

7.1. Occupational disease
Chapter Contents
Silicates 337
Inert dusts 337
Mixed-dust pneumoconiosis 337
Aluminium 351
Rare earth (cerium) pneumoconiosis 351
Hard-metal disease (cobalt lung) 351
Berylliosis 352
Polyvinyl chloride pneumoconiosis 354
Flock worker's lung 354
Popcorn worker's lung 355
Paint spraying 355
Mineral oils and petroleum 355
Welding 355
Toxic fumes and gases 355
Anoxic asphyxia 357
Occupational fevers 358
References 358
This chapter deals with the pneumoconioses, occupational asthma, occupational fevers and the effects o£ toxic fume and gases. Occupational diseases of the lung considered elsewhere include the effects of atmospheric pressure changes (see p. 368), extrinsic allergic alveolitis (see p. 279), carcinoma of the lung (see p. 534) and asbestos-induced pleural disease (see pp. 714–729). Dusty occupations in general are also associated with an increased risk of chronic obstructive pulmonary disease.1, 2
Pneumoconiosis: general features
Definition of pneumoconiosis
The term ‘pneumoconiosis’ is an abbreviation (and etymological corruption) of Zenker's pneumonokoniosis,3 which derives from pneumon (lung) and konis (dust) and therefore translates as dusty lung; in practice the term is confined to the effects of mineral dust on the lungs. Diseases caused by organic dusts are not included among the pneumoconioses and, in medicolegal practice at least, the presence of dust alone is insufficient to indicate pneumoconiosis: for compensation to be considered, the mineral dust must alter the structure of the lung and cause disability. The British Industrial Injuries Advisory Council defined pneumoconiosis as ‘permanent alteration of lung structure due to the inhalation of mineral dust and the tissue reactions of the lung to its presence, excluding bronchitis and emphysema’.4 Parkes recommends that cancer and asthma caused by mineral dust should also be excluded from the definition, an opinion with which we concur.5
Dust deposition in the lung
To reach the lung, dust particles have to be very small. Particle density and shape also affect the aerodynamic properties of dust. Host factors such as airflow characteristics, airway branching patterns and airway disease also affect dust deposition. Three deposition mechanisms are recognised (Fig. 7.1.1 ):
-
1
Inertial impaction: When air streams change direction or velocity, the inertia of the entrained particles causes them to maintain their original direction for a distance that depends upon their density and the square of their diameter. The same rules govern a car approaching a bend too fast: the car crashes into the outside of the bend.
-
2
Sedimentation (gravitational settlement): Under the influence of gravity, particles settle with a speed that is proportional to their density and the square of their diameter.
-
3
Diffusion: Very small airborne particles acquire a random motion as a result of bombardment by the surrounding gas molecules.
Figure 7.1.1.
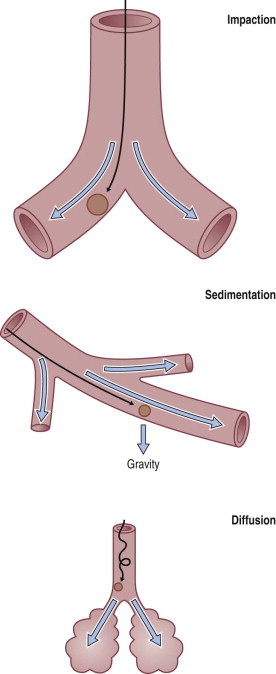
Mechanisms of particle deposition in the respiratory tract.
Inhaled dust particles are liable to sediment out in the alveoli if they have a diameter in the range of 1–5 µm, are roughly spherical in shape, and in density approximate to that of water. Larger or denser particles impact or precipitate on the walls of the conductive airways and are rapidly removed by ciliary action. Smaller particles may reach the alveoli but do not sediment so readily and many are therefore exhaled. Very small particles are deposited on the walls of alveoli by diffusion but because they are so small the total amount of dust deposited in this way is insignificant compared with that deposited by sedimentation (Fig. 7.1.2 ). Direct measurement shows that most lung dust (96%) has a particle diameter less than 2.5 µm.6
Figure 7.1.2.
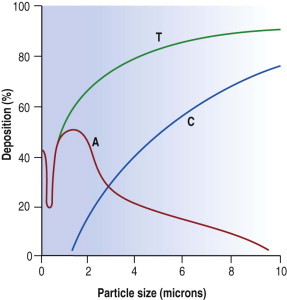
Percentage dust deposition in the respiratory tract according to particle diameter. The major alveolar impact is that of particles 1–3 µm diameter. Submicron particles are of negligible mass. T, total dust deposition; C, deposition on the ciliated epithelium; A, alveolar deposition.
Fibrous dust particles behave differently. Fibres over 100 µm in length may reach the alveoli if they are very thin and remain aligned with the air stream. Fibre penetration is inversely related to path length and the number of bifurcations.7 Tall people have longer conductive airways and experience more deposition in these sites than short people who have greater alveolar deposition for the same level of exposure.8
Slightly more dust is deposited in the right lung than the left, probably because the right main bronchus is more in line with the trachea, and is broader and shorter than the left, and carries 55% of the inhaled air.9, 10
Dust clearance from the lung
Inhaled dust that settles in the conductive airways is removed within a day or two by ciliary action. Only dust that reaches the alveoli is liable to cause pneumoconiosis and much of this is also removed, but the clearance rate here is much slower: many coalminers continue to expectorate mine dust years after retirement. Alveolar clearance is largely effected by macrophages, principally via the airways to the pharynx but also via lymphatics to the regional lymph nodes. The airway and interstitial routes interconnect at the bronchiolar level11 where some dust-laden macrophages leave the interstitium for the air space.12 This interconnection is probably the route utilised by circulating macrophages clearing other parts of the body of endogenous or exogenous particulate matter via the lung.13 Long asbestos fibres present a particular problem to macrophage clearance. Some minerals, notably chrysotile asbestos, undergo slow physicochemical dissolution in the lungs.
Only a small fraction of the inhaled dust gains access to the interstitium, a necessary step if it is to cause pneumoconiosis. Some free dust enters through the bronchus-associated lymphoid tissue11, 12 and some is taken up by, or pierces, the alveolar epithelium (Fig. 1.34, p. 21).14, 15, 16 Some of this is transported within hours to the hilar lymph nodes.17 So rapid is this translocation that it is thought not to involve phagocytes, although interstitial macrophages are undoubtedly important in continuing the transportation of dust to the nodes. Ultrafine dust particles are particularly liable to be transported across the alveolar epithelium.11 The integrity of the alveolar epithelium is very important to dust translocation from the air spaces to the interstitium. Much more dust reaches the interstitium if the epithelium is damaged.18, 19
It is widely thought that macrophages that have left the interstitium for the alveolar space never return,17, 20 but this is probably untrue.21 Heavily laden macrophages accumulate in alveoli bordering the terminal and respiratory bronchioles, eventually filling them completely. Erosion of the alveolar epithelium permits re-entry of these macrophages into the interstitium,22 very close to foci of bronchial mucosa-associated lymphoid tissue (MALT), which are found near the terminal bronchioles.23 These aggregates guard the mouths of lymphatics, which commence at this point; alveoli are devoid of lymphatics. Dust-laden interstitial macrophages accumulate in and around the bronchial MALT, which Macklin therefore referred to as dust sumps.24 Most pneumoconiotic lesions are found in the region of the dust sumps and are therefore focal. Asbestosis is diffuse rather than focal because the long asbestos fibres are not readily mobilised and cannot be concentrated in the centriacinar dust sumps. This is also seen on occasion with platy non-fibrous dusts such as talc, mica, kaolinite and feldspar.25, 26, 27, 28, 29, 30, 31, 32, 33 Within the dust sumps the dust particles are not static. They are constantly being freed and reingested by interstitial macrophages and, because these cells are mobile, successively inhaled dusts soon become intimately mixed.34 Macrophages play an important role in pneumoconiosis and if the dust is fibrogenic the repeated phagocytosis of indestructible mineral particles results in constant fibroblast stimulation.
The zonal distribution of pneumoconiosis
Pneumoconiosis affects both lungs but seldom evenly and some pneumoconioses show characteristic patterns of lung involvement. In most, the lesions are more numerous and better developed in the upper lobes than the bases but the reverse is true of asbestosis. The reasons for this are complex but undoubtedly involve the dust deposition:clearance ratio for the effect of the dust will depend upon both its amount and the duration of its stay in the lungs. There are well-recognised regional differences in the distribution and clearance of inhaled material, which in turn are dependent upon man's upright posture, the consequent gravitational forces being maximal at the apices.35 When standing at rest, the apices of the lungs are hardly perfused, so that lymph formation and clearance are much better at the bases.36, 37, 38 Similarly, the apices are relatively less well aerated; alveoli in the lower lobes receive more air than those in the upper lobes.37, 39 The greater respiratory excursions at the bases are thought to promote macrophage mobility there. It is to be expected therefore that the bases would both receive and clear more dust than the apices, rendering it difficult to predict on theoretical grounds which parts of the lungs carry the heaviest dust burden. In fact, more dust of all types is found in the upper lobes, the part most severely affected by every type of pneumoconiosis except asbestosis.40, 41 The predilection of asbestos to affect the periphery of the lower lobes is attributed to the dangerous long asbestos fibres preponderating there.41, 42
Pulmonary reactions to mineral dust
The main tissue reaction to mineral dust is fibrosis. Silica is highly fibrogenic and is therefore very likely to cause pneumoconiosis. Carbon is non-fibrogenic and therefore, unless there are complications, coal pneumoconiosis causes little disability. Tin too is harmless, and stannosis therefore unimportant, although the chest radiograph is highly abnormal because tin is very radiopaque. Stannosis is one of several terms that specify pneumoconiosis due to a particular mineral, the best known being silicosis, asbestosis and anthracosis. Table 7.1.1 summarises the various pulmonary reactions to mineral dust.
Table 7.1.1.
Pulmonary reactions to mineral dust
| Pulmonary reaction | Examples |
|---|---|
| Macrophage accumulation with a little reticulin deposition |
|
| Nodular or massive fibrosis |
|
| Diffuse fibrosis |
|
| Epithelioid and giant cell granulomas | Chronic berylliosis |
| Alveolar lipoproteinosis | ‘Acute’ silicosis, but also seen with heavy exposure to other dusts (see p. 317) |
| Small-airway disease | Various dusts (see p. 123) |
Identification of the dust
The blackness of carbon and red-brown colour of iron give ample evidence, both naked-eye and microscopically, of the type and amount of these dusts when they are present in the lung (Fig. 7.1.3 ), but other inorganic dusts may be more difficult to identify. However, a flick-out substage condenser and Polaroid filters to test for refractility and birefringence respectively are useful adjuncts that are too often neglected by the histopathologist. Crystalline silica is traditionally regarded as being only weakly birefringent, in contrast to silicates which generally show up brightly with simple crossed Polaroid filters.43 However, with modern microscope lamps, if the light source is set at high intensity when using Polaroid filters, both silica and silicates are birefringent.44 Mineralogists use polarising microscopy for analysis, but only by studying large polished crystals with controlled orientation of the light. The small dust particles found in tissue sections are too small to permit analysis by this technique but it is nevertheless very useful for detecting their presence (Fig. 7.1.4 ).
Figure 7.1.3.
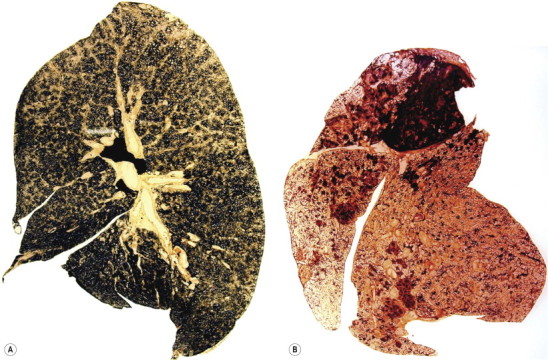
(A) Coal and (B) haematite miner's lungs. The respective black and red colours of these lungs give a good indication of their mineral content. Paper-mounted whole-lung sections.
(Courtesy of WGJ Edwards, London, UK.)
Figure 7.1.4.
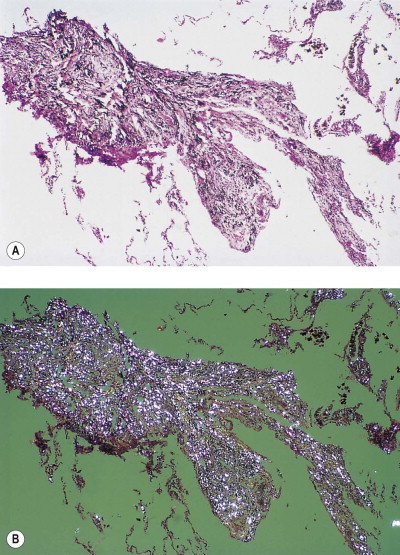
Talc pneumoconiosis. Only carbon is evident when the lesions are viewed with (A) non-polarised light, whereas the abundant talc particles are readily seen when (B) polarised light is employed.
Particle shape gives a useful indication of mineral type but appearances are sometimes deceptive: the plate-like crystals of talc are seldom observed as such, usually being viewed edge-on, when they appear to be needle-shaped. Occasionally, stains can be used to identify minerals, e.g. a modified Perls’ reaction for inhaled iron, and Irwin's aluminon stain for aluminium, but these too have largely been replaced by modern analytical techniques.
Microincineration combined with dark-field microscopy can also be used to demonstrate small particles. Incombustible mineral particles that cannot be seen with bright-field or polarising microscopy are rendered visible by this technique and their position on the slide can be compared with tissue reactions evident in a serial section that has not been incinerated. Microincineration has, however, also been largely replaced by modern analytical techniques that will now be considered.
Analytical electron microscopy is very helpful in identifying minerals, whether applied to lung digests or tissue sections.45, 46, 47, 48 Scanning electron microscopy permits the examination of thicker sections than transmission electron microscopy but does not detect very small particles. However, scanning electron microscopy allows more tissue to be examined and avoids the difficulty of cutting mineral particles with an ultramicrotome.
Mineral particles in a 5-µm thick deparaffinised section can be recognised in a scanning electron microscope set to collect the back-scattered electrons.46 The instrument can then be focused on points of potential interest and switched to X-ray diffraction, which provides information on crystal structure (Fig. 7.1.5 ). Alternatively, elemental analysis may be undertaken with either energy-dispersive or wavelength-dispersive X-ray spectroscopy. With energy-dispersive X-ray spectroscopy, all elements of atomic number above 11 are identified, whilst with wavelength-dispersive X-ray spectroscopy the section can be scanned for one particular element. With the former technique different elements are shown graphically as individual peaks, the heights of which are proportional to the amounts of the different elements within the particle studied, thereby giving information on probable molecular formula (Fig. 7.1.6 ). Thus, different silicates can be distinguished from each other and also from silica, which registers as pure silicon, oxygen (atomic number 8) not being detected. The fact that the elements of low atomic number that constitute organic chemicals are not detected means that any minerals present (except beryllium, atomic number 4) can be recognised easily in tissue sections. Only particles can be analysed however: elements present in only molecular amounts cannot be detected by X-ray analysis.
Figure 7.1.5.
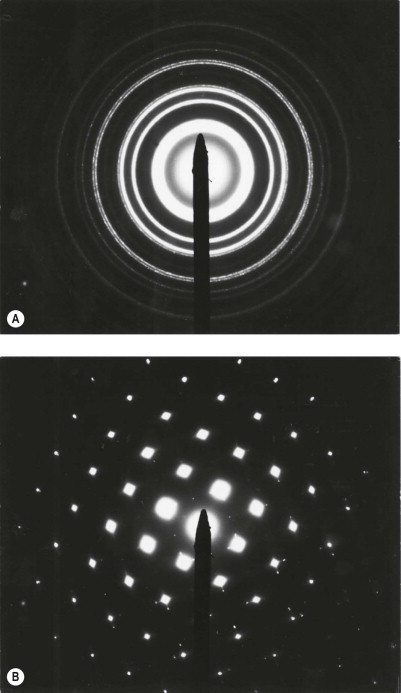
Electron diffraction patterns of gold, used for calibration purposes. The ring pattern (A) indicates that the material is polycrystalline and the spot pattern (B) indicates that it is a single crystal: amorphous materials give no regular pattern. The spacing of the rings gives information on crystalline structure and can be usefully applied to distinguish the various crystalline forms of silica (quartz, tridymite, cristobalite) for example.
(Courtesy of Dr M Wineberg, London, UK.)
Figure 7.1.6.
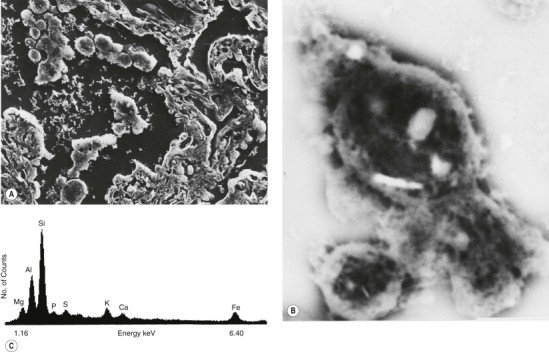
Dust particles in the lung: electron microprobe analysis. (A) The secondary (scanning) electron microscopic image of a deparaffinised 5-µm-thick section of lung showing clumps of macrophages in an alveolar lumen. (B) Back-scattered scanning electron microscopic image showing three of the macrophages at higher power: several bright particles worthy of further attention are evident. (C) X-ray energy spectrum of one of the particles showing it to be a silicate. The section had been transferred from a glass to a Perspex slide to avoid background signals, glass being siliceous. Each element emits a unique energy pattern when bombarded by electrons while the number of counts is in proportion to the amount of the particular element that is present in the particle.
(Courtesy of Professor DA Levison, Dundee, UK.)
The detection of trace amounts of substances such as beryllium requires bulk chemical analysis or techniques that are not widely available such as atomic absorption spectrometry, neutron activation analysis and microprobe mass spectrometry.49, 50 The last of these techniques can also provide molecular (as opposed to elemental) analysis of organic as well as inorganic particles.51 Another analytical technique of interest is microscopic infrared spectroscopy which provides data on the compound nature of microscopic particles in tissue sections (Fig. 7.1.7 ). Micro-Raman spectroscopy is also useful in this respect. Some metals cause hypersensitivity, which can be identified by exposing the patient's lymphocytes to metals and measuring their reaction in vitro.52
Figure 7.1.7.
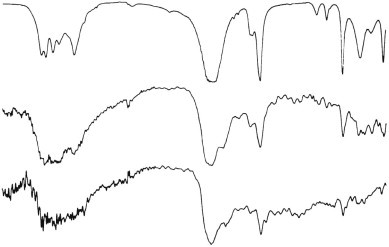
Infrared spectra of calcium oxalate. Top, reference standard; centre and bottom, crystals in human tissue sections.
(Courtesy of Professor DA Levison, Dundee, UK.)
Radiological grading of pneumoconiosis
A scheme for grading pneumoconiosis radiologically by comparison with standard radiographs has been adopted by the International Labour Organisation (ILO) and is widely used.53 Small opacities (up to 1 cm diameter) are graded by their profusion, 1, 2 and 3 indicating increasing numbers, and by their size, increasing through p, q and r if rounded and s, t and u if irregular. Type p opacities are described as punctiform and measure up to 1.5 mm in diameter; larger lesions up to 3 mm in diameter (type q) are described as micronodular or miliary; and those over 3 mm and up to 1 cm in diameter (type r) are described as nodular. Irregular opacities cannot be sized so accurately, s, t and u indicating fine, medium and coarse respectively. Large opacities (over 1 cm diameter) are graded by their combined size, increasing through A, an opacity measuring between 1 and 5 cm in diameter; B, one or more opacities whose combined area does not exceed the equivalent of one-third of the area of the right lung field (when they are regrouped in the mind's eye or measured with a transparent ruler); and C, one or more opacities whose combined area exceeds one-third of the area of the right lung field (when similarly regrouped). In coalworkers, small opacities (up to 1 cm diameter) correspond to simple coalworker's pneumoconiosis and large opacities (over 1 cm diameter) to complicated coalworker's pneumoconiosis, which is also known as progressive massive fibrosis.
Silicosis43
Mineralogy
Silicosis is caused by the inhalation of silica (silicon dioxide, SiO2), which is to be distinguished from the silicates, these being more complex compounds in which silicon and oxygen form an anion combined with cations such as aluminium and magnesium: talc, for example, is a hydrated magnesium silicate with the formula Mg3Si4O10(OH)2. The element silicon is also to be distinguished from the synthetic organic polymer silicone, used in implants.
Crystalline silica is highly fibrogenic whereas amorphous silica and silicates other than asbestos are relatively inert. Silica exists in several crystalline forms, of which quartz, cristobalite and tridymite are the most important: tridymite is the most fibrogenic and cristobalite more so than quartz.43, 54
Occupations at risk
Silicotic lesions have been identified in the lungs of Egyptian mummies, and the injurious effects on the lungs of inhaling mine dust have been recognised for more than 400 years. As long ago as the sixteenth century in Joachimsthal, Bohemia (now Jachymov, Czech Republic), diseases of miners’ lungs were attributed to the dust the miners breathed. Silicosis, tuberculosis and lung cancer are all now known to have been prevalent among the miners in this region, the cancer being largely attributable to the high level of radioactivity in the mines.
Silicosis was recognised in the UK soon after the discovery in 1720 that the addition of calcined flint to the clay from which china is made produced a finer, whiter and tougher ware. The preparation and use of this flint powder were highly dangerous, causing the condition known as potter's rot, one of the first of the many trade names by which silicosis has since been known. Aluminium oxide (alumina) now provides a safe, effective substitute for flint in this industry.
In 1830 it was noted that Sheffield fork grinders who used a dry grindstone died early, and amongst other preventive measures it was recommended that the occupation should be confined to criminals: fortunately for them, the substitution of carborundum (silicon carbide) for sandstone was effective enough. However, silicosis still occurs in some miners, tunnellers, quarrymen, stone dressers and metal workers.
Silica in one form or another is used in many trades – in the manufacture of glass and pottery, in the moulds used in iron foundries, as an abrasive in grinding and sandblasting, and as a furnace lining that is refractory to high temperatures. Rocks such as granite and sandstone are siliceous and their dusts are encountered in many mining and quarrying operations. In coal mining in the UK the highest incidence of the disease was in pits where the thinness of the coal seams required the removal of a large amount of siliceous rock, a process known as ‘hard heading’. In South Africa, silicosis causes a high mortality among the gold miners on the Witwatersrand, where the metallic ore is embedded in quartz. Slate is a metamorphic rock that contains both silica and silicates, and slateworkers develop both silicosis and mixed-dust pneumoconiosis.55, 56 Nor are rural industries immune from the disease, particularly if ventilation is inadequate, as it is in certain African huts where stone implements are used to pound meal and the occupants develop mixed-dust pneumoconiosis.57 Silicosis and mixed-dust pneumoconiosis have also been reported in dental technicians.58
Desert sand is practically pure silica but the particles are generally too large to reach the lungs. However, silicosis has been reported in inhabitants of the Sahara, Libyan and Negev deserts and those living in windy valleys high in the Himalayan mountains,59, 60, 61, 62, 63, 64, 65 whilst in California the inhalation of dust raised from earth has led to silicate pneumoconiosis in farm workers,66 horses67 and a variety of zoo animals.68
The silica in rocks such as granite, slate and sandstone is largely in the form of quartz and this is therefore the type of silica encountered in most of the industries considered above. Cristobalite and tridymite, which are possibly even more fibrogenic than quartz, are more likely to be encountered in the ceramic, refractory and diatomaceous earth industries where processing involves high temperatures.
Clinical features
Many workers with silicosis are asymptomatic. As a general rule, exposure to silica dust extends over many years, often 20 or more, before the symptoms of silicosis first appear: by the time the disease becomes overt clinically, much irreparable damage has been inflicted on the lungs. The initial symptoms are cough and breathlessness. From then onwards, respiratory disability progresses, even if the patient is no longer exposed to silica dust. Ultimately, there may be distressing dyspnoea with even the slightest exercise.
Silicosis sometimes develops more rapidly, perhaps within a year or so of first exposure. Such ‘acute silicosis’ was observed in the scouring powder industry in the 1930s when these cleansing agents consisted of ground sandstone mixed with a little soap and washing soda.69, 70 The additives were considered to have rendered the silica in the sandstone more dangerous but it is possible that the rapidity of onset of the disease merely reflected the intensity of the dust cloud to which the packers were exposed. Confusingly, the term ‘acute silicosis’ has since been applied to a further effect of heavy dust exposure in tunnellers, sand blasters and silica flour workers, namely pulmonary alveolar lipoproteinosis (see below),71, 72 whilst the terms ‘accelerated silicosis’ or ‘cellular phase silicosis’ have been substituted for ‘acute silicosis’ in referring to the rapid development of early cellular lesions.43, 73
The time from first exposure to the development of symptoms (the latency period) is inversely proportional to the exposure level. However, it is evident that a certain amount of silica can be tolerated in the lungs without fibrosis developing, indicating either a time factor in the pathogenetic process or a threshold dust load that has to be reached before fibrosis develops.
Pathological findings
Silica particles that are roughly spherical in shape and of a diameter in the range of 1–5 µm sediment out in the alveoli and are concentrated within macrophages at Macklin's dust sumps, as explained previously (see p. 27). Early lesions, as seen in so-called accelerated or cellular phase silicosis, consist of collections of macrophages separated by only an occasional wisp of collagen. The early lesions have been likened to granulomas and on occasion have been mistaken for Langerhans cell histiocytosis or a storage disorder, but Langerhans cells are scanty and the histiocytes contain dust particles rather than accumulated lipid or polysaccharide. The macrophages of the early lesion are gradually replaced by fibroblasts and collagen is laid down in a characteristic pattern. The mature silicotic nodule is largely acellular and consists of hyaline collagen arranged in a whorled pattern, the whole lesion being well demarcated (Fig. 7.1.8 ) and sometimes calcified. Small numbers of birefringent crystals are generally evident within the nodules when polarising filters are used, but these mainly represent silicates such as mica and talc, inhaled with the silica. Silica particles are generally considered to be only weakly birefringent,43 but fairly strong birefringence is evident in strong light (see above).44
Figure 7.1.8.
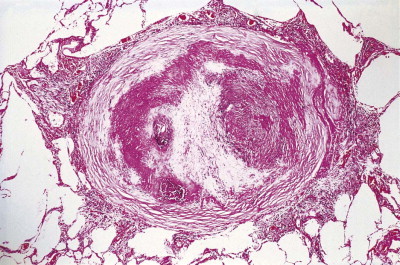
A silicotic nodule consisting of hyaline collagen arranged in a whorled pattern.
The silicotic nodules are situated in the centres of the pulmonary acini and are more numerous in the upper zones than the bases (Fig. 7.1.9A). They measure up to 5 mm across and are hard and easily palpable. They are grey if caused by relatively pure silica but black in coalminers and red in haematite miners.
Figure 7.1.9.
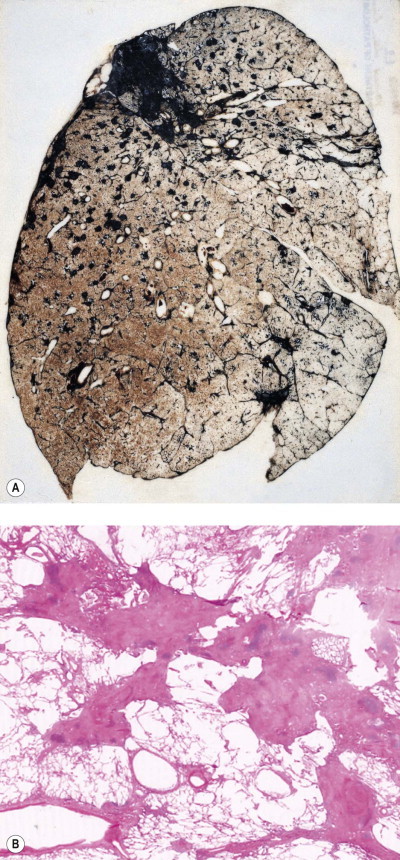
Silicosis. (A) The nodules are most numerous in the upper part of the lung where there is a conglomerate silicotic mass at one point. Paper-mounted whole-lung section. (B) Microscopy shows silicotic nodules scattered throughout otherwise normal lung tissue.
Silicotic nodules develop first in the hilar lymph nodes and are generally better developed there than in the lungs.74, 75, 76 Indeed, silicotic nodules are occasionally found in the hilar lymph nodes of persons who have no occupational history of exposure to silica and whose lungs are free of such lesions, the silica in the nodes being presumed to represent inhaled particles derived from quartz-rich soil.77 Severely affected lymph nodes often calcify peripherally, giving a characteristic eggshell-like radiographic pattern. This is sometimes the only radiological abnormality.76 Such enlarged lymph nodes may occasionally press upon and obstruct adjacent large bronchi78 or result in a left recurrent laryngeal nerve palsy,79 so simulating malignancy.80 Sometimes the nodules develop within the walls of major bronchi, occasionally causing a middle-lobe syndrome (see p. 92).81 Silicotic nodules are also found along the lines of the pleural lymphatics75, 82 where they have been likened to drops of candle wax on the visceral pleura. Very rarely, silica-induced fibrosis is more pronounced in the pleura than in the lungs.83
Lung tissue between the nodules is often quite normal and not until the process is very advanced is there any disability (Fig. 7.1.9B). In severe cases large masses of fibrous tissue are formed, which may undergo central necrosis and cavitation (Fig. 7.1.10 ).84 On close inspection it is evident that these consist of conglomerations of many silicotic nodules closely packed together. In such severe cases cor pulmonale develops. Occasionally, silicotic nodules develop in the abdominal as well as the thoracic lymph nodes, and in the liver, spleen, peritoneum and bone marrow.85, 86, 87, 88, 89
Figure 7.1.10.
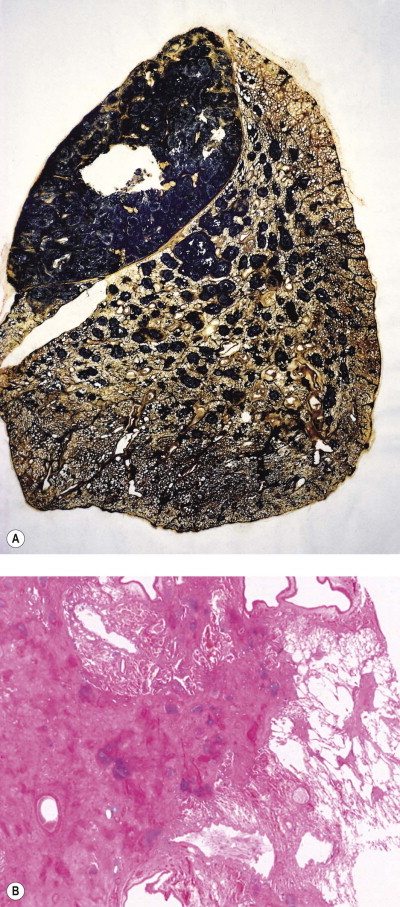
Silicosis. (A) The nodules are larger than in Figure 7.1.9a and have fused together. Cavitating conglomerate silicosis destroys most of the upper lobe. Paper-mounted whole-lung section. (B) Microscopy of conglomerate silicosis.
In about 10% of cases, the typical pulmonary nodules that predominantly affect the upper lobes are accompanied by diffuse fibrosis that is maximal in the lower lobes.27, 33, 90, 91, 92 The latter may show ‘honeycombing’ and closely resemble idiopathic pulmonary fibrosis. The association is too common to be explained by chance and the diffuse fibrosis is therefore regarded as a further manifestation of the pneumoconiosis, possibly due to an interaction between the dust and the immunological factors discussed below.
Pathogenesis
The pathogenesis of silicosis has excited much interest and many different theories have been advanced over the years. An early theory held that the hardness of the silica was responsible, but this was discounted by the observation that silicon carbide (carborundum) is harder than silica but is non-fibrogenic. Theories based on the piezoelectric property and on the solubility of silica were successively abandoned although the latter had a long period of popularity. It gained support from Kettle's experiments which showed that fibrosis developed about chambers placed in an animal's peritoneal cavity if the chambers contained silica powder sealed in by a collodion membrane through which solutes such as silicic acid could pass. However, it was later shown that the pores in a collodion membrane are quite irregular in size and when the experiments were repeated using chambers guarded by millipore membranes, no fibrosis developed, despite solutes being able to diffuse out.93 The solubility theory also fails to take account of the differing fibrogenicity of the various forms of silica despite them being of similar solubility.54 Furthermore, if the outer, more soluble layer of the particles is removed by etching, fibrogenicity is increased although solubility is decreased. In line with this, freshly fractured crystalline silica is more pathogenic in every respect than its aged equivalent,94 which may partly explain the severity of silicosis in trades such as sandblasting. These observations suggest that the fibrogenicity of silica is connected with its surface configuration.
It is now known that uptake of the silica by macrophages is necessary for silicosis to develop. If silica and macrophages are enclosed together in peritoneal millipore chambers, a soluble product of the macrophages diffuses out and causes fibrosis. This observation led to the realisation that the fibrogenicity of the various crystalline forms of silica correlated well with their toxicity to macrophages and for a time macrophage death was thought to be necessary.95 It is now considered that before the macrophages are killed by the ingested silica, they are stimulated to secrete factors that both damage other constituents of the lung and promote fibrosis.96, 97, 98, 99, 100, 101, 102 Transforming growth factor-β is one fibrogenic factor that has been implicated in the pathogenesis of silicosis.103, 104, 105
Toxic damage to macrophages is due to silica particles injuring the phagolysosomal membranes, so releasing acid hydrolases into the cytoplasm.95 It is important in the pathogenesis of the disease indirectly because when the macrophage crumbles, the silica particles are taken up by fresh macrophages and the fibrogenic process continues. It has been suggested that early involvement of the hilar lymph nodes in the fibrogenic process promotes the development of the disease in the lung by delaying dust clearance.74
Immunological aspects of silicosis
Immunological factors have been implicated in the pathogenesis of silicosis because many patients with silicosis have polyclonal hypergammaglobulinaemia, rheumatoid factor or antinuclear antibodies, and because there is a well-recognised association between autoimmune diseases such as systemic sclerosis and rheumatoid disease and exposure to silica.43, 106, 107, 108, 109 The relation of immunity to dust exposure appears to be a reciprocal one: on the one hand, the presence of dust results in rheumatoid lesions in the lungs being more florid (see Caplan's syndrome, p. 341), whilst on the other, non-specific immunisation of rabbits with horse serum results in experimental silicotic lesions being larger and more collagenous.110 It is doubtful whether pneumoconiosis and autoimmune disease play a causative role in each other but one seems to aggravate the other and may lead to its earlier development.
Tuberculosis complicating silicosis (silicotuberculosis)
One of the commonest and most feared complications of silicosis is chronic respiratory tuberculosis.109 Once this infection has been added to the silicosis, the prognosis rapidly worsens. It is thought that in the presence of silica, the tubercle bacilli proliferate more rapidly because the ingested silica particles damage phagolysosomal membranes and thereby interfere with the defensive activity of the macrophages. The synergistic action of silica dust has long been held responsible for the inordinately high incidence of respiratory tuberculosis in mining communities. Many former South African gold miners now have acquired immunodeficiency syndrome (AIDS) as well as silicosis and tuberculosis has consequently reached almost epidemic proportions amongst these men. Phagocyte damage by ingested dust particles may also cause some cases of chronic necrotising aspergillosis complicating pneumoconiosis.111
Silica-induced lung cancer
A series of studies suggesting that there might be a link between silica inhalation and lung cancer was reviewed by the International Agency for Research on Cancer in 1987, leading to the conclusion that the evidence for carcinogenicity of crystalline silica in experimental animals was sufficient, while in humans it was limited.112 Subsequent epidemiological publications were reviewed in 1996, when it was concluded that the epidemiological evidence linking exposure to silica to the risk of lung cancer had become somewhat stronger113 but that in the absence of lung fibrosis remained scanty.113 The pathological evidence in humans is also weak in that premalignant changes around silicotic nodules are seldom evident.114 Nevertheless, on this rather insubstantial evidence, lung cancer in the presence of silicosis (but not coal or mixed-dust pneumoconiosis) has been accepted as a prescribed industrial disease in the UK since 1992.115 Some subsequent studies have provided support for this decision.116 In contrast to the sparse data on classic silicosis, the evidence linking carcinoma of the lung to the rare diffuse pattern of fibrosis attributed to silica and mixed dusts is much stronger and appears incontrovertible.33, 92
Alveolar lipoproteinosis in response to heavy dust exposure
A further complication of exposure to silica is the development of alveolar lipoproteinosis (see p. 317).71, 72, 117, 118 Very heavy experimental exposure to silica, and indeed other dusts, stimulates hypersecretion of alveolar surfactant to such an extent that the normal clearance mechanism is overwhelmed.119, 120, 121, 122, 123, 124, 125 Alveolar macrophages are enlarged by numerous phagolysosomes distended by lamellar bodies that represent ingested surfactant. The alveoli are filled by such cells and, having a foamy cytoplasm, they produce the appearances of endogenous lipid pneumonia, similar to that more usually encountered as part of an obstructive pneumonitis distal to a bronchial tumour. The macrophages gradually disintegrate and the free denatured surfactant slowly becomes compacted, during which time its staining with both eosin and the periodic acid–Schiff reagents intensifies until the appearances are finally those of alveolar lipoproteinosis. This process prevents the aggregation and concentration of the dust in the usual foci and thereby hinders the development of silicosis. Lipoproteinosis and silicosis may be seen in conjunction but, more often, different areas of the lung show one or the other. The lipoproteinosis has its own severe impact on lung function, but, unlike silicosis, is potentially reversible (by massive alveolar lavage).
Silica-induced renal disease
Occasional patients exposed to silica develop renal disease.126, 127, 128, 129 Two mechanisms appear to operate. First, translocation of silica particles from the lungs leads to their deposition in the renal interstitium with resultant nephrotoxity. Second, silica stimulates an autoimmune response characterised by the formation of various antibodies, notably rheumatoid factor and antinuclear antibodies, which leads to the development of immune complex-mediated glomerulonephritis.128, 129
Amorphous silica
Manmade submicron forms of silica, variously known as amorphous, vitreous, colloidal, synthetic or precipitated silica, are widely used in industry. They consist of pure non-crystalline silicon dioxide. Particle size ranges from 5 to 200 nm but aggregates of the particles measure from 1 to 10 µm. Industrial surveys suggest that inhalation of such dust is harmless, observations that are in accord with the results of animal experiments.54
Diatomaceous earth (keiselguhr)
An amorphous silica is the principal component of the fossilised remains of diatoms that constitute the sedimentary rock, diatomite (Fig. 7.1.11 ). This is generally obtained by open-cast mining, following which the rock is crushed and calcined. The calcined product is used in filters, insulation material and as a filler. Being amorphous, the silica in diatomite is harmless, but calcining (>1000°C) results in its conversion to crystalline forms of silica. Diatomaceous earth pneumoconiosis is unusual and its risk appears to be related to the amount of cristobalite and tridymite (two forms of crystalline silica) produced in the calcining process.130
Figure 7.1.11.

Diatomaceous earth. (A) Scanning electron micrograph showing the calcified diatoms, ×15 000. (B) Electron microprobe analysis shows only silicon (Si); the oxygen with which silicon is combined in silica (silicon dioxide, SiO2) is of too low an atomic number to register. The titanium (Ti) peak derives from the sample holder.
(Courtesy of Dr D Dinsdale, Leicester, UK and Professor B Nemery, Leuven, Belgium.)
Silicates43
The silicates are complex compounds in which silicon and oxygen form an anion combined with cations such as aluminium and magnesium: talc, for example, is a hydrated magnesium silicate with the formula Mg3Si4O10(OH)2. Silicates include fibrous forms (asbestos and the zeolites), plate-like forms (talc and mica) and clays (kaolinite and fuller's earth). In histological sections, the platy talc and mica particles are generally cut tangentially and therefore appear needle-shaped (see Fig. 7.1.4). They are strongly birefringent whereas the clays are only weakly so. Talc particles in the lung exceeding 5 µm in length should arouse suspicion of intravenous drug abuse.131
Of the fibrous silicates, zeolite is used as a building material in certain communities, notably in central Turkey. Pneumoconiosis is not a problem but zeolites are of medical interest because, like asbestos, they present a mesothelioma risk. Asbestos is dealt with separately (see below).
Pneumoconiosis has been described with various non-fibrous silicates, notably in the rubber industry, which uses talc and, less commonly, mica as lubricants. Other occupations posing a risk include the extraction of kaolinite from china clay (kaolin),27, 132, 133 and in the open-cast and underground mining of fuller's earth (montmorillonite, bentonite and attapulgite clays, which were originally used in ‘fulling’ (degreasing) wool).134, 135 However, all these substances are commonly contaminated with silica, asbestos or both, and it has been questioned whether in pure state they are at all fibrogenic. The modifying effect of inert substances such as iron on that of silica is well known (see mixed-dust pneumoconiosis, below) and it has been suggested that talc, mica and fuller's earth act in a similar way in regard to their more fibrogenic contaminants, the pneumoconioses attributed to them in reality representing mixed-dust pneumoconiosis or asbestosis. Contrary evidence comes from reports of pulmonary fibrosis in persons heavily exposed to pure talc, mica or kaolin.32
All these silicates are evident in the tissues as plate-like birefringent crystals which often provoke a foreign-body giant cell reaction (see Fig. 7.1.3) and may result in fibrotic nodules. Large focal lesions resembling the progressive massive fibrosis of coalworkers may be produced, and also a diffuse ‘asbestosis-like’ form of pneumoconiosis, the latter attributed to poor macrophage mobilisation of the plate-like particles.27, 28, 29, 30, 31, 32, 132, 133, 134, 135, 136, 137, 138 It would appear therefore that silicates are indeed fibrogenic if enough is inhaled; they appear to vary in fibrogenicity but in all cases they are less fibrogenic than silica.
Inert dusts
Inert dusts are non-fibrogenic and therefore of little clinical consequence, although elements of high atomic number can give rise to a striking chest radiograph.139 It should be noted however that inert or lowly fibrogenic materials may be associated with substances of medical importance, for example, kaolin, bentonite and barytes (barite) may all be contaminated with silica27, 134, 140 and talc may be contaminated with asbestos.
The best known of the inhaled inert mineral dusts is carbon while, of the remainder, iron is the most widespread. Others include tin and barium. With all these dusts, particles retained in the lung are gathered at Macklin's dust sumps by heavily laden macrophages which are lightly bound together there by a few reticulin fibres. Collagen is not formed and the worker suffers no ill-effects. The lungs take on the colour of the dust and in siderosis assume a deep brick-red hue.
Carbon deposition is commonly found in the lungs, particularly those of city dwellers and tobacco smokers. It is also the principal constituent of coal, which is dealt with separately below, and large amounts of pure carbon may be inhaled by workers involved in the manufacture of carbon black, carbon electrodes and charcoal.141, 142, 143, 144 Although carbon is regarded as being non-fibrogenic, the very heavy lung burdens encountered in industries such as these may lead to the complicated form of pneumonconiosis known as progressive massive fibrosis that is more commonly encountered in coal workers (see p. 340). Heavy pure carbon deposition may also be acquired domestically when wood is burnt in buildings devoid of a chimney, so-called ‘hut lung’,145 a term that is also applied to the domestic acquisition of carbon mixed with silica or silicates, resulting in forms of mixed-dust pneumoconiosis.57, 64, 65
Anthracofibrosis is a term introduced by Chinese bronchoscopists for bronchial stenosis or obliteration associated with carbon pigmentation of the mucosa.146 Although the original description incriminated tuberculosis, mixtures of various mineral dusts acquired at work or domestically are a more likely cause.147, 148, 149, 150
Iron dust in the lungs was first described by Zenker in 1867, when he also introduced the terms siderosis and pneumonokoniosis.3 Zenker was describing a woman who coloured paper with iron oxide powder (‘rouge’), a substance which is still encountered by some workers engaged in polishing silver, glass, stone and cutlery. Siderosis is also found in welders, iron foundry fettlers, steel workers, boiler scalers and haematite miners and crushers. Iron dust particles are reddish-brown but in the lung may be masked by carbon151: when evident, or revealed by microincineration, they resemble haemosiderin and generally give a positive Perls’ reaction, but particularly with haematite, heat (60–80oC) and concentrated (12N) hydrochloric acid may be necessary.152
Haematite miners in both the UK (Cumbria) and France (Lorraine) have an increased risk of bronchial carcinoma, but radon gas rather than haematite is the suspected carcinogen. Radon is a decay product of uranium. Minute amounts are present in all rocks but local concentrations occur and these are liable to build up in mines if ventilation is limited.
Silver, as well as iron, is found in the lungs of silver polishers, where it stains elastin in alveolar walls and pulmonary vessels grey. Such argyrosiderosis is as harmless as siderosis.
Tin miners are subject to silicosis but not stannosis because the ore, which is found in association with siliceous rocks, contains only low concentrations of the metal. Tin smelters, on the other hand, and factory workers exposed to high concentrations of tin dust or fume, are liable to inhale large amounts of this inert metal and develop the striking chest radiograph of stannosis. They remain in good health however for tin is completely non-fibrogenic. Tin particles in the lung resemble carbon but are strongly birefringent and remain after microincineration: microprobe analysis provides positive identification.
Other inert dusts include barium, which also has a high atomic number and is therefore radiopaque,139 and minerals of low radiodensity such as limestone, marble and cement (all chiefly composed of calcium carbonate) and gypsum (hydrated calcium sulphate). However, the extraction of barium ore (almost entirely in the form of barium sulphate, which is known as barytes in Europe and barite in the USA) may entail exposure to silica and silicates. Pure baritosis resembles stannosis and siderosis.
Mixed-dust pneumoconiosis
The term ‘mixed-dust pneumoconiosis’ refers to the changes brought about by inhaling a mixture of silica and some other less fibrogenic substance such as iron, carbon, kaolin or mica.33, 151, 153, 154, 155 The proportion of silica is usually less than 10%. Typical occupations include foundry work and welding and the mining of coal, haematite, slate, shale and china clay.
The action of the silica is modified and, although fibrotic nodules are formed, they lack the well-demarcated outline and concentric pattern of classic silicosis. The lesions are found in a centriacinar position and are stellate in outline with adjacent scar emphysema. They are firm and generally measure no more than 5 mm in diameter. They closely resemble the fibrotic nodules of simple coal pneumoconiosis (see below). Confluent lesions also occur on occasions. These resemble the progressive massive fibrosis of coalworkers and appear to represent a single large lesion rather than a conglomeration of individual nodules, as in advanced silicosis. Abundant dust is generally evident in lesions of all sizes; this consists of black carbon or brown iron mixed with crystals of varying degrees of birefringence, silicates generally being strongly birefringent and silica weakly so. Calcification is unusual. Mixed-dust pneumoconiosis carries an increased risk of pulmonary tuberculosis, but not to the same degree as silicosis. In some cases the stellate nodules are accompanied by diffuse fibrosis, as in silicosis and again possibly involving interactions between the dust and immunological factors. Involvement of the bronchi with consequent stenosis (so-called anthracofibrosis) is described above.
Coal pneumoconiosis156
The term ‘anthracosis’ was initially applied to changes observed in a coalminer's lung157 but is now often extended to include the common carbon pigmentation of city dwellers’ lungs, and the term ‘coal pneumoconiosis’ is more appropriate to a special form of pneumoconiosis to which coalworkers are subject, particularly those who work underground. The principal constituent of coal, carbon, is non-fibrogenic, so suspicion has naturally fallen on the ash content of mine dust, some of which derives from the coal, some from adjacent rock strata and some from stone dust laid in the roadways to minimise the risk of coal dust explosions. Coal itself appears to be the responsible agent because coal-trimmers, working in the docks and not exposed to rock dust, also develop the disease.158 Coalminers encountering siliceous rock are, of course, also liable to develop silicosis like other underground workers.
Mineralogy
Coal consists largely of elemental carbon, oxygen and hydrogen with traces of iron ore and clays such kaolinite, muscovite and illite, but no silica. The mineral content varies with the type and rank (calorific value) of the coal. All coal derives from peat, the youngest type being lignite and the oldest anthracite, with bituminous (house) coal in between. As it ages, the oxygen and mineral constituents diminish and the coal hardens. Lignite is soft and said to be of low rank, anthracite hard and of high rank, with bituminous coal intermediate.
Although high-rank coal is of low mineral content, its dust is more toxic to macrophages in vitro and is cleared more slowly in vivo. This observation may explain why, in the UK, high-rank coal is associated with a higher prevalence of coal pneumoconiosis.
The low mineral content of high-rank coal is reflected in the mineral content of the lungs of those who hew such coal in the UK, but in the Ruhr, in Germany, and in Pennsylvania, in the USA, anthracite miners’ lungs contain more silica than those who hew bituminous coal, the silica presumably deriving from other sources. Not surprisingly, the presence of silica is reflected in the tissue reaction to the inhaled dust, resulting in a more fibrotic reaction very analogous to mixed-dust pneumoconiosis. A spectrum of changes is therefore encountered in coalminers’ lungs, ranging from coal pneumoconiosis through mixed-dust pneumoconiosis to silicosis; the findings in any individual depend upon the nature of the coal being mined and the type of work undertaken.
In high-rank British collieries the development of coal pneumoconiosis appears to depend on the total mass of dust inhaled, whereas in low-rank British collieries the mineral content of the lung dust appears to be more important.159 This may explain apparently contrary data drawn from different coalfields – data based on coals of different composition that are not strictly comparable. Some workers have stressed the importance of silica in the dust whereas others, particularly in the high-rank coalfields of south Wales, have been unable to detect any association between silica and the level of pneumoconiosis. Both findings may be correct, but only for the particular group of miners examined in each case.160
Pathology
The lesions of coal pneumoconiosis are generally focal and fall into one or other of two major types, simple and complicated, depending upon whether the lesions measure up to or over 1 cm; simple corresponds to categories 1–3 of the ILO grading system (see p. 331) and complicated, which is also known as progressive massive fibrosis, to ILO categories A–C. More diffuse interstitial fibrosis has been reported in about 16% of Welsh and West Virginian coalminers, usually involving those carrying a particularly heavy dust burden; it runs a more benign course than non-occupational interstitial fibrosis (idiopathic pulmonary fibrosis).161 Similar findings have been reported from France.162
Simple coal pneumoconiosis consists of focal dust pigmentation of the lungs, which may be associated with a little fibrosis and varying degrees of emphysema. Its clinical effects are relatively minor. Some degree of black pigmentation (anthracosis) of the lungs is common in the general urban population, especially in industrial areas, but much denser pigmentation is seen in coalminers, whose lungs at necropsy are black or slate-grey. Black pigment is evident in the visceral pleura along the lines of the lymphatics and on the cut surface where it outlines the interlobular septa and is concentrated in Macklin's centriacinar dust sumps (Fig. 7.1.12 ). The dust is generally more plentiful in the upper parts of the lungs and in the hilar lymph nodes, possibly due to poorer perfusion and consequently poorer lymphatic drainage there (see p. 21).162
Figure 7.1.12.

A coalminer's lung showing heavy dust deposition in the centres of the acini (Macklin's dust sumps). On palpation the dust deposits felt soft and were consequently described as macules rather than nodules. Despite the heavy dust deposition, there is minimal pneumoconiosis. Part of a paper-mounted whole-lung section.
Two forms of coal dust foci are recognised, macules and nodules, the former being soft and impalpable and the latter hard due to substantial amounts of collagen. Both lesions are typically stellate but the more fibrotic the nodules, the more rounded they become, until it is difficult to distinguish them macroscopically from those of silicosis. In these circumstances reliance has to be placed on the whorled pattern of the collagen that is evident microscopically in silicosis. The stellate nodules are analogous to those seen in mixed-dust pneumoconiosis caused by mixtures of silica and inert dusts other than carbon (see above). With polarising filters, small numbers of birefringent crystals may be seen in both macules and nodules, usually representing mica or kaolinite derived from rock that bordered the coal.
Macules consist of closely packed dust particles, free or within heavily laden macrophages, so that the lesion appears black throughout (Fig. 7.1.13 ). Appropriate stains show that the dust-laden macrophages and free dust are lightly bound by reticulin. Very little collagen is evident. Although striking in their appearance, dust macules are thought to have little effect on lung function.
Figure 7.1.13.
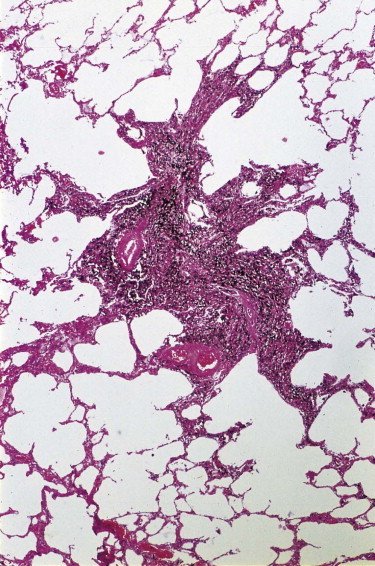
Coal macule. A centriacinar dust deposit of stellate outline consisting of macrophages heavily laden with coal dust and bound together by only a little reticulin. Collagen is not a feature.
Nodules contain substantial amounts of collagen and are thought to have an adverse, but limited, effect on respiration. They vary from a heavily pigmented, stellate lesion, which apart from its collagen content resembles the dust macule (Fig. 7.1.14 ), to one that is less pigmented and more circumscribed. The stellate, heavily pigmented type of nodule is seen in lungs that have a relatively low ash content whilst the more rounded and less pigmented nodule is seen in lungs with relatively high ash loads.163
Figure 7.1.14.
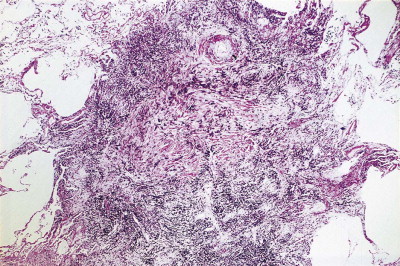
Coal nodule. A centriacinar dust nodule of stellate outline in which coal dust-laden macrophages are mixed with moderate amounts of collagen.
Radiologically (see p. 321), p-type opacities correspond to macules, q-type opacities to the stellate nodules that resemble those of mixed-dust pneumoconiosis and r-type opacities to the rounded nodules that resemble those of silicosis.53, 164 Thus, the radiological changes of simple coalworker's pneumoconiosis are due to the dust and the small amount of collagen present and do not reflect any emphysema that may also be present. However, pulmonary dust foci are often associated with emphysema (Fig. 7.1.15 ) and the severity of the emphysema appears to correlate with the dust load. The prevalence of chronic bronchitis and emphysema is high in the coal industry and it has long been debated whether occupation or cigarette smoking is the major factor contributing to emphysema in coalminers.165, 166, 167, 168 As well as mineral dust, nitrous fumes from shot-firing form another occupational hazard of coal mining.
Figure 7.1.15.
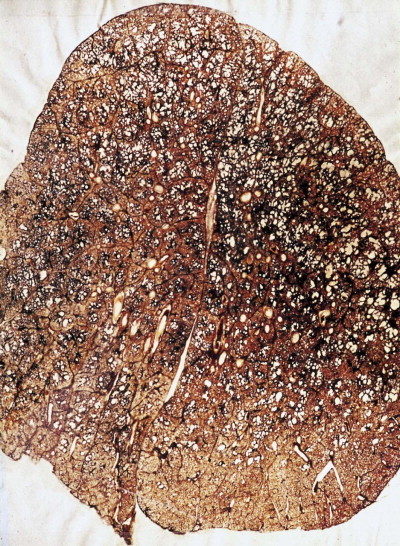
Coalworker's pneumoconiosis – simple type, consisting of coal macules and nodules associated with focal emphysema. Paper-mounted whole-lung section.
Heppleston made a special study of the emphysema found in coalminers, claiming that it differs from centriacinar emphysema, as seen in smokers in the general population, and attributing it to the dust.169 He introduced the term ‘focal emphysema of coalworkers’ to describe this special process. Others find it very difficult to identify any convincing difference between the emphysema of coalworkers and that encountered outside the industry but Heppleston based his claims on the study of serial sections. By this means he showed that, although both forms affect respiratory bronchioles, the focal emphysema of coalworkers affects more proximal orders of these airways and is not associated with the bronchiolitis seen with centriacinar emphysema. Furthermore, focal emphysema is a dilatation lesion whereas centriacinar emphysema involves destruction of adjacent alveolar walls. By definition, therefore, focal emphysema is not a true emphysema at all (see p. 102). However, it has been shown that mineral dusts cause elastin and collagen breakdown in the rat lung.170 Focal emphysema may progress to the destructive centriacinar form and this has strengthened claims that mine dust plays a causal role in centriacinar emphysema.1, 171, 172, 173, 174, 175, 176 In the UK, these claims have been accepted and chronic bronchitis and emphysema in coalminers and metal production workers have been accepted as prescribed industrial diseases since 1992.177 In Germany too, chronic obstructive pulmonary disease is now compensatable as an occupational disease. The conditions for compensation in the UK were initially:
-
•
underground coal mining for a minimum of 20 years in aggregate
-
•
forced expiratory volume in 1 second at least 1 litre below that expected or less than 1 litre in total
-
•
radiological category of at least 1/1.
However the last of these criteria has now been dropped. The inclusion of a time element and the omission of some estimate of dust load (such as radiological category) have been criticised, with some justification.178 As with lung cancer caused by chromates benefit is paid irrespective of smoking habits.
Whereas simple coal pneumoconiosis, particularly the macular variety, has little effect on lung function, complicated coal pneumoconiosis, also known as progressive massive fibrosis, can have very serious consequences. Particularly when the lesions are large, it is associated with productive cough, breathlessness, significant impairment of lung function and premature death. The major factor accounting for the development of progressive massive fibrosis appears to be the sheer bulk of coal dust in the lung, rather than coal rank or the silica content of the mine dust.179 Progressive massive fibrosis has occasionally been recorded in dockers loading silica-free coal into the holds of ships158 and in workers exposed to pure carbon in the manufacture of carbon black and carbon electrodes.141, 142, 143
Progressive massive fibrosis is characterised by large (over 1 cm) black masses, situated anywhere in the lungs but most common in the upper lobes. The lesions may be solitary or multiple and very large, occupying most of the lobe and even crossing an interlobar fissure to involve an adjacent lobe (Figs 7.1.3B, 7.1.16 ). They cut fairly easily, often with the release from a central cavity of black fluid flecked by cholesterol crystals. For many years it was believed that the condition was the result of synergism between mycobacterial infection and dust but the failure of the attack rate to decrease as tuberculosis declined negated this view.180 Today, more emphasis is placed on total dust load for the lesions tend to affect lungs that carry an unduly heavy dust burden. If the remainder of the lung shows little evidence of dust accumulation, the possibility of the masses representing Caplan-type lesions (see below) should be considered.
Figure 7.1.16.
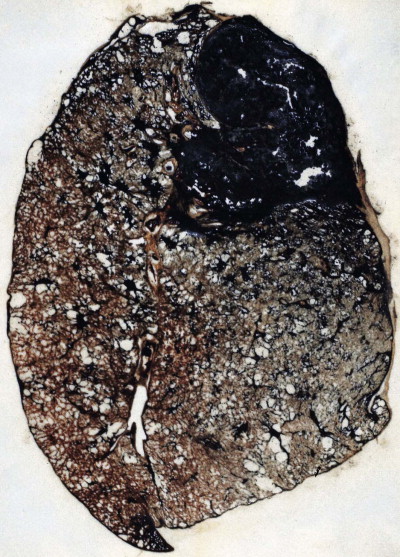
Coal pneumoconiosis – complicated type. In addition to the focal dust deposits there is a large area of progressive massive fibrosis in the upper lobe. Paper-mounted whole-lung section.
Microscopically, the lesions consist of dust and connective tissue intermixed in a random fashion. Central necrosis and cavitation commonly occur. The necrosis is thought to be ischaemic.181 It is amorphous or finely granular, and eosinophilic apart from abundant dust particles and cholesterol crystal clefts. The fibrotic component in a complicated pneumoconiotic lesion is rich in fibronectin, with collagen only more abundant at the periphery.182 Two types of progressive massive fibrosis are recognisable, corresponding to the two types of nodule described in simple coal pneumoconiosis.163 The first appears to have arisen by enlargement of a single nodule, whereas the second is a conglomeration of individual lesions, each of which corresponds to the more circumscribed type of nodule seen in simple coal pneumoconiosis. The ash content of the lungs bearing these two types of progressive massive fibrosis varies in the same way as with the two types of simple pneumoconiotic nodules, the enlarged single lesion being found in lungs with a relatively low ash content, and the conglomerate lesion in lungs with a relatively high ash content. The second type resembles the conglomerate nodules of large silicotic lesions but lacks the characteristic whorled pattern of the latter.
The diffuse interstitial fibrosis found in a minority of coalworkers is associated with heavy dust deposition. It may progress to honeycombing but, as with the focal forms and unlike idiopathic interstitial fibrosis, it is better developed in the upper zones, the reasons for which are discussed above (see the zonal distribution of pneumoconiosis, p. 329).
Pathogenesis
The pathogenesis of coal pneumoconiosis has much in common with that of silicosis, and indeed many other pneumoconioses. It involves the promotion of fibrogenic factor synthesis and release by cells phagocytosing the inhaled dust. Several such factors have now been identified, the degree of fibrosis produced varying with the amount of dust inhaled and the ability of its constituents to promote the production of the responsible cytokines. These include platelet-derived growth factor, insulin-like growth factors 1 and 6, transforming growth factor-β and tumour necrosis factor-α.101, 183, 184 As with other minerals, the indestructability of the dust perpetuates the process.
As in silicosis, immunological factors appear to be involved, for there is an increased prevalence of rheumatoid arthritis185 and of circulating autoantibodies186, 187, 188 in miners with coal pneumoconiosis. Rheumatoid factor has also been demonstrated within the lung lesions.189 These abnormalities are generally more pronounced in miners with complicated pneumoconiosis but are also found in those with the simple variety. It is also possibly pertinent to the immunological basis of coal pneumoconiosis that some of the pulmonary manifestations of rheumatoid disease are more pronounced in coalminers. This was first pointed out by Caplan and will be considered next.
Pneumoconiosis and rheumatoid disease (Caplan's syndrome)
Caplan described distinctive radiographic opacities in the lungs of coalminers with rheumatoid disease,190 and it is now recognised that similar lesions may develop in rheumatoid patients exposed to siliceous dusts. The development of such rheumatoid pneumoconiosis does not correlate with the extrapulmonary or serological activity of the rheumatoid process. Nor is there a strong relation to dust burden: Caplan lesions are characteristically seen in chest radiographs that show little evidence of simple coal pneumoconiosis.
Pathologists recognise the lesions as particularly large necrobiotic nodules similar to those seen in rheumatoid patients who are not exposed to dust (Fig. 7.1.17 ). However, because of their large size (up to 5 cm diameter) they may be confused with progressive massive fibrosis undergoing central ischaemic necrosis (see above) or silicosis complicated by caseating tuberculosis. Such errors will be less likely if the radiological evolution of the lesions is considered for they tend to cavitate and undergo rapid remission, only to be succeeded by others. They are also well demarcated radiologically. Pathologically, they resemble rheumatoid nodules in showing peripheral palisading but differ in their large size and the presence of dust.191 The dust accumulates in circumferential bands or arcs within the necrotic centres of the lesion (Fig. 7.1.18 ), an arrangement that suggests periodic episodes of inflammatory activity. Caplan lesions differ from tuberculosis in lacking satellite lesions and tubercle bacilli, and from progressive massive fibrosis in showing characteristic bands of dust pigmentation (Table 7.1.2 ) and only light dust deposition in the surrounding lung.
Figure 7.1.17.
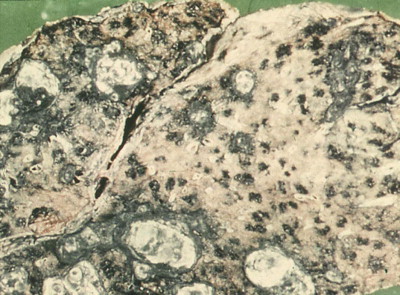
Caplan lesions in a coalminer's lung, which characteristically shows only light dust deposition.
(Courtesy of Dr R Seal, formerly of Penarth, UK.)
Figure 7.1.18.
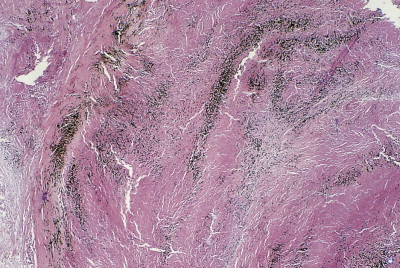
A Caplan lesion, characterised by successive bands of dust within the centre of a large necrobiotic nodule.
Table 7.1.2.
Histological features of Caplan lesions, silicotuberculosis and progressive massive fibrosis
| Caplan lesions | Silicotuberculosis | Progressive massive fibrosis | |
|---|---|---|---|
| Palisading | + | – | – |
| Dust banding | + | – | – |
| Satellite tubercles | – | + | – |
| Necrosis | + | + | + |
| Cholesterol crystals | + | ± | + |
| Calcification | + | + | ± |
+ present; ± poorly represented; – absent.
Asbestosis
Asbestosis is defined as diffuse interstitial fibrosis of the lung caused by exposure to asbestos dust.192, 193 It does not cover asbestos-induced carcinoma of the lung or asbestos-induced pleural disease. The development of asbestosis depends on the presence of fairly large dust burdens: this is in contrast to mesothelioma and other forms of asbestos-induced pleural disease, which, although also dose-related, occur following the inhalation of far smaller amounts of asbestos dust.
Asbestos types and production
Asbestos is a generic term for more than 30 naturally occurring fibrous silicates, fibre being defined as an elongated particle with a length-to-breadth (aspect) ratio of at least 3. Asbestos fibres have a high aspect ratio, generally over 8. Based on their physical configuration they can be divided into two major groups, serpentine and amphibole. The physical dimensions and configuration of asbestos fibres are strongly linked to their pathogenicity.
Chrysotile (white asbestos) is the only important serpentine form. It accounts for most of the world production of asbestos of all types (Table 7.1.3 ).194 Being a serpentine mineral, chrysotile consists of long, curly fibres that can be carded, spun and woven like cotton (Fig. 7.1.19 ). The curly chrysotile fibres are carried into the lungs less readily than the straight amphibole asbestos fibres, and once there undergo physicochemical dissolution and are cleared more readily. They readily fragment into short particles that are easily ingested by macrophages and in the acidic environment of the macrophage phagolysosome they are particularly unstable. The half-life of chrysotile in the lungs is estimated to be in the order of only a few months.195, 196 Not surprisingly therefore chrysotile is the least harmful type of asbestos in respect of all forms of asbestos-induced pleuropulmonary disease.197, 198, 199 It may nevertheless cause pulmonary fibrosis if sufficient is inhaled.200, 201
Table 7.1.3.
Asbestos production by country, 2000194
| Country | Tons |
|---|---|
| Russia | 752 000 |
| China | 350 000 |
| Canada | 320 000 |
| Brazil | 209 000 |
| Kazakhstan | 179 000 |
| Zimbabwe | 152 000 |
| Others | 88 000 |
Figure 7.1.19.
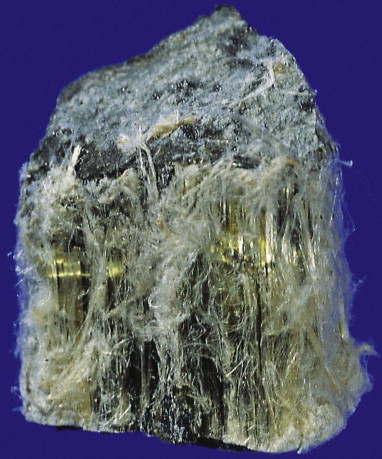
A sample of Quebec chrysotile showing the fibrous nature of the ore.
In contrast to chrysotile, amphibole forms of asbestos consist of straight rigid fibres that are stable within the lung. They do not fragment, they are insensitive to chemical attack and their clearance half-lives are in the order of decades rather than months.196 The main amphibole forms of asbestos of commercial importance are crocidolite (blue asbestos) and amosite (brown asbestos). Crocidolite, reputedly the most dangerous in regard to all forms of asbestos-related disease, was formerly mined in Western Australia (Wittenoom) and South Africa (Cape Province and the Transvaal); it was the principal amphibole used in the UK. Amosite, the name of which derives from the acronym for the former Asbestos Mines of South Africa company in the Transvaal, was the principal amphibole used in North America. Amphiboles are no longer imported by the developed countries but much remains in old lagging and presents a considerable dust hazard when this is removed. Tremolite, a further amphibole asbestos, contaminates Quebec chrysotile deposits, Montana vermiculite and many forms of commercial (non-cosmetic) talc and is responsible for much of the asbestos-related disease in chrysotile miners and millers.202 Another amphibole asbestos, anthophyllite, was formerly mined in Finland. It causes pleural plaques (see p. 716) but not lung disease, possibly because its fibres are relatively thick (Fig. 7.1.20 ).203
Figure 7.1.20.
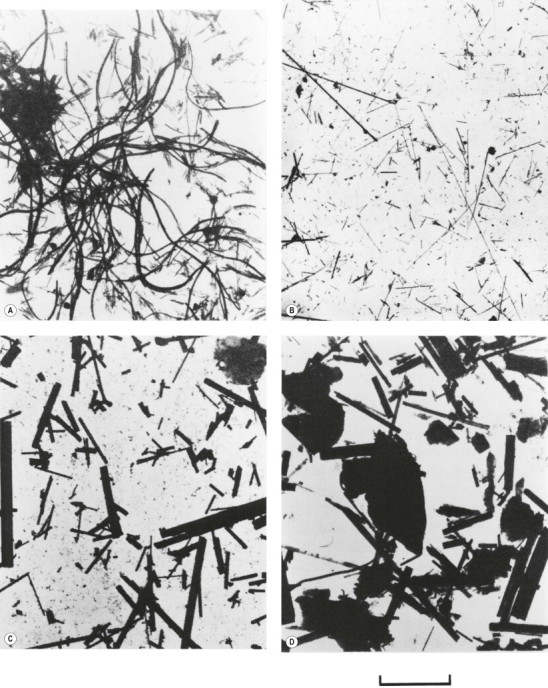
Electron micrographs of dispersed samples of asbestos. (A) Chrysotile; (B) crocidolite; (C) amosite; (D) anthophyllite, all ×2800, the bar representing 10 µm.
(Reproduced from Wagner et al. (1980)203by permission of Professor FD Pooley and the British Medical Bulletin.)
Erionite is a zeolite rather than a type of asbestos but is comparable in form to amphibole asbestos and is also biopersistent. It is found in parts of central Turkey where it causes both mesothelioma and a pattern of interstitial pulmonary fibrosis that is comparable to asbestosis.204, 205
Asbestos use and exposure
Exposure to asbestos occurs in countries where it is extracted (Table 7.1.3), which is mostly by the open-cast method, and in those countries that continue to use this mineral (Table 7.1.4 ). The developed nations have largely ceased to use asbestos but countries keen to develop their industrial base and less concerned with health issues continue to use considerable amounts in a vast range of trades. Asbestos is used particularly for fireproofing, in heat and sound insulation and for strengthening plastics and cement. Thus, unless adequate precautions are taken, exposure is experienced by dockers unloading asbestos in the close confines of a ship's hold, by thermal insulation workers (laggers and strippers) in shipyards, power stations, train maintenance depots, factories and other large buildings, by construction workers such as carpenters cutting asbestos building panels, and by workers making asbestos products such as fireproof textiles, brake and clutch linings, and specialised cement.
Table 7.1.4.
Asbestos consumption by country, 2000194
| Country | Tons | kg/capita/year |
|---|---|---|
| Russia | 447 000 | 3.4 |
| China | 410 000 | 0.4 |
| Brazil | 182 000 | 1.3 |
| India | 125 000 | 0.2 |
| Thailand | 121 000 | 3.0 |
| Japan | 99 000 | 1.5 |
| Indonesia | 55 000 | 0.3 |
| South Korea | 29 000 | 1.9 |
| Mexico | 27 000 | 0.4 |
| Others | 178 000 | |
As well as such direct exposure, exposure may also be:
-
•
indirect, as experienced by the families of asbestos workers
-
•
paraoccupational, as experienced by those working alongside an asbestos worker
-
•
neighbourhood, as experienced by those living downwind of an asbestos works or mine
-
•
ambient, as experienced by those living or working in a building containing asbestos.
Exposure to asbestos incorporated in the structure of a building carries a negligible health risk if the asbestos material is well maintained to prevent shedding of dust. Stripping asbestos out is more dangerous than maintaining it in situ, but maintenance is sometimes neglected. The near indestructibility of asbestos accentuates the health problems that its ubiquity poses.
Asbestos bodies
Because of their aerodynamic properties, fibres of 100 µm or more in length may reach the finer bronchioles and alveoli. Once impacted, the sharp asbestos fibres become coated with a film of protein that is rich in iron. The coating is thickest at the ends of the fibres, giving a dumb-bell appearance. In time, a distinctive segmentation of this outer layer develops (Fig. 7.1.21 ). These coated structures are termed ‘asbestos bodies’. Because other fibres may gain a similar coat, the non-specific term ‘ferruginous body’ has been advocated. However, coated carbon fibres (so-called coal bodies) are easily recognised as such by their black core.206 In practice, ferruginous bodies with the appearance of asbestos bodies almost always prove to have an asbestos core.207, 208 Long fibres are more likely to be coated than short ones, which are cleared more quickly: in one study few fibres less than 5 µm in length were coated and few fibres over 40 µm in length were uncoated.209 Amphiboles form bodies more readily than chrysotile. A comparison of light and electron microscopic fibre counts found that 0.14% of chrysotile, 5% of crocidolite and 26.5% of amosite formed bodies.210 Nevertheless, sufficient chrysotile fibres are coated to permit recognition of asbestosis by standard histological criteria (diffuse fibrosis and asbestos bodies), even if chrysotile is the only asbestos present.211 Despite the biodegradability of chrysotile, asbestos body numbers do not materially diminish with time.212 Very occasionally however a patient with diffuse pulmonary fibrosis and a history of asbestos exposure has no evident asbestos bodies but analysis shows a fibre burden within the range found in asbestosis, justifying fibre analysis in such cases.212a
Figure 7.1.21.
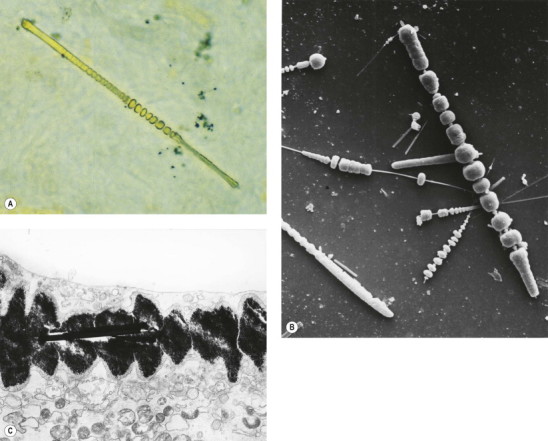
Asbestos bodies seen by light microscopy in (A) an unstained 30-µm-thick paraffin section; (B) by scanning electron microscopy in the digest of an asbestos worker's lung and (C) by transmission electron microscopy in lung tissue. The asbestos fibres have acquired the iron-protein coating that characterises an asbestos body. In most places the coating has become segmented, giving rise to bead-like formations, a change accompanying ageing of the bodies.
((B) Courtesy of Dr B Fox, London, UK and (C) courtesy of Miss A Dewar, Brompton, UK.)
There is evidence that alveolar macrophages are involved in the coating of asbestos fibres to form asbestos bodies and that the bodies are less harmful to the macrophages than uncoated fibres.213 Asbestos bodies give a Prussian blue reaction for iron when stained by Perls’ method and their yellow-brown colour makes them easily recognisable in unstained films of sputum or in unstained histological sections. Sections may be cut 30 µm thick to increase the yield and help identify bodies that lie at an angle to the microtome blade. There is a good correlation between the numbers of asbestos bodies seen in lung sections and those in tissue digests.214, 215 The bodies may be found singly or in irregular clumps or stellate clusters. They are unevenly distributed but in well-established asbestosis they are easily found. If they are not evident, asbestos burden may be assessed quantitatively in tissue digests (see below). Their presence in lung tissue, sputum or bronchoalveolar lavage fluid merely confirms exposure, not the presence of disease. However, the number of asbestos bodies in lavage fluid correlates well with lung asbestos burden216, 217 and the number in sputum correlates with the duration and intensity of exposure.216, 217, 218
Fibre counts193, 208, 219, 220, 221, 222, 223
Quantitation is desirable in certain circumstances (Box 7.1.1 ), in which case it is best effected on 2-cm3 blocks of fixed or fresh lung tissue obtained from three different sites, avoiding tumour and thickened pleura. The tissue blocks are digested with caustic soda or bleach, following which the fibres may be collected on a millipore membrane or viewed in suspension in a red blood cell-counting chamber. If phase contrast optics are used both coated and uncoated fibres can be assessed.219 Alternatively, dark ground illumination can be used to demonstrate uncoated fibres.221 However, electron microscopy is to be preferred as it detects far more fibres than are visible by light microscopy and can also provide information on fibre type. It is important that the laboratory is well practised in fibre analysis and has established its own control range for the general population as well as asbestosis as most lungs contain some asbestos.
Box 7.1.1. Circumstances in which asbestos fibre counts are desirable.
| Quantitation desirable | Quantitation unnecessary |
|---|---|
| Mesothelioma if exposure is disputed and asbestos bodies are not evident | Mesothelioma if there is a history of exposure or asbestos bodies are evident |
| Pulmonary fibrosis (with or without carcinoma of the lung) in an asbestos worker but asbestos bodies are not evident | If there is obvious asbestosis |
| Carcinoma of the lung in an asbestos worker if there is no fibrosis |
Ambient fibres are generally shorter than 5 µm and some workers therefore confine their counts to fibres that are at least as long as this.224 Justification for this comes from animal experiments demonstrating that long fibres cause more inflammation, chromosomal damage, fibrosis, lung tumours and mesotheliomas than short fibres,225, 226, 227, 228 and from studies in humans suggesting that long fibres are responsible for asbestosis.229 Other human studies have shown that, although asbestos load is maximal in the upper lobes, more long fibres are found at the bases, where fibrosis is most marked.41, 230 A further reason for limiting attention to the longer fibres is that the shorter ones are cleared more easily and their number therefore varies with the time lapsed since last exposure. For these reasons asbestos regulations in many countries now limit attention to fibres that are over 5 µm in length and have a length-to-diameter (aspect) ratio greater than 3: such fibres have become known as regulatory or World Health Organization (WHO) fibres.
Values are best expressed as fibres/g dry lung. By light microscopy, normal values range up to 50 000: over 20 000 is seen with mesotheliomas, and over 1 000 000 in asbestosis (Table 7.1.5 ).216, 224, 231, 232 However, compared with electron microscopy, light microscopy is relatively insensitive, showing only 26.5% of the amosite, 5% of the crocidolite and 0.14% of the chrysotile.210 Light microscopic counts correlate poorly with severity of asbestosis219 and electron microscopy is better in this respect.197, 198, 199 By transmission electron microscopy, values may range up to 5 000 000 in controls, with asbestosis generally above 100 000 000 and mesotheliomas found at any level down to 1 000 000, all these figures representing amphibole fibres/g dried lung (see Table 7.1.5).216, 231, 232 It should be noted that counts from different parts of the same lung may vary widely;42, 231, 232, 233, 234, 235 caution should therefore be exercised in interpreting a count obtained on a single sample. There is also wide discrepancy between laboratories, even when analysing the same sample.234 Results obtained in an individual case therefore have to be evaluated against a standard set of values unique to that laboratory.
Table 7.1.5.
Lung asbestos fibre counts per gram of dried lung
| Light microscopy | Electron microscopy | |
|---|---|---|
| Normal city dwellers | Up to 50 000 | Up to 5 000 000 |
| Mesothelioma | Over 20 000 | Over 1 000 000 |
| Asbestosis (minimal) | Over 100 000 | Over 10 000 000 |
| Asbestosis (established) | Over 1 000 000 | Over 100 000 000 |
The light microscopic counts include total fibres (coated and uncoated). The electron microscopic counts include only amphibole asbestos. Results from different laboratories vary and these figures, derived from several sources,216, 231, 232 provide only a general guide. Reliable results depend upon counts being made regularly and the normal range from that laboratory being ascertained. Ratios of counts obtained by electron and light microscopy vary greatly but approximate to 100.
Electron microscopy also provides valuable information on the type of fibre. Chrysotile differs physically from the amphiboles in two respects: its fibres are both curved and hollow (Figure 7.1.20, Figure 7.1.22 ). With an electron microscope equipped for microprobe analysis, the various forms of asbestos may also be distinguished from other fibres and from each other (Box 7.1.2 ),230, 236 an important point as the amphibole forms of asbestos are far more dangerous than chrysotile (Table 7.1.6 ).197, 198, 199
Figure 7.1.22.
At high magnification chysotile fibres are seen to be tubular. Transmission electron micrograph ×51 000.
(Courtesy of Miss A Dewar, Brompton, UK.)
Box 7.1.2. Molecular formulae of various forms of asbestos. When subjected to microprobe analysis, the total counts recorded for each element (see Fig. 7.1.6c) are proportional to the numbers of their atoms in the molecule. Thus, with tremolite the silicon peak would be four times as high as that for calcium and a little less than twice as high as that for magnesium.
| Chrysotile | Mg3(Si2O5)(OH)4 |
| Crocidolite | Na2Fe5(Si8O22)(OH)2 |
| Amosite | (Fe,Mg)7(Si8O22)(OH)2 |
| Tremolite | Ca2Mg5(Si8O22)(OH)2 |
Table 7.1.6.
Mean percentage lung fibre type by diagnostic category in a group of British asbestos factory workers compared to local controls199
| Fibre type | Controls | Asbestosis | Carcinoma of the lung | Pleural mesothelioma |
|---|---|---|---|---|
| Crocidolite | 2.8 | 60.1 | 56.7 | 20.2 |
| Amosite | 2.6 | 17.7 | 30.4 | 39.3 |
| Chrysotile | 25.9 | 4.0 | 6.9 | 17.0 |
| Other (mainly fly ash) | 68.7 | 18.2 | 6.0 | 23.5 |
Non-asbestos fibres commonly found in the lung include mullite, which derives from fly ash. This may constitute up to 70% of the total fibre burden (see Table 7.1.6) and is thought to be harmless. There is no firm evidence that manmade fibres present a health hazard237 but in certain localities natural non-asbestos mineral fibres, zeolites for example, are important causes of mesothelioma (see p. 718) and also cause interstitial pulmonary fibrosis.204
Clinical and radiological features
In contrast to the first half of the twentieth century, much of the asbestosis encountered today is asymptomatic, identified radiologically or histologically in lungs resected for carcinoma or removed at autopsy. Symptomatic cases are characterised by an insidious onset of breathlessness, a dry cough and crackles over the lower lung fields. Finger clubbing is a variable feature. Lung function tests show a restrictive respiratory defect. Radiology initially shows small irregular basal opacities that gradually coalesce to become linear, coarsen and eventually progress to a honeycomb pattern of small cysts.
The principal differential diagnosis, both clinically and pathologically, is from idiopathic pulmonary fibrosis. This is aided by the slow progression of asbestosis, which often extends over 20 years, as opposed to an average course of 2–3 years from presentation to death for the idiopathic condition.
Most cases of asbestosis are diagnosed solely on the occupational history and these clinicoradiological features. Recourse to histology is unusual but biopsy (preferably as a wedge of lung) may be undertaken if the clinical features are atypical. Histology also arises when the pathologist samples lung parenchyma remote from a resected carcinoma (the universal importance of which cannot be overemphasised). Coroners require autopsy verification of the diagnosis in all suspected cases and this also necessitates hystology.
Morbid anatomy
When the lungs from a patient with asbestosis are seen at autopsy, pleural fibrosis is often found, and although this may also be attributable to asbestos exposure it is to be regarded as an independent process and not part of the asbestosis: it is dealt with separately on page 715.
Slicing the lung affected by asbestosis shows a fine subpleural fibrosis, especially of the lower lobes (Fig. 7.1.23 ). In severe cases the fibrosis often extends upwards to involve the middle lobe and lingula, and sometimes the upper lobes also. Microcystic change associated with the fibrosis develops in advanced cases and in severe disease there may be cysts over 1 cm in diameter. However, these classic changes are seldom seen in developed countries today. Following decades of dust suppression in asbestos factories, current patients have mild to moderate asbestosis and are dying of related cancer or of non-pulmonary disease.238 In some of these cases the asbestosis is only detectable microscopically. Fixation of the lungs through the bronchi and the use of Heard's barium sulphate impregnation technique facilitate demonstration of the fibrosis (see p. 757 and Fig. 7.1.23). The mild degree of asbestosis currently encountered is of little functional significance but is often critical in determining whether an associated carcinoma of the lung should be attributed to asbestos exposure (see below).
Figure 7.1.23.
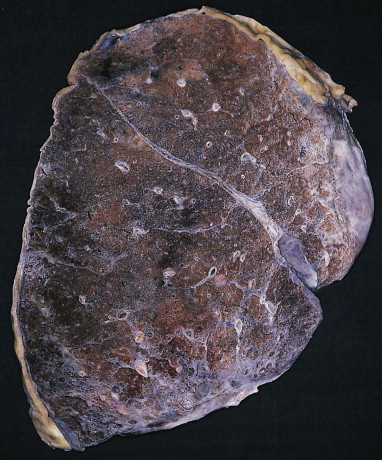
Asbestosis. Fine interstitial fibrosis is evident beneath the pleura at the base of the lung. Barium sulphate-impregnated specimen.
Histological appearances
The histological diagnosis of asbestosis requires an appropriate pattern of interstitial fibrosis associated with the presence of asbestos bodies. Both components must be present. The fibrosis is paucicellular, lacking any significant degree of inflammation and being collagenous rather than fibroblastic.
It is generally considered that asbestosis begins about the respiratory bronchioles and alveolar ducts where most of the asbestos fibres impact.15 Alveolar walls attached to these bronchioles show fine interstitial fibrosis. However, this early lesion has to be interpreted with caution because it is not specific to asbestos, being found with other inhaled mineral dusts239, 240 and even in many cigarette smokers who have not been so exposed.241 It more likely represents a non-specific reaction to a variety of inhaled particles. It may cause mild airflow obstruction but is not associated with the radiographic, clinical or restrictive changes of classic asbestosis.
As the disease progresses, the focal changes join up so that the basal subpleural regions show widespread interstitial fibrosis and eventually complete destruction of the alveolar architecture. In severe cases there may be honeycombing and metaplastic changes in the alveolar and bronchiolar epithelium. Apart from the presence of asbestos bodies the changes resemble those of non-specific interstitial pneumonia, or more rarely usual interstitial pneumonia. Fibroblastic foci may be found but they are uncommon. There is often an increase in alveolar macrophages but the desquamative interstitial pneumonia that has been reported in association with asbestos242, 243 is not to be regarded as a variant of asbestosis192; concomitant smoking is a more likely cause.241 A variety of other non-specific inflammatory processes such as organising pneumonia have been reported in asbestos workers and if localised some have been suspected of representing malignancy until biopsied.244
Several schemes have been proposed for grading the extent and severity of asbestosis. These are of value in epidemiological studies but should only be applied to cases meeting the histopathological criteria for a diagnosis of asbestosis. One such scheme is shown in Box 7.1.3 .193, 245, 246
Box 7.1.3. Criteria for grading asbestosis193, 245, 246.
| Extent | |
| A | None |
| B | Less than 25% of the lung substance involved |
| C | 25–50% of the lung affected |
| D | More than 50% of the lung diseased |
| Severitya | |
| 0 | No appreciable peribronchiolar fibrosis, or fibrosis confined to the bronchiolar walls |
| 1b | Fibrosis confined to the walls of respiratory bronchioles and the first tier of adjacent alveoli |
| 2b | Extension of fibrosis to involve alveolar ducts and/or two or more tiers of alveoli adjacent to the respiratory bronchiole, with sparing of at least some alveoli between adjacent bronchioles |
| 3 | Fibrotic thickening of the walls of all alveoli between at least two adjacent respiratory bronchioles |
| 4 | Honeycomb change |
An average score is obtained for an individual case by adding the scores for each slide (0–4), then dividing by the number of slides examined
Grade 1 and, to a lesser extent, grade 2 need to be distinguished from smoking-induced peribronchiolar fibrosis and mixed-dust pneumoconiosis.
In well-established asbestosis asbestos bodies are numerous and easy to find, aggregates of them sometimes forming clumps (Fig. 7.1.24 ). In earlier lesions a detailed search may be necessary, in which case the examination of unstained or Perls-stained sections facilitates their identification. Minimum criteria for the diagnosis of asbestosis require the identification of diffuse interstitial fibrosis in well-inflated lung tissue remote from a lung cancer or other mass lesion and the presence of either two or more asbestos bodies in tissue with a section area of 1 cm2 or a count of uncoated asbestos fibres that falls in the range recorded for asbestosis by the same laboratory.192, 193 There are marked variations in the concentration of asbestos fibres between samples from the same lung216, 235 and it is therefore recommended that at least three areas be sampled, the apices of the upper and lower lobes and the base of the lower lobe.223
Figure 7.1.24.
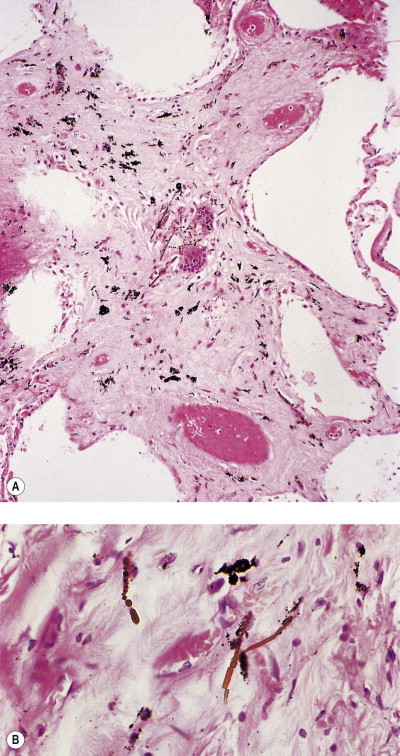
Asbestosis. Interstitial fibrosis is associated with many asbestos bodies (A), which are shown in detail in (B).
The equivalent of Mallory's alcoholic hyalin of the liver has been described in the lungs in asbestosis,193, 247 and subsequently in other pulmonary conditions.248, 249, 250, 251It is seen as small eosinophilic cytoplasmic inclusions within hyperplastic type II alveolar epithelial cells (Fig. 7.1.25A ). Electron microscopy shows that the inclusions consist of a tangle of tonofilaments (Fig. 7.1.25B) and by immunocytochemistry a positive reaction is obtained with antibodies to cytokeratin, both these features being typical of Mallory's hyalin in the liver. The inclusions also react for ubiquitin, the accumulation of which is indicative of cellular damage, in particular faulty proteinolysis.251
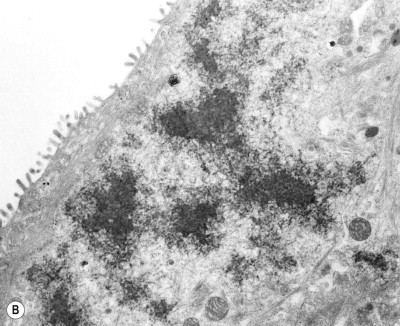
(B) Electron microscopy shows that the inclusions represent a tangle of microfilaments.
(Courtesy of Professor D Henderson, Adelaide, Australia.) (Both images reproduced by permission of the publishers of Archives of Pathology and Laboratory Medicine.193)
Figure 7.1.25.
Cytoplasmic hyaline in asbestosis. (A) Eosinophilic inclusions are seen in the cytoplasm of several hyperplastic type II pneumocytes. Haematoxylin & eosin.
(Courtesy of Professor VL Roggli, Durham, USA.)
Differential diagnosis
The differential diagnosis of asbestosis includes pulmonary fibrosis due to many other causes, any of which may of course affect an asbestos worker as much as members of the general population. The proportion of diffuse pulmonary fibrosis in asbestos workers that is not attributable to asbestos has been estimated to be as high as 5% and likely to rise as the risk of asbestosis diminishes with better industrial hygiene.252
The principal differential diagnosis of asbestosis is from idiopathic pulmonary fibrosis. Both diseases affect the bases and periphery of the lungs predominantly. In the late stages, cystic change is more evident in idiopathic pulmonary fibrosis but this criterion is not totally reliable. Nor is the presence of pleural fibrosis, although it is usually present in asbestosis and is seldom found in idiopathic pulmonary fibrosis. Asbestosis seldom progresses or does so very slowly after exposure ceases253, 254 whereas idiopathic pulmonary fibrosis typically proves fatal within 3–4 years from onset. The fibrosis of asbestosis is generally paucicellular: inflammation is not a feature and the fibroblastic foci that characterise the usual interstitial pneumonia pattern of fibrosing alveolitis are seldom observed in asbestosis.
Very often the distinction from idiopathic pulmonary fibrosis has to be based on the amount of asbestos in the lung and, if asbestos bodies are not readily identifiable, this has to depend on fibre counts. Errors are made both by overlooking substantial numbers of asbestos bodies completely and by ascribing undue importance to scanty bodies. If considering the possibility of minimal asbestosis (to justify attributing carcinoma of the lung to asbestos, for example) it should be remembered that a little peribronchiolar fibrosis is also characteristic of smokers’ lungs, centriacinar emphysema and early mixed-dust pneumoconiosis.239, 240, 241, 255 As described above, at least two asbestos bodies/cm2 in the presence of interstitial fibrosis distant from any lung cancer or other mass lesion is required for a diagnosis of asbestosis.192
Pathogenesis
Although the causes of asbestosis and idiopathic pulmonary fibrosis are very different, they resemble each other in several ways, suggesting that similar pathogenetic mechanisms may operate.105, 256, 257, 258 In both these diseases there is degeneration of the alveolar epithelium and capillary endothelium, with patchy loss of the former,256 and bronchoalveolar lavage shows an increase in macrophages that might perpetuate the damage by releasing lysosomal enzymes, nitric oxide and hydroxyl radicals.257, 259, 260, 261 Both diseases are also characterised by an increased prevalence of circulating non-organ-specific autoantibodies.262 Experimentally, asbestos exposure leads to the activation of a variety of fibrogenic cytokines at sites of lung injury.105, 263, 264, 265, 266, 267, 268
Inhaled asbestos activates a complement-dependent chemoattractant for macrophages269 and macrophage stimulation involves the secretion of fibroblast stimulating factors,270, 271, 272 asbestos being intermediate between haematite and silica in regard to macrophage-mediated fibrogenicity.273 The epithelial damage could be mediated directly by the needle-like asbestos fibres or indirectly through enhanced phagocyte generation of free radicals (which is much greater with amphibole asbestos than with either chrysotile or silica).268, 274 Fibrogenic cytokines released by activated pulmonary phagocytes and regenerating alveolar epithelial cells in asbestosis include tumour necrosis factor-α and transforming growth factor-β,268 as in idiopathic pulmonary fibrosis.
Asbestos-induced lung cancer
As a result of better industrial hygiene, asbestosis is less severe today than in earlier years when it followed much heavier exposure, with the consequence that death from respiratory failure and cor pulmonale is less common and sufferers are surviving longer. There is therefore now a greater risk of asbestos-related cancer eventually developing. Asbestos exposure predisposes to two varieties of malignant neoplasm, carcinoma of the lung and mesothelioma of the pleura and peritoneum. The risk of malignancy increases with dose but the relative risk of carcinoma is much smaller than that of mesothelioma. For example, with heavy exposure, as in lagging, the risk of mesothelioma is increased 1000-fold whereas it is increased only fivefold for lung cancer. Hence, with light exposure there is a substantial risk of mesothelioma but only a small risk of lung cancer. Asbestosis requires heavy exposure and in one group of patients with asbestosis, 39% died of pulmonary carcinoma, 10% of mesothelioma and 19% of other respiratory diseases.238 Although there were many earlier reports, the link with carcinoma of the lung may be considered to have been firmly established by 1955,275 that between crocidolite asbestos and mesothelioma by 1960,276 and that between amosite asbestos and mesothelioma by 1972.277 Mesothelioma is considered on page 717.
In regard to carcinoma of the lung, asbestos is not such a potent pulmonary carcinogen as cigarette smoke but together their effects are multiplicative rather than additive (Table 7.1.7 ).278, 279 However, the risk attributable to asbestos is the same regardless of smoking history, being increased fivefold in both smokers and non-smokers. There is usually a latent period in excess of 20 years between first exposure to asbestos and the development of lung cancer and the risk increases the greater the cumulative exposure. The increased risk involves carcinomas of all the histological types encountered in the lung, although adenocarcinoma has been disproportionately overrepresented.245, 280, 281, 282, 283, 284, 285
Table 7.1.7.
Excess mortality from carcinoma of the lung attributable to asbestos exposure and cigarette smoking in insulation workers.278 Together, these factors have a multiplicative rather than additive effect
| Group | Mortality ratio |
|---|---|
| Non-smoking controls | 1 |
| Non-smoking asbestos workers | 5 |
| Cigarette-smoking controls | 11 |
| Cigarette-smoking asbestos workers | 53 |
It is uncertain114 whether the increased risk of carcinoma is caused by the asbestos192, 286, 287, 288, 289, 290, 291 or the asbestosis.292, 293, 294, 295, 296, 297, 298, 299 The latter view envisages the carcinoma arising in the foci of alveolar epithelial hyperplasia and dysplasia that commonly accompany any interstitial fibrosis (see carcinoma complicating idiopathic pulmonary fibrosis, p. 275). However, most carcinomas complicating asbestosis arise in the bronchi rather than the alveolar tissue. On the other hand, more arise in the sites worst affected by asbestosis, the lower lobes and the periphery of the lung, than in the general population (Fig. 7.1.26 ).238, 245, 281, 282, 283, 284, 285
Figure 7.1.26.

Asbestosis associated with carcinoma of the lung. The asbestosis has been highlighted by barium sulphate impregnation and is seen as a grey subpleural band to the right of the picture. Although the carcinoma has arisen in the same lobe as the asbestosis it has not obviously arisen in an area affected by asbestosis.
The majority view has been that asbestosis is a necessary precursor of the carcinoma but evidence to the contrary is finding increasing support (Table 7.1.8 ).192 In the UK, industrial compensation was formerly only awarded to an asbestos worker for carcinoma of the lung if there was also asbestosis or diffuse pleural fibrosis but new rules were introduced in 2006. Asbestosis remains a sufficient criterion but diffuse pleural thickening is not and asbestosis is no longer a necessary criterion: asbestos is deemed to have been responsible if the patient worked in asbestos textile manufacture, spraying, lagging or gas mask manufacture for at least 5 years before 1975 or 10 years after 1975. The basis for these changes is the premise that heavy asbestos exposure is sufficient in itself to account for carcinoma of the lung. Lung fibre counts in the asbestosis range (see Table 7.1.5) provide valuable evidence of such exposure. Compensation standards for asbestos-associated lung cancer in different countries are shown in Box 7.1.4 .192, 289
Table 7.1.8.
Requirements for attributing carcinoma of the lung to asbestos
| The traditional view that the carcinoma complicates asbestosis | The Helsinki criteria,192 which are based upon substantial exposure |
|---|---|
|
|
Box 7.1.4. Compensation standards for asbestos-associated lung cancer in different countries289.
Government standards vary considerably and in the civil courts claims are often based on lesser evidence. Some examples of government standards are:
UK
Asbestosis or employment in a specified industry for a specified time (see text)
USA
The presence of asbestos-related bilateral pleural plaques or asbestos-related bilateral pleural thickening and occupational exposure and a lag time of at least 12 years
Germany
The presence of asbestosis or pleural plaques or diffuse pleural thickening or fibre-years of exposure
Denmark
Only fibre-years of exposure are taken into account
Finland
Exposure, at least 10 years’ latency and asbestos-related pleural or parenchymal changes
Sweden
Asbestosis is not required but smoking is taken into consideration
Norway
Attempts are made to quantify separately the attributability to asbestos, smoking and other factors (e.g. radon)
Asbestos-induced airway disease
Although asbestosis causes a restrictive respiratory defect, airflow limitation is also seen in this disease. Much of the airflow limitation is attributable to cigarette smoking but it is also seen in non-smoking asbestos workers and is worse in those with asbestosis.300 The pathological basis of this appears to be small-airways disease (see p. 123).301 It is possibly a non-specific reaction to inhaled dust or cigarette smoke.302 Because it is not established that this lesion progresses to interstitial alveolar fibrosis (asbestosis) the term ‘asbestos airways disease’ is suggested.302 Fibrosis limited to the bronchioles is specifically excluded from the definition of asbestosis in the latest guidelines (although these retain grade 0 for fibrosis limited to the bronchiolar walls).193 It should also be noted that, although emphysema is considered to be a destructive rather than fibrotic condition, a little focal fibrosis is generally evident in this common condition255 and does not necessarily indicate early asbestosis.
Aluminium
Aluminium has been implicated in the development of respiratory disease during the refining of its principal ore, bauxite, to yield various aluminium oxides (aluminas), in the preparation of the metal by smelting alumina, in the production of corundum abrasive and in the production of special aluminium powders used in explosives.
Refining of bauxite
Bauxite is a mixture of various aluminium oxides, hydroxides and silicates, iron oxide and titanium dioxide. The oxides of aluminium are obtained by differential heating of the ore and the respiratory effects of this work appear to be no more than mild airway irritation. It is generally accepted that aluminium oxide is inert.
Aluminium smelting
Aluminium is prepared by the electrolytic reduction of its oxide dissolved in sodium aluminium fluoride (cryolite), a process releasing a considerable amount of fluoride-rich effluent. Exposed workers have complained of what is termed pot-room asthma. The pathology of this condition is not well described but the pathogenesis is thought to involve irritation rather than allergy.303
Abrasive manufacture
The abrasive corundum is formed from bauxite mixed with coke and iron heated in an electric arc furnace, a process in which workers may be exposed to the fumes of alumina and free silica. In the past some of these workers developed diffuse pulmonary fibrosis (Shaver's disease)369 and, although this was initially attributed to the aluminium, it is now agreed that the free silica was the responsible agent. The exposure to free silica has been reduced and the disease is now regarded as historic.
Aluminium powder
Aluminium powder holds a paradoxical position in regard to lung disease. In certain industries it has caused very severe pulmonary fibrosis, yet in others it has proved harmless. Indeed, at one time Canadian miners breathed aluminium dust before work, in the belief that this would reduce the danger of silica in the mine dust304 and more recently silicosis has been treated by such means in France.305 It is questionable whether this practice is effective but it at least appears to cause no harm. The explanation for these contradictory observations probably lies in differing methods of manufacture of aluminium powder.
Aluminium metal appears to be an inert substance but this is only because it has a high affinity for oxygen and the surface layer of aluminium oxide so formed is firmly bound to the underlying metal, unlike ferric oxide which permits further rusting of iron. Granular aluminium powders, produced in a ball mill or from a jet of molten aluminium, therefore acquire a protective coat of surface oxide and are inert. With stamped aluminium powders, however, surface oxidation is prevented by lubricants added to aid the separation of these flake-like particles. The usual lubricant (stearin) contains stearic acid and this polar compound combines with the underlying metal, which is thereby protected from both atmospheric oxidation and the action of body fluids when such dust is inhaled. In certain circumstances, however, non-polar lubricants in the form of mineral oils have been substituted for stearin. This happened in Germany during the Second World War when munition production was stepped up but stearin was difficult to obtain,306, 307 and in the UK in the 1950s to make the powder darker for purely commercial reasons.308 In vitro, oil-coated stamped aluminium powder reacts with water to produce aluminium hydroxide, which affords the underlying metal no protection against further attack, so that aluminium hydroxide continues to be formed.309 This substance is a protein denaturant, once used in the tanning industry, and it is believed that this property underlies the very exceptional cases of severe pulmonary fibrosis that have occurred in connection with stamped aluminium powder produced with mineral oil rather than stearin.309, 310 The fibrosis has a very characteristic pattern, affecting the upper lobes and progressing rapidly, the interval from onset of symptoms to death being as short as 2 years.308 There is marked shrinkage of the lungs with gross elevation of the diaphragm and buckling of the trachea (Fig. 7.1.27 ). The lungs are grey (Fig. 7.1.28 ) and microscopically, numerous small black jagged particles are seen. These can be shown to contain aluminium with Irwin's aluminon stain or by microprobe analysis.311
Figure 7.1.27.
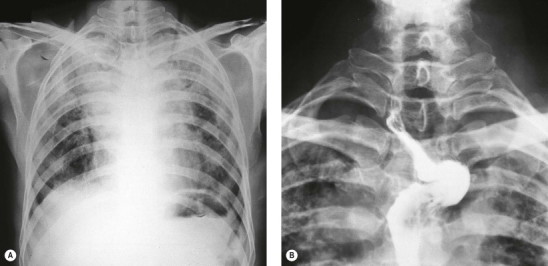
Aluminium pneumoconiosis. (A) A chest radiograph showing opacification of the lung apices. (B) A barium swallow showing severe buckling of the oesophagus due to fibrous contracture of the lung apices and adjacent mediastinal tissues, as seen in Figure 7.1.B.
(Courtesy of the late Professor RE Lane, Manchester, UK.)
Figure 7.1.28.
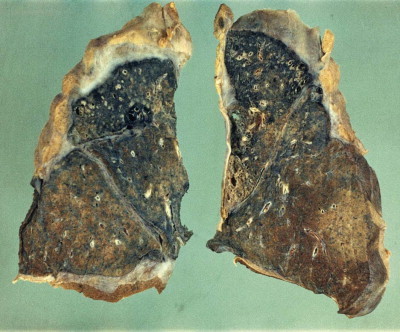
Aluminium pneumoconiosis. The lungs postmortem show severe fibrosis of the apex and overlying pleura.
(Courtesy of the late Dr G Manning, Bolton, UK.)
What appears to be a different pathological effect of aluminium dust on the lungs is the rare development of granulomatous disease resembling sarcoidosis and berylliosis.312, 313 This represents hypersensitivity to the metal, amenable to confirmation with a lymphocyte transformation test similar to that used to diagnosis berylliosis (see below).
Aluminium welding
Rare cases of desquamative interstitial pneumonia and pulmonary fibrosis have been reported in aluminium welders.311, 314
Rare earth (cerium) pneumoconiosis
Elements with atomic numbers from 57 (lanthanum) to 71 (lutetium) are known as the lanthanides or rare earth metals. They are used in many manufacturing processes, including the production of high-temperature ceramics and the grinding of optical lenses. Carbon arc lamps used in reproduction photography emit appreciable quantities of oxidised lanthanides, particularly cerium oxide, and there are reports of pneumoconiosis in exposed individuals.315 The pathological changes reported have varied from granulomatous nodules to diffuse interstitial fibrosis indistinguishable from the idiopathic variety except for the presence of rare earth elements (usually cerium) detected by polarising light microscopy and electron microprobe analysis.315
Hard-metal disease (cobalt lung)
Hard metal is a tungsten alloy containing small amounts of cobalt, titanium, molybdenum and nickel. It is exceptionally tough and once formed can only be worked with diamond. It is used in the tips of drill bits, on abrasive wheels and discs, and in armaments. Interstitial lung disease is liable to arise in its manufacture or in those using hard metal as an abrasive.316 Experimental work suggests that cobalt is the dangerous constituent317 but this element is soluble and, unless industrial contact has been recent, analysis of lung tissue usually shows tungsten and titanium but no cobalt. The role of cobalt is also indicated by the development of similar interstitial lung disease in diamond polishers using high-speed polishing discs made with a diamond-cobalt surface that lacked tungsten carbide and the other constituents of hard metal.318, 319
Hard-metal lung disease and cobalt lung take two forms, an industrial asthma and interstitial fibrosis. The latter has a diffuse lower zonal distribution and the appearances mimic idiopathic pulmonary fibrosis. However, an unusual feature is the presence of moderate, or perhaps only small numbers, of giant cells (Fig. 7.1.29A, B ).316, 320 Not only are there multinucleate alveolar macrophages but syncytial cell forms develop in the alveolar epithelium. Electron microscopy confirms that these are multinucleate type II pneumocytes (Fig. 7.1.29C).316 Such epithelial changes are well known in measles pneumonia but the viral inclusion bodies that characterise this infection are not found in hard-metal pneumoconiosis. The changes are those initially described as a particular pattern of idiopathic interstitial pneumonia termed giant cell interstitial pneumonia or GIP (see p. 264). Elemental analysis shows that many, but not all, cases of GIP represent hard-metal disease. The exceptions seldom give a history of cobalt exposure and must be presumed to represent true idiopathic cases. Conversely, epithelial giant cells are not always found in hard-metal pneumoconiosis and so their presence, although highly characteristic, is neither totally specific nor totally sensitive.
Figure 7.1.29.
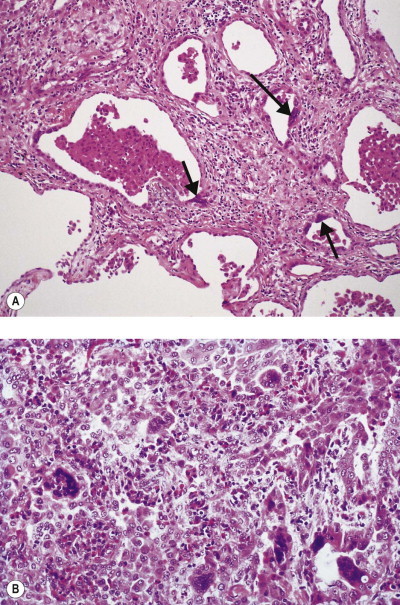
Giant cell interstitial pneumonia in hard-metal workers. (A) There is interstitial pneumonia and fibrosis and several alveolar epithelial cells are multinucleate (arrows). (B) Higher magnification shows a light lymphocytic infiltrate, numerous alveolar macrophages and several multinucleate giant cells.
Berylliosis
Beryllium is the lightest of metals. It has an atomic weight of 4 and special properties that make it especially useful in many applications. It is more rigid than steel, has a high melting point and is an excellent conductor of heat and elecricity. Unfortunately, the inhalation of beryllium dust or fume is exceedingly dangerous.321, 322 Those who worked with beryllium compounds before precautionary measures were taken suffered a high morbidity and mortality. Sometimes, the escape of dangerous fumes from the factories was on such a scale that people living in nearby houses, downwind from the places in which these materials were being worked, contracted and occasionally died from berylliosis (‘neighbourhood cases’).323 Alternatively, contamination of a beryllium worker's clothes might lead to berylliosis in a relative.324 Beryllium compounds may also cause contact dermatitis and conjunctivitis.325 Beryllium is also classified as a probable pulmonary carcinogen,326 but this is controversial.
Two forms of berylliosis are recognised, acute and chronic. Acute berylliosis was first reported in Germany in 1933327 and is now largely of historical interest, being only encountered as a result of rare accidental or unexpected exposure. It follows the inhalation of a soluble beryllium salt and represents chemical injury, the pathology being that of diffuse alveolar damage (see p. 136). Further consideration will be confined to chronic berylliosis, which is allergic in nature.
Uses of beryllium and occupations at risk
Chronic berylliosis was first reported in 1946 in the fluorescent lamp industry.328 Beryllium has now been replaced in this application but it has since proved to be of great value in the nuclear, electronic, computer and aerospace industries and the production of refractory materials and crucibles that are to be subjected to particularly high temperatures. The alloys of beryllium are also now widely used, especially those with copper, on which it confers elasticity and resistance to fatigue. Alloy manufacture and the machining of beryllium alloys are therefore further activities that entail a risk of berylliosis, as is the recovery of the metal in the recycling of scrapped electronic and computer parts. Seemingly innocuous occupations such as dental laboratory technician are not without risk of chronic berylliosis.329
Pathogenesis
There are good grounds for regarding chronic berylliosis as being an allergic condition. Many of those affected react strongly to skin tests with dilute solutions of beryllium salts, although these must be undertaken with care: occasionally in a highly sensitised person even so small an exposure may evoke a systemic reaction. The skin reaction is of the delayed type, occurs in only 5% of exposed individuals, is not associated with a clear-cut dose–response curve and represents a granulomatous response. Further evidence for the disease having an allergic basis derives from bronchoalveolar lavage, which demonstrates an excess of T-helper lymphocytes that proliferate in vitro on exposure to beryllium salts.330 A positive transformation test given by these lymphocytes is a more reliable indicator of disease than in vitro blood lymphocyte transformation testing,52 which is safe but not wholly reliable and indicates only sensitization, rather than berylliosis.
Susceptibility to berylliosis varies widely from person to person and it is notable that chronic pulmonary disease is strongly associated with the HLA antigen DPβ1 and the Glu69 gene.331, 332 The importance of genetic factors is supported by a report of the disease in identical twins.333
Chronic berylliosis is thought to be initiated by the metal binding to tissue proteins and acting as a hapten to initiate a delayed hypersensitivity response characterised by a proliferation of T-helper lymphocytes. These sensitised cells in turn secrete a variety of cytokines (e.g. interleukin-2, tumour necrosis factor-α and interferon-γ) that recruit and activate macrophages, which mature into epithelioid cells. The resultant epithelioid cell granulomas destroy the lung tissue and lead to pulmonary fibrosis.
Pathological changes
If beryllium enters the subcutaneous tissues through a cut or abrasion, as often happened in the earlier days of fluorescent lamp manufacture, a sarcoid-like granuloma soon appears at the site; in time, the overlying epidermis may break down to form an ulcer.
Even more serious are the lesions produced by the inhalation of beryllium. Chronic pulmonary berylliosis takes the form of a widespread granulomatous pneumonia with a histological picture identical to that of sarcoidosis (Fig. 7.1.30A ). Both berylliosis and sarcoidosis affect the upper lobes more than the lower (Fig. 7.1.30B) and in both diseases the granulomas are preferentially distributed along lymphatics and may involve adjacent blood vessels. In neither condition is there widespread necrosis but in both diseases the granulomas occasionally display a little central necrosis or hyalinisation. As in sarcoidosis, the hilar lymph nodes may be involved but, unlike sarcoidosis, not in isolation.
Figure 7.1.30.
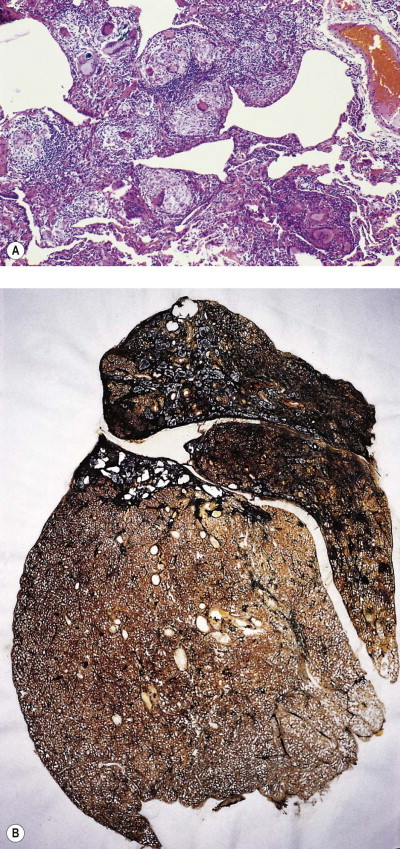
Chronic berylliosis. (A) Numerous non-necrotising epithelioid and giant cell granulomas are seen in the lungs. (B) The upper and middle lobes and the apex of the lower lobe are contracted by extensive fibrosis. Paper mounted whole lung section.
Over a period of many years, the sarcoid-like granulomas gradually undergo progressive fibrosis, with consequent impairment of pulmonary function. In the later stages, when the disease has become chronic, dispersal of beryllium from its site of initial absorption may lead to generalisation of the disease and to the appearance of similar granulomas elsewhere, particularly in the liver, kidneys, spleen and skin, but this is unusual.
Clinical features
Chronic berylliosis is characterised by the gradual onset of cough, shortness of breath, chest pain, night sweats and fatigue. These symptoms may develop within a few weeks of exposure or many years later. Once the worker is exposed, the beryllium is retained in the tissues and there is a lifelong risk of disease. Progression often entails alternating exacerbations and remissions, long after exposure has ceased.
Analysis
In keeping with the view that berylliosis is a hypersensitivity reaction, very little beryllium is necessary to cause the disease. Particulate beryllium is so scanty in the affected tissues and the atomic number of beryllium so low that electron microprobe analysis is generally unsuitable for its detection. Furthermore conventional detectors are protected by a beryllium window. However, the substitution of a polymeric window has enabled beryllium to be detected by electron microprobe analysis, presumably in a patient with fairly heavy exposure.334 Ion or laser microprobe mass spectroscopy can also detect very small amounts of beryllium in tissue sections but these techniques are not widely available.
Differential diagnosis
The differential diagnosis of chronic berylliosis is from sarcoidosis, to which it is identical morphologically.335, 336, 337 However, as noted above, it is unusual for berylliosis to cause significant hilar lymphadenopathy in the absence of pulmonary disease, which is a common feature of sarcoidosis. Extrathoracic granulomas, erythema nodosum and uveitis, which are all common in sarcoidosis, are unusual in berylliosis. However, one group found that 6% of patients initially diagnosed as having sarcoidosis actually had chronic berylliosis.338 Similar findings have been reported by others.324, 339 Any patient thought to have sarcoidosis who has worked with or near metals should be offered a beryllium lymphocyte transformation test. A list of laboratories performing this test can be found at www.dimensional.com/~mhj/medical_testing.html.
Polyvinyl chloride pneumoconiosis
Although polyvinyl is not a mineral and the reaction of the lungs to its presence is therefore not a true pneumoconiosis, it is generally so termed and is dealt with here for convenience. Workers are exposed to polyvinyl chloride dust in the milling and bagging of this plastic and micronodular opacities may be detected in their lungs radiologically. However, the material is non-fibrogenic and histology merely shows a foreign-body reaction to the dust particles.340 The radiological opacities may abate when exposure ceases.341 Nevertheless, one polyvinyl chloride worker developed systemic sclerosis,342 which is a recognised complication of silicosis (see p. 335). Polyvinyl chloride is produced from vinyl chloride monomer, which has a causal association with angiosarcoma of the liver and probably other forms of cancer, including carcinoma of the lung (see p. 534).
Flock worker's lung
In the late 1990s a characteristic lung disease was identified in workers at several factories producing plush material by spraying nylon flock on to an adhesive backing material.343, 344, 345, 346 The flock fibres are too large to be inspired but may be mixed with smaller nylon shards of respirable size. The workers complained of cough and breathlessness and were found to have a restrictive ventilatory defect with interstitial markings on radiography. Their symptoms improved on removal from the workplace but relapsed on return to work. Pathologically, there was lymphocytic bronchiolitis and peribronchiolitis with widespread lymphoid hyperplasia represented by lymphoid aggregates. Granulomas were not identified. The histological appearances suggest a severe immunological reaction and raise possibilities such as rheumatoid disease and Sjögren's syndrome but consideration of the clinical and serological setting and the occupation should permit recognition of the cause.
Popcorn worker's lung
The industrial production of popcorn and other foodstuffs appears to carry a risk of obstructive airway disease.347, 348, 349, 350 Biopsy of affected workers has shown peribronchiolar fibrosis and granulomas and air sampling has identified many volatile organic compounds, of which the flavouring agent diacetyl (2,3-butanedione) is suspected of being responsible for the bronchiolitis.
Paint spraying
It is difficult to continue paint spraying (air brushing, aerographics) without adequate respiratory protection but in the early 1990s several small aerographic factories operated in the neighbourhood of Alicante, southeastern Spain without any concern for the workers’ health. The workers were required to paint patterns on textiles using a hand-held spray gun. The atmospheric pollution was intense but complaints of respiratory difficulties were met with reassurances and the workers urged to continue. This they did because of the otherwise poor economy, often returning to work when disabling breathlessness had settled down. A change of paint (to Acramin F) may have contributed because the worst-affected workers were employed at two plants that had made this switch. Their illness has been described as the ‘Ardystil syndrome’ after the name of one of these factories. Some workers were left with permanent respiratory disability. One required a lung transplant and 6 others died.351, 352, 353, 354 Transbronchial biopsy showed organising pneumonia, which in the fatal cases had progressed to irreversible interstitial fibrosis. A similar outbreak of respiratory disease was subsequently reported in Algerian textile factories where Acromin F was applied by the same technique.355, 356 Acromin F is marketed as a paste and used as such without ill-effect. Its use in heavy spray form appears to be responsible for the ‘Ardystil syndrome’.
Mineral oils and petroleum
Workers in engineering workshops may be exposed to the prolonged inhalation of fine sprays or mists of the longer-chain hydrocarbons that constitute many mineral oils. This may result in exogenous lipid pneumonia,357 which is described on page 314, or extrinsic allergic alveolitis.358, 359, 360 The vapour of shorter-chain hydrocarbons such as paraffin oil (kerosene: C10–16) and petrol (gasoline: C4–12) and gaseous hydrocarbons such as propane may act as acute asphyxiants or central nervous system depressants but have negligible pulmonary toxicity. However, if they are ingested or aspirated in their liquid form they are acutely toxic to the lungs, producing a chemical pneumonitis with the features of diffuse alveolar damage. Ingestion may be accidental or deliberate (see Fig. 4.19, p. 142) whereas aspiration is generally inadvertent, occurring in siphoning accidents, such as those experienced by fairground operatives who ‘breath or eat fire’ (‘fire-eater's lung’).361, 362 Animal experiments involving the intratracheal injection of kerosene resulted in acute pulmonary exudates, which cleared except for residual bronchiolitis.363
Welding
Welder's pneumoconiosis, first recognised in 1936,364 essentially represents the fairly harmless deposition of iron in the lungs (siderosis – see p. 337). However, welders may suffer various ill-effects from the inhalation of substances other than iron (Table 7.1.9 ). Some of these are para-occupational risks, that is, encountered by welders because they work near another process and are inadvertently exposed: thus, shipyard welders may be exposed to asbestos,365 and those in foundries to silica. Welders may therefore develop a mixed-dust pneumoconiosis (see p. 337), rather than just siderosis. However, one analytical investigation identified excess amounts of iron alone in association with pulmonary fibrosis; the silicon content did not differ from that in controls.366
Table 7.1.9.
Principal respiratory risks of welding
| Source | Derivative | Effect |
|---|---|---|
| Metals being welded |
|
|
| Burning of adjacent surfaces |
|
|
| Electrode insulation Protective clothes |
Asbestos | Mesothelioma |
| Gas used to prevent oxidation | Carbon dioxide | Asphyxia |
| Combustive gas | Carbon monoxide | Carbon monoxide poisoning |
| Ultraviolet light | Ozone | Bronchitis |
| Various | Silica, silicates | Silicosis, mixed-dust pneumoconiosis |
More directly, welders may be exposed to asbestos insulation that they themselves use, while welders of special steel alloys run the risk of metal-induced asthma, metal fume fever, polymer fume fever and the consequences of toxic metal fume inhalation,367 all of which are described separately in this chapter, as is lung disease in aluminium welders. Chronic bronchitis has been attributed to the inhalation of low concentrations of irritants such as ozone and nitrogen dioxide by welders but this risk is unproven and the subject of much controversy. Welders may also inhale carcinogenic hexavalent chromium compounds in the course of their work and therefore develop lung cancer. The term ‘welder's lung’ is often applied indiscriminately to any of these diseases and, as it has no specific meaning, is best avoided.
Toxic fumes and gases
Dust, fume and gas are some of the terms used to describe different physical forms of respirable agents. They are defined in Table 7.2.1 on page 372. The parts of the respiratory tract at which they exert their maximal effect are influenced by particle size and solubility (see Table 7.2.2 and Fig. 7.2.2, p. 372).
Table 7.2.1.
Definitions of respirable agents by physical form
| Gas | A formless compressible fluid in which all molecules of the agent move freely at room temperature (25°C) and standard pressure (760 mmHg) to fill the space available |
| Vapour | Gaseous state of an agent which is normally liquid or solid at room temperature and standard pressure |
| Aerosol | Dispersion of solid or liquid particles of microscopic size in a gaseous medium. The following are all examples: |
| Dust | Dispersion of solid particles. Those of respirable size are not readily seen with the naked eye unless they are bathed in bright light |
| Fog | Dispersion of liquid particles generated by condensation from the vapour state |
| Fume | Dispersion of solid particles generated by condensation from the vapour state |
| Mist | Dispersion of liquid particles generated by condensation or mechanical means (e.g. nebulisation). The droplets are generally larger than those of a fog and may be visible individually to the naked eye |
| Smog | Mixture of smoke and fog, the former being the result of industrial pollution, the latter of natural climatic factors |
| Smoke | Dispersion of small particles (usually less than 0.1 µm diameter) resulting from incomplete combustion of organic substances |
Table 7.2.2.
Relation of solubility of an inhaled gas to its major site of absorption or toxicity41
| Gas | Henry's constant at 37°C | Major site of absorption or toxicity |
|---|---|---|
| Ammonia | 0.0011 | Upper respiratory tract |
| Sulphur dioxide | 0.05 | Upper respiratory tract and trachea |
| Formaldehyde | 0.56 | Upper respiratory tract and trachea |
| Ozone | 6.4 | Tracheobronchial |
| Nitrogen dioxide | 8.8 | Tracheobronchial and pulmonary |
| Oxygen | 42 | Pulmonary |
| Nitrogen | 77 | Pulmonary |
Henry's constant: moles/l(air)/moles/l(water).
Figure 7.2.2.
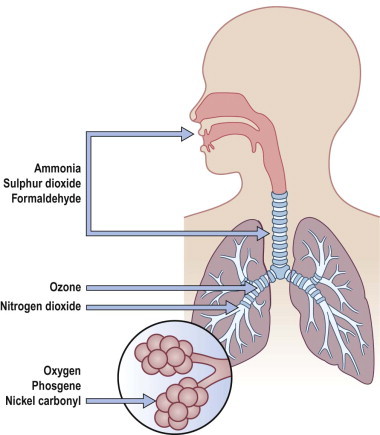
The site of maximum uptake of an inhaled gas is dependent upon its solubility. Thus, ammonia, sulphur dioxide and formaldehyde, which are highly soluble, have their major impact on the upper respiratory tract and trachea whereas oxygen, phosgene (COCl2) and nickel carbonyl (Ni(CO)4), which are less soluble, are taken up in the alveoli, where their toxicity is mainly experienced.
Toxic metal fume368
The finely divided fume of several metals is highly toxic to the lungs and capable of producing severe acute and chronic damage to both the conductive airways and the alveoli, resulting in acute tracheobronchitis and bronchiolitis, diffuse alveolar damage, obliterative bronchiolitis and pulmonary fibrosis. Important metal fumes in this respect include aluminium, which is released together with silica fume in bauxite smelting (see Shaver's disease,369 above), cadmium from welding or cutting special steels, chromium from cutting its alloys or in the manufacture of chromates, cobalt released in the production and use of its alloys (see hard-metal disease, above), mercury released in various industries and in the home,370 nickel carbonyl released during the purification of metallic nickel or the manufacture of nickel alloys371 and beryllium (see above).
Toxic gases
Many irritant gases cause severe acute and chronic damage to both the conductive airways and alveoli. The changes are non-specific and similar to those wrought by toxic metal fumes (see above) and viruses amongst other agents. They consist of acute tracheobronchitis and bronchiolitis, obliterative bronchiolitis, diffuse alveolar damage and pulmonary fibrosis.
The gases liable to produce such damage include oxides of nitrogen, sulphur dioxide, ozone, phosgene, chlorine, ammonia and various constituents of smoke, notably acrolein. Some of these are also touched upon in Chapter 7.2 because they are of general as well as occupational importance, although there is no rigid difference between general and occupational pollution.
Ozone, sulphur dioxide and nitrogen dioxide are oxidising gases that may be found together as industrial atmospheric pollutants. Each is capable of producing diffuse alveolar damage by means of its oxidising properties and the release of free active radicals. In addition, they cause damage to distal airways, particularly terminal and respiratory bronchioles, with resulting bronchiolitis.
Oxides of nitrogen may be encountered with fatal consequences by farmhands seeking to free a blockage in a silo when they encounter pockets of this gas that have accumulated on top of the fermenting silage: the term ‘silo-filler's disease’ is generally applied to the initial haemorrhagic oedema or the obliterative bronchiolitis that develops in those who survive the initial chemical injury.372, 373, 374, 375 Asphyxia due to the farmhand encountering pockets of carbon dioxide is a further hazard within agricultural silos. Other farmhands have suffered from the inhalation of toxic gases or bacteria when handling liquid manure.376, 377, 378, 379 Welding, which is considered below, may also involve exposure to toxic gases such as oxides of nitrogen.
Ozone, the principal oxidant gas of photochemical smog, produces pulmonary changes at relatively low levels and may be encountered at higher concentrations in various industries. Potentially dangerous levels of ozone are produced from atmospheric oxygen by ultraviolet radiation given off in welding while ozone is used in industry to sterilise water, bleach paper, flour and oils, and mask the odour of organic effluents. The damage wrought by ozone is predominantly centriacinar in distribution, affecting terminal and respiratory bronchiolar epithelium and proximal alveolar epithelium.380, 381, 382 There is loss of cilia and necrosis of centriacinar alveolar type I epithelial cells. The changes are dose-dependent and, in one study, the youngest animals were most sensitive.383 In long-term experiments, hyperplastic bronchiolar Clara and ciliated cells extended peripherally to line alveolar ducts.384 The role of granulocytes is stressed in some experimental studies385 and it is notable that neutrophil migration is prominent when the human lungs are damaged by ozone.386
Aldehydes such as acetaldehyde, formaldehyde and acrylic aldehyde (acrolein) are widely used in the plastics and chemical industries. The first is a liquid and the others are water-soluble gases. Pathologists are of course familiar with formaldehyde solution from its use as a disinfectant and histological fixative. All these aldehydes are intensely irritant and their acute effects generally prevent prolonged exposure to high concentrations. Chronic effects include skin sensitivity and asthma, and in rats nasal carcinoma. However, the doses to which these experimental animals were exposed far exceed any that are likely to be encountered by humans, in whom there is no convincing evidence of aldehyde-induced cancer.387
Ammonia gas is extensively used in industry as a raw material, notably in the manufacture of nitrogenous products such as fertilisers and plastics. It is highly soluble and its acute irritative effects are mainly felt in the eyes, nose and throat, but high levels affect the major airways, possibly leading to them being blocked by exudates. Survival usually brings full recovery but bronchiectasis and obliterative bronchiolitis have been described.
Chlorine gas is widely used in the chemical industry. It is transported and stored under pressure in liquid form. Heavy exposure through its accidental release or use as a war gas has proved fatal through its acute toxicity causing exudative airway occlusion and pulmonary oedema. Survivors usually recover completely but, as with nitrogen dioxide and ammonia, there is a risk of obliterative bronchiolitis.
Phosgene (carbonyl chloride, COCl2) is a poisonous, colourless gas that was responsible for thousands of deaths during World War I, when it was used in chemical warfare. It is used industrially in the preparation of some organic chemical compounds and is formed, perhaps inadvertently,388 by the combustion of methylene chloride in products such as paint strippers. Phosgene causes injury to terminal bronchioles and alveoli, with resulting oedema and hyaline membrane formation. The mechanism of cell damage is uncertain but it may depend on inactivation of intracellular enzymes by the gas. Long-term problems are rare but chronic bronchitis and emphysema have been described in survivors.
Mustard gas (bichloroethyl sulphide, C4H8Cl2S) is a further agent that has been used in chemical warfare. It is primarily a skin vesicant but when inhaled it results in widespread epithelial destruction and pulmonary oedema. Survivors may be left with irritant-induced asthma (reactive airways dysfunction), chronic bronchitis, tracheobronchomalacia, bronchiectasis and bronchiolitis obliterans.389, 390, 391
Thionyl chloride is used in the manufacture of lithium batteries where it is liable to result in the release of sulphur dioxide and hydrochloric acid fumes. Workers in such factories have developed lung injury varying from mild, reversible interstitial disease to severe obliterative bronchiolitis.392
Hydrogen sulphide is the principal chemical hazard of natural gas production. High levels of the gas also buid up in sheds housing large numbers of pigs, the source here being the pig manure. Once inhaled the gas is rapidly absorbed into the blood stream. The effects are therefore widespread but include the usual respiratory effects of irritant gases, varying from sneezing to pulmonary oedema and acute respiratory distress, depending upon the exposure. In Alberta 221 cases were identified over a 5-year period. The overall mortality was 6%; 5% of victims were dead on arrival at hospital. Most required admission to hospital but the survivors experienced no long-term adverse effects.392a
Anoxic asphyxia
The danger of asphyxia from the inhalation of gases devoid of oxygen is fairly widespread in industry.393 It generally arises from the use of inert gases, which, being non-toxic, give a false sense of security. Pockets of these gases tend to form in confined spaces. Anoxic death from the accumulation of methane is well known in mines and has also occurred in slurry pits and sewers. Anoxic asphyxia in diving (and anaesthesia) has resulted from the incorrect connection of gas cylinders or failure to notice that a mixed gas contains insufficient oxygen. Deaths have occurred in welding when argon or carbon dioxide has been used to shield the weld and prevent oxidation of the metals at the high temperatures employed. Deaths have also resulted from inadvertent entry to discharged oil tanks filled with nitrogen to reduce the risk of explosions, or from the formation of pockets of nitrogen gas applied in liquid form to freeze the contents of damaged pipes so that they can be repaired without the necessity to drain down.
The respiration of a gas devoid of oxygen causes loss of consciousness within seconds because it not only fails to provide oxygen but removes that present in the pulmonary arterial blood. The changes at autopsy are those common to cellular hypoxia. They include cerebral and serosal petechiae and pulmonary congestion and haemorrhage but these features are not specific and are not always present. The cause of death can generally only be surmised from the circumstances surrounding the death.
Occupational asthma
Occupational asthma is the commonest cause of work-related respiratory disease in many western countries (Table 7.1.10 ).394, 395, 396 The reported incidence ranges from 13 per million workers in South Africa to 174 per million workers in Finland.395, 396 It occurs in many industries (Table 7.1.11 )397 and occupational factors can be identified as contributing to asthma in about 15% of adult cases.398 Over 250 aetiological agents have been identified.399 In the UK a third are organic, a third chemical, 6% metallic and the rest miscellaneous. The commonest, in descending order, are isocyanates, flour and grain, laboratory animals, glutaraldehyde, solder or colophony and hardening agents.400
Table 7.1.10.
Work-related respiratory disease in the UK394
| Disease | Estimated annual number of cases |
|---|---|
| Occupational asthma | 941 |
| Non-malignant pleural disease | 730 |
| Mesothelioma | 644 |
| Pneumoconiosis | 341 |
| Inhalation accidents | 280 |
| Lung cancer | 70 |
| Infectious disease | 59 |
| Extrinsic allergic alveolitis | 46 |
| Bronchitis | 38 |
| Byssinosis | 1 |
| Other diagnoses | 117 |
| Total | 3207 |
Table 7.1.11.
Agents that cause occupational asthma and examples of the occupations involved397
| Causative agent | Occupations involved |
|---|---|
| High-molecular-weight agents (patients are usually atopic) | |
| Laboratory animals | Laboratory animal handling |
| Flour and grain | Baking, milling, farming |
| Enzymes | Detergent and drug manufacture, baking |
| Seafoods | Food processing |
| Gums | Carpet and drug manufacture |
| Low-molecular-weight agents (patients are not necessarily atopic) | |
| Isocyanates | Foam and plastic manufacture, spray painting, insulation |
| Anhydrides | Plastic and epoxy resin handling |
| Wood dusts | Forestry, carpentry |
| Soldering fluxes | Electronics |
| Formaldehyde, glutaraldehyde | Histopathology, nursing, radiography |
| Amines | Shellac and lacquer handling, soldering |
| Chloramine | Cleaning |
| Dyes | Textiles |
| Acrylates | Adhesive application |
| Metals | Solderers, refiners |
| Drugs | Pharmaceutical work |
Atopy appears to predispose to occupational asthma when the allergen is of high molecular weight but not when it is of low molecular weight. For example, atopic individuals are particularly prone to develop asthma if employed in the manufacture of biological detergents, whereas atopy does not increase the risk of asthma from sensitisation to toluene di-isocyanate, which is a serious health problem in the manufacture of polyurethane. Similarly, platinum salts are such potent sensitising agents that nearly all those exposed to them develop asthma. Asthma-provoking metals other than platinum include chromium, cobalt, nickel and vanadium, all of which are used in steel alloys, and possibly aluminium (see pot-room asthma, p. 357). Other asthma-inducing factors encountered in industry include grain and flour dust, certain wood dusts, soldering fluxes containing colophony (pine resin), epoxy resin hardeners such as phthallic anhydride, isocyanate-containing foams and paints, formaldehyde and the excreta of laboratory animals. Contaminated humidifiers may cause occupational asthma as well as humidifier fever and extrinsic allergic alveolitis.401 Pathologically, occupational asthma is identical to non-occupational asthma (see p. 109).
Byssinosis
Byssinosis is a further form of occupational asthma,402 one encountered in the cotton industry. The sensitising agent is a component of the cotton bract, which is the part of the cotton harvest other than the cotton fibre. Bract consists of dried leaf, other plant debris and soil particles and contains a variety of fungal and bacterial residues, including lipopolysaccharide endotoxin, but the exact nature of the sensitising agent remains unknown.403 The endotoxin is unlikely to be responsible for byssinosis but may be the cause of so-called mill fever, a self-limiting illness characterised by malaise, fever and leukocytosis that is experienced by many people on first visiting a cotton mill.
Dust levels and the risk of byssinosis are particularly high in the carding rooms where the raw cotton is teased out before it is spun. Affected workers are worse when they return to work after the weekend break, a feature attributed to antibody levels having built up during this brief respite from the cotton dust. There is no link with atopy and the fluctuating antibodies are precipitins of the immunoglobulin G class. Complement activation by both arms of the complement cascade has been reported.404, 405
When the Lancashire economy was largely cotton-based, necropsies on workers suffering from byssinosis generally showed gross emphysema, and this came to be accepted as evidence of byssinosis. However, it is now realised that in this heavily industrialised part of the UK, emphysema is as common in the general population as in cotton workers and it can no longer be considered a component of byssinosis. Other findings in byssinosis are more commensurate with asthma, namely an increase in bronchial muscle and mucous cells.406 No granulomas or other evidence of extrinsic allergic alveolitis are found.
Occupational fevers
Fever may be the predominant feature in a variety of occupational illnesses and the unifying term ‘inhalation fever’ has been proposed.407 However, the individual occupations are of interest and these conditions will therefore be considered separately. Mill fever has been mentioned above under byssinosis.
Humidifier fever408
Humidifier fever is an acute illness characterised by malaise, fever, myalgia, cough, tightness in the chest and breathlessness, all of which are worse on Monday mornings if the humidifier responsible is at work rather than home. The chest complaints, and their aggravation on return to work after the weekend, are features shared with byssinosis (see above) but the general complaints fit better with extrinsic allergic alveolitis (see p. 279). Humidifier fever develops in circumstances that also lead to the development of a form of extrinsic allergic alveolitis, and not surprisingly the same name has been extended to this latter condition, with inevitable confusion. Both diseases are caused by microbiological contamination of humidifiers or air conditioners so that a fine spray of microorganisms is emitted into the office, factory or home. Investigations have generally shown the baffle plates of the air conditioner to be covered with a slime of bacteria, fungi or protozoa (mainly amoeba and ciliates), and extracts of this have been used to identify precipitins in the patient's sera, as in extrinsic allergic alveolitis. However, unlike extrinsic allergic alveolitis, humidifier fever resolves within a day and leaves no permanent injury. For this reason there is seldom the opportunity to study the tissue changes, and partly for this reason it remains unclear whether the disease is mediated by immune complexes, as in extrinsic allergic alveolitis, or by endotoxins derived from the contaminants.
Pulmonary mycotoxicosis
A febrile illness occurring in precipitin-negative farm-workers after heavy exposure to fungi in their silos was attributed to inhaled fungal toxins and named pulmonary mycotoxicosis.409 It is also known as precipitin test-negative farmer's lung and organic dust toxic syndrome.410 The condition is generally self-limiting and is seldom biopsied but desquamative interstitial pneumonia and diffuse alveolar damage have been reported.411, 412
Metal fume fever
This is a self-limiting acute illness characterised by fever, sweating, myalgia, chest pain, headache and nausea, that comes on Monday mornings when occupational exposure is experienced after a weekend's respite, as with bysinnosis and humidifier fever; during the week tolerance develops.368, 413 The disease involves the release of cytokines such as tumour necrosis factor and is presumed to have an allergic basis.414 The metals involved are chiefly zinc, copper and magnesium, and, to a lesser extent, aluminium, antimony, iron, manganese and nickel. Occupations at risk include any that generate such metal fumes, but particularly welding. It is most commonly associated with welding zinc-coated surfaces. If the symptoms persist, alternative diagnoses, such as acute cadmium poisoning and other specific toxic metal fume diseases, should be suspected: these are not self-limiting and may cause severe bronchiolitis or diffuse alveolar damage (see above).
Polymer fume fever
This illness resembles metal fume fever except that it occurs without regard to previous exposure: no tolerance develops and there is therefore no particular susceptibility on Mondays. The polymers concerned are quite inert, except when heated to produce fume: polytetrafluorethylene (PTFE, Teflon, Fluon, Halon) is a notable example. As with other self-limiting diseases, little is known of the tissue changes.
References
Pneumoconiosis: general features
- 1.Oxman AD, Muir DCF, Shannon HS, et al. Occupational dust exposure and chronic obstructive pulmonary disease – a systematic overview of the evidence. Am Rev Respir Dis. 1993;148:38–48. doi: 10.1164/ajrccm/148.1.38. [DOI] [PubMed] [Google Scholar]
- 2.Blanc PD, Iribarren C, Trupin L, et al. Occupational exposures and the risk of COPD: dusty trades revisited. Thorax. 2009;64:6–12. doi: 10.1136/thx.2008.099390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meiklejohn A. The origin of the term ‘pneumonokoniosis’. Br J Ind Med. 1960;17:155–160. [Google Scholar]
- 4.Industrial Injuries Advisory Council . HMSO; London: 1973. Pneumoconiosis and byssinosis. p. 1. [Google Scholar]
- 5.Parkes WR. In: Occupational Lung Disorders. 3rd ed. Parkes WR, editor. Butterworth Heinemann; Oxford: 1994. Aerosols: their deposition and clearance; p. 35. [Google Scholar]
- 6.Churg A, Brauer M. Human lung parenchyma retains PM2.5. Amer J Respir Crit Care Med. 1997;155:2109–2111. doi: 10.1164/ajrccm.155.6.9196123. [DOI] [PubMed] [Google Scholar]
- 7.Pinkerton KE, Plopper CG, Mercer RR, et al. Airway branching patterns influence asbestos fiber location and the extent of tissue injury in the pulmonary parenchyma. Lab Invest. 1986;55:688–695. [PubMed] [Google Scholar]
- 8.Becklake MR, Toyota B, Stewart M, et al. Lung structure as a risk factor in adverse pulmonary responses to asbestos exposure. A case-referent study in Quebec chrysotile miners and millers. Am Rev Respir Dis. 1983;128:385–388. doi: 10.1164/arrd.1983.128.3.385. [DOI] [PubMed] [Google Scholar]
- 9.Schlesinger MR, Lippman M. Particle deposition in casts of the human upper tracheobronchial tree. Am Ind Hyg Assoc J. 1972;33:237–250. doi: 10.1080/0002889728506636. [DOI] [PubMed] [Google Scholar]
- 10.Pityn P, Chamberlain MJ, King ME, et al. Differences in particle deposition between the two lungs. Respir Med. 1995;89:15–19. doi: 10.1016/0954-6111(95)90065-9. [DOI] [PubMed] [Google Scholar]
- 11.Ferin J, Oberdorster G, Penney DP. Pulmonary retention of ultrafine and fine particles in rats. Am J Respir Cell Mol Biol. 1992;6:535–542. doi: 10.1165/ajrcmb/6.5.535. [DOI] [PubMed] [Google Scholar]
- 12.Brundelet PJ. Experimental study of the dust-clearance mechanism of the lung. Acta Path Microbiol Scand. 1965:7–141. [PubMed] [Google Scholar]
- 13.Cordingley JL, Nicol T. The lung: an excretory route for macromolecules and particles. J Physiol (London) 1967;190:7. [PubMed] [Google Scholar]
- 14.Corrin B. Phagocytic potential of pulmonary alveolar epithelium with particular reference to surfactant metabolism. Thorax. 1970;25:110–115. doi: 10.1136/thx.25.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brody AR, Hill LH, Adkins B, et al. Chrysotile asbestos inhalation in rats: deposition pattern and reaction of alveolar epithelium and pulmonary macrophages. Am Rev Respir Dis. 1981;123:670–678. doi: 10.1164/arrd.1981.123.6.670. [DOI] [PubMed] [Google Scholar]
- 16.Churg A. The uptake of mineral particles by pulmonary epithelial cells. Amer J Respir Crit Care Med. 1996;154:1124–1140. doi: 10.1164/ajrccm.154.4.8887617. [DOI] [PubMed] [Google Scholar]
- 17.Lehnert BE, Valdez YE, Stewart CC. Translocation of particles to the tracheobronchial lymph nodes after lung deposition: kinetics and particle-cell relationships. Exp Lung Res. 1986;10:245–266. doi: 10.3109/01902148609061496. [DOI] [PubMed] [Google Scholar]
- 18.Adamson IYR, Hedgecock C. Patterns of particle deposition and retention after instillation to mouse lung during acute injury and fibrotic repair. Exp Lung Res. 1995;21:695–709. doi: 10.3109/01902149509050837. [DOI] [PubMed] [Google Scholar]
- 19.Adamson IYR, Prieditis H. Silica deposition in the lung during epithelial injury potentiates fibrosis and increases particle translocation to lymph nodes. Exp Lung Res. 1998;24:293–306. doi: 10.3109/01902149809041536. [DOI] [PubMed] [Google Scholar]
- 20.Gross P, Westrick M. The permeability of lung parenchyma to particulate matter. Am J Pathol. 1954;30:195–207. [PMC free article] [PubMed] [Google Scholar]
- 21.Corry D, Kulkarni P, Lipscomb MF. The migration of bronchoalveolar macrophages into hilar lymph nodes. Am J Pathol. 1984;115:321–328. [PMC free article] [PubMed] [Google Scholar]
- 22.Policard A, Collet A, Pregermain S, et al. Etude au microscope electronique du granulome pulmonaire silicotique experimental. Presse Med. 1957;65:121–124. [PubMed] [Google Scholar]
- 23.Emery JL, Dinsdale F. The postnatal development of lymphoreticular aggregates and lymph nodes in infants’ lungs. J Clin Pathol. 1973;26:539–545. doi: 10.1136/jcp.26.7.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Macklin CS. Pulmonary sumps, dust accumulations, alveolar fluid and lymph vessels. Acta Anat. 1955;23:1–33. doi: 10.1159/000140979. [DOI] [PubMed] [Google Scholar]
- 25.O'Driscoll BRC, Cooke RDP, Mamtora H, et al. Candida lung abscesses complicating parenteral nutrition. Thorax. 1988;43:418–419. doi: 10.1136/thx.43.5.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pintar K, Funahashi A, Siegesmund KA. A diffuse form of pulmonary silicosis in foundry workers. Arch Pathol Lab Med. 1976;100:535–538. [PubMed] [Google Scholar]
- 27.Wagner JC, Pooley FD, Gibbs A, et al. Inhalation of china stone and china clay dusts: relationship between mineralogy of dust retained in the lungs and pathological changes. Thorax. 1986;41:190–196. doi: 10.1136/thx.41.3.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kleinfeld M, Giel CP, Majeranowski JF, et al. Talc pneumoconiosis. A report of six patients with postmortem findings. Arch Env Health. 1963;7:101–115. doi: 10.1080/00039896.1963.10663500. [DOI] [PubMed] [Google Scholar]
- 29.Ruttner JR, Spycher MA, Sticher H. Diffuse ‘asbestosis-like’ interstitial fibrosis of the lung. Pathol Microbiol. 1972;38:250–257. [PubMed] [Google Scholar]
- 30.Vallyathan NV, Craighead JE. Pulmonary pathology in workers exposed to nonasbestiform talc. Hum Pathol. 1981;12:28–35. doi: 10.1016/s0046-8177(81)80239-0. [DOI] [PubMed] [Google Scholar]
- 31.Davies D, Cotton R. Mica pneumoconiosis. Br J Ind Med. 1983;40:22–27. doi: 10.1136/oem.40.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gibbs AE, Pooley FD, Griffiths DM, et al. Talc pneumoconiosis – a pathologic and mineralogic study. Hum Pathol. 1992;23:1344–1354. doi: 10.1016/0046-8177(92)90053-6. [DOI] [PubMed] [Google Scholar]
- 33.Honma K, Chiyotani K. Diffuse interstitial fibrosis in nonasbestos pneumoconiosis – a pathological study. Respiration. 1993;60:120–126. doi: 10.1159/000196185. [DOI] [PubMed] [Google Scholar]
- 34.Heppleston AG. The disposal of coal and haematite dusts inhaled successively. J Pathol Bacteriol. 1958;75:113–126. doi: 10.1002/path.1700750114. [DOI] [PubMed] [Google Scholar]
- 35.Glazier JB, Hughes JMB, Maloney JE, et al. Vertical gradient of alveolar size in lungs of dogs frozen intact. J Appl Physiol. 1967;23:694–705. doi: 10.1152/jappl.1967.23.5.694. [DOI] [PubMed] [Google Scholar]
- 36.Dock W. Apical localization of phthisis. Am Rev Tuberc. 1946;53:297–305. doi: 10.1164/art.1946.53.4.297. [DOI] [PubMed] [Google Scholar]
- 37.West JB, Dollery CT. Distribution of blood flow and ventilation perfusion ratio in the lung, measured with radioactive CO2. J Appl Physiol. 1960;15:405–410. doi: 10.1152/jappl.1960.15.3.405. [DOI] [PubMed] [Google Scholar]
- 38.Goodwin RA, Des Prez RM. Apical localization of pulmonary tuberculosis, chronic pulmonary histoplasmosis, and progressive massive fibrosis of the lung. Chest. 1983;83:801–805. doi: 10.1378/chest.83.5.801. [DOI] [PubMed] [Google Scholar]
- 39.Bake B, Wood L, Murphy B, et al. Effect of inspiratory flow rate on the regional distribution of inspired gas. J Appl Physiol. 1974;37:8–17. doi: 10.1152/jappl.1974.37.1.8. [DOI] [PubMed] [Google Scholar]
- 40.Davson J, Susman W. Apical scars and their relationship to siliceous dust accumulation in non-silicotic lungs. J Path Bact. 1937;45:597–612. [Google Scholar]
- 41.Sebastien P, Fondimare A, Bignon J, et al. In: Inhaled Particles IV. Walton WH, editor. Pergamon; Oxford: 1977. Topographic distribution of asbestos fibres in human lung in relation to occupational and non-occupational exposure; pp. 435–446. [PubMed] [Google Scholar]
- 42.Churg A. The distribution of amosite asbestos in the periphery of the normal human lung. Br J Ind Med. 1990;47:677–681. doi: 10.1136/oem.47.10.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Silicosis and Silicate Disease Committee Diseases associated with exposure to silica and nonfibrous silicate minerals. Arch Pathol Lab Med. 1988;112:673–720. [PubMed] [Google Scholar]
- 44.McDonald JW, Roggli VL. Detection of silica particles in lung tissue by polarizing light microscopy. Arch Pathol Lab Med. 1995;119:242–246. [PubMed] [Google Scholar]
- 45.Crocker PR, Doyle DV, Levison DA. A practical method for the identification of particulate and crystalline material in paraffin-embedded tissue specimens. J Pathol. 1980;131:165–173. doi: 10.1002/path.1711310209. [DOI] [PubMed] [Google Scholar]
- 46.Crocker PR, Toulson E, Levison DA. Particles in paraffin sections demonstrated in the backscattered electron image [BEI] Micron. 1982;13:437–446. [Google Scholar]
- 47.Abraham JL, Burnett BR. Quantitative analysis of inorganic particulate burden in situ in tissue sections. Scanning Electron Microsc. 1983;II:681–696. [PubMed] [Google Scholar]
- 48.Levison DA. Microanalysis in histopathology. J Pathol. 1989;157:95–97. doi: 10.1002/path.1711570203. [DOI] [PubMed] [Google Scholar]
- 49.Abraham JL, Rossi R, Marquez N, et al. Ion microprobe mass analysis of beryllium in situ in human lung: preliminary results. Scanning Electron Microsc. 1976;III:501–506. [Google Scholar]
- 50.Jones-Williams W, Wallach ER. Laser probe mass spectrometry (LAMMS) analysis of beryllium, sarcoidosis and other granulomatous diseases. Sarcoidosis. 1991;6:111–117. [PubMed] [Google Scholar]
- 51.DeNollin S, Poels K, VanVaeck L, et al. Molecular identification of foreign inclusions in inflammatory tissue surrounding metal implants by Fourier transform laser microprobe mass spectrometry. Pathol Res Pract. 1997;193:313–318. doi: 10.1016/S0344-0338(97)80009-X. [DOI] [PubMed] [Google Scholar]
- 52.Jones Williams W, Williams WR. Value of beryllium lymphocyte transformation test in chronic beryllium disease and in potentially exposed workers. Thorax. 1983;38:41–44. doi: 10.1136/thx.38.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vallyathan V, Brower PS, Green FHY, et al. Radiographic and pathologic correlation of coal workers’ pneumoconiosis. Amer J Respir Crit Care Med. 1996;154:741–748. doi: 10.1164/ajrccm.154.3.8810614. [DOI] [PubMed] [Google Scholar]
Silicosis
- 54.King EJ, Mohanty GP, Harrison CV, et al. The action of different forms of pure silica on the lungs of rats. Br J Ind Med. 1953;10:9–17. doi: 10.1136/oem.10.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gibbs AR, Craighead JE, Pooley FD, et al. The pathology of slate workers’ pneumoconiosis in North Wales and Vermont. Ann Occup Hyg. 1988;32:273–278. [Google Scholar]
- 56.Craighead JE, Emerson RJ, Stanley DE. Slateworker's pneumoconiosis. Hum Pathol. 1992;23:1098–1105. doi: 10.1016/0046-8177(92)90027-z. [DOI] [PubMed] [Google Scholar]
- 57.Palmer PES, Daynes G. Transkei silicosis. S Afr Med J. 1967;41:1182–1188. [PubMed] [Google Scholar]
- 58.Selden A, Sahle W, Johansson L, et al. Three cases of dental technician's pneumoconiosis related to cobalt-chromium-molybdenum dust exposure: diagnosis and follow-up. Chest. 1996;109:837–842. doi: 10.1378/chest.109.3.837. [DOI] [PubMed] [Google Scholar]
- 59.Policard A, Collet A. Deposition of silicosis dust in the lungs of the inhabitants of the Sahara regions. Arch Ind Hyg Occup Med. 1952;5:527–534. [PubMed] [Google Scholar]
- 60.Fossati C. Sulla possibilita e sulla frequenza della silicosi pulmonare tra gli abitanti del deserto libico. Med Lav. 1969;60:144–149. [PubMed] [Google Scholar]
- 61.Hirsch M, Bar-Ziv J, Lehmann E, et al. Simple siliceous pneumoconiosis of Bedouin females in the Negev desert. Clin Radiol. 1974;25:507–510. doi: 10.1016/s0009-9260(74)80135-2. [DOI] [PubMed] [Google Scholar]
- 62.Bar-Ziv J, Goldberg G. Simple siliceous pneumoconiosis in Negev Bedouins. Arch Environ Health. 1974;29:121–126. doi: 10.1080/00039896.1974.10666548. [DOI] [PubMed] [Google Scholar]
- 63.Fennerty A, Hunter AM, Smith AP, et al. Silicosis in a Pakistani farmer. BMJ. 1983;ii:648. doi: 10.1136/bmj.287.6393.648-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Norboo T, Angchuk PT, Yahya M, et al. Silicosis in a Himalayan village population – role of environmental dust. Thorax. 1991;46:341–343. doi: 10.1136/thx.46.5.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saiyed HN, Sharma YK, Sadhu HG, et al. Non-occupational pneumoconiosis at high altitude villages in central Ladakh. Br J Ind Med. 1991;48:825–829. doi: 10.1136/oem.48.12.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sherwin RP, Barman ML, Abraham JL. Silicate pneumoconiosis of farm workers. Lab Invest. 1979;40:576–582. [PubMed] [Google Scholar]
- 67.Schwartz LW, Knight HD, Malloy RL, et al. Silicate pneumoconiosis and pulmonary fibrosis in horses from the Monterey-Carmel peninsula. Chest. 1981;80:82S–85S. doi: 10.1378/chest.80.1_supplement.82s. [DOI] [PubMed] [Google Scholar]
- 68.Brambilla C, Abraham J, Brambilla E, et al. Comparative pathology of silicate pneumoconiosis. Am J Pathol. 1979;96:149–170. [PMC free article] [PubMed] [Google Scholar]
- 69.MacDonald G, Piggot AP, Gilder FW. Two cases of acute silicosis – with a suggested theory of causation. Lancet. 1930;2:846–848. [Google Scholar]
- 70.Chapman EM. Acute silicosis. JAMA. 1932;98:1439–1441. [Google Scholar]
- 71.Vincent M, Arthaud Y, Crettet G, et al. Silicose aigue fatale par inhalation volontaire de poudre à recurer. Rev Mal Resp. 1995;12:499–502. [PubMed] [Google Scholar]
- 72.Suratt PM, Winn WC, Brody AR, et al. Acute silicosis in tombstone sandblasters. Am Rev Respir Dis. 1977;115:521–529. doi: 10.1164/arrd.1977.115.3.521. [DOI] [PubMed] [Google Scholar]
- 73.Seaton A, Legge JS, Henderson J, et al. Accelerated silicosis in Scottish stonemasons. Lancet. 1991;337:341–344. doi: 10.1016/0140-6736(91)90956-p. [DOI] [PubMed] [Google Scholar]
- 74.Murray J, Webster I, Reid G, et al. The relation between fibrosis of hilar lymph glands and the development of parenchymal silicosis. Br J Ind Med. 1991;48:267–269. doi: 10.1136/oem.48.4.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hessel PA, Sluis-Cremer GK, Lee SL. Distribution of silicotic collagenization in relation to smoking habits. Am Rev Respir Dis. 1991;144:297–301. doi: 10.1164/ajrccm/144.2.297. [DOI] [PubMed] [Google Scholar]
- 76.Baldwin DR, Lambert L, Pantin CFA, et al. Silicosis presenting as bilateral hilar lymphadenopathy. Thorax. 1996;51:1165–1167. doi: 10.1136/thx.51.11.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tosi P, Franzinelli A, Miracco C, et al. Silicotic lymph node lesions in non-occupationally exposed lung carcinoma patients. Eur J Respir Dis. 1986;68:362–369. [PubMed] [Google Scholar]
- 78.Kampalath BN, McMahon JT, Cohen A, et al. Obliterative central bronchitis due to mineral dust in patients with pneumoconiosis. Arch Pathol Lab Med. 1998;122:56–62. [PubMed] [Google Scholar]
- 79.Lardinois D, Gugger M, Balmer MC, et al. Left recurrent laryngeal nerve palsy associated with silicosis. Eur Resp J. 1999;14:720–722. doi: 10.1034/j.1399-3003.1999.14c37.x. [DOI] [PubMed] [Google Scholar]
- 80.Argani P, Ghossein R, Rosai J. Anthracotic and anthracosilicotic spindle cell pseudotumors of mediastinal lymph nodes: Report of five cases of a reactive lesion that simulates malignancy. Hum Pathol. 1998;29:851–855. doi: 10.1016/s0046-8177(98)90456-7. [DOI] [PubMed] [Google Scholar]
- 81.Chien HP, Lin TP, Chen HL, et al. Right middle lobe atelectasis associated with endobronchial silicotic lesions. Arch Pathol Lab Med. 2000;124:1619–1622. doi: 10.5858/2000-124-1619-RMLAAW. [DOI] [PubMed] [Google Scholar]
- 82.Rashid AMH, Green FHY. Pleural pearls following silicosis: a histological and electronmicroscopic study. Histopathology. 1995;26:84–87. doi: 10.1111/j.1365-2559.1995.tb00627.x. [DOI] [PubMed] [Google Scholar]
- 83.Zeren EH, Colby TV, Roggli VL. Silica-induced pleural disease: An unusual case mimicking malignant mesothelioma. Chest. 1997;112:1436–1438. doi: 10.1378/chest.112.5.1436. [DOI] [PubMed] [Google Scholar]
- 84.Ng TP, Chan SL. Factors associated with massive fibrosis in silicosis. Thorax. 1991;46:229–232. doi: 10.1136/thx.46.4.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lynch KM. Silicosis of systemic distribution. Am J Pathol. 1942;18:313–331. [PMC free article] [PubMed] [Google Scholar]
- 86.Langlois SLEP, Sterrett GF, Henderson DW. Hepatosplenic silicosis. Australas Radiol. 1977;21:143–149. doi: 10.1111/j.1440-1673.1977.tb03185.x. [DOI] [PubMed] [Google Scholar]
- 87.Carmichael GP, Targoff C, Pintar K, et al. Hepatic silicosis. Am J Clin Pathol. 1980;73:720–722. doi: 10.1093/ajcp/73.5.720. [DOI] [PubMed] [Google Scholar]
- 88.Eide J, Gylseth B, Skaug V. Silicotic lesions of the bone marrow: histopathology and microanalysis. Histopathology. 1984;8:693–703. doi: 10.1111/j.1365-2559.1984.tb02381.x. [DOI] [PubMed] [Google Scholar]
- 89.Miranda RN, McMillan PN, Pricolo VE, et al. Peritoneal silicosis. Arch Pathol Lab Med. 1996;120:300–302. [PubMed] [Google Scholar]
- 90.Harding HE, McLaughlin AIG. Pulmonary fibrosis in non-ferrous foundry workers. Br J Ind Med. 1955;12:92–99. doi: 10.1136/oem.12.2.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cockcroft AE, Wagner JC, Seal EM, et al. Irregular opacities in coalworkers’ pneumoconiosis – correlation with pulmonary function and pathology. Ann Occup Hyg. 1982;26:767–787. [PubMed] [Google Scholar]
- 92.Katabami M, Dosakaakita H, Honma K, et al. Pneumoconiosis-related lung cancers – Preferential occurrence from diffuse interstitial fibrosis-type pneumoconiosis. Amer J Respir Crit Care Med. 2000;162:295–300. doi: 10.1164/ajrccm.162.1.9906138. [DOI] [PubMed] [Google Scholar]
- 93.Rowsell EV, Nagelschmidt G, Curran RC. The effects of dusts on peritoneal cells within diffusion chambers. J Pathol Bacteriol. 1960;80:337–344. doi: 10.1002/path.1700800217. [DOI] [PubMed] [Google Scholar]
- 94.Vallyathan V, Castranova V, Pack D, et al. Freshly fractured quartz inhalation leads to enhanced lung injury and inflammation: potential role of free radicals. Am J Respir Crit Care Med. 1995;152:1003–1009. doi: 10.1164/ajrccm.152.3.7663775. [DOI] [PubMed] [Google Scholar]
- 95.Allison AC, Harington JS, Birbeck M. An examination of the cytotoxic effects of silica on macrophages. J Exp Med. 1966;124:141–154. doi: 10.1084/jem.124.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Heppleston AG, Styles JA. Activity of macrophage factor in collagen formation by silica. Nature. 1967;214:521–522. doi: 10.1038/214521a0. [DOI] [PubMed] [Google Scholar]
- 97.Aalto M, Kulonen E. Fractionation of connective-tissue-activating factors from the culture medium of silica-treated macrophages. Acta Path Microbiol Scand Sect C. 1979;87:241–250. [PubMed] [Google Scholar]
- 98.Aalto M, Turakainen H, Kulonen E. Effect of SiO2-liberated macrophage factor on protein synthesis in connective tissue in vitro. Scand J Clin Lab Invest. 1979;39:205–213. doi: 10.1080/00365517909106095. [DOI] [PubMed] [Google Scholar]
- 99.Lugano EM, Dauber JH, Elias JA, et al. The regulation of lung fibroblast proliferation by alveolar macrophages in experimental silicosis. Am Rev Respir Dis. 1984;129:767–771. doi: 10.1164/arrd.1984.129.5.767. [DOI] [PubMed] [Google Scholar]
- 100.Donaldson K, Slight J, Brown GM, et al. The ability of inflammatory bronchoalveolar leucocyte populations elicited with microbes or mineral dust to injure alveolar epithelial cells and degrade extracellular matrix in vitro. Br J Exp Pathol. 1988;69:327–338. [PMC free article] [PubMed] [Google Scholar]
- 101.Vanhee D, Gosset P, Boitelle A, et al. Cytokines and cytokine network in silicosis and coal workers’ pneumoconiosis. Eur Respir J. 1995;8:834–842. [PubMed] [Google Scholar]
- 102.Claudio E, Segade F, Wrobel K, et al. Activation of murine macrophages by silica particles in vitro is a process independent of silica-induced cell death. Am J Respir Cell Mol Biol. 1995;13:547–554. doi: 10.1165/ajrcmb.13.5.7576690. [DOI] [PubMed] [Google Scholar]
- 103.Mariani TJ, Roby JD, Mecham RP, et al. Localization of type I procollagen gene expression in silica-induced granulomatous lung disease and implication of transforming growth factor-beta as a mediator of fibrosis. Am J Pathol. 1996;148:151–164. [PMC free article] [PubMed] [Google Scholar]
- 104.Jagirdar J, Begin R, Dufresne A, et al. Transforming growth factor-beta (TGF-beta) in silicosis. Amer J Respir Crit Care Med. 1996;154:1076–1081. doi: 10.1164/ajrccm.154.4.8887610. [DOI] [PubMed] [Google Scholar]
- 105.Mossman BT, Churg A. Mechanisms in the pathogenesis of asbestosis and silicosis. Amer J Respir Crit Care Med. 1998;157:1666–1680. doi: 10.1164/ajrccm.157.5.9707141. [DOI] [PubMed] [Google Scholar]
- 106.Erasmus LD. Scleroderma in gold-miners on the Witwatersrand with particular reference to pulmonary manifestations. S Afr J Lab Clin Med. 1957;3:209–231. [PubMed] [Google Scholar]
- 107.Vigliani EC, Pernis B. Immunological factors in the pathogenesis of the hyaline tissue of silicosis. Br J Ind Med. 1958;15:8–14. doi: 10.1136/oem.15.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Scheule RK, Holian A. Mini-review – immunologic aspects of pneumoconiosis. Exp Lung Res. 1991;17:661–685. doi: 10.3109/01902149109062872. [DOI] [PubMed] [Google Scholar]
- 109.Beckett W, Abraham J, Becklake M, et al. Adverse effects of crystalline silica exposure. Amer J Respir Crit Care Med. 1997;155:761–768. doi: 10.1164/ajrccm.155.2.9032226. [DOI] [PubMed] [Google Scholar]
- 110.Powell DEB, Gough J. Br J Exp Pathol. 1959;40:40–43. [PMC free article] [PubMed] [Google Scholar]
- 111.Kato T, Usami I, Morita H, et al. Chronic necrotizing pulmonary aspergillosis in pneumoconiosis – clinical and radiologic findings in 10 patients. Chest. 2002;121:118–127. doi: 10.1378/chest.121.1.118. [DOI] [PubMed] [Google Scholar]
- 112.International Agency for Research on Cancer (Lyons) Silica and some silicates. Monographs on the evaluation of the carcinogenic risk of chemicals to humans. 1987;42 [PubMed] [Google Scholar]
- 113.Weill H, McDonald JC. Occupational lung disease: 1. exposure to crystalline silica and risk of lung cancer: the epidemiological evidence. Thorax. 1996;51:97–102. doi: 10.1136/thx.51.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Craighead JE. Do silica and asbestos cause lung cancer? Arch Pathol Lab Med. 1992;116:16–20. [PubMed] [Google Scholar]
- 115.Department of Social Security . HMSO; London: 1992. Lung cancer in relation to occupational exposure to silica. Report by the Industrial Injuries Advisory Council in accordance with Section 171 of the Social Security Administration Act 1992 on the question whether lung cancer in relation to occupational exposure to silica should be prescribed. p. 1–5. [Google Scholar]
- 116.Checkoway H, Hughes JM, Weill H, et al. Crystalline silica exposure, radiological silicosis, and lung cancer mortality in diatomaceous earth industry workers. Thorax. 1999;54:56–59. doi: 10.1136/thx.54.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Buechner HA, Ansari A. Acute silico-proteinosis. Dis Chest. 1969;55:274–284. doi: 10.1378/chest.55.4.274. [DOI] [PubMed] [Google Scholar]
- 118.Xipell JM, Ham KN, Price CG, et al. Acute silicolipoproteinosis. Thorax. 1977;32:104–111. doi: 10.1136/thx.32.1.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Heppleston AG. Atypical reaction to inhaled silica. Nature. 1967;213:199. doi: 10.1038/213199a0. [DOI] [PubMed] [Google Scholar]
- 120.Gross P, De Treville RTP. Alveolar proteinosis. Its experimental production in rodents. Arch Pathol. 1968;86:255–261. [PubMed] [Google Scholar]
- 121.Corrin B, King E. Experimental endogenous lipid pneumonia and silicosis. J Pathol. 1969;97:325–330. doi: 10.1002/path.1710970218. [DOI] [PubMed] [Google Scholar]
- 122.Corrin B, King E. Pathogenesis of experimental pulmonary alveolar proteinosis. Thorax. 1970;25:230–236. doi: 10.1136/thx.25.2.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Heppleston AG, Fletcher K, Wyatt I. Changes in the composition of lung lipids and the ‘turnover’ of dipalmitoyl lecithin in experimental alveolar lipo-proteinosis induced by inhaled quartz. Br J Exp Pathol. 1974;55:384–395. [PMC free article] [PubMed] [Google Scholar]
- 124.Miller RR, Churg AM, Hutcheon M, et al. Case report: pulmonary alveolar proteinosis and aluminum dust exposure. Am Rev Respir Dis. 1984;130:312–315. doi: 10.1164/arrd.1984.130.2.312. [DOI] [PubMed] [Google Scholar]
- 125.Miller BE, Bakewell WE, Katyal SL, et al. Induction of surfactant protein (SP-A) biosynthesis and SP-A mRNA in activated type II cells during acute silicosis in rats. Am J Respir Cell Mol Biol. 1990;3:217–226. doi: 10.1165/ajrcmb/3.3.217. [DOI] [PubMed] [Google Scholar]
- 126.Hauglustaine D, Van Damme B, Daenens P, et al. Silicon nephropathy: a possible occupational hazard. Nephron. 1980;26:219–224. doi: 10.1159/000181988. [DOI] [PubMed] [Google Scholar]
- 127.Bolton WK, Suratt PM, Strugill BC. Rapidly progressive silicon nephropathy. Am J Med. 1981;71:823–828. doi: 10.1016/0002-9343(81)90374-0. [DOI] [PubMed] [Google Scholar]
- 128.Osorio AM, Thun MJ, Novak RF, et al. Silica and glomerulonephritis: case report and review of the literature. Am J Kidney Dis. 1987;9:224–230. doi: 10.1016/s0272-6386(87)80059-8. [DOI] [PubMed] [Google Scholar]
- 129.Sherson D, Jorgensen F. Rapidly progressive crescenteric glomerulonephritis in a sandblaster with silicosis. Br J Ind Med. 1989;46:675–676. doi: 10.1136/oem.46.9.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dutra FR. Diatomaceous earth pneumoconiosis. Arch Env Health. 1965;11:613–619. doi: 10.1080/00039896.1965.10664269. [DOI] [PubMed] [Google Scholar]
Silicates
- 131.Abraham JL, Brambilla C. Particle size for differentiation between inhalation and injection pulmonary talcosis. Environ Res. 1980;21:94–96. doi: 10.1016/0013-9351(80)90011-0. [DOI] [PubMed] [Google Scholar]
- 132.Lapenas DJ, Gale PN. Kaolin pneumoconiosis. A case report. Arch Pathol Lab Med. 1983;107:650–653. [PubMed] [Google Scholar]
- 133.Lapenas D, Gale P, Kennedy T, et al. Kaolin pneumoconiosis. Radiologic, pathologic and mineralogic findings. Am Rev Respir Dis. 1984;130:282–288. doi: 10.1164/arrd.1984.130.2.282. [DOI] [PubMed] [Google Scholar]
- 134.Phibbs BP, Sundin RE, Mitchell RS. Silicosis in Wyoming bentonite workers. Am Rev Respir Dis. 1971;103:1–17. doi: 10.1164/arrd.1971.103.1.1. [DOI] [PubMed] [Google Scholar]
- 135.Sakula A. Pneumoconiosis due to Fuller's earth. Thorax. 1961;16:176–179. doi: 10.1136/thx.16.2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wells IP, Dubbins PA, Whimster WF. Pulmonary disease caused by the inhalation of cosmetic talcum powder. Br J Radiol. 1979;52:586–588. doi: 10.1259/0007-1285-52-619-586. [DOI] [PubMed] [Google Scholar]
- 137.Berner A, Gylseth B, Levy F. Talc dust pneumoconiosis. Acta Path Microbiol Scand. 1981;89:17–21. doi: 10.1111/j.1699-0463.1981.tb00181.x. [DOI] [PubMed] [Google Scholar]
- 138.Landas SK, Schwartz DA. Mica-associated pulmonary interstitial fibrosis. Am Rev Respir Dis. 1991;144:718–721. doi: 10.1164/ajrccm/144.3_Pt_1.718. [DOI] [PubMed] [Google Scholar]
Inert dusts
- 139.Doig AT. Baritosis: a benign pneumoconiosis. Thorax. 1976;31:30–39. doi: 10.1136/thx.31.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Seaton A, Ruckley VA, Addison J, et al. Silicosis in barium miners. Thorax. 1986;41:591–595. doi: 10.1136/thx.41.8.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Watson AJ, Black J, Doig AT, et al. Pneumoconiosis in carbon electrode makers. Br J Ind Med. 1959;16:274. doi: 10.1136/oem.16.4.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Miller AA, Ramsden F. Carbon pneumoconiosis. Br J Ind Med. 1961;18:103–113. doi: 10.1136/oem.18.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gaensler EA, Cadigan JB, Sasahara AA, et al. Graphite pneumoconiosis of electrotypers. Am J Med. 1966;41:864–882. doi: 10.1016/0002-9343(66)90046-5. [DOI] [PubMed] [Google Scholar]
- 144.Diaz JV, Koff J, Gotway MB, et al. Case report: a case of wood-smoke-related pulmonary disease. Environ Health Perspect. 2006;114:759–762. doi: 10.1289/ehp.8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gold JA, Jagirdar J, Hay JG, et al. Hut lung. A domestically acquired particulate lung disease. Medicine (Baltimore) 2000;79:310–317. doi: 10.1097/00005792-200009000-00004. [DOI] [PubMed] [Google Scholar]
- 146.Chung MP, Kim H, Rhee CH, et al. Bronchial stenosis due to anthracofibrosis. Chest. 1998;113:344–350. doi: 10.1378/chest.113.2.344. [DOI] [PubMed] [Google Scholar]
- 147.Naccache JM, Monnet I, Nunes H, et al. Anthracofibrosis attributed to mixed mineral dust exposure: report of three cases. Thorax. 2008;63:655–657. doi: 10.1136/thx.2006.070243. [DOI] [PubMed] [Google Scholar]
- 148.Wynn GJ, Turkington PM, O'Driscoll BR. Anthracofibrosis, bronchial stenosis with overlying anthracotic mucosa: possibly a new occupational lung disorder. Chest. 2008;134:1069–1073. doi: 10.1378/chest.08-0814. [DOI] [PubMed] [Google Scholar]
- 149.Naccache JM, Monnet I, Guillon F, et al. Occupational anthracofibrosis. Chest. 2009;135:1694. doi: 10.1378/chest.09-0291. [DOI] [PubMed] [Google Scholar]
- 150.Wynn GJ, Turkington PM, O'Driscoll BR. Response. Chest. 2009;135:1694–1695. doi: 10.1378/chest.09-0291. [DOI] [PubMed] [Google Scholar]
- 151.McLaughlin AIG, Harding HE. Pneumoconiosis and other causes of death in iron and steel foundry workers. Arch Ind Health. 1956;14:350–378. [PubMed] [Google Scholar]
- 152.Highman B. Histochemical study of certain iron ore dusts. Bull Internat Assoc Med museums. 1951;32:97–99. [Google Scholar]
Mixed-dust pneumoconiosis
- 153.Honma K, Abraham JL, Chiyotani K, et al. Proposed criteria for mixed-dust pneumoconiosis: definition, descriptions, and guidelines for pathologic diagnosis and clinical correlation. Hum Pathol. 2004;35:1515–1523. doi: 10.1016/j.humpath.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 154.Arakawa H, Johkoh T, Honma K, et al. Chronic interstitial pneumonia in silicosis and mix-dust pneumoconiosis: its prevalence and comparison of CT findings with idiopathic pulmonary fibrosis. Chest. 2007;131:1870–1876. doi: 10.1378/chest.06-2553. [DOI] [PubMed] [Google Scholar]
- 155.Honma K, Abraham JL, Chiyotani K, et al. Proposed criteria for mixed-dust pneumoconiosis: Definition, descriptions, and guidelines for pathologic diagnosis and clinical correlation. Human Pathology. 2004;35:1515–1523. doi: 10.1016/j.humpath.2004.09.008. [DOI] [PubMed] [Google Scholar]
Coal pneumoconiosis
- 156.Green FHY, Laqueur WA. Coal workers’ pneumoconiosis. Pathol Annu. 1980;15:333–410. [PubMed] [Google Scholar]
- 157.Stratton TML. Edin Med Surg J. 1838;49:490. [PMC free article] [PubMed] [Google Scholar]
- 158.Gough J. Pneumoconiosis in coal trimmers. J Pathol Bacteriol. 1940;51:277–285. [Google Scholar]
- 159.Douglas AN, Robertson A, Chapman JS, et al. Dust exposure, dust recovered from the lung, and associated pathology in a group of British coalminers. Br J Ind Med. 1986;43:795–801. doi: 10.1136/oem.43.12.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Davis JMG. In: Health Implication of New Energy Technologies. Rom WN, Archer VE, editors. Butterworth; Ann Arbor: 1980. The relationship between the mass and composition of coal mine dust and the development of pneumoconiosis; pp. 283–292. [Google Scholar]
- 161.McConnochie K, Green FHY, Vallyathan V, et al. Interstitial fibrosis in coal miners – experience in Wales and West Virginia. Ann Occup Hyg. 1988;32:553–560. [Google Scholar]
- 162.Remy-Jardin M, Degreef JM, Beuscart R, et al. Coal worker's pneumoconiosis: CT assessment in exposed workers and correlation with radiographic findings. Radiology. 1990;177:363–371. doi: 10.1148/radiology.177.2.2217770. [DOI] [PubMed] [Google Scholar]
- 163.Davis JMG, Chapman J, Collings P, et al. Variations in the histological patterns of the lesions of coal workers’ pneumoconiosis in Britain and their relationship to lung dust content. Am Rev Respir Dis. 1983;128:118–124. doi: 10.1164/arrd.1983.128.1.118. [DOI] [PubMed] [Google Scholar]
- 164.Ruckley VA, Fernie JM, Chapman JS, et al. Comparison of radiographic appearances with associated pathology and lung dust content in a group of coalworkers. Br J Ind Med. 1984;41:459–467. doi: 10.1136/oem.41.4.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Churg A, Wright JL, Wiggs B, et al. Small airways disease and mineral dust exposure. Prevalence, structure and function. Am Rev Respir Dis. 1985;131:139–143. doi: 10.1164/arrd.1985.131.1.139. [DOI] [PubMed] [Google Scholar]
- 166.Morgan WKC. Coal mining, emphysema, and compensation revisited. Br J Ind Med. 1993;50:1051–1052. doi: 10.1136/oem.50.11.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Seaton A. Coal mining, emphysema, and compensation revisited – reply. Br J Ind Med. 1993;50:1052–1053. doi: 10.1136/oem.50.11.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Coggon D, Taylor AN. Coal mining and chronic obstructive pulmonary disease: a review of the evidence. Thorax. 1998;53:398–407. doi: 10.1136/thx.53.5.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Heppleston AG. The pathogenesis of simple pneumokoniosis in coal workers. J Path Bact. 1954;67:51–63. doi: 10.1002/path.1700670106. [DOI] [PubMed] [Google Scholar]
- 170.Li K, Keeling B, Churg A. Mineral dusts cause elastin and collagen breakdown in the rat lung: a potential mechanism of dust-induced emphysema. Am J Respir Crit Care Med. 1996;153:644–649. doi: 10.1164/ajrccm.153.2.8564112. [DOI] [PubMed] [Google Scholar]
- 171.Ryder R, Lyons JP, Campbell H, et al. Emphysema in coal workers’ pneumoconiosis. BMJ. 1970;3:481–487. doi: 10.1136/bmj.3.5721.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lyons JP, Ryder R, Campbell H, et al. Pulmonary disability in coal workers’ pneumoconiosis. BMJ. 1972:713–716. doi: 10.1136/bmj.1.5802.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Leigh J, Outhred KG, McKenzie HI, et al. Quantified pathology of emphysema, pneumoconiosis, and chronic bronchitis in coal workers. Br J Ind Med. 1983;40:258–263. doi: 10.1136/oem.40.3.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ruckley VA, Gauld SJ, Chapman JS, et al. Emphysema and dust exposure in a group of coal workers. Am Rev Respir Dis. 1984;129:528–532. [PubMed] [Google Scholar]
- 175.Nemery B. Coal worker's lung: not only black, but also full of holes. Am J Respir Crit Care Med. 2009;180:199–200. doi: 10.1164/rccm.200905-0772ED. [DOI] [PubMed] [Google Scholar]
- 176.Kuempel ED, Wheeler MW, Smith RJ, et al. Contributions of dust exposure and cigarette smoking to emphysema severity in coal miners in the United States. Am J Respir Crit Care Med. 2009;180:257–264. doi: 10.1164/rccm.200806-840OC. [DOI] [PubMed] [Google Scholar]
- 177.Department of Social Security . HMSO; London: 1992. Chronic bronchitis and emphysema. Report by the Industrial Injuries Advisory Council in accordance with Section 171 of the Social Security Administration Act 1992 on the question whether bronchitis and emphysema in coal miners and metal production workers should be prescribed. p. 1. [Google Scholar]
- 178.Seaton A. The new prescription: industrial injuries benefits for smokers? Thorax. 1998;53:335–336. doi: 10.1136/thx.53.5.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hurley JF, Alexander WP, Hazledine DJ, et al. Exposure to respirable coalmine dust and incidence of progressive massive fibrosis. Br J Ind Med. 1987;44:661–672. doi: 10.1136/oem.44.10.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Cochrane AL. The attack rate of progressive massive fibrosis. Br J Ind Med. 1962;19:52–64. doi: 10.1136/oem.19.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Theodos PA, Cathcart RT, Fraimow W. Ischemic necrosis in anthracosilicosis. Arch Env Health. 1961;2:609–619. doi: 10.1080/00039896.1961.10662915. [DOI] [PubMed] [Google Scholar]
- 182.Wagner JC, Wusterman FS, Edwards JH, et al. The composition of massive lesions in coal miners. Thorax. 1975;30:382–388. doi: 10.1136/thx.30.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Vanhee D, Gosset P, Wallaert B, et al. Mechanisms of fibrosis in coal workers’ pneumoconiosis: increased production of platelet-derived growth factor, insulin-like growth factor type I, and transforming growth factor beta and relationship to disease severity. Am J Respir Crit Care Med. 1994;150:1049–1055. doi: 10.1164/ajrccm.150.4.7921435. [DOI] [PubMed] [Google Scholar]
- 184.Vanhee D, Gosset P, Marquette CH, et al. Secretion and mRNA expression of TNF alpha and IL-6 in the lungs of pneumoconiosis patients. Am J Respir Crit Care Med. 1995;152:298–306. doi: 10.1164/ajrccm.152.1.7599837. [DOI] [PubMed] [Google Scholar]
- 185.Caplan A, Payne RB, Withey JL. A broader concept of Caplan's syndrome related to rheumatoid factors. Thorax. 1962;17:205–212. doi: 10.1136/thx.17.3.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ball J. Differential agglutination test in rheumatoid arthritis complicated by pneumoconiosis. Ann Rheum Dis. 1955;14:159–161. doi: 10.1136/ard.14.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Soutar CA, Turner-Warwick M, Parkes WR. Circulating antinuclear antibody and rheumatoid factor in coal pneumoconiosis. BMJ. 1974;3:145–152. doi: 10.1136/bmj.3.5924.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Pearson DJ, Mentnech MS, Elliott JA, et al. Serologic changes in pneumoconiosis and progressive massive fibrosis of coal workers. Am Rev Respir Dis. 1981;124:696–699. doi: 10.1164/arrd.1981.124.6.696. [DOI] [PubMed] [Google Scholar]
- 189.Wagner JC, McCormick JN. Immunological investigations of coalworkers’ disease. J R Coll Physicians Lond. 1967;2:49–56. [PMC free article] [PubMed] [Google Scholar]
- 190.Caplan A. Certain unusual radiological appearances in the chest of coal-miners suffering from rheumatoid arthritis. Thorax. 1953;8:29–37. doi: 10.1136/thx.8.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Gough J, Rivers D, Seal RME. Pathological studies of modified pneumoconiosis in coal-miners with rheumatoid arthritis (Caplan's syndrome) Thorax. 1955;10:9–18. doi: 10.1136/thx.10.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Asbestosis
- 192.Asbestos, asbestosis, and cancer: the Helsinki criteria for diagnosis and attribution. Scand J Work Environ Health. 1997;23:311–316. [PubMed] [Google Scholar]
- 193.Roggli VL, Gibbs AR, Attanoos R, et al. Pathology of asbestosis: an update of the diagnostic criteria. Report of the Asbestosis Committee of the College of American Pathologists and Pulmonary Pathology Society. Arch Pathol Lab Med. 2010;134:462–480. doi: 10.5858/134.3.462. [DOI] [PubMed] [Google Scholar]
- 194.LaDou J. The asbestos cancer epidemic. Environ Health Perspect. 2004;112:285–290. doi: 10.1289/ehp.6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Churg A, Wright JL, Gilks B, et al. Rapid short-term clearance of chrysotile compared with amosite asbestos in the guinea pig. Am Rev Respir Dis. 1989;139:885–890. doi: 10.1164/ajrccm/139.4.885. [DOI] [PubMed] [Google Scholar]
- 196.Churg A, Wright JL. Persistence of natural mineral fibers in human lungs: an overview. Environ Health Perspect. 1994;102:229–233. doi: 10.1289/ehp.94102s5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wagner JC, Pooley FD, Berry G, et al. A pathological and mineralogical study of asbestos-related deaths in the United Kingdom in 1977. Ann Occup Hyg. 1982;26:423–431. [PubMed] [Google Scholar]
- 198.Wagner JC, Moncrieff CB, Coles R, et al. Correlation between fibre content of the lungs and disease in naval dockyard workers. Br J Ind Med. 1986;43:391–395. doi: 10.1136/oem.43.6.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wagner JC, Newhouse ML, Corrin B, et al. Correlation between fibre content of the lung and disease in east London asbestos factory workers. Br J Ind Med. 1988;45:305–308. doi: 10.1136/oem.45.5.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Wagner JC, Berry G, Skidmore JW, et al. The effects of the inhalation of asbestos in rats. Br J Cancer. 1974;29:252–269. doi: 10.1038/bjc.1974.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Davis JMG, Beckett ST, Bolton RE, et al. Mass and number of fibres in the pathogenesis of asbestos related lung disease in rats. Br J Cancer. 1978;37:673–688. doi: 10.1038/bjc.1978.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Churg A, Wright JL, Vedal S. Fiber burden and patterns of asbestos-related disease in chrysotile miners and millers. Am Rev Respir Dis. 1993;148:25–31. doi: 10.1164/ajrccm/148.1.25. [DOI] [PubMed] [Google Scholar]
- 203.Wagner JC, Berry G, Pooley FD. Carcinogenesis and mineral fibres. Br Med Bull. 1980;36:53–56. doi: 10.1093/oxfordjournals.bmb.a071614. [DOI] [PubMed] [Google Scholar]
- 204.Emri S, Demir A, Dogan M, et al. Lung diseases due to environmental exposures to erionite and asbestos in Turkey. Toxicol Lett. 2002;127:251–257. doi: 10.1016/s0378-4274(01)00507-0. [DOI] [PubMed] [Google Scholar]
- 205.Baris YI, Grandjean P. Prospective study of mesothelioma mortality in Turkish villages with exposure to fibrous zeolite. J Natl Cancer Inst. 2006;98:414–417. doi: 10.1093/jnci/djj106. [DOI] [PubMed] [Google Scholar]
- 206.Dodson RF, O'Sullivan M, Corn CJ, et al. Analysis of ferruginous bodies in bronchoalveolar lavage from foundry workers. Br J Ind Med. 1993;50:1032–1038. doi: 10.1136/oem.50.11.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Warnock ML. Analysis of the cores of ferruginous (asbestos) bodies from the general population. Lab Invest. 1979;40:622–626. [PubMed] [Google Scholar]
- 208.Churg A. Fiber counting and analysis in the diagnosis of asbestos-related disease. Hum Pathol. 1982;13:381–392. doi: 10.1016/s0046-8177(82)80227-x. [DOI] [PubMed] [Google Scholar]
- 209.Morgan A, Holmes A. Concentrations and dimensions of coated and uncoated asbestos fibres in the human lung. Br J Ind Med. 1980;37:25–32. doi: 10.1136/oem.37.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Pooley FD, Ranson DL. Comparison of the results of asbestos fibre dust counts in lung tissue obtained by analytical electron microscopy and light microscopy. J Clin Pathol. 1986;39:313–317. doi: 10.1136/jcp.39.3.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Holden J, Churg A. Asbestos bodies and the diagnosis of asbestosis in chrysotile workers. Environ Res. 1986;39:232–236. doi: 10.1016/s0013-9351(86)80024-x. [DOI] [PubMed] [Google Scholar]
- 212.Johansson LG, Albin MP, Jakobsson KM, et al. Ferruginous bodies and pulmonary fibrosis in dead low to moderately exposed asbestos cement workers: histological examination. Br J Ind Med. 1987;44:550–558. doi: 10.1136/oem.44.8.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider F, Sporn TA, Roggli VL. Asbestos fiber content of lungs with diffuse interstitial fibrosis: an analytical scanning electron microscopic analysis of 249 cases. Archives of Pathology & Laboratory Medicine. 2010;134:457–461. doi: 10.5858/134.3.457. [DOI] [PubMed] [Google Scholar]
- 213.McLemore TL, Roggli V, Marshall MV, et al. Comparison of phagocytosis of uncoated versus coated asbestos fibers by cultured human pulmonary alveolar macrophages. Chest. 1981;80:39S–42S. doi: 10.1378/chest.80.1_supplement.39s. [DOI] [PubMed] [Google Scholar]
- 214.Roggli VL, Pratt PC. Numbers of asbestos bodies on iron-stained tissue sections in relation to asbestos body counts in lung tissue digests. Hum Pathol. 1983;14:355–361. doi: 10.1016/s0046-8177(83)80122-1. [DOI] [PubMed] [Google Scholar]
- 215.Roggli VL, Pratt PC, Brody AR. Asbestos content of lung tissue in asbestos associated diseases: a study of 110 cases. Br J Ind Med. 1986;43:18–28. doi: 10.1136/oem.43.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Gibbs AR, Pooley FD. Analysis and interpretation of inorganic mineral particles in ‘lung’ tissues. Thorax. 1996;51:327–334. doi: 10.1136/thx.51.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Karjalainen A, Piipari R, Mantyla T, et al. Asbestos bodies in bronchoalveolar lavage in relation to asbestos bodies and asbestos fibres in lung parenchyma. Eur Respir J. 1996;9:1000–1005. doi: 10.1183/09031936.96.09051000. [DOI] [PubMed] [Google Scholar]
- 218.Paris C, GalateauSalle F, Creveuil C, et al. Asbestos bodies in the sputum of asbestos workers: correlation with occupational exposure. Eur Resp J. 2002;20:1167–1173. doi: 10.1183/09031936.02.00262102. [DOI] [PubMed] [Google Scholar]
- 219.Ashcroft T, Heppleston AG. The optical and electron microscopic determination of pulmonary asbestos fibre concentration and its relation to human pathology reaction. J Clin Pathol. 1973;26:224–234. doi: 10.1136/jcp.26.3.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Davis JMG, Glyseth B, Morgan A. Assessment of mineral fibres from human lung tissue. Thorax. 1986;41:167–175. doi: 10.1136/thx.41.3.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.James KR, Bull TB, Fox B. Detection of asbestos fibres by dark ground microscopy. J Clin Pathol. 1987;40:1259–1260. doi: 10.1136/jcp.40.10.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Churg A. Analysis of lung asbestos content. Br J Ind Med. 1991;48:649–652. doi: 10.1136/oem.48.10.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.De Vuyst P, Karjalainen A, Dumortier P, et al. Guidelines for mineral fibre analyses in biological samples: report of the ERS Working Group. Eur Resp J. 1998;11:1416–1426. doi: 10.1183/09031936.98.11061416. [DOI] [PubMed] [Google Scholar]
- 224.Whitwell F, Scott J, Grimshaw M. Relationship between occupations and asbestos-fibre content of the lungs in patients with pleural mesothelioma, lung cancer, and other diseases. Thorax. 1977;32:377–386. doi: 10.1136/thx.32.4.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Donaldson K, Brown GM, Brown DM, et al. Inflammation-generating potential of long and short-fibre amosite asbestos samples. Br J Ind Med. 1989;46:271–276. doi: 10.1136/oem.46.4.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Donaldson K, Li XY, Dogra S, et al. Asbestos-stimulated tumour necrosis factor release from alveolar macrophages depends on fibre length and opsonization. J Pathol. 1992;168:243–248. doi: 10.1002/path.1711680214. [DOI] [PubMed] [Google Scholar]
- 227.Davis JMG, Addison J, Bolton RE, et al. The pathogenicity of long versus short fibre amosite asbestos administered to rats by inhalation and intra-peritoneal injection. Br J Exp Pathol. 1986;67:415–430. [PMC free article] [PubMed] [Google Scholar]
- 228.Donaldson K, Golyasnya N. Cytogenetic and pathogenic effects of long and short amosite asbestos. J Pathol. 1995;177:303–307. doi: 10.1002/path.1711770313. [DOI] [PubMed] [Google Scholar]
- 229.Green FHY, Harley R, Vallyathan V, et al. Exposure and mineralogical correlates of pulmonary fibrosis in chrysotile asbestos workers. Occup Environ Medicine. 1997;54:549–559. doi: 10.1136/oem.54.8.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Pooley FD, Clark NJ. Quantitative assessment of inorganic fibrous particulates in dust samples with an analytical transmission electron microscope. Ann Occup Hyg. 1979;22:253–271. doi: 10.1093/annhyg/22.3.253. [DOI] [PubMed] [Google Scholar]
- 231.Gibbs AR, Stephens M, Griffiths DM, et al. Fibre distribution in the lungs and pleura of subjects with asbestos related diffuse pleural fibrosis. Br J Ind Med. 1991;48:762–770. doi: 10.1136/oem.48.11.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Dawson A, Gibbs AR, Pooley FD, et al. Malignant mesothelioma in women. Thorax. 1993;48:269–274. doi: 10.1136/thx.48.3.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Churg A, Wood P. Observations on the distribution of asbestos fibres in human lungs. Environ Res. 1983;31:374–380. doi: 10.1016/0013-9351(83)90015-4. [DOI] [PubMed] [Google Scholar]
- 234.Gylseth B, Churg A, Davis JMG, et al. Analysis of asbestos fibres and asbestos bodies in tissue samples from human lung: an international interlaboratory trial. Scand J Work Environ Health. 1985;11:107–110. doi: 10.5271/sjweh.2246. [DOI] [PubMed] [Google Scholar]
- 235.Morgan A, Holmes A. Distribution and characteristics of amphibole asbestos fibres in the left lung of an insulation worker measured with the light microscope. Br J Ind Med. 1983;40:45–50. doi: 10.1136/oem.40.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Pooley FD. The identification of asbestos dust with an electron microprobe analyser. Ann Occup Hyg. 1975;13:181–186. doi: 10.1093/annhyg/18.3.181. [DOI] [PubMed] [Google Scholar]
- 237.Doll R. Symposium on man-made mineral fibres, Copenhagen, October 1986: overview and conclusions. Ann Occup Hyg. 1987;31:805–820. doi: 10.1093/annhyg/31.4b.805. [DOI] [PubMed] [Google Scholar]
- 238.Coutts II, Gilson JC, Kerr IH, et al. Mortality in cases of asbestosis diagnosed by a pneumoconiosis medical panel. Thorax. 1987;42:111–116. doi: 10.1136/thx.42.2.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Churg A, Wright JL. Small-airway lesions in patients exposed to nonasbestos mineral dusts. Hum Pathol. 1983;14:688–693. doi: 10.1016/s0046-8177(83)80140-3. [DOI] [PubMed] [Google Scholar]
- 240.Churg A, Wright JL. Small airways disease and mineral dust exposure. Pathol Annu. 1983;18:233–251. [PubMed] [Google Scholar]
- 241.Auerbach O, Garfinkel L, Hammond EC. Relation of smoking and age to findings in lung parenchyma: a microscopic study. Chest. 1974;65:29–35. doi: 10.1378/chest.65.1.29. [DOI] [PubMed] [Google Scholar]
- 242.Corrin B, Price AB. Electron microscopic studies in desquamative interstitial pneumonia associated with asbestos. Thorax. 1972;27:324–331. doi: 10.1136/thx.27.3.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Freed JA, Miller A, Gordon RE, et al. Desquamative interstitial pneumonia associated with chrysotile asbestos fibres. Br J Ind Med. 1991;48:332–337. doi: 10.1136/oem.48.5.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Hammar SP, Hallman KO. Localized inflammatory pulmonary disease in subjects occupationally exposed to asbestos. Chest. 1993;103:1792–1799. doi: 10.1378/chest.103.6.1792. [DOI] [PubMed] [Google Scholar]
- 245.Hinson KFW, Otto H, Webster I, et al. In: Biological Effects of Asbestos IARC Lyon Scientific publications no8. Bogovski P, Gilson JC, Timbrell V, et al., editors. Pergamon; Oxford: 1973. Criteria for the diagnosis and grading of asbestosis; pp. 54–57. [Google Scholar]
- 246.Craighead JE, Abraham JL, Churg A, et al. The pathology of asbestos-associated diseases of the lung and pleural cavities: diagnostic criteria and proposed grading schema. Report of the Pneumoconiosis Committee of the College of American Pathologists and the National Institute for Occupational Safety and Health. Arch Pathol Lab Med. 1982;106:544–596. [PubMed] [Google Scholar]
- 247.Kuhn C, Kuo TT. Cytoplasmic hyalin in asbestosis. Arch Pathol. 1973;95:190–194. [PubMed] [Google Scholar]
- 248.Warnock ML, Press M, Churg A. Further observations on cytoplasmic hyaline in the lung. Hum Pathol. 1980;11:59–65. doi: 10.1016/s0046-8177(80)80106-7. [DOI] [PubMed] [Google Scholar]
- 249.Franks TJ, Chong PY, Chui P, et al. Lung pathology of severe acute respiratory syndrome (SARS): A study of 8 autopsy cases from Singapore. Hum Pathol. 2003;34:743–748. doi: 10.1016/S0046-8177(03)00367-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Nicholls JM, Poon LL, Lee KC, et al. Lung pathology of fatal severe acute respiratory syndrome. Lancet. 2003;361:1773–1778. doi: 10.1016/S0140-6736(03)13413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Yamada T, Uehara K, Kawanishi R, et al. Immunohistochemical detection of ubiquitin-positive intracytoplasmic eosinophilic inclusion bodies in diffuse alveolar damage. Histopathology. 2006;48:846–854. doi: 10.1111/j.1365-2559.2006.02445.x. [DOI] [PubMed] [Google Scholar]
- 252.Gaensler EA, Jederlinic PJ, Churg A. Idiopathic pulmonary fibrosis in asbestos-exposed workers. Am Rev Respir Dis. 1991;144:689–696. doi: 10.1164/ajrccm/144.3_Pt_1.689. [DOI] [PubMed] [Google Scholar]
- 253.Berry G. Mortality of workers certified by pneumoconiosis medical panels as having asbestosis. Br J Ind Med. 1981;38:130–137. doi: 10.1136/oem.38.2.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Jones RN, Diem JE, Ziskand MM, et al. Radiographic evidence of asbestos effects in American marine engineers. J Occup Med. 1984;26:281–284. [PubMed] [Google Scholar]
- 255.Thurlbeck WM. In: Chronic airflow obstruction in lung disease. Thurlbeck WM, editor. Saunders; Philadelphia: 1976. Morphology of emphysema and emphysema-like conditions; pp. 98–99. [Google Scholar]
- 256.Corrin B, Dewar A, Rodriguez-Roisin R, et al. Fine structural changes in cryptogenic fibrosing alveolitis and asbestosis. J Pathol. 1985;147:107–119. doi: 10.1002/path.1711470206. [DOI] [PubMed] [Google Scholar]
- 257.Gadek J, Hunninghake G, Schoenberger C, et al. Pulmonary asbestosis and idiopathic pulmonary fibrosis: pathogenetic parallels. Chest. 1981;80:63S–64S. doi: 10.1378/chest.80.1_supplement.63s. [DOI] [PubMed] [Google Scholar]
- 258.Adamson IYR, Bowden DH. Crocidolite-induced pulmonary fibrosis in mice. Cytokinetic and biochemical studies. Am J Pathol. 1986;122:261–267. [PMC free article] [PubMed] [Google Scholar]
- 259.Schapira RM, Ghio AJ, Effros RM, et al. Hydroxyl radicals are formed in the rat lung after asbestos instillation invivo. Am J Respir Cell Mol Biol. 1994;10:573–579. doi: 10.1165/ajrcmb.10.5.8179922. [DOI] [PubMed] [Google Scholar]
- 260.Thomas G, Ando T, Verma K, et al. Asbestos fibers and interferon-gamma up-regulate nitric oxide production in rat alveolar macrophages. Am J Respir Cell Mol Biol. 1994;11:707–715. doi: 10.1165/ajrcmb.11.6.7524571. [DOI] [PubMed] [Google Scholar]
- 261.Kinnula VL. Oxidant sand antioxidant mechanisms of lung disease caused by asbestos fibres. Eur Resp J. 1999;14:706–716. doi: 10.1034/j.1399-3003.1999.14c35.x. [DOI] [PubMed] [Google Scholar]
- 262.Turner-Warwick M, Haslam P. Antibodies in some chronic fibrosing lung diseases. Clin Allergy. 1971;1:83–95. doi: 10.1111/j.1365-2222.1971.tb02450.x. [DOI] [PubMed] [Google Scholar]
- 263.Janssen YMW, Driscoll KE, Howard B, et al. Asbestos causes translocation of p65 protein and increases NF- kappa B DNA binding activity in rat lung epithelial and pleural mesothelial cells. Am J Pathol. 1997;151:389–401. [PMC free article] [PubMed] [Google Scholar]
- 264.Liu JY, Morris GF, Lei WH, et al. Rapid activation of PDGF-A and -B expression at sites of lung injury in asbestos-exposed rats. Am J Respir Cell Molec Biol. 1997;17:129–140. doi: 10.1165/ajrcmb.17.2.2956. [DOI] [PubMed] [Google Scholar]
- 265.Lasky JA, Tonthat BH, Liu JY, et al. Upregulation of the PDGF-alpha receptor precedes asbestos-induced lung fibrosis in rats. Amer J Respir Crit Care Med. 1998;157:1652–1657. doi: 10.1164/ajrccm.157.5.9704116. [DOI] [PubMed] [Google Scholar]
- 266.Liu JY, Brass DM, Hoyle GW, et al. TNF-alpha receptor knockout mice are protected from the fibroproliferative effects of inhaled asbestos fibers. Am J Pathol. 1998;153:1839–1847. doi: 10.1016/s0002-9440(10)65698-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Tsuda A, Stringer BK, Miljailovich SM, et al. Alveolar cell stretching in the presence of fibrous particles induces interleukin-8 responses. Am J Respir Cell Molec Biol. 1999;21:455–462. doi: 10.1165/ajrcmb.21.4.3351. [DOI] [PubMed] [Google Scholar]
- 268.Kamp DW, Weitzman SA. The molecular basis of asbestos induced lung injury. Thorax. 1999;54:638–652. doi: 10.1136/thx.54.7.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Warheit DB, George G, Hill LH, et al. Inhaled asbestos activates a complement-dependent chemoattractant for macrophages. Lab Invest. 1985;52:505–514. [PubMed] [Google Scholar]
- 270.Lemaire I, Beaudoin H, Masse S, et al. Alveolar macrophage stimulation of lung fibroblast growth in asbestos-induced pulmonary fibrosis. Am J Pathol. 1986;122:205–211. [PMC free article] [PubMed] [Google Scholar]
- 271.Rom WN, Travis WD, Brody AR. Cellular and molecular basis of the asbestos-related diseases. Am Rev Respir Dis. 1991;143:408–422. doi: 10.1164/ajrccm/143.2.408. [DOI] [PubMed] [Google Scholar]
- 272.Liu JY, Morris GF, Lei WH, et al. Up-regulated expression of transforming growth factor-α in the bronchiolar-alveolar duct regions of asbestos-exposed rats. Am J Pathol. 1996;149:205–217. [PMC free article] [PubMed] [Google Scholar]
- 273.Bateman ED, Emerson RJ, Cole PJ. A study of macrophage-mediated initiation of fibrosis by asbestos and silica using a diffusion chamber technique. Br J Exp Pathol. 1982;63:414–425. [PMC free article] [PubMed] [Google Scholar]
- 274.Vallyathan V, Mega JF, Shi XL, et al. Enhanced generation of free radicals from phagocytes induced by mineral dusts. Am J Respir Cell Mol Biol. 1992;6:404–413. doi: 10.1165/ajrcmb/6.4.404. [DOI] [PubMed] [Google Scholar]
- 275.Doll R. Mortality from lung cancer in asbestos workers. Br J Ind Med. 1955;12:81–86. doi: 10.1136/oem.12.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Wagner JC, Sleggs CA, Marchand P. Diffuse pleural mesotheliomas and asbestos exposure in Northwestern Cape Province. Br J Ind Med. 1960;17:260–271. doi: 10.1136/oem.17.4.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Selikoff IJ, Hammond EC, Churg J. Carcinogenicity of amosite asbestos. Arch Env Health. 1972;25:183–186. doi: 10.1080/00039896.1972.10666158. [DOI] [PubMed] [Google Scholar]
- 278.Hammond EC, Selikoff IJ, Seidman H. Asbestos exposure, cigarette smoking and death rates. Ann N Y Acad Sci. 1979;330:473–490. doi: 10.1111/j.1749-6632.1979.tb18749.x. [DOI] [PubMed] [Google Scholar]
- 279.de Klerk NH, Musk AW, Armstrong BK, et al. Smoking, exposure to crocidolite, and the incidence of lung cancer and asbestosis. Br J Ind Med. 1991;48:412–417. doi: 10.1136/oem.48.6.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Kannerstein M, Churg J. Pathology of carcinoma of the lung associated with asbestos exposure. Cancer. 1972;30:14–21. doi: 10.1002/1097-0142(197207)30:1<14::aid-cncr2820300104>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 281.Whitwell F, Newhouse ML, Bennett DR. A study of the histological cell types of lung cancer in workers suffering from asbestosis in the United Kingdom. Br J Ind Med. 1974;31:298–303. doi: 10.1136/oem.31.4.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Churg A. Lung cancer cell type and asbestos exposure. JAMA. 1985;253:2984–2985. [PubMed] [Google Scholar]
- 283.Johansson L, Albin M, Jakobsson K, et al. Histological type of lung carcinoma in asbestos cement workers and matched controls. Br J Ind Med. 1992;49:626. doi: 10.1136/oem.49.9.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Raffn E, Lynge E, Korsgaard B. Incidence of lung cancer by histological type among asbestos cement workers in Denmark. Br J Ind Med. 1993;50:85–89. doi: 10.1136/oem.50.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Raffn E, Villadsen E, Engholm G, et al. Lung cancer in asbestos cement workers in Denmark. Occup Environ Med. 1996;53:399–402. doi: 10.1136/oem.53.6.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.De Vos Irvine H, Lamont DW, Hole DJ, et al. Asbestos and lung cancer in Glasgow and the west of Scotland. BMJ. 1993;306:1503–1506. doi: 10.1136/bmj.306.6891.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.de Klerk NH, Musk AW, Eccles JL, et al. Exposure to crocidolite and the incidence of different histological types of lung cancer. Occup Environ Med. 1996;53:157–159. doi: 10.1136/oem.53.3.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Rudd R. Asbestos and lung cancer. Thorax. 1997;52:306. [PubMed] [Google Scholar]
- 289.Henderson DW, de Klerk NH, Hammar SP, et al. In: Pathology of Lung Tumours. Corrin B, editor. Churchill Livingstone; London: 1997. Asbestos and lung cancer: is it attributable to asbestosis or to asbestos fibre burden? pp. 83–118. [Google Scholar]
- 290.Pohlabeln H, Wild P, Schill W, et al. Asbestos fibreyears and lung cancer: a two phase case-control study with expert exposure assessment. Occup Environ Med. 2002;59:410–414. doi: 10.1136/oem.59.6.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Hodgson JT, Darnton A. The quantitative risks of mesothelioma and lung cancer in relation to asbestos exposure. Ann Occup Hyg. 2000;44:565–601. [PubMed] [Google Scholar]
- 292.Browne K. Is asbestos or asbestosis the cause of the increased risk of lung cancer in asbestos workers? Br J Ind Med. 1986;43:145–149. doi: 10.1136/oem.43.3.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Sluis-Cremer GK, Bezuidenhout BN. Relation between asbestosis and bronchial cancer in amphibole asbestos miners. Br J Ind Med. 1989;46:537–540. doi: 10.1136/oem.46.8.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Hughes JM, Weill H. Asbestosis as a precursor of asbestos related lung cancer – results of a prospective mortality study. Br J Ind Med. 1991;48:229–233. doi: 10.1136/oem.48.4.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Jones RN, Hughes JM, Weill H. Asbestos exposure, asbestosis, and asbestos-attributable lung cancer. Thorax. 1996;51:S9–S15. doi: 10.1136/thx.51.suppl_2.s9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Jones RN. Asbestos and lung cancer – Reply. Thorax. 1997;52:306. [PubMed] [Google Scholar]
- 297.Cagle PT. Criteria for attributing lung cancer to asbestos exposure. Am J Clin Pathol. 2002;117:9–15. doi: 10.1309/KGRX-TC9T-PPHM-1J46. [DOI] [PubMed] [Google Scholar]
- 298.Weiss W. Asbestosis: A marker for the increased risk of lung cancer among workers exposed to asbestos. Chest. 1999;115:536–549. doi: 10.1378/chest.115.2.536. [DOI] [PubMed] [Google Scholar]
- 299.Hessel PA, Gamble JF, McDonald JC. Asbestos, asbestosis, and lung cancer: a critical assessment of the epidemiological evidence. Thorax. 2005;60:433–436. doi: 10.1136/thx.2004.037267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Begin R, Cantin A, Berthiaume Y, et al. Airway function in lifetime-nonsmoking older asbestos workers. Am J Med. 1983;75:631–638. doi: 10.1016/0002-9343(83)90449-7. [DOI] [PubMed] [Google Scholar]
- 301.Wright JL, Churg A. Severe diffuse small airways abnormalities in long term chrysotile asbestos miners. Br J Ind Med. 1985;42:556. doi: 10.1136/oem.42.8.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Wright JL, Churg A. Morphology of small-airway lesions in patients with asbestos exposure. Hum Pathol. 1984;15:68–74. doi: 10.1016/s0046-8177(84)80332-9. [DOI] [PubMed] [Google Scholar]
Aluminium
- 303.Abramson MJ, Wlodarczyk JH, Saunders NA, et al. Does aluminum smelting cause lung disease? Am Rev Respir Dis. 1989;139:1042–1057. doi: 10.1164/ajrccm/139.4.1042. [DOI] [PubMed] [Google Scholar]
- 304.Crombie DW, Blaischell JL, MacPherson G. The treatment of silicosis by aluminum powder. Can Med Assoc J. 1944;50:318–328. [PMC free article] [PubMed] [Google Scholar]
- 305.Duchange L, Brichet A, Lamblin C, et al. Silicose aigue. Caractéristiques cliniques, radiologiques, fonctionnelles et cytologiques du liquide broncho-alveolaire. A propos de 6 observations. Rev Mal Resp. 1998;15:527–534. [PubMed] [Google Scholar]
- 306.Goralewski G, Jaeger R. Zur Klinik, Pathologie and Pathogenesis der Aluminiumlunge. Arch Gewerbepath Hyg. 1941;11:102–105. [Google Scholar]
- 307.Goralewski G. Die Aluminiumlunge – eine neue Gewerbeerkrankung. Z f d ges Inn Med. 1947;2:665–673. [Google Scholar]
- 308.Mitchell J, Manning GB, Molyneux M, et al. Pulmonary fibrosis in workers exposed to finely powdered aluminium. Br J Ind Med. 1961;18:10–20. doi: 10.1136/oem.18.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Corrin B. Aluminium pneumoconiosis I. in vitro comparison of stamped aluminium powders containing different lubricating agents and a granular aluminium powder. Br J Ind Med. 1963;20:264–267. doi: 10.1136/oem.20.4.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Corrin B. Aluminium pneumoconiosis II. Effect on the rat lung of intratracheal injections of stamped aluminium powders containing different lubricating agents and of a granular aluminium powder. Br J Ind Med. 1963;20:268–276. doi: 10.1136/oem.20.4.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Herbert A, Sterling G, Abraham J, et al. Desquamative interstitial pneumonia in an aluminum welder. Hum Pathol. 1982;13:694–699. doi: 10.1016/s0046-8177(82)80291-8. [DOI] [PubMed] [Google Scholar]
- 312.Vuyst P, Dumortier P, Schandene L, et al. Sarcoidlike lung granulomatosis induced by aluminum dusts. Am Rev Respir Dis. 1987;135:493–497. doi: 10.1164/arrd.1987.135.2.493. [DOI] [PubMed] [Google Scholar]
- 313.Chen W-J, Monnat RJ, Chen M, et al. Aluminum induced pulmonary granulomatosis. Hum Pathol. 1978;9:705–711. doi: 10.1016/s0046-8177(78)80053-7. [DOI] [PubMed] [Google Scholar]
- 314.Hull MJ, Abraham JL. Aluminum welding fume-induced pneumoconiosis. Hum Pathol. 2002;33:819–825. doi: 10.1053/hupa.2002.125382. [DOI] [PubMed] [Google Scholar]
Rare earth (cerium) pneumoconiosis
- 315.McDonald JW, Ghio AJ, Sheehan CE, et al. Rare earth (cerium oxide) pneumoconiosis: analytical scanning electron microscopy and literature review. Mod Pathol. 1995;8:859–865. [PubMed] [Google Scholar]
Hard metal disease (cobalt lung)
- 316.Davison AG, Haslam PL, Corrin B, et al. Interstitial lung disease and asthma in hard-metal workers: bronchoalveolar lavage, ultrastructural, and analytical findings and results of bronchial provocation tests. Thorax. 1983;38:119–128. doi: 10.1136/thx.38.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Schepers GWH. The biological action of tungsten carbide and cobalt: studies on experimental pulmonary histopathology. Arch Ind Health. 1955;12:140–146. [PubMed] [Google Scholar]
- 318.Demedts M, Gheysens B, Nagels J, et al. Cobalt lung in diamond polishers. Am Rev Respir Dis. 1984;130:130–135. doi: 10.1164/arrd.1984.130.1.130. [DOI] [PubMed] [Google Scholar]
- 319.Nemery B, Nagels J, Verbeken E, et al. Rapidly fatal progression of cobalt lung in a diamond polisher. Am Rev Respir Dis. 1990;141:1373–1378. doi: 10.1164/ajrccm/141.5_Pt_1.1373. [DOI] [PubMed] [Google Scholar]
- 320.Rolfe MW, Paine R, Davenport RB, et al. Hard metal pneumoconiosis and the association of tumor necrosis factor-alpha. Am Rev Respir Dis. 1992;146:1600–1602. doi: 10.1164/ajrccm/146.6.1600. [DOI] [PubMed] [Google Scholar]
Berylliosis
- 321.Kriebel D, Brain JD, Sprince NL, et al. The pulmonary toxicity of beryllium. Am Rev Respir Dis. 1988;137:464–473. doi: 10.1164/ajrccm/137.2.464. [DOI] [PubMed] [Google Scholar]
- 322.Meyer KC. Beryllium and lung disease. Chest. 1994;106:942–946. doi: 10.1378/chest.106.3.942. [DOI] [PubMed] [Google Scholar]
- 323.Maier LA, Martyny JW, Liang J, et al. Recent chronic beryllium disease in residents surrounding a beryllium facility. Am J Respir Crit Care Med. 2008;177:1012–1017. doi: 10.1164/rccm.200607-1042OC. [DOI] [PubMed] [Google Scholar]
- 324.Newman LS, Kreiss K. Nonoccupational beryllium disease masquerading as sarcoidosis – identification by blood lymphocyte proliferative response to beryllium. Am Rev Respir Dis. 1992;145:1212–1214. doi: 10.1164/ajrccm/145.5.1212. [DOI] [PubMed] [Google Scholar]
- 325.Williams WJ. Beryllium disease. Postgrad Med J. 1988;64:511–516. doi: 10.1136/pgmj.64.753.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.International Agency for Research on Cancer . IARC; Lyons: 1987. An evaluation of carcinogenic risk to humans. Overall evaluation of carcinogenicity: an updating of IARC monographs vols 1–42. Supplement 7. [PubMed] [Google Scholar]
- 327.Weber HH, Englehardt WE. Uber eine Apparatur zur Erzeugung niedriger Staubkonzentrationen von grosser Konstanz und eine Methode zur mikrogravinctrischen Staubbestemmung. Anwendung bei der Untersuchung von Stauben aus der Berylliumsgewinnung. Zentbl GewHyg Unfallerhut. 1933;10:41–47. [Google Scholar]
- 328.Hardy HL, Tabershaw IR. Delayed chenical pneumonitis occurring in workers exposed to beryllium compounds. J Ind Hyg Toxicol. 1946;28:197–211. [PubMed] [Google Scholar]
- 329.Kotloff RM, Richman PS, Greenacre JK, et al. Chronic beryllium disease in a dental laboratory technician. Am Rev Respir Dis. 1993;147:205–207. doi: 10.1164/ajrccm/147.1.205. [DOI] [PubMed] [Google Scholar]
- 330.Saltini C, Winestock K, Kirby M, et al. Maintenance of alveolitis in patients with chronic beryllium disease by beryllium-specific helper T cells. N Engl J Med. 1989;320:1103–1109. doi: 10.1056/NEJM198904273201702. [DOI] [PubMed] [Google Scholar]
- 331.Richeldi L, Sorrentino R, Saltini C. HLA-DPB1 glutamate 69: a genetic marker of beryllium disease. Science. 1993;262:242–248. doi: 10.1126/science.8105536. [DOI] [PubMed] [Google Scholar]
- 332.Saltini C, Amicosante M, Franchi A, et al. Immunogenetic basis of environmental lung disease: lessons from the berylliosis model. Eur Resp J. 1998;12:1463–1475. doi: 10.1183/09031936.98.12061463. [DOI] [PubMed] [Google Scholar]
- 333.McConnochie K, Williams WR, Kilpatrick GS, et al. Beryllium disease in identical twins. Br J Dis Chest. 1988;82:431–435. doi: 10.1016/0007-0971(88)90101-5. [DOI] [PubMed] [Google Scholar]
- 334.Butnor KJ, Sporn TA, Ingram P, et al. Beryllium detection in human lung tissue using electron probe X-ray microanalysis. Mod Pathol. 2003;16:1171–1177. doi: 10.1097/01.MP.0000094090.90571.ED. [DOI] [PubMed] [Google Scholar]
- 335.Jones Williams W. Diagnostic criteria for chronic beryllium disease (CBD) based on the UK Registry 1945–1991. Sarcoidosis. 1993;10:41–43. [PubMed] [Google Scholar]
- 336.Jones Williams W. United Kingdom beryllium registry: mortality and autopsy study. Environ Health Perspect. 1996;104:949–951. doi: 10.1289/ehp.96104s5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Alberts WM. Lung disease and the lightest of metals. Chest. 2004;126:1730–1732. doi: 10.1378/chest.126.6.1730. [DOI] [PubMed] [Google Scholar]
- 338.Fireman E, Haimsky E, Noiderfer M, et al. Misdiagnosis of sarcoidosis in patients with chronic beryllium disease. Sarcoidosis Vasc Diffuse Lung Dis. 2003;20:144–148. [PubMed] [Google Scholar]
- 339.Muller-Quernheim J, Gaede KI, Fireman E, et al. Diagnoses of chronic beryllium disease within cohorts of sarcoidosis patients. Eur Respir J. 2006;27:1190–1195. doi: 10.1183/09031936.06.00112205. [DOI] [PubMed] [Google Scholar]
Polyvinyl chloride pneumoconiosis
- 340.Arnaud A, Pommier de Santi P, Garbe L, et al. Polyvinyl chloride pneumoconiosis. Thorax. 1978;33:19–25. doi: 10.1136/thx.33.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.White NW, Ehrlich RI. Regression of polyvinylchloride polymer pneumoconiosis. Thorax. 1997;52:748–749. doi: 10.1136/thx.52.8.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Studnicka MJ, Menzinger G, Drlicek M, et al. Pneumoconiosis and systemic sclerosis following 10 years of exposure to polyvinyl chloride dust. Thorax. 1995;50:583–585. doi: 10.1136/thx.50.5.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Flock workers’ lung
- 343.Eschenbacher WL, Kreiss K, Lougheed MD, et al. Nylon flock-associated interstitial lung disease. Am J Respir Crit Care Med. 1999;159:2003–2008. doi: 10.1164/ajrccm.159.6.9808002. [DOI] [PubMed] [Google Scholar]
- 344.Boag AH, Colby TV, Fraire AE, et al. The pathology of interstitial lung disease in nylon flock workers. Am J Surg Pathol. 1999;23:1539–1545. doi: 10.1097/00000478-199912000-00012. [DOI] [PubMed] [Google Scholar]
- 345.Kern DG, Kuhn C, Ely EW, et al. Flock worker's lung: broadening the spectrum of clinicopathology, narrowing the spectrum of suspected etiologies [see comments] Chest. 2000;117:251–259. doi: 10.1378/chest.117.1.251. [DOI] [PubMed] [Google Scholar]
- 346.Barroso E, Ibanez MD, Aranda FI, et al. Polyethylene flock-associated interstitial lung disease in a Spanish female. Eur Resp J. 2002;20:1610–1612. doi: 10.1183/09031936.02.00030102. [DOI] [PubMed] [Google Scholar]
Popcorn workers’ lung
- 347.Kreiss K, Gomaa A, Kullman G, et al. Clinical bronchiolitis obliterans in workers at a microwave-popcorn plant. N Engl J Med. 2002;347:330–338. doi: 10.1056/NEJMoa020300. [DOI] [PubMed] [Google Scholar]
- 348.Kreiss K, Gomaa A, Kullman G, et al. Clinical bronchiolitis obliterans in workers at a microwave-popcorn plant. N Engl J Med. 2002;347:330–338. doi: 10.1056/NEJMoa020300. [DOI] [PubMed] [Google Scholar]
- 349.Akpinar-Elci M, Travis WD, Lynch DA, et al. Bronchiolitis obliterans syndrome in popcorn production plant workers. Eur Respir J. 2004;24:298–302. doi: 10.1183/09031936.04.00013903. [DOI] [PubMed] [Google Scholar]
- 350.Hendrick DJ. ‘Popcorn worker's lung’ in Britain in a man making potato crisp flavouring. Thorax. 2008;63:267–268. doi: 10.1136/thx.2007.089607. [DOI] [PubMed] [Google Scholar]
Paint spraying
- 351.Sanz P, Prat A. Toxicity in textile air-brushing in Spain. Lancet. 1993;342:240. doi: 10.1016/0140-6736(93)92330-v. [DOI] [PubMed] [Google Scholar]
- 352.Moya C, Anto JM, Taylor AJN, et al. Outbreak of organising pneumonia in textile printing sprayers. Lancet. 1994;344:498–502. doi: 10.1016/s0140-6736(94)91896-1. [DOI] [PubMed] [Google Scholar]
- 353.Sole A, Cordero PJ, Morales P, et al. Epidemic outbreak of interstitial lung disease in aerographics textile workers – the ‘Ardystil syndrome’: a first year follow up. Thorax. 1996;51:94–95. doi: 10.1136/thx.51.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Romero S, Hernandez L, Gil J, et al. Organizing pneumonia in textile printing workers: a clinical description. Eur Resp J. 1998;11:265–271. doi: 10.1183/09031936.98.11020265. [DOI] [PubMed] [Google Scholar]
- 355.Kadi OF, Mohammed-Brahim B, Fyad A, et al. Outbreak of pulmonary disease in textile dye sprayers in Algeria. Lancet. 1994;344:962–963. doi: 10.1016/s0140-6736(94)92323-x. [DOI] [PubMed] [Google Scholar]
- 356.Kadi FO, Abdesslam T, Nemery B. Five-year follow-up of Algerian victims of the ‘‘Ardystil syndrome’’. Eur Resp J. 1999;13:940–941. doi: 10.1034/j.1399-3003.1999.13d41.x. [DOI] [PubMed] [Google Scholar]
Mineral oils and petroleum
- 357.Skorodin MS, Chandrasekhar AJ. An occupational cause of exogenous lipoid pneumonia. Arch Pathol Lab Med. 1983;107:610–611. [PubMed] [Google Scholar]
- 358.Bernstein DI, Lummus ZL, Santilli G, et al. Machine operator's lung. A hypersensitivity pneumonitis disorder associated with exposure to metalworking fluid aerosols. Chest. 1995;108:636–641. doi: 10.1378/chest.108.3.636. [DOI] [PubMed] [Google Scholar]
- 359.Kreiss K, Cox-Ganser J. Metalworking fluid-associated hypersensitivity pneumonitis: a workshop summary. Am J Ind Med. 1997;32:423–432. doi: 10.1002/(sici)1097-0274(199710)32:4<423::aid-ajim16>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 360.Bracker A, Storey E, Yang C, et al. An outbreak of hypersensitivity pneumonitis at a metalworking plant: a longitudinal assessment of intervention effectiveness. Appl Occup Environ Hyg. 2003;18:96–108. doi: 10.1080/10473220301436. [DOI] [PubMed] [Google Scholar]
- 361.Brander PE, Taskinen E, Stenius-Aarniala B. Fire-eater's lung. Eur Respir J. 1992;5:112–114. [PubMed] [Google Scholar]
- 362.Gentina T, Tillie-Leblond I, Birolleau S, et al. Fire-eater's lung: seventeen cases and a review of the literature. Medicine (Baltimore) 2001;80:291–297. doi: 10.1097/00005792-200109000-00002. [DOI] [PubMed] [Google Scholar]
- 363.Scharf SM, Prinsloo I. Pulmonary mechanics in dogs given different doses of kerosene intratracheally. Am Rev Resp Dis. 1982;126:695–700. doi: 10.1164/arrd.1982.126.4.695. [DOI] [PubMed] [Google Scholar]
Welding
- 364.Doig MB, McLaughlin ALG. X-ray appearance of the lungs of electric arc welders. Lancet. 1936;i:771–775. [Google Scholar]
- 365.McMillan GH. The health of welders in naval dockyards: the risk of asbestos-related diseases occurring in welders. J Soc Occup Med. 1983;25:727–730. doi: 10.1097/00043764-198310000-00011. [DOI] [PubMed] [Google Scholar]
- 366.Funahashi A, Schlueter DP, Pintar K, et al. Welders’ pneumoconiosis: tissue elemental microanalysis by energy dispersive X ray analysis. Br J Ind Med. 1988;45:14–18. doi: 10.1136/oem.45.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Sferlazza SJ, Beckett WS. The respiratory health of welders. Am Rev Respir Dis. 1991;143:1134–1148. doi: 10.1164/ajrccm/143.5_Pt_1.1134. [DOI] [PubMed] [Google Scholar]
Toxic fumes and gases
- 368.Nemery B. Metal toxicity and the respiratory tract. Eur Respir J. 1990;3:202–219. [PubMed] [Google Scholar]
- 369.Shaver CG, Riddell AR. Lung changes associated with the manufacture of alumina abrasives. Am J Med Sci. 1947;29:145–157. [PubMed] [Google Scholar]
- 370.Asano S, Eto K, Kurisaki E, et al. Acute inorganic mercury vapor inhalation poisoning. Pathol Int. 2000;50:169–174. doi: 10.1046/j.1440-1827.2000.01032.x. [DOI] [PubMed] [Google Scholar]
- 371.Sunderman FW, Kincaid JF. Nickel poisoning II. Studies on patients suffering from acute exposure to vapors of nickel carbonyl. JAMA. 1954;155:889–894. doi: 10.1001/jama.1954.03690280013003. [DOI] [PubMed] [Google Scholar]
- 372.Lowry T, Schuman LM. Silo-filler's disease – a syndrome caused by nitrogen dioxide. JAMA. 1956;162:153. doi: 10.1001/jama.1956.02970200001001. [DOI] [PubMed] [Google Scholar]
- 373.Ramirez RJ, Dowell AR. Silo-filler's disease: nitrogen dioxide-induced lung injury. Ann Intern Med. 1971;74:569–576. doi: 10.7326/0003-4819-74-4-569. [DOI] [PubMed] [Google Scholar]
- 374.Douglas WW, Hepper NGG, Colby TV. Silo filler's disease. Mayo Clin Proc. 1989;64:291–304. doi: 10.1016/s0025-6196(12)65249-5. [DOI] [PubMed] [Google Scholar]
- 375.Zwemer FL, Pratt DS, May JJ. Silo-filler's disease in New York state. Am Rev Respir Dis. 1992;146:650–653. doi: 10.1164/ajrccm/146.3.650. [DOI] [PubMed] [Google Scholar]
- 376.Osbern L, Crapo R. Dung lung: a report of toxic exposure to liquid manure. Ann Intern Med. 1981;95:312–314. doi: 10.7326/0003-4819-95-3-312. [DOI] [PubMed] [Google Scholar]
- 377.Morse DL, Woodbury MA, Rentmeester K, et al. Death caused by fermenting manure. JAMA. 1981;245:63–64. [PubMed] [Google Scholar]
- 378.Donham KJ, Knapp LW, Monson R, et al. Acute toxic exposure to gases from liquid manure. J Occup Med. 1982;24:142–145. [PubMed] [Google Scholar]
- 379.Fahy JV, Walley T, Gibney RTN, et al. ‘Slurry lung’: a report of three cases. Thorax. 1991;46:394–395. doi: 10.1136/thx.46.5.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Plopper CG, Dungworth DL, Tyler WS. Pulmonary lesions in rats exposed to ozone. Am J Pathol. 1973;71:375–394. [PMC free article] [PubMed] [Google Scholar]
- 381.Pratt PC. In: The Lung. Thurlbeck WM, Abell MR, editors. Williams and Wilkins; Baltimore: 1978. Pathology of adult respiratory distress syndrome; pp. 43–57. [Google Scholar]
- 382.Harkema JR, Plopper CG, Hyde DM, et al. Response of macaque bronchiolar epithelium to ambient concentrations of ozone. Am J Pathol. 1993;143:857–866. [PMC free article] [PubMed] [Google Scholar]
- 383.Bils RF. Ultrastructural alterations of alveolar tissue of mice. Arch Env Health. 1970;20:468–480. doi: 10.1080/00039896.1970.10665624. [DOI] [PubMed] [Google Scholar]
- 384.Pinkerton KE, Dodge DE, Cederdahl-Demmler J, et al. Differentiated bronchiolar epithelium in alveolar ducts of rats exposed to ozone for 20 months. Am J Pathol. 1993;142:947–956. [PMC free article] [PubMed] [Google Scholar]
- 385.Hyde DM, Hubbard WC, Wong V, et al. Ozone-induced acute tracheobronchial epithelial injury – relationship to granulocyte emigration in the lung. Am J Respir Cell Mol Biol. 1992;6:481–497. doi: 10.1165/ajrcmb/6.5.481. [DOI] [PubMed] [Google Scholar]
- 386.Aris RM, Christian D, Hearne PQ, et al. Ozone-induced airway inflammation in human subjects as determined by airway lavage and biopsy. Am Rev Respir Dis. 1993;148:1363–1372. doi: 10.1164/ajrccm/148.5.1363. [DOI] [PubMed] [Google Scholar]
- 387.Gardner MJ, Pannett B, Winter PD, et al. A cohort study of workers exposed to formaldehyde in the British chemical industry – an update. Br J Ind Med. 1993;50:827–834. doi: 10.1136/oem.50.9.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Snyder RW, Mishel HS, Christensen GC. Pulmonary toxicity following exposure to methylene chloride and its combustion product, phosgene. Chest. 1992;102:1921. doi: 10.1378/chest.102.6.1921-b. [DOI] [PubMed] [Google Scholar]
- 389.Emad A, Rezaian GR. The diversity of the effects of sulfur mustard gas inhalation on respiratory system 10 years after a single, heavy exposure: analysis of 197 cases. Chest. 1997;112:734–738. doi: 10.1378/chest.112.3.734. [DOI] [PubMed] [Google Scholar]
- 390.Ghanei M, Moqadam FA, Mohammad MM, et al. Tracheobronchomalacia and air trapping after mustard gas exposure. Am J Respir Crit Care Med. 2006;173:304–309. doi: 10.1164/rccm.200502-247OC. [DOI] [PubMed] [Google Scholar]
- 391.Ghanei M, Tazelaar HD, Chilosi M, et al. An International collaborative pathologic study of surgical lung biopsies from mustard gas-exposed patients. Respiratory Medicine. 2008;102:825–830. doi: 10.1016/j.rmed.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 392.Konichezky S, Schattner A, Ezri T, et al. Thionyl-chloride-induced lung injury and bronchiolitis obliterans. Chest. 1993;104:971–973. doi: 10.1378/chest.104.3.971. [DOI] [PubMed] [Google Scholar]
- Burnett WW, King EG, Grace M, Hall WF. Hydrogen sulfide poisoning: review of 5 years’ experience. Can Med Assoc J. 1977;117:1277–1280. [PMC free article] [PubMed] [Google Scholar]
Anoxic asphyxia
- 393.James PB, Calder IM. Anoxic asphyxia – a cause of industrial fatalities: a review. J R Soc Med. 1991;84:493. doi: 10.1177/014107689108400815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Occupational asthma
- 394.Ross DJ, Sallie BA, McDonald JC. SWORD ‘94: surveillance of work-related and occupational respiratory disease in the UK. Occup Med. 1995;45:175–178. doi: 10.1093/occmed/45.4.175. [DOI] [PubMed] [Google Scholar]
- 395.Hnizdo E, Esterhuizen TM, Rees D, et al. Occupational asthma as identified by the Surveillance of Work-related and Occupational Respiratory Diseases programme in South Africa. Clin Exp Allergy. 2001;31:32–39. doi: 10.1046/j.1365-2222.2001.00981.x. [DOI] [PubMed] [Google Scholar]
- 396.Karjalainen A, Kurppa K, Virtanen S, et al. Incidence of occupational asthma by occupation and industry in Finland. Am J Ind Med. 2000;37:451–458. doi: 10.1002/(sici)1097-0274(200005)37:5<451::aid-ajim1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 397.Sallie BA, Ross DJ, Meredith SK, et al. SWORD ‘93: surveillance of work-related and occupational respiratory disease in the UK. Occup Med. 1994;44:177–182. doi: 10.1093/occmed/44.4.177. [DOI] [PubMed] [Google Scholar]
- 398.Balmes J, Becklake M, Blanc P, et al. American Thoracic Society Statement: Occupational contribution to the burden of airway disease. Am J Respir Crit Care Med. 2003;167:787–797. doi: 10.1164/rccm.167.5.787. [DOI] [PubMed] [Google Scholar]
- 399.Chanyeung M, Malo JL. Aetiological agents in occupational asthma. Eur Respir J. 1994;7:346–371. doi: 10.1183/09031936.94.07020346. [DOI] [PubMed] [Google Scholar]
- 400.McDonald JC, Keynes HL, Meredith SK. Reported incidence of occupational asthma in the United Kingdom, 1989–97. Occup Environ Med. 2000;57:823–829. doi: 10.1136/oem.57.12.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Burge PS, Finnegan M, Horsefield N, et al. Occupational asthma in a factory with a contaminated humidifier. Thorax. 1985;40:248–254. doi: 10.1136/thx.40.4.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Rooke GB. The pathology of byssinosis. Chest. 1981;79:67S–71S. doi: 10.1378/chest.79.4_supplement.67s. [DOI] [PubMed] [Google Scholar]
- 403.Niven RM, Pickering CAC. Byssinosis: a review. Thorax. 1996;51:632–637. doi: 10.1136/thx.51.6.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Mundie TG, Boackle RJ, Ainsworth SK. In vitro alternative and classical activation of complement by extracts of cotton mill dust: a possible mechanism in the pathogenesis of byssinosis. Environ Res. 1983;32:47–56. doi: 10.1016/0013-9351(83)90190-1. [DOI] [PubMed] [Google Scholar]
- 405.Kutz SA, Olenchok SA, Elliot JA, et al. Antibody independent complement activation by card-room cotton dust. Environ Res. 1979;19:405–414. doi: 10.1016/0013-9351(79)90065-3. [DOI] [PubMed] [Google Scholar]
- 406.Edwards C, Macartney J, Rooke G, et al. The pathology of the lung in byssinotics. Thorax. 1975;30:612–623. doi: 10.1136/thx.30.6.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Occupational fevers
- 407.Raskandersen A, Pratt DS. Inhalation fever – a proposed unifying term for febrile reactions to inhalation of noxious substances. Br J Ind Med. 1992;49:40. doi: 10.1136/oem.49.1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.MRC Symposium Humidifier fever. Thorax. 1977;32:653–663. [Google Scholar]
- 409.Emanuel DA, Wenzel FJ, Lawton BR. Pulmonary mycotoxicosis. Chest. 1975;67:293–297. doi: 10.1378/chest.67.3.293. [DOI] [PubMed] [Google Scholar]
- 410.May JJ, Stallones L, Darrow D, et al. Organic dust toxicity (pulmonary mycotoxicosis) associated with silo unloading. Thorax. 1986;41:919–923. doi: 10.1136/thx.41.12.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Lougheed MD, Roos JO, Waddell WR, et al. Desquamative interstitial pneumonitis and diffuse alveolar damage in textile workers: potential role of mycotoxins. Chest. 1995;108:1196–1200. doi: 10.1378/chest.108.5.1196. [DOI] [PubMed] [Google Scholar]
- 412.Perry LP, Iwata M, Tazelaar HD, et al. Pulmonary mycotoxicosis: A clinicopathologic study of three cases. Modern Pathol. 1998;11:432–436. [PubMed] [Google Scholar]
- 413.Vogelmeier C, Konig G, Bencze K, et al. Pulmonary involvement in zinc fume fever. Chest. 1987;92:946–948. doi: 10.1378/chest.92.5.946. [DOI] [PubMed] [Google Scholar]
- 414.Blanc PD, Boushey HA, Wong H, et al. Cytokines in metal fume fever. Am Rev Respir Dis. 1993;147:134–138. doi: 10.1164/ajrccm/147.1.134. [DOI] [PubMed] [Google Scholar]
7.2. Environmental lung disease
Chapter Contents
Environmental irradiation
Environmental irradiation chiefly affects the skin but in some parts of the world rocks near the surface release significant amounts of radon gas. This carcinogen is liable to accumulate in buildings and be inhaled, so subjecting the occupants to an increased risk of lung cancer. The installation of underfloor ventilation is therefore advocated in such areas. This subject is explored more fully on page 533.
Atmospheric pressure changes
The body is vulnerable to both increases and decreases in pressure and it is the lungs that often bear the brunt of the damage. Increased pressure may result in blast injury or crushing of the chest while decreased pressure may result in the lungs literally bursting or dissolved gases being released within the blood (caisson disease), or the vascular alterations that underlie mountain sickness developing. Some of these pressure changes entail a risk of pneumothorax and it is essential that this is properly investigated postmortem by the chest being opened under a water seal. Loud music has been incriminated as a specific form of air pressure change causing pneumothorax and metereologists have shown that ‘spontaneous’ pneumothoraces tend to occur in clusters associated with natural drops in atmospheric pressure.1, 2
Blast injury
Explosions may cause injury by the body being violently thrown against a less moveable object, by objects being thrown against the body or by the blast wave hitting the body. These mechanisms often act together but sometimes there is only blast injury, to which the lungs are particularly vulnerable. For a time it was considered that the damage was direct, the blast wave travelling down the airways to injure the lungs. However, at the start of the Second World War, experiments conducted in the UK showed that the lungs were injured indirectly, the blast wave being transmitted to them through the chest wall: pulmonary blast injury is worst on the side of the body towards the explosion, and can be reduced by protective clothing.3 Underwater explosions are particularly dangerous because water is incompressible. There may be severe internal injury but no external evidence of damage other than a trickle of blood from the mouth or nose. This is because the injury is rate-dependent. Quite small thoracic deformation may produce severe pulmonary damage if peak compression is attained very quickly, typically in less than 5 ms. Conversely, severe chest wall distortion may produce only minor pulmonary contusion if this time is extended beyond 20 ms.4
At necropsy, the lungs are contused, with blood evident in the airways and parenchyma. Depending on the force of the blast, the haemorrhage may be pinpoint, patchy or confluent. It tends to follow the lines of the ribs and may be accompanied by pleuropulmonary lacerations having the same distribution. In this case there will also be haemothorax, pneumothorax and possibly air embolism. Patchy pulmonary haemorrhages cuff the blood vessels.5, 6 In patients who survive for a few days, the lungs resemble the liver macroscopically and histologically show chronic interstitial inflammation and fibrosis as well as haemorrhage.7 Other injuries are often present and fat embolism, aspiration pneumonia, fluid overload and infection may all be added to the effects of the blast wave.
Chest squeeze
‘Chest squeeze’ is another form of barotrauma caused by high pressure but here the body is compressed rather than subject to a sudden wave of pressure as in blast injury. It is experienced by divers who descend very deeply, thereby subjecting their bodies to such high pressure that their chest walls are literally crushed, so that their ribs break and their lungs are severely compressed. More common mishaps experienced by divers include drowning and decompression sickness, both of which are dealt with below, and neurological syndromes such as nitrogen narcosis, which will not be considered further.8
Burst lung
‘Burst lung’ is the most acute form of decompression sickness.9 It is experienced by divers and submariners making rapid ascents from depth and by aviators who ascend too rapidly in unpressurised aeroplanes, experience failure of a plane's pressure system or have to eject at high altitudes. Injury to the lung is caused by trapped alveolar gas expanding so rapidly that it exceeds total lung capacity before it can escape through the trachea. The lungs literally burst: the alveolar walls rupture and blood mixes directly with alveolar air. The victim experiences chest pain and there may be blood-stained froth at the mouth or frank haemoptysis. Air may enter the alveolar walls to cause interstitial emphysema or air embolism. Asthmatics may be at particular risk because of regional air-trapping.10. Diving mammals such as porpoises and whales are protected from such dangers of peripheral air-trapping by cartilage extending far out into the finest conductive airways so that these passages never close, even at the end of full expiration (Fig. 7.2.1 ).11, 12
Figure 7.2.1.
Sea lion lung. The smallest airways of deep-diving mammals such as the sea lion are buttressed by cartilage to ensure that all air is evacuated from the lungs when the animal is at depth to prevent the lungs bursting on rapid ascent. In humans there are several generations of airways that lack cartilage (the bronchioles) and consequently close before exhalation is complete. The trapped gas (the residual volume) is normally of no consequence but it poses a danger that the lungs may burst or of decompression sickness if ascent is rapid, whether in the sea or by air.
(Courtesy of Professor D Denison, Brompton, UK.)
Patients requiring positive-pressure artificial respiration are also at risk of burst lung, but the complications of the resultant interstitial emphysema differ from those experienced by divers. In divers, the chest wall is buttressed by the surrounding water and air in the interstitium is liable to track towards the hilum of the lungs and enter pulmonary veins, with resultant cerebral and coronary air embolism, either of which may prove fatal.13 Iatrogenic burst lung, on the other hand, takes place in patients whose chest wall is not so buttressed, and then outward rupture of the interstitial air is more likely, resulting in pneumothorax. Extension of the interstitial emphysema to the mediastinum, neck and chest wall is also more likely in such patients, resulting in surgical emphysema at these sites. However, there are exceptional cases marked by both cerebral embolism and extensive air tracking.14
Decompression sickness (caisson disease)
The same circumstances that lead to burst lung may also cause decompression sickness, which is also known as caisson disease.9 In this condition there is a sudden release of nitrogen gas that has gone into solution in the lipids of adipose tissue and of myelinated nervous tissue at the higher pressure: the released nitrogen gains access to the blood stream in which it forms bubbles.15 Doppler ultrasound techniques show that this is quite customary when divers ascend from depth,16 but the lungs generally provide an effective filter so that there are no untoward systemic effects, although there may be sudden chest pain on deep inspiration ('the chokes’). Gradual decompression permits the nitrogen to diffuse across the alveolar membranes and be exhaled. If, however, substantial amounts of nitrogen are released from solution, sufficient pulmonary arteries may be blocked to cause pulmonary hypertension, with resultant opening of arteriovenous communications or a patent foraman ovale, so permitting the gas to enter the systemic circulation. This is often followed by limb pains ('the bends’) and perhaps cerebral symptoms ('the staggers’). Fatal cases are characterised by gas bubbles within blood vessels throughout the body and froth in the heart chambers. Delayed effects include ischaemic necrosis of bones and other tissues.17
Deep-diving mammals are protected by the same mechanism that prevents them suffering from burst lung. They exhale before diving and during the dive the chest is compressed to the extent that virtually all the gas in the lungs passes into the cartilage-buttressed non-respiratory airways (see Fig. 7.2.1), resulting in very little to be absorbed by the blood. The pulmonary collapse also serves to reduce buoyancy. The distribution of the little gas that is absorbed is minimised by bradycardia.18 Many viscera experience anaerobic respiration but hypoxia is minimised in the heart and musculature by high levels of haemoglobin and myoglobin. The brain is further protected by the supplying arteries drawing on oxygen stored in an unusual sponge-like cervical organ known as the rete mirabilis.
Mountain sickness
Mountain sickness is due to reduced atmospheric pressure brought about more slowly than that responsible for decompression sickness.19, 20 It may be acute or chronic.
Acute mountain sickness
Acute mountain sickness is likely to be experienced by anyone who ascends above 3000–4000 m without a period of acclimatisation at intermediate levels. Symptoms are as liable to occur in people born at high altitude who return after a few weeks spent at sea level as in those who go to the mountains for the first time: acclimatisation is obviously short-lived and is therefore necessary whenever an ascent is to be made. The ill-effects are commonly precipitated by exercise. In the susceptible, acute mountain sickness commonly appears within 3 days of ascent.
The basis of acute mountain sickness is tissue hypoxia. It results in deteriorating intellectual and psychological function, headache, nausea, vomiting, and more rarely pulmonary and cerebral oedema. High-altitude pulmonary oedema is characterised by increasing dyspnoea, cyanosis and a dry cough, and later the production of copious, frothy sputum, which sometimes becomes blood-stained.21 The pulmonary artery pressure is markedly raised but wedge pressures are normal, indicating that the left side of the heart is unaffected and that pulmonary venous constriction is unlikely to be an important contributory factor.
The pulmonary oedema fluid has a high protein content22 and the condition has been characterised as a non-cardiogenic high-permeability oedema associated with excessive pulmonary hypertension.23, 24 Hypoxia is a well-known cause of pulmonary arteriolar constriction but in acute mountain sickness the vascular response appears to be exaggerated for the pulmonary artery pressure is considerably higher than is usual for the altitude. An association with certain HLA complexes (HLA-DR6 and HLA-DQ4) suggests that this has a genetic basis.25 Although arteriolar constriction only tends to protect the pulmonary capillaries, it could explain the oedema if the process was patchy – as is the resultant oedema – for patchy arteriolar constriction would subject the rest of the lung to abnormally high pressures and lead to capillary stress failure in these areas (see pp. 402 and 448).26, 27 Measurements of capillary pressure suggest that this is indeed the case.28 Furthermore, vasodilators such as calcium channel blocking agents and inhaled nitric oxide gas23, 29, 30 have been used with success to counter acute mountain sickness, supporting the idea that hypoxic vasoconstriction plays a central role.
Autopsy shows the lungs to be heavy and firm. The cut surface weeps oedema fluid, which is often blood-stained, but a striking feature is the patchy distribution of the changes. Areas of haemorrhagic oedema alternate with others that contain clear oedema fluid and others that are normal apart from overinflation. Pulmonary arterial thrombi are commonly found. Microscopy confirms the presence of haemorrhagic oedema and may show neutrophils and hyaline membranes in the alveoli. The alveolar capillaries are congested and may contain thrombi. There may also be an increase in mast cells and rarely pulmonary infarction. The right ventricle is commonly dilated whereas the left ventricle is normal. Highlanders generally show right ventricular hypertrophy and increased muscle in their pulmonary arteriesm, changes that are not apparent in lowlanders.31, 32
Chronic mountain sickness
Prolonged residence at high altitude leads to hypoxic pulmonary hypertension (see p. 424), an increase in red cell mass and cor pulmonale. Livestock taken from lowland plains to high-altitude pastures suffer similarly but the natural stock of the Himalayas and Ethiopian highlands are apparently immune. So too are other species long established at high altitude such as the llama and yak. These species are said to have adapted to their climate, that is, the forces of natural selection have bred out the pulmonary vasoconstrictive response to hypoxia. Cattle of European origin and humans acclimatise to high altitude by processes such as increasing their red cell mass but generally they are not adapted like native species and suffer hypoxic pulmonary hypertension at altitudes in excess of 3000 m. Certain Himalayan highlanders may be an exception to this in that their small pulmonary arteries are reported not to show the muscularisation that characterises hypoxic pulmonary hypertension.33
In cattle of European origin, the dependent oedema of right-sided cardiac failure caused by hypoxic pulmonary hypertension affects the breast (brisket) particularly and in the Rocky Mountains of North America such cattle are said to have ‘brisket disease’.34 A human counterpart of this has been described in children of Chinese ancestry who have been taken to reside in Tibet and who have developed a fatal form of subacute infantile mountain sickness.35
A small minority of permanent residents in the Andes develop the changes of chronic mountain sickness to a marked degree and are said to suffer from Monge's disease.36 The basis of this is alveolar hypoventilation, which leads to a progressive fall in systemic arterial oxygen saturation and elevation of haemoglobin concentration to an unusually severe degree. The latter averages about 25 g/dl, which exceeds even the 20 g/dl found in healthy high-altitude residents. Patients with Monge's disease are so deeply cyanosed that their lips are virtually black. Their pulmonary artery resistance is also markedly raised. The cause of the alveolar hypoventilation is uncertain but the only cases of Monge's disease that have come to necropsy had conditions such as kyphoscoliosis that predispose to alveolar hypoxia.
Drowning
Drowning is defined as suffocation by submersion, and usually occurs in water. It is the commonest cause of accidental death among divers but 96% of drowning accidents do not involve deep descents. Falling into quite shallow water is a particularly common cause of drowning in young children. In adults, men outnumber women by 4 to 1. More die in fresh water than the sea, not because it is more hazardous to the lungs than sea water, but because unguarded inland waters and swimming pools are visited more frequently. Alcohol consumption contributes to many deaths by drowning.
Drowning is not simply a matter of being unable to keep one's head above water. This may be merely a secondary event. For example, the entry dive may result in underwater head injury, or the exertion of swimming may precipitate a heart attack. Furthermore, the struggling swimmer going down for the third time ('drowning not waving’) is the exception: most drowning is characterised by the swimmer failing to surface or quietly dropping beneath the surface without anyone noticing.
Swimming underwater can be extremely hazardous if it is preceded by hyperventilation, a danger that needs to be more widely appreciated. Hyperventilation results in undue loss of carbon dioxide so that instead of hypercapnia forcing the swimmer to surface to breathe, progress under water may be continued until hypoxia causes sudden loss of consciousness.
Panic contributes to many swimming accidents and is often precipitated by the inadvertent aspiration of just a little water. Most people are naturally buoyant, but only slightly so. With the lungs fully expanded the average adult has a positive buoyancy of about 2.5 kg, which is sufficient to keep the head out of the water if the rest of the body is submerged. If an arm (weight about 3 kg) is raised to wave for help, the head will go down. If the swimmer shouts, exhalation reduces buoyancy to neutral at normal end-expiration and to negative at residual volume. Buoyancy cannot be regained when the head is submerged and unless able to swim to the surface, the person will continue to sink.
Autopsy generally shows that the lungs are full of water, but some victims die of ‘dry drowning’ due to laryngospasm. Events may also be modified by the temperature of the water. Sudden immersion in cold water may result in tachycardia, hypertension and hyperventilation, making it difficult for the victim to keep the airways free of water. It may also result in sudden death due to ventricular fibrillation. Even a good swimmer loses consciousness within an hour of immersion in very cold water. Drowning is then inevitable unless a correctly fitted life jacket is worn, in which case there is a danger of death from hypothermia. However, as in open heart surgery, cold prolongs the interval before there is irreversible brain damage.
If the person is rescued, water in the lungs is quickly absorbed, even if it is saline, and therefore hyperosmolar:aspirated sea water is quickly equilibrated by pure water joining it from the blood but the alveolar epithelial barrier remains impermeable to protein and once osmotic equilibrium is reached, all is quickly reabsorbed.37, 38, 39 Fresh water is absorbed even more quickly. It is unnecessary to tip the patient to hasten this process. Any water recovered in this way comes from the stomach and time that should be devoted to mouth-to-mouth breathing and cardiac massage is lost. These resuscitative efforts may need to be prolonged as fresh water in particular inactivates alveolar surfactant, leading to alveolar collapse which persists until the surfactant is replenished. Very few victims who are resuscitated on site fail to survive, and very few who cannot be resuscitated on site recover later.
Interchange of fluid between the blood and air spaces may cause major fluctuations in plasma volume with consequent changes in ionic concentrations and haemolysis. Hypervolaemia may cause circulatory problems but hyperkalaemia consequent upon the haemolysis is not thought to be as important as was formerly believed: ventricular fibrillation following submersion is more likely to be a complication of hypothermia than of electrolyte imbalance.
Circulatory collapse may ensue shortly after rescue. This is due to loss of the circulatory support provided by the pressure the water exerts on the body, which results in a considerable increase in cardiac output while the body is immersed. On leaving the water the loss of this support results in a tendency to venous pooling. Although this is countered by baroreceptor responses, these are reduced by prolonged immersion in cold water. Circulatory collapse is believed to be the cause of death in many persons who perish within minutes of rescue. To counter this effect, patients should be lifted out of the water in the prone position.
It can be seen that, in fatal cases, the pathologist is faced with several possibilities. Thus, death may have been due to:
-
•
natural causes before the body entered the water
-
•
unnatural causes before entry, the body merely being disposed of in the water
-
•
natural causes in the water
-
•
injuries received in the water from impact with rocks, a boat or a ship's propeller, or in tropical waters from predators such as a crocodile or a shark (any of which may also be incurred after death, as may disfigurement by fish and rats)
-
•
'dry drowning’
-
•
true drowning
-
•
hypothermia
-
•
circulatory failure after rescue.
True drowning is indicated by froth in the airways and heavy water-filled lungs. Both fresh and salt water contain numerous microscopic algae known as diatoms and those representative of the water in which the drowning occurred are found in the lungs. Unless death occurred before submersion, diatoms are also found in other viscera because these tiny life forms easily enter the circulation. Thus, the presence of diatoms in digests of organs such as the kidneys, liver, brain and bone marrow suggests that death was due to drowning. Because they have a siliceous capsule, diatoms are resistant to putrefaction as well as digestion and can be identified in the body long after death. However, a positive test is not always accepted as proof of drowning and a negative test does not exclude drowning.
Inhaled toxic agents
The various physical forms in which respirable environmental agents may be encountered are defined in Table 7.2.1 . Some effects of inhalant lung injury are recognised as distinct disease entities and are dealt with elsewhere: for example, the pneumoconioses on page 327, extrinsic allergic alveolitis on page 279, chronic bronchitis on page 98 and lung cancer on page 532. Other respirable agents, such as lead fume and carbon monoxide gas, exert their harmful effects elsewhere in the body and will not be considered further. This section is concerned with toxic substances that may be inhaled by the general public. Those that are more likely to be encountered in the workplace or in war zones are considered on page 355.
The lungs have a rather stereotyped pattern of response to inhaled toxins, displaying degenerative changes and inflammation of varying degree, the former sometimes amounting to necrosis. In general, the site of maximal absorption or injury is related to solubility (for gases and vapours) and particle size (for aerosols such as dusts, fog, fumes, mists, smog and smoke): the less water-soluble and the smaller the particle size, the further down the respiratory tract the agent will penetrate (Fig. 7.2.2 and Table 7.2.2 ).40, 41, 42 Thus, ammonia produces intense congestion of the upper respiratory passages and laryngeal oedema whereas phosgene has little effect on these sites but causes pulmonary oedema.40
Air pollution43, 44, 45, 46, 47
The toxic (as opposed to allergenic) air pollutants thought to pose the greatest threat to the lungs comprise smoke particles, sulphur dioxide, oxides of nitrogen, various aldehydes and ozone. Smoke and sulphur dioxide derive particularly from the combustion of fossil fuels in domestic fires and power stations, nitrogen dioxide is an important car exhaust and domestic gas appliance pollutant and ozone is the principal photochemical product of smog. Aldehydes such as formaldehyde and acrylic aldehyde (acrolein) also contribute to general air pollution because they are released in the combustion of diesel oil and petrol. Collectively, these pollutants have been incriminated in the exacerbation (rather than causation) of asthma. They also predispose to respiratory infection and result in airway inflammation and hypersecretion.48, 49 Their effect on children is of particular concern because development of the lungs is known to continue well into childhood and damage to the lungs before their growth is complete is likely to be irreparable. At the other extreme of life episodes of severe air pollution are known to hasten the deaths of many patients with chronic airway disease. Particularly high concentrations of the agents responsible for air pollution may be encountered in industry and their effects are therefore also considered in Chapter 7.1, on occupational diseases of the lung. Many of the polycyclic hydrocarbons found in polluted air are carcinogenic (see p. 532) and it is therefore not surprising that urban air pollution has been found to be associated with excess mortality from lung cancer.50
Domestic air pollution is rife in many of the poorer parts of the world due to the burning of biomass (wood, dried cow dung, bagasse, straw) in unventilated living rooms for heating and cooking. The women are particularly at risk of developing chronic bronchitis while their children have an increased incidence of acute respiratory infections.51, 52, 52a
Volcanic ash (tephra) irritates the eyes, skin and respiratory tract and in some eruptions may contain much free silica (e.g. Montserrat in 1995 and Mount St Helens, Washington state, USA in 1980) or be associated with the release of radon gas (e.g. the Azores in 1957).53 The destruction of the World Trade Center in 2001 caused massive air pollution of New York city that had lasting respiratory effects on survivors, rescue workers and local residents.54, 55, 56 At the time of the disaster there was much smoke from combustion of aeroplane fuel and flammable materials in the building while the collapse of the twin towers released dust from cement and dry-wall partitions that was highly alkaline.57, 58, 59 This caused considerable irritation of the eyes and the conductive airways. A year later many victims were still suffering from bronchial hyperreactivity and poor ventilatory function, in a so-called reactive airways dysfunction syndrome54, 55 and there was continuing spirometric decline 5 years later.60 The respirable portion of the dust formed only a small fraction of the whole but given the level of exposure its future effects cannot be discounted, particularly as it contained substances such as asbestos. Unusual effects attributed to the disaster include acute eosinophilic pneumonia and granulomatous pneumonitis.61, 62
Allergenic air pollutants are dealt with in detail in the sections on asthma (see p. 109) and extrinsic allergic alveolitis (see p. 279). Allergenic air pollution is generally occupational or domestic but periodic widespread air pollution was responsible for the epidemics of asthma seen in Barcelona in the 1980s, which were eventually traced to ships discharging cargoes of soya flour (see p. 114).
Tobacco smoke
Smoking-related diseases figure large throughout this book and in this section they are merely summarised collectively. Of the greatest importance, both in the number of patients they affect and in their clinical effects on the individual, are the various forms of chronic obstructive lung disease and lung cancer, but there are many other respiratory diseases associated with smoking, and a few that are less common in smokers (Box 7.2.1 ).63 Not surprisingly, these diseases are often encountered in combination and sometimes one may obscure another. For example, a cigarette smoker may have emphysema in the upper lobes and idiopathic pulmonary fibrosis in the lower lobes.64, 65 Alternatively, Langerhans cell histiocytosis and desquamative interstitial pneumonia may affect the same parts of the lungs, in which case the focal lesions of the former may be masked by the latter condition.63
Box 7.2.1. Diseases related to smoking.
Respiratory diseases caused by smoking
Carcinoma
- Chronic obstructive lung disease
- Chronic bronchitis
- ‘Small-airways disease’
- Emphysema
Respiratory bronchiolitis
Desquamative interstitial pneumonia
Respiratory diseases that are commoner or worse in smokers
Adult respiratory distress syndrome
- Respiratory infections
- Common cold
- Influenza
- Varicella pneumonia
- Bacterial pneumonia
- Tuberculosis
Pneumothorax
Cryptogenic fibrosing alveolitis
Langerhans cell histiocytosis
Asbestosis
Goodpasture's disease
Respiratory diseases that are less common or less severe in smokers
Extrinsic allergic alveolitis
Sarcoidosis
The term ‘smoking-related interstitial lung disease’ has been introduced to cover a spectrum of interstitial diseases related to smoking63, 66, 67 as well as being used in a more restricted sense to describe a combination of air space enlargement and interstitial fibrosis predominantly affecting the lower lobes.68, 69, 69a
Quite advanced interstitial fibrosis has been reported in smokers with no clinical evidence of interstitial lung disease.69b Early changes detectable in smokers include chronic bronchiolitis, fibrosis of the bronchiolar wall and mild peribronchiolar interstitial fibrosis.70, 71 Even earlier changes are detectable at the molecular level: as many as 152 smoking-responsive genes that are significantly up-regulated or down-regulated have been identified in normal cigarette smokers.72 There is marked individual variation, which may explain why many lifelong heavy smokers experience no respiratory problems.
Histological evidence that a patient smokes is provided by an increase in the number of alveolar macrophages and a characteristic brown discoloration of cytoplasm due to the phagocytosis of tar and other particulate matter derived from tobacco smoke (Fig. 7.2.3 ).
Figure 7.2.3.
Macrophages of a heavy cigarette smoker. (A) Numerous brown macrophages fill the alveoli. Electron microscopy shows that the lysosomal dense bodies are increased in number and contain lipidic ‘tar bodies’ (B) or the needle-shaped crystals of kaolinite (C).
(Transmission electron micrographs, courtesy of Miss A Dewar, Brompton, UK.)
Cigarette smokers are at greater risk of lung disease than cigar and pipe smokers, probably because they inhale more deeply. They do this because cigarette smoke is more acid than cigar and pipe smoke and its nicotine content is therefore absorbed more easily through the lungs than the buccal mucosa. Smokers obviously put their own health at greatest risk but the lesser hazards of passive smoking are now well recognized (see p. 532). Passive smoking involves both the smoke exhaled by others and that coming from smouldering tobacco between puffs, the latter being known as sidestream smoke. The harmful effects of maternal smoking on the unborn child also come in this category. They include increased airway responsiveness and reduced lung function during the neonatal period and an increased risk of sudden infant death syndrome. Reduced numbers of alveolar attachments to the bronchioles have been demonstrated in such infants.73 Smoking is also associated with disease of other organs (e.g. carcinoma of the oesophagus and bladder) but these are outwith the remit of this text.
Tobacco smoking by waterpipe (shisha, hubble-bubble) is enjoying a rise in popularity, both in its heartland, the Middle East, and western countries, and wherever it is practised it is widely perceived as being less dangerous than smoking cigarettes.74 This is probably a misconception. What evidence there is suggests that waterpipe tobacco smoking is just as harmful as cigarette smoking, if not more so.75
Burns and smoke inhalation
The lungs may be injured in burned patients in many ways (Box 7.2.2 )76, 77 but an important consideration when a body is recovered from a fire is whether death was due to the fire or took place beforehand, the latter raising the possibility of foul play. A vital reaction to the skin burns and the presence of soot in the lower airways provide evidence that death occurred in the fire but an absence of soot from the airways may be due to death occurring rapidly, from asphyxia or poisoning by gases released in the conflagration. Soot is cleared rapidly and if the patient survives a few days an absence of soot from the airways is to be expected.76
Box 7.2.2. Possible pulmonary insults in burned patients, arranged in approximate sequential order.
Blast injury
Asphyxia
Poisoning by combustion products (e.g. carbon monoxide, cyanide)
Direct thermal injury (largely limited to the trachea)
Irritant smoke, fume and gas (e.g. oxides of nitrogen, ammonia, acrolein, sulphur dioxide)
Hypovolaemic shock secondary to skin loss
- Septicaemic shock from:
- Infected skin burns
- Infected central lines
Secondary viral and bacterial pneumonia
Fluid overload
Tracheostomy complications, including tracheobronchitis, pneumonia and barotrauma
Oxygen toxicity
Absorption of toxic topical disinfectants
Thromboembolism
Uraemia
Lung injury may result directly from heat and smoke inhalation or indirectly from the release of mediators associated with blast injury or shock. Although air temperature in a fire may reach very high temperatures thermal injury seldom extends beyond the carina but more extensive injury from heat alone was seen in men exposed to steam escaping from a fractured boiler pipe.78 Those dying immediately showed coagulative necrosis of the respiratory mucosa down to the level of the alveolar ducts and alveolar congestion and oedema, while those surviving a little longer exhibited diffuse alveolar damage. The diffuse alveolar damage probably represented a manifestation of shock from their extensive cutaneous scalding whereas the mucosal necrosis is directly attributable to heat. Diffuse alveolar damage is usually part of systemic multiorgan failure in these patients, and is the leading cause of death in burns.79
The ubiquity of plastics today means that smoke contains numerous irritants, including isocyanates, aldehydes and fluorinated organic chemicals. Irritant smoke products have two principal effects. Firstly, they cause an immediate painful stimulation of the eyes and respiratory tract which at low concentrations may prevent escape and at high concentrations may cause laryngeal spasm and death. Secondly, they cause bronchopulmonary injury some hours after exposure. Burned patients dying within 4–12 days often show tracheobronchial necrosis and diffuse alveolar damage with prominent hyaline membranes.76, 77, 80 Secondary herpesvirus infection is often present.81, 82
The respiratory changes caused by heat and smoke are non-specific and careful consideration of the many causes of lung injury in burned patients listed in Box 7.2.2 and of the clinical circumstances and management is generally required. Often it will be concluded that the cause of the lung injury is multifactorial. Long-term consequences of smoke inhalation include bronchiectasis and obliterative bronchiolitis.83
Methyl isocyanate, the chemical released at Bhopal
The Bhopal catastrophe of 1984 was caused by the accidental release of 30 tons of methyl isocyanate gas (CH3 —N C O) from a pesticide plant.84 Over 200 000 people were exposed, of whom 2500 died, mostly within hours of exposure, and 60 000 were seriously injured. The victims complained of intense ocular and respiratory irritation. Some survivors were left with persistent respiratory impairment, which was thought to be due to obliterative bronchiolitis.85, 86
Methyl isocyanate is an extremely potent respiratory irritant, destroying the epithelium throughout the conducting airways, with comparatively less parenchymal injury. In survivors, epithelial regeneration, often involving squamous metaplasia, quickly commences, but not before endobronchial granulation tissue projections have developed, resulting in obliterative bronchiolitis.
Tear gas
Tear gases are chemical irritants delivered as an aerosol for the purpose of riot control. They react with mucocutaneous sensory nerve receptors causing intense irritation of the eyes, mucous membranes and skin. The respiratory effects are mainly concentrated on the upper tract so that there is violent sneezing, severe rhinorrhoea and cough but there may also be tracheobronchitis and rarely pulmonary oedema.87 Patients with pre-existent asthma or chronic obstructive pulmonary disease are most severely affected while others may be left with reactive airways dysfunction.
Ingested toxic agents
Toxins reaching the lungs via the blood stream may be drugs, food contaminants, metabolites produced elsewhere in the body, or chemicals ingested intentionally or accidentally, either in the home or the workplace.
The lungs are selectively damaged by certain blood-borne toxins for a variety of reasons. For example, the herbicide paraquat is preferentially taken up by the lungs because of its molecular homology with certain endogenous substances. As detailed below, the type I alveolar epithelial cells are the cells that bear the brunt of the damage in paraquat poisoning. On the other hand, the alveolar capillary endothelium has its own selective uptake mechanisms (see Metabolic functions of the pulmonary endothelium, p. 23) which may be responsible for it being selectively damaged by other chemicals.
The bronchiolar Clara cells are selectively injured by some ingested chemicals because they are equipped to deal with inhaled xenobiotics, but occasionally this activity results in metabolites that are extremely toxic. An example of this from veterinary medicine is provided by the furan-derivative 4-ipomeanol, which is found in mouldy sweet potatoes and results in acute pulmonary oedema in cattle fed such a diet. When this chemical is injected into mice, the bronchioles are denuded of Clara cells whereas the intervening ciliated cells are completely unaffected. The selective damage to the bronchiolar Clara cells appears to stem from the oxidative efficiency of their P-450 cytochromes,88 which is much higher than those of the liver. Chemicals having a similarly selective effect on bronchiolar Clara cells include 3-methylfuran, carbon tetrachloride, naphthalene and 1,1-dichloroethylene, the last of which is a volatile compound that is widely used in the plastics industry. Procarcinogens may be activated in the airways by similar mechanisms.
Paraquat
Paraquat is a dipyridylium compound that is widely used in agriculture as a herbicide. It kills all green plants but is inactivated on contact with the soil. It is applied as a spray and if the manufacturer's instructions are followed there is no danger to health. Most fatal cases of paraquat poisoning, both accidental and suicidal, have been due to ingestion of the 20% aqueous solution Gramoxone. The less concentrated granular form Weedol is unlikely to be ingested accidentally but may be taken suicidally.89 Paraquat is not absorbed by the intact skin but repeated or prolonged application damages the epidermis so that absorption into the blood stream with consequent systemic effects is possible, but rare.90
Although paraquat has toxic effects on the liver, kidneys and myocardium, these are transient and attention has centred on the pulmonary changes, which are usually fatal. Following suicidal ingestion of large amounts of paraquat, death from multiorgan failure and pulmonary haemorrhage occurs within a few days, whereas most victims of accidental paraquat poisoning die from progressive pulmonary fibrosis between 10 and 14 days after ingestion. In those who survive longer, a honeycomb pattern of pulmonary fibrosis may be apparent.91
Paraquat is a powerful oxidant and owes its toxicity to the production of active oxygen radicals. The lungs are particularly susceptible because paraquat is concentrated there by an active uptake mechanism in the alveolar epithelium. The inadvertent uptake of paraquat probably stems from a similarity between the molecular arrangement of its quaternary nitrogen atoms and the amine groups of endogenous oligoamines such as putrescine, spermidine and spermine, which are concerned in alveolar epithelial cell division and differentiation (Fig. 7.2.4 ).92 This results in paraquat levels being 6–10 times higher in the lung than in the plasma. Once taken up by the lung, paraquat is not metabolised but participates in redox cycling so that superoxide radicals are constantly produced. Epithelial injury is proportional to the concentration of paraquat, while it is lessened by hypoxia and antioxidants such as superoxide dismutase, and potentiated by increased concentrations of oxygen.93, 94, 95, 96 The high concentration of oxygen in the alveoli is a further reason why the lungs are particularly vulnerable to paraquat.
Figure 7.2.4.
Formulae of paraquat and the endogenous oligoamines putrescine, spermine and spermidine showing the molecular similarities. The name paraquat derives from the chemical's para-methyl groups and its quaternary nitrogen atoms.
Knowledge of the toxic effects of paraquat comes from observations on autopsy series89, 97, 98 and from experimental studies that have enabled the sequence of pulmonary changes to be observed.99, 100, 101, 102 In accordance with paraquat being taken up by the alveolar epithelium, electron microscopy shows that these cells suffer more profound damage than the endothelium.99 Type I epithelial cells swell and undergo necrosis (Fig. 7.2.5 ),103 whilst type II cells, although remaining capable of proliferation, show ultrastructural evidence of damage with derangement of cell organelles.99, 100 Histological changes in the lungs follow the pattern of diffuse alveolar damage, with a characteristic feature of the early exudative phase being intense vascular congestion and alveolar haemorrhage.89, 97, 104 Hyaline membranes are most clearly seen by about 5 days (Fig. 7.2.6 ) and epithelial proliferation and fibrosis are conspicuous by about 14 days.
Figure 7.2.5.
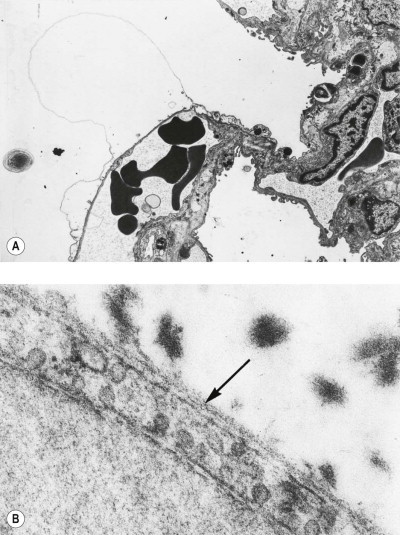
Paraquat poisoning. After initial swelling of the cytoplasm (A) the type I alveolar epithelial cells proceed to complete necrosis (B), exposing the basement membrane (arrow) they share with the alveolar capillary endothelium to alveolar air. Flecks of fibrin are seen in the alveolus. Intact endothelium and part of an erythrocyte are seen beneath the denuded basement membrane.
(Reproduced by permission of the editors of the Journal of Pathology99and Progress in Respiratory Research.103)
Figure 7.2.6.
Suicidal paraquat poisoning. The ingestion of large amounts of the chemical has led to death from pulmonary haemorrhage and diffuse alveolar damage within 3 days. Note the hyaline membrane formation and early interstitial fibrosis.
(Courtesy of Dr D Melcher, Brighton, UK.)
The pattern of pulmonary fibrosis in paraquat poisoning has been disputed. Some authors have stressed its interstitial position, whereas others have clearly demonstrated that it is intra-alveolar.98, 102, 104, 105, 106, 107 However, as described on page 148, it generally assumes an obliterative pattern of intra-alveolar fibrosis in which the lumina of several adjacent alveoli are totally effaced, rendering them completely airless (see Fig. 4.24, p. 148).
Toxic oil syndrome
A new multisystem disease appeared abruptly in the environs of Madrid in 1981.108, 109, 110 Over 20 000 people were affected and about 1 in 60 died. The disease was initially thought to be Mycoplasma pneumonia but was soon found to be associated with the use of adulterated oil sold illicitly by door-to-door salesmen. Although it was sold for culinary purposes the oil had been produced for industrial use in steel manufacture. It consisted of rapeseed and olive oil mixed with liquified animal fat, aniline and other organic chemicals. It has not been possible to identify the exact chemical responsible for the disease or to reproduce the changes in other species but the later induction of similar pathological changes by another substance contaminated with an aniline derivative is possibly relevant (see l-tryptophan-induced eosinophilia–myalgia syndrome, p. 389).111 Some clinical and pathological features of the disease suggest that immune mechanisms may also be involved.
The initial clinical features included fever, respiratory distress, cough, haemoptysis, skin eruptions and marked eosinophilia. Radiographs suggested pulmonary oedema and sometimes showed pleural effusion. About 5% of patients died at this stage but most recovered quickly. However, within a few weeks many were readmitted to hospital with nausea, vomiting, diarrhoea and abdominal pain. About a quarter then proceeded to develop weakness, myalgia, weight loss, scleroderma-like skin signs and pulmonary hypertension.112, 113 Many of these patients died after a long, wasting illness or are permanently disabled with neurological and hepatic disorders.
In the early phase the lungs showed the most severe changes, which consisted of a combination of diffuse alveolar damage, eosinophilic infiltrates and arterial luminal narrowing by endothelial swelling and vacuolation, intimal foam cell infiltration and a non-necrotising vasculitis.109, 112, 114 There was also capillary thrombosis, which later extended into arteries and veins, culminating in fibrosing obliteration of these blood vessels. In some patients dying of haemoptysis, dilated thin-walled blood vessels were identified in the mucosa of major blood-filled airways. Late features in the lungs included plexogenic arteriopathy (see p. 420), possibly secondary to changes in the liver. Similar inflammatory and vascular changes were seen in many other tissues. Notable extrapulmonary features included fasciitis, vasculitis, neuronal degeneration, perineuritis, hepatic injury and tissue eosinophilia.
Sauropus androgynus
Sauropus androgynus is a vegetable that is widely cultivated for the table in many south-eastern Asian countries. It is apparently harmless when cooked but recently there has been a vogue in Taiwan for consuming large amounts of its unprocessed juice, blended with that of guavas or pineapple, because of its supposed efficacy as a slimming aid and in blood pressure control. Coincident with this fad there has been an upsurge in patients with symptoms of obstructive lung disease. Within a 4-month period more than 60 such patients were seen at one hospital.115, 116, 117 They had four features in common: recent consumption of uncooked S. androgynus juice, fixed ventilatory obstruction, radiological evidence of bilateral bronchiectasis and an absence of any previous chronic respiratory disease. Four patients agreed to undergo open-lung biopsy. This showed chronic bronchiolitis or obliterative bronchiolitis of constrictive pattern. The lymphocytes were mainly T cells but immunofluorescent and electron microscopy showed no evidence of an immune process. Four patients underwent single-lung transplantation. The excised lungs showed sclerotic obliteration of bronchial arteries in the walls of bronchi 4–5 mm in diameter with segmental necrosis of bronchi 2–4 mm in diameter. The changes were considered to fit best with segmental ischaemic necrosis of bronchi at the watershed zone of the bronchial and pulmonary vasculature.118 Further patients have required lung transplantation but public education of the dangers of this herbal medicine now appears to have been successful.119
Recreational drugs
Alcohol and nicotine outstrip all other recreational drugs in popularity and their effects are of course well known. Those of tobacco smoking are summarised above and dealt with in detail in the chapters on obstructive lung disease (Chapter 3) and carcinoma of the lung (Chapter 12.1). Less well known is the lung disease that results from smoking Blackfat tobacco, a practice popular with Guyanese Indians. Blackfat is the trade name of a type of tobacco that is flavoured with mineral oil, some of which vaporises and is inhaled when the tobacco is smoked, to cause exogenous lipid pneumonia (see p. 314 [Ch8]).120 In recent years the smoking of two other substances, marijuana and cocaine, has gained in popularity. It would not be surprising if the long-term effects of smoking these substances were similar to those of cigarette smoking but as yet it is too early to judge. However, the short-term effects are similar to those of tobacco smoking and this bodes badly for their ultimate effects.
Marijuana
Marijuana consists of the dried leaves of the cannabis plant, also known as hemp, as opposed to hashish, which is the plant's resin, and a further extract known as ‘weed oil’. All these substances are smoked because they contain cannabis alkaloids which have psychoactive effects. However, this habit also exposes the lungs to many of the same respiratory irritants that are found in tobacco smoke. Initial exposure to marijuana smoke often results in coughing while habitual smokers produce black sputum. Bronchial biopsy shows inflammation and squamous metaplasia and bronchoalveolar lavage demonstrates increased numbers of cells, which are predominantly macrophages but also include neutrophils.121, 122, 123, 124, 125 These changes are virtually identical to the short-term effects of tobacco smoke and are therefore likely to be similarly followed by the development of chronic obstructive lung disease and lung cancer. Indeed, the dangers of smoking marijuana are probably greater than those of smoking tobacco as compared with tobacco smoking it is associated with a fivefold greater increase in blood carboxyhaemoglobin and a threefold increase in the amount of tar inhaled.126 It is estimated that three cannabis cigarettes result in the same degree of bronchial damage as 20 tobacco cigarettes.127 There is also evidence that the effects of smoking marijuana and tobacco are additive.128
Not surprisingly therefore, epidemiological studies report a dose-related impairment of large-airway function in marijuana smokers.129 There are also several reports attributing pneumothorax to marijuana smoking (Fig. 7.2.7 ).130, 131 The pneumothorax may be spontaneous or develop during the deep, sustained inspiratory effort involved in smoking marijuana (or cocaine), which may be enhanced by a partner applying positive ventilatory pressure by mouth-to-mouth contact. Thoracoscopy in such cases has shown predominantly apical, irregular bullous emphysema, while lung biopsy has demonstrated widespread alveolar filling by heavily pigmented macrophages.131, 132 Evidence is also beginning to accumulate that long-term cannabis use increases the risk of lung cancer.133 Smoking cannabis in the form of weed oil is also reported to result in exogenous lipid pneumonia.134
Figure 7.2.7.
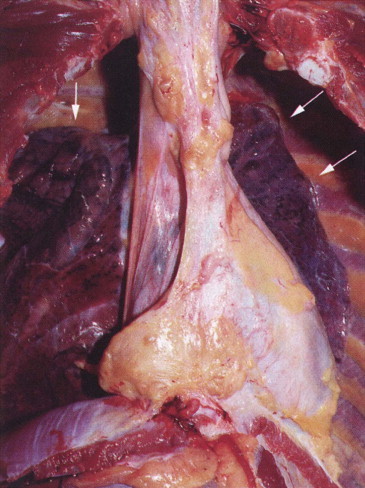
Bilateral pneumothoraces in a 23-year-old man who died suddenly while smoking marijuana. The collapsed lungs (arrows) have retracted towards the mediastinum.
(Courtesy of Dr JF Tomashefski Junior, Cleveland, USA.131)
Cocaine
Cocaine hydrochloride is a fine white powder derived from the leaves of the plant Erythroxolon coca by a complex chemical process. It is heat-labile and therefore cannot be smoked. Users inject it intravenously or inhale it unheated through the nose, the latter practice being known as ‘snorting’. However, a heat-stable free-base form that can be smoked is easily prepared from the hydrochloride with baking powder and a solvent such as ether. This process results in a crystalline deposit that is known as ‘rock’ because of its appearance or ‘crack’ because of the crackling sound it emits when heated. When smoked, the cocaine is readily absorbed and an intense surge of euphoria is experienced within 8 seconds. The intravenous route takes twice as long and ‘snorting’ several minutes. The hard addict therefore prefers to smoke ‘crack’.
A variety of pulmonary complications of smoking free-base cocaine has been reported.128, 135, 136, 137, 138, 139, 140, 141, 142, 143, 144 Acute effects include cough, shortness of breath, chest pain and haemoptysis. Asthma may be aggravated, black sputum is produced, and pneumothorax and interstitial emphysema have resulted from Valsalva manoeuvres undertaken in the belief that they promote even more rapid absorption. Biopsy has shown pulmonary congestion and oedema, organising pneumonia, haemorrhage, haemosiderosis, diffuse alveolar damage and interstitial pneumonia or fibrosis. Less common effects include eosinophilic pneumonia, extrapulmonary eosinophilic angiitis, medial thickening of pulmonary arteries and the barotrauma described above (see Fig. 7.2.7). Severe burning of the airways has also been seen due to ‘crack’ being smoked before all the ether used in its preparation has evaporated.
'Snorting’ unheated cocaine has its own complications: substances such as cellulose or talc with which the drug is ‘cut’ (mixed as a diluent) are liable to provoke a foreign-body giant cell reaction in the lungs (Fig. 7.2.8 ).145 However, particles of foreign material larger than those in the usual respirable range (allowing for the fibrous shape of substances such as cellulose) should suggest intravenous use (see ‘Filler embolism’, below).
Figure 7.2.8.
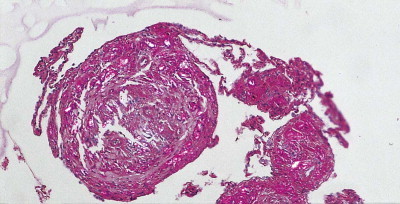
A foreign-body granulomatous response in the lungs to cellulose filler inhaled by a cocaine sniffer. Section viewed by partially polarised light microscopy.
Heroin
Heroin is usually injected, but it may be smoked, when, as with marijuana, it is liable to lead to a very pronounced macrophage response. Intravenous heroin abuse sometimes causes the sudden onset of a potentially fatal high-permeability pulmonary oedema (Fig. 7.2.9 ). Intravenous abuse of heroin and other drugs is also liable to cause ‘filler embolism’, which will now be considered.
Figure 7.2.9.
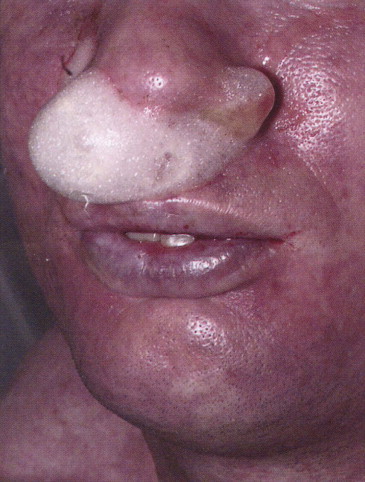
Frothy oedema fluid protrudes from the nostrils of a person who died while injecting heroin intravenously. Similar froth filled the whole respiratory tract.
(Courtesy of Dr JF Tomashefski Junior, Cleveland, USA.131)
‘Filler embolism’
‘Filler embolism’ is the result of illicit drug usage in which compounds designed for oral use are injected intravenously to heighten their effects. Oral preparations consist largely of fillers such as talc or starch and this insoluble particulate matter accumulates in the pulmonary capillaries. It provokes a foreign-body giant cell reaction, thrombosis and fibrosis and may cause pulmonary hypertension (Fig. 7.2.10 and see Fig. 8.1.13, p. 412).146, 147, 148, 149, 150, 151, 152, 153 The various materials may be distinguished by their morphology, staining characteristics (see Fig. 7.2.10 and Table 7.2.3 ) and elemental composition, as studied by X-ray spectroscopy (see p. 330).
Figure 7.2.10.
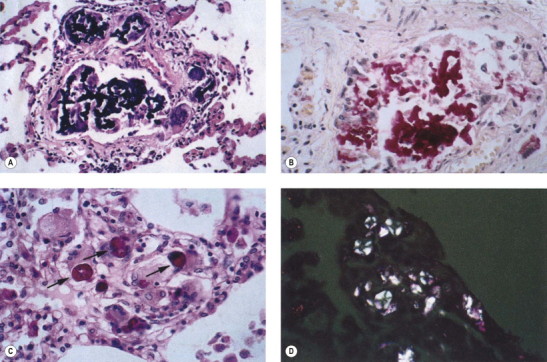
Tablet filler materials in the lungs. (a) Intravascular and perivascular deposits of the tablet dispersant crospovidone eliciting a foreign-body giant cell reaction. (b) A crosprovidone granuloma stained with mucicarmine. (c) A starch granuloma stained with periodic acid–Schiff reagents. (d) The Maltese cross birefringence of starch viewed by polarized light.
(Courtesy of Dr JF Tomashefski Junior, Cleveland, USA.131)
Table 7.2.3.
Tablet filler materials
| Filler | Shape | Size | Polarisation | Histochemistry |
|---|---|---|---|---|
| Starch | Round | 8–12 µm | Maltese cross | d-PAS |
| Talc | Platy but seen edge-on as needles | 5–15 µm | Strong | None |
| Cellulose | Fibrous | 25–200 µm | Strong | PAS, Congo red |
| Crosprovidone | Globular or coral-like | 100 µm | Negative | Mucicarmine, Congo red |
| Magnesium stearate | Irregular | 5–10 µm | Positive | None |
| Silica | Irregular | 10–20 µm | Positive | None |
d-PAS, diastase-controlled periodic acid–Schiff.
4-methyl-aminorex
This ‘designer’ drug, taken for its central stimulant activity (street names ‘ice’ or ‘U-4-E-uh’, pronounced euphoria), is related to the appetite suppressor aminorex, discussed on page 424, and has similarly been associated with pulmonary hypertension.154
References
Atmospheric pressure changes
- 1.Noppen M, Verbanck S, Harvey J, et al. Music: a new cause of primary spontaneous pneumothorax. Thorax. 2004;59:722–724. doi: 10.1136/thx.2003.007385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alifano M, Forti Parri SN, Bonfanti B, et al. Atmospheric pressure influences the risk of pneumothorax: beware of the storm! Chest. 2007;131:1877–1882. doi: 10.1378/chest.06-2206. [DOI] [PubMed] [Google Scholar]
- 3.Zuckerman S. Experimental study of blast injuries to the lungs. Lancet. 1940;2:219–224. [Google Scholar]
- 4.Cooper GJ, Taylor DEM. Biophysics of impact injury to the chest and abdomen. J R Army Med Corps. 1989;135:58–67. doi: 10.1136/jramc-135-02-04. [DOI] [PubMed] [Google Scholar]
- 5.Tsokos M, Paulsen F, Petri S, et al. Histologic, immunohistochemical, and ultrastructural findings in human blast lung injury. Amer J Respir Crit Care Med. 2003;168:549–555. doi: 10.1164/rccm.200304-528OC. [DOI] [PubMed] [Google Scholar]
- 6.Tsokos M, Paulsen F, Petri S, et al. Histologic, immunohistochemical, and ultrastructural findings in human blast lung injury. Am J Respir Crit Care Med. 2003;168:549–555. doi: 10.1164/rccm.200304-528OC. [DOI] [PubMed] [Google Scholar]
- 7.Brown RFR, Cooper GJ, Maynard RL. The ultrastructure of rat lung following acute primary blast injury. Int J Exp Pathol. 1993;74:151–162. [PMC free article] [PubMed] [Google Scholar]
- 8.Nadel JA, Denison D. In: Textbook of Respiratory Medicine. 2nd ed. Murray JF, Nadel JA, editors. Saunders; Philadelphia: 1994. Disorders associated with diving; pp. 2099–2116. [Google Scholar]
- 9.Kidd DJ, Elliott DH. In: The Physiology and Medicine of Diving and Compressed Air Work. 2nd ed. Bennett PB, Elliott DH, editors. Bailliere Tindall; London: 1975. Decompression disorders in divers; pp. 471–495. [Google Scholar]
- 10.Liebow AA, Stark JE, Vogel J, et al. Intrapulmonary air trapping in submarine escape training casualties. U S Armed Forces Med J. 1959;10:265–289. [PubMed] [Google Scholar]
- 11.Denison DM, Warrell DA, West JB. Airway structure and alveolar emptying in the lungs of sea lions and dogs. Respir Physiol. 1971;13:253–260. doi: 10.1016/0034-5687(71)90029-6. [DOI] [PubMed] [Google Scholar]
- 12.Denison DM, Kooyman GL. The structure and function of the small airways in pinniped and sea otter lungs. Respir Physiol. 1973;17:1–10. doi: 10.1016/0034-5687(73)90105-9. [DOI] [PubMed] [Google Scholar]
- 13.Cooperman EM, Hogg J, Thurlbeck WM. Mechanisms of death in shallow-water scuba diving. Can Med Assoc J. 1968;99:1128–1131. [PMC free article] [PubMed] [Google Scholar]
- 14.Broome CR, Jarvis LJ, Clark RJ. Pulmonary barotrauma in submarine escape training. Thorax. 1994;49:186–187. doi: 10.1136/thx.49.2.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elliott DH, Hallenbeck JM. In: The Physiology and Medicine of Diving and Compressed Air Work. 2nd ed. Bennett PB, Elliott DH, editors. Bailliere Tindall; London: 1975. The pathophysiology of decompression sickness; pp. 435–455. [Google Scholar]
- 16.Boussuges A, Blanc F, Carturan D. Hemodynamic changes induced by recreational scuba diving. Chest. 2006;129:1337–1343. doi: 10.1378/chest.129.5.1337. [DOI] [PubMed] [Google Scholar]
- 17.McCallum RI. In: The Physiology and Medicine of Diving and Compressed Air Work. 2nd ed. Bennett PB, Elliott DH, editors. Bailliere Tindall; London: 1975. Dysbaric osteonecrosis: aseptic necrosis of bone; pp. 504–521. [Google Scholar]
- 18.Fahlman A, Olszowka A, Bostrom B, et al. Deep diving mammals: Dive behavior and circulatory adjustments contribute to bends avoidance. Respir Physiol Neurobiol. 2006;153:66–77. doi: 10.1016/j.resp.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 19.Hackett PH, Roach RC. Current concepts: High-altitude illness. N Engl J Med. 2001;345:107–114. doi: 10.1056/NEJM200107123450206. [DOI] [PubMed] [Google Scholar]
- 20.Schoene RB. Illnesses at high altitude. Chest. 2008;134:402–416. doi: 10.1378/chest.07-0561. [DOI] [PubMed] [Google Scholar]
- 21.Bartsch P. High altitude pulmonary edema. Respiration. 1997;64:435–443. doi: 10.1159/000196720. [DOI] [PubMed] [Google Scholar]
- 22.Schoene RB, Hackett PH, Henderson WR, et al. High altitude pulmonary oedema; characteristics of lung lavage fluid. JAMA. 1986;256:63–69. [PubMed] [Google Scholar]
- 23.Bartsch P, Maggiorini M, Ritter M, et al. Prevention of high-altitude pulmonary edema by nifedipine. N Engl J Med. 1991;325:1284–1289. doi: 10.1056/NEJM199110313251805. [DOI] [PubMed] [Google Scholar]
- 24.Naeije R. Pulmonary circulation at high altitude. Respiration. 1997;64:429–434. doi: 10.1159/000196719. [DOI] [PubMed] [Google Scholar]
- 25.Hanaoka M, Kubo K, Yamazaki Y, et al. Association of high-altitude pulmonary edema with the major histocompatibility complex. Circulation. 1998;97:1124–1128. doi: 10.1161/01.cir.97.12.1124. [DOI] [PubMed] [Google Scholar]
- 26.West JB, Colice GL, Lee YJ, et al. Pathogenesis of high-altitude pulmonary oedema: direct evidence of stress failure of pulmonary capillaries. Eur Respir J. 1995;8:523–529. [PubMed] [Google Scholar]
- 27.Hultgren HN. High-altitude pulmonary edema: current concepts. Annu Rev Med. 1996;47:267–284. doi: 10.1146/annurev.med.47.1.267. [DOI] [PubMed] [Google Scholar]
- 28.Maggiorini M, Melot C, Pierre S, et al. High-altitude pulmonary edema is initially caused by an increase in capillary pressure. Circulation. 2001;103:2078–2083. doi: 10.1161/01.cir.103.16.2078. [DOI] [PubMed] [Google Scholar]
- 29.Reeves JT, Schoene RB. When lungs on mountains leak – studying pulmonary edema at high altitudes. N Engl J Med. 1991;325:1306–1307. doi: 10.1056/NEJM199110313251810. [DOI] [PubMed] [Google Scholar]
- 30.Scherrer U, Vollenweider L, Delabays A, et al. Inhaled nitric oxide for high-altitude pulmonary edema. N Engl J Med. 1996;334:624–629. doi: 10.1056/NEJM199603073341003. [DOI] [PubMed] [Google Scholar]
- 31.Hultgren HN, Wilson R, Kosek JC. Lung pathology in high-altitude pulmonary edema. Wilderness Environ Med. 1997;8:218–220. doi: 10.1580/1080-6032(1997)008[0218:lpihap]2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 32.Droma Y, Hanaoka M, Hotta J, et al. Pathological features of the lung in fatal high altitude pulmonary edema occurring at moderate altitude in Japan. High Alt Med Biol. 2001;2:515–523. doi: 10.1089/152702901753397081. [DOI] [PubMed] [Google Scholar]
- 33.Gupta ML, Rao KS, Anand IS, et al. Lack of smooth muscle in the small pulmonary arteries of the native Ladakhi – is the Himalayan highlander adapted? Am Rev Respir Dis. 1992;145:1201–1204. doi: 10.1164/ajrccm/145.5.1201. [DOI] [PubMed] [Google Scholar]
- 34.Harris P. In: Aspects of Hypoxia. Heath D, editor. Liverpool University Press; Liverpool: 1986. Evolution,hypoxia and high altitude; pp. 207–216. [Google Scholar]
- 35.Heath D, Harris P, Sui GJ, et al. Pulmonary blood vessels and endocrine cells in subacute infantile mountain sickness. Respir Med. 1989;83:77–81. doi: 10.1016/s0954-6111(89)80064-2. [DOI] [PubMed] [Google Scholar]
- 36.Harris P, Heath D. 3rd ed. Churchill Livingstone; Edinburgh: 1986. The Human Pulmonary Circulation: Its Form and Function in Health and Disease. [Google Scholar]
Drowning
- 37.Cohen DS, Matthay MA, Cogan MG, et al. Pulmonary edema associated with salt water near-drowning – new insights. Am Rev Respir Dis. 1992;146:794–796. doi: 10.1164/ajrccm/146.3.794. [DOI] [PubMed] [Google Scholar]
- 38.Folkesson HG, Kheradmand F, Matthay MA. The effect of salt water on alveolar epithelial barrier function. Am J Respir Crit Care Med. 1994;150:1555–1563. doi: 10.1164/ajrccm.150.6.7952614. [DOI] [PubMed] [Google Scholar]
- 39.Gregorakos L, Markou N, Psalida V, et al. Near-drowning: clinical course of lung injury in adults. Lung. 2009;187:93–97. doi: 10.1007/s00408-008-9132-4. [DOI] [PubMed] [Google Scholar]
Inhaled toxic agents
- 40.Haggard HW. Action of irritant gases upon the respiratory tract. J Indust Hyg. 1924;5:390. [Google Scholar]
- 41.Miller FJ, Overton JH, Kimbell JS, et al. In: Toxicology of the Lung. 2nd ed. Gardner DE, Crapo JD, McClellan RO, editors. Raven Press; New York: 1993. Regional respiratory tract absorption of inhaled reactive gases. [Google Scholar]
- 42.Newman Taylor AJ. Respiratory irritants encountered at work. Thorax. 1996;51:541–545. doi: 10.1136/thx.51.5.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bascom R, Bromberg PA, Hill C, et al. Health effects of outdoor air pollution. Am J Respir Crit Care Med. 1996;153:3–50. doi: 10.1164/ajrccm.153.1.8542133. 477–98. [DOI] [PubMed] [Google Scholar]
- 44.Committee of the Environmental and Occupational Health Assembly of the American Thoracic Society Health effects of outdoor air pollution. Am J Respir Crit Care Med. 1996;153:3–50. doi: 10.1164/ajrccm.153.1.8542133. [DOI] [PubMed] [Google Scholar]
- 45.Committee of the Environmental and Occupational Health Assembly of the American Thoracic Society Health effects of outdoor air pollution. Part 2. Am J Respir Crit Care Med. 1996;153:477–498. doi: 10.1164/ajrccm.153.2.8564086. [DOI] [PubMed] [Google Scholar]
- 46.Samet JM, Dominici F, Curriero FC, et al. Fine particulate air pollution and mortality in 20 US Cities, 1987–1994. N Engl J Med. 2000;343:1742–1749. doi: 10.1056/NEJM200012143432401. [DOI] [PubMed] [Google Scholar]
- 47.Sydbom A, Blomberg A, Parnia S, et al. Health effects of diesel exhaust emissions. Eur Resp J. 2001;17:733–746. doi: 10.1183/09031936.01.17407330. [DOI] [PubMed] [Google Scholar]
- 48.Souza MB, Saldiva PHN, Pope CA, et al. Respiratory changes due to long-term exposure to urban levels of air pollution: A histopathologic study in humans. Chest. 1998;113:1312–1318. doi: 10.1378/chest.113.5.1312. [DOI] [PubMed] [Google Scholar]
- 49.Sherwin RP, Richters V, Everson RB, et al. Chronic glandular bronchitis in young individuals residing in a metropolitan area. Virchows Archiv. 1998;433:341–348. doi: 10.1007/s004280050258. [DOI] [PubMed] [Google Scholar]
- 50.Dockery DW, Pope CA, Xu XP, et al. An association between air pollution and mortality in 6 United States cities. N Engl J Med. 1993;329:1753–1759. doi: 10.1056/NEJM199312093292401. [DOI] [PubMed] [Google Scholar]
- 51.Akhtar T, Ullah Z, Khan MH, et al. Chronic bronchitis in women using solid biomass fuel in rural peshawar, Pakistan. Chest. 2007;132:1472–1475. doi: 10.1378/chest.06-2529. [DOI] [PubMed] [Google Scholar]
- 52.Emmelin A, Wall S. Indoor air pollution: a poverty-related cause of mortality among the children of the world. Chest. 2007;132:1615–1623. doi: 10.1378/chest.07-1398. [DOI] [PubMed] [Google Scholar]
- Hu G, Zhou Y, Tian J, et al. Risk of COPD from exposure to biomass smoke. Chest. 2010;138:20–31. doi: 10.1378/chest.08-2114. [DOI] [PubMed] [Google Scholar]
- 53.Weinstein P, Cook A. Human health impacts of volcanic activity. Histopathology. 2002;41:329–333. [Google Scholar]
- 54.Banauch GI, Alleyne D, Sanchez R, et al. Persistent hyperreactivity and reactive airway dysfunction in firefighters at the World Trade Center. Am J Respir Crit Care Med. 2003;168:54–62. doi: 10.1164/rccm.200211-1329OC. [DOI] [PubMed] [Google Scholar]
- 55.Reibman J, Lin S, Hwang SA, et al. The World Trade Center residents’ respiratory health study: new-onset respiratory symptoms and pulmonary function. Environ Health Perspect. 2005;113:406–411. doi: 10.1289/ehp.7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Banauch GI, Hall C, Weiden M, et al. Pulmonary function after exposure to the World Trade Center collapse in the New York City Fire Department. Am J Respir Crit Care Med. 2006;174:312–319. doi: 10.1164/rccm.200511-1736OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lioy PJ, Weisel CP, Millette JR, et al. Characterization of the dust/smoke aerosol that settled east of the World Trade Center (WTC) in lower Manhattan after the collapse of the WTC 11 September 2001. Environ Health Perspect. 2002;110:703–714. doi: 10.1289/ehp.02110703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McGee JK, Chen LC, Cohen MD, et al. Chemical analysis of World Trade Center fine particulate matter for use in toxicologic assessment. Environ Health Perspect. 2003;111:972–980. doi: 10.1289/ehp.5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Landrigan PJ, Lioy PJ, Thurston G, et al. Health and environmental consequences of the World Trade Center disaster. Environ Health Perspect. 2004;112:731–739. doi: 10.1289/ehp.6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Skloot GS, Schechter CB, Herbert R, et al. Longitudinal assessment of spirometry in the World Trade Center medical monitoring program. Chest. 2009;135:492–498. doi: 10.1378/chest.08-1391. [DOI] [PubMed] [Google Scholar]
- 61.Rom WN, Weiden M, Garcia R, et al. Acute eosinophilic pneumonia in a New York City firefighter exposed to World Trade Center dust. Am J Respir Crit Care Med. 2002;166:797–800. doi: 10.1164/rccm.200206-576OC. [DOI] [PubMed] [Google Scholar]
- 62.Safirstein BH, Klukowicz A, Miller R, et al. Granulomatous pneumonitis following exposure to the World Trade Center collapse. Chest. 2003;123:301–304. doi: 10.1378/chest.123.1.301. [DOI] [PubMed] [Google Scholar]
- 63.Ryu JH, Colby TV, Hartman TE, et al. Smoking-related interstitial lung diseases: a concise review. Eur Resp J. 2001;17:122–132. doi: 10.1183/09031936.01.17101220. [DOI] [PubMed] [Google Scholar]
- 64.Grubstein A, Bendayan D, Schactman I, et al. Concomitant upper-lobe bullous emphysema, lower-lobe interstitial fibrosis and pulmonary hypertension in heavy smokers: report of eight cases and review of the literature. Respir Med. 2005;99:948–954. doi: 10.1016/j.rmed.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 65.Jankowich MD, Polsky M, Klein M, et al. Heterogeneity in combined pulmonary fibrosis and emphysema. Respiration. 2008;75:411–417. doi: 10.1159/000107048. [DOI] [PubMed] [Google Scholar]
- 66.Nagai S, Hoshino Y, Hayashi M, et al. Smoking-related interstitial lung diseases. Curr Opin Pulm Med. 2000;6:415–419. doi: 10.1097/00063198-200009000-00005. [DOI] [PubMed] [Google Scholar]
- 67.Wells AU, Nicholson AG, Hansell DM. Challenges in pulmonary fibrosis 4: Smoking-induced diffuse interstitial lung diseases. Thorax. 2007;62:904–910. doi: 10.1136/thx.2004.031021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cottin V, Nunes H, Brillet PY, et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J. 2005;26:586–593. doi: 10.1183/09031936.05.00021005. [DOI] [PubMed] [Google Scholar]
- 69.Kawabata Y, Hoshi E, Murai K, et al. Smoking-related changes in the background lung of specimens resected for lung cancer: a semiquantitative study with correlation to postoperative course. Histopathology. 2008;53:707–714. doi: 10.1111/j.1365-2559.2008.03183.x. [DOI] [PubMed] [Google Scholar]
- Balbi B, Cottin V, Singh S, et al. Clinical Assembly contribution to the celebration of 20 years of the ERS. Smoking-related lung diseases: a clinical perspective. Eur Respir J. 2010;35:231–233. doi: 10.1183/09031936.00189309. [DOI] [PubMed] [Google Scholar]
- Katzenstein ALA, Mukhopadhyay S, Zanardi C, et al. Clinically occult interstitial fibrosis in smokers: classification and significance of a surprisingly common finding in lobectomy specimens. Hum Path. 2010;41:316–325. doi: 10.1016/j.humpath.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 70.Adesina AM, Vallyathan V, McQuillen EN, et al. Bronchiolar inflammation and fibrosis associated with smoking – a morphologic cross-sectional population analysis. Am Rev Respir Dis. 1991;143:144–149. doi: 10.1164/ajrccm/143.1.144. [DOI] [PubMed] [Google Scholar]
- 71.Auerbach O, Garfinkel L, Hammond EC. Relation of smoking and age to findings in lung parenchyma: a microscopic study. Chest. 1974;65:29–35. doi: 10.1378/chest.65.1.29. [DOI] [PubMed] [Google Scholar]
- 72.Ammous Z, Hackett NR, Butler MW, et al. Variability in small airway epithelial gene expression among normal smokers. Chest. 2008;133:1344–1353. doi: 10.1378/chest.07-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Elliot JG, Carroll NG, James AL, et al. Airway alveolar attachment points and exposure to cigarette smoke in utero. Am J Respir Crit Care Med. 2003;167:45–49. doi: 10.1164/rccm.2110005. [DOI] [PubMed] [Google Scholar]
- 74.Warren CW, Jones NR, Eriksen MP, et al. Patterns of global tobacco use in young people and implications for future chronic disease burden in adults. The Lancet 367:749–53. [DOI] [PubMed]
- 75.Maziak W, Ward KD, Afifi Soweid RA, et al. Tobacco smoking using a waterpipe: a re-emerging strain in a global epidemic. Tob Control. 2004;13:327–333. doi: 10.1136/tc.2004.008169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Foley FD, Moncrief JA, Mason AD. Pathology of the lung in fatally burned patients. Ann Surg. 1968;167:251–264. doi: 10.1097/00000658-196802000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Toor AH, Tomashefski JF, Kleinerman J. Respiratory tract pathology in patients with severe burns. Hum Pathol. 1990;21:1212–1220. doi: 10.1016/s0046-8177(06)80033-x. [DOI] [PubMed] [Google Scholar]
- 78.Brinkmann B, Puschel K. Heat injuries to the respiratory system. Virchows Arch A Pathol Anat Histopathol. 1978;379:299–311. doi: 10.1007/BF00464473. [DOI] [PubMed] [Google Scholar]
- 79.Sheridan RL, Ryan CM, Yin LM, et al. Death in the burn unit: sterile multiple organ failure. Burns. 1998;24:307–311. doi: 10.1016/s0305-4179(97)00062-4. [DOI] [PubMed] [Google Scholar]
- 80.Nash G, Foley FD, Langlinais PC. Pulmonary interstitial edema and hyaline membranes in adult burn patients. Hum Pathol. 1975;5:149–160. doi: 10.1016/s0046-8177(74)80062-6. [DOI] [PubMed] [Google Scholar]
- 81.Hayden FG, Himel HN, Heggers JP. Herpesvirus infections in burn patients. Chest. 1994;106:S15–S21. doi: 10.1378/chest.106.1_supplement.15s. [DOI] [PubMed] [Google Scholar]
- 82.Byers RJ, Hasleton PS, Quigley A, et al. Pulmonary herpes simplex in burns patients. Eur Respir J. 1996;9:2313–2317. doi: 10.1183/09031936.96.09112313. [DOI] [PubMed] [Google Scholar]
- 83.Tasaka S, Kanazawa M, Mori M, et al. Long-term course of bronchiectasis and bronchiolitis obliterans as late complication of smoke inhalation. Respiration. 1995;62:40–42. doi: 10.1159/000196386. [DOI] [PubMed] [Google Scholar]
- 84.Weill H. Disaster at Bhopal: the accident, early findings and respiratory health outlook in those injured. Bull Eur Physiopath Resp. 1987;23:587–590. [PubMed] [Google Scholar]
- 85.Vijayan VK, Sankaran K, Sharma SK, et al. Chronic lung inflammation in victims of toxic gas leak at Bhopal. Respir Med. 1995;89:105–111. doi: 10.1016/0954-6111(95)90192-2. [DOI] [PubMed] [Google Scholar]
- 86.Cullinan P, Acquilla S, Ramana Dhara V. Respiratory morbidity 10 years after the Union Carbide gas leak at Bhopal: a cross sectional survey. BMJ. 1997;314:338–342. doi: 10.1136/bmj.314.7077.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Carron PN, Yersin B. Management of the effects of exposure to tear gas. BMJ. 2009;338:b2283. doi: 10.1136/bmj.b2283. [DOI] [PubMed] [Google Scholar]
Ingested toxic agents
- 88.Boyd MR. Evidence for the Clara cell as a site of cytochrome P450-dependent mixed-function oxidase activity in lung. Nature. 1977;269:713–715. doi: 10.1038/269713a0. [DOI] [PubMed] [Google Scholar]
- 89.Rebello G, Mason JK. Pulmonary histological appearances in fatal paraquat poisoning. Histopathology. 1977;2:53–66. doi: 10.1111/j.1365-2559.1978.tb01693.x. [DOI] [PubMed] [Google Scholar]
- 90.Papiris SA, Maniati MA, Kyriakidis V, et al. Pulmonary damage due to paraquat poisoning through skin absorption. Respiration. 1995;62:101–103. doi: 10.1159/000196400. [DOI] [PubMed] [Google Scholar]
- 91.Hudson M, Patel SB, Ewen SWB, et al. Paraquat induced pulmonary fibrosis in three survivors. Thorax. 1991;46:201–204. doi: 10.1136/thx.46.3.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hoet PHM, Dinsdale D, Lewis CPL, et al. Kinetics and cellular localisation of putrescine uptake in human lung tissue. Thorax. 1993;48:1235–1241. doi: 10.1136/thx.48.12.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fisher HK, Clements JA, Wright RR. Enhancement of oxygen toxicity by the herbicide paraquat. Am Rev Respir Dis. 1973;107:246–252. doi: 10.1164/arrd.1973.107.2.246. [DOI] [PubMed] [Google Scholar]
- 94.Rhodes ML, Zavala DC, Brown D. Hypoxic protection in paraquat poisoning. Lab Invest. 1976;35:496–500. [PubMed] [Google Scholar]
- 95.Raffin TA, Simon LM, Douglas WHJ, et al. The effects of variable O2 tension and of exogenous superoxide dismutase on type II pneumocytes exposed to paraquat. Lab Invest. 1980;42:205. [PubMed] [Google Scholar]
- 96.Skillrud DM, Martin WJ. Paraquat-induced injury of type II alveolar cells. Am Rev Respir Dis. 1984;129:995–999. doi: 10.1164/arrd.1984.129.6.995. [DOI] [PubMed] [Google Scholar]
- 97.Parkinson C. The changing pattern of paraquat poisoning in man. Histopathology. 1980;4:171–183. doi: 10.1111/j.1365-2559.1980.tb02910.x. [DOI] [PubMed] [Google Scholar]
- 98.Takahashi T, Takahashi Y, Nio M. Remodeling of the alveolar structure in the paraquat lung of humans: a morphometric study. Hum Pathol. 1994;25:702–708. doi: 10.1016/0046-8177(94)90304-2. [DOI] [PubMed] [Google Scholar]
- 99.Vijeyaratnam GS, Corrin B. Experimental paraquat poisoning. J Pathol. 1971;103:123–129. doi: 10.1002/path.1711030207. [DOI] [PubMed] [Google Scholar]
- 100.Smith P, Heath D, Kay JM. The pathogenesis and structure of paraquat-induced pulmonary fibrosis in rats. J Pathol. 1974;114:57–67. doi: 10.1002/path.1711140202. [DOI] [PubMed] [Google Scholar]
- 101.Sykes BI, Purchase IFH, Smith LL. Pulmonary ultrastructure after oral and intravenous dosage of paraquat to rats. J Pathol. 1977;121:233–241. doi: 10.1002/path.1711210407. [DOI] [PubMed] [Google Scholar]
- 102.Fukuda Y, Ferrans VJ, Schoenberger CI, et al. Patterns of pulmonary structural remodeling after experimental paraquat toxicity. Am J Pathol. 1985;118:452–475. [PMC free article] [PubMed] [Google Scholar]
- 103.Corrin B, Vijeyaratnam GS. Experimental models of interstitial pneumonia: paraquat, iprindole. Prog Resp Res. 1975;8:107–120. [Google Scholar]
- 104.Toner PG, Vetters JM, Spilg WGS, et al. Fine structure of the lung lesion in a case of paraquat poisoning. J Pathol. 1970;102:182–183. doi: 10.1002/path.1711020311. [DOI] [PubMed] [Google Scholar]
- 105.Copland GM, Kolin A, Shulman HS. Fatal pulmonary intra-alveolar fibrosis after paraquat ingestion. N Engl J Med. 1974;291:290–292. doi: 10.1056/NEJM197408082910607. [DOI] [PubMed] [Google Scholar]
- 106.Smith P, Heath D. Paraquat lung: a reappraisal. Thorax. 1974;29:643–653. doi: 10.1136/thx.29.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hara H, Manabe T, Hayashi T. An immunohistochemical study of the fibrosing process in paraquat lung injury. Virchows Arch A Pathol Anat Histopathol. 1989;415:357–366. doi: 10.1007/BF00718638. [DOI] [PubMed] [Google Scholar]
- 108.Tabuenca JM. Toxic-allergic syndrome caused by ingestion of rapeseed oil denatured with aniline. Lancet. 1981;2:567–568. doi: 10.1016/s0140-6736(81)90949-1. [DOI] [PubMed] [Google Scholar]
- 109.Martinez-Tello FJ, Navas-Palacios JJ, Ricoy JR, et al. Pathology of a new toxic syndrome caused by ingestion of adulterated oil in Spain. Virchows Arch A Pathol Anat Histopathol. 1982;397:261–285. doi: 10.1007/BF00496569. [DOI] [PubMed] [Google Scholar]
- 110.Kilbourne EM, Rigaue-Perez JG, Heath CW, et al. Clinical epidemiology of toxic oil syndrome. N Engl J Med. 1983;309:1408–1414. doi: 10.1056/NEJM198312083092302. [DOI] [PubMed] [Google Scholar]
- 111.Mayeno AN, Belongia EA, Lin F, et al. 3-(phenylamino)alanine, a novel aniline-derived amino acid associated with the eosinophilia-myalgia syndrome – a link to the toxic oil syndrome. Mayo Clin Proc. 1992;67:1134–1139. doi: 10.1016/s0025-6196(12)61142-2. [DOI] [PubMed] [Google Scholar]
- 112.Gomez-Sanchez MA, de Juan MJM, Gomez-Pajuelo C, et al. Pulmonary hypertension due to toxic oil syndrome. A clinicopathologic study. Chest. 1989;95:325–331. doi: 10.1378/chest.95.2.325. [DOI] [PubMed] [Google Scholar]
- 113.Cheng TO. Pulmonary hypertension in patients with eosinophilia–myalgia syndrome or toxic oil syndrome. Mayo Clin Proc. 1993;68:823. doi: 10.1016/s0025-6196(12)60645-4. [DOI] [PubMed] [Google Scholar]
- 114.Fernandez-Segoviano P, Esteban A, Martinez-Cabruja R. Pulmonary vascular lesions in the toxic oil syndrome in Spain. Thorax. 1983;38:724–729. doi: 10.1136/thx.38.10.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lai R-S, Chiang AA, Wu M-T, et al. Outbreak of bronchiolitis obliterans associated with consumption of Sauropus androgynus in Taiwan. Lancet. 1996;348:83–85. doi: 10.1016/s0140-6736(96)00450-3. [DOI] [PubMed] [Google Scholar]
- 116.Chang H, Wang JS, Tseng HH, et al. Histopathological study of Sauropus androgynus-associated constrictive bronchiolitis obliterans: a new cause of constrictive bronchiolitis obliterans. Am J Surg Pathol. 1997;21:35–42. doi: 10.1097/00000478-199701000-00004. [DOI] [PubMed] [Google Scholar]
- 117.Wang JS, Tseng HH, Lai RS, et al. Sauropus androgynus-constrictive obliterative bronchitis/bronchiolitis – histopathological study of pneumonectomy and biopsy specimens with emphasis on the inflammatory process and disease progression. Histopathology. 2000;37:402–410. doi: 10.1046/j.1365-2559.2000.00990.x. [DOI] [PubMed] [Google Scholar]
- 118.Chang YL, Yao YT, Wang NS, et al. Segmental necrosis of small bronchi after prolonged intakes of Sauropus androgynus in Taiwan. Am J Respir Crit Care Med. 1998;157:594–598. doi: 10.1164/ajrccm.157.2.9704040. [DOI] [PubMed] [Google Scholar]
- 119.Wang JS, Tseng HH, Lai RS. Sauropus bronchiolitis – Reply. Am J Surg Pathol. 1998;22:380–381. doi: 10.1097/00000478-199803000-00017. [DOI] [PubMed] [Google Scholar]
Recreational drugs
- 120.Miller GJ, Ashcroft MT, Beadnell HMSG, et al. The lipoid pneumonia of blackfat tobacco smokers in Guyana. Q J Med. 1971;40:457–470. [PubMed] [Google Scholar]
- 121.Hyashi M, Sornberger GC, Huber GL. A morphometric analysis of the male and female tracheal epithelium after experimental exposure to marijuana smoke. Lab Invest. 1980;42:65–69. [PubMed] [Google Scholar]
- 122.Barbers RG, Gong H, Tashkin DP, et al. Differential examination of bronchoalveolar lavage cells in tobacco cigarette and marijuana smokers. Am Rev Respir Dis. 1987;135:1271–1275. doi: 10.1164/arrd.1987.135.6.1271. [DOI] [PubMed] [Google Scholar]
- 123.Gong H, Fligiel S, Tashkin DP, et al. Tracheobronchial changes in habitual, heavy smokers of marijuana with and without tobacco. Am Rev Respir Dis. 1987;136:142–149. doi: 10.1164/ajrccm/136.1.142. [DOI] [PubMed] [Google Scholar]
- 124.Roth MD, Arora A, Barsky SH, et al. Airway inflammation in young marijuana and tobacco smokers. Am J Respir Crit Care Med. 1998;157:928–937. doi: 10.1164/ajrccm.157.3.9701026. [DOI] [PubMed] [Google Scholar]
- 125.Just N, Delourme J, Delattre C, et al. An unusual cause of patchy ground-glass opacity. Thorax. 2009;64:12. doi: 10.1136/thx.2008.096875. & 74. [DOI] [PubMed] [Google Scholar]
- 126.Wu TC, Tashkin DP, Djahed B, et al. Pulmonary hazards of smoking marijuana as compared with tobacco. N Engl J Med. 1988;318:347–351. doi: 10.1056/NEJM198802113180603. [DOI] [PubMed] [Google Scholar]
- 127.British Lung Foundation A smoking gun? 2003. www.lunguk.org/news/index.html Available online at.
- 128.Fligiel SEG, Roth MD, Kleerup EC, et al. Tracheobronchial histopathology in habitual smokers of cocaine, marijuana, and/or tobacco. Chest. 1997;112:319–326. doi: 10.1378/chest.112.2.319. [DOI] [PubMed] [Google Scholar]
- 129.Aldington S, Williams M, Nowitz M, et al. Effects of cannabis on pulmonary structure, function and symptoms. Thorax. 2007;62:1058–1063. doi: 10.1136/thx.2006.077081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Johnson MK, Smith RP, Morrison D, et al. Large lung bullae in marijuana smokers. Thorax. 2000;55:340–342. doi: 10.1136/thorax.55.4.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tomashefski JF, Jr, Felo JA. The pulmonary pathology of illicit drug and substance abuse. Current Diagnostic Pathology. 2004;10:413–426. [Google Scholar]
- 132.Gill AF. Bong lung: regular smokers of cannabis show relatively distinctive histologic changes that predispose to pneumothorax. American Journal of Surgical Pathology. 2005;29:980–982. doi: 10.1097/01.pas.0000157998.68800.cb. [DOI] [PubMed] [Google Scholar]
- 133.Aldington S, Harwood M, Cox B, et al. Cannabis use and risk of lung cancer: a case-control study. Eur Respir J. 2008;31:280–286. doi: 10.1183/09031936.00065707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Vethanayagam D, Pugsley S, Dunn EJ, et al. Exogenous lipid pneumonia related to smoking weed oil following cadaveric renal transplantation. Can Respir J. 2000;7:338–342. doi: 10.1155/2000/248915. [DOI] [PubMed] [Google Scholar]
- 135.Ettinger NA, Albin RJ. A review of the respiratory effects of smoking cocaine. Am J Med. 1989;87:664–668. doi: 10.1016/s0002-9343(89)80401-2. [DOI] [PubMed] [Google Scholar]
- 136.Jentzen J. Medical complications of cocaine abuse. Am J Clin Pathol. 1993;100:475–476. doi: 10.1093/ajcp/100.5.475. [DOI] [PubMed] [Google Scholar]
- 137.Greenebaum E, Copeland A, Grewal R. Blackened bronchoalveolar lavage fluid in crack smokers – a preliminary study. Am J Clin Pathol. 1993;100:481–487. doi: 10.1093/ajcp/100.5.481. [DOI] [PubMed] [Google Scholar]
- 138.Bailey ME, Fraire AE, Greenberg SD, et al. Pulmonary histopathology in cocaine abusers. Hum Pathol. 1994;25:203–207. doi: 10.1016/0046-8177(94)90279-8. [DOI] [PubMed] [Google Scholar]
- 139.Haim DY, Lippmann ML, Goldberg SK, et al. Pulmonary complications of crack cocaine: a comprehensive review. Chest. 1995;107:233–240. doi: 10.1378/chest.107.1.233. [DOI] [PubMed] [Google Scholar]
- 140.Orriols R, Munoz X, Ferrer J, et al. Cocaine-induced Churg–Strauss vasculitis. Eur Respir J. 1996;9:175–177. doi: 10.1183/09031936.96.09010175. [DOI] [PubMed] [Google Scholar]
- 141.Murray RJ, Smialek JE, Golle M, et al. Pulmonary artery medial hypertrophy in cocaine users without foreign particle microembolization. Chest. 1989;96:1050–1053. doi: 10.1378/chest.96.5.1050. [DOI] [PubMed] [Google Scholar]
- 142.Forrester JM, Steele AW, Waldron JA, et al. Crack lung: an acute pulmonary syndrome with a spectrum of clinical and histopathologic findings. Am Rev Respir Dis. 1990;142:462–467. doi: 10.1164/ajrccm/142.2.462. [DOI] [PubMed] [Google Scholar]
- 143.Tashkin DP, Kleerup EC, Hoh CK, et al. Effects of ‘crack’ cocaine on pulmonary alveolar permeability. Chest. 1997;112:327–335. doi: 10.1378/chest.112.2.327. [DOI] [PubMed] [Google Scholar]
- 144.Perez GMGR, Bragado FG, Gil AMP. Pulmonary hemorrhage and antiglomerular basement membrane antibody-mediated glomerulonephritis after exposure to smoked cocaine (crack): A case report and review of the literature. Pathol Int. 1997;47:692–697. doi: 10.1111/j.1440-1827.1997.tb04443.x. [DOI] [PubMed] [Google Scholar]
- 145.Cooper CB, Bai TR, Heyderman E, et al. Cellulose granulomas in the lungs of a cocaine sniffer. BMJ. 1983;286:2021–2022. doi: 10.1136/bmj.286.6383.2021-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Arnett EN, Battle WE, Russo JV, et al. Intravenous injection of talc-containing drugs intended for oral use. A cause of pulmonary granulomatosis and pulmonary hypertension. Am J Med. 1976;60:711–718. doi: 10.1016/0002-9343(76)90508-8. [DOI] [PubMed] [Google Scholar]
- 147.Gross EM. Autopsy findings in drug addicts. Pathol Annu. 1978;13:35–67. [PubMed] [Google Scholar]
- 148.Pare JAP, Fraser RG, Hogg JC, et al. Pulmonary ‘mainline’ granulomatosis: talcosis of intravenous methadone abuse. Medicine (Baltimore) 1979;58:229–239. [PubMed] [Google Scholar]
- 149.Waller BF, Brownlee WJ, Roberts WC. Self induced pulmonary granulomatosis. A consequence of intravenous injection of drugs intended for oral use. Chest. 1980;78:90–94. doi: 10.1378/chest.78.1.90. [DOI] [PubMed] [Google Scholar]
- 150.Tomashefski JF, Jr, Hirsch CS. The pulmonary vascular lesions of intravenous drug abuse. Hum Pathol. 1980;11:133–145. doi: 10.1016/s0046-8177(80)80130-4. [DOI] [PubMed] [Google Scholar]
- 151.Tomashefski JF, Hirsch CS, Jolly PN. Microcrystalline cellulose pulmonary embolism and granulomatosis. Arch Pathol Lab Med. 1981;105:89–93. [PubMed] [Google Scholar]
- 152.Farber HW, Fairman RP, Glauser FL. Talc granulomatosis: laboratory findings similar to sarcoidosis. Am Rev Respir Dis. 1982;125:258–261. doi: 10.1164/arrd.1982.125.2.258. [DOI] [PubMed] [Google Scholar]
- 153.Rajs J, Harm T, Ormstad K. Postmortem findings of pulmonary lesions of older datum in intravenous drug addicts. A forensic-pathologic study. Virchows Arch A Pathol Anat Histopathol. 1984;402:405–414. doi: 10.1007/BF00734637. [DOI] [PubMed] [Google Scholar]
- 154.Gaine SP, Rubin LJ, Kmetzo JJ, et al. Recreational use of Aminorex and pulmonary hypertension. Chest. 2000;118:1496–1500. doi: 10.1378/chest.118.5.1496. [DOI] [PubMed] [Google Scholar]
7.3. Iatrogenic lung disease
Chapter Contents
- Adverse drug reactions 383
- Reduced respiratory drive 384
- Drug-induced bronchospasm 384
- Oblitereative bronchiolitis 385
- Cytotoxic effects of drugs 385
- Phospholipidosis 387
- Alveolar proteinosis 388
- Eosinophilic pneumonia 388
- Churg–Strauss syndrome 389
- Eosinophilia–myalgia syndrome 389
- Granulomatous alveolitis 389
- Aspiration lesions 389
- Pulmonary hypertension 390
- Pulmonary embolism 390
- Diffuse pulmonary haemorrhage 390
- Opportunistic infection 391
- Metastatic calcification 391
- Carcinoma of the lung 391
- Pleural disease 391
Radiation injury 391
Respirator lung and oxygen toxicity 391
Blood transfusion 392
Cardiopulmonary bypass 392
Complications of cardiac injury 393
Complications of radiofrequency ablation 393
Complications of central vascular cannulation 393
Complications of tracheal manipulations 393
Complications of bronchoscopy 393
Complications of thoracic drainage tubes 394
Pneumonectomy 394
References 394
Adverse drug reactions
It is estimated that 5% of all hospital admissions are due to effects of therapeutic drugs, that 10–18% of inpatients experience a drug reaction and that 3% of deaths in hospital may be related to drug therapy.1, 2, 3, 4 The lungs are often involved in these adverse reactions.
Mechanisms of adverse drug reactions5
The mechanism of an adverse drug reaction may be based on:
-
•
overdosage: toxicity linked to excess dose, or impaired excretion, or both
-
•
side-effect: undesirable pharmacological effect at recommended dose
-
•
interaction with other drugs
and in susceptible individuals only:
-
•
intolerance (representing a low threshold to the normal action of the drug)
-
•
idiosyncrasy (an abnormal reaction based on a genetically determined metabolic or enzymic deficiency)
-
•allergy in the form of any of the four main hypersensitivity reactions:
-
1immediate IgE-mediated hypersensitivity
-
2IgG- or IgM-mediated cytotoxicity
-
3IgG- or IgM-mediated immune complex disease
-
4T-cell-mediated cellular hypersensitivity
-
1
-
•
pseudoallergic reaction: A reaction with the same clinical manifestations as an allergic reaction (e.g. as a result of histamine release) but lacking immunological specificity.
Classification of adverse drug reactions
One classification of adverse drug reactions is that based upon the type of drug (Box 7.3.1 ).6 This is not adopted here but in passing it is worth noting that pharmacists are generally very helpful in supplying details of adverse reactions to specific drugs. Alternatively, information on the long list of potentially pneumotoxic drugs may be obtained at http://www.pneumotox.com. A useful scheme for assessing whether a particular clinical manifestation represents an adverse drug reaction considers previous experience with the drug, alternative aetiological agents, the timing of events, drug levels, and the effect of withdrawing the drug and rechallenge with the drug.7 It is worth bearing in mind that:
-
•
One drug may cause several patterns of disease.
-
•
One pattern of disease may be produced by a variety of drugs.
-
•
A drug reaction may develop long after the drug has been withdrawn.
-
•
A drug reaction may develop suddenly even though the dose of the drug has not been altered.
-
•
Drug effects may be augmented by factors such as age, previous radiotherapy and elevated oxygen levels.
-
•
Drug reactions may be localised.
-
•
Many drugs cross the placenta to affect the fetus.
Box 7.3.1. Principal therapeutic agents known to cause pulmonary disease6.
Chemotherapeutic drugs
Antibiotics
Anti-inflammatory drugs
Immunosuppressive drugs
|
Analgesics
Cardiovascular drugs
Fibrinolytic agents
Inhalants
Intravenous agents
Miscellaneous
|
Particularly common agents are italicized.
An alternative classification of adverse drug reactions, which is more appropriate to pathology practice and which will be followed here, is one based on the pattern of disease. Some pathological patterns of drug-induced lung disease are shown in Table 7.3.1 .
Table 7.3.1.
Pathological patterns of drug-induced lung disease
| Pathological pattern | Prototypic drug(s) |
|---|---|
|
Chemotherapy |
| Phospholipidosis | Amiodarone |
| Alveolar proteinosis | Chemotherapy |
| Eosinophilic pneumonia | Nitrofurantoin |
| Eosinophilia–myalgia syndrome | l-tryptophan |
| Granulomatous alveolitis | Methotrexate |
| Aspiration lesions | Liquid paraffin |
| Pulmonary vascular disease | |
| Hypertension | Aminorex |
| Thromboembolism | Oestrogen |
| Haemorrhage | Anticoagulants |
| Opportunistic infection | Immunosuppressive agents |
| Metastatic calcification | Vitamin D |
| Carcinoma of the lung | Arsenicals |
| Pleural disease | Practolol |
NSIP, non-specific interstitial pneumonia; UIP, usual interstitial pneumonia.
Reduced respiratory drive
Central depression of respiration occurs as a side-effect of barbiturates, morphine and its derivatives, and even mild sedatives, and may be particularly troublesome in patients suffering from chronic obstructive lung disease. Ventilation in such patients may be largely dependent on hypoxic respiratory drive and treatment with oxygen may therefore also have an adverse effect on respiration by lowering the degree of hypoxia and so diminishing the stimulation of the respiratory centre. Peripheral impairment of the respiratory drive may be brought about by aminosides and other antibiotics, while corticosteroids may result in a myopathy affecting the respiratory muscles. Other iatrogenic hazards affecting the peripheral nerves controlling respiration include nerve root disease complicating immunisation and surgical damage to the spinal and phrenic nerves.
Drug-induced bronchospasm
Asthmatic patients are particularly susceptible to exacerbations of their disease by drugs (Box 7.3.2 ). This effect may occur either as a predictable pharmacological side-effect of the drug or as an idiosyncratic response. Examples of the former include β-adrenergicic antagonists and cholinergic agents while examples of the latter include sensitivity to the colouring agent tartrazine, for which reason many manufacturers have eliminated tartrazine from their red, orange and yellow tablets. Allergic bronchoconstriction also forms part of generalised anaphylactic reactions induced by vaccines and antisera and occurs as a localised response to penicillin, iodine-containing contrast media, iron dextran and other medicaments. Bronchospasm may also be initiated by the non-specific irritant effect of inhaling nebulised drugs if they are prepared as a hypotonic solution, a side-effect that is prevented by using isotonic solutions.
Box 7.3.2. Drugs known to cause or aggravate bronchoconstriction.
Non-specific
Hypotonic nebulised preparations
Pharmacological
β-sympathetic antagonists
Cholinergic agents, e.g. pilocarpine
Idiosyncratic
Penicillin
Iodine-containing contrast media
Iron dextran
Tartrazine
Prostaglandin potentiation
Aspirin and other non-steroidal anti-inflammatory agents
Occupational allergy
Penicillin
Cephalosporin
Aspirin-induced asthma has been recognised for many years and more recently several of the newer anti-inflammatory drugs have been found to exacerbate asthma in certain sensitive individuals. The basis for this is uncertain but the likelihood of an individual anti-inflammatory drug provoking an asthmatic response is related to its potency as an inhibitor of prostaglandin cyclooxygenase pathway, resulting in the production of leukotrienes.8, 9, 10
As well as asthma being exacerbated by drugs, the disease has been caused by occupational exposure in the pharmaceutical industry to certain drugs which can be inhaled during manufacture, notably penicillin, cephalosporin, methyldopa, cimetidine and piperazine.
Obliterative bronchiolitis
Obliterative bronchiolitis of the constrictive type has been reported with penicillamine11, 12 and gold13, 14 but in many cases it is possibly the underlying condition rather than the drug that is responsible (see p. 123). This is often rheumatoid disease, which is sometimes complicated by bronchiolitis obliterans whether the patient is under treatment or not.15
Organising pneumonia extending into peripheral bronchioles (see p. 120) may be seen with a variety of drugs but results in a restrictive rather than obstructive lung defect and is to be regarded as a cytotoxic effect of the drug acting primarily at the alveolar level (see below).
Raw Sancropus androgyns taken as a slimming aid causes severe obliterative bronchiolitis (see p. 376).
Cytotoxic effects of drugs
The cytotoxic effects of drugs may be acute or chronic, leading to changes as varied as pulmonary oedema, diffuse alveolar damage, pulmonary haemorrhage and haemosiderosis, organising pneumonia, interstitial pneumonitis and interstitial fibrosis.16, 17 Some of the most severe acute effects are seen with the chemotherapeutic agents used in malignant disease18 but they are also recorded with drugs that are not traditionally thought to be cytotoxic, e.g. desferrioxamine administered as a prolonged intravenous infusion in acute iron poisoning.19
Pulmonary toxicity due to busulphan was first described in 1961,20 and has been the subject of several subsequent studies.21, 22, 23, 24 It remains the mainstay of treatment for chronic myeloid leukaemia. Like other alkylating agents, it acts by cross-linking DNA strands. Clinical estimates of the incidence of pulmonary toxicity vary around 4% but subclinical damage is thought to be much more common. Although not strictly dose-dependent, toxicity is rarely seen with a total cumulative dose of less than 500 mg. Synergy with radiation and other cytotoxic drugs occurs.25 Similar effects have been reported for most cytotoxic agents, particularly bleomycin.26 Pulmonary toxicity is seen less commonly with other alkylating agents, such as cyclophosphamide and melphalan.27, 28, 29, 30
Bleomycin is a cytotoxic antibiotic derived from Streptomyces species. It is widely used in the treatment of neoplasms such as lymphomas and germ cell tumours, and is thought to produce its therapeutic and toxic effects by altering the normal balance between oxidants (active oxygen radicals) and antioxidant systems.26 Bleomycin produces superoxide radicals when incubated with oxygen and iron in vitro. Oxygen enhances its effects,31 a fact well known to anaesthetists who accordingly take care to limit concentrations of inspired oxygen to 30% in patients on bleomycin who are undergoing surgery.32, 33, 34 Radiotherapy and cytotoxic agents such as bleomycin are also synergistic. Bleomycin is preferentially concentrated in the lungs and pulmonary fibrosis can be produced in animals when it is administered intravenously, intraperitoneally or by intratracheal instillation. Electron microscopy shows that the early changes consist of swelling and vesiculation of endothelial cells, interstitial oedema and type I epithelial cell necrosis.35, 36 The reported incidence of bleomycin toxicity varies from 2 to 40% depending on the type of patient being treated and on dosage. In general, toxic effects increase with age and cumulative dose: above a total dose of about 500 units they rise significantly.
The acute morphological changes attributable to drugs include pulmonary oedema and diffuse alveolar damage. Acute pulmonary oedema is seen in heroin addicts who die while injecting themselves intravenously but it is also seen in patients administered a variety of drugs therapeutically, for example hydrochlorothiazide, salicylate, opiates, vinorelbine,and desferrioxamine. The oedema is of the high permeability type (see p. 402), rich in protein, and is occasionally haemorrhagic or accompanied by the hyaline membranes of diffuse alveolar damage.
Diffuse alveolar damage has alveolar epithelial necrosis as its basis (Figure 7.3.1, Figure 7.3.2 ). However, the continuing action of many cytotoxic drugs affects the regeneration process so that atypical type II epithelial cells develop, a characteristic feature that was first described with busulphan and subsequently with bleomycin.21, 37 These two drugs differ chemically but both act (by different mechanisms) on DNA. The atypical cells have abundant deeply eosinophilic or amphophilic cytoplasm and large nuclei, which may be multiple but are usually single. The nuclei measure up to 12 µm and are densely stained throughout or contain either large homogeneous deeply eosinophilic inclusions or clear vacuoles (Fig. 7.3.3 ). Electron microscopy distinguishes the inclusions from nucleoli and shows them to consist of tubular aggregates derived from the internal nuclear membrane.37 Airway epithelium shows similar nuclear changes and often undergoes squamous metaplasia. The presence of such cells in sputum specimens submitted for cytology can lead to a misdiagnosis of malignancy.
Figure 7.3.1.
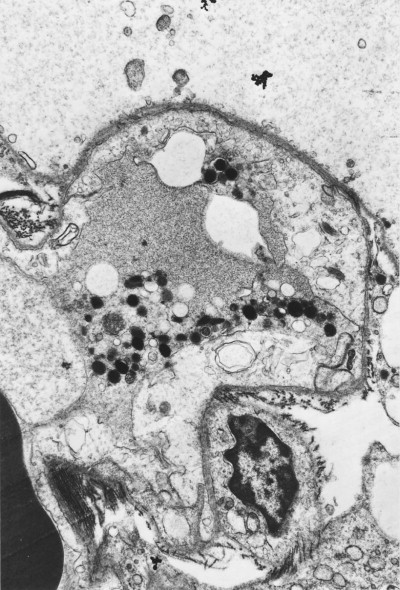
Drug toxicity. A cancer patient administered a cocktail of cytotoxic drugs developed acute respiratory distress and biopsy showed loss of the type I alveolar epithelial cells when examined by electron microscopy. The alveolar basement membrane is bare on its alveolar aspect (above). Capillary endothelial cells show cytoplasmic swelling.
(Courtesy of Miss A Dewar, Brompton, UK.)
Figure 7.3.2.
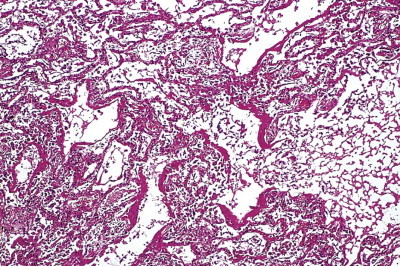
Amiodarone toxicity resulting in diffuse alveolar damage characterised by hyaline membrane formation.
Figure 7.3.3.

Busulphan toxicity. The alveoli are lined by regenerating epithelial cells and the central cell has a very prominent nucleus.
Fibrosis may follow diffuse alveolar damage or develop insidiously, perhaps many years after drug therapy ceased (Fig. 7.3.4 ).38 It may be both interstitial and intra-alveolar. The interstitial component is often accompanied by a non-specific chronic inflammatory infiltrate. The proportions of inflammation, which is potentially reversible, and fibrosis, which when collagenous is irreversible, obviously bear on the prognosis. However, most case reports antedate the recent classification of interstitial pneumonia described in Chapter 6 and it is uncertain how their pathological appearances would now be classified. The majority lack the classic features of usual interstitial pneumonia and fibrotic non-specific interstitial pneumonia. Many show overlapping patterns of intersitital pneumonia and this alone should arouse suspicion that a drug may have been responsible. However, there are drugs that undoubtedly cause a usual interstitial pneumonia pattern, for example the chemotherapeutic agents and nitrofurantoin (Fig. 7.3.5 ), while others, for example the statins, are recorded as having induced a non-specific interstitial pneumonia pattern.39 A drug history is therefore imperative when assessing any patient with diffuse parenchymal lung disease.
Figure 7.3.4.

Fatal pulmonary fibrosis resulting from busulphan therapy, which had been administered for 2 years several years previously.
Figure 7.3.5.
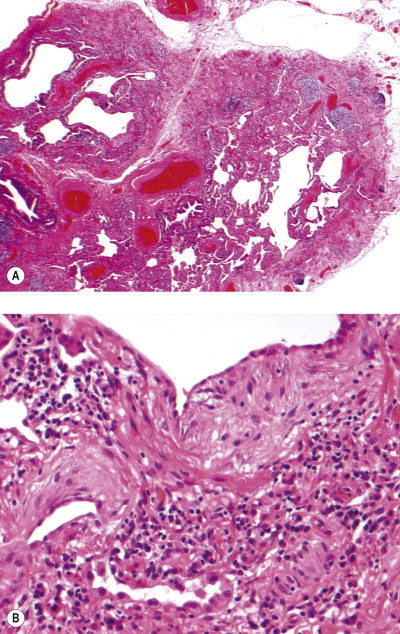
Nitrofurantoin toxicity. The lung shows patchy subpleural fibrosis (A) with fibroblastic foci (B). These are the classic features of usual interstitial pneumonia but lung function improved after withdrawal of the drug.
Organising pneumonia similar to the cryptogenic condition described on page 308, and probably similarly reversible with steroids, has been encountered with a variety of drugs, including amiodarone, sulphasalazine and pencillamine.40 Penicillamine has also been incriminated in the development of both diffuse alveolitis and bronchiolitis obliterans, but both these changes could well be due to the underlying rheumatoid disease for which the pencillamine is administered.15 In busulphan lung there may be an organising intra-alveolar fibrinous exudate,21 which at its most extreme results in irreversible effacement of the alveolar architecture by sheets of loose connective tissue (see p. 148).
Some cytotoxic drugs result in pulmonary changes by more than one mechanism: for example, methotrexate may produce hypersensitivity reactions with granuloma formation41, 42, 43, 44 or pulmonary eosinophilia45 as well as diffuse alveolar damage. Pulmonary toxicity is also occasionally seen in patients undergoing treatment with gold salts for rheumatoid disease: in addition to diffuse alveolar damage, there may be eosinophilia and dermatitis in these cases, again indicating possible hypersensitivity.46 Nitrofurantoin is another example of a drug resulting in a variety of patterns of alveolar injury: diffuse alveolar damage, desquamative interstitial pneumonia, giant cell interstitial pneumonia, organising pneumonia and eosinophilic pneumonia have all been recorded in association with this drug.47, 48, 49
It should also be noted that in patients with neoplastic disease, clinical features suggestive of a pulmonary drug reaction may be due to factors other than drugs. In leukaemic patients, for example, these include direct infiltration of the lungs by leukaemic cells, opportunist infection and, if bone marrow transplantation has been undertaken, the effects of irradiation and possibly graft-versus-host disease.
Phospholipidosis
Phospholipidosis is encountered with drugs such as the antidysrhythmic agent amiodarone,50 which block lysosomal enzymes involved in the breakdown of complex lipids. This leads to their accumulation throughout the body but the effect is most marked in tissues that take up the drug and contain cells rich in lysosomes. The lung fulfils both these requirements through its rich complement of alveolar macrophages. These cells accumulate the enzyme substrate (phospholipid) in their cytoplasm with the result that large foam cells fill the alveoli (Fig. 7.3.6 ). The appearances are those of endogenous lipid pneumonia, similar to that seen in obstructive pneumonitis. However, with amiodarone cytoplasmic vacuolation is also seen in epithelial and interstitial cells. The phospholipid inclusions contained within the vacuoles are particularly well seen in unstained frozen sections viewed by polarised light.51 Identical changes to those induced by amiodarone were seen in the lungs of rats exposed to very high levels of the antidepressant drug iprindole52 and the anorectic drug chlorphentermine.53 These three compounds, iprindole, chlorphentermine and amiodarone, all belong to the amphiphilic group of drugs which block lysosomal phospholipase and sphingomyelinase. Although their pharmacological actions are very different, a molecular homology is apparent (Fig. 7.3.7 ).
Figure 7.3.6.
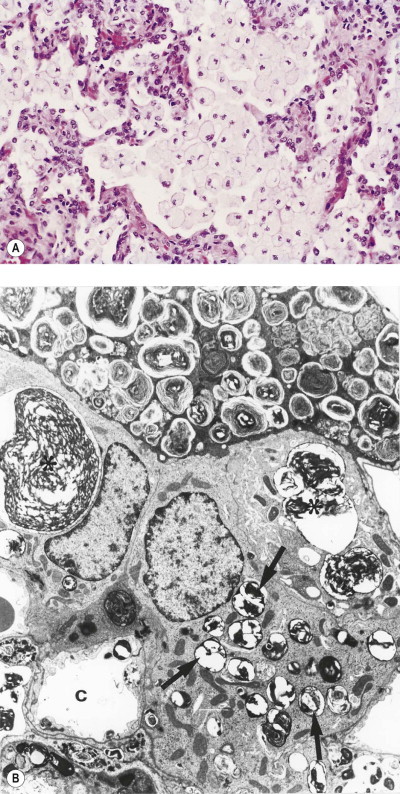
Phospholipidosis due to prolonged amiodarone administration. Amiodarone blocks lysosomal phospholipases, leading to the accumulation of phospholipids in many organs. In the lung this is manifest as endogenous lipid pneumonia. (A) By light microscopy the alveoli are filled with foamy macrophages. (B) Electron micrograph showing an alveolar macrophage (top) packed with osmiophilic lamellar bodies. The macrophage covers several type II pneumocytes that, in addition to their normal surfactant inclusions (arrows), contain large lamellated drug-induced inclusions (asterisks), which are also seen in capillary endothelial cells. C, capillary.
(Reproduced with permission from Costa-Jussa et al. (1984).50)
Figure 7.3.7.
Formulae of several amphiphilic drugs, all of which block lysosomal phospholipases and cause endogenous lipid pneumonia. The pharmacological actions of these drugs differ but a molecular homology is evident.
It is likely that all patients receiving substantial amounts of amiodarone develop phospholipidosis throughout the body, but this is generally well tolerated. Only a minority experience respiratory impairment and in these there is also evidence of pulmonary inflammation and fibrosis, which is possibly mediated immunologically.54 These patients generally have a restrictive lung deficit, the onset of which may be acute or chronic. Bronchoalveolar lavage shows foamy macrophages but these cells indicate exposure to the drug rather than drug toxicity; nor are they specific to amiodarone, being observed on occasion with other drugs. Lymphocytes of suppressor type may also be detected on lavage.54 Histologically, amiodarone toxicity is diagnosed on a combination of phospholipidosis and interstitial pneumonia and fibrosis. Occasionally the hyaline membranes of diffuse alveolar damage are superimposed on the interstitial changes (see Fig. 7.3.2).55, 56, 57 In some patients the fibrosis is intra-alveolar rather than interstitial and the appearances are those of organising pneumonia.58 The process may be localised and mimic a neoplasm radiologically.59, 60
Amiodarone toxicity is probably dose-dependent but there is considerable individual variation in the amount required,61, 62 which appears to be under genetic control.63 Amiodarone toxicity is uncommon in patients taking daily doses of 200 mg or less whereas the prevalence of the disease exceeds 50% in patients treated with doses of 1200 mg/day. Duration is also important: it may require 2–3 years for a patient on 200 mg/day to develop symptoms but only 10 months for one on 400 mg/day. The drug has a long half-life and may take weeks to clear the body completely. Previous pulmonary injury renders the lung unduly sensitive to amiodarone. Acute toxicity has been encountered in patients on moderate doses who have experienced recent or even concomitant pulmonary procedures such as intubation, lobectomy or ventilation with high concentrations of oxygen.64
Alveolar proteinosis
With continued experimental administration of the drug iprindole mentioned above, the phospholipidosis it produced gradually evolved into alveolar proteinosis (more properly called lipoproteinosis; see p. 316),65 but this has not been reported as a drug effect in humans. Alveolar proteinosis has however been recognised in a number of patients receiving chemotherapy for conditions such as leukaemia. The mechanism here is probably based on the cytotoxic action of the drug and the material filling the alveoli may represent the detritus of degenerate alveolar cells rather than excess pulmonary surfactant, as in the primary auto-immune form of alveolar proteinosis.
Eosinophilic pneumonia
Eosinophilic pneumonia, the pathology of which is described on page 461, may be caused by several drugs, including nitrofurantoin, para-aminosalicylic acid, sulphasalazine, phenylbutazone, gold compounds, aspirin and penicillin (see Box 9.3, p. 460).66, 67 It may also follow radiation to the chest.68 The tissue eosinophilia is generally accompanied by a rise in the number of eosinophils in the blood. The clinical picture varies from transient asymptomatic opacities on a chest radiograph to a life-threatening illness with severe respiratory distress and hypoxaemia, so-called acute eosinophilic pneumonia (see p. 462). The reaction is often associated with a florid rash. Withdrawal of the drug may be all that is required to effect resolution but corticosteroids are usually given as they produce a marked improvement.
Churg–Strauss syndrome
This syndrome of necrotising granulomatosis, vasculitis and eosinophilia in asthmatic patients, which is described more fully on page 465, has been reported when leukotriene receptor antagonists have been used to treat asthma. However, it is likely that the syndrome has been merely unmasked by the antileukotriene permitting a reduction in corticosteroid dose rather than representing a direct effect of the antileukotriene.69, 70 Mesalazine has also been implicated in inducing a vasculitis during treatment for inflammatory bowel disease.71
Eosinophilia–myalgia syndrome
The eosinophilia–myalgia syndrome was identified in the USA in 1989 and quickly identified as being due to the ingestion of l-tryptophan from one particular Japanese supplier. Withdrawal of this substance led to the virtual elimination of the disease, but not before 2000 patients had been affected, 1 in 60 fatally.72, 73, 74, 75, 76 Cases were subsequently described in Europe where there were further fatalities.
l-tryptophan is an essential amino acid that is freely available to the public: its purchase does not require a medical prescription. It has been promoted as a dietary supplement and as an agent against insomnia and premenstrual tension. Women in the reproductive years preponderated in the patients affected by the resultant eosinophilia–myalgia. The clinicopathological features of the syndrome are similar to those of the Spanish toxic oil syndrome (see p. 375) and differ more in degree than type. The discovery of an aniline-derived contaminant in the tryptophan-induced condition is a further link connecting these two syndromes.77 An immune basis is suggested by the identification of T lymphocytes activated against fibroblasts in the eosinophilia–myalgia syndrome.78
The illness is a multisystem disorder and besides blood eosinophilia and myalgia there may be arthralgia, fever, rash and involvement of the lungs, liver and central nervous system. As in the toxic oil syndrome, there is fasciitis, wasting and muscle pain associated with blood and tissue eosinophilia. The lungs are affected in 60% of cases. Pulmonary symptoms have included cough, dyspnoea and chest pain. Radiographs have shown diffuse bilateral infiltrates and pulmonary hypertension has been documented in a few cases.79
Histology of the lungs shows an oedematous myxoid intimal thickening affecting small pulmonary blood vessels and a diffuse interstitial lymphocytic and eosinophilic infiltrate.72, 73, 75, 76, 80 These cells may also be seen within the walls of the thickened blood vessels (Fig. 7.3.8 ).72, 76 Massive ingestion of l-tryptophan has resulted in the appearances of an organising pneumonia.81
Figure 7.3.8.
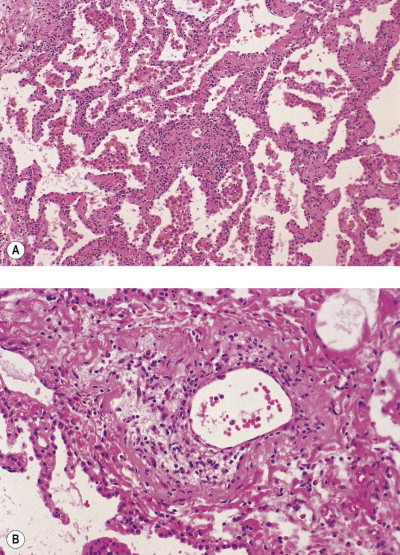
l-tryptophan toxicity. (A) There is a diffuse interstitial infiltrate of lymphocytes with smaller numbers of eosinophils. (B) The same infiltrate involves pulmonary blood vessels.
(Courtesy of Dr TV Colby, Scottsdale, USA.)
Granulomatous alveolitis
As an adverse drug reaction, granulomatous alveolitis is best exemplified by the extrinsic allergic alveolitis of pituitary snuff-takers, but it is also encountered on rare occasions with cytotoxic and other drugs, including methotrexate, bacille Calmette–Guérin (BCG) immunisation, interferons, ciprofloxacin, antiviral therapy and tumour necrosis factor antagonists.42, 43, 44, 48, 82, 83, 84, 85, 86, 87, 88, 89, 90 The histological appearances may suggest extrinsic allergic alveolitis or sarcoidosis but the centriacinar or lymphangitic concentration of these conditions is usually lacking. However, unless an infective agent can be demonstrated the diagnosis generally requires consideration of the clinical and environmental details, including any drug regimen.
Aspiration lesions
Exogenous lipid pneumonia may result from the unintentional aspiration of various fat-based medicaments such as liquid paraffin, oily nose drops and petroleum jelly or of fat-rich dietary supplements in the form of ghee.91, 92, 93, 94, 95, 96, 97 The consumption of liquid paraffin as an aperient is common in some countries and may be taking place without the knowledge of the patient's medical practitioner. Regurgitation and aspiration of ingested oil are especially likely to happen during sleep in the presence of a hiatus hernia or when the oesophagus fails to empty completely into the stomach because of achalasia of the cardia. The aspiration of vegetable oil occurred in the past from the use of menthol in olive oil for the treatment of tuberculous laryngitis, and occasionally from the use of iodinated vegetable oils for bronchography.98, 99, 100, 101 More recently exogenous lipid pneumonia has developed from the constant sucking of lollipops formulated for the administration of the analgesic fentanyl but also containing a stearate component.102 The treatment of epistaxis by nasal packing with paraffin gauze has also led to exogenous lipid pneumonia. The pathology of exogenous lipid pneumonia is described on page 314. Other medicines may also be aspirated unwittingly, for example a ferrous sulphate tablet may cause brown iron staining and necrosis of the bronchus at the point of impact, progressing to bronchial stenosis.103, 104, 105 Distal infection is then likely, as with any foreign body. Barium sulphate aspiration may complicate gastrointestinal radiography.106 Large amounts may impair ventilation but being inert there is no permanent injury to the lungs, although the striking changes are evident on the chest radiograph.
Pulmonary hypertension
An outbreak of pulmonary hypertension affecting many Swiss, Austrian and German patients in the period 1966–68 was probably due to the anorectic drug aminorex,107 which was accordingly withdrawn with regression in the number of new cases. The pathology in these patients was identical to that of primary pulmonary hypertension (see p. 420) and it proved impossible to reproduce the condition in laboratory animals but the epidemiological evidence that aminorex was to blame is very strong. Fenfluramine and phentermine, further anorectic drugs that are chemically similar to aminorex, have also been associated with such plexogenic pulmonary hypertension,108, 109, 110, 111, 112 and with fibroproliferative plaque on the tricuspid valve and pulmonary arteries.113
Pulmonary hypertension due to pulmonary veno-occlusive disease has sometimes complicated the use of cytotoxic chemotherapeutic agents114 or followed bone marrow transplantation.115
Non-steroidal anti-inflammatory agents such as indomethacin and diclofenac cross the placenta and, if given in late pregnancy, may cause premature closure of the ductus arteriosus, resulting in severe neonatal pulmonary hypertension.116, 117
Pulmonary hypertension is a well-recognised association of human immunodeficiency virus (HIV) infection but until recently has been unexplained. Now, however, evidence is emerging that the highly active antiretroviral therapy administered to HIV-positive patients might be responsible for the pulmonary hypertension.118
Pulmonary embolism
The older high-oestrogen contraceptive drugs carried a slight risk of thromboembolism but this is not seen with the newer preparations. Pulmonary thromboembolism has also occurred with a drug-induced lupus syndrome associated with anticardiolipin antibodies. Chemotherapeutic drugs such as mitomycin may cause widespread small-vessel thrombosis resulting in the haemolytic–uraemic (thrombotic microangiopathic) syndrome. There is prominent involvement of pulmonary vessels and patients often suffer from respiratory as well as renal insufficiency, and pulmonary hypertension. The syndrome can develop during treatment or up to several months after the drug has been withdrawn. Pulmonary thromboembolism is also recorded as a complication of immunoglobulin infusion.119
Non-traumatic fat embolism has resulted from the agglutination or ‘creaming’ of fat emulsions administered intravenously as a source of calories to debilitated patients.120, 121, 122, 123, 124, 125 The agglutinated liposomes occlude fine blood vessels throughout the body, causing effects such as priapism, osteonecrosis and pancreatitis. They may be demonstrated in the pulmonary capillaries but the lungs have considerable vascular reserve and it is uncertain what effect the vascular occlusion has on pulmonary function. Agglutination of these fat emulsions is particularly common in severely ill patients and this has been attributed to the elevated blood levels of acute-phase proteins, especially C-reactive protein, that are found in the very ill. The agglutination is also induced by calcium and may be brought about by administering calcium and other mineral supplements through the same venous line as the fat. Once agglutinated, the fat is less soluble and may be demonstrated in paraffin sections. Sudan black is especially useful for this purpose (Fig. 7.3.9 ). Microvascular crystal embolism is a further risk of parenteral nutrition, the crystals representing various calcium salts that may precipitate in the circulation.126
Figure 7.3.9.
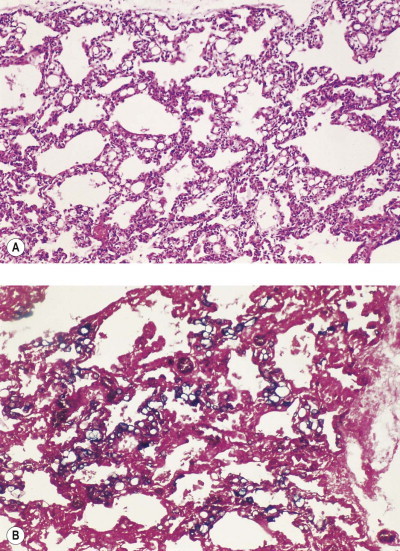
Agglutination of emulsified fat administered intravenously. (A) With haematoxylin and eosin staining, seemingly empty vacuoles appear to occupy the alveolar capillaries. (B) The agglutinated fat has not been dissolved in processing and can be demonstrated by Sudan black.
(Courtesy of Dr G Hulman, Nottinghamshire, UK.)
Transient diffusion abnormalities attributed to oil embolism are very common in patients undergoing lymphangiography but serious respiratory impairment is limited to those patients with pre-existing lung disease or in whom substantial amounts of contrast medium are injected rapidly.127, 128, 129, 130
Other emboli of an iatrogenic nature described in pulmonary arteries include the broken-off ends of intravenous catheters and cannulas, particles from dialysis tubing,131 prosthetic implants of substances such as Teflon and silicone88, 132, 133, 134, 135 and various materials injected to occlude abnormal blood vessels.136, 137
Diffuse pulmonary haemorrhage
Diffuse pulmonary haemorrhage may result from interference with the clotting mechanism by anticoagulants138 or from widespread pulmonary capillaritis, the latter reported in leukaemic patients treated with retinoic acid.139 Pulmonary haemorrhage has also been reported as an idiosyncratic reaction to lymphangiography media140 and as a complication of immunoglobulin infusion,141 while the development of anti-basement membrane antibodies resulting in Goodpasture's syndrome has been attributed to penicillamine.141a
Opportunistic infection
Infection is a common pulmonary hazard in any patient receiving corticosteroids, chemotherapy or any other immunosuppressant drug. Viral, bacterial, fungal and protozoal infections, often in combination, may all develop in the lungs of such patients and tissue reactions may be atypical. Pneumocystis jiroveci, for example, may elicit a granulomatous reaction or cause diffuse alveolar damage rather than the usual foamy alveolar exudate (see p. 226).
Metastatic calcification
Metastatic calcification, described on page 489, may result from any drug causing hypercalcaemia, e.g. high doses of vitamin D, calcium and inorganic phosphate or excessive alkali intake in the treatment of peptic ulceration.
Carcinoma of the lung
Carcinoma of the lung may be promoted by drugs. Arsenicals cause squamous metaplasia of the bronchi and occasionally squamous carcinoma, while peripheral scar cancers, usually adenocarcinomas, have developed in lungs showing fibrosis due to drugs such as busulphan.
Pleural disease
Drugs may result in a variety of pleural diseases.142 Common examples include effusions, chronic inflammation and fibrosis. These are usually encountered in isolation but may be associated with chronic interstitial pneumonia or fibrosis. Sometimes there is also serological evidence of systemic lupus erythematosus: many drugs, including hydantoin, practolol, procainamide, hydralazine and sulphonamides, are associated with the development of a syndrome resembling systemic lupus erythematosus that includes pleural disease. Whether the drugs are directly responsible for the syndrome or merely promote the development of latent natural disease is uncertain.
Ergotamine derivatives such as methysergide and bromocriptine are notable for the production of pleural fibrosis, which is sometimes associated with mediastinal and retroperitoneal fibrosis large amounts or prolonged treatment are generally required to produce this effect.143, 144, 145 In patients given practolol, pleural thickening has become evident several years after the drug was discontinued. This shows the need for a careful drug history in any patient with unexplained pleural fibrosis.
Radiation injury
Reports of radiation-induced lung damage began to appear soon after ionising radiation became widely used in the treatment of malignant disease.146, 147, 148 Despite refinements in radiotherapy techniques it is often impossible to avoid irradiating small areas of lung when treating cancer of the lung, breast, spine, thymus and oesophagus. Parts of the lungs are also included in ‘mantle’ irradiation of mediastinal lymph nodes affected by lymphoma. Occasionally, the whole of both lungs is irradiated, as in the treatment of widespread pulmonary metastases or as part of whole-body irradiation prior to marrow transplantation for the treatment of leukaemia. Radiation pneumonitis, usually localised, is estimated to affect about 8% of patients.149
Therapeutic irradiation is given as divided doses over several weeks in order to minimise damage to adjacent tissue. The effects of such fractionated treatment are cumulative. In the lungs an early exudative phase soon passes and progressive damage becomes apparent only after months or even years.150, 151 The changes are generally confined to the area of lung that is irradiated but are widespread when the whole body is irradiated prior to bone marrow transplantation or there is accidental whole-body irradiation. However, localised irradiation of the lung has been followed by abnormalities in non-irradiated areas. These include bilateral alveolar exudates,152 migratory organising pneumonia affecting both lungs153, 154 and fulminant bilateral interstitial pneumonia.155 The likelihood of lung injury is increased by the simultaneous use of cytotoxic drugs and oxygen therapy.156 Furthermore, chemotherapy following irradiation may result in exacerbation of the injury in areas previously irradiated, a phenomenon termed ‘recall pneumonitis’.157, 158 In the long term, irradiation also results in an increased incidence of lung carcinoma. This was seen in patients given therapeutic irradiation to the spine for ankylosing spondylitis159 and is still encountered on occasion following irradiation for breast cancer.160 The pathogenesis of radiation injury is described on page 146.
Radiation damage to the lung is traditionally separated into fulminant acute injury coming on within days, subacute pneumonitis developing within several weeks (typically 2–3 months) and interstitial fibrosis slowly evolving from the subacute stage or making itself apparent years later. The migratory organising pneumonia referred to above is an unusual further effect, as is chronic eosinophilic pneumonia.68 In the pleura, radiation causes fibrinous effusions and adhesions. Pleural effusion and pulmonary oedema may be augmented by the long-term effects of radiation on the heart.
Fulminant acute injury is an unusual and unexpected effect of therapeutic radiation but one that is likely to come to the attention of the pathologist as an autopsy is often requested. The clinical features are those of acute lung injury and the pathological changes are those of diffuse alveolar damage. The cause is likely to be accidental overdosage, augmentation of the radiation damage by accompanying oxygen therapy or treatment with cytotoxic drugs. Occasionally however these factors can be excluded, in which case the damage has to be ascribed to ‘hypersensitivity’.
Subacute radiation pneumonitis is encountered more commonly. After an interval of about 2–3 months the patient complains of shortness of breath and a non-productive cough. The chest radiograph shows hazy opacification proceeding to more dense consolidation. Lung biopsy shows alveolar and interstitial oedema, possibly with residual hyaline membranes, proliferation of atypical alveolar epithelial cells and interstitial fibroblasts and organising thrombosis. Later, as the process advances, there is widespread fibrosis comparable to that illustrated in Figure 4.24 on page 148 and ultimately dense scarring (Fig. 7.3.10 ).
Figure 7.3.10.

Radiation damage. A dense scar is seen in an area of the lung that had been irradiated previously.
Tracheal and aortic injury may complicate radiation treatment of tracheal lesions, sometimes resulting in an aortotracheal fistula.161
Respirator lung and oxygen toxicity
Patients requiring mechanical ventilation are liable to suffer lung injury in a number of ways. In addition to effects of barotrauma such as pneumothorax and surgical emphysema, they often develop diffuse alveolar damage. The high oxygen tension that is often combined with mechanical ventilation is a major factor162, 163, 164 but mechanical forces other than the high pressures responsible for barotrauma can also contribute to this form of lung injury, notably by resulting in excessive end-expiratory stretch and repeated collapse/recruitment of the alveolar walls.165, 166 Low tidal volume ventilation is therefore a fundamental part of the management of diffuse alveolar damage.
Although oxygen is necessary to life, it is cytotoxic in high concentrations. Severe hyperoxia damages DNA, inhibits cellular proliferation and ultimately kills cells. Its toxicity is thought to be due to the intracellular production of active oxygen radicals, some of which derive from activated neutrophils attracted to the site of injury.167, 168, 169, 170 Under normal conditions most of the oxygen is reduced to water by cytochrome oxidase, and any active radicals produced are eliminated by superoxide dismutase, catalase and other antioxidants. However, these defence mechanisms may prove inadequate when active radicals are produced in excess.171
Problems are likely to arise in clinical practice when lung disease necessitates the concentration of oxygen in the inspired air being raised in order to maintain normal blood levels of oxygen and prevent cerebral hypoxia.172, 173, 174 A ‘safe’ level for oxygen administration is not firmly established and, because of species differences in susceptibility to oxygen, caution is needed in extrapolating from animal studies. However, animal experiments have shown that previous damage to the lungs renders them unduly sensitive to oxygen175, 176 and conversely that prior exposure to high levels of oxygen confers some resistance to subsequent oxygen exposure.177 Clinical studies suggest that less than 50% oxygen (at atmospheric pressure) can be tolerated for long periods without ill effect. Little, if any, serious lung damage results from administration of 100% oxygen for up to 48 hours but concentrations between 50% and 100% carry a risk of damage if this period is exceeded.171, 178 Extracorporeal oxygenation of the blood circumvents the problem but if it is to be prolonged it becomes a major undertaking that poses its own hazards; it is therefore generally reserved for patients who remain hypoxaemic despite other measures.179 Intravenous blood oxygenators are employed to minimise the supplementation of inspired oxygen and partial liquid ventilation utilising perfluorocarbon has also been used.180 Experimentally, disruption of CD40 binding to reduce the release of proinflammatory cytokines has shown promising results in blunting oxygen-induced lung injury.181
None of the morphological changes attributable to oxygen toxicity is specific.174 The earliest ultrastructural change in experimental oxygen poisoning is swelling of endothelial cells, the cytoplasm of which becomes grossly oedematous and vacuolated. Swelling and fragmentation of type I epithelial cells follow and these cells become separated from their basement membrane, which is then coated by thin strands of protein.178 This coating is replaced by proliferating type II cells by the 12th day. With recovery in room air the lungs practically return to normal.182 The full clinical picture of oxygen poisoning is the acute respiratory distress syndrome and the corresponding pathological changes are those of diffuse alveolar damage,174 as described on page 136.
Blood transfusion
Patients with hypovolaemic shock or undergoing major surgery often require massive blood transfusions and this provides another possible cause of pulmonary damage. Although hypervolaemia is the commonest cause of pulmonary oedema after blood transfusion, transfusion-related acute lung injury is more often fatal. Platelet and white cell aggregates are known to develop in stored blood, but a relationship between the number of microaggregates transfused and the degree of respiratory impairment has not been convincingly demonstrated. Leukocyte antibodies are a more likely cause of lung injury in these patients. Such antibodies are often found in multiparous female donors as a result of sensitisation by fetal white cells during pregnancy. Alternatively, the recipient may have developed them during pregnancy or as a result of previous blood transfusions. The implicated antibodies are thought to initiate alveolar capillary damage within hours of transfusion by stimulating granulocyte aggregation.183, 184 Electron microscopy has shown capillary endothelial damage with activated granulocytes in contact with alveolar basement membranes.185
Cardiopulmonary bypass
Cardiopulmonary bypass entails oxygenation and circulation of the blood by extracorporeal devices, so permitting major heart surgery. In the early days of such surgery it was not unusual for patients to develop fatal respiratory insufficiency in the postoperative period. This led to the term ‘postperfusion lung’. Electron microscopic studies showed alveolar damage with degranulation of neutrophils in pulmonary capillaries.186, 187 The syndrome is now less common but infants remain susceptible.188
The most likely explanation is that the synthetic materials with which blood comes into contact during the bypass procedure are able to activate complement. This is mediated by Hageman factor (factor XII) and the alternative pathway. Aggregation of neutrophils leads to their sequestration in the lungs and damage results from their release of lysosomal enzymes and active radicals.188, 189, 190 The process is delayed by hypothermia.189
Complications of cardiac injury
A postcardiac injury syndrome develops after a variety of myocardial or pericardial injuries: it has been described after cardiac surgery (postpericardiotomy syndrome), myocardial infarction (Dressler's syndrome), blunt trauma to the chest, percutaneous puncture of the heart and implantation of a pacemaker.191 There is a delay of anything between a few days and a few months between the cardiac injury and the onset of symptoms, which comprise chest pain, breathlessness, dyspnoea and fever. Examination usually reveals haemorrhagic pleural or pericardial effusions and pulmonary infiltrates. The syndrome usually resolves spontaneously and few pathological studies have therefore been conducted. However, the changes of diffuse alveolar damage have been reported, principally hyaline membrane formation and type II pneumocyte hyperplasia.192 The pathogenesis is obscure. Antibodies reacting with myocardial antigens often develop after cardiac surgery but there is no relationship between these and the development of the syndrome.192, 193, 194
Complications of radiofrequency ablation
This minimally invasive technique is used to destroy lesions as varied as pulmonary metastases and the connection between the left atrium and ectopic foci in the muscular sleeves that surround the terminations of the pulmonary veins (see p. 76). The former may be complicated by pneumothorax and the latter by pulmonary vein stenosis.195, 196
Complications of central vascular cannulation
Central venous cannulation (synonym: catheterisation) is widely used in treating seriously ill patients and may give rise to serious complications. The commonest early complications related to the respiratory tract are caused by local trauma: they include pneumothorax, subcutaneous emphysema, haemothorax and air embolism. Infection occurs later, causing endocarditis, septic emboli and lung abscesses.197 Thrombosis is another common late complication: one autopsy study of patients with central venous lines showed that 15% had major pulmonary emboli and 65% had microscopic emboli in their pulmonary arteries.198 Pulmonary artery cannulation, for example with a Swan–Ganz catheter, may result in pulmonary infarction or any of the traumatic complications of central venous catheterisation mentioned above.
Complications of tracheal manipulations199, 200
Tracheotomy entails a small immediate risk of haemorrhage from damaged subthyroidal arteries, while an endotracheal tube predisposes to infection, as with all foreign bodies. Infection is also promoted by the filtering action of the upper respiratory air passages being bypassed. The latter factor also necessitates humidification of the inspired air and on occasion the humidifier or ventilator has become contaminated so that an aerosol of bacteria is introduced directly into the lower respiratory tract.201 High-pressure ventilation may also lead to interstitial emphysema, pneumothorax and surgical emphysema.
Asphyxia may follow an endotracheal tube becoming blocked by secretions or through it being badly positioned. Secretions need to be constantly removed yet repeated suctioning to achieve this has led to cardiac dysrrythmia and even cardiac arrest.202
If the balloon on the endotracheal tube is too near the tracheostomy it may act as a fulcrum, causing the tip of the tube to press into the tracheal wall. Pressure necrosis and perforation may follow, leading to mediastinitis, tracheo-oesophageal fistula or erosion of a large blood vessel. These are also complications of tracheobronchial laser therapy.
Pressure from the balloon may lead to a tracheal diverticulum and after the tube is withdrawn the trachea may become narrowed at either the site of the incision or further down where the balloon on the tracheal tube causes pressure. Small, shallow ulcers generally heal quickly but deeper ulcers cause necrosis of the tracheal cartilage, and healing is then often accompanied by fibrous stenosis (Fig. 7.3.11 ) or web formation. This results in wheezing and dyspnoea but not before the trachea has narrowed to 30% of its original size, which may take months. Earlier narrowing may be caused by oedema or a fibrinous pseudomembrane.203, 204 Sometimes the stenosis takes the form of a large mass of granulation tissue at the tracheostomy site, a so-called granuloma ball. In children especially, intubation may lead to tracheomalacia so that after the tube is removed the airway collapses.205 Necrotising sialometaplasia is a further complication of prolonged intubation.206 The incidence of such posttracheostomy complications can be minimised by careful placement of the stoma and tube, avoidance of large apertures and high cuff pressures, elimination of heavy connecting equipment and meticulous care of the tracheostomy.
Figure 7.3.11.
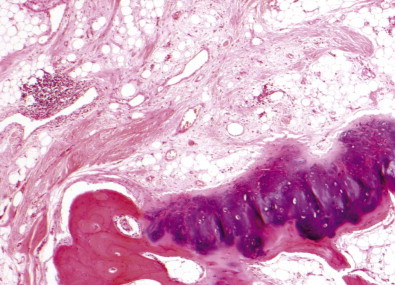
Tracheal stenosis following prolonged intubation. The tracheal wall shows fibrosis while the tracheal cartilage is dysplastic and shows osseous metaplasia, appearances similar to those seen in relapsing polychondritis (compare with Fig. 3.4, p. 95).
Nasogastric feeding tubes may of course lead to aspiration lesions in the lungs and even fatal asphyxia if they are inadvertently allowed to enter the trachea rather than the oesophagus.
Complications of bronchoscopy
Bronchoscopy is generally a safe, almost routine procedure. A review of 23 862 patients who underwent bronchoscopy identified severe complications in 152 (0.637%), of whom three died.207 The fatal cases comprised a 78-year-old with coronary heart disease who developed cardiac arrest and two patients who had had tracheal transplantation for oesophageal cancer and required bronchoscopic laser treatment but died of airway obstruction.
Complications of thoracic drainage tubes
The pleural cavity is intubated in the treatment of pneumothorax and pleural effusions the tube being placed anteriorly to drain air and posteriorly to drain fluid. Complications include laceration of an intercostal artery or vein, the lung, the diaphragm and the heart.
Pneumonectomy208
Pneumonectomy has been practised since the 1930s, since when the mortality associated with this operation has dropped from over 50% to near zero in the best hospitals. Risk factors include underlying lung disease, other medical conditions and more extensive procedures such as pleuropneumonectomy and pneumonectomy combined with chest wall resection.
The anatomical changes that take place soon after pneumonectomy have been extensively studied by radiologists who describe the air-filled postpneumonectomy space gradually filling with fluid and contracting as the mediastinum shifts and the ipsilateral dome of the diaphragm rises.209 Much of the space is filled by fluid within 2 weeks but complete opacification may take up to 6 months. Rapid filling in the immediate postoperative period suggests haemorrhage or chylothorax. However, fluid accumulation is normally rapid after pleuropneumonectomy and may compromise the function of the other lung.
Pathologists conducting autopsies long after the operation may find complete fibrous obliteration of the postpneumonectomy space, coupled with mediastinal shift and elevation of the hemidiaphragm, but often there is persistent brown fluid, which may be clear, cloudy or occasionally purulent.210 The remaining lung is generally enlarged, with its volume greater than predicted. Animal studies have shown that if one lung is excised early in life the enlargement is partly due to enhanced growth but later it represents only dilatation of existing air spaces. Hepatocyte growth factor is thought to be involved in the proliferation of residual lung cells following pneumonectomy.211
Pulmonary complications include those typically seen after other thoracic procedures, such as haemorrhage and infection, and those unique to the postpneumonectomy state, namely anastomotic dehiscence and postpneumonectomy pulmonary oedema. The latter presents as the acute respiratory distress syndrome and represents the early stages of diffuse alveolar damage. It follows severe shift of the heart and mediastinum, which is commoner in children and young adults, in whom the tissues are more compliant.212, 213, 214, 215 The condition complicates up to 4% of lung resections216, 217 and is commoner following excision of the right lung when severe herniation of the left lung into the postpneumonectomy space stretches the trachea and left main bronchus and the latter is compressed between the left pulmonary artery in front and the arch of the aorta behind. In the long term the compression can result in bronchomalacia and postobstructive bronchiectasis. If postpneumonectomy oedema develops the immediate postoperative mortality is high – 50% following pneumonectomy, 42% following lobectomy and 22% following sublobar resections.217, 218 The pathogenesis is probably multifactorial but apart from factors such as fluid overload and high inspired oxygen concentrations there is probably an element of alveolar wall injury, induced by oxidant generation secondary to lung stretching and general surgical trauma.219, 220
References
Adverse drug reactions
- 1.Shapiro S, Slone D, Lewis GP, et al. Fatal drug reactions among medical inpatients. JAMA. 1971;216:467–472. [PubMed] [Google Scholar]
- 2.Roughead EE, Gilbert AL, Primrose JG, et al. Drug-related hospital admissions: a review of Australian studies published 1988–1996. Med J Aust. 1998;168:405–408. doi: 10.5694/j.1326-5377.1998.tb138996.x. [DOI] [PubMed] [Google Scholar]
- 3.Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies (see comments) JAMA. 1998;279:1200–1205. doi: 10.1001/jama.279.15.1200. [DOI] [PubMed] [Google Scholar]
- 4.Wilson RM, Runciman WB, Gibberd RW, et al. The Quality in Australian Health Care Study. Med J Aust. 1995;163:458–471. doi: 10.5694/j.1326-5377.1995.tb124691.x. [DOI] [PubMed] [Google Scholar]
- 5.Vervloet D, Durham S. Adverse reactions to drugs. BMJ. 1998;316:1511–1514. doi: 10.1136/bmj.316.7143.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenow EC, Myers JL, Swensen SJ, et al. Drug-induced pulmonary disease – an update. Chest. 1992;102:239–250. doi: 10.1378/chest.102.1.239. [DOI] [PubMed] [Google Scholar]
- 7.Hutchinson TA, Leventhal JM, Kramer MS, et al. An algorithm for the operational assessment of adverse drug reactions. II. Demonstration of reproducibility and validity. JAMA. 1979;242:633–638. [PubMed] [Google Scholar]
- 8.Van Arsdel PP. Aspirin idiosyncrasy and tolerance. J Allergy Clin Immunol. 1984;73:431–434. doi: 10.1016/0091-6749(84)90351-8. [DOI] [PubMed] [Google Scholar]
- 9.Ameisen JC, Capron A, Joseph M, et al. Aspirin-sensitive asthma: abnormal platelet response to drugs inducing asthmatic attacks; diagnostic and physiopathological implications. Int Arch Allergy Appl Immunol. 1985;78:438–448. doi: 10.1159/000233927. [DOI] [PubMed] [Google Scholar]
- 10.Lee TH. Mechanism of aspirin sensitivity. Am Rev Respir Dis. 1992;145:S34–S36. doi: 10.1164/ajrccm/145.2_Pt_2.S34. [DOI] [PubMed] [Google Scholar]
- 11.Epler GR, Snider GL, Gaensler EA, et al. Bronchiolitis and bronchitis in connective tissue disease. A possible relationship to the use of penicillamine. JAMA. 1979;242:528–532. [PubMed] [Google Scholar]
- 12.Boehler A, Vogt P, Speich R, et al. Bronchiolitis obliterans in a patient with localized scleroderma treated with d-penicillamine. Eur Respir J. 1996;9:1317–1319. doi: 10.1183/09031936.96.09061317. [DOI] [PubMed] [Google Scholar]
- 13.Schwartzman KJ, Bowie DM, Yeadon C, et al. Constrictive bronchiolitis obliterans following gold therapy for psoriatic arthritis. Eur Respir J. 1995;8:2191–2193. doi: 10.1183/09031936.95.08122191. [DOI] [PubMed] [Google Scholar]
- 14.Tomioka H, King TE. Gold-induced pulmonary disease: clinical features, outcome, and differentiation from rheumatoid lung disease. Amer J Respir Crit Care Med. 1997;155:1011–1020. doi: 10.1164/ajrccm.155.3.9116980. [DOI] [PubMed] [Google Scholar]
- 15.Geddes DM, Corrin B, Brewerton DA, et al. Progressive airway obliteration in adults and its association with rheumatoid disease. Q J Med. 1977;46:427–444. [PubMed] [Google Scholar]
- 16.Camus P, Foucher P, Bonniaud P, et al. Drug-induced infiltrative lung disease. Eur Resp J. 2001;18:93S–100S. [PubMed] [Google Scholar]
- 17.Camus P, Bonniaud P, Fanton A, et al. Drug-induced and iatrogenic infiltrative lung disease. Clin Chest Med. 2004;25:479–519. doi: 10.1016/j.ccm.2004.05.006. vi. [DOI] [PubMed] [Google Scholar]
- 18.Camus P, Kudoh S, Ebina M. Interstitial lung disease associated with drug therapy. Br J Cancer. 2004;91:S18–S23. doi: 10.1038/sj.bjc.6602063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tenenbein M, Kowalski S, Sienko A, et al. Pulmonary toxic effects of continuous desferrioxamine administration in acute iron poisoning. Lancet. 1992;339:699–701. doi: 10.1016/0140-6736(92)90598-w. [DOI] [PubMed] [Google Scholar]
- 20.Oliner H, Schwartz R, Rubio R, et al. Interstitial pulmonary fibrosis following busulfan therapy. Am J Med. 1961;31:134–139. doi: 10.1016/0002-9343(61)90229-7. [DOI] [PubMed] [Google Scholar]
- 21.Heard BE, Cooke RA. Busulphan lung. Thorax. 1968;23:187–193. doi: 10.1136/thx.23.2.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burns WA, McFarland W, Matthews MJ. Busulphan-induced pulmonary disease. Report of a case and review of the literature. Am Rev Respir Dis. 1970;101:408–413. doi: 10.1164/arrd.1970.101.3.408. [DOI] [PubMed] [Google Scholar]
- 23.Podoll LN, Winkler SS. Busulfan lung: report of two cases and review of the literature. AJR Am J Roentgenol. 1974;120:151–156. doi: 10.2214/ajr.120.1.151. [DOI] [PubMed] [Google Scholar]
- 24.Elias AD, Mark EJ, Trotman-Dickenson B. A 60-year-old man with pulmonary infiltrates after a bone marrow transplantation – Busulfan pneumonitis. N Engl J Med. 1997;337:480–489. doi: 10.1056/NEJM199708143370708. [DOI] [PubMed] [Google Scholar]
- 25.Cooper JAD, White DA, Matthay RA. Drug-induced pulmonary disease. Am Rev Respir Dis. 1986;133:321–340. doi: 10.1164/arrd.1986.133.2.321. [DOI] [PubMed] [Google Scholar]
- 26.Moseley PL, Shasby DM, Brady M, et al. Lung parenchymal injury induced by bleomycin. Am Rev Respir Dis. 1984;130:1082–1086. doi: 10.1164/arrd.1984.130.6.1082. [DOI] [PubMed] [Google Scholar]
- 27.Slavin RE, Millan JC, Mullins CM. Pathology of high dose intermittent cyclophosphamide therapy. Hum Pathol. 1975;6:693–709. doi: 10.1016/s0046-8177(75)80078-5. [DOI] [PubMed] [Google Scholar]
- 28.Mark GJ, Lehimgar-Zaden A, Ragsdale BD. Cyclophosphamide pneumonitis. Thorax. 1978;33:89–93. doi: 10.1136/thx.33.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taetle R, Dickman PS, Feldman PS. Pulmonary histopathologic changes associated with melphalan therapy. Cancer. 1978;42:1239–1245. doi: 10.1002/1097-0142(197809)42:3<1239::aid-cncr2820420332>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 30.Goucher G, Rowland V, Hawkins J. Melphalan-induced pulmonary interstitial fibrosis. Chest. 1980;77:805–806. doi: 10.1378/chest.77.6.805. [DOI] [PubMed] [Google Scholar]
- 31.Tryka AF, Skornik WA, Godlaski JJ, et al. Potentiation of bleomycin-induced lung injury by exposure to 70% oxygen. Am Rev Respir Dis. 1982;126:1074–1079. doi: 10.1164/arrd.1982.126.6.1074. [DOI] [PubMed] [Google Scholar]
- 32.Goldiner PL, Carlon GC, Cvitkovik E, et al. Factors influencing postoperative morbidity and mortality in patients treated with bleomycin. BMJ. 1978;i:1664–1667. doi: 10.1136/bmj.1.6128.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alan SC, Riddell GS, Butchart EG. Bleomycin therapy and anaesthesia. Anaesthesia. 1981;36:60–63. doi: 10.1111/j.1365-2044.1981.tb08602.x. [DOI] [PubMed] [Google Scholar]
- 34.Hulbert JC, Grossman JE, Cummings KB. Risk factors of anesthesia and surgery in bleomycin-treated patients. J Urol. 1983;130:163–164. doi: 10.1016/s0022-5347(17)51014-9. [DOI] [PubMed] [Google Scholar]
- 35.Adamson IYR, Bowden DH. The pathogenesis of bleomycin-induced pulmonary damage in mice. Am J Pathol. 1974;77:185–197. [PMC free article] [PubMed] [Google Scholar]
- 36.Bedrossian CWM, Greenberg SD, Yawn DH, et al. Experimentally induced bleomycin sulfate toxicity. Arch Pathol Lab Med. 1977;101:248–254. [PubMed] [Google Scholar]
- 37.Gyorkey F, Gyorkey P, Sinkovics JG. Origin and significance of intranuclear tubular inclusions in type II pulmonary alveolar epithelial cells of patients with bleomycin and busulfan toxicity. Ultrastruct Pathol. 1980;1:211–221. doi: 10.3109/01913128009141418. [DOI] [PubMed] [Google Scholar]
- 38.Hasleton PS, O'Driscoll BR, Lynch P, et al. Late BCNU lung – a light and ultrastructural study on the delayed effect of BCNU on the lung parenchyma. J Pathol. 1991;164:31–36. doi: 10.1002/path.1711640106. [DOI] [PubMed] [Google Scholar]
- 39.Lantuejoul S, Brambilla E, Brambilla C, et al. Statin-induced fibrotic nonspecific interstitial pneumonia. Eur Resp J. 2002;19:577–580. doi: 10.1183/09031936.02.00258802. [DOI] [PubMed] [Google Scholar]
- 40.Camus P, Lombard JN, Perrichon M, et al. Bronchiolitis obliterans organizing pneumonia during treatment with acebutolol and amiodarone. Thorax. 1989;44:711–715. doi: 10.1136/thx.44.9.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sastman HD, Matthay RA, Putman CE. Methotrexate-induced pneumonitis (Baltimore) Medicine (Baltimore) 1976;55:371–388. doi: 10.1097/00005792-197609000-00002. [DOI] [PubMed] [Google Scholar]
- 42.White DA, Rankin JA, Stover DE, et al. Methotrexate pneumonitis. Bronchoalveolar lavage findings suggest an immunologic disorder. Am Rev Respir Dis. 1989;139:18–21. doi: 10.1164/ajrccm/139.1.18. [DOI] [PubMed] [Google Scholar]
- 43.Leduc D, Devuyst P, Lheureux P, et al. Pneumonitis complicating low-dose methotrexate therapy for rheumatoid arthritis – discrepancies between lung biopsy and bronchoalveolar lavage findings. Chest. 1993;104:1620–1623. doi: 10.1378/chest.104.5.1620. [DOI] [PubMed] [Google Scholar]
- 44.Imokawa S, Colby TV, Leslie KO, et al. Methotrexate pneumonitis: review of the literature and histopathological findings in nine patients. Eur Resp J. 2000;15:373–381. doi: 10.1034/j.1399-3003.2000.15b25.x. [DOI] [PubMed] [Google Scholar]
- 45.Ginsberg SJ, Comis RL. The pulmonary toxicity of antineoplastic agents. Semin Oncol. 1982;9:34–35. [PubMed] [Google Scholar]
- 46.Scott DL, Bradby GVH, Aitman TJ, et al. Relationship of gold and penicillamine therapy to diffuse interstitial lung disease. Proc Soc Exp Biol Med. 1981;40:136–141. doi: 10.1136/ard.40.2.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bone RC, Wolfe J, Sobonya RE, et al. Desquamative interstitial pneumonia following long-term nitrofurantoin therapy. Am J Med. 1976;60:697–701. doi: 10.1016/0002-9343(76)90505-2. [DOI] [PubMed] [Google Scholar]
- 48.Magee F, Wright JL, Chan N, et al. Two unusual pathological reactions to nitrofurantoin: case reports. Histopathology. 1986;10:701–706. doi: 10.1111/j.1365-2559.1986.tb02523.x. [DOI] [PubMed] [Google Scholar]
- 49.Cameron RJ, Kolbe J, Wilsher ML, et al. Bronchiolitis obliterans organising pneumonia associated with the use of nitrofurantoin. Thorax. 2000;55:249–251. doi: 10.1136/thorax.55.3.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Costa-Jussa FR, Corrin B, Jacobs JM. Amiodarone lung toxicity: a human and experimental study. J Pathol. 1984;144:73–79. doi: 10.1002/path.1711440202. [DOI] [PubMed] [Google Scholar]
- 51.Jacobson W, Stewart S, Gresham GA, et al. Effect of amiodarone on the lung shown by polarized light microscopy. Arch Pathol Lab Med. 1997;121:1269–1271. [PubMed] [Google Scholar]
- 52.Vijeyaratnam GS, Corrin B. Fine structural alterations in the lungs of iprindole-treated rats. J Pathol. 1974;114:233–239. doi: 10.1002/path.1711140408. [DOI] [PubMed] [Google Scholar]
- 53.Heath D, Smith P, Hasleton PS. Effects of chlorphentermine on the rat lung. Thorax. 1973;28:551–558. doi: 10.1136/thx.28.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Akoun GM, Gauthier-Rahman S, Milleron BJ, et al. Amiodarone-induced hypersensitivity pneumonitis. Evidence of an immunological cell-mediated mechanism. Chest. 1984;85:133–135. doi: 10.1378/chest.85.1.133. [DOI] [PubMed] [Google Scholar]
- 55.Darmanata JI, van Zandwijk N, Duren DR, et al. Amiodarone pneumonitis: three further cases with a review of published reports. Thorax. 1984;39:57–64. doi: 10.1136/thx.39.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dean PJ, Groshart KD, Porterfield JG, et al. Amiodarone-associated pulmonary toxicity. A clinical and pathologic study of eleven cases. Am J Clin Pathol. 1987;87:7–13. doi: 10.1093/ajcp/87.1.7. [DOI] [PubMed] [Google Scholar]
- 57.Myers JL, Kennedy JI, Plumb VJ. Amiodarone lung: pathologic findings in clinically toxic patients. Hum Pathol. 1987;18:349–354. doi: 10.1016/s0046-8177(87)80164-8. [DOI] [PubMed] [Google Scholar]
- 58.Oren S, Turkot S, Golzman B, et al. Amiodarone-induced bronchiolitis obliterans organizing pneumonia (BOOP) Respir Med. 1996;90:167–169. doi: 10.1016/s0954-6111(96)90159-6. [DOI] [PubMed] [Google Scholar]
- 59.Kimura T, Kuramochi S, Katayama T, et al. Amiodarone-related pulmonary mass and unique membranous glomerulonephritis in a patient with valvular heart disease: Diagnostic pitfall and new findings. Pathol Int. 2008;58:657–663. doi: 10.1111/j.1440-1827.2008.02286.x. [DOI] [PubMed] [Google Scholar]
- 60.Ruangchira-Urai R, Colby TV, Klein J, et al. Nodular amiodarone lung disease. Am J Surg Pathol. 2008;32:1654–1660. doi: 10.1097/PAS.0b013e31816d1cbc. [DOI] [PubMed] [Google Scholar]
- 61.Polkey MI, Wilson POG, Rees PJ. Amiodarone pneumonitis: no safe dose. Respir Med. 1995;89:233–235. doi: 10.1016/0954-6111(95)90254-6. [DOI] [PubMed] [Google Scholar]
- 62.Hargreaves MR, Benson MK. Amiodarone pneumonitis: no safe dose. Respir Med. 1996;90:119. doi: 10.1016/s0954-6111(96)90212-7. [DOI] [PubMed] [Google Scholar]
- 63.Wilson BD, Lippmann ML. Susceptibility to amiodarone-induced pulmonary toxicity: relationship to the uptake of amiodarone by isolated lung cells. Lung. 1996;174:31–41. doi: 10.1007/BF00167949. [DOI] [PubMed] [Google Scholar]
- 64.Handschin AE, Lardinois D, Schneiter D, et al. Acute amiodarone-induced pulmonary toxicity following lung resection. Respiration. 2003;70:310–312. doi: 10.1159/000072016. [DOI] [PubMed] [Google Scholar]
- 65.Vijeyaratnam GS, Corrin B. Pulmonary alveolar proteinosis developing from desquamative interstitial pneumonia in long term toxicity studies of iprindole in the rat. Virchows Arch A Pathol Anat Histopathol. 1973;358:1–10. doi: 10.1007/BF00555550. [DOI] [PubMed] [Google Scholar]
- 66.Allen JN, Davis WB. Eosinophilic lung diseases. Am J Respir Crit Care Med. 1994;150:1423–1438. doi: 10.1164/ajrccm.150.5.7952571. [DOI] [PubMed] [Google Scholar]
- 67.Tanigawa K, Sugiyama K, Matsuyama H, et al. Mesalazine-induced eosinophilic pneumonia. Respiration. 1999;66:69–72. doi: 10.1159/000029341. [DOI] [PubMed] [Google Scholar]
- 68.Cottin V, Frognier R, Monnot H, et al. Chronic eosinophilic pneumonia after radiation therapy for breast cancer. Eur Resp J. 2004;23:9–13. doi: 10.1183/09031936.03.00071303. [DOI] [PubMed] [Google Scholar]
- 69.Wechsler ME, Finn D, Gunawardena D, et al. Churg–Strauss syndrome in patients receiving montelukast as treatment for asthma. Chest. 2000;117:708–713. doi: 10.1378/chest.117.3.708. [DOI] [PubMed] [Google Scholar]
- 70.LeGall C, Pham S, Vignes S, et al. Inhaled corticosteroids and Churg–Strauss syndrome: a report of five cases. Eur Resp J. 2000;15:978–981. doi: 10.1034/j.1399-3003.2000.15e29.x. [DOI] [PubMed] [Google Scholar]
- 71.Faller M, Gasser B, Massard G, et al. Pulmonary migratory infiltrates and pachypleuritis in a patient with Crohn's disease. Respiration. 2000;67:459–463. doi: 10.1159/000029550. [DOI] [PubMed] [Google Scholar]
- 72.Tazelaar HD, Myers JL, Drage CW, et al. Pulmonary disease associated with l-tryptophan-induced eosinophilic myalgia syndrome. Clinical and pathologic features. Chest. 1990;97:1032–1036. doi: 10.1378/chest.97.5.1032. [DOI] [PubMed] [Google Scholar]
- 73.Flannery MT, Wallach PM, Espinoza LR, et al. A case of the eosinophilia-myalgia syndrome associated with use of an l-tryptophan product. Ann Intern Med. 1990;112:300–301. doi: 10.7326/0003-4819-112-4-300. [DOI] [PubMed] [Google Scholar]
- 74.Belongia EA, Hedberg CW, Gleich GJ, et al. An investigation of the cause of the eosinophilia–myalgia syndrome associated with tryptophan use. N Engl J Med. 1990;323:357–365. doi: 10.1056/NEJM199008093230601. [DOI] [PubMed] [Google Scholar]
- 75.Herrick MK, Chang Y, Horoupian DS, et al. l-tryptophan and the eosinophilia–myalgia syndrome: pathologic findings in eight patients. Hum Pathol. 1991;22:12–21. doi: 10.1016/0046-8177(91)90055-t. [DOI] [PubMed] [Google Scholar]
- 76.Winkelmann RK, Connolly SM, Quimby SR, et al. Histopathologic features of the l-tryptophan-related eosinophilia–myalgia (fasciitis) syndrome. Mayo Clin Proc. 1991;66:457–463. doi: 10.1016/s0025-6196(12)62384-2. [DOI] [PubMed] [Google Scholar]
- 77.Mayeno AN, Belongia EA, Lin F, et al. 3-(phenylamino)alanine, a novel aniline-derived amino acid associated with the eosinophilia–myalgia syndrome – a link to the toxic oil syndrome. Mayo Clin Proc. 1992;67:1134–1139. doi: 10.1016/s0025-6196(12)61142-2. [DOI] [PubMed] [Google Scholar]
- 78.Illa I, Dinsmore S, Dalakas MC. Immune-mediated mechanisms and immune activation of fibroblasts in the pathogenesis of eosinophilia-myalgia syndrome induced by l-tryptophan. Hum Pathol. 1993;24:702–709. doi: 10.1016/0046-8177(93)90005-2. [DOI] [PubMed] [Google Scholar]
- 79.Cheng TO. Pulmonary hypertension in patients with eosinophilia–myalgia syndrome or toxic oil syndrome. Mayo Clin Proc. 1993;68:823. doi: 10.1016/s0025-6196(12)60645-4. [DOI] [PubMed] [Google Scholar]
- 80.Tazelaar HD, Myers JL, Strickler JG, et al. Tryptophan-induced lung disease – an immunophenotypic, immunofluorescent, and electron microscopic study. Mod Pathol. 1993;6:56–60. [PubMed] [Google Scholar]
- 81.Mar KE, Sen P, Tan K, et al. Bronchiolitis obliterans organizing pneumonia associated with massive l-tryptophan ingestion. Chest. 1993;104:1924–1926. doi: 10.1378/chest.104.6.1924. [DOI] [PubMed] [Google Scholar]
- 82.Clarysse AM, Cathey WJ, Cartwright GE, et al. Pulmonary disease complicating intermittent therapy with methotrexate. JAMA. 1969;209:1861–1864. [PubMed] [Google Scholar]
- 83.Hasan FM, Mark EJ. Case records of the Massachusetts General Hospital. A 28-year-old man with increasing dyspnea, dry cough, and fever after chemotherapy for lymphoma. N Engl J Med. 1990;323:737–747. doi: 10.1056/NEJM199009133231108. [DOI] [PubMed] [Google Scholar]
- 84.Pietropaoli A, Modrak J, Utell M. Interferon-alpha therapy associated with the development of sarcoidosis. Chest. 1999;116:569–572. doi: 10.1378/chest.116.2.569. [DOI] [PubMed] [Google Scholar]
- 85.Ayers MM, Jeffery PK. Proliferation and differentiation in mammalian airway epithelium. Eur Respir J. 1988;1:58–80. [PubMed] [Google Scholar]
- 86.Naccache JM, Antoine M, Wislez M, et al. Sarcoid-like pulmonary disorder in human immunodeficiency virus-infected patients receiving antiretroviral therapy. Amer J Respir Crit Care Med. 1999;159:2009–2013. doi: 10.1164/ajrccm.159.6.9807152. [DOI] [PubMed] [Google Scholar]
- 87.Steiger D, Bubendorf L, Oberholzer M, et al. Ciprofloxacin-induced acute interstitial pneumonitis. Eur Resp J. 2004;23:172–174. doi: 10.1183/09031936.03.00057903. [DOI] [PubMed] [Google Scholar]
- 88.Vavricka SR, Wettstein T, Speich R, et al. Pulmonary granulomas after tumour necrosis factor alpha antagonist therapy. Thorax. 2003;58:278–279. doi: 10.1136/thorax.58.3.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.O'Shea FD, Marras TK, Inman RD. Pulmonary sarcoidosis developing during infliximab therapy. Arthritis Rheum. 2006;55:978–981. doi: 10.1002/art.22351. [DOI] [PubMed] [Google Scholar]
- 90.Kudrin A, Chilvers ER, Ginawi A, et al. Sarcoid-like granulomatous disease following etanercept treatment for RA. J Rheumatol. 2007;34:648–649. [PubMed] [Google Scholar]
- 91.Elston CW. Pneumonia due to liquid paraffin: with chemical analysis. Arch Dis Child. 1966;41:428–434. doi: 10.1136/adc.41.218.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Salm R, Hughes EW. A case of chronic paraffin pneumonitis. Thorax. 1970;25:762–768. doi: 10.1136/thx.25.6.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fox B. Liquid paraffin pneumonia – with chemical analysis and electron microscopy. Virchows Arch A Pathol Anat Histopathol. 1979;382:339–346. doi: 10.1007/BF00430409. [DOI] [PubMed] [Google Scholar]
- 94.Corrin B, Crocker PR, Hood BJ, et al. Paraffinoma confirmed by infrared spectrophotometry. Thorax. 1987;42:389–390. doi: 10.1136/thx.42.5.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wagner JC, Adler DI, Fuller DN. Foreign body granulomata of the lungs due to liquid paraffin. Thorax. 1955;10:157–170. doi: 10.1136/thx.10.2.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brown AC, Slocum PC, Putthoff SL, et al. Exogenous lipoid pneumonia due to nasal application of petroleum jelly. Chest. 1994;105:968–969. doi: 10.1378/chest.105.3.968. [DOI] [PubMed] [Google Scholar]
- 97.Annobil SH, Morad NA, Khurana P, et al. Reaction of human lungs to aspirated animal fat (ghee): a clinicopathological study. Virchows Arch. 1995;426:301–305. doi: 10.1007/BF00191368. [DOI] [PubMed] [Google Scholar]
- 98.Rayl JE. Clinical reactions following bronchography. Ann Otol Rhinol Laryngol. 1965;74:1120–1132. doi: 10.1177/000348946507400418. [DOI] [PubMed] [Google Scholar]
- 99.Felton WL. The reaction of pulmonary tissue to lipiodol. J Thorac Surg. 1953;25:530–542. [PubMed] [Google Scholar]
- 100.Greenberg SD, Spjut HJ, Hallman GL. Experimental study of bronchographic media on lung. Arch Otolaryngol Head Neck Surg. 1966;83:276–282. doi: 10.1001/archotol.1966.00760020278019. [DOI] [PubMed] [Google Scholar]
- 101.Felton WL. A method for the identification of lipiodol in tissue sections. Lab Invest. 1952;1:364–367. [PubMed] [Google Scholar]
- 102.Mathai SK, Rubinowitz AN, Homer RJ, et al. Of Lungs, Lipids, and Lollipops. Chest. 2009;136:1420–1423. doi: 10.1378/chest.08-2133. [DOI] [PubMed] [Google Scholar]
- 103.Lamaze R, Trechot P, Martinet Y. Bronchial necrosis and granuloma induced by the aspiration of a tablet of ferrous sulphate. Eur Respir J. 1994;7:1710–1711. doi: 10.1183/09031936.94.07091710. [DOI] [PubMed] [Google Scholar]
- 104.Lee P, Culver DA, Farver C, et al. Syndrome of iron pill aspiration. Chest. 2002;121:1355–1357. doi: 10.1378/chest.121.4.1355. [DOI] [PubMed] [Google Scholar]
- 105.Sundar KM, Elliott CG, Thomsen GE. Tetracycline aspiration – Case report and review of the literature. Respiration. 2001;68:416–419. doi: 10.1159/000050538. [DOI] [PubMed] [Google Scholar]
- 106.Tamm I, Kortsik C. Severe barium sulfate aspiration into the lung: Clinical presentation, prognosis and therapy. Respiration. 1999;66:81–84. doi: 10.1159/000029344. [DOI] [PubMed] [Google Scholar]
- 107.Kay JM, Smith P, Heath D. Aminorex and the pulmonary circulation. Thorax. 1971;26:262–270. doi: 10.1136/thx.26.3.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Douglas JG, Munro JF, Kitchin AH, et al. Pulmonary hypertension and fenfluramine. BMJ. 1981;283:881–883. doi: 10.1136/bmj.283.6296.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McMurray J, Bloomfield P, Miller HC. Irreversible pulmonary hypertension after treatment with fenfluramine. BMJ. 1986;292:239–240. doi: 10.1136/bmj.292.6515.239-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kay JM. Dietary pulmonary hypertension. Thorax. 1994;49:S33–S38. doi: 10.1136/thx.49.suppl.s33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Abenhaim L, Moride Y, Brenot F, et al. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. N Engl J Med. 1996;335:609–616. doi: 10.1056/NEJM199608293350901. [DOI] [PubMed] [Google Scholar]
- 112.Mark EJ, Patalas ED, Chang HT, et al. Fatal pulmonary hypertension associated with short-term use of fenfluramine and phentermine. N Engl J Med. 1997;337:602–606. doi: 10.1056/NEJM199708283370904. [DOI] [PubMed] [Google Scholar]
- 113.Tomita T, Zhao Q. Autopsy findings of heart and lungs in a patient with primary pulmonary hypertension associated with use of fenfluramine and phentermine. Chest. 2002;121:649–652. doi: 10.1378/chest.121.2.649. [DOI] [PubMed] [Google Scholar]
- 114.Lombard CM, Churg A, Winokur S. Pulmonary veno-occlusive disease following therapy for malignant neoplasms. Chest. 1987;92:871–876. doi: 10.1378/chest.92.5.871. [DOI] [PubMed] [Google Scholar]
- 115.Williams LM, Fussell S, Veith RW, et al. Pulmonary veno-occlusive disease in an adult following bone marrow transplantation: case report and review of the literature. Chest. 1996;109:1388–1391. doi: 10.1378/chest.109.5.1388. [DOI] [PubMed] [Google Scholar]
- 116.Moise KJ, Jr, Huhta JC, Sharif DS, et al. Indomethacin in the treatment of premature labor. Effects on the fetal ductus arteriosus. N Engl J Med. 1988;319:327–331. doi: 10.1056/NEJM198808113190602. [DOI] [PubMed] [Google Scholar]
- 117.Auer M, Brezinka C, Eller P, et al. Prenatal diagnosis of intrauterine premature closure of the ductus arteriosus following maternal diclofenac application. Ultrasound Obstet Gynecol. 2004;23:513–516. doi: 10.1002/uog.1038. [DOI] [PubMed] [Google Scholar]
- 118.Wang X, Chai H, Lin PH, et al. Roles and mechanisms of human immunodeficiency virus protease inhibitor ritonavir and other anti-human immunodeficiency virus drugs in endothelial dysfunction of porcine pulmonary arteries and human pulmonary artery endothelial cells. Am J Pathol. 2009;174:771–781. doi: 10.2353/ajpath.2009.080157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Alliot C, Rapin JP, Besson M, et al. Pulmonary embolism after intravenous immunoglobulin. J R Soc Med. 2001;94:187–188. doi: 10.1177/014107680109400412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Barson AJ, Chiswick ML, Doig CM. Fat embolism in infancy after intravenous fat infusions. Arch Dis Child. 1978;53:218–223. doi: 10.1136/adc.53.3.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Levene MI, Wigglesworth JS, Desai R. Pulmonary fat accumulation after Intralipid infusion in the preterm infant. Lancet. 1980;II:815–818. doi: 10.1016/s0140-6736(80)90170-1. [DOI] [PubMed] [Google Scholar]
- 122.Hulman G, Levene M. Intralipid microemboli. Arch Dis Child. 1986;61:702–703. doi: 10.1136/adc.61.7.702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hulman G. The pathogenesis of fat embolism. J Pathol. 1995;176:3–9. doi: 10.1002/path.1711760103. [DOI] [PubMed] [Google Scholar]
- 124.Kitchell CC, Balogh K. Pulmonary lipid emboli in association with long-term hyperalimentation. Hum Pathol. 1986;17:83–85. doi: 10.1016/s0046-8177(86)80158-7. [DOI] [PubMed] [Google Scholar]
- 125.Lekka ME, Liokatis S, Nathanail C, et al. The impact of intravenous fat emulsion administration in acute lung injury. Am J Resp Crit Care Med. 2004;169:638–644. doi: 10.1164/rccm.200305-620OC. [DOI] [PubMed] [Google Scholar]
- 126.Reedy JS, Kuhlman JE, Voytovich M. Microvascular pulmonary emboli secondary to precipitated crystals in a patient receiving total parenteral nutrition – A case report and description of the high-resolution CT findings. Chest. 1999;115:892–895. doi: 10.1378/chest.115.3.892. [DOI] [PubMed] [Google Scholar]
- 127.Gough JH, Gough MH, Thomas ML. Pulmonary complications following lymphangiography with a note on technique. Br J Radiol. 1964;37:416–421. doi: 10.1259/0007-1285-37-438-416. [DOI] [PubMed] [Google Scholar]
- 128.Fraimow W, Wallace S, Lewis P, et al. Changes in pulmonary function due to lymphangiography. Radiology. 1965;85:231–241. doi: 10.1148/85.2.231. [DOI] [PubMed] [Google Scholar]
- 129.Davidson JW. Pulmonary complications of lymphangiography. N Engl J Med. 1971;285:237. doi: 10.1056/NEJM197107222850421. [DOI] [PubMed] [Google Scholar]
- 130.Silvestri RC, Hyseby JS, Rughani I, et al. Respiratory distress syndrome from lymphangiography contrast medium. Am Rev Respir Dis. 1980;122:543–549. doi: 10.1164/arrd.1980.122.4.543. [DOI] [PubMed] [Google Scholar]
- 131.Leong AS-Y, Disney APS, Gove DW. Spallation and migration of silicone from blood-pump tubing in patients on hemodialysis. N Engl J Med. 1982;306:135–140. doi: 10.1056/NEJM198201213060303. [DOI] [PubMed] [Google Scholar]
- 132.Chastre J, Basset F, Viau F, et al. Acute pneumonitis after subcutaneous injections of silicone in transsexual men. N Engl J Med. 1983;308:764–767. doi: 10.1056/NEJM198303313081307. [DOI] [PubMed] [Google Scholar]
- 133.Robinson MJ, Nestor M, Rywlin AM. Pulmonary granulomas secondary to embolic prosthetic valve material. Hum Pathol. 1981;12:759–762. doi: 10.1016/s0046-8177(81)80181-5. [DOI] [PubMed] [Google Scholar]
- 134.Mittleman RE, Marraccini JV. Pulmonary Teflon granulomas following periurethral Teflon injection for urinary incontinence. Arch Pathol Lab Med. 1983;107:611–612. [PubMed] [Google Scholar]
- 135.Lai YF, Chao TY, Wong SL. Acute pneumonitis after subcutaneous injections of silicone for augmentation mammaplasty. Chest. 1994;106:1152–1155. doi: 10.1378/chest.106.4.1152. [DOI] [PubMed] [Google Scholar]
- 136.Fairfax AJ, Ball J, Batten JC, et al. A pathological study following bronchial artery embolization for haemoptysis in cystic fibrosis. Br J Dis Chest. 1980;74:345–352. [PubMed] [Google Scholar]
- 137.Coard K, Silver MD, Perkins G, et al. Isobutyl-2-cyanoacrylate pulmonary emboli associated with occlusive embolotherapy of cerebral arteriovenous malformations. Histopathology. 1984;8:917–926. doi: 10.1111/j.1365-2559.1984.tb02410.x. [DOI] [PubMed] [Google Scholar]
- 138.Lena H, Desrues B, Quinquenel ML, et al. Hemorragie alveolaire diffuse secondaire a l’utilisation d’anticoagulants oraux. Rev Mal Resp. 1995;12:496–498. [PubMed] [Google Scholar]
- 139.Nicolls MR, Terada LS, Tuder RM, et al. Diffuse alveolar hemorrhage with underlying pulmonary capillaritis in the retinoic acid syndrome. Amer J Respir Crit Care Med. 1998;158:1302–1305. doi: 10.1164/ajrccm.158.4.9709085. [DOI] [PubMed] [Google Scholar]
- 140.Wiertz LM, Gagnon JH, Anthonisen NR. Intrapulmonary hemorrhage with anemia after lymphangiography. N Engl J Med. 1971;285:1364–1365. doi: 10.1056/NEJM197112092852409. [DOI] [PubMed] [Google Scholar]
- 141.Kalra S, Bell MR, Rihal CS. Alveolar hemorrhage as a complication of treatment with abciximab. Chest. 2001;120:126–131. doi: 10.1378/chest.120.1.126. [DOI] [PubMed] [Google Scholar]
- Sternlick I, Bennett B, Scheinberg JH. D-penicillamine induced Goodpasture's syndrome in Wilson's disease. Ann Intern Med 82:673–6. [DOI] [PubMed]
- 142.Morelock SY, Sahn SA. Drugs and the pleura. Chest. 1999;116:212–221. doi: 10.1378/chest.116.1.212. [DOI] [PubMed] [Google Scholar]
- 143.Pfitzenmeyer P, Foucher P, Dennewald G, et al. Pleuropulmonary changes induced by ergoline drugs. Eur Respir J. 1996;9:1013–1019. doi: 10.1183/09031936.96.09051013. [DOI] [PubMed] [Google Scholar]
- 144.Comet R, Domingo C, Such JJ, et al. Pleuropulmonary disease as a side-effect of treatment with bromcriptine. Resp Med. 1998;92:1172–1174. doi: 10.1016/s0954-6111(98)90415-2. [DOI] [PubMed] [Google Scholar]
- 145.Danoff SK, Grasso ME, Terry PB, et al. Pleuropulmonary disease due to pergolide use for restless legs syndrome. Chest. 2001;120:313–316. doi: 10.1378/chest.120.1.313. [DOI] [PubMed] [Google Scholar]
Radiation injury
- 146.Hines LE. Fibrosis of the lung following roentgen-ray treatments for tumor. JAMA. 1922;79:720–722. [Google Scholar]
- 147.Warren S, Spencer J. Radiation reaction in the lung. AJR Am J Roentgenol. 1940;43:682–701. [Google Scholar]
- 148.Warren S, Gates O. Radiation pneumonitis: experimental and pathologic observations. Arch Pathol Lab Med. 1940;30:440–460. [Google Scholar]
- 149.Roach M, III, Gandara DR, Yuo HS, et al. Radiation pneumonitis following combined modality therapy for lung cancer: analysis of prognostic factors. J Clin Oncol. 1995;13:2606–2612. doi: 10.1200/JCO.1995.13.10.2606. [DOI] [PubMed] [Google Scholar]
- 150.Gross NJ. The pathogenesis of radiation-induced lung damage. Lung. 1981;159:115–125. doi: 10.1007/BF02713907. [DOI] [PubMed] [Google Scholar]
- 151.Chandler Smith J. Radiation pneumonitis: a review. Am Rev Respir Dis. 1963;87:647–655. doi: 10.1164/arrd.1963.87.5.647. [DOI] [PubMed] [Google Scholar]
- 152.Fulkerson WJ, McLendon RE, Proznitz LR. Adult respiratory distress syndrome after limited thoracic radiotherapy. Cancer. 1986;57:1941–1946. doi: 10.1002/1097-0142(19860515)57:10<1941::aid-cncr2820571009>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 153.Crestani B, Kambouchner M, Soler P, et al. Migratory bronchiolitis obliterans organizing pneumonia after unilateral radiation therapy for breast carcinoma. Eur Respir J. 1995;8:318–321. doi: 10.1183/09031936.95.08020318. [DOI] [PubMed] [Google Scholar]
- 154.Bayle JY, Nesme P, Bejui-Thivolet F, et al. Migratory organizing pneumonitis ‘primed’ by radiation therapy. Eur Respir J. 1995;8:322–326. doi: 10.1183/09031936.95.08020322. [DOI] [PubMed] [Google Scholar]
- 155.Wharton SP, Rogers TK. Hamman–Rich syndrome ‘primed’ by radiation? Resp Med. 1999;93:136–137. doi: 10.1016/s0954-6111(99)90304-9. [DOI] [PubMed] [Google Scholar]
- 156.Ma LD, Taylor GA, Wharam MD, et al. ‘Recall’ pneumonitis: adriamycin potentiation of radiation pneumonitis in two children. Radiology. 1993;187:465–467. doi: 10.1148/radiology.187.2.8475291. [DOI] [PubMed] [Google Scholar]
- 157.Thomas PS, Agrawal S, Gore M, et al. Recall lung pneumonitis due to carmustine after radiotherapy. Thorax. 1995;50:1116–1118. doi: 10.1136/thx.50.10.1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Movsas B, Raffin TA, Epstein AH, et al. Pulmonary radiation injury. Chest. 1997;111:1061–1076. doi: 10.1378/chest.111.4.1061. [DOI] [PubMed] [Google Scholar]
- 159.Court-Brown WM, Doll R. Mortality from cancer and other causes after radiotherapy for ankylosing spondylitis. BMJ. 1965;2:1327–1332. doi: 10.1136/bmj.2.5474.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Neugut AI, Murray T, Santos J, et al. Increased risk of lung cancer after breast cancer radiation therapy in cigarette smokers. Cancer. 1994;73:1615–1620. doi: 10.1002/1097-0142(19940315)73:6<1615::aid-cncr2820730612>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 161.Allende DS, Rodriguez ER, Tan CD. Aortotracheal fistula secondary to bacterial aortitis. Arch Pathol Lab Med. 2009;133:983–986. doi: 10.5858/133.6.983. [DOI] [PubMed] [Google Scholar]
Respirator lung and oxygen toxicity
- 162.Nash G, Bowen JA, Langlinais PC. Respirator lung – a misnomer. Arch Pathol Lab Med. 1972;21:234–238. [PubMed] [Google Scholar]
- 163.Pratt PC. In: The Lung. Thurlbeck WM, Abell MR, editors. Williams and Wilkins; Baltimore: 1978. Pathology of adult respiratory distress syndrome; pp. 43–57. [Google Scholar]
- 164.Pratt PC, Vollmer RT, Shelburne JD, et al. Pulmonary morphology in a multihospital collaborative extracorporeal membrane oxygenation project. Am J Pathol. 1979;95:191–214. [PMC free article] [PubMed] [Google Scholar]
- 165.Slutsky AS. Lung injury caused by mechanical ventilation. Chest. 1999;116:9S–15S. doi: 10.1378/chest.116.suppl_1.9s-a. [DOI] [PubMed] [Google Scholar]
- 166.Ricard JD, Dreyfuss D, Saumon G. Ventilator-induced lung injury. Eur Resp J. 2003;22:2S–9S. doi: 10.1183/09031936.03.00420103. [DOI] [PubMed] [Google Scholar]
- 167.Sacks T, Moldow CF, Craddock PR, et al. Oxygen radicals mediate endothelial cell damage by complement-stimulated granulocytes. J Clin Invest. 1978;61:1161–1167. doi: 10.1172/JCI109031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Johnson KJ, Fantone JC, Daplan J, et al. In vitro damage of rat lungs by oxygen metabolites. J Clin Invest. 1981;67:983–993. doi: 10.1172/JCI110149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wegner CD, Wolyniec WW, Laplante AM, et al. Intercellular adhesion molecule-1 contributes to pulmonary oxygen toxicity in mice – role of leukocytes revised. Lung. 1992;170:267–279. doi: 10.1007/BF00566679. [DOI] [PubMed] [Google Scholar]
- 170.Kang BH, Crapo JD, Wegner CD, et al. Intercellular adhesion molecule-1 expression on the alveolar epithelium and its modification by hyperoxia. Am J Respir Cell Mol Biol. 1993;9:350–355. doi: 10.1165/ajrcmb/9.4.350. [DOI] [PubMed] [Google Scholar]
- 171.Deneke SM, Fanburg BL. Normobaric oxygen toxicity of the lung. N Engl J Med. 1980;303:76–86. doi: 10.1056/NEJM198007103030204. [DOI] [PubMed] [Google Scholar]
- 172.Kapanci Y, Tosco R, Eggermann J, et al. Oxygen pneumonitis in man. Chest. 1972;62:162–169. doi: 10.1378/chest.62.2.162. [DOI] [PubMed] [Google Scholar]
- 173.Pratt PC. Pathology of pulmonary oxygen toxicity. Am Rev Respir Dis. 1974;110:51–57. doi: 10.1164/arrd.1974.110.6P2.51. [DOI] [PubMed] [Google Scholar]
- 174.Katzenstein ALA, Bloor CM, Liebow AA. Diffuse alveolar damage – the role of oxygen, shock and related factors. Am J Pathol. 1976;85:210–222. [PMC free article] [PubMed] [Google Scholar]
- 175.Haschek WM, Brody AR, Klein-Szanto AJP, et al. Diffuse interstitial pulmonary fibrosis. Pulmonary fibrosis in mice induced by treatment with butylated hydroxytoluene and oxygen. Am J Pathol. 1981;105:334–335. [PMC free article] [PubMed] [Google Scholar]
- 176.Witschi HR, Haschek WM, Klein-Szanto AJP, et al. Potentiation of diffuse lung damage by oxygen: determining values. Am Rev Respir Dis. 1981;123:98–103. doi: 10.1164/arrd.1981.123.1.98. [DOI] [PubMed] [Google Scholar]
- 177.Yamamoto E, Wittner M, Rosenbaum RM. Resistance and susceptibility to oxygen toxicity by cell types of the gas–blood barrier of the rat lung. Am J Pathol. 1970;59:409–436. [PMC free article] [PubMed] [Google Scholar]
- 178.Gould VE, Tosco R, Wheelis RF, et al. Oxygen pneumonitis in man. Ultrastructural observations on the development of the alveolar lesions. Lab Invest. 1972;26:499–508. [PubMed] [Google Scholar]
- 179.Peek GJ, Moore HM, Moore N, et al. Extracorporeal membrane oxygenation for adult respiratory failure. Chest. 1997;112:759–764. doi: 10.1378/chest.112.3.759. [DOI] [PubMed] [Google Scholar]
- 180.Bruch LA, Flint A, Hirschl RB. Pulmonary pathology of patients treated with partial liquid ventilation. Mod Pathol. 1997;10:463–468. [PubMed] [Google Scholar]
- 181.Adawi A, Zhang Y, Baggs R, et al. Disruption of the CD40-CD40 ligand system prevents an oxygen-induced respiratory distress syndrome. Am J Pathol. 1998;152:651–657. [PMC free article] [PubMed] [Google Scholar]
- 182.Kapanci Y, Weibel ER, Kaplan HP, et al. Pathogenesis and reversibility of the pulmonary lesions of oxygen toxicity in monkeys. II Ultrastructural and morphometric studies. Lab Invest. 1969;20:101–118. [PubMed] [Google Scholar]
Blood transfusion
- 183.Popovsky MA, Abel MD, Moore SB. Transfusion-related acute lung injury associated with passive transfer of antileukocyte antibodies. Am Rev Respir Dis. 1983;128:185–189. doi: 10.1164/arrd.1983.128.1.185. [DOI] [PubMed] [Google Scholar]
- 184.Divertie MB. Diffuse alveolar damage, respiratory failure and blood transfusion. Mayo Clin Proc. 1984;59:643–644. doi: 10.1016/s0025-6196(12)62419-7. [DOI] [PubMed] [Google Scholar]
- 185.Dry SM, Bechard KM, Milford EL, et al. The pathology of transfusion-related acute lung injury. Amer J Clin Pathol. 1999;112:216–221. doi: 10.1093/ajcp/112.2.216. [DOI] [PubMed] [Google Scholar]
Cardiopulmonary bypass
- 186.Ratliff NB, Youg WG, Hackol DB, et al. Pulmonary injury – secondary to extracorporeal circulation. J Thorac Cardiovasc Surg. 1973;65:425–432. [PubMed] [Google Scholar]
- 187.Asada S, Yamaguchi M. Fine structural changes in the lungs following cardiopulmonary bypass. Chest. 1971;59:478–483. doi: 10.1378/chest.59.5.478. [DOI] [PubMed] [Google Scholar]
- 188.Westaby S. Complement and the damaging effects of cardiopulmonary bypass. Thorax. 1983;38:321–325. doi: 10.1136/thx.38.5.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Tonz M, Mihaljevic T, Vonsegesser LK, et al. Acute lung injury during cardiopulmonary bypass: are the neutrophils responsible? Chest. 1995;108:1551–1556. doi: 10.1378/chest.108.6.1551. [DOI] [PubMed] [Google Scholar]
- 190.Wan S, LeClerc JL, Vincent JL. Inflammatory response to cardiopulmonary bypass: Mechanisms involved and possible therapeutic strategies. Chest. 1997;112:676–692. doi: 10.1378/chest.112.3.676. [DOI] [PubMed] [Google Scholar]
Complications of cardiac injury
- 191.Stelzner TJ, King TE, Antony VB, et al. The pleuropulmonary manifestations of the postcardiac injury syndrome. Chest. 1983;84:383. doi: 10.1378/chest.84.4.383. [DOI] [PubMed] [Google Scholar]
- 192.Weiser NJ, Kantor M, Russell HK, et al. The postmyocardial infarction syndrome. The nonspecificity of the pulmonary manifestations. Circulation. 1962;25:643–650. doi: 10.1161/01.cir.25.4.643. [DOI] [PubMed] [Google Scholar]
- 193.Khan AH. The postcardiac injury syndromes. Clin Cardiol. 1992;15:67–72. doi: 10.1002/clc.4960150203. [DOI] [PubMed] [Google Scholar]
- 194.Akl ES, Latif N, Dunn MJ, et al. Antiheart antibodies following open heart surgery: incidence and correlation with postpericardiotomy syndrome. Eur J Cardiothorac Surg. 1992;6:503–507. doi: 10.1016/1010-7940(92)90249-w. [DOI] [PubMed] [Google Scholar]
Complications of radiofrequency ablation
- 195.Gillams AR, Lees WR. Analysis of the factors associated with radiofrequency ablation-induced pneumothorax. Clin Radiol. 2007;62:639–644. doi: 10.1016/j.crad.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Yang HM, Lai CK, Patel J, et al. Irreversible intrapulmonary vascular changes after pulmonary vein stenosis complicating catheter ablation for atrial fibrillation. Cardiovasc Pathol. 2007;16:51–55. doi: 10.1016/j.carpath.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 196.Kojodjojo P, Wong T, Wright AR, et al. Pulmonary venous stenosis after treatment for atrial fibrillation. BMJ. 2008;336:830–832. doi: 10.1136/bmj.39457.764942.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Complications of central vascular cannulation
- 197.Rowley KM, Clubb BSS, Smith GJW, et al. Right sided infective endocarditis as a consequence of flow directed pulmonary artery catheterisation. N Engl J Med. 1984;311:1152–1156. doi: 10.1056/NEJM198411013111804. [DOI] [PubMed] [Google Scholar]
- 198.Connors AF, Jr, Castele RJ, Farhat NZ, et al. Complication of right heart catherisation. Chest. 1985;88:567–572. doi: 10.1378/chest.88.4.567. [DOI] [PubMed] [Google Scholar]
Complications of tracheal manipulations
- 199.Stauffer JL, Olson DE, Petty TL. Complications and consequences of endotracheal intubation and tracheotomy. A prospective study of 150 critically ill adult patients. Am J Med. 1981;70:65–75. doi: 10.1016/0002-9343(81)90413-7. [DOI] [PubMed] [Google Scholar]
- 200.Vanheurn LWE, Theunissen PHMH, Ramsay G, et al. Pathologic changes of the trachea after percutaneous dilatational tracheotomy. Chest. 1996;109:1466–1469. doi: 10.1378/chest.109.6.1466. [DOI] [PubMed] [Google Scholar]
- 201.Phillips I. Pseudomonas aeruginosa respiratory tract infections in patients receiving mechanical ventilation. J Hyg. 1967;65:229–235. doi: 10.1017/s002217240004571x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Skim C. Cardiac arrhythmias resulting from tracheal suctioning. Ann Intern Med. 1969;71:1149–1153. doi: 10.7326/0003-4819-71-6-1149. [DOI] [PubMed] [Google Scholar]
- 203.Deslee G, Brichet A, Lebuffe G, et al. Obstructive fibrinous tracheal pseudomembrane – A potentially fatal complication of tracheal intubation. Amer J Respir Crit Care Med. 2000;162:1169–1171. doi: 10.1164/ajrccm.162.3.9910047. [DOI] [PubMed] [Google Scholar]
- 204.Francois B, Bellissant E, Gissot V, et al. 12-h pretreatment with methylprednisolone versus placebo for prevention of postextubation laryngeal oedema: a randomised double-blind trial. Lancet. 2007;369:1083–1089. doi: 10.1016/S0140-6736(07)60526-1. [DOI] [PubMed] [Google Scholar]
- 205.Jacobs IN, Wetmore RF, Tom LW, et al. Tracheobronchomalacia in children. Arch Otolaryngol Head Neck Surg. 1994;120:154–158. doi: 10.1001/archotol.1994.01880260026006. [DOI] [PubMed] [Google Scholar]
- 206.Romagosa V, Bella MR, Truchero C, et al. Necrotizing sialometaplasia (adenometaplasia) of the trachea. Histopathology. 1992;21:280–282. doi: 10.1111/j.1365-2559.1992.tb00389.x. [DOI] [PubMed] [Google Scholar]
Complications of bronchoscopy
- 207.Jin F, Mu D, Chu D, et al. Severe complications of bronchoscopy. Respiration. 2008;76:429–433. doi: 10.1159/000151656. [DOI] [PubMed] [Google Scholar]
Pneumonectomy
- 208.Kopec SE, Irwin RS, UmaliTorres CB, et al. The postpneumonectomy state. Chest. 1998;114:1158–1184. doi: 10.1378/chest.114.4.1158. [DOI] [PubMed] [Google Scholar]
- 209.Biondetti PR, Fiore D, Sartori F, et al. Evaluation of post-pneumonectomy space by computed tomography. J Comput Assist Tomogr. 1982;6:238–242. doi: 10.1097/00004728-198204000-00002. [DOI] [PubMed] [Google Scholar]
- 210.Suarez J, Clagett T, Brown AL., Jr The postpneumonectomy space: factors influencing its obliteration. J Thorac Cardiovasc Surg. 1969;57:539–542. [PubMed] [Google Scholar]
- 211.Sakamaki Y, Matsumoto K, Mizuno S, et al. Hepatocyte growth factor stimulates proliferation of respiratory epithelial cells during postpneumonectomy compensatory lung growth in mice. Amer J Respir Cell Molec Biol. 2002;26:525–533. doi: 10.1165/ajrcmb.26.5.4714. [DOI] [PubMed] [Google Scholar]
- 212.Adams HD, Junod F, Aberdeen E, et al. Severe airway obstruction caused by mediastinal displacement after right pneumonectomy in a child. A case report. J Thorac Cardiovasc Surg. 1972;63:534–539. [PubMed] [Google Scholar]
- 213.Grillo HC, Shepard JA, Mathisen DJ, et al. Postpneumonectomy syndrome: diagnosis, management, and results. Ann Thorac Surg. 1992;54:638–650. doi: 10.1016/0003-4975(92)91006-u. [DOI] [PubMed] [Google Scholar]
- 214.Cordova FC, Travaline JM, O’Brien GM, et al. Treatment of left pneumonectomy syndrome with an expandable endobronchial prosthesis. Chest. 1996;109:567–570. doi: 10.1378/chest.109.2.567. [DOI] [PubMed] [Google Scholar]
- 215.Boiselle PM, Shepard JA, McLoud TC, et al. Postpneumonectomy syndrome: another twist. J Thorac Imaging. 1997;12:209–211. doi: 10.1097/00005382-199707000-00007. [DOI] [PubMed] [Google Scholar]
- 216.Kutlu CA, Williams EA, Evans TW, et al. Acute lung injury and acute respiratory distress syndrome after pulmonary resection. Ann Thorac Surg. 2000;69:376–380. doi: 10.1016/s0003-4975(99)01090-5. [DOI] [PubMed] [Google Scholar]
- 217.Tang SS, Redmond K, Griffiths M, et al. The mortality from acute respiratory distress syndrome after pulmonary resection is reducing: a 10-year single institutional experience. Eur J Cardiothorac Surg. 2008;34:898–902. doi: 10.1016/j.ejcts.2008.06.020. [DOI] [PubMed] [Google Scholar]
- 218.Dulu A, Pastores SM, Park B, et al. Prevalence and mortality of acute lung injury and ards after lung resection. Chest. 2006;130:73–78. doi: 10.1378/chest.130.1.73. [DOI] [PubMed] [Google Scholar]
- 219.Williams EA, Quinlan GJ, Anning PB, et al. Lung injury following pulmonary resection in the isolated, blood-perfused rat lung. Eur Resp J. 1999;14:745–750. doi: 10.1034/j.1399-3003.1999.14d04.x. [DOI] [PubMed] [Google Scholar]
- 220.Jordan S, Mitchell JA, Quinlan GJ, et al. The pathogenesis of lung injury following pulmonary resection. Eur Resp J. 2000;15:790–799. doi: 10.1034/j.1399-3003.2000.15d26.x. [DOI] [PubMed] [Google Scholar]