occupational or employed as a weapon of war and is of course also part of the therapeutic armamentarium. Environmental and occupational exposure carries an increased risk of lung cancer and is dealt with on page 533 while the iatrogenic effects of radiation treatment are dealt with on page 391. This section is concerned with the high-level radiation experienced by the victims of nuclear attack or accident.
Nuclear explosions cause significant acute and long-term damage to the victims. The immediate mortality is high and in such cases it is difficult to separate the direct effects of radiation from those of thermal injury and the secondary effects of bone marrow failure. Nevertheless, by studying the victims of the atomic bombs dropped on Japan in 1945 much was learnt of the effects of large doses of whole-body irradiation. The lungs of victims dying within 2 weeks showed focal collapse and oedema, while those dying between 2 and 6 weeks after exposure had centriacinar areas of necrosis and haemorrhage; and those dying more than 6 weeks afterwards showed broader areas of necrosis with a heavy infiltrate of neutrophils.127
Pathogenesis
Radiation generates free radicals in the tissues and its effects are potentiated by the presence of oxygen. Free radicals result in DNA damage and chromosomal abnormalities.128 At the cellular level, alveolar capillary endothelial cells and type I epithelial cells are most susceptible.
Endothelial injury is believed to be of prime importance in radiation pneumonitis.129 Severe or prolonged endothelial damage disturbs the normal endothelial–mesenchymal relationships and allows uncoordinated fibroblast proliferation. Endothelial changes are detectable within days of exposure. In rats they are seen within 48 hours after 1100 rads and within 5 days after 650 rads.130 Endothelial cells become swollen and vacuolated and separate from the basement membrane, resulting in increased capillary permeability and interstitial oedema. Adhesion of platelets to the denuded basement membrane initiates thrombosis and occlusion of the vascular lumen. Proliferation of endothelial cells follows and endothelial basement membrane is reduplicated.128, 130, 131 By light microscopy the early vascular changes are evident as thrombosis, which is followed by cellular intimal thickening or even complete occlusion of small arteries and arterioles. In the later fibrotic stage vessels are thick-walled and hyaline.132
Epithelial cell damage is also well described in radiation injury. Within 10 days of exposure type I cells become swollen and undergo necrosis and sloughing with the formation of hyaline membranes. Type II cells proliferate and appear swollen and vacuolated.131 Pleomorphism of regenerating alveolar lining cells is apparent and similar changes are seen in bronchial and bronchiolar epithelium.133 In many respects therefore the appearances closely resemble those due to cytotoxic drugs (see p. 385).
Pathology
Early radiation changes in human lung include oedematous thickening of the alveolar walls, hyperplasia and swelling of type II cells, and alveolar exudates. The changes are essentially those of DAD, as described above. Fibrosis develops later and is typically interstitial. Type II cells and fibroblasts are often atypical, with large nuclei and prominent nucleoli. Blood vessels are sclerosed and the vasculature consequently reduced. Irradiation also impairs the bactericidal properties of pulmonary macrophages and predisposes to infection.
Idiopathic acute alveolar injury
In some cases of rapidly progressive DAD no cause is evident. Such patients were described by Hamman and Rich,134 since when these authors’ names have often been applied eponymously to rapidly progressive idiopathic pulmonary fibrosis.135 Others favour the term ‘acute interstitial pneumonia’ for such cases and emphasise the differences from chronic interstitial pneumonia,135, 136 but all intermediate stages are encountered. Furthermore, some patients with idiopathic pulmonary fibrosis that for the most part has run a typically chronic course exhibit acute exacerbations. If they die of such a flare-up of their disease autopsy shows the hyaline membranes of DAD superimposed upon long-established collagenous fibrosis (see Fig. 6.1.10, p. 274).137, 138 In a series of 58 patients with DAD diagnosed by surgical biopsy, 21% had no identifiable cause or predisposing condition (i.e. acute interstitial pneumonia) and in 12% the changes represented an acute exacerbation of idiopathic pulmonary fibrosis.139 Patients with idiopathic pulmonary fibrosis of non-specific interstitial pneumonia pattern may similarly experience acute exacerbations characterised by DAD, while patients with connective tissue disease may develop DAD ab initio or have it complicate pre-existent interstitial fibrosis.140, 141, 142
Treatment of acute respiratory distress
The treatment of ARDS is based upon minimising whichever of the several causes of the condition are deemed to be operating and achieving a balance between maintaining blood oxygen levels and affording the lungs the rest that all tissues recovering from injury require.4, 143 Ideally, the lungs would be rested completely and the blood oxygenated in some other way. Indeed, this is occasionally attempted by a process of extracorporeal oxygenation in which the systemic blood is diverted through an artificial lung in the form of a membrane oxygenator that sits alongside the patient.143a, 143b, 143c This is a major procedure that generally occupies a surgical operating theatre and is often attended by problems with haemostasis and, if prolonged, haemolysis. Lesser procedures involve the insertion of an intravenous oxygenator or the use of an extracorporeal device that removes carbon dioxide from the blood passing through a cannula connecting a femoral artery to its vein, but these only treat a fraction of the blood leaving the heart.
More often, blood oxygenation is maintained by artificial ventilation, sacrificing the optimal conditions for lung recovery in favour of supplying other organs, particularly the brain, with oxygen. Simply allowing the mechanically expanded lung to collapse again several times a minute would establish a pattern of respiration quite unlike the normal and one that would incur further damage to the lungs. With a normal lining of surfactant the alveoli do not collapse completely at the end of expiration. A considerable residual volume of gas is normally retained but, when the alveolar lining film is lost, as in DAD, the alveoli collapse completely at the end of each respiratory excursion and the inspiratory phase commences from a much lower baseline. It is now recognised that this exerts considerable mechanical stress upon the delicate alveolar epithelial lining,144 the integrity of which is already severely compromised in ARDS. These purely mechanical forces result in the generation and release of a variety of injurious cytokines (e.g. tumour necrosis factor-α) and reactive oxygen species without necessarily involving any inflammation.145, 146 To minimise this, positive pressure is usually maintained throughout the respiratory cycle, a form of ventilation termed positive end-expiratory pressure, frequently referred to as PEEP.147 If, despite PEEP, hypoxaemia approaches dangerous levels the intensivist has no choice but to raise the concentration of oxygen in the inspired air, although it is recognised that high concentrations of oxygen are themselves injurious to the lung. With severe lung damage a situation is often reached where to prevent cerebral injury increasingly higher concentrations of oxygen have to be employed. The initial damage to the lung is then compounded by a combination of mechanical and chemical injury, resulting in an aggravated form of DAD to which the term ‘respirator lung’ is often applied.
Nitric oxide, a selective pulmonary vasodilator with anti-inflammatory properties, has been used in acute lung injury but a meta-analysis found that despite limited improvement in oxygenation it confers no mortality benefit and may cause harm.147a
A promising technique that is now under experimental investigation follows the recent recognition that bone marrow stem cells are capable of differentiating into a variety of mature cell types, including those that constitute the alveolar epithelium.148 The successful application of this would hasten the healing process and minimise the likelihood of the damaged lung developing irreversible fibrosis.
Outcome
DAD carries a high mortality rate, around 50% overall but reaching 94% when aspiration of gastric acid is the cause.124 The DAD associated with septic shock also carries a particularly high mortality rate.149 Certain polymorphisms of the vascular endothelial growth factor (VEGF) gene have been associated with a higher mortality.150 Survivors may appear to recover completely but tests of lung function often show that they have a mild restrictive or diffusion defect.151, 152
Pulmonary fibrosis
The healing of DAD by fibrosis leads to a consideration of pulmonary fibrosis in general. This is not an inevitable consequence of injury for, if the damaged tissue is capable of regeneration, healing by resolution is possible and normality is regained. However, if tissue is irretrievably lost, healing can only take place by repair, entailing the replacement of the lost tissue by fibrous tissue and resulting in scar formation. Various patterns of fibrosis are encountered in the lungs and these will now be described.
Focal scars are quite commonly found in the lungs at necropsy, particularly at the apices of the upper lobes where they consist of narrow bands of contracted, often blackened, lung covered by thickened pleura, the so-called apical cap.153, 154, 155, 155a When such apical scars are accompanied by calcification and pleural adhesions, they have probably followed tuberculosis, but this is now unusual in developed countries. Most apical scars in these countries are probably attributable to the relative ischaemia of the apices of the lungs, which, due to our upright posture, are barely perfused at all for much of the day. Quite minor apical scars are often associated with bullae and rupture of these underlies many spontaneous pneumothoraces (see p. 711). Apical scarring also develops in ankylosing spondylitis (see p. 478). In other parts of the lungs, a focal subpleural scar may be the result of a primary tuberculous lesion or the corresponding primary lesions of fungal infections such as histoplasmosis. Focal scars also result from embolic infarction and pneumonia. In such scars combined stains for elastin and collagen (such as the elastin-van Gieson stain) often show that the alveolar framework of the lung is completely lost, reflecting total destruction of the affected area. Such scars are generally rich in elastin, a feature common to organs such as the lungs and the heart that are subject to repeated movement and one that is not seen to the same degree in scars of organs such as the liver and kidneys that are subjected to less movement.
With more widespread pulmonary fibrosis, elastin stains often show that the framework of the alveolar walls is maintained,156 and one of three patterns of fibrosis may then be recognised: intra-alveolar, interstitial and obliterative.157, 158 These patterns are not mutually exclusive. For example, interstitial fibrosis may result from the incorporation of organising air space exudates into the alveolar wall,159, 160 as described above in the proliferative phase of DAD. This is particularly likely if the epithelium is lost on a broad front and its regeneration is delayed. Whether the fibrosis has an intra-alveolar, interstitial or obliterative pattern largely depends on the severity and duration of the initial injury. To some extent therefore these patterns are of prognostic significance.
Intra-alveolar fibrosis (organising pneumonia)
Intra-alveolar fibrosis represents organisation of an alveolar exudate.161 It is characterised by the presence within the alveoli of polypoid knots of myxoid granulation tissue, rich in glycosaminoglycans, fibroblasts and myofibroblasts162 but containing little polymerised collagen. These intra-alveolar knots of granulation tissue are known as Masson bodies163 or bourgeons conjonctifs (see Figs 4.17B, p. 142, 5.2.4, p. 180 and 6.2.2, p. 310). This is the classic pattern of postpneumonic carnification, found particularly when bacterial pneumonia fails to resolve. It is familiar to all pathologists conducting autopsies and must have been well known to the great morbid anatomists of the nineteenth century. Twentieth-century descriptions date back at least to 1912.164, 165, 166
Organising pneumonia may also represent incomplete resolution of eosinophilic pneumonia or the fibrin-rich transudate of severe left ventricular failure, or be caused by inhaled irritants,167 viral infection, including human immunodeficiency virus,168 drugs,169, 170 radiation171, 172, 173 and connective tissue disease.174, 175 Organising pneumonia is also found in transplanted lungs176 and is commonly seen around tumours or other localised lung lesions. It is also one of the minor features of extrinsic allergic alveolitis (Box 4.3 ). Although organising pneumonia is readily recognisable in transbronchial biopsies, such specimens may not include these underlying lesions, which may therefore remain undetected unless a surgical biopsy is obtained.
Box 4.3. Causes of intra-alveolar fibrosis.
Bacterial pneumonia
Aspiration pneumonia
Eosinophilic pneumonia
Severe left-sided cardiac failure (‘congestive consolidation’)
Inhaled irritants
Viral infection
Drugs
Radiation
Connective tissue disease
Inflammatory bowel disease
Extrinsic allergic alveolitis
Adjacent lesions such as tumour, abscess, Wegener's granulomatosis
Idiopathic (cryptogenic organising pneumonia)
There is also an idiopathic variety of organising pneumonia, known as cryptogenic organising pneumonia (formerly known as idiopathic bronchiolitis obliterans-organising pneumonia). This is described in Chapter 6.2 (p. 308).
Obliterative fibrosis
Pulmonary fibrosis sometimes effaces the lumen of several adjacent alveoli completely, rendering them totally airless (Fig. 4.24 ). This obliterative pattern of fibrosis is the result of severe lung injury due to any of the causes of DAD considered above. Intra-alveolar and obliterative pulmonary fibrosis have many causes in common. The pattern of fibrosis depends not so much on the nature of the damage as its severity. With very severe injury the alveolar lumen is flooded with a fibrin-rich exudate, organisation of which completely obliterates the air spaces over broad tracts of lung. Within these areas, however, the framework of the alveolar walls can often still be appreciated, particularly with elastin or basement membrane stains. It is as if the architecture has undergone a form of petrification. The parts of the lung affected by obliterative fibrosis are completely non-functioning. This pattern of fibrosis is unlikely to resolve.
Figure 4.24.
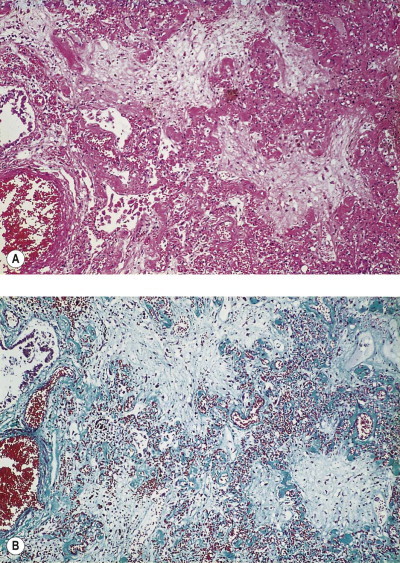
Accidental paraquat poisoning. The patient died from pulmonary fibrosis within 10 days of ingesting a relatively small amount of the chemical. The fibrosis is of the obliterative pattern, resulting from organisation of exudate that flooded many alveoli so that the air spaces are completely obliterated although the alveolar walls can still be identified. (A) Haematoxylin and eosin. (B) Masson stain.
Interstitial fibrosis
This pattern of pulmonary fibrosis involves the interstitial compartment of the alveolar walls and largely spares the air spaces (Fig. 4.25 ). It often entails the laying down of connective tissue within the alveolar walls but it may also be brought about by an accretive process involving incorporation into the interstitium of exudates or connective tissue first formed in air spaces.159, 160, 177 The causes are varied but may be divided into two broad groups, one involving the formation of exudates and transudates and the other involving the formation of what may be loosely termed granulomas (Box 4.4 ). It is particularly notable that the ‘exudate and transudate’ group of diseases predominantly affects the basal portion of the lungs and the ‘granulomatous’ group more the upper parts. The reason for this is not well understood but it can be a helpful diagnostic pointer in advanced disease. It requires no great skill in interpreting chest radiographs to assign a patient with widespread reticulonodular opacities to one or other of these two broad groups on the basis of the distribution of the disease. This criterion is also useful when assessing widespread pulmonary fibrosis postmortem.
Figure 4.25.
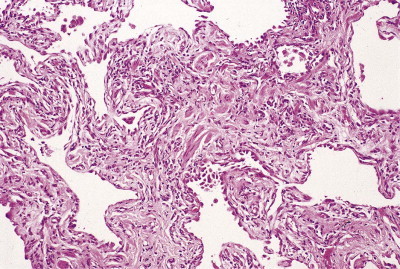
Interstitial pulmonary fibrosis.
Box 4.4. Causes of predominantly interstitial pulmonary fibrosis.
| A ‘transudate/exudate’ group that affects the dependent parts of the lung | A ‘granulomatous’ group that tends to spare the bases |
|---|---|
| Organising diffuse alveolar damage | Sarcoidosis |
| Idiopathic pulmonary fibrosis (usual interstitial pneumonia) | Extrinsic allergic alveolitis Langerhans cell histiocytosis |
| Chronic oedema | Silicosis |
| Asbestosis | Chronic berylliosis |
The conditions in the left-hand column are ranked on a time scale from organising diffuse alveolar damage, which may progress to fibrosis within 10 days, to asbestosis, which evolves over many years.
In the ‘exudate and transudate’ group many cases are unexplained but some represent the outcome of DAD caused by agents that range from fumes to viruses, irradiation, the aspiration of regurgitated gastric acid and the ingestion of various chemicals (see Box 4.1, p. 136). These causes may also lead to an obliterative pattern of pulmonary fibrosis (see above) but in the present context, instead of exudates flooding alveoli, they line the denuded alveolar walls as hyaline membranes, organisation of which leads to their incorporation into the alveolar interstitium (see Figure 4.12, Figure 4.13). This augments the activity of interstitial fibroblasts, the two processes combining to cause fibrosis of the alveolar walls. A similar process, albeit at a slower tempo, is envisaged in idiopathic pulmonary fibrosis (usual interstitial pneumonia), which is dealt with in Chapter 6.1. Most of these conditions entail damage to the delicate lining cells of the alveoli and capillaries with consequent exudation. It is perhaps because of this that the subsequent interstitial fibrosis is most marked in the dependent parts of the lungs. This distribution is also seen in the interstitial fibrosis that follows long-standing interstitial oedema in conditions such as mitral stenosis and pulmonary veno-occlusive disease.
The other major group of causes of interstitial fibrosis may be termed ‘granulomatous’ because focal collections of activated macrophages are involved in the development of the fibrosis. This group includes sarcoidosis, extrinsic allergic alveolitis and Langerhans cell histiocytosis (eosinophilic granuloma), all of which are described in Chapter 6. The fibrosis they cause is predominantly mid- or upper zonal.
The pneumoconioses constitute an important group of diseases causing interstitial pulmonary fibrosis. They are dealt with separately in Chapter 7.1 but it is possible to allocate individual pneumoconioses to one or other of the above two groups (see Box 4.2). Thus, asbestosis resembles the idiopathic cases in having a predominantly basal distribution, whereas others, e.g. silicosis and chronic berylliosis, resemble the granulomatous diseases morphologically (given that early silicotic nodules resemble granulomas) and in their upper zone distribution.
‘Honeycomb lung’
In advanced cases of pulmonary fibrosis the normal alveolar architecture is lost and the three patterns just described can no longer be distinguished. At this stage the lung is replaced by a series of cystic spaces giving an appearance that has been termed ‘honeycomb lung’. The spaces represent a combination of disrupted alveoli showing bronchiolisation and bronchiolectasis (see Figs 6.1.5 and 6.1.10AFig 6.1.5Fig 6.1.12A, pp. 269 and 274). ‘Honeycomb lung’ is not a specific disease but represents the final result of many, being an end-stage pattern of injury comparable to the granular contracted kidney and cirrhosis of the liver. Idiopathic pulmonary fibrosis is its commonest cause, particularly those cases with the pattern of usual interstitial pneumonia, and pathologists may see this remodelling microscopically in the absence of changes on high-resolution computed tomography. Other causes include extrinsic allergic alveolitis, Langerhans cell histiocytosis, sarcoidosis and berylliosis. Lymphangioleiomyomatosis also produces widespread cystic change but generally lacks the extensive fibrosis and bronchiolisation seen in these other conditions.
Dystrophic calcification and ossification
Dystrophic calcification is very common in pulmonary scars, particularly those resulting from tuberculosis, chickenpox and histoplasmosis. Pulmonary calcification in the absence of hypercalcaemia also occurs in the tracheobronchial cartilages of the elderly, the cartilaginous nodules of tracheobronchopathia osteochondroplastica and bronchopulmonary amyloid tumours. Pulmonary calcification also accompanies haemosiderin deposition in the lungs and is therefore found in chronic haemorrhagic conditions such as idiopathic haemosiderosis and the postcapillary pulmonary hypertension of mitral stenosis, lymphangioleiomyomatosis and veno-occlusive disease. Pulmonary calcification secondary to hypercalcaemia (metastatic calcification) is described on page 489.
Dystrophic pulmonary ossification takes place in similar circumstances to dystrophic pulmonary calcification: it is found with scarring, ageing of the bronchial cartilages, tracheobronchopathia osteochondroplastica and amyloid tumour formation. Lamellar bone, readily recognisable as such, is laid down, and marrow spaces are often evident. Sometimes branching spicules of bone extend through the lung in a racemose or dendriform manner.178, 179, 180, 181, 182, 183 Isolated foci of laminated bone may also be found within alveoli of otherwise normal appearance (see Fig. 6.2.25, p. 322).
References
Acute respiratory distress syndrome
- 1.Petty TL, Ashbaugh DG. The adult respiratory distress syndrome. Clinical features, factors influencing prognosis and principles of management. Chest. 1971;60:233–239. doi: 10.1378/chest.60.3.233. [DOI] [PubMed] [Google Scholar]
- 2.Petty TL. The acute respiratory distress syndrome – historic perspective. Chest. 1994;105:S44–S47. doi: 10.1378/chest.105.3_supplement.44s. [DOI] [PubMed] [Google Scholar]
- 3.Ware LB, Matthay MA. Medical progress – The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 4.Rubenfeld GD, Caldwell E, Peabody E. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 5.Bernard GR, Artigas A, Brigham KL. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149:818–824. doi: 10.1164/ajrccm.149.3.7509706. [DOI] [PubMed] [Google Scholar]
- 6.Desai SR. Acute respiratory distress syndrome: imaging of the injured lung. Clin Radiol. 2002;57:8–17. doi: 10.1053/crad.2001.0889. [DOI] [PubMed] [Google Scholar]
- 7.Desai SR, Wells AU, Suntharalingam G. Acute respiratory distress syndrome caused by pulmonary and extrapulmonary injury: a comparative CT study. Radiology. 2001;218:689–693. doi: 10.1148/radiology.218.3.r01mr31689. [DOI] [PubMed] [Google Scholar]
- 8.Bone RC, Balk R, Slotman G. Adult Respiratory Distress Syndrome – sequence and importance of development of multiple organ failure. Chest. 1992;101:320–326. doi: 10.1378/chest.101.2.320. [DOI] [PubMed] [Google Scholar]
- 9.Bulpa PA, Dive AM, Mertens L. Combined bronchoalveolar lavage and transbronchial lung biopsy: safety and yield in ventilated patients. Eur Resp J. 2003;21:489–494. doi: 10.1183/09031936.03.00298303. [DOI] [PubMed] [Google Scholar]
- 10.Patel SR, Karmpaliotis D, Ayas NT. The role of open-lung biopsy in ARDS. Chest. 2004;125:197–202. doi: 10.1378/chest.125.1.197. [DOI] [PubMed] [Google Scholar]
Diffuse alveolar damage
- 11.Liebow AA. The Lung. Williams & Wilkins; Baltimore: 1967. New concepts and entities in pulmonary disease; pp. 332–365. International Academy of Pathology monograph No.8., editor. [Google Scholar]
- 12.Liebow AA. Definition and classification of interstitial pneumonias in human pathology. Prog Resp Res. 1975;8:1–33. [Google Scholar]
- 13.Katzenstein ALA, Bloor CM, Liebow AA. Diffuse alveolar damage – the role of oxygen, shock and related factors. Am J Pathol. 1976;85:210–222. [PMC free article] [PubMed] [Google Scholar]
- 14.Corrin B. Diffuse alveolar damage. In: Evans TW, Haslett C, editors. ARDS Acute Respiratory Distress in Adults. Chapman & Hall Medical; London: 1996. pp. 37–46. [Google Scholar]
- 15.Matsubara O. Pathogenesis of diffuse alveolar damage. Histopathology. 2002;41:438–441. [Google Scholar]
- 16.Vijeyaratnam GS, Corrin B. Experimental paraquat poisoning. J Pathol. 1971;103:123–129. doi: 10.1002/path.1711030207. [DOI] [PubMed] [Google Scholar]
- 17.Bachofen M, Weibel ER. Alterations of the gas exchange apparatus in adult respiratory insufficiency associated with septicaemia. Am Rev Respir Dis. 1977;116:589–615. doi: 10.1164/arrd.1977.116.4.589. [DOI] [PubMed] [Google Scholar]
- 18.Corrin B. Lung pathology in septic shock. J Clin Pathol. 1980;33:891–894. doi: 10.1136/jcp.33.9.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yazdy AM, Tomashefski JF, Yagan R. Regional alveolar damage (RAD). A loocalized counterpart of diffuse alveolar damage. Am J Clin Pathol. 1989;92:10–15. doi: 10.1093/ajcp/92.1.10. [DOI] [PubMed] [Google Scholar]
- 20.Barth PJ, Holtermann W, Muller B. The spatial distribution of pulmonary lesions in severe ARDS – An autopsy study of 35 cases. Pathol Res Pract. 1998;194:465–471. doi: 10.1016/s0344-0338(98)80115-5. [DOI] [PubMed] [Google Scholar]
- 21.Hill JD, Ratcliff JL, Parrott JCW. Pulmonary pathology in acute respiratory insufficiency: lung biopsy as a diagnostic tool. J Thorac Cardiovasc Surg. 1976;71:64–69. [PubMed] [Google Scholar]
- 22.Pratt PC, Vollmer RT, Shelburne JD. Pulmonary morphology in a multihospital collaborative extracorporeal membrane oxygenation project. Am J Pathol. 1979;95:191–214. [PMC free article] [PubMed] [Google Scholar]
- 23.Evans MJ, Cabral LJ, Stephens RJ. Renewal of alveolar epithelium in the rat following exposure to NO2. Am J Pathol. 1973;70:175–198. [PMC free article] [PubMed] [Google Scholar]
- 24.Adamson IYR, Bowden DH. The type 2 cells as progenitor of alveolar epithelial regeneration. Lab Invest. 1974;30:25–42. [PubMed] [Google Scholar]
- 25.Warnock ML, Press M, Churg A. Further observations on cytoplasmic hyaline in the lung. Hum Pathol. 1980;11:59–65. doi: 10.1016/s0046-8177(80)80106-7. [DOI] [PubMed] [Google Scholar]
- 26.Yamada T, Uehara K, Kawanishi R. Immunohistochemical detection of ubiquitin-positive intracytoplasmic eosinophilic inclusion bodies in diffuse alveolar damage. Histopathology. 2006;48:846–854. doi: 10.1111/j.1365-2559.2006.02445.x. [DOI] [PubMed] [Google Scholar]
- 27.Ogino S, Franks TJ, Yong M. Extensive squamous metaplasia with cytologic atypia in diffuse alveolar damage mimicking squamous cell carcinoma: A report of 2 cases. Hum Pathol. 2002;33:1052–1054. doi: 10.1053/hupa.2002.128246. [DOI] [PubMed] [Google Scholar]
- 28.Katzenstein ALA. Pathogenesis of ‘fibrosis’ in interstitial pneumonia: an electron microscopic study. Hum Pathol. 1985;16:1015–1024. doi: 10.1016/s0046-8177(85)80279-3. [DOI] [PubMed] [Google Scholar]
- 29.Burkhardt A. Pathogenesis of pulmonary fibrosis. Hum Pathol. 1986;17:971–973. doi: 10.1016/s0046-8177(86)80653-0. [DOI] [PubMed] [Google Scholar]
- 30.Burkhardt A. Alveolitis and collapse in the pathogenesis of pulmonary fibrosis. Am Rev Respir Dis. 1989;140:513–524. doi: 10.1164/ajrccm/140.2.513. [DOI] [PubMed] [Google Scholar]
- 31.Reidy MA, Schwartz SM. Endothelial regeneration. III. Time course of intimal changes after small defined injury to rat aortic endothelium. Lab Invest. 1981;44:301. [PubMed] [Google Scholar]
- 32.Kapanci Y, Weibel ER, Kaplan HP. Pathogenesis and reversibility of the pulmonary lesions of oxygen toxicity in monkeys. II Ultrastructural and morphometric studies. Lab Invest. 1969;20:101–118. [PubMed] [Google Scholar]
- 33.Kapanci Y, Tosco R, Eggermann J. Oxygen pneumonitis in man. Chest. 1972;62:162–169. doi: 10.1378/chest.62.2.162. [DOI] [PubMed] [Google Scholar]
- 34.Tomashefski JF, Davies P, Boggis C. The pulmonary vascular lesions of the adult respiratory distress syndrome. Am J Pathol. 1983;112:112–116. [PMC free article] [PubMed] [Google Scholar]
- 35.Pache JC, Christakos PG, Gannon DE. Myofibroblasts in diffuse alveolar damage of the lung. Modern Pathol. 1998;11:1064–1070. [PubMed] [Google Scholar]
- 36.Guarino M, Tosoni A, Nebuloni M. Direct contribution of epithelium to organ fibrosis: epithelial–mesenchymal transition. Human Pathology. 2009;40:1365–1376. doi: 10.1016/j.humpath.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 37.Hackett TL, Warner SM, Stefanowicz D. Induction of epithelial–mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-β1. Am J Respir Crit Care Med. 2009;180:122–133. doi: 10.1164/rccm.200811-1730OC. [DOI] [PubMed] [Google Scholar]
- 38.Borthwick LA, Parker SM, Brougham KA. Epithelial to mesenchymal transition (EMT) and airway remodelling after human lung transplantation. Thorax. 2009;64:770–777. doi: 10.1136/thx.2008.104133. [DOI] [PubMed] [Google Scholar]
- 39.Bachofen M, Weibel ER. Basic pattern of tissue repair in human lungs following unspecific injury. Chest. 1974;65:14s–19s. doi: 10.1378/chest.65.4_supplement.14s. [DOI] [PubMed] [Google Scholar]
- 40.Meduri GU, Chinn A. Fibroproliferation in late adult respiratory distress syndrome – pathophysiology, clinical and laboratory manifestations, and response to corticosteroid rescue treatment. Chest. 1994;105:S127–S129. doi: 10.1378/chest.105.3_supplement.127s. [DOI] [PubMed] [Google Scholar]
- 41.Brody AR, Craighead JE. Interstitial associations of cells lining air spaces in human pulmonary fibrosis. Virchows Arch A Pathol Anat Histopathol. 1976;372:39–49. doi: 10.1007/BF00429715. [DOI] [PubMed] [Google Scholar]
- 42.Brody AR, Soler P, Basset F. Epithelial–mesenchymal associations of cells in human pulmonary fibrosis and in BHT-oxygen-induced fibrosis in mice. Exp Lung Res. 1981;2:207–220. doi: 10.3109/01902148109052316. [DOI] [PubMed] [Google Scholar]
- 43.Adamson IYR, Young L, Bowden DH. Relationship of alveolar epithelial injury and repair to the induction of pulmonary fibrosis. Am J Pathol. 1988;130:377–383. [PMC free article] [PubMed] [Google Scholar]
- 44.Adamson IYR, Hedgecock C, Bowden DH. Epithelial cell–fibroblast interactions in lung injury and repair. Am J Pathol. 1990;137:385–392. [PMC free article] [PubMed] [Google Scholar]
- 45.Matsubara O, Tamura A, Ohdama S. Alveolar basement membrane breaks down in diffuse alveolar damage: an immunohistochemical study. Pathol Int. 1995;45:473–482. doi: 10.1111/j.1440-1827.1995.tb03488.x. [DOI] [PubMed] [Google Scholar]
- 46.Nash JRG, McLaughlin PJ, Hoyle C. Immunolocalization of tumour necrosis factor-alpha in lung tissue from patients dying with adult respiratory distress syndrome. Histopathology. 1991;19:395–402. doi: 10.1111/j.1365-2559.1991.tb00228.x. [DOI] [PubMed] [Google Scholar]
- 47.Pan L-H, Ohtani H, Yamauchi K. Co-expression of TNF alpha and IL-1 beta in human acute pulmonary fibrotic diseases: an immunohistochemical analysis. Pathol Int. 1996;46:91–99. doi: 10.1111/j.1440-1827.1996.tb03584.x. [DOI] [PubMed] [Google Scholar]
- 48.Fukuda Y, Ferrans VJ, Schoenberger CI. Patterns of pulmonary structural remodeling after experimental paraquat toxicity. Am J Pathol. 1985;118:452–475. [PMC free article] [PubMed] [Google Scholar]
- 49.Fukuda Y, Ishizaki M, Masuda Y. The role of intraalveolar fibrosis in the process of pulmonary structural remodeling in patients with diffuse alveolar damage. Am J Pathol. 1987;126:171–182. [PMC free article] [PubMed] [Google Scholar]
- 50.Mandal RV, Mark EJ, Kradin RL. Organizing pneumonia and pulmonary lymphatic architecture in diffuse alveolar damage. Hum Pathol. 2008;39:1234–1238. doi: 10.1016/j.humpath.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 51.Kang D, Nakayama T, Togashi M. Two forms of diffuse alveolar damage in the lungs of patients with acute respiratory distress syndrome. Human Pathology. 2009;40:1618–1627. doi: 10.1016/j.humpath.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 52.Zapol WM, Trelstad RL, Coffey JW. Pulmonary fibrosis in severe acute respiratory failure. Am Rev Respir Dis. 1979;119:547–554. doi: 10.1164/arrd.1979.119.4.547. [DOI] [PubMed] [Google Scholar]
- 53.Martin C, Papazian L, Payan MJ. Pulmonary fibrosis correlates with outcome in adult respiratory distress syndrome: a study in mechanically ventilated patients. Chest. 1995;107:196–200. doi: 10.1378/chest.107.1.196. [DOI] [PubMed] [Google Scholar]
- 54.Slavin G, Nunn J, Crow J. Bronchiolectasis: a complication of artificial respiration. BMJ. 1982;285:931–934. doi: 10.1136/bmj.285.6346.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Churg A, Golden J, Fligiel S. Bronchopulmonary dysplasia in the adult. Am Rev Respir Dis. 1983;127:117–120. doi: 10.1164/arrd.1983.127.1.117. [DOI] [PubMed] [Google Scholar]
- 56.Wohl MEB. Bronchopulmonary dysplasia in adulthood. N Engl J Med. 1990;323:1834–1836. doi: 10.1056/NEJM199012273232609. [DOI] [PubMed] [Google Scholar]
- 57.Hert R, Albert RK. Sequelae of the adult respiratory distress syndrome. Thorax. 1994;49:8–13. doi: 10.1136/thx.49.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Katzenstein ALA, Fiorelli RF. Nonspecific interstitial pneumonia/fibrosis – histologic features and clinical significance. Am J Surg Pathol. 1994;18:136–147. [PubMed] [Google Scholar]
- 59.Hudson LD, Milberg JA, Anardi D. Clinical risks for development of the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1995;151:293–301. doi: 10.1164/ajrccm.151.2.7842182. [DOI] [PubMed] [Google Scholar]
- 60.Stapleton RD, Wang BM, Hudson LD. Causes and timing of death in patients with ARDS. Chest. 2005;128:525–532. doi: 10.1378/chest.128.2.525. [DOI] [PubMed] [Google Scholar]
- 61.Markowicz P, Wolff M, Djedaini K. Multicenter prospective study of ventilator-associated pneumonia during acute respiratory distress syndrome. Incidence, prognosis, and risk factors. ARDS Study Group. Am J Respir Crit Care Med. 2000;161:1942–1948. doi: 10.1164/ajrccm.161.6.9909122. [DOI] [PubMed] [Google Scholar]
- 62.Frevert CW, Warner AE, Kobzik L. Defective pulmonary recruitment of neutrophils in a rat model of endotoxemia. Am J Respir Cell Mol Biol. 1994;11:716–723. doi: 10.1165/ajrcmb.11.6.7524572. [DOI] [PubMed] [Google Scholar]
- 63.Parrillo JE. Mechanisms of disease – pathogenetic mechanisms of septic shock. N Engl J Med. 1993;328:1471–1477. doi: 10.1056/NEJM199305203282008. [DOI] [PubMed] [Google Scholar]
- 64.Evans TJ, Krausz T. Pathogenesis and pathology of shock. In: Anthony PP, MacSween RNM, editors. Recent Advances in Histopathology, volume 16. Churchill Livingstone; Edinburgh: 1994. pp. 21–47. [Google Scholar]
- 65.McGovern VJ. Shock. Pathol Annu. 1971;6:279–298. [PubMed] [Google Scholar]
- 66.McGovern VJ. The pathophysiology of Gram-negative septicaemia. Pathology. 1972;4:265–271. doi: 10.3109/00313027209068952. [DOI] [PubMed] [Google Scholar]
- 67.McGovern VJ. Hypovolaemic shock with particular reference to the myocardial and pulmonary lesions. Pathology. 1980;12:63–72. doi: 10.3109/00313028009060054. [DOI] [PubMed] [Google Scholar]
- 68.Moon VH. The pathology of secondary shock. Am J Pathol. 1948;24:235–273. [PMC free article] [PubMed] [Google Scholar]
- 69.Martin AM, Soloway HB, Simmons RL. Pathologic anatomy of the lungs following shock and trauma. J Trauma. 1968;8:687–699. doi: 10.1097/00005373-196809000-00007. [DOI] [PubMed] [Google Scholar]
- 70.Bredenburg CE, James PM, Collins J. Respiratory failure in shock. Ann Surg. 1969;169:392–403. doi: 10.1097/00000658-196903000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stergiopoulou T, Meletiadis J, Roilides E. Host-dependent patterns of tissue injury in invasive pulmonary aspergillosis. Am J Clin Pathol. 2007;127:349–355. doi: 10.1309/UJRV9DLC11RM3G8R. [DOI] [PubMed] [Google Scholar]
- 72.Thiemermann C, Szabo C, Mitchell JA. Vascular hyporeactivity to vasoconstrictor agents and hemodynamic decompensation in hemorrhagic shock is mediated by nitric oxide. Proc Natl Acad Sci USA. 1993;90:267–271. doi: 10.1073/pnas.90.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Clowes GHA. Pulmonary abnormalities in sepsis. Surg Clin North Am. 1974;54:993–1013. doi: 10.1016/s0039-6109(16)40433-0. [DOI] [PubMed] [Google Scholar]
- 74.Stephens KE, Ishizaka A, Larrick JW. Tumor necrosis factor causes increased pulmonary permeability and edema. Am Rev Respir Dis. 1988;137:1364–1370. doi: 10.1164/ajrccm/137.6.1364. [DOI] [PubMed] [Google Scholar]
- 75.Millar AB, Singer M, Meager A. Tumour necrosis factor in bronchopulmonary secretions of patients with adult respiratory distress syndrome. Lancet. 1989;ii:712–714. doi: 10.1016/s0140-6736(89)90772-1. [DOI] [PubMed] [Google Scholar]
- 76.Johnson J, Brigham KL, Jesmok G. Morphologic changes in lungs of anesthetized sheep following intravenous infusion of recombinant tumor necrosis factor-alpha. Am Rev Respir Dis. 1991;144:179–186. doi: 10.1164/ajrccm/144.1.179. [DOI] [PubMed] [Google Scholar]
- 77.Monick MM, Hunninghake GW. Activation of second messenger pathways in alveolar macrophages by endotoxin. Eur Resp J. 2002;20:210–222. doi: 10.1183/09031936.02.00252001. [DOI] [PubMed] [Google Scholar]
- 78.Xing Z, Kirpalani H, Torry D. Polymorphonuclear leukocytes as a significant source of tumor necrosis factor-alpha in endotoxin-challenged lung tissue. Am J Pathol. 1993;143:1009–1015. [PMC free article] [PubMed] [Google Scholar]
- 79.Vannhieu JT, Misset B, Lebargy F. Expression of tumor necrosis factor-alpha gene in alveolar macrophages from patients with the adult respiratory distress syndrome. Am Rev Respir Dis. 1993;147:1585–1589. doi: 10.1164/ajrccm/147.6_Pt_1.1585. [DOI] [PubMed] [Google Scholar]
- 80.Martin TR. Lung cytokines and ARDS. Chest. 1999;116:2S–8S. doi: 10.1378/chest.116.suppl_1.2s. [DOI] [PubMed] [Google Scholar]
- 81.Lui SF, Adcock IM, Old RW. Lipopolysaccharide treatment in vivo induces widespread tissue expression of inducible nitric oxide synthase mRNA. Biochemistry and Biophysical Research Communications. 1993;196:1208–1213. doi: 10.1006/bbrc.1993.2380. [DOI] [PubMed] [Google Scholar]
- 82.Buttery LDK, Evans TJ, Springall DR. Immunochemical localization of inducible nitric oxide synthase in endotoxin-treated rats. Lab Invest. 1994;71:755–764. [PubMed] [Google Scholar]
- 83.Ermert M, Ruppert C, Gunther A. Cell-specific nitric oxide synthase-isoenzyme expression and regulation in response to endotoxin in intact rat lungs. Lab Invest. 2002;82:425–441. doi: 10.1038/labinvest.3780436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ward PA. Role of complement in lung inflammatory injury. Am J Pathol. 1996;149:1079. [PMC free article] [PubMed] [Google Scholar]
- 85.Roberts JC, Gulino SP, Peak KK. Fatal necrotizing pneumonia due to a Panton–Valentine leukocidin positive community-associated methicillin-sensitive Staphylococcus aureus and influenza co-infection: a case report. Ann Clin Microbiol Antimicrob. 2008;7:5. doi: 10.1186/1476-0711-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tanaka N, Kita T, Kasai K. The immunocytochemical localization of tumour necrosis factor and leukotriene in the rat heart and lung during endotoxin shock. Virchows Arch. 1994;424:273–277. doi: 10.1007/BF00194611. [DOI] [PubMed] [Google Scholar]
- 87.Hollenberg SM, Cunnion RE, Parrillo JE. The effect of tumour necrosis factor on vascular smooth muscle: in vitro studies using rat aorta rings. Chest. 1991;100:1133–1137. doi: 10.1378/chest.100.4.1133. [DOI] [PubMed] [Google Scholar]
- 88.Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathophysiology and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 89.Tsokos M. Immunohistochemical detection of sepsis-induced lung injury in human autopsy material. Leg Med (Tokyo) 2003;5:73–86. doi: 10.1016/s1344-6223(03)00010-5. [DOI] [PubMed] [Google Scholar]
- 90.Freeman B. Free radical chemistry of nitric oxide – looking at the dark side. Chest. 1994;105:S79–S84. doi: 10.1378/chest.105.3_supplement.79s. [DOI] [PubMed] [Google Scholar]
- 91.Gongora MC, Lob HE, Landmesser U. Loss of extracellular superoxide dismutase leads to acute lung damage in the presence of ambient air: a potential mechanism underlying adult respiratory distress syndrome. Am J Pathol. 2008;173:915–926. doi: 10.2353/ajpath.2008.080119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rinaldo JE, Rogers RM. Adult respiratory distress syndrome: changing concepts of lung injury and repair. N Engl J Med. 1982;306:900–909. doi: 10.1056/NEJM198204153061504. [DOI] [PubMed] [Google Scholar]
- 93.Ratliff NB, Wilson JW, Hackel DB. The lung in hemorrhagic shock. II. Observations on alveolar and vascular ultrastructure. Am J Pathol. 1970;58:353–373. [PMC free article] [PubMed] [Google Scholar]
- 94.Kasajima K, Wax SD, Webb WR. Effects of methylprednisolone on pulmonary microcirculation. Surg Gynecol Obstet. 1974;139:1–5. [PubMed] [Google Scholar]
- 95.Craddock PR, Hammerschmidt DE, White JG. Complement (C5a)-induced granulocyte aggregation in vitro: a possible mechanism of complement-mediated leukostasis and leukopenia. J Clin Invest. 1977;60:260–264. doi: 10.1172/JCI108763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tate RM, Repine JE. Neutrophils and the adult respiratory distress syndrome. Am Rev Respir Dis. 1983;128:552–559. doi: 10.1164/arrd.1983.128.3.552. [DOI] [PubMed] [Google Scholar]
- 97.Yodice PC, Astiz ME, Kurian BM. Neutrophil rheologic changes in septic shock. Am J Respir Crit Care Med. 1997;155:38–42. doi: 10.1164/ajrccm.155.1.9001286. [DOI] [PubMed] [Google Scholar]
- 98.Drost EM, Kassabian G, Meiselman HJ. Increased rigidity and priming of polymorphonuclear leukocytes in sepsis. Am J Respir Crit Care Med. 1999;159:1696–1702. doi: 10.1164/ajrccm.159.6.9803061. [DOI] [PubMed] [Google Scholar]
- 99.Wegner CD, Wolyniec WW, Laplante AM. Intercellular adhesion molecule-1 contributes to pulmonary oxygen toxicity in mice – role of leukocytes revised. Lung. 1992;170:267–279. doi: 10.1007/BF00566679. [DOI] [PubMed] [Google Scholar]
- 100.Seekamp A, Mulligan MS, Till GO. Role of beta-2 integrins and ICAM-1 in lung injury following ischemia–reperfusion of rat hind limbs. Am J Pathol. 1993;143:464–472. [PMC free article] [PubMed] [Google Scholar]
- 101.Zimmerman GA, Albertine KH, Carveth HJ. Endothelial activation in ARDS. Chest. 1999;116:18S–24S. doi: 10.1378/chest.116.suppl_1.18s. [DOI] [PubMed] [Google Scholar]
- 102.Doerschuk CM, Mizgerd JP, Kubo H. Adhesion molecules and cellular biomechanical changes in acute lung injury. Chest. 1999;116:37S–43S. doi: 10.1378/chest.116.suppl_1.37s-a. [DOI] [PubMed] [Google Scholar]
- 103.Laudes IJ, Guo RF, Riedemann NC. Disturbed homeostasis of lung intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 during sepsis. Am J Pathol. 2004;164:1435–1445. doi: 10.1016/S0002-9440(10)63230-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sacks T, Moldow CF, Craddock PR. Oxygen radicals mediate endothelial cell damage by complement-stimulated granulocytes. J Clin Invest. 1978;61:1161–1167. doi: 10.1172/JCI109031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Repine JE. Scientific perspectives on adult respiratory distress syndrome. Lancet. 1992;339:466–469. doi: 10.1016/0140-6736(92)91067-i. [DOI] [PubMed] [Google Scholar]
- 106.Donnelly SC, Haslett C. Cellular mechanisms of acute lung injury – implications for future treatment in the adult respiratory distress syndrome. Thorax. 1992;47:260–263. doi: 10.1136/thx.47.4.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Williams JH, Patel SK, Hatakeyama D. Activated pulmonary vascular neutrophils as early mediators of endotoxin-induced lung inflammation. Am J Respir Cell Mol Biol. 1993;8:134–144. doi: 10.1165/ajrcmb/8.2.134. [DOI] [PubMed] [Google Scholar]
- 108.Varani J, Fligiel SEG, Till GO. Pulmonary endothelial cell killing by human neutrophils: possible involvement of hydroxyl radical. Lab Invest. 1985;53:656–663. [PubMed] [Google Scholar]
- 109.Donnelly SC, Macgregor I, Zamani A. Plasma elastase levels and the development of the adult respiratory distress syndrome. Am J Respir Crit Care Med. 1995;151:1428–1433. doi: 10.1164/ajrccm.151.5.7735596. [DOI] [PubMed] [Google Scholar]
- 110.Torii K, Iida KI, Miyazaki Y. Higher concentrations of matrix metalloproteinases in bronchoalveolar lavage fluid of patients with adult respiratory distress syndrome. Am J Respir Crit Care Med. 1997;155:43–46. doi: 10.1164/ajrccm.155.1.9001287. [DOI] [PubMed] [Google Scholar]
- 111.Bone RC, Francis PB, Pierce AK. Intravascular coagulation associated with the adult respiratory distress syndrome. Am J Med. 1976;61:585–589. doi: 10.1016/0002-9343(76)90135-2. [DOI] [PubMed] [Google Scholar]
- 112.Bleyl U, Rossner JA. Globular hyaline microthrombi – their nature and morphogenesis. Virchows Arch A Pathol Anat Histopathol. 1976;370:113–128. doi: 10.1007/BF00430808. [DOI] [PubMed] [Google Scholar]
- 113.Hardaway RM. Disseminated intravascular coagulation as possible cause of acute respiratory failure. Surg Gynecol Obstet. 1973;137:1–5. [PubMed] [Google Scholar]
- 114.Schneider RC, Zapol WM, Carvalho AC. Platelet consumption and sequestration in severe acute respiratory failure. Am Rev Respir Dis. 1980;122:445–451. doi: 10.1164/arrd.1980.122.3.445. [DOI] [PubMed] [Google Scholar]
- 115.Aabo K, Hansen KB. Megakarocytes in pulmonary blood vessels. 1. Incidences at autopsy, clinicopathological relations especially to disseminated intravascular coagulation. Acta Path Microbiol Scand. 1978;86:285–291. [PubMed] [Google Scholar]
- 116.Shimamura K, Oka K, Nakazawa M. Distribution patterns of microthrombi in disseminated intravascular coagulation. Arch Pathol Lab Med. 1983;107:543–547. [PubMed] [Google Scholar]
- 117.Blaisdell FW, Lim RC, Stallone RJ. The mechanism of pulmonary damage following traumatic shock. Surg Gynecol Obstet. 1970;130:15–22. [PubMed] [Google Scholar]
- 118.Sankey EA, Crow J, Mallett SV. Pulmonary platelet aggregates – possible cause of sudden peroperative death in adults undergoing liver transplantation. J Clin Pathol. 1993;46:222–227. doi: 10.1136/jcp.46.3.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Haynes JB, Hyers TM, Giclas PC. Elevated fibrin(ogen) degradation products in adult respiratory distress syndrome. Am Rev Respir Dis. 1980;122:841–847. doi: 10.1164/arrd.1980.122.6.841. [DOI] [PubMed] [Google Scholar]
- 120.Manwaring D, Thorning D, Currer PW. Mechanisms of acute pulmonary dysfunction induced by fibrinogen degradation product D. Surgery. 1978;84:45–53. [PubMed] [Google Scholar]
- 121.Mendelson CL. The aspiration of stomach contents into the lungs during obstetric anesthesia. Am J Obstet Gynecol. 1946;52:191–205. doi: 10.1016/s0002-9378(16)39829-5. [DOI] [PubMed] [Google Scholar]
- 122.Ribando CA, Grace WJ. Pulmonary aspiration. Am J Med. 1971;50:510–520. doi: 10.1016/0002-9343(71)90339-1. [DOI] [PubMed] [Google Scholar]
- 123.Matthay MA, Rosen GD. Acid aspiration induced lung injury: new insights and therapeutic options. Am J Respir Crit Care Med. 1996;154:277–278. doi: 10.1164/ajrccm.154.2.8756794. [DOI] [PubMed] [Google Scholar]
- 124.Fowler AA, Hamman RF, Good JT. Adult respiratory distress syndrome: risk with common predispositions. Ann Intern Med. 1983;98:593–597. doi: 10.7326/0003-4819-98-5-593. [DOI] [PubMed] [Google Scholar]
- 125.Alexander IGS. The ultrastructure of the pulmonary alveolar vessels in Mendelson's (acid pulmonary aspiration) syndrome. Br J Anaesth. 1968;40:408–414. doi: 10.1093/bja/40.6.408. [DOI] [PubMed] [Google Scholar]
- 126.du Moulin GC, Paterson DG, Hedley-Whyte J. Aspiration of gastric bacteria in antacid-treated patients: a frequent cause of postoperative colonisation of the airway. Lancet. 1982;i:242–245. doi: 10.1016/s0140-6736(82)90974-6. [DOI] [PubMed] [Google Scholar]
- 127.Liebow AA, Warren S, DeCowsey E. Pathology of atomic bomb casualties. Am J Pathol. 1949;25:853–940. [PMC free article] [PubMed] [Google Scholar]
- 128.Gross NJ. The pathogenesis of radiation-induced lung damage. Lung. 1981;159:115–125. doi: 10.1007/BF02713907. [DOI] [PubMed] [Google Scholar]
- 129.Adamson IYR, Bowden DH. Endothelial injury and repair in radiation-induced pulmonary fibrosis. Am J Pathol. 1983;112:224–230. [PMC free article] [PubMed] [Google Scholar]
- 130.Adamson IYR, Bowden DH, Wyatt JP. A pathway to pulmonary fibrosis: an ultrastructural study of mouse and rats following radiation to the whole body and hemithorax. Am J Pathol. 1970;58:481–498. [PMC free article] [PubMed] [Google Scholar]
- 131.Madrazo A, Suzuki Y, Churg J. Radiation pneumonitis: ultrastructural changes in the pulmonary alveoli following high doses of radiation. Arch Pathol Lab Med. 1973;96:262–268. [PubMed] [Google Scholar]
- 132.Warren S, Spencer J. Radiation reaction in the lung. AJR Am J Roentgenol. 1940;43:682–701. [Google Scholar]
- 133.Warren S, Gates O. Radiation pneumonitis: experimental and pathologic observations. Arch Pathol Lab Med. 1940;30:440–460. [Google Scholar]
- 134.Hamman L, Rich AR. Acute diffuse interstitial fibrosis of the lungs. Bull Johns Hopkins Hospital. 1944;74:177–212. [Google Scholar]
- 135.Olson J, Colby TV, Elliott CG. Hamman–Rich syndrome revisited. Mayo Clin Proc. 1990;65:1538–1548. doi: 10.1016/s0025-6196(12)62187-9. [DOI] [PubMed] [Google Scholar]
- 136.Katzenstein A-LA, Myers JL, Mazur MT. Acute interstitial pneumonia. A clinicopathologic, ultrastructural, and cell kinetic study. Am J Surg Pathol. 1986;10:256–267. [PubMed] [Google Scholar]
- 137.Kondo A, Saiki S. Acute exacerbation in idiopathic interstitial pneumonia. In: Harasawa M, Fukuchi Y, Morinari H, editors. Interstitial Pneumonia of Unknown Etiology. University of Tokyo Press; Tokyo: 1989. pp. 33–42. [Google Scholar]
- 138.Parambil JG, Myers JL, Ryu JH. Histopathologic features and outcome of patients with acute exacerbation of idiopathic pulmonary fibrosis undergoing surgical lung biopsy. Chest. 2005;128:3310–3315. doi: 10.1378/chest.128.5.3310. [DOI] [PubMed] [Google Scholar]
- 139.Parambil JG, Myers JL, Aubry MC. Causes and prognosis of diffuse alveolar damage diagnosed on surgical lung biopsy. Chest. 2007;132:50–57. doi: 10.1378/chest.07-0104. [DOI] [PubMed] [Google Scholar]
- 140.Rice AJ, Wells AU, Bouros D. Terminal diffuse alveolar damage in relation to interstitial pneumonias. An autopsy study. Am J Clin Pathol. 2003;119:709–714. doi: 10.1309/UVAR-MDY8-FE9F-JDKU. [DOI] [PubMed] [Google Scholar]
- 141.Parambil JG, Myers JL, Ryu JH. Diffuse alveolar damage: uncommon manifestation of pulmonary involvement in patients with connective tissue diseases. Chest. 2006;130:553–558. doi: 10.1378/chest.130.2.553. [DOI] [PubMed] [Google Scholar]
- 142.Park IN, Kim DS, Shim TS. Acute exacerbation of interstitial pneumonia other than idiopathic pulmonary fibrosis. Chest. 2007;132:214–220. doi: 10.1378/chest.07-0323. [DOI] [PubMed] [Google Scholar]
- 143.Leaver SK, Evans TW. Acute respiratory distress syndrome. BMJ. 2007;335:389–394. doi: 10.1136/bmj.39293.624699.AD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapol WM, Snider MT, Schneider RC. Extracorporeal membrane oxygenation for acute respiratory failure. Anesthesiology. 1977;46:272–285. doi: 10.1097/00000542-197704000-00008. [DOI] [PubMed] [Google Scholar]
- Peek GJ, Moore HM, Moore N. Extracorporeal membrane oxygenation for adult respiratory failure. Chest. 1977;112:759–764. doi: 10.1378/chest.112.3.759. [DOI] [PubMed] [Google Scholar]
- Peek GJ, Mugford M, Tiruvoipati R. Efficacy and economic assessment of conventional ventilatory support versus extracorporeal membrane oxygenation for severe adult respiratory failure (CESAR): a multicentre randomized controlled trial. Lancet. 2009;374:1351–1363. doi: 10.1016/S0140-6736(09)61069-2. [DOI] [PubMed] [Google Scholar]
- 144.Muscedere JG, Mullen JB, Gan K. Tidal ventilation at low airway pressures can augment lung injury. Am J Respir Crit Care Med. 1994;149:1327–1334. doi: 10.1164/ajrccm.149.5.8173774. [DOI] [PubMed] [Google Scholar]
- 145.Steinberg JM, Schiller HJ, Halter JM. Alveolar instability causes early ventilator-induced lung injury independent of neutrophils. Am J Resp Crit Care Med. 2004;169:57–63. doi: 10.1164/rccm.200304-544OC. [DOI] [PubMed] [Google Scholar]
- 146.Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol. 2004;202:145–156. doi: 10.1002/path.1491. [DOI] [PubMed] [Google Scholar]
- 147.Halter JM, Steinberg JM, Schiller HJ. Positive end-expiratory pressure after a recruitment maneuver prevents both alveolar collapse and recruitment/ derecruitment. Am J Respir Crit Care Med. 2003;167:1620–1626. doi: 10.1164/rccm.200205-435OC. [DOI] [PubMed] [Google Scholar]
- Adhikari NKJ, Burns KEA, Friedrich JO. Effect of nitric oxide on oxygenation and mortality in acute lung injury: systemiatic review and meta-analysis. BMJ. 2007;334:779–782. doi: 10.1136/bmj.39139.716794.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ali NN, Edgar AJ, Samadikuchaksaraei A. Derivation of type II alveolar epithelial cells from murine embryonic stem cells. Tissue Eng. 2002;8:541–550. doi: 10.1089/107632702760240463. [DOI] [PubMed] [Google Scholar]
- 149.Martin GS, Mangialardi RJ, Wheeler AP. Albumin and furosemide therapy in hypoproteinemic patients with acute lung injury. Crit Care Med. 2002;30:2175–2182. doi: 10.1097/00003246-200210000-00001. [DOI] [PubMed] [Google Scholar]
- 150.Zhai R, Gong MN, Zhou W. Genotypes and haplotypes of the VEGF gene are associated with higher mortality and lower VEGF plasma levels in patients with ARDS. Thorax. 2007;62:718–722. doi: 10.1136/thx.2006.069393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hudson LD. What happens to survivors of the adult respiratory distress syndrome. Chest. 1994;105:S123–S126. doi: 10.1378/chest.105.3_supplement.123s. [DOI] [PubMed] [Google Scholar]
- 152.Neff TA, Stocker R, Frey HR. Long-term assessment of lung function in survivors of severe ARDS. Chest. 2003;123:845–853. doi: 10.1378/chest.123.3.845. [DOI] [PubMed] [Google Scholar]
Pulmonary fibrosis
- 153.Davson J, Susman W. Apical scars and their relationship to siliceous dust accumulation in non-silicotic lungs. J Path Bact. 1937;45:597–612. [Google Scholar]
- 154.Butler C, Kleinerman J. The pulmonary apical cap. Am J Pathol. 1970;60:205–216. [PMC free article] [PubMed] [Google Scholar]
- 155.Yousem SA. Pulmonary apical cap – A distinctive but poorly recognized lesion in pulmonary surgical pathology. Am J Surg Pathol. 2001;25:679–683. doi: 10.1097/00000478-200105000-00018. [DOI] [PubMed] [Google Scholar]
- Yi E, Aubry MC. Pulmonary pseudoneoplasms. Arch Pathol Lab Med. 2010;134:417–426. doi: 10.5858/134.3.417. [DOI] [PubMed] [Google Scholar]
- 156.Negri EM, Montes GS, Saldiva PHN. Architectural remodelling in acute and chronic interstitial lung disease: fibrosis or fibroelastosis? Histopathology. 2000;37:393–401. doi: 10.1046/j.1365-2559.2000.00992.x. [DOI] [PubMed] [Google Scholar]
- 157.Basset F, Ferrans VJ, Soler P. Intraluminal fibrosis in interstitial lung disorders. Am J Pathol. 1986;122:443–461. [PMC free article] [PubMed] [Google Scholar]
- 158.Usuki J, Fukuda Y. Evolution of three patterns of intra-alveolar fibrosis produced by bleomycin in rats. Pathol Int. 1995;45:552–564. doi: 10.1111/j.1440-1827.1995.tb03503.x. [DOI] [PubMed] [Google Scholar]
- 159.Hogg JC. Chronic interstitial lung disease of unknown cause – a new classification based on pathogenesis. Am J Roentgenol. 1991;156:225–233. doi: 10.2214/ajr.156.2.1898791. [DOI] [PubMed] [Google Scholar]
- 160.Cohen AJ, King TE, Jr, Downey GP. Rapidly progressive bronchiolitis obliterans with organizing pneumonia. Am J Respir Crit Care Med. 1994;149:1670–1675. doi: 10.1164/ajrccm.149.6.8004328. [DOI] [PubMed] [Google Scholar]
- 161.Cordier JF. Organising pneumonia. Thorax. 2000;55:318–328. doi: 10.1136/thorax.55.4.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kuhn C, McDonald JA. The roles of the myofibroblast in idiopathic pulmonary fibrosis – ultrastructural and immunohistochemical features of sites of active extracellular matrix synthesis. Am J Pathol. 1991;138:1257–1265. [PMC free article] [PubMed] [Google Scholar]
- 163.Masson P, Riopelle JL, Martin P. Poumon rheumatismal. Ann Anat Path. 1937;14:359–382. [Google Scholar]
- 164.Kidd P. Some moot points in the pathology and clinical history of pneumonia. Lancet. 1912;i:1665–1670. [Google Scholar]
- 165.Floyd R. Organization of pneumonic exudates. Am J Med Sci. 1922;163:527–548. [Google Scholar]
- 166.Auerbach SH, Mims OM, Goodpasture EW. Pulmonary fibrosis secondary to pneumonia. Am J Pathol. 1952;28:69–87. [PMC free article] [PubMed] [Google Scholar]
- 167.Moya C, Anto JM, Taylor AJN. Outbreak of organising pneumonia in textile printing sprayers. Lancet. 1994;344:498–502. doi: 10.1016/s0140-6736(94)91896-1. [DOI] [PubMed] [Google Scholar]
- 168.Allen JN, Wewers MD. HIV-associated bronchiolitis obliterans organising pneumonia. Chest. 1989;96:197–198. doi: 10.1378/chest.96.1.197. [DOI] [PubMed] [Google Scholar]
- 169.Camus P, Lombard JN, Perrichon M. Bronchiolitis obliterans organizing pneumonia during treatment with acebutolol and amiodarone. Thorax. 1989;44:711–715. doi: 10.1136/thx.44.9.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mar KE, Sen P, Tan K. Bronchiolitis obliterans organizing pneumonia associated with massive l-tryptophan ingestion. Chest. 1993;104:1924–1926. doi: 10.1378/chest.104.6.1924. [DOI] [PubMed] [Google Scholar]
- 171.Crestani B, Kambouchner M, Soler P. Migratory bronchiolitis obliterans organizing pneumonia after unilateral radiation therapy for breast carcinoma. Eur Respir J. 1995;8:318–321. doi: 10.1183/09031936.95.08020318. [DOI] [PubMed] [Google Scholar]
- 172.Bayle JY, Nesme P, Bejui-Thivolet F. Migratory organizing pneumonitis ‘‘primed’’ by radiation therapy. Eur Respir J. 1995;8:322–326. doi: 10.1183/09031936.95.08020322. [DOI] [PubMed] [Google Scholar]
- 173.Stover DE, Milite F, Zakowski M. A newly recognized syndrome – Radiation related bronchiolitis obliterans and organizing pneumonia. Respiration. 2001;68:540–544. doi: 10.1159/000050566. [DOI] [PubMed] [Google Scholar]
- 174.Yousem SA, Colby TV, Carrington CB. Lung biopsy in rheumatoid arthritis. Am Rev Respir Dis. 1985;131:770–777. doi: 10.1164/arrd.1985.131.5.770. [DOI] [PubMed] [Google Scholar]
- 175.Rees JH, Woodhead MA, Sheppard MN. Rheumatoid arthritis and cryptogenic organising pneumonitis. Respir Med. 1991;85:243–246. doi: 10.1016/s0954-6111(06)80088-0. [DOI] [PubMed] [Google Scholar]
- 176.Yousem SA, Duncan SR, Griffith BP. Interstitial and airspace granulation tissue reactions in lung transplant recipients. Am J Surg Pathol. 1992;16:877–884. doi: 10.1097/00000478-199209000-00006. [DOI] [PubMed] [Google Scholar]
- 177.Spencer H. Interstitial pneumonia. Ann Rev Med. 1967;18:423–442. doi: 10.1146/annurev.me.18.020167.002231. [DOI] [PubMed] [Google Scholar]
- 178.Muller KM, Kriemann J, Stichroth E. Dendriform pulmonary ossification. Pathol Res Pract. 1980;168:163–172. doi: 10.1016/s0344-0338(80)80215-9. [DOI] [PubMed] [Google Scholar]
- 179.Pounder DJ, Pieterse AS. Dendriform pulmonary ossification. Arch Pathol Lab Med. 1987;111:1062–1064. [PubMed] [Google Scholar]
- 180.Joines RW, Roggli VL. Dendriform pulmonary ossification: report of two cases with unique findings. Am J Clin Pathol. 1989;91:398–402. doi: 10.1093/ajcp/91.4.398. [DOI] [PubMed] [Google Scholar]
- 181.Chow LTC, Shum BSF, Chow WH. Diffuse pulmonary ossification – a rare complication of tuberculosis. Histopathology. 1992;20:435–437. doi: 10.1111/j.1365-2559.1992.tb01016.x. [DOI] [PubMed] [Google Scholar]
- 182.Ikeda Y, Yamashita H, Tamura T. Diffuse pulmonary ossification and recurrent spontaneous pneumothorax in a patient with bronchial asthma. Resp Med. 1998;92:887–889. doi: 10.1016/s0954-6111(98)90397-3. [DOI] [PubMed] [Google Scholar]
- 183.Lara JF, Catroppo JF, Kim DU. Dendriform pulmonary ossification, a form of diffuse pulmonary ossification: report of a 26-year autopsy experience. Arch Pathol Lab Med. 2005;129:348–353. doi: 10.5858/2005-129-348-DPOAFO. [DOI] [PubMed] [Google Scholar]