

Abstract
Purpose of review
In 2013, the association between T-Box factor 4 (TBX4) variants and pulmonary arterial hypertension (PAH) has first been described. Now – in 2020 – growing evidence is emerging indicating that TBX4 variants associate with a wide spectrum of lung disorders.
Recent findings
TBX4 variants are enriched in both children and adults with PAH. The clinical phenotype associated with a TBX4 variant seems to be milder than that in other PAH-associated gene mutations. Further, TBX4 variants have increasingly been associated with a variety of clinical and histopathological phenotypes, including lethal developmental parenchymal lung diseases such as not only acinar dysplasia in neonates, but also less outspoken parenchymal lung diseases in children and adults.
Summary
The clinical phenotype of a TBX4 variant has recently been recognised to expand from bone disorders to different types of lung diseases. Recent data suggest that variants of TBX4, a transcription factor known to be an important regulator in embryonic development, are not rare in both children and adults with PAH and/or developmental parenchymal lung diseases.
Keywords: lung disease, pulmonary arterial hypertension, T-Box factor 4
INTRODUCTION
In 1927, Nadezhda Alexandrovna Dobrovolskaya-Zavadskaya described a heterozygous mutation in mice that affected both tail length and the sacral vertebrae [1]. This mutation was called brachyury (short tail, Greek) mutation. Later, because of the T(tail)-locus of the gene product, the mutation was renamed into T-Box mutation [2,3]. In 1996, T-Box transcription factor 4 (TBX4) was identified, in a series of other T-Box genes (TBX1–5), as a gene highly expressed during organogenesis [4,5].
This review deepens into the TBX4 gene and its variants in pulmonary diseases, since in the recent years, the spectrum of clinical phenotypes associated with variants of the TBX4 gene [including both mutations and copy number variations (CNVs)] is expanding from bone abnormalities to conditions affecting other organs, especially the lungs. We aim to discuss the current knowledge on the role of TBX4 variants in human lung diseases.
Box 1.

no caption available
EMBRYOGENESIS
T-Box genes are DNA-binding transcription factors that play a crucial role during embryogenesis [6]. The TBX4 gene is located on chromosome 17q23.2 and has shown to be important for hindlimb and pelvic development in both animals and humans [5,7,8]. Animal studies have also suggested an important role for TBX4 in the formation of the body wall, development of external genitalia and anorectal development [5,9,10].
In addition, TBX4 is expressed in the lung mesenchyme, already at an early stage during embryogenesis [10]. Together with TBX5, TBX4 regulates the process of lung branching by controlling the expression of the secreted fibroblast growth factor (FGF)10 and activation of FGF10 signalling [11,12]. FGF10 is expressed in the mesenchyme surrounding the distal epithelial tips that mark the site of future bud formation and acts by mainly binding to the FGF receptor 2b [13–15]. FGF10 is involved in the initial production of paired buds of endoderm from the ventral foregut and their eventual outgrowth forming the precursors of the main bronchi [14]. Second, FGF10 induces new bud formation and therefore plays a key role in branching morphogenesis [14,16].
Another important regulatory gene that is affected by TBX4 and TBX5 signalling is canonical wingless-2 (WNT2), a mesenchymal protein that is essential for lung specification [11,17].
Reduction of TBX4 or TBX5 leads to a severe decrease in branching by a loss of expression of both FGF10 and WNT2. This results in a defect of formation of lung lobes and failure of lung lobe separation, eventually leading to a smaller overall lung size [11,18]. Absence of both TBX4 and TBX5 leads to complete branching arrest [19].
In addition, TBX4 and TBX5, via SOX9 and independently of the FGF10 signalling pathway, regulate the condensation of cells to form the cartilage rings of the trachea and bronchi, and also the development of the smooth muscle cells in the trachea [11,20]. Reduction of both TBX4 and TBX5 causes a decreased number of tracheal cartilage rings and can therefore result in tracheomalacia or tracheal stenosis [11]. TBX4 has also been shown to be present in pulmonary veins and arteries, suggesting that pulmonary vessels arise from the surrounding lung mesenchyme [10]. These experimental data, derived from cell and animal studies, indicate rather convincingly that TBX4 plays an important role in foetal development, including both parenchymal and vascular lung development.
CLINICAL PHENOTYPES
Clinical consequences of TBX4 variants in humans were first revealed in 2004, when the association between a spectrum of limb and skeletal abnormalities, known as small patella syndrome (SPS) or ischiocoxopodopatellar syndrome, was identified (Fig. 1) [22]. This syndrome causes aplasia or hypoplasia of the patella and developmental anomalies of the pelvis and feet.
FIGURE 1.
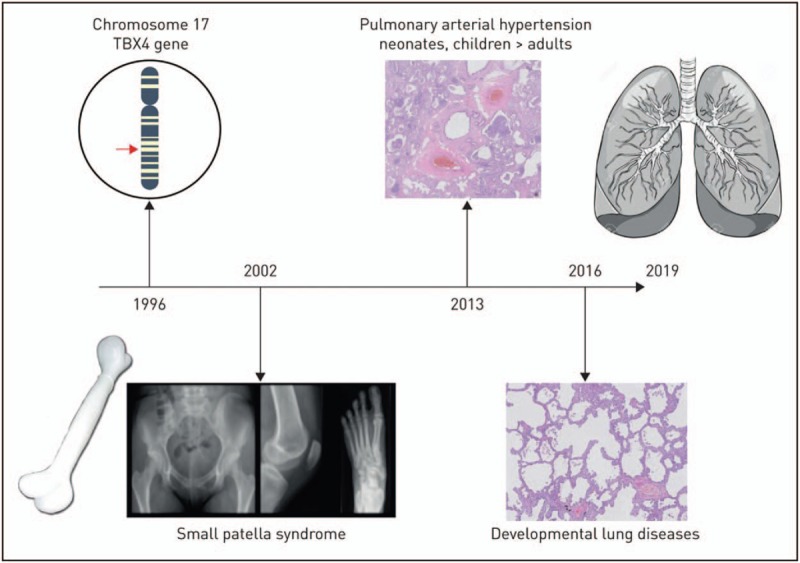
Timeline of the expanding clinical phenotypes of TBX4 variants since the discovery of this gene in 1996. Reproduced with permission from [21].
In 2013, Kerstjens-Frederikse et al.[23] discovered that mutations in the TBX4 gene or deletions encompassing the TBX4 gene were associated with pulmonary arterial hypertension (PAH) in childhood. Pulmonary hypertension, an increased pulmonary arterial pressure, is a haemodynamic symptom, and its underlying diseases are characterised by a clinical classification (Table 1) [24,25]. PAH is a specific form of PH and is a rare, progressive and often lethal disease of the pulmonary vasculature, characterised by an increased pulmonary vascular resistance, leading to right ventricular failure and eventually death [26,27]. It may be associated with a variety of underlying diseases, including connective tissue diseases, congenital heart diseases, portal hypertension, HIV infection and drugs. In the absence of associated conditions, it is classified as idiopathic PAH (IPAH) or it can be caused by a genetic defect (hereditary PAH; HPAH) (Table 1) [24,25]. In recent years, a progressive number of gene mutations is being been identified as cause for PAH, including mutations in bone morphogenetic protein receptor type II (BMPR2) being the most frequently found gene mutation in both children and adults with PAH [28–30]. Further, mutations in activin receptor-like kinase 1 (ACVRL1), endoglin (ENG), potassium channel subfamily K member 3 (KCNK3), eukaryotic translation initiation factor 2-alpha kinase 4 (EIF2AK4), Caveolin-1 (CAV1) and SMAD9 have currently been identified as associated with the occurrence of PAH, and this list is growing [31–36].
Table 1.
| 1 PAH 1.1 Idiopathic PAH 1.2 Heritable PAH 1.3 Drug- and toxin-induced PAH 1.4 PAH associated with: 1.4.1 Connective tissue disease 1.4.2 HIV infection 1.4.3 Portal hypertension 1.4.4 Congenital heart disease 1.4.5 Schistosomiasis 1.5 PAH longterm responders to calcium channel blockers 1.6 PAH with overt features of venous/capillaries (PVOD/PCH) involvement 1.7 Persistent PH of the newborn syndrome |
| 2 PH due to left heart disease 2.1 PH due to heart failure with preserved LVEF 2.2 PH due to heart failure with reduced LVEF 2.3 Valvular heart disease 2.4 Congenital/acquired cardiovascular conditions leading to postcapillary PH |
| 3 PH due to lung diseases and/or hypoxia 3.1 Obstructive lung disease 3.2 Restrictive lung disease 3.3 Other lung disease with mixed restrictive/obstructive pattern 3.4 Hypoxia without lung disease 3.5 Developmental lung disorders |
| 4 PH due to pulmonary artery obstructions 4.1 Chronic thromboembolic PH 4.2 Other pulmonary artery obstructions |
| 5 PH with unclear and/or multifactorial mechanisms 5.1 Haematological disorders 5.2 Systemic and metabolic disorders 5.3 Others 5.4 Complex congenital heart disease |
LVEF, left ventricular ejection fraction; PAH, pulmonary arterial hypertension; PCH, pulmonary capillary haemangiomatosis; PVOD, pulmonary veno-occlusive disease.
In the study of Kerstjens-Frederikse et al.[23], genetic analyses in a cohort of 20 children with IPAH revealed deletions at 17q23.2 with an overlapping region harbouring the TBX4 gene in three children with PAH and mental retardation, and an intragenic TBX4 mutation in three children with PAH with normal intelligence. All appeared to have skeletal malformations characteristic for SPS.
Since the discovery of the association between TBX4 variants and paediatric PAH, different cohorts confirmed an enrichment of TBX4 variants in childhood P(A)H. Levy et al.[33] reported three TBX4 truncating mutations in a subgroup of 40 paediatric IPAH/HPAH patients (8%). Zhu et al.[37▪] found in a cohort of 155 patients with paediatric-onset IPAH/HPAH that TBX4 variants were present in a substantial proportion of children (8%). Of these, 5% were pathogenic and likely pathogenic (LP) variants in TBX4, while another 3% were variants of unknown significance (VUS).
In summary, the prevalence of TBX4 variants in children with IPAH/HPAH is estimated 7–8%, which makes TBX4, after BMPR2, the second most frequently affected gene in these children [33,37▪,38].
A lower number of TBX4 variants seemed to be present in the adult population with IPAH/HPAH. Kerstjens-Frederikse et al.[23] in the same study, as previously mentioned, identified two TBX4 variants (one LP, one VUS) in a cohort of 49 adults with IPAH. In a larger cohort of 257 adults with IPAH/HPAH, Zhu et al.[37▪] identified three adult patients with a TBX4 variant (one LP, two VUS) (1.2%). Navas et al.[39] reported a prevalence of 2.4% of TBX4 variants in patients with IPAH/HPAH. In a cohort of 966 adult patients with IPAH/HPAH, Gräf et al.[40] reported that 1.4% of the patients had a TBX4 variant. Finally, in a French cohort, Eyries et al.[38] reported that 2.6% of the patients with IPAH/HPAH had a TBX4 variant, concluding that also in adulthood PAH, TBX4 may be the most frequently mutated gene after BMPR2. In summary, the overall prevalence of TBX4 variants in adults with IPAH/HPAH is estimated around 1.7%.
The reason why TBX4 variants seem to be more frequent in childhood-onset compared with adulthood-onset IPAH/HPAH is insufficiently clear. A detrimental disease course of TBX4-associated HPAH with early decease before reaching adulthood would have explained such phenomenon, however currently available, preliminary outcome data invalidate such speculation. The clinical implications of the presence of TBX4 variants in children with PAH have been scarcely studied. Where BMPR2 mutation carriers have shown to have a worse survival in both children and adults with HPAH when compared with noncarriers, children with TBX4 variants in a Dutch national cohort of 70 children with PAH showed less severe clinical presentation and more favourable survival compared with BMPR2 mutation carriers, similar to nonmutation carriers (own data). Similar findings have been reported by Levy et al.[33] in a French cohort of 66 children with PAH.
Navas et al.[39] confirmed these findings in adults with IPAH/HPAH and showed that patients with a TBX4 variant tended to have a milder clinical presentation compared with patients with IPAH, BMPR2 mutation carriers, KCNK3 mutation carriers and patients with familial PAH without a mutation. Also, survival in patients with a TBX4 variant was better compared with patients with a BMPR2 mutation or a KCNK3 mutation.
Zhu et al.[37▪] reported that HPAH patients carrying a TBX4 variant had a 20-year younger age-of-onset than BMPR2 mutation carriers (mean ± SD: 7.9 ± 9.0 compared with 28.2 ± 15.4, respectively, P < 0.0001). Although the clinical phenotype of PAH in TBX4 variant carriers in general seems to be milder than in BMPR2 mutation carriers, it is not necessarily favourable, and varies from mild/moderate PAH to also more severe forms of PAH, eventually fatal or requiring lung transplantation [37▪].
Until today, available clinical data do not justify patients with HPAH harbouring a TBX4 variant to be treated differently than patients with other forms of PAH-associated conditions.
TBX4 AND PARENCHYMAL LUNG DISEASES
After the first discovery of a role for TBX4 variants in the development of PAH, Szafranski et al.[41] described in 2016 a neonate with signs of acinar dysplasia (AcDys), a severe diffuse developmental lung disease, who showed to have a de novo TBX4 mutation. This was the first report suggesting a relation between a TBX4 variant and developmental parenchymal lung disease. Subsequently, in 2019, German et al.[42] described another case of a preterm born neonate with respiratory failure due to a diagnosis of AcDys, harbouring a deletion from chromosome 17q23.2 to 17q23.2 involving 14 genes including TBX4 (Fig. 2). In the same year, Karolak et al.[43▪▪] described a cohort of 26 children with AcDys, congenital alveolar dysplasia (CAD) or other rare pulmonary hypoplasias and reported in 16 children, a TBX4 variant or FGF10 variant. In almost 50% of these cases, the developmental lung disease was accompanied by PH. Suhrie et al.[44] identified pathogenic TBX4 mutations in two cases of infants presenting with respiratory failure. One child had abnormal lung lobulation with marked deficiency in acinar/alveolar development, and eventually deceased. The other child had a lung biopsy that demonstrated areas of the lung with near normal alveolarisation adjacent to regions with deficient alveolar growth, characterised by simplified, cystically dilated alveolar spaces [45].
FIGURE 2.
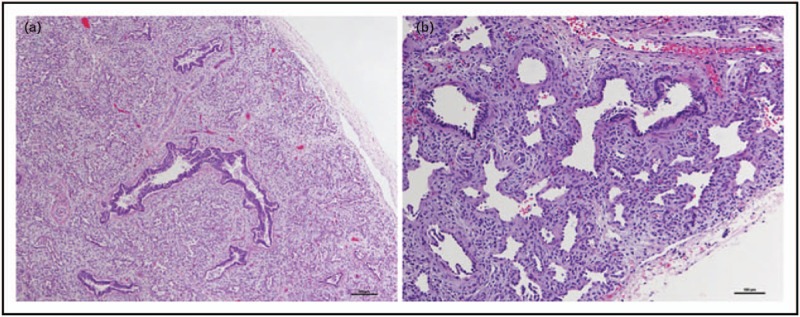
Lung histopathology of a patient with acinar dysplasia. (a) Lung from the infant's autopsy, showing an immature appearance for gestational age with no saccular structures or alveoli. (b) Higher power demonstrating abnormal acinar structures with abundant intervening mesenchyme (hematoxylin and eosin). Reproduced with permission from [42].
Galambos et al.[46▪▪] subsequently further extended the spectrum of paediatric diffuse parenchymal lung disease associated with TBX4 variants. The authors described a cohort of 19 children with TBX4 variants, including six children with loss of the TBX4 gene due to 17q23 deletions and thirteen children with intragenic mutations. The majority of the children (63%) presented with a biphasic clinical course of neonatal hypoxic respiratory failure and persistent pulmonary hypertension of the neonate (PPHN), which seemed to resolve around the age of 1 month, but followed by chronic PH later in infancy or early childhood. Histopathological assessment was available in seven children and showed mainly signs of severe disruption of all compartments of distal lung development similar to CAD or AcDys, or signs of pulmonary interstitial glycogenosis (PIG). The children with a severe presentation of PPHN in the beginning of their life also showed more severe disruption of the distal lung development compared with children who were diagnosed later in childhood. Some children had prominent interstitial lung disease. This series unequivocally shows that TBX4 variants are not only associated with lethal developmental lung diseases such as AcDys or CAD, but may also present with milder growth abnormallities or nonspecific interstitial lung diseases. Interestingly, a recent case report of Maurac et al.[47▪] suggests that such developmental growth anomalies are not necessarily associated with neonatal presentation. The authors describe a 34-year-old woman presenting with unexplained PAH and right heart failure [47▪]. On x-rays of the feet and the pelvis, the patient showed signs of SPS. Chest computed tomography (CT) revealed peripheral reticular lines, centrilobular nodules, thin-walled cysts in the upper lobes and an appearance resembling diverticula in the trachea and large bronchi. Lung biopsy from the left upper lobe showed densification foci of the lung parenchyma with cholesterol crystals. Although the histopathological abnormalities were not typical for developmental distal lung abnormalities, the tracheal abnormalities in this patient highlight a potential role of TBX4 in a disturbed development of the larger airways [11].
The studies described here strongly suggest that TBX4 variants can be associated with a broad spectrum of lung diseases with a wide age range of clinical presentations. Neonates and infants with TBX4 variants are clearly overrepresented in the reports on parenchymal lung diseases and authors have frequently used the terminology ‘developmental lung disease’. Different classifications for children's interstitial and diffuse lung disease (chILD) have been proposed. The American Thoracic Society published a classification of chILD in 2013 where AcDys, CAD, alveolar capillary dysplasia with misalignment of pulmonary veins (ACDMPV) are classified as diffuse developmental disorders, whereas primary pulmonary hypoplasia is classified under growth abnormalities (Table 2) [48,49▪]. The structural changes seen in these diseases are associated with the different stages during lung embryogenesis in which growth arrest occurs; the pseudoglandular (AcDys), canalicular (CAD) and saccular/alveolar (both ACDMPV and primary pulmonary hypoplasia) phase [50▪▪]. Also, PIG, characterised by interstitial expansion due to increased mesenchymal cellularity, is regarded a disease of altered lung development [49▪,51]. Also, other diffuse lung diseases such as chronic neonatal lung disease (i.e. bronchopulmonary dysplasia), or surfactant dysfunction disorders (i.e. nonspecific interstitial pneumonia) are associated with disturbed or disrupted vascular and parenchymal lung development [46▪▪,47▪,48].
Table 2.
Classification scheme for paediatric diffuse lung disease [48]
| I. Disorders more prevalent in infancy A. Diffuse developmental disorders 1. Acinar dysplasia 2. Congenital alveolar dysplasia 3. Alveolar-capillary dysplasia with pulmonary vein misalignment B. Growth abnormalities 1. Pulmonary hypoplasia 2. Chronic neonatal lung disease A. Prematurity-related chronic lung disease (bronchopulmonary dysplasia) B. Acquired chronic lung disease in term infants 3. Structural pulmonary changes with chromosomal abnormalities A. Trisomy 21 B. Others 4. Associated with congenital heart disease in chromosomally normal children C. Specific conditions of undefined aetiology 1. Pulmonary interstitial glycogenosis 2. Neuroendocrine cell hyperplasia of infancy D. Surfactant dysfunction mutations and related disorders 1. SPFTB genetic mutations – PAP and variant dominant histologic pattern 2. SPFTC genetic mutations – CPI dominant histologic pattern; also DIP and NSIP 3. ABCA3 genetic mutations – PAP variant dominant pattern; also CPI, DIP, NSIP 4. Others with histology consistent with surfactant dysfunction disorder without a yet recognised genetic disorder |
| II. Disorders not specific to infancy A. Disorders of the normal host 1. Infectious and postinfectious processes 2. Disorders related to environmental agents: hypersensitivity pneumonia, toxic inhalation 3. Aspiration syndromes 4. Eosinophilic syndromes B. Disorders related to systemic disease processes 1. Immune-related disorders 2. Storage disease 3. Sarcoidosis 4. Langerhans cell histiocytosis 5. Malignant infiltrates C. Disorders of the immunocompromised host 1. Opportunistic infection 2. Disorders related to therapeutic intervention 3. Disorders related to transplantation and rejection syndromes 4. Diffuse alveolar damage of unknown aetiology D. Disorders masquerading as interstitial disease 1. Arterial hypertensive vasculopathy 2. Congestive vasculopathy, including veno-occlusive disease 3. Lymphatic disorders 4. Congestive changes related to cardiac dysfunction |
| III. Unclassified – includes end-stage disease, nondiagnostic biopsies and those with inadequate material |
CPI, chronic pneumonitis of infancy; DIP, desquamative cell interstitial pneumonia; NISP, nonspecific interstitial pneumonia; PAP, pulmonary alveolar proteinosis.
ChILD is a complex spectrum of disorders with a high variety in clinical phenotypes for which the associated pathophysiology is insufficiently understood. Therefore, although the notable role of TBX4 in embryonic development may be tempting, the term ‘developmental lung disease’ as a common denominator in the phenotypical spectrum of TBX4-associated lung diseases should be used with caution.
The reason for the variety in clinical presentation in patients carrying a TBX4 variant remains unclear. It has been hypothesised that the type of mutation may determine both phenotype and its severity, but currently, there does not seem to be a pattern when comparing all described different mutations [41,46▪▪,50▪▪]. Genetic modifiers – coding or noncoding – may play a role in the variability of the phenotype. Karolak et al. demonstrated that all infants with developmental lung disease with either a TBX4 variant or FGF10 variant also harboured at least one noncoding mutation in BCAS3 (microtubule associated cell migration factor), which has overlap with the predicted regulatory elements that have been identified in the human foetal lung fibroblast cell line [43▪▪,52]. However, children with deletions of chromosome 17q23.2, harbouring both TBX4 and BCAS3, do not necessarily present with lung disease or PAH [45]. Karolak et al.[53] recently published a case report on an infant with PIG, harbouring both a TBX4 variant and a CTNNB1 mutation (associated with intellectual disability and neurodevelopmental abnormalities). The authors state that a heterozygous TBX4 variant alone may not be sufficient to cause lung disease and suggest that mixed phenotypes may be due to complex compound inheritance [54]. In addition, environmental and maternal/pregnancy-related factors may also influence the severity of the clinical phenotype [55].
DIAGNOSTICS
Characterising chILD is difficult especially in neonates [48]. Symptoms are nonspecific and neonatal presentation will complicate the diagnostic process. For a long time, lung biopsy for histopathological analyses has been the ultimate diagnostic, limited by high procedural risks and often low diagnostic yield. Genetic testing is gaining ground due to the identification of an increasing number of genetic causes for several interstitial lung diseases [48]. The presence of a certain mutation can have profound effects for clinical decision making: the dismal prognosis of a FOXF1 mutation causing ACDMPV will affect treatment decisions in a neonate dependent on intensive ventilatory support and will avoid the otherwise required lung biopsy [50▪▪]. Standard testing for TBX4 variants in neonates with respiratory insufficiency has been proposed: Vincent et al.[50▪▪] proposed to screen infants with AcDys, CAD or other lung hypoplasias on TBX4 variants when there is a family history of PAH. However, as long as the prevalence and incidence of TBX4 variants in neonates and the consequences in terms of treatment strategies are not defined, the role of standard testing on TBX4 variants is unclear.
CLASSIFICATION OF PULMONARY ARTERIAL HYPERTENSION ASSOCIATED WITH TBX4
During the most recent World Symposium on Pulmonary Hypertension in 2018, the clinical classification of PH was updated [25]. In this classification, unexplained PAH-associated TBX4 mutations are assigned to group 1 PAH (HPAH). The Pediatric Task Force introduced in Group 3: PH due to lung diseases and/or hypoxia, a subclass ‘PH associated with developmental lung disease’, in order to accommodate neonatal PH (other than PPHN) in the classification. Now, as TBX4 variants are frequently associated with developmental lung diseases, the appropriate classification of an infant or child with a TBX4 variant and PH becomes more complicated and may be hazardous. This forces the diagnosing physician to a meticulous assessment of any lung disease and, if present, its causal role in the presence of PH. This is obviously a challenging task in small infants, but crucial in defining optimal treatment strategy.
CONCLUSION
Animal studies have demonstrated an important role of the TBX4 gene in the embryonic development of limbs and tail mesoderm, the lung mesenchyme and the genital papilla [5,9]. The clinical phenotype of TBX4 variants in humans has expanded from bone disorders to vascular and parenchymal lung diseases and is likely to expand further. The enrichment of TBX4 variants in PAH justifies inclusion of this gene in genetic analyses in patients with PAH. Further, TBX4 variants have shown to contribute to diffuse lung diseases, especially but not exclusively in neonates, and seems to be multifaced. Importantly, the exact genotype-phenotype correlations have to be further ‘unboxed’.
Acknowledgements
None.
Financial support and sponsorship
This work was supported by the Sebald Fund.
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1.Dobrovolskaïa-Zavadskaïa N. On spontaneous tail mortification in newborn mice and on the existence of a ’non-viable’ hereditary trait (factor). Soc Biol 1927; 97:116–118. [Google Scholar]
- 2.Herrmann BG, Labeit S, Poustka A, et al. Cloning of the T gene required in mesoderm formation in the mouse. Nature 1990; 343:617–622. [DOI] [PubMed] [Google Scholar]
- 3.Bollag RJ, Siegfried Z, Cebra-Thomas JA, et al. An ancient family of embryonically expressed mouse genes sharing a conserved protein motif with the T locus. Nat Genet 1994; 7:383–389. [DOI] [PubMed] [Google Scholar]
- 4.Agulnik SI, Garvey N, Hancock S, et al. Evolution of mouse T-box genes by tandem duplication and cluster dispersion. Genetics 1996; 144:249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chapman DL, Garvey N, Hancock S, et al. Expression of the T-box family genes, Tbx1-Tbx5, during early mouse development. Dev Dyn 1996; 206:379–390. [DOI] [PubMed] [Google Scholar]
- 6.Naiche LA, Harrelson Z, Kelly RG, et al. T-box genes in vertebrate development. Annu Rev Genet 2005; 39:219–239. [DOI] [PubMed] [Google Scholar]
- 7.Yi CH, Russ A, Brook JD. Virtual cloning and physical mapping of a human T-box gene, TBX4. Genomics 2000; 67:92–95. [DOI] [PubMed] [Google Scholar]
- 8.Gibson-Brown JJ, Agulnik I, Silver S, et al. Expression of T-box genes Tbx2-Tbx5 during chick organogenesis. Mech Dev 1998; 74:165–169. [DOI] [PubMed] [Google Scholar]
- 9.Li M, Zhang H, Liu H, et al. Abnormal expression of TBX4 during anorectal development in rat embryos with ethylenethiourea-induced anorectal malformations. Biol Res 2019; 52:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naiche LA, Arora R, Kania A, et al. Identity and fate of Tbx4-expressing cells reveal developmental cell fate decisions in the allantois, limb, and external genitalia. Dev Dyn 2011; 240:2290–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arora R, Metzger RJ, Papaioannou VE. Multiple roles and interactions of Tbx4 and Tbx5 in development of the respiratory system. PLoS Genet 2012; 8:e1002866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cebra-Thomas JA, Bromer J, Gardner R, et al. T-box gene products are required for mesenchymal induction of epithelial branching in the embryonic mouse lung. Dev Dyn 2003; 226:82–90. [DOI] [PubMed] [Google Scholar]
- 13.Abler LL, Mansour SL, Sun X. Conditional gene inactivation reveals roles for Fgf10 and Fgfr2 in establishing a normal pattern of epithelial branching in the mouse lung. Dev Dyn 2008; 238:1999–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bellusci S, Grindley J, Emoto H, et al. Fibroblast Growth Factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development 1997; 124:4867–4878. [DOI] [PubMed] [Google Scholar]
- 15.Ohuchi H, Hori Y, Yamasaki M, et al. FGF10 acts as a major ligand for FGF receptor 2 IIIb in mouse multiorgan development. Biochem Biophys Res Commun 2000; 277:643–649. [DOI] [PubMed] [Google Scholar]
- 16.Sakiyama JI, Yamagishi A, Kuroiwa A. Tbx4-Fgf10 system controls lung bud formation during chicken embryonic development. Development 2003; 130:1225–1234. [DOI] [PubMed] [Google Scholar]
- 17.Goss AM, Tian Y, Tsukiyama T, et al. Wnt2/2b and β-catenin signaling are necessary and sufficient to specify lung progenitors in the foregut. Dev Cell 2009; 17:290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Itoh N. FGF10: a multifunctional mesenchymal-epithelial signaling growth factor in development, health, and disease. Cytokine Growth Factor Rev 2016; 28:63–69. [DOI] [PubMed] [Google Scholar]
- 19.Takahashi T, Friedmacher F, Zimmer J, et al. Expression of T-box transcription factors 2, 4 and 5 is decreased in the branching airway mesenchyme of nitrofen-induced hypoplastic lungs. Pediatr Surg Int 2017; 33:139–143. [DOI] [PubMed] [Google Scholar]
- 20.Akiyama H, Chaboissier MC, Martin JF, et al. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev 2002; 16:2813–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haarman MG, Kerstjens-Frederikse WS, Berger RMF. The ever-expanding phenotypical spectrum of human TBX4 mutations: from toe to lung. Eur Respir J 2019; 54:pii: 1901504. [DOI] [PubMed] [Google Scholar]
- 22.Bongers EMHF, Duijf PHG, van Beersum SEM, et al. Mutations in the human TBX4 gene cause small patella syndrome. Am J Hum Genet 2004; 74:1239–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kerstjens-Frederikse WS, Bongers EMHF, Roofthooft MTR, et al. TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J Med Genet 2013; 50:500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53:pii: 1801913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenzweig EB, Abman SH, Adatia I, et al. Paediatric pulmonary arterial hypertension: updates on definition, classification, diagnostics and management. Eur Respir J 2019; 53:pii: 1801916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barst RJ, Ertel SI, Beghetti M, et al. Pulmonary arterial hypertension: a comparison between children and adults. Eur Respir J 2011; 37:665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Loon RLE, Roofthooft MTR, van Osch-Gevers M, et al. Clinical characterization of pediatric pulmonary hypertension: complex presentation and diagnosis. J Pediatr 2009; 155:176–182. e1. [DOI] [PubMed] [Google Scholar]
- 28.Harrison RE, Berger R, Haworth SG, et al. Transforming growth factor-β receptor mutations and pulmonary arterial hypertension in childhood. Circulation 2005; 111:435–441. [DOI] [PubMed] [Google Scholar]
- 29.Deng Z, Morse JH, Slager SL, et al. Familial primary pulmonary hypertension (Gene PPH1) is caused by mutations in the bone morphogenetic protein receptor–II gene. Am J Hum Genet 2000; 67:737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lane KB, Machado RD, Pauciulo MW, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nat Genet 2000; 26:81–84. [DOI] [PubMed] [Google Scholar]
- 31.Austin ED, Ma L, LeDuc C, et al. Whole exome sequencing to identify a novel gene (Caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet 2012; 5:336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eyries M, Montani D, Girerd B, et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat Genet 2014; 46:65–69. [DOI] [PubMed] [Google Scholar]
- 33.Levy M, Eyries M, Szezepanski I, et al. Genetic analyses in a cohort of children with pulmonary hypertension. Eur Respir J 2016; 48:1118–1126. [DOI] [PubMed] [Google Scholar]
- 34.Ma L, Roman-Campos D, Austin ED, et al. A novel channelopathy in pulmonary arterial hypertension. N Engl J Med 2013; 369:351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Girerd B, Weatherald J, Montani D, Humbert M. Heritable pulmonary hypertension: from bench to bedside. Eur Respir Rev 2017; 26:170037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ivy DD, Abman SH, Barst RJ, et al. Pediatric pulmonary hypertension. J Am Coll Cardiol 2013; 62: Suppl 25: D117–D126. [DOI] [PubMed] [Google Scholar]
- 37▪.Zhu N, Gonzaga-Jauregui C, Welch CL, et al. Exome sequencing in children with pulmonary arterial hypertension demonstrates differences compared with adults. Circ Genomic Precis Med 2018; 11:e001887. [DOI] [PMC free article] [PubMed] [Google Scholar]; A large study on exome sequencing in patients with pulmonary arterial hypertension (PAH) that demonstrates that TBX4 variants are enriched in children with idiopathic/heritable PAH (IPAH/HPAH) compared with adults with IPAH/HPAH. This study also shows that TBX4 variant carriers have a 20-younger earlier age of disease onset compared with BMPR2 mutation carriers.
- 38.Eyries M, Montani D, Nadaud S, et al. Widening the landscape of heritable pulmonary hypertension mutations in paediatric and adult cases. Eur Respir J 2018; 53:pii: 1801371. [DOI] [PubMed] [Google Scholar]
- 39.Navas P, Tenorio J, Quezada CA, et al. Molecular analysis of BMPR2, TBX4, and KCNK3 and genotype-phenotype correlations in Spanish patients and families with idiopathic and hereditary pulmonary arterial hypertension. Rev Española Cardiol (Engl Ed) 2016; 69:1011–1019. [DOI] [PubMed] [Google Scholar]
- 40.Gräf S, Haimel M, Bleda M, et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat Commun 2018; 9:1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szafranski P, Coban-Akdemir ZH, Rupps R, et al. Phenotypic expansion of TBX4 mutations to include acinar dysplasia of the lungs. Am J Med Genet Part A 2016; 170:2440–2444. [DOI] [PubMed] [Google Scholar]
- 42.German K, Deutsch GH, Freed AS, et al. Identification of a deletion containing TBX4 in a neonate with acinar dysplasia by rapid exome sequencing. Am J Med Genet Part A 2019; 179:842–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43▪▪.Karolak JA, Vincent M, Deutsch G, et al. Complex compound inheritance of lethal lung developmental disorders due to disruption of the TBX-FGF pathway. Am J Hum Genet 2019; 104:213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports on a fairly large group of neonates with developmental lung diseases in whom TBX4 and FGF10 variants were found. The results of this study address the important role of the TBX4-FGF10-FGFR2 epithelial-mesenchymal signalling in human lung organogenesis and in the pathogenesis of this rare entity of lung diseases.
- 44.Suhrie K, Pajor NM, Ahlfeld SK, et al. Neonatal lung disease associated with TBX4 mutations. J Pediatr 2019; 206:286–292. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ballif BC, Theisen A, Rosenfeld JA, et al. Identification of a recurrent microdeletion at 17q23.1q23 2 flanked by segmental duplications associated with heart defects and limb abnormalities. Am J Hum Genet 2010; 86:454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46▪▪.Galambos C, Mullen MP, Shieh JT, et al. Phenotype characterisation of TBX4 mutation and deletion carriers with neonatal and pediatric pulmonary hypertension. Eur Respir J 2019; 54:pii: 1801965. [DOI] [PubMed] [Google Scholar]; This study describes a group of 19 children with TBX4 variants and lung diseases, including developmental lung diseases. This study demonstrates that the spectrum of lung diseases caused by TBX4 variants not only includes severe neonatal developmental lung diseases but also milder developmental abnormalities or nonspecific interstitial lung diseases.
- 47▪.Maurac A, Lardenois É, Eyries M, et al. T-box protein 4 mutation causing pulmonary arterial hypertension and lung disease. Eur Respir J 2019; 54:pii: 1900388. [DOI] [PubMed] [Google Scholar]; This case report highlights the role of a TBX4 variant in an adult with PAH and lung disease, demonstrating that the combination of a TBX4 variant and lung disease not only occurs in children but also in adults.
- 48.Kurland G, Deterding RR, Hagood JS, et al. An official american thoracic society clinical practice guideline: classification, evaluation, and management of childhood interstitial lung disease in infancy. Am J Respir Crit Care Med 2013; 188:376–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49▪.Cunningham S, Jaffe A, Young LR. Children's interstitial and diffuse lung disease. Lancet Child Adolesc Heal 2019; 3:568–577. [DOI] [PubMed] [Google Scholar]; This review deepens into the progress that has already been made in the diagnostics and treatment of chILD. However, it also addresses the many difficulties of this rare group of disease that need to be overcome.
- 50▪▪.Vincent M, Karolak JA, Deutsch G, et al. Clinical, histopathological, and molecular diagnostics in lethal lung developmental disorders. Am J Respir Crit Care Med 2019; 200:1093–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]; This review sheds light on the new insights of genetic causes of lethal developmental lung diseases. The authors propose genetic testing of children with a developmental lung disease, which may help in identifying new candidate genes responsible for developmental lung diseases.
- 51.Canakis AM, Cutz E, Manson D, O’Brodovich H. Pulmonary interstitial glycogenosis: a new variant of neonatal interstitial lung disease. Am J Respir Crit Care Med 2002; 165:1557–1565. [DOI] [PubMed] [Google Scholar]
- 52.Hnisz D, Abraham BJ, Lee TI, et al. Super-enhancers in the control of cell identity and disease. Cell 2013; 155:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karolak JA, Szafranski P, Kilner D, et al. Heterozygous CTNNB1 and TBX4 variants in a patient with abnormal lung growth, pulmonary hypertension, microcephaly, and spasticity. Clin Genet 2019; 96:366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Posey JE, Harel T, Liu P, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med 2017; 376:21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yue H, Ji X, Li G, et al. Maternal exposure to PM2.5 affects fetal lung development at sensitive windows. Environ Sci Technol 2019; 54:316–324. [DOI] [PubMed] [Google Scholar]