

Abstract
Purpose of review
Crescents are classical histopathological lesions found in severe forms of rapidly progressive glomerulonephritis, also referred to as crescentic glomerulonephritis (CGN). Crescent formation is a consequence of diverse upstream pathomechanisms and unraveling these mechanisms is of great interest for improving the management of patients affected by CGN. Thus, in this review, we provide an update on the latest insight into the understanding on how crescents develop and how they resolve.
Recent findings
Cellular crescents develop from activated parietal epithelial cells (PECs) residing along Bowman's capsule and their formation has as a consequence the decline in glomerular filtration rate (GFR). Cellular crescents can be reversible, but when multilevel growth of PECs associate with an epithelial--mesenchymal transition-like change in cell phenotype, fibrous crescents form, and crescents become irreversible also in terms of GFR recovery. Different molecular pathways trigger the activation of PECs and are a prime therapeutics target in CGN. First, crescent formation requires also vascular injury causing ruptures in the glomerular basement membrane that trigger plasmatic coagulation within Bowman's space. This vascular necrosis can be triggered by different upstream mechanisms, such as small vessel vasculitides, immune complex glomerulonephritis, anti-GBM disease, and C3 glomerulonephritis, that all share complement activation but involve diverse upstream immune mechanisms outside the kidney accessible for therapeutic intervention.
Summary
Knowing the upstream mechanisms that triggered crescent formation provides a tool for the development of therapeutic interventions for CGN.
Keywords: cellular crescents, crescentic glomerulonephritis, parietal epithelial cells
INTRODUCTION
The term ‘glomerular crescent’ is used for hyperplastic lesions involving 10% or more of the circumference of Bowman‘s capsule. Crescents can be composed of a variable mixture of cells, fibrin, and fibrous matrix [1]. A composition of more than 75% cells and fibrin and less than 25% fibrous matrix is referred to as ‘cellular crescent’, a composition of more 25–75% cells and fibrin and the remainder fibrous matrix is a ‘fibrocellular crescent’, and more than 75% fibrous matrix and less than 25% cells and fibrin is a ‘fibrous crescent’ [1].
Diagnostic kidney biopsies reveal the occasional presence of crescents in a wide range of glomerular disorders but crescents are the predominant histopathological lesions found in severe forms of rapidly progressive glomerulonephritis (RPGN), frequently in association with acute or subacute renal failure, also referred to as ‘crescentic glomerulonephritis’ (CGN). However, RPGN and CGN are only descriptive clinical or morphological terms, respectively, and no diagnoses [2▪]. Only integrating all results from clinical phenotyping, standard laboratory tests, and specific immunological exams can ultimately clarify the diagnosis underlying CGN.
As such, crescent formation is the consequence of diverse upstream pathomechanisms that fuel into shared cellular responses. Attempts to unravel these mechanisms involve different tools on the clinical and experimental research domains (Table 1). Here we provide an update on the latest insights into the understanding on how crescents develop, how they resolve, and how this knowledge may help to improve the management of patients affected by CGN.
Table 1.
Tools how to study crescentic GN: advantages and disadvantages
| Model systems |
| Humans: histological manifestation of severe glomerular damage characterized by crescents in ≥50% glomeruli and loss of kidney function. CGN can be classified according to immunopathologic features: type I (produced by anti-GBM antibodies); type II (immune complexes deposited in glomeruli), and type III (without deposits of immunoglobulings or complement). A fourth type has been proposed for those cases in which there is a coexistence of anti-GBM disease and ANCA-associated GN. |
| Animal models: species used are mainly mice and rats. Induction with nephrotoxic serum nephritis in mice and rats that can effectively mimic an immune complex-mediated GN. Good availability and controllable experimental settings. There are also some spontaneous animal models developing CGN, such as the spontaneous crescentic glomerulonephritis-forming/Kinjoh (SCG/Kj) mice. However, these animal models do not totally mimic human CGN, which complicates translation of experimental therapies into practice. |
| In-vitro studies: good availability and controllable experimental settings, using both cell lines and primary cells. Isolation of capsulated glomeruli as a model of glomerular crescentic lesion formation and use of immortalized or primary murine PECs for studying the activation of PECs. |
| Analytical tools |
| Kidney biopsy: to perform a differential diagnosis for glomerulonephritis. |
| Imaging tools: immunofluorescence and confocal microscopy. |
| Laser capture microdissection: performed to isolate glomerular hyperplastic lesions and healthy glomerular tufts. |
ANCA, antineutrophil cytoplasmic antibodies; CGN, crescentic glomerulonephritis; GBM, glomerular basement membrane; PEC, parietal epithelial cell.
Box 1.

no caption available
THE EVOLUTION AND RESOLUTION OF GLOMERULAR CRESCENTS
The dynamics of crescent formation can be assumed from different stages in single patient biopsies, serial biopsies, or animal models (Fig. 1).
FIGURE 1.
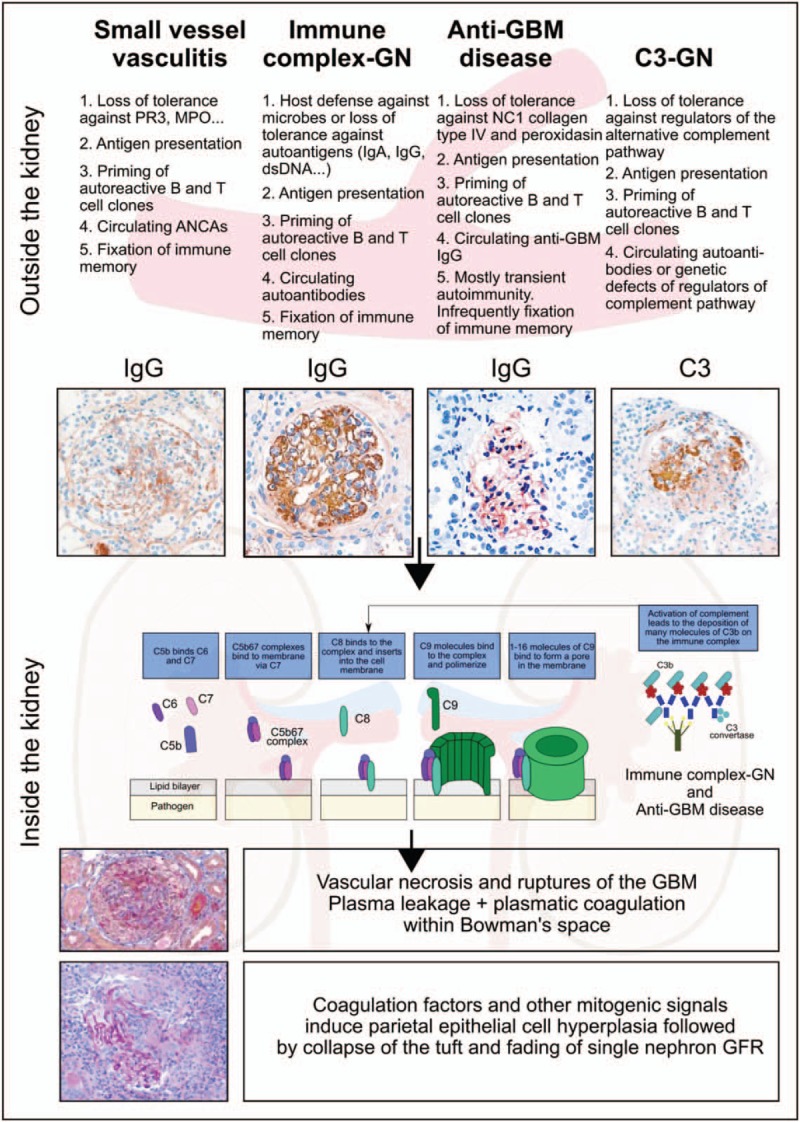
Evolution of crescent formation. The crescent is an unspecific histopathological lesion that can be triggered by a variety of different underlying disorders. This figure illustrates four of those and highlights first the extrarenal pathomechanisms leading to glomerular injury. The different patterns of immunoglobulin and complement deposits help to dissect these entities. Indeed, all of the aforementioned extrarenal drivers of injury to the glomerular microvasculature involve local complement activation as a shared molecular target for therapeutic interventions. Whenever, microvascular injury leads to rupture of the glomerular basement membrane (GBM), the leakage of plasma proteins drives parietal epithelial cell hyperplasia as the key cellular component of the crescent. Single nephron GFR decreases because of tuft collapse, rupture of the Bowman capsule, and influx of immune cells and fibroblasts are all secondary events that may or may not occur in individual glomeruli. Periglomerular immune cell infiltrates or fibrotic encasting of the activated parietal cells (fibrocellular crescents) are subsequent events that may affect the dynamics and prognosis of the disease. ANCA, antineutrophil cytoplasmic antibody; GFR = glomerular filtration rate; Ig, immunoglobulin; MPO, myeloperoxidase; PR3, proteinase 3.
Cellular crescents
Lineage tracing studies in transgenic mice clearly document that the cellular crescents develop from the parietal epithelial cells (PECs) residing along the inner aspect of Bowman‘s capsule [3]. The cell of origin may be a subset of less differentiated PECs that maintained an immature progenitor-like phenotype with a higher capacity for hyperplastic expansion [4]. Whether glomerular crescents are monoclonal or polyclonal lesions is unknown and would require experimental tools allowing clonal lineage tracing [5]. Adjacent podocytes may become part of the crescent but do not contribute to the primary lesion [3]. The functional consequence of crescent formation is a decline in single nephron glomerular filtration rate (GFR) for two reasons. First, the increasing intra-glomerular mass increases the counter-pressure that together with oncotic pressure counterbalances arterial filtration pressure. Already slight increases in counter-pressure will cease the physiological net filtration pressure across the glomerular filtration barrier of 10 mmHg and may endorse the collapse of the glomerular tuft [6]. Second, once a glomerular crescent involves and obstructs the urinary pole, the entire nephron no longer contributes to total GFR [7]. However, the fortunate clinical course of some patients with severe CGN suggests that cellular crescents are reversible lesions. Indeed, treatment with steroids and cyclophosphamide elicits anti-inflammatory and antiproliferative effects not only on the immune system but also on hyperplastic epithelial lesions. Whether cells of a cellular crescent are cleared by detachment and shedding of cells into the urine or by apoptosis and phagocytic clearance in situ is not known.
Fibrocellular and fibrous crescents
Multilevel growth of PECs can be associated with an epithelial--mesenchymal transition-like change in cell phenotype characterized by a loss of polarization and release of extracellular matrix towards all directions. The histomorphological hallmark of this process is a gradual encasement of PECs with extracellular matrix leading to honeycomb-like structures within Bowman‘s space [8]. The expanding extracellular matrix can make up a majority of the crescent area, whereas the cellular components succumb, that is, a fibrous crescent. These structures are considered irreversible in terms of a potential recovery of single nephron GFR, and the nephron ultimately undergoes atrophy and phagocytic clearance accompanied and followed by interstitial fibrosis. This sequence of events argues against interstitial fibrosis being a suitable target for therapeutic intervention as reducing interstitial fibrosis in this setting neither recovers lost nephrons nor their function.
Periglomerular inflammation and ruptures of Bowman‘s capsule
Glomerular crescents can be accompanied by a strong periglomerular inflammatory response, potentially triggered by proinflammatory mediators released from activated parietal epithelial cells across Bowman‘s capsule. Indeed, immune cells more easily adhere and transmigrate from the low flow, low shear stress, postcapillary venules rather than from the high flow, high pressure glomerular capillaries. Particularly animal models of RPGN show prominent periglomerular T-cell infiltrates but Bowman‘s capsule forms a barrier preventing T cells from entering the glomerulus and contributing fibrous organization of crescents and nonrecoverable injury [9]. Indeed, Bowman's capsule acts as an active immunologic shield that protects the glomerular integrity in glomerulonephritis. In contrast, the well-described role of T-cell subsets to experimental CGN may rather relate to extrarenal roles in regulating systemic (auto-) immunity upstream to renal manifestations [10,11▪▪,12]. Ruptures of Bowman‘s capsule are sometimes seen, which allows periglomerular immune cells to populate the crescent [13].
MOLECULAR PATHWAYS OF PARIETAL EPITHELIAL CELL HYPERPLASIA IN CRESCENTIC GLOMERULONEPHRITIS
Activation of PECs is an important pathomechanism and prime therapeutic target in crescentic glomerulonephritis, given its association with cellular crescent formation [3]. Several molecular pathways are involved in the hyperplasia of PECs (Fig. 1).
CD44 and CD9
A recent study in experimental CGN found that CD44, a cell surface glycoprotein that plays a key role in various cellular processes, is expressed in activated PECs and that its deficiency was associated with reduced presence of PECs in Bowman‘s space [14▪▪]. In addition, CD44 deficiency reduced glomerular cell proliferation and reduced albuminuria, indicating a link among CD44-expresing activated PECs, the formation of crescents, and the development of albuminuria. In association with CD44 expression, CD9, a tetraspanin involved in cell proliferation, migration, adhesion, and survival was found in PECs of a CGN-like rodent model [15▪▪]. Silencing CD9 attenuated the ability of PECs to proliferate and migrate and attenuated glomerulosclerosis. One possible mechanism of PEC activation via CD9 relates to the activation of epidermal growth factor receptor, a key driver of kidney damage in early stages of glomerulonephritis [15▪▪]. Thus, suppressing the local expression of CD9 can alleviate glomerular damage and could be a therapeutic option for crescentic glomerulonephritis.
Glucocorticoids
Glucocorticoids have remained in use for the treatment of glomerulonephritis since decades. A recent study investigated the effects of glucocorticoids in glomerulonephritis and found that glucocorticoid receptor inhibition was associated with decreased cellular crescent formation and inhibition of proliferation and migration of PECs [16]. This therapeutic approach also reduced the inflammatory infiltrate within the kidney, suggesting intracellular steroid receptors may contribute to inflammation in CGN [16].
Heparin-binding epidermal growth factor-like growth factor
Heparin-binding epidermal growth factor-like growth factor (HB-EGF), a member of the EGF family, increases phosphorylation of the EGFR. In an animal model of CGN, proHB-EGF was induced in PECs and podocytes even before the first appearance of crescents [17]. Similar results were obtained in kidney biopsies from subjects with crescentic RPGN, in which HB-EGF staining was noted in all epithelia of the nephron, that is, podocytes, PECs, and tubules [17]. HB-EGF deficiency attenuated CGN as did conditional deletion of EGFR only in podocytes, suggesting autocrine and paracrine feedback loops. Indeed, even delayed pharmacological inhibition of the EGFR mice could still attenuate CGN, which may be of translational relevance for human RPGN.
THE SHARED UPSTREAM TRIGGER OF CRESCENT FORMATION: VASCULAR NECROSIS
Why do parietal epithelial cells start to proliferate in some but not in all forms of glomerulonephritis? It is of note that isolated podocyte loss and tuft--capsule adhesions lead to migration of parietal epithelial cells on the tuft. Some parietal epithelial cells have an intrinsic (progenitor-like) capacity to differentiate into podocytes and hence to regenerate lost podocytes, a condition that can be pharmaceutically enhanced in superficial nephrons [18]. In contrast, once parietal epithelial cells that lack such progenitor capacity migrate on the tuft they contribute to focal segmental glomerulosclerosis [19]. Neither of these conditions involves crescent formation. Crescent formation requires an additional level of injury to the glomerular filtration barrier, that is, vascular injury with holes or major breaks of the glomerular basement membrane (GBM) that trigger the plasmatic coagulation cascade within Bowman‘s space (Fig. 1) [6]. Usually, GBM ruptures occur upon massive immunological injuries affecting the GBM and only rarely upon GBM degeneration [8,20]. Fibrin deposits devoid of platelets, neutrophils, and red blood cells indicate the plasmatic coagulation process within the crescent [1,6,8], which is a direct stimulus for parietal cell hyperplasia [8], as for re-epithelialization of other bleeding wounds [21]. Indeed, urokinase and heparin abrogate crescent formation in mice [8,22]. In the following, we discuss the upstream mechanisms that lead to the GBM ruptures as the upstream event in CGN.
SPECIFIC TRIGGERS FOR VASCULAR NECROSIS IN DIFFERENT FORMS OF CRESCENTIC GLOMERULONEPHRITIS
Different upstream mechanisms trigger glomerular loop necrosis in the different forms of CGN (Fig. 1).
Small vessel vasculitides
Most biopsy studies name the different forms of pauci-immune small vessel vasculitis as the most frequent cause underlying CGN. These diseases involve antineutrophil cytoplasmic antibodies (ANCA), which render neutrophils more susceptible to the release of neutrophil extracellular traps (NETs) along the endothelial interface of the renal microvasculature [23▪]. NETs are composed of nuclear DNA that by itself contributes few to vascular necrosis [24]. Rather the nuclear, granular, and cytoplasmic proteins, such as elastase, myeloperoxidase, cathepsins, and histones decorating the NETs confer severe endothelial cell and vascular wall injury [23▪,25–27,28▪]. NETs also deposit neutrophil autoantigens along the capillary wall that are picked up by antigen-presenting cells that can trigger local T-cell responses [29▪]. Local complement dysregulation is an essential component in the vascular injury process [30,31] and blocking elements of the terminal complement cascade is a promising therapeutic avenue for small vessel vasculitis [32]. However, upstream of NET formation the pathogenesis of small vessel vasculitis involves loss of tolerance to neutrophil antigens and all components of adaptive immunity that contribute to the secretion of ANCA and the priming of autoantigen-specific T cells [23▪,33▪▪]. Global immunosuppressants attenuate autoimmunity but recently B-cell-targeting therapies have been found to be as potent in abrogating vasculitis and CGN [23▪,34▪]. This may not only relate to the role of B cells as precursors for ANCA-secreting plasma cells but also to their role as antigen-presenting cells [34▪].
Immune complex glomerulonephritis
IC-GNs are frequent, in particular, in low-income and middle-income countries, and occasionally present as CGN [35]. In crescentic IC-GN, the proportion of glomeruli affected by crescents is lower and more frequently shows signs of chronicity, for example, in IgA nephropathy [36,37]. The presence of crescents in IgA nephropathy predicts poor renal outcome [35,36,38▪▪,39,40]. Crescents were not included as a criterion into the prognostic score of the initial Oxford classification but the original study did not include those with RPGN who reached end-stage kidney disease within 1 year, and thus was underrepresented to study impact of crescents. [41]. Its further additional expansion indeed does include crescents as a prognostic morphology marker [42]. The new data show that crescents either cellular or fibrocellular involving less than 25% of glomeruli are not associated with worse prognosis if patents received immunosuppression.
In contrast, patients with more than 25%, crescents prognosis was worse even with immunosuppression [42]. The reasons why some patients with IgA nephropathy develop crescents whereas most other do not are uncertain and one may speculate that additional pathogenic factors that accelerate injury to the glomerular microvasculature coincide, for example, genetic variants in complement genes or additional pathogenic autoantibodies. Some patients with crescentic IgA nephropathy show concomitant positivity for ANCA and clinical signs of systemic autoimmunity [43,44]. Proliferative lupus nephritis presents as CGN in 40% of the cases and concomitant ANCA positivity increases this percentage to 60% together with an increase in the prevalence of vascular necrosis [45]. Noncrescentic and crescentic lupus nephritis responded similarly to induction therapy when the analysis was corrected for disease severity [45]. Nevertheless, vascular necrosis and crescents are an important element of the histopathological score that is used to grade the activity of lupus nephritis [1,46]. As IC-GNs develop from adaptive immune responses during host defense of systemic autoimmunity, global immunosuppressants are currently used to attenuate the disease process. However, as immune complexes trigger glomerular injury via complement activation, complement inhibitors are upcoming options to be tested in clinical trials on patients with IC-GN.
Antiglomerular basement membrane disease
Anti-GBM disease and Goodpasture syndrome develop from an autoimmune response against antigens located in the NC1 domain of collagen type IV and the peroxidase peroxidasin [47]. The disease is rare and the trigger unknown but its occurrence in small clusters of unrelated patients suggest yet undefined environmental factors [48]. The circulating ‘anti-GBM’ antibodies are considered pathogenic as the appearance and disappearance of these antibodies coincides with CGN disease activity, although occasionally antibodies reacting in GBM bioassays may not bind to patient‘s GBM [49] or not all linear IgG deposits in CGN may relate to ‘GBM antibodies’ [50]. Nevertheless, these pathogenic autoantibodies are the central therapeutic target in anti-GBM disease and either depleting circulating IgG by plasma exchange or by a specific IgG-degrading enzyme improves disease activity [51▪]. GBM antisera are frequently used to induce CGN in rodents to study more downstream mechanisms of antibody-mediated injury to the glomerular microvasculature using crescent formation as a readout parameter. For example, glomerular IgG deposits promote glomerular neutrophil recruitment via Fc gamma receptor IIA on neutrophils [52]. In addition, the pro-inflammatory roles of murine double minute-2, NETs, and extracellular histones all contribute to anti-GBM IgG-induced CGN [22,53]. In contrast, regulatory T cells only contribute to the afferent phase of lymphocyte priming but not to intrarenal inflammation [12].
C3 glomerulonephritis
C3 glomerulonephritis (C3GN) arises from abnormal regulation of the alternative complement pathway leading to electron dense deposits in the mesangium or along the glomerular filtration barrier that contain complement but no immunoglobulins. Although C3GN most frequently presents with membranoproliferative lesions, C3GN has been reported to present as severe necrotizing and crescentic glomerulonephritis in humans and mice [54,55]. When acquired antibodies against factor H, B or other complement regulators are the cause of a C3GN, drugs that suppress the adaptive immune system such as steroids, mycophenolate mofetil or rituximab may be effective. In contrast, patients with genetic defects in complement regulators may (in the future) benefit from therapy with C3 convertase inhbitors or other downstream elements of the complement cascade [55,56].
CONCLUSION
The glomerular crescent is a histomorphological indicator of a rupture of glomerular capillaries, usually as a consequence of an intense immune attack involving cytotoxic elements, such as complement membrane attack complex and extracellular histones, that is, necroinflammation. The vascular leakage of coagulation factors and fibrin formation within Bowman‘s space devoid of platelets and other cellular components of blood clots triggers hyperplasia of parietal epithelial cells, the crescent. Crescent formation leads to fading of single nephron GFR by increasing counter pressure and collapse of the glomerular tuft or by obstructing the tubular outflow. Fibrotic transformation, periglomerular immune cell infiltrates, rupture of Bowman‘s capsule, and influx of immune cells are inconstant downstream features of crescent formation, and hence less suitable therapeutic targets. Therapeutic interventions should target the upstream mechanisms of vascular necrosis, such as extrarenal autoimmunity, pathogenic autoantibodies, and intrarenal complement activation and inflammation. Despite the growing knowledge about CGN, numerous open questions remain some of which are listed in Table 2. As a conceptual conclusion, the crescent, like other unspecific histopathological lesions frequently noted on diagnostic kidney biopsies, provides a mechanistic clue to the underlying disease pathomechanisms, here: rupture of glomerular capillaries. As such, CGN is no diagnosis and only points towards a set of differential diagnoses that require further diagnostic workup.
Table 2.
Open questions deserving further research
| Are glomerular crescents monoclonal or polyclonal lesions? |
| How are cellular crescents cleared in those patients where cellular crescent lesions are reversed? Is this achieved by detachment and shedding of cells into the urine or by apoptosis and phagocytic clearance in situ. |
| Why do parietal epithelial cells start to proliferate in some but not all forms of GN? |
| Why some patients with IgA nephropathy develop crescents while most other do not? |
| Do those have concomitant pathogenic variants in complement regulator genes? |
| In anti-GBM disease, which is the trigger of the disease? Are environmental factors associated with the occurrence in small clusters of unrelated patients? |
GBM, glomerular basement membrane; GN, glomerulonephritis.
Acknowledgements
None.
Financial support and sponsorship
H.J.A. was supported by the Deutsche Forschungs-gemeinschaft (AN372/14-3, 16-2, 23-1, 24-1, 27-1). H.J.A. and R.K. were supported by European Union's Horizon 2020 research and innovation programme under grant agreement No. 668036 (project RELENT). The materials presented and views expressed here are the responsibility of the author(s) only. The EU Commission takes no responsibility for any use made of the information set out.
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1.Bajema IM, Wilhelmus S, Alpers CE, et al. Revision of the International Society of Nephrology/Renal Pathology Society classification for lupus nephritis: clarification of definitions, and modified National Institutes of Health activity and chronicity indices. Kidney Int 2018; 93:789–796. [DOI] [PubMed] [Google Scholar]
- 2▪.Sethi S, Fervenza FC. Standardized classification and reporting of glomerulonephritis. Nephrol Dial Transplant 2019; 34:193–199. [DOI] [PubMed] [Google Scholar]; This consensus statement presents a novel approach how to standardize a kidney biopsy report to determine the cause of the glomerulonephritis starting from the pattern of immune deposits.
- 3.Smeets B, Uhlig S, Fuss A, et al. Tracing the origin of glomerular extracapillary lesions from parietal epithelial cells. J Am Soc Nephrol 2009; 20:2604–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smeets B, Angelotti ML, Rizzo P, et al. Renal progenitor cells contribute to hyperplastic lesions of podocytopathies and crescentic glomerulonephritis. J Am Soc Nephrol 2009; 20:2593–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Romagnani P, Rinkevich Y, Dekel B. The use of lineage tracing to study kidney injury and regeneration. Nat Rev Nephrol 2015; 11:420–431. [DOI] [PubMed] [Google Scholar]
- 6.Fogo AB, Lusco MA, Najafian B, et al. AJKD atlas of renal pathology: pauci-immune necrotizing crescentic glomerulonephritis. Am J Kidney Dis 2016; 68:e31–e32. [DOI] [PubMed] [Google Scholar]
- 7.Puelles VG, Fleck D, Ortz L, et al. Novel 3D analysis using optical tissue clearing documents the evolution of murine rapidly progressive glomerulonephritis. Kidney Int 2019; 96:505–516. [DOI] [PubMed] [Google Scholar]
- 8.Ryu M, Migliorini A, Miosge N, et al. Plasma leakage through glomerular basement membrane ruptures triggers the proliferation of parietal epithelial cells and crescent formation in non-inflammatory glomerular injury. J Pathol 2012; 228:482–494. [DOI] [PubMed] [Google Scholar]
- 9.Chen A, Lee K, D’Agati VD, et al. Bowman's capsule provides a protective niche for podocytes from cytotoxic CD8+ T cells. J Clin Invest 2018; 128:3413–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krebs CF, Schmidt T, Riedel JH, et al. T helper type 17 cells in immune-mediated glomerular disease. Nat Rev Nephrol 2017; 13:647–659. [DOI] [PubMed] [Google Scholar]
- 11▪▪.Gan PY, Chan A, Ooi JD, et al. Biologicals targeting T helper cell subset differentiating cytokines are effective in the treatment of murine antimyeloperoxidase glomerulonephritis. Kidney Int 2019; 96:1047–1246. [DOI] [PubMed] [Google Scholar]; This study describes the biphasic autoimmunity in a murine model of antimyeloperoxidase glomerulonephritis and how defining dominant T-helper differentiating subsets by renal CD4+ T-cell cytokine gene expression allows effective proper phase monoclonal antibody treatment.
- 12.Klinge S, Yan K, Reimers D, et al. Role of regulatory T cells in experimental autoimmune glomerulonephritis. Am J Physiol Renal Physiol 2019; 316:F572–F581. [DOI] [PubMed] [Google Scholar]
- 13.Chen A, Lee K, Guan T, et al. Role of CD8+ T cells in crescentic glomerulonephritis. Nephrol Dial Transplant 2019; pii: gfz043. doi: 10.1093/ndt/gfz043. [Epub ahead of print] PMID: 30879039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14▪▪.Eymael J, Sharma S, Loeven MA, et al. CD44 is required for the pathogenesis of experimental crescentic glomerulonephritis and collapsing focal segmental glomerulosclerosis. Kidney Int 2018; 93:626–642. [DOI] [PubMed] [Google Scholar]; This study defines that acquired glomerular CD44 expression by activated parietal epithelial cells is necessary for the pathogenesis of experimental crescentic glomerulonephritis.
- 15▪▪.Lazareth H, Henique C, Lenoir O, et al. The tetraspanin CD9 controls migration and proliferation of parietal epithelial cells and glomerular disease progression. Nat Commun 2019; 10:3303. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports that pathogenic expression of CD9 by glomerular parietal epithelial cells drives glomerular damage during crescentic glomerulonephritis and focal segmental glomerulosclerosis.
- 16.Kuppe C, van Roeyen C, Leuchtle K, et al. Investigations of glucocorticoid action in GN. J Am Soc Nephrol 2017; 28:1408–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bollee G, Flamant M, Schordan S, et al. Epidermal growth factor receptor promotes glomerular injury and renal failure in rapidly progressive crescentic glomerulonephritis. Nat Med 2011; 17:1242–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Romoli S, Angelotti ML, Antonelli G, et al. CXCL12 blockade preferentially regenerates lost podocytes in cortical nephrons by targeting an intrinsic podocyte-progenitor feedback mechanism. Kidney Int 2018; 94:1111–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuppe C, Leuchtle K, Wagner A, et al. Novel parietal epithelial cell subpopulations contribute to focal segmental glomerulosclerosis and glomerular tip lesions. Kidney Int 2019; 96:80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mulay SR, Linkermann A, Anders HJ. Necroinflammation in kidney disease. J Am Soc Nephrol 2016; 27:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suarez-Alvarez B, Liapis H, Anders HJ. Links between coagulation, inflammation, regeneration, and fibrosis in kidney pathology. Lab Invest 2016; 96:378–390. [DOI] [PubMed] [Google Scholar]
- 22.Kumar SV, Kulkarni OP, Mulay SR, et al. Neutrophil extracellular trap-related extracellular histones cause vascular necrosis in severe GN. J Am Soc Nephrol 2015; 26:2399–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23▪.Nakazawa D, Masuda S, Tomaru U, et al. Pathogenesis and therapeutic interventions for ANCA-associated vasculitis. Nat Rev Rheumatol 2019; 15:91–101. [DOI] [PubMed] [Google Scholar]; This review summarizes the pathogenesis of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis, focusing on the excessive activation of neutrophils and the involvement of neutrophil extracellular traps (NETs).
- 24.Westhorpe CL, Bayard JE, O'Sullivan KM, et al. In vivo imaging of inflamed glomeruli reveals dynamics of neutrophil extracellular trap formation in glomerular capillaries. Am J Pathol 2017; 187:318–331. [DOI] [PubMed] [Google Scholar]
- 25.Nakazawa D, Marschner JA, Platen L, Anders HJ. Extracellular traps in kidney disease. Kidney Int 2018; 94:1087–1098. [DOI] [PubMed] [Google Scholar]
- 26.Boeltz S, Amini P, Anders HJ, et al. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ 2019; 26:395–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masuda S, Nonokawa M, Futamata E, et al. Formation and disordered degradation of neutrophil extracellular traps in necrotizing lesions of anti-neutrophil cytoplasmic antibody-associated vasculitis. Am J Pathol 2019; 189:839–846. [DOI] [PubMed] [Google Scholar]
- 28▪.Silvestre-Roig C, Braster Q, Wichapong K, et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019; 569:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows a role for histone-induced cell death at the core of chronic vascular disease orchestrated leading to destabilization of atherosclerotic plaques.
- 29▪.Westhorpe CLV, Norman MU, Hall P, et al. Effector CD4(+) T cells recognize intravascular antigen presented by patrolling monocytes. Nat Commun 2018; 9:747. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows the process of intravascular antigen presentation by MHCII+ monocytes to CD4+ T cells within glomerular capillaries, which leads to antigen-dependent inflammation.
- 30.Hamano Y, Ito F, Suzuki O, et al. Vasculitis and crescentic glomerulonephritis in a newly established congenic mouse strain derived from ANCA-associated vasculitis-prone SCG/Kj mice. Autoimmunity 2019; 52:208–219. [DOI] [PubMed] [Google Scholar]
- 31.Dick J, Gan PY, Ford SL, et al. C5a receptor 1 promotes autoimmunity, neutrophil dysfunction and injury in experimental antimyeloperoxidase glomerulonephritis. Kidney Int 2018; 93:615–625. [DOI] [PubMed] [Google Scholar]
- 32.Jayne DRW, Bruchfeld AN, Harper L, et al. Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated vasculitis. J Am Soc Nephrol 2017; 28:2756–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33▪▪.Bashford-Rogers RJM, Bergamaschi L, McKinney EF, et al. Analysis of the B cell receptor repertoire in six immune-mediated diseases. Nature 2019; 574:122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study compares the B cell receptor repertoire in different immune-mediated diseases and determines that there is a complex B-cell architecture that can help understanding the pathological mechanisms of the diseases and design proper treatment strategies.
- 34▪.McClure M, Gopaluni S, Jayne D, Jones R. B cell therapy in ANCA-associated vasculitis: current and emerging treatment options. Nat Rev Rheumatol 2018; 14:580–591. [DOI] [PubMed] [Google Scholar]; This review discusses the therapeutic approach of B-cell-targeted immunotherapy that combines B-cell depletion and B-cell-activating factor blockade in ANCA-associated vasculitis.
- 35.Rampelli SK, Rajesh NG, Srinivas BH, et al. Clinical spectrum and outcomes of crescentic glomerulonephritis: a single center experience. Indian J Nephrol 2016; 26:252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang W, Zhou Q, Hong L, et al. Clinical outcomes of IgA nephropathy patients with different proportions of crescents. Medicine (Baltimore) 2017; 96:e6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sevillano AM, Diaz M, Caravaca-Fontan F, et al. IgA nephropathy in elderly patients. Clin J Am Soc Nephrol 2019; 14:1183–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38▪▪.Park S, Baek CH, Cho H, et al. Glomerular crescents are associated with worse graft outcome in allograft IgA nephropathy. Am J Transplant 2019; 19:145–155. [DOI] [PubMed] [Google Scholar]; This retrospective cohort study on kidney transplant recipients diagnosed with IgA nephropathy in allograft biopsy describes that presence of glomerular crescents in IgA nephropathy is associated with a worse graft prognosis, suggesting that the degree of glomerular crescent formation may be an important risk factor for a poor prognosis.
- 39.Park S, Baek CH, Park SK, et al. Clinical significance of crescent formation in IgA nephropathy - a multicenter validation study. Kidney Blood Press Res 2019; 44:22–32. [DOI] [PubMed] [Google Scholar]
- 40.Peng W, Tang Y, Tan L, Qin W. Crescents and global glomerulosclerosis in Chinese IgA nephropathy patients: a five-year follow-up. Kidney Blood Press Res 2019; 44:103–112. [DOI] [PubMed] [Google Scholar]
- 41.Cattran DC, Coppo R, Cook HT, et al. The Oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classification. Kidney Int 2009; 76:534–545. [DOI] [PubMed] [Google Scholar]
- 42.Haas M, Verhave JC, Liu ZH, et al. A multicenter study of the predictive value of crescents in IgA nephropathy. J Am Soc Nephrol 2017; 28:691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yasukawa M, Kitagawa S, Togashi R, et al. A patient with MPO-ANCA-positive IgA nephropathy diagnosed with the clinical onset of macrohematuria. Intern Med 2019; 58:2051–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie L, He J, Liu X, et al. Clinical value of systemic symptoms in IgA nephropathy with ANCA positivity. Clin Rheumatol 2018; 37:1953–1961. [DOI] [PubMed] [Google Scholar]
- 45.Turner-Stokes T, Wilson HR, Morreale M, et al. Positive antineutrophil cytoplasmic antibody serology in patients with lupus nephritis is associated with distinct histopathologic features on renal biopsy. Kidney Int 2017; 92:1223–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Obrisca B, Jurubita R, Andronesi A, et al. Histological predictors of renal outcome in lupus nephritis: the importance of tubulointerstitial lesions and scoring of glomerular lesions. Lupus 2018; 27:1455–1463. [DOI] [PubMed] [Google Scholar]
- 47.McCall AS, Bhave G, Pedchenko V, et al. Inhibitory anti-peroxidasin antibodies in pulmonary-renal syndromes. J Am Soc Nephrol 2018; 29:2619–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lingaraj U, Mallappa SS, Neminah RE, et al. A ‘Mini-Epidemic’ of antiglomerular basement membrane disease: clinical and epidemiological study. Saudi J Kidney Dis Transpl 2017; 28:1057–1063. [DOI] [PubMed] [Google Scholar]
- 49.Sadeghi-Alavijeh O, Henderson S, Bass P, et al. Crescentic glomerulonephritis with anti-GBM antibody but no glomerular deposition. BMC Nephrol 2018; 19:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adapa S, Konala VM, Hou J, et al. Seronegative atypical antiglomerular basement membrane crescentic glomerulonephritis. Ann Transl Med 2019; 7:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51▪.Soveri I, Molne J, Uhlin F, et al. The IgG-degrading enzyme of Streptococcus pyogenes causes rapid clearance of antiglomerular basement membrane antibodies in patients with refractory antiglomerular basement membrane disease. Kidney Int 2019; 96:1234–1238. [DOI] [PubMed] [Google Scholar]; This study describes the treatment effect of endopeptidase IgG-degrading enzyme of Streptococcus pyogenes in three patients with refractory antiglomerular basement membrane nephritis. This treatment is able to clear circulating and kidney-bound anti-GBM antibodies without a positive effect in renal function.
- 52.Nishi H, Furuhashi K, Cullere X, et al. Neutrophil FcgammaRIIA promotes IgG-mediated glomerular neutrophil capture via Abl/Src kinases. J Clin Invest 2017; 127:3810–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mulay SR, Romoli S, Desai J, et al. Murine double minute-2 inhibition ameliorates established crescentic glomerulonephritis. Am J Pathol 2016; 186:1442–1453. [DOI] [PubMed] [Google Scholar]
- 54.Ravindran A, Fervenza FC, Smith RJH, Sethi S. C3 glomerulonephritis with a severe crescentic phenotype. Pediatr Nephrol 2017; 32:1625–1633. [DOI] [PubMed] [Google Scholar]
- 55.Wang X, Van Lookeren Campagne M, Katschke KJ, Jr, et al. Prevention of fatal C3 glomerulopathy by recombinant complement receptor of the Ig superfamily. J Am Soc Nephrol 2018; 29:2053–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith RJH, Appel GB, Blom AM, et al. C3 glomerulopathy - understanding a rare complement-driven renal disease. Nat Rev Nephrol 2019; 15:129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]