

A high proportion of observed fetal sex discordances between single-nucleotide polymorphism–based noninvasive prenatal screening tests and prenatal or newborn examinations are attributable to disorders of sexual development.
OBJECTIVE:
To assess the causes of reported discordance between noninvasive prenatal testing (NIPT) and ultrasound or other clinical information.
METHODS:
In this retrospective, observational study, all cases in which single-nucleotide polymorphism (SNP)–based NIPT reported normal sex chromosomes and the laboratory was notified by the patient or health care provider of discordance between NIPT and observed or expected fetal sex from clinical information were reviewed. When discordances were unresolved after internal and external laboratory clerical data review or repeat ultrasound imaging, additional clinical records, genetic testing results and pregnancy outcomes were reviewed.
RESULTS:
Of the 1,301,117 eligible NIPT cases, fetal sex discordances were reported in 91 (0.007%; 1:14,300; 95% CI 1:11,600–1:17,800); partial or complete outcome information was available for 83 of 91 cases. In 30 of 83 (36%) cases, karyotyping was performed, and sufficient clinical information was provided to establish the diagnosis of disorders of sexual development. The disorders of sexual development were classified into three categories: 46,XY disorders of sexual development (n=19), 46,XX disorders of sexual development (n=4), and sex chromosome disorders of sexual development (n=7). In 28 of 83 (34%) cases, the cause of the apparent discrepancy was attributable to human error, predominantly phlebotomy labeling or ultrasound misassignment. In 25 of 83 cases, a diagnosis was not possible; the outcome reported was either abnormal (18/83, 22%) or no abnormalities were reported (7/83, 8%). When normal sex chromosomes were predicted by SNP-based NIPT and clinical information was discordant, disorders of sexual development were common. Internal laboratory clerical data review and re-imaging confirmed the NIPT fetal sex reports in 34% cases, providing reassurance that no further evaluation was necessary.
CONCLUSION:
Identification of apparent fetal sex discordances with NIPT results, and reporting this suspicion to the laboratory, provides an opportunity for further evaluation to identify the cause of apparent discordances and the involvement of a multi-disciplinary team, as necessary to prepare for postnatal care. We propose a protocol for evaluation of these cases.
FUNDING SOURCE:
This study was funded by Natera, Inc.
Lo et al1 first reported the presence of cell-free fetal DNA in the plasma of pregnant women. This led to the development of noninvasive prenatal testing (NIPT), which involves analysis of cell-free DNA in maternal plasma to evaluate the risk for common fetal aneuploidies by quantifying the fetal chromosome complement.2,3 Fetal sex can be reported from NIPT, although not considered a primary medical indication.4 Before NIPT, fetal sex was historically reported from ultrasound analysis of fetal external genitalia with high accuracy.5 Single-nucleotide polymorphism (SNP)–based NIPT uses relative SNP allele frequency patterns to determine chromosome copy number. Studies have reported the performance of cell-free DNA analysis for analysis of fetal sex chromosomes with sensitivities of greater than 99% (99.0–100%).4,6,7 Analytic validation of the SNP-based NIPT methodology reported correct fetal sex assignment in 100% (99.0–100%) of cases.6
In our experience, approximately 80% of the NIPT requisitions request information on fetal sex.4 Fetal sex discordance rates of 0.0–0.9% have been reported.6,8,9 Although case reports exist,10,11 a PubMed search using the terms “sex discordance or fetal sex discordance” and “NIPT” did not identify any large-scale studies evaluating the frequency and etiology for health care provider- or patient-reported discordance.
Discrepant results between NIPT and prenatal ultrasound or other clinical information may be explained by human error and biological mechanisms. Sources of human error include blood sample mislabeling, laboratory methodologic limitations, transcription errors and the limited performance of ultrasound imaging at early gestational age or with suboptimal visualization of the external genitalia. Biological reasons for discordance include the presence of fetoplacental mosaicism for sex chromosome abnormalities, the presence of a vanishing twin, maternal transplant recipient from a male donor, disorders of sexual development or other fetal abnormalities associated with anomalous or ambiguous external genitalia. The objective of this study was to analyze the causes for discordance between fetal sex predicted by a SNP-based NIPT, and fetal sex based on reported phenotype or genotype, so as to guide clinicians in the counseling, evaluation, and management of these pregnancies.
ROLE OF THE FUNDING SOURCE
All data were obtained from the sponsor's quality assurance database. The authors had access to relevant aggregated study data and other information (such as study protocol, analytic plan and report, validated data table, and clinical study report) required to understand and report research findings. The authors take responsibility for the presentation and publication of the research findings, have been fully involved at all stages of publication and presentation development, and are willing to take public responsibility for all aspects of the work. All aspects of the study were funded by Natera, Inc., including the design, execution, analysis, and manuscript development. All individuals included as authors and contributors who made substantial intellectual contributions to the research, data analysis, and publication or presentation development are listed appropriately. The role of the sponsor in the design, execution, analysis, reporting, and funding is fully disclosed. The authors' personal interests, financial or nonfinancial, relating to this research and its publication have been disclosed.
METHODS
This was a retrospective analysis of prospectively collected data for SNP-based NIPT cases received from March 1, 2013, to December 31, 2018. Samples were eligible for inclusion if fetal sex reporting was requested on the requisition, testing results reported a low-risk result for aneuploidy and the presence of two sex chromosomes (XX or XY), and contact was initiated by the patient or health care provider regarding an apparent discordance between the NIPT results and clinical or ultrasound findings. Descriptive analyses were provided for the fetal sex discordance cases based on the outcomes reported by the health care provider. Cases were excluded if fetal sex reporting was not requested on the requisition. High-risk results for any aneuploidy, multiple gestations and pregnancies involving an egg donor or surrogate were also excluded. If no results were obtained from NIPT for autosomes, or one or both sex chromosomes, the case was excluded.
Samples were analyzed at a Clinical Laboratory Improvement Act–certified, College of American Pathologists–accredited laboratory using previously described methodology.6,12
Review of patient identifiers from photographs of the kits and samples taken at accessioning, and review of DNA-based molecular barcode tracing and SNP patterns was completed by the laboratory. To rule out a sample swap, the health care provider was informed that the laboratory would accept a repeat sample at no charge to the patient or the payor. Repeat NIPT testing allowed for the comparison of maternal SNP-allele frequencies with that from the original NIPT, to confirm that both samples were from the same patient. If the patient was still pregnant at the time of reporting, the health care provider was encouraged to ensure that the fetal genitalia be assessed using ultrasound imaging at a minimum gestational age of 16 weeks. As part of laboratory quality assurance, health care providers were informed that the laboratory would accept a placental sample after delivery for SNP-based microarray testing, including evaluation for maternal cell contamination. Placental tissue or products of conception received for confirmatory testing were evaluated at the same laboratory using the Illumina CytoSNP-12 genotyping microarray platform.13,14 It was estimated that mosaicism of at least 20% would be detected by the array.
For each case, the laboratory attempted to collect information regarding the clinical data used in assigning the fetal sex by the health care provider, including results of preimplantation genetic testing for aneuploidy, if performed, the timing and ultrasound appearance of the fetal genitalia, the presence of additional abnormal findings and the gestational age at which the ultrasound scans were performed. Genetic testing of amniotic fluid, fetal blood or tissue, or neonatal blood or tissue was considered “truth.” If a pregnancy loss occurred, results of genetic analysis from products of conception (origin of tissue recorded) and autopsy examination were requested. Information regarding pregnancy outcome, including results of the newborn examination, were also requested. A minimum of three attempts were made to contact the referring provider using telephone, email or fax to obtain results of further fetal evaluation and outcome.
Cases were categorized into five groups: 1) “Apparent discordance resolved, normal outcome” if repeat ultrasound examination was consistent with NIPT result, sample mislabeling was identified and ultrasound imaging was consistent with a correctly labeled, maternally concordant NIPT sample, or if a clerical or laboratory error was identified and NIPT results were confirmed to be concordant with corrected clinical information; 2) disorders of sexual development were considered confirmed if a fetal or neonatal karyotype result was not consistent with clinical evaluation of the external genitalia: disorders of sexual development were further classified as 46,XY disorders of sexual development, 46,XX disorders of sexual development, or sex chromosome disorders of sexual development. Sex chromosome disorders of sexual development was defined as the presence of a fetus with an abnormal sex chromosome cell line and discordance between the sex chromosomes and the genitalia. This group includes those cases in which two normal sex chromosomes were suspected by NIPT but the fetus or neonate had a cell line with sex chromosome aneuploidy; 45,X and variants; 47,XXY and variants; 46,XX/46,XY based on clinical information and additional genetic or laboratory evaluation. Cases with insufficient outcome data to include in groups 1 or 2 were assigned into three additional groups based on known information and pregnancy outcome: 3) abnormal outcome without diagnosis, 4) no reported abnormalities; or 5) “lost to follow-up,” if no additional information was provided beyond a single ultrasound examination or newborn examination.
Reported discordant cases and all relevant information were digitally stored in an internal password-protected database accessible only to study personnel. This observational study was reported as per the STROBE (Strengthening the Reporting of Observational Studies in Epidemiology) guidelines.15 The data collected and reported represent routine quality assurance practices by the laboratory, therefore the study was deemed to be exempt from independent review board approval. Publication of these data was confirmed as exempt by an independent review board (Ethical and Independent ID 19040-01).
RESULTS
A total of 1,301,117 cases were reported as low risk for aneuploidy and normal sex chromosomes, in singleton, nonegg donor or nonsurrogate pregnancies during the study period. Outcome data were not available for most; a report from a health care provider or patient of a discordance between NIPT sex prediction and other clinical information was received in 91 (0.007%; 1:14,300; 95% CI 1:11,600–1:17,800) cases. Sufficient follow-up information was obtained for classification for 83 of 91 (91%) cases; eight (9%) were lost to follow-up (Fig. 1).
Fig. 1. Summary of case classification. *Confined to cases receiving low-risk results for all evaluated chromosomes, fetal sex requested and reported XX or XY. NIPT, noninvasive prenatal testing.
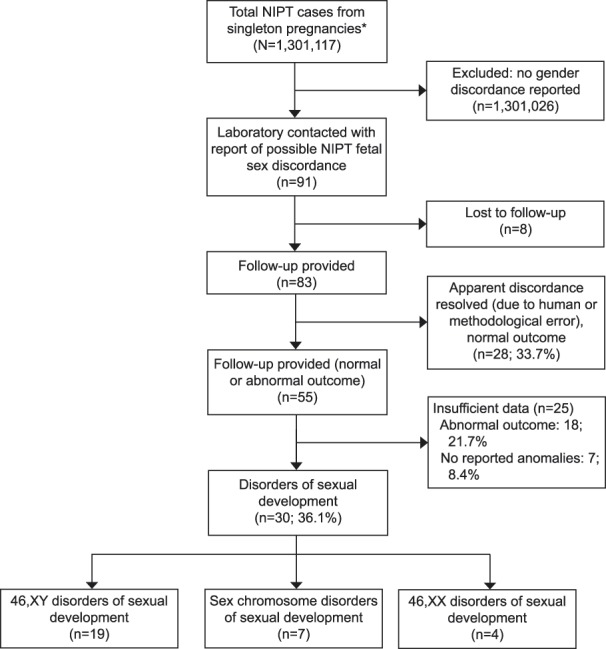
Dhamankar. Sex Discordance and Noninvasive Prenatal Testing. Obstet Gynecol 2020.
In 28 of 83 (34%) cases, the apparent discordance was the result of human or methodologic error, most commonly sample mislabeling (n=14) or fetal sex misassignment by ultrasonography (n=11, Table 1). Initial ultrasonograms were performed between 13 and 20 weeks of gestation; re-imaging and newborn examination (when reported) were consistent with NIPT results. Four women chose to repeat NIPT before re-imaging, which confirmed maternal sample concordance and the first NIPT result. Follow-up ultrasound imaging at later gestational ages showed fetal external genitalia consistent with the NIPT results. Amniocentesis was requested by 4 of 28 (14%) women without waiting for re-imaging or repeat NIPT; all amniocentesis results were consistent with the NIPT results. Two cases were attributable to the same, identified laboratory error. Sample identifications were swapped in the course of laboratory processing for these two cases. The laboratory standard operating procedures were revised to prevent re-occurrence of this error. For the third case considered to be an error attributable to the laboratory methodology, fetal sex from the NIPT results was initially reported as XY. Noninvasive prenatal testing was repeated when female external genitalia were seen on ultrasound imaging and increased risk for XXY was reported. Postnatal blood karyotype indicated 46,XX. A placental sample was not submitted to rule out confined placental mosaicism, which is a recognized cause for discordance between results of NIPT and fetal testing.
Table 1.
Cases Resolved (Corrected Report of External Genitalia–Karyotype and NIPT Result Concordant)

Of the 83 cases for which sufficient clinical information allowed outcome assignment, 30 (36%) were due to disorders of sexual development. These cases could be further categorized, as presented in Table 2. The most common disorders of sexual development category was 46,XY disorders of sexual development (n=19), of which androgen insensitivity syndrome was observed in seven cases. In all seven cases of androgen insensitivity syndrome, NIPT reported fetal sex as male. The discrepancy for fetal sex was identified by observation of female genitalia either on ultrasound imaging (n=6) or by phenotypic examination after birth (n=1). In three of these cases, karyotype from amniocentesis revealed 46,XY and genetic testing for androgen insensitivity syndrome was then performed on amniocytes. The remaining four cases had postnatal karyotypes of 46,XY with subsequent genetic testing for androgen insensitivity syndrome.
Table 2.
Cases With Disorders of Sexual Development

A single diagnosis of Smith-Lemli-Opitz syndrome was confirmed after delivery. Noninvasive prenatal testing reported the fetal sex to be male. Biochemical screening for aneuploidy indicated an increased risk for either trisomy 18 or Smith-Lemli-Opitz syndrome. The ultrasonography reported normal female genitalia at 18 and 20 weeks of gestation; however, at 21 weeks, the genitalia could not be categorized as either normal male or female and, therefore, were reported as ambiguous. Postnatal testing confirmed the diagnosis of Smith-Lemli-Opitz syndrome. Outcomes consistent with 46,XY disorders of sexual development were reported in an additional 11 cases however a specific diagnosis could not be established; details are provided in Appendix 1, available online at http://links.lww.com/AOG/B804.
There were four cases for which a diagnosis of 46,XX disorder of sexual development was confirmed. Congenital adrenal hyperplasia was confirmed in one case; the NIPT result indicated fetal sex to be female. Ultrasound examination at 20 weeks of gestation reported the fetal sex as male or ambiguous, and amniocentesis confirmed a normal female karyotype, 46,XX. An extended carrier screening panel was performed for the patient and her partner, which identified them both to be carriers for 21-hydroxylase deficiency. Subsequent DNA testing from the amniocentesis confirmed congenital adrenal hyperplasia in the fetus. A case of 46,XX testicular disorder of sexual development was identified secondary to a chromosomal duplication and rearrangement. Noninvasive prenatal testing reported fetal sex as female; however, ultrasonography at 20 weeks of gestation reported the fetal sex as male. Prenatal diagnosis was pursued and confirmed the fetal karyotype as 46,XX. Chromosome microarray analysis revealed a 487 kb duplication of 4q13.3. Fluorescent in situ hybridization of metaphase chromosomes determined the duplication was inserted on Xq (exact breakpoints were not determined), upstream of the SOX3 gene. This insertion was hypothesized to alter SOX3 expression, leading to 46,XX testicular disorder of sexual development. A similar case was described in the literature by Haines et al in 2015.16 The remaining two cases with 46,XX disorders of sexual development are described in Table 2.
Seven cases were found to have sex chromosome disorders of sexual development (specific information provided in Table 3). Six of the seven were due to Turner syndrome, and karyotyping (prenatal or postnatal) confirmed the presence of a 46,XY or 47,XYY cell line in addition to the 45,X cell line. Ultrasonography findings were normal in two of six Turner syndrome cases, and prenatal resolution of hydrops was noted in one additional case. Postnatal karyotyping of the seventh case showed the presence of two normal cell lines. Results of SNP genotyping were not provided; therefore, a specific mechanism for this abnormality cannot be established. In two of the Turner syndrome cases, placental testing was requested, results indicated 46,XY, consistent with NIPT.
Table 3.
Details of Cases With Sex Chromosome Disorder of Sexual Development

An additional 25 cases with limited evaluation and outcome information were classified based on the pregnancy outcome being abnormal (n=18) or without abnormalities reported (n=7). The specific details for these cases are provided in Appendixes 2 and 3, available online at http://links.lww.com/AOG/B804. In the 18 cases with abnormal outcomes, only two had repeat NIPT to rule out sample mislabeling (repeat NIPT result unchanged, maternal concordance confirmed). Ten of the 18 (56%) pregnancies resulted in intrauterine fetal death before 24 weeks of gestation, and another pregnancy ended for unknown reasons at 16 weeks of gestation. Only three of these underwent some type of genetic evaluation; one was consistent with NIPT, and the other two were found to be XY after NIPT suggested XX. In several of these cases, different health care providers assigned the fetal sex differently based on observation after delivery. Although the information provided suggests that there may have been an underlying biological cause for the apparent discordant results, we hesitate to speculate in the absence of definitive testing. Hypospadias or micropenis were frequently described in the postnatal evaluations for those cases for which a disorder of sexual development or other abnormal outcomes were reported (Appendixes 1 and 2, http://links.lww.com/AOG/B804).
There were seven cases in which no genital or other abnormalities were reported (Appendix 3, http://links.lww.com/AOG/B804). In four of seven cases, NIPT predicted XY and postnatal examination was reported as normal male. In three cases, NIPT predicted XX. In two of these, ultrasound and postnatal examinations were reported as male, the third case did not include a postnatal examination. No genetic confirmatory studies were reported for any of these cases.
DISCUSSION
Results of NIPT may be different from either the ultrasound appearance of the external genitalia or results of preimplantation genetic testing for aneuploidy for multiple reasons. In 34% of cases (28/83), the apparent discordance was related to human or methodologic error; and was resolved by identifying sample mislabeling and clerical errors and repeating the ultrasonography. After ruling out human and methodologic errors, the most important diagnoses to consider are disorders of sexual development, which were confirmed in 30 of 83 (36%) cases of reported discordance. The outcome information in these cases were sufficient to assign a diagnosis of disorder of sexual development. More than half of the cases with fetal sex discordance (48/83, 58%) experienced some abnormal pregnancy outcome.
Understanding the various etiologies of discordant cases provides a framework for evaluation (Fig. 2). Individuals with 46,XY disorders of sexual development may demonstrate “under-virilization” owing to decreased testosterone production or sensitivity. Complete androgen insensitivity was the second most common disorder of sexual development in this cohort. Complete androgen insensitivity has been traditionally diagnosed during puberty when girls fail to menstruate but has also been reported as a cause of an incorrect prenatal fetal sex assignment.17 The necessity or timing of gonadectomy remains controversial in cases of complete androgen insensitivity; review of the current medical literature is advised before nonreversible management decisions.18–20 In cases of 46,XX disorders of sexual development, the fetus becomes “virilized” owing to exposure to endogenous or exogenous (maternal or placental) androgens. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency is one of the most common causes of 46,XX disorders of sexual development and may be associated with life-threatening salt wasting which can be prevented with early diagnosis and treatment.
Fig. 2. Generalized protocol for the clinical management of noninvasive prenatal testing (NIPT) results with fetal sex discordance. PGT-A, preimplantation genetic testing for aneuploidies; SNP, single-nucleotide polymorphism; MFM, maternal–fetal medicine.
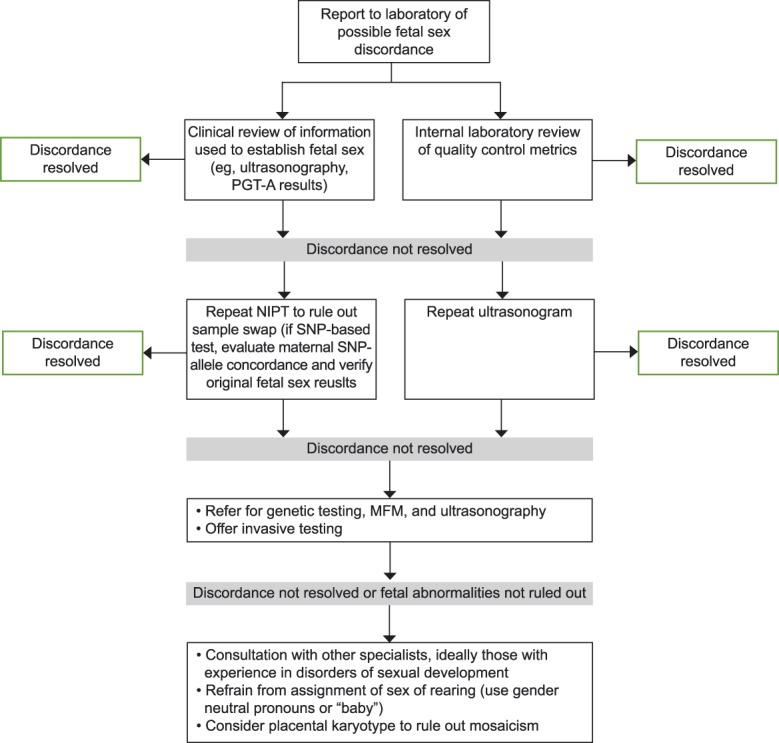
Dhamankar. Sex Discordance and Noninvasive Prenatal Testing. Obstet Gynecol 2020.
This study has several limitations; primarily that only cases in which apparent fetal sex discordance was reported to the laboratory could be included. Under-ascertainment of the outcomes observed is likely, as most of 1,301,117 cases reported during the study period did not have outcome data. Our minimum disorders of sexual development incidence of 1 in 43,371 is significantly lower than the hypothesized 1 in 1,000 frequency reported by Ostrer.21 This may be for many reasons: all patients or health care providers may not have volunteered abnormal outcomes to a laboratory. Excluding cases in which NIPT suspected a sex chromosome abnormality reduced the ascertainment of this type of disorders of sexual development. Many disorders of sexual development cases are not identified until later childhood or adulthood. The true population frequency for disorders of sexual development is unknown, as the criteria used for diagnosis of a disorder of sexual development is highly variable and dependent on the age of the cohort being evaluated. Another limitation of the study is that we chose to focus only on cases with NIPT results indicating two sex chromosomes because we anticipate that an increased risk for a sex chromosome aneuploidy by NIPT would result in an automatic referral to a maternal–fetal medicine specialist. Finally, the ability to generalize our findings to other cell-free DNA screening tests using different laboratory methodologies is unknown. Use of SNP-based NIPT allows the identification of many conditions that may be associated with fetal sex discordance, for example, vanishing twin or maternal chromosome abnormality.
After human error has been ruled out as a cause for NIPT and clinical fetal sex discordance, the family should be counseled by individuals with experience in both genetics and disorders of sexual development to provide early evaluation and preparation for the birth of a child with a disorder of sexual development or other abnormal outcome.22 If possible, this counseling would have the benefit of a prenatal (ideally amniotic fluid) karyotype. It is important that health care providers discuss with the family that assignment of the “sex of rearing” for the child is typically not possible until days or weeks after birth, particularly without karyotype evaluation and additional investigations. Speculation by health care providers about the sex of rearing should be discouraged until the cause of the discordance is known. In our experience, we have found that it can be very difficult for the family to adjust to the potential need for a change in sex assignment, especially if there was a prolonged period of time between initial fetal sex assignment and confirmed diagnosis of the underlying condition. The opportunity to involve members of a multidisciplinary disorders of sexual development team should reduce misconceptions and guide further work-up and management. If a multidisciplinary disorders of sexual development team is not available, a pediatric endocrinologist, pediatric gynecologist or reproductive endocrinologist may best assist with initiating evaluation and care. Finally, as these events can be quite traumatic for the parents, it is important to involve a mental health provider early and provide emotional support in addition to honest and clear medical information. The Accord Alliance (https://www.accordalliance.org/) has excellent resources for both clinicians and parents.
In summary, for a prenatally reported fetal sex discrepancy, human error must be ruled out by confirming correct sample labeling. This is performed when a SNP-based NIPT method is used by repeating the sample and verifying maternal SNP concordance. All available clinical data should be reviewed. If ultrasound examination is discordant with the NIPT finding, despite performance by an experienced examiner after 16 weeks of gestation with optimal visualization, the option of invasive prenatal diagnosis by amniocentesis should be discussed. We do not encourage chorionic villus sampling, owing to the frequency of confined placental mosaicism associated with sex chromosome abnormalities. If a discrepancy persists, we suggest that a multidisciplinary team meet to review all of the available information and develop a care pathway for the family. This team is essential to guide the transition from prenatal to postnatal life for the child and family, regardless of whether the family elects to pursue prenatal diagnostic evaluation or delay until after delivery. It is critical that at least one member of this team remain involved with the child and family after delivery. Not only does this provide continuity of care for the family, but perhaps even more importantly, it ensures that all of the information obtained during the prenatal period is communicated completely and accurately to the pediatric care team.
Footnotes
Editorial and medical writing support was provided by Aparna Shetty, PhD, ISMPP CMPP from Natera, Inc.
Financial Disclosure Rupin Dhamankar disclosed that he is an employee of Natera Inc., and holds stock options in Natera. Wendy DiNonno disclosed that she is an employee of Natera, Inc. and holds stock options in Natera, Inc. Kimberly Martin disclosed that she was an employee of Natera, holds stock options in Natera, and is currently a consultant to Natera (effective May 2, 2019). Zachary Demko disclosed that he is an employee of Natera, and holds stock and options to buy stock in Natera. The other author did not report any potential conflicts of interest.
Presented at the National Society of Genetic Counselors' Annual Conference, November 14–17, 2018, Atlanta, Georgia, and at the European Society of Human Genetics Conference, June 15–18, 2019, Gothenburg, Sweden.
The authors thank Dusan Kijacic, MS, Akshita Kalyan, MS, Allison Ryan, PhD, and Varsha Vijayan, MS, all from Natera, Inc., for their role in internal data reviews.
Each author has confirmed compliance with the journal's requirements for authorship.
Peer reviews are available at http://links.lww.com/AOG/B805.
REFERENCES
- 1.Lo ES, Lo YM, Hjelm NM, Thilaganathan B. Transfer of nucleated maternal cells into fetal circulation during the second trimester of pregnancy. Br J Haematol 1998;100:605–6. [DOI] [PubMed] [Google Scholar]
- 2.Allyse M, Minear MA, Berson E, Sridhar S, Rote M, Hung A, et al. Non-invasive prenatal testing: a review of international implementation and challenges. Int J Wom Health 2015;7:113–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wapner RJ, Babiarz JE, Levy B, Stosic M, Zimmermann B, Sigurjonsson S, et al. Expanding the scope of noninvasive prenatal testing: detection of fetal microdeletion syndromes. Am J Obstet Gynecol 2015;212:332.e1–9. [DOI] [PubMed] [Google Scholar]
- 4.Dar P, Curnow KJ, Gross SJ, Hall MP, Stosic M, Demko Z, et al. Clinical experience and follow-up with large scale single-nucleotide polymorphism-based noninvasive prenatal aneuploidy testing. Am J Obstet Gynecol 2014;211:527.e1–17. [DOI] [PubMed] [Google Scholar]
- 5.Colmant C, Morin-Surroca M, Fuchs F, Fernandez H, Senat MV. Non-invasive prenatal testing for fetal sex determination: is ultrasound still relevant? Eur J Obstet Gynecol Reprod Biol 2013;171:197–204. [DOI] [PubMed] [Google Scholar]
- 6.Pergament E, Cuckle H, Zimmermann B, Banjevic M, Sigurjonsson S, Ryan A, et al. Single-nucleotide polymorphism-based noninvasive prenatal screening in a high-risk and low-risk cohort. Obstet Gynecol 2014;124:210–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Porreco RP, Garite TJ, Maurel K, Marusiak B, Ehrich M, van den Boom D, et al. ; Obstetrix Collaborative Research N. Noninvasive prenatal screening for fetal trisomies 21, 18, 13 and the common sex chromosome aneuploidies from maternal blood using massively parallel genomic sequencing of DNA. Am J Obstet Gynecol 2014;211:365.e1–12. [DOI] [PubMed] [Google Scholar]
- 8.Nicolaides KH, Syngelaki A, del Mar Gil M, Quezada MS, Zinevich Y. Prenatal detection of fetal triploidy from cell-free DNA testing in maternal blood. Fetal Diagn Ther 2014;35:212–7. [DOI] [PubMed] [Google Scholar]
- 9.Moise KJ, Jr, Boring NH, O'Shaughnessy R, Simpson LL, Wolfe HM, Baxter JK, et al. Circulating cell-free fetal DNA for the detection of RHD status and sex using reflex fetal identifiers. Prenatal Diagn 2013;33:95–101. [DOI] [PubMed] [Google Scholar]
- 10.Byers HM, Neufeld-Kaiser W, Chang EY, Tsuchiya K, Oehler ES, Adam MP. Discordant sex between fetal screening and postnatal phenotype requires evaluation. J Perinatol 2019;39:28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richardson EJ, Scott FP, McLennan AC. Sex discordance identification following non-invasive prenatal testing. Prenatal Diagn 2017;37:1298–304. [DOI] [PubMed] [Google Scholar]
- 12.Zimmermann B, Hill M, Gemelos G, Demko Z, Banjevic M, Baner J, et al. Noninvasive prenatal aneuploidy testing of chromosomes 13, 18, 21, X, and Y, using targeted sequencing of polymorphic loci. Prenatal Diagn 2012;32:1233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lathi RB, Massie JA, Loring M, Demko ZP, Johnson D, Sigurjonsson S, et al. Informatics enhanced SNP microarray analysis of 30 miscarriage samples compared to routine cytogenetics. PLoS One 2012;7:e31282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lathi RB, Gustin SL, Keller J, Maisenbacher MK, Sigurjonsson S, Tao R, et al. Reliability of 46,XX results on miscarriage specimens: a review of 1,222 first trimester miscarriage specimens. Fertil Steril 2014;101:178–82. [DOI] [PubMed] [Google Scholar]
- 15.von Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Ann Intern Med 2007;147:573–7. [DOI] [PubMed] [Google Scholar]
- 16.Haines B, Hughes J, Corbett M, Shaw M, Innes J, Patel L, et al. Interchromosomal insertional translocation at Xq26.3 alters SOX3 expression in an individual with XX male sex reversal. J Clin Endocrinol Metab 2015;100:E815–20. [DOI] [PubMed] [Google Scholar]
- 17.Zilberman D, Parikh LI, Skinner M, Landy HJ. Prenatal diagnosis of androgen insensitivity syndrome using cell-free fetal DNA testing. Ultrasound Obstet Gynecol 2015;45:114–5. [DOI] [PubMed] [Google Scholar]
- 18.Deans R, Creighton SM, Liao LM, Conway GS. Timing of gonadectomy in adult women with complete androgen insensitivity syndrome (CAIS): patient preferences and clinical evidence. Clin Endocrinol 2012;76:894–8. [DOI] [PubMed] [Google Scholar]
- 19.Weidler EM, Linnaus ME, Baratz AB, Goncalves LF, Bailey S, Hernandez SJ, et al. A management protocol for gonad preservation in patients with androgen insensitivity syndrome. J Pediatr Adolesc Gynecol 2019;32:605–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lanciotti L, Cofini M, Leonardi A, Bertozzi M, Penta L, Esposito S. Different clinical presentations and management in complete androgen insensitivity syndrome (CAIS). Int J Environ Res Publ Health 2019;16:E1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ostrer H. Disorders of sex development (DSDs): an update. J Clin Endocrinol Metab 2014;99:1503–9. [DOI] [PubMed] [Google Scholar]
- 22.Ayala NK, Kole MB, Forcier M, Halliday J, Russo ML. Sex discordance between cell-free fetal DNA and mid-trimester ultrasound: a modern conundrum. Prenat Diagn 2019. Oct 16 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]