

Abstract
Background
Disease‐modifying pharmacological agents for transthyretin (TTR)‐related familial amyloid polyneuropathy (FAP) have become available in the last decade, but evidence on their efficacy and safety is limited. This review focuses on disease‐modifying pharmacological treatment for TTR‐related and other FAPs, encompassing amyloid kinetic stabilisers, amyloid matrix solvents, and amyloid precursor inhibitors.
Objectives
To assess and compare the efficacy, acceptability, and tolerability of disease‐modifying pharmacological agents for familial amyloid polyneuropathies (FAPs).
Search methods
On 18 November 2019, we searched the Cochrane Neuromuscular Specialised Register, the Cochrane Central Register of Controlled Trials, MEDLINE, and Embase. We reviewed reference lists of articles and textbooks on peripheral neuropathies. We also contacted experts in the field. We searched clinical trials registries and manufacturers' websites.
Selection criteria
We included randomised clinical trials (RCTs) or quasi‐RCTs investigating any disease‐modifying pharmacological agent in adults with FAPs.
Disability due to FAP progression was the primary outcome. Secondary outcomes were severity of peripheral neuropathy, change in modified body mass index (mBMI), quality of life, severity of depression, mortality, and adverse events during the trial.
Data collection and analysis
We followed standard Cochrane methodology.
Main results
The review included four RCTs involving 655 people with TTR‐FAP. The manufacturers of the drugs under investigation funded three of the studies. The trials investigated different drugs versus placebo and we did not conduct a meta‐analysis.
One RCT compared tafamidis with placebo in early‐stage TTR‐FAP (128 randomised participants). The trial did not explore our predetermined disability outcome measures. After 18 months, tafamidis might reduce progression of peripheral neuropathy slightly more than placebo (Neuropathy Impairment Score (NIS) in the lower limbs; mean difference (MD) ‐3.21 points, 95% confidential interval (CI) ‐5.63 to ‐0.79; P = 0.009; low‐certainty evidence). However, tafamidis might lead to little or no difference in the change of quality of life between groups (Norfolk Quality of Life‐Diabetic Neuropathy (Norfolk QOL‐DN) total score; MD ‐4.50 points, 95% CI ‐11.27 to 2.27; P = 0.19; very low‐certainty evidence). No clear between‐group difference was found in the numbers of participants who died (risk ratio (RR) 0.65, 95% CI 0.11 to 3.74; P = 0.63; very low‐certainty evidence), who dropped out due to adverse events (RR 1.29, 95% CI 0.30 to 5.54; P = 0.73; very low‐certainty evidence), or who experienced at least one severe adverse event during the trial (RR 1.16, 95% CI 0.37 to 3.62; P = 0.79; very low‐certainty evidence).
One RCT compared diflunisal with placebo (130 randomised participants). At month 24, diflunisal might reduce progression of disability (Kumamoto Score; MD ‐4.90 points, 95% CI ‐7.89 to ‐1.91; P = 0.002; low‐certainty evidence) and peripheral neuropathy (NIS plus 7 nerve tests; MD ‐18.10 points, 95% CI ‐26.03 to ‐10.17; P < 0.001; low‐certainty evidence) more than placebo. After 24 months, changes from baseline in the quality of life measured by the 36‐Item Short‐Form Health Survey score showed no clear difference between groups for the physical component (MD 6.10 points, 95% CI 2.56 to 9.64; P = 0.001; very low‐certainty evidence) and the mental component (MD 4.40 points, 95% CI ‐0.19 to 8.99; P = 0.063; very low‐certainty evidence). There was no clear between‐group difference in the number of people who died (RR 0.46, 95% CI 0.15 to 1.41; P = 0.17; very low‐certainty evidence), in the number of dropouts due to adverse events (RR 2.06, 95% CI 0.39 to 10.87; P = 0.39; very low‐certainty evidence), and in the number of people who experienced at least one severe adverse event (RR 0.77, 95% CI 0.18 to 3.32; P = 0.73; very low‐certainty evidence) during the trial.
One RCT compared patisiran with placebo (225 randomised participants). After 18 months, patisiran reduced both progression of disability (Rasch‐built Overall Disability Scale; least‐squares MD 8.90 points, 95% CI 7.00 to 10.80; P < 0.001; moderate‐certainty evidence) and peripheral neuropathy (modified NIS plus 7 nerve tests ‐ Alnylam version; least‐squares MD ‐33.99 points, 95% CI ‐39.86 to ‐28.13; P < 0.001; moderate‐certainty evidence) more than placebo. At month 18, the change in quality of life between groups favoured patisiran (Norfolk QOL‐DN total score; least‐squares MD ‐21.10 points, 95% CI ‐27.20 to ‐15.00; P < 0.001; low‐certainty evidence). There was little or no between‐group difference in the number of participants who died (RR 0.61, 95% CI 0.21 to 1.74; P = 0.35; low‐certainty evidence), dropped out due to adverse events (RR 0.33, 95% CI 0.13 to 0.82; P = 0.017; low‐certainty evidence), or experienced at least one severe adverse event (RR 0.91, 95% CI 0.64 to 1.28; P = 0.58; low‐certainty evidence) during the trial.
One RCT compared inotersen with placebo (172 randomised participants). The trial did not explore our predetermined disability outcome measures. From baseline to week 66, inotersen reduced progression of peripheral neuropathy more than placebo (modified NIS plus 7 nerve tests ‐ Ionis version; MD ‐19.73 points, 95% CI ‐26.50 to ‐12.96; P < 0.001; moderate‐certainty evidence). At week 65, the change in quality of life between groups favoured inotersen (Norfolk QOL‐DN total score; MD ‐10.85 points, 95% CI ‐17.25 to ‐4.45; P < 0.001; low‐certainty evidence). Inotersen may slightly increase mortality (RR 5.94, 95% CI 0.33 to 105.60; P = 0.22; low‐certainty evidence) and occurrence of severe adverse events (RR 1.48, 95% CI 0.85 to 2.57; P = 0.16; low‐certainty evidence) compared to placebo. More dropouts due to adverse events were observed in the inotersen than in the placebo group (RR 8.57, 95% CI 1.16 to 63.07; P = 0.035; low‐certainty evidence).
There were no studies addressing apolipoprotein AI‐FAP, gelsolin‐FAP, and beta‐2‐microglobulin‐FAP.
Authors' conclusions
Evidence on the pharmacological treatment of FAPs from RCTs is limited to TTR‐FAP. No studies directly compare disease‐modifying pharmacological treatments for TTR‐FAP. Results from placebo‐controlled trials indicate that tafamidis, diflunisal, patisiran, and inotersen may be beneficial in TTR‐FAP, but further investigations are needed. Since direct comparative studies for TTR‐FAP will be hampered by sample size and costs required to demonstrate superiority of one drug over another, long‐term non‐randomised open‐label studies monitoring their efficacy and safety are needed.
Plain language summary
Drug treatment for familial amyloid polyneuropathy
What is the aim of this review?
The aim of this review was to assess whether drugs to treat familial amyloid polyneuropathies (FAPs) reduce disability due to nerve damage, reduce the severity of nerve damage, and improve nutritional status, quality of life, and depression. We also reviewed evidence on side effects.
Key messages
Studies of drug treatments for people with FAP are so far limited to transthyretin‐FAP (also called TTR‐FAP). Four trials have compared a drug with a placebo (an inactive, dummy compound), but none have directly compared drugs with each other. The studies provided evidence that all four drugs studied (tafamidis, diflunisal, patisiran, and inotersen) are probably of benefit for people with TTR‐FAP, but it remains possible that their true effects are different from these results. As research comparing drugs with each other is limited by study size required to demonstrate the superiority of one drug and costs, studies monitoring their effects for longer are needed.
What was studied in the review?
FAPs are a group of hereditary progressive conditions in which insoluble protein deposits (fibrils) affect mainly peripheral nerves (nerves outside the brain and spinal cord). Fibrils are deposited because they are made up of genetically abnormal protein that does not fold properly. This process also occurs in many other organs including the heart, kidneys, and eyes, thus these disorders are complex. The type of protein fibril deposited and the exact genetic defect determine the type of FAP. TTR‐FAP is the most common FAP by far. Although liver transplantation was the only treatment, done in highly selected cases, drugs which may affect the disease course have recently become available for people with TTR‐FAP.
What are the main results of the review?
The review authors found four relevant studies, which involved 655 adults with TTR‐FAP.
In one study, people with early‐stage TTR‐FAP received tafamidis or placebo for 18 months. Disability was not measured. The evidence suggested that tafamidis may reduce both the proportion of people with progression of peripheral neuropathy and the mean change (worsening) of nerve damage (based on a score of strength and sensation) compared to placebo. It is uncertain whether tafamidis has an effect on quality of life and the number of deaths, dropouts due to harmful effects or any severe side effects.
Evidence from a 24‐month study in people with TTR‐FAP suggested that compared to placebo, diflunisal may slightly decrease disability due to FAP progression and lessen worsening of peripheral neuropathy. It is uncertain whether diflunisal has an effect on quality of life and number of deaths, dropouts due to side effects, and people who experience severe side effects.
Results from an 18‐month study in people with TTR‐FAP suggested that compared to placebo, patisiran probably decreases disability due to FAP progression, and lessens worsening of peripheral neuropathy. Quality of life may decline slightly less with patisiran than placebo. Patisiran may lead to little or no difference in mortality, dropouts due to side effects or number of people experiencing severe side effects.
In the fourth study, people with TTR‐FAP received inotersen or placebo for 66 weeks. Disability was not measured. This study indicated that inotersen probably reduces worsening of peripheral neuropathy, but may have little effect on the change in quality of life compared to placebo. Inotersen may be associated with numbers of adverse events above the placebo rate, illustrated by an increase in the number of dropouts due to side effects compared to placebo. There was little or no difference in mortality or number of people experiencing severe side effects.
The review authors did not find any study addressing drug therapy for people with other types of FAP.
Three of the four studies were funded by the manufacturer of the drug investigated.
How up‐to‐date is this review?
The evidence is current to November 2019.
Summary of findings
Background
Description of the condition
Familial amyloid polyneuropathies (FAPs) are a group of relentless, disabling and life‐threatening hereditary polyneuropathies affecting the somatic and autonomic components of the peripheral nervous system. FAPs are due to endoneurial deposition of amyloid, an insoluble substance constituted from misfolded mutated proteins that aggregate in nonbranching fibrils oriented in a β‐pleated sheet structure. Extracellular deposition of amyloid fibrils usually also occurs in many organs, including the heart, kidneys and eyes. Therefore, FAPs fall into the category of multisystem diseases (Planté‐Bordeneuve 2011). According to the precursor proteins involved in amyloidogenesis, FAPs are classified as (Lunn 2016):
transthyretin (TTR)‐FAP, formerly termed FAP type I (Portuguese–Swedish–Japanese type) and type II (Indiana‐Swiss or Maryland‐German type);
apolipoprotein AI (ApoAI)‐FAP, also referred to as FAP type III, Van Allen type or Iowa type;
gelsolin (Gel)‐FAP, previously known as FAP type IV, Finnish type or Meretoja type; and
beta‐2‐microglobulin (B2M)‐FAP.
Symptoms suggestive of peripheral neuropathy were reported in a single case of hereditary amyloidosis due to a mutation in the gene encoding lysozyme, but electrodiagnostic tests and nerve biopsy were not performed to confirm the diagnosis (Nasr 2017).
FAPs have an autosomal dominant pattern of inheritance. Age at onset, symptomatology, pattern of organ involvement and disease progression of these conditions can be highly variable (Planté‐Bordeneuve 2011; Sipe 2014).
TTR‐FAP
TTR‐FAP was originally described by Andrade in Portuguese families and then recognised in Sweden, Japan, Ireland, and worldwide (Andrade 1952; Rowczenio 2015). Its overall prevalence is estimated to be 0.87 to 1.1 per one million people (Adams 2014). TTR is a plasma protein which is mainly synthesised by the liver and involved in the transport of thyroid hormones and retinol (vitamin A). The clinical spectrum of TTR‐related amyloidosis encompasses FAP, familial amyloid cardiomyopathy, ocular and leptomeningeal amyloidosis, whereas renal involvement is uncommon (Rapezzi 2013). To date, more than 120 amyloidogenic mutations in the TTR gene have been described (Rowczenio 2014). Some mutations are associated with particular phenotypes, with prominent neurological or cardiac involvement (Rapezzi 2010; Rapezzi 2013). However, a single gene mutation does not necessarily account for a single phenotype and a significant intramutation and intrafamilial variability, as well as differences in natural history within and among mutations are reported (Mariani 2015). In addition to genetic factors, environmental and epigenetic determinants may influence phenotypic expression and/or variability of age at onset (Rapezzi 2010; Planté‐Bordeneuve 2018). The Val30Met substitution is the most frequent mutation causing TTR‐FAP and is present in large foci in Portugal, Japan and Sweden, thus giving rise to the so called “endemic Val30Met TTR‐FAP phenotype”. Cases of Val30Met TTR‐related amyloidosis reported outside the endemic geographic foci are referred to as “nonendemic Val30Met TTR‐FAP phenotypes”. Endemic Val30Met TTR‐FAP typically begins in the fourth decade of life (Portuguese and Japanese cases) or later (Swedish cases), manifesting as a slowly progressive, length‐dependent, sensorimotor polyneuropathy, often associated with autonomic involvement. Clinical manifestations include loss of superficial sensation, such as nociception and thermal sensations, neuropathic pain, orthostatic hypotension, sexual impotence, neurogenic bladder and gastrointestinal dysfunction. Restrictive cardiomyopathy progressing to congestive heart failure, arrhythmias and conduction blocks is a later manifestation. Nonendemic Val30Met TTR‐FAP usually shows higher age at onset (sixth or seventh decade of life), extreme male predominance, broader phenotypic heterogeneity and markedly low age‐related penetrance, so that a positive family history is less frequent and the diagnosis often delayed. Cardiac involvement can be very prominent already at presentation and autonomic dysfunction is typically milder. Age at onset of TTR‐FAP is quite variable, particularly for the Val30Met‐related form in endemic or nonendemic geographic areas, so that early‐onset (< 50 years) and late‐onset (> 50 years) phenotypes have been defined. Phenotypes related to non‐Val30Met mutations vary considerably, encompassing selective and nonselective neurological, cardiac and oculoleptomeningeal involvement (Rapezzi 2010). Walking difficulties requiring aid (stage 2) occur after a mean disease duration of six years, with confinement to a wheelchair (stage 3) on average after 10 years of disease (Coutinho 1980; Adams 2014). Studies in Portuguese people with TTR‐FAP indicate that death occurs within a mean interval of 10.8 years after the onset of symptoms (Hund 2012).
ApoAI‐FAP
ApoAI‐FAP was first recognised in Iowa. ApoAI is the major protein constituent of plasma high‐density lipoprotein. ApoAI is synthesised in the liver and small intestine in approximately the same proportions. ApoAI‐FAP usually begins in the fourth decade of life and is characterised by amyloid deposition in major organs, including the liver, gastrointestinal tract and kidneys, leading to severe gastric ulcer disease and renal failure. Although a length‐dependent polyneuropathy with slow progression can occur in ApoAI‐related amyloidosis, it is not a major feature of the disease. Four mutations in the APOAI gene are associated with the neuropathic pattern of ApoAI‐related amyloidosis (Gly26Arg, Leu174Ser, His155Metfs*46, Ala154Glyfs*48) (Rowczenio 2014).
Gel‐FAP
Gel‐FAP was first identified in Finland, but sporadic cases are recognised worldwide. Gel is a calcium‐dependent actin‐binding protein. Two point mutations in the GEL gene (Asp187Asn, Asp187Tyr) are known to cause neuropathic forms of Gel‐related amyloidosis (Planté‐Bordeneuve 2011; Rowczenio 2014). The first manifestations of Gel‐amyloidosis occur at age 25 to 30 years and include corneal lattice dystrophy, cranial neuropathies (typically unilateral or bilateral facial paralysis), peripheral sensory neuropathy, and abnormal skin laxity. Cardiac, renal and pharyngeal abnormalities are less common. The clinical course of Gel‐FAP is slow and quite benign, since life‐threatening cardiac and renal complications are rare.
B2M‐FAP
A single French family has been reported with an Asp76Asn mutation in the B2M gene causing gastrointestinal involvement (chronic diarrhoea with weight loss, autonomic and sensorimotor neuropathy, and persistent sicca syndrome) (Valleix 2012; Rowczenio 2014).
ApoAI‐FAP and Gel‐FAP are very rare and cases of B2M‐FAP are anecdotal, so the worldwide prevalence of these conditions is unknown.
The diagnosis of FAP is often challenging because of phenotypic and genotypic heterogeneity, especially in the absence of family history and when the clinical presentation is atypical. Amyloidosis is diagnosed by demonstration of amyloid in tissue biopsy specimens, where it appears as Congo red–binding material with characteristic apple‐green birefringence under cross‐polarised light. DNA testing to identify an amyloidogenic gene mutation is necessary to confirm the diagnosis of FAP (Planté‐Bordeneuve 2011). In recent years, a number of diagnostic tools have been investigated in people with TTR‐FAP. These include magnetic resonance neurography (Kollmer 2015; Kollmer 2017), skin biopsy for the evaluation of intraepidermal nerve fiber density (Masuda 2017), and Sudoscan technology for the measurement of electrochemical skin conductance (Castro 2016). Timely detection of clinical manifestations of TTR‐FAP and prompt definitive diagnosis are critical for appropriate treatment and optimal outcomes. Asymptomatic gene carriers should be monitored regularly for early recognition of symptoms and signs. The new diagnostic tools look set to permit an earlier and more reliable diagnosis of this condition (Planté‐Bordeneuve 2018).
Description of the intervention
The treatment of FAPs requires a multidisciplinary approach, including:
disease‐modifying treatments for stopping or slowing down the progression of amyloidogenesis;
pharmacological agents to manage the symptoms of peripheral and autonomic neuropathy (e.g. neuropathic pain, orthostatic hypotension, bladder and gastrointestinal disturbances); and
treatment of complications due to severe organ involvement by amyloidosis (i.e. effects on the heart, eye, or kidney).
Overall, TTR gene mutations account for the majority of FAP cases (Rowczenio 2014), and clinical trials have focused on TTR‐FAP to date.
Liver transplantation was the only treatment option in TTR‐FAP in the pre‐pharmacological era. Since TTR is synthesised mainly in the liver, liver transplantation suppresses the main source of mutant TTR. Liver transplantation surgery has shown a favourable effect on the progression of peripheral neuropathy in Val30Met TTR‐FAP even in the long term. However, autonomic dysfunction is unchanged after liver transplantation, and cardiac, renal, and oculoleptomeningeal manifestations of the disease are influenced to a lesser degree, if at all. Indeed, long‐term disease‐related complications of liver transplantation include TTR‐related cerebral amyloid angiopathy and cardiomyopathy (Planté‐Bordeneuve 2018), as well as resultant TTR‐FAP in recipients of explanted FAP liver after a “domino” procedure. People with TTR‐FAP and severe renal or heart failure may benefit from a combined kidney‐liver or heart‐liver transplant (Adams 2013). However, large numbers of people with TTR‐FAP are not suitable transplant candidates, and the development of pharmacological disease‐modifying options has been an active field of research over the last decades, with a number of completed and ongoing trials (Dubrey 2015). Our review focused on disease‐modifying pharmacological agents for FAP, which are expected to be the only treatment options for the majority of the FAP population. We did not consider symptomatic agents for neuropathic pain, orthostatic hypotension, or bladder and gastrointestinal disturbances, as these are covered in other reviews (Maule 2007; Kempler 2011; Chiang 2015), nor did we discuss treatments for complications of severe organ involvement.
How the intervention might work
Pharmacologic disease‐modifying strategies for TTR‐FAP (some of which are approved and some are still under investigation) may involve a number of classes of drugs:
amyloid kinetic stabilisers, such as tafamidis and diflunisal, which bind mutant misfolded TTR, preventing its aggregation in amyloid fibrils;
amyloid matrix solvents, such as doxycycline and taurodeoxycholic acid, which act to disrupt deposited amyloid fibrils; and
amyloid precursor inhibitors (i.e. gene therapy with antisense oligonucleotides and small interfering RNA ), which block expression of both mutant and wild type TTR reducing amyloid precursor protein synthesis.
In future, people with TTR‐FAP might benefit from immunisation against amyloid precursors (Dubrey 2015). A number of antibodies, including monoclonal antibodies against TTR and antibodies against serum amyloid P, are undergoing testing in people with various forms of amyloidosis (Planté‐Bordeneuve 2018).
No specific disease‐modifying agent for ApoAI‐FAP, Gel‐FAP, and B2M‐FAP is currently available.
Why it is important to do this review
The purpose of this review was firstly to evaluate the current level of evidence for pharmacologic disease‐modifying treatments for FAPs, secondly to compare the efficacy of different disease‐modifying treatments for FAPs, and thirdly to highlight gaps in knowledge that require further investigation. The review is intended for people with FAPs, healthcare professionals, and researchers. We hope that it will draw attention to, and be a stimulus for, more research in this area.
Objectives
To assess and compare the efficacy, acceptability, and tolerability of disease‐modifying pharmacological agents for familial amyloid polyneuropathies (FAPs).
Methods
Criteria for considering studies for this review
Types of studies
As recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), we included all identified randomised clinical trials (RCTs) or quasi‐RCTs of pharmacological disease‐modifying agents for familial amyloid polyneuropathies (FAPs), compared to placebo or to other disease‐modifying agents. Quasi‐RCTs are studies in which participants are allocated to groups using a method that is partially systematic (e.g. by alternation, case record number, or date of birth). We reported evidence for pharmacological agents only investigated in non‐randomised studies, such as cohort studies, case‐control studies or case reports, in the Discussion section. We included studies reported as full‐text, those published as abstract only, and unpublished data. We did not impose any restrictions as to language of publication.
Types of participants
We included studies of people aged 18 years or older, of either gender, with a diagnosis of FAP based on clinical or neurophysiological evidence of polyneuropathy, or both, and positive DNA testing for TTR, APOAI, GEL, or B2M gene mutations, irrespective of biopsy confirmation of amyloid deposits. We included people with FAP as the leading cause of their neuropathy. We excluded people whose neuropathy was attributable to another cause but not those who had comorbidities that may be associated with a neuropathy, where the presenting neuropathy was FAP‐related.
Types of interventions
We considered any disease‐modifying pharmacological intervention for FAP in any dose and by any route, compared to placebo, no intervention, or any other active comparator. We allowed any previous or concomitant treatment except other FAP disease‐modifying agents.
Types of outcome measures
As with many rare diseases, there were no validated outcome measures specific for FAP in the era preceding the first clinical trials in this area. Therefore, measures of disease progression and nerve impairment with demonstrated sensitivity and specificity in other axonal neuropathies (e.g. Charcot‐Marie Tooth disease (CMT) or diabetic polyneuropathy) were included. The tools used to assess the progression of transthyretin (TTR)‐FAP have been refined after the first trials on TTR stabilisers. The Neuropathy Impairment Score (NIS) (Dyck 1995), the NIS of the lower limbs (NIS‐LL) (Dyck 1997; Bril 1999), the Norfolk Quality of Life‐Diabetic Neuropathy Questionnaire (Norfolk QOL‐DN) (Vinik 2005), and composite clinical and neurophysiological evaluations of nerve fiber function, such as the NIS plus 7 nerves test (NIS+7) (Berk 2013), have appeared valid and reliable measures of TTR‐FAP severity (Coelho 2017).
Primary outcomes
-
Disability due to FAP progression
-
Expressed as the change from baseline in the:
clinical staging of TTR‐FAP (FAP stage) (Coutinho 1980);
Polyneuropathy Disability Score (PDS) (Steen 1983);
Modified Norris Test Score (MNT) (Lacomblez 1989);
Portuguese classification system (PCS) (Sales‐Luís 1990);
Kumamoto Score (KS) (Tashima 1999);
Yamamoto Score (YS) (Yamamoto 2007); and
Rasch‐built Overall Disability Scale (R‐ODS) (van Nes 2011).
-
Secondary outcomes
-
Severity of peripheral neuropathy
-
Expressed as the change from baseline in the:
NIS (Dyck 1995);
NIS of the Upper Limbs (NIS‐UL) (Lozeron 2013);
NIS+7 (Berk 2013)
modified versions of NIS+7 (mNIS+7 Alnylam version and mNIS+7 Ionis version) (Suanprasert 2014; Dyck 2017; Adams 2018; Benson 2018);
CMT Neuropathy Score (CMTNS) (Shy 2005);
CMT Neuropathy Score second version (CMTNS2) (Murphy 2011)
Neuropathy Disability Score revised version (NDS) (Abbott 2002); and
Compound Autonomic Dysfunction Test (CADT) (Denier 2007).
-
-
Change in modified body mass index (mBMI)
A measure of wasting and autonomic gastrointestinal function, calculated as the product of the BMI and serum albumin concentration (g/L) (Suhr 1984), expressed as change from baseline
-
Quality of life
-
Expressed as the change from baseline in a validated scale or patient‐reported questionnaire, including the:
36‐Item Short‐Form Health Survey (SF‐36) scale (Ware 1992);
Norfolk QOL‐DN (Vinik 2005);
EuroQoL Quality of Life Scale (EQ‐5D) (Rabin 2001); and
Karnofsky Performance Status (KPS) (Yates 1980).
-
-
Severity of depression
-
Expressed as the change from baseline in a validated scale or clinical diagnostic interview, including the:
Beck Depression Inventory (BDI‐II) (Beck 1988); and
Hamilton Rating Scale for Depression (HAM‐D) (Hamilton 1960).
-
Number of participants who died during the trial
-
Adverse events analysed as:
number of participants experiencing at least one adverse event;
number of participants experiencing mild adverse events;
number of dropouts due to adverse events;
number of participants experiencing at least one severe adverse event.
All outcomes were assessed after 12 and 24 months of treatment (where possible) and at the end of the blinded follow‐up period. We have summarised characteristics of the scores included as primary and secondary outcome measures in Table 5. In order not to miss any side effects, in the data extraction phase, we collected all side effects data reported in the literature, and discussed ways to summarise them post hoc.
1. Scores included as outcome measures.
| Instrument name | Abbreviation | Score (range) | Direction of response | References | Data analysed as |
| Stage of disease based on disability or disability | |||||
| Clinical staging of TTR‐FAP | FAP stage | 1 to 3 | Higher scores indicate greater disease severity | Coutinho 1980 | Ordinal |
| Polyneuropathy Disability Score | PDS | 0 to 5 | Higher scores indicate greater walking disability | Steen 1983 | Ordinal |
| Modified Norris Test Score | MNT | 75 to 0 | Lower scores indicate greater disability | Lacomblez 1989 | Continuous |
| Portuguese classification system | PCS | 0 to 6 | Higher scores indicate greater disease severity | Sales‐Luís 1990 | Ordinal |
| Kumamoto Score | KS | 0 to 96 | Higher scores indicate greater disease severity | Tashima 1999 | Continuous |
| Yamamoto Score | YS | 0 to 4 | Higher scores indicate greater disease severity | Yamamoto 2007 | Ordinal |
| Rasch‐built Overall Disability Scale | R‐ODS | 0 to 48 | Lower scores indicate greater disability | van Nes 2011 | Continuous |
| Impairment due to nerve function | |||||
| Neuropathy Impairment Score | NIS | 0 to 244 | Higher scores indicate greater deficits | Dyck 1995 | Continuous |
| Neuropathy Impairment Score in the lower limbs | NIS‐LL | 0 to 88 | Higher scores indicate greater deficits | Bril 1999; Dyck 1997 | Continuous |
| Neuropathy Impairment Score in the upper limbs | NIS‐UL | 0 to 116 | Higher scores indicate greater deficits | Lozeron 2013 | Continuous |
| Neuropathy Impairment Score plus 7 nerve tests | NIS+7 | 0 to 270 | Higher scores indicate greater deficits | Berk 2013 | Continuous |
| Modified Neuropathy Impairment Score plus 7 nerve tests (Alnylam version) | mNIS+7Alnylam | 0 to 304 | Higher scores indicate greater deficits | Suanprasert 2014; Adams 2018 | Continuous |
| Modified Neuropathy Impairment Score plus 7 nerve tests (Ionis version) | mNIS+7Ionis | ‐22.3 to 346.3 | Higher scores indicate greater deficits | Suanprasert 2014; Dyck 2017; Benson 2018 | Continuous |
| Charcot‐Marie Tooth Neuropathy Score | CMTNS | 0 to 36 | Higher scores indicate greater deficits | Shy 2005 | Continuous |
| Charcot‐Marie Tooth Neuropathy Score 2nd version | CMTNS2 | 0 to 36 | Higher scores indicate greater deficits | Murphy 2011 | Continuous |
| Neuropathy Disability Score revised version | rNDS | 0 to 10 | Higher scores indicate greater deficits | Abbott 2002 | Continuous |
| Compound Autonomic Dysfunction Test | CADT | 0 to 16 | Higher scores indicate greater autonomic impairment | Denier 2007 | Continuous |
| Wasting and autonomic gastrointestinal function | |||||
| Modified body mass index | mBMI | Product of the BMI (kg/m2) and serum albumin concentration (g/L) |
Lower scores indicate greater wasting (worse nutritional status) | Suhr 1984 | Count |
| Quality of life | |||||
| Short Form 36 Health Survey Questionnaire | SF‐36 | 0 to 100 | Lower scores indicate worse status |
Ware 1992 | Continuous |
| Norfolk Quality of Life–Diabetic Neuropathy Questionnairea | Norfolk QOL‐DN | ‐2 to 138 ‐4 to 136 |
Higher scores indicate worse status |
Vinik 2005 Adams 2018 Benson 2018 |
Continuous |
| EuroQoL Quality of Life Scale | EQ‐5D | 5 to 15 | Higher scores indicate worse status |
Rabin 2001 | Continuous |
| Karnofsky Performance Status | Karnofsky | 0 to 100 | Lower scores indicate worse status |
Yates 1980 | Continuous |
| Depression | |||||
| Beck Depression Inventory 2nd version | BDI‐II | 0 to 63 | Higher total scores indicate more severe depressive symptoms | Beck 1988 | Continuous |
| Hamilton Depression Rating Scale for Depression | HAM‐D | 0 to 50 | Higher total scores indicate more severe depressive symptoms | Hamilton 1960 | Continuous |
a Slightly different score ranges are reported in different RCTs (range (‐2 to 138) in Coelho 2012 and range (‐4 to 136) in Adams 2018 and Benson 2018), although the direction of the scores is the same.
Search methods for identification of studies
Electronic searches
The Cochrane Neuromuscular Information Specialist searched the following databases on 18 November 2019.
Cochrane Neuromuscular Specialised Register via the Cochrane Register of Studies (CRS‐Web; Appendix 1).
Cochrane Central Register of Controlled Trials (CENTRAL) via CRS‐Web (Appendix 2).
MEDLINE (1946 to 15 November 2019; Appendix 3).
Embase (1974 to 15 November 2019; Appendix 4).
We did not impose any language restrictions.
Searching other resources
We reviewed reference lists of all included studies, narrative reviews, and major textbooks on peripheral neuropathies (written in English) for published reports and citations of unpublished research. We also conducted a citation search via the Web of Science (included studies only) on 18 November 2019 to identify additional studies. We contacted known experts in the field via email between 2017 and 2018. We conducted complementary searches on the World Health Organization International Clinical Trials Registry Platform (www.who.int/ictrp), ClinicalTrials.gov (www.clinicaltrials.gov), and ClinicalTrialsRegister.eu (www.clinicaltrialsregister.eu) (Appendix 5). We reviewed pharmaceutical companies' websites ‐ www.pfizer.com, www.merck.com, www.ionispharma.com, and www.alnylam.com ‐ to identify ongoing trials and additional published or unpublished data on 18 November 2019.
Data collection and analysis
Selection of studies
Three review authors (FM, GZ, and ST) independently screened titles and abstracts identified by the electronic searches using Covidence. The same review authors obtained the full‐text reports of all potentially eligible studies for independent assessment and suggested studies to be included. All review authors (FM, GMF, LS, FioM, GZ, TC, and ST) agreed on which studies met the inclusion criteria. We resolved any disagreement about inclusion criteria by discussion and consensus. We identified and excluded duplicates and collated multiple reports of the same study so that each study, rather than each report, was the unit of interest in the review. We recorded the selection process in sufficient detail to complete a PRISMA flow diagram and a 'Characteristics of excluded studies' table (Moher 2009).
Data extraction and management
Two review authors (FM and ST) independently extracted data from studies selected for inclusion, using a tailored data collection form. We used 'Characteristics of included studies' tables to present the essential features of the included studies. Two other review authors (GMF and TC) checked the data extraction. There were no disagreements on data extraction and entry, which would have been discussed with the remaining authors (LS, FioM, and GZ) if necessary. We contacted trial authors and sponsors for missing data when possible. We extracted the following study characteristics.
Methods: study design, total duration of study, details of any 'run in' period, number of study centres and location, study setting, withdrawals, and date of study
Participants: N, mean age, age range, gender, severity of condition, diagnostic criteria, baseline characteristics, inclusion criteria, and exclusion criteria
Interventions: intervention, comparison, concomitant medications, and excluded medications
Outcomes: primary and secondary outcomes specified and collected, and time points reported
Notes: funding for trial, notable conflicts of interest of trial authors
One review author (FM) transferred data into Review Manager 5 (Review Manager 2014). A second review author (ST) checked the outcome data entries. A third review author (GZ) spot‐checked study characteristics for accuracy against the trial report.
Assessment of risk of bias in included studies
Two review authors (FM and ST) independently assessed risk of bias for each study using the Cochrane 'Risk of bias' tool, as described in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). All review authors (FM, GMF, LS, FioM, GZ, TC, and ST) resolved disagreements by discussion until we reached consensus. We assessed the risk of bias according to the following domains.
Random sequence generation
Allocation concealment
Blinding of participants and personnel
Blinding of outcome assessment
Incomplete outcome data
Selective outcome reporting
Other sources of bias
We graded studies as having high, low, or unclear risk of bias in each of these domains and provided justifications for our judgements in the 'Risk of bias' tables, with a quote from the study, when appropriate. Where we obtained information on risk of bias from unpublished data, we noted this in the 'Risk of bias' table.
Measures of treatment effect
The primary and secondary outcomes comprised both continuous and dichotomous data. As predetermined in the protocol for this review (Magrinelli 2016), we analysed ordinal or continuous data results from measurement scales (Table 1) as continuous variables (disability due to FAP progression, severity of peripheral neuropathy, mBMI, quality of life, severity of depression, and results for mortality and adverse events as dichotomous variables.
Continuous data
For continuous data, we provided mean differences (MDs) and 95% confidence intervals (CIs).
Dichotomous data
We reported dichotomous data as risk ratios (RRs) with 95% CIs.
Skewed and non‐quantitative data
We presented skewed data and non‐quantitative data descriptively.
Unit of analysis issues
The unit of analysis was the participant, which was also the unit of randomisation in the type of trials that we considered for this review. We took into account the level at which randomisation occurred.
Dealing with missing data
We contacted principal study investigators and sponsors in order to request information about missing data. Where possible, we analysed all outcome measures using an intention‐to‐treat analysis, following the principle 'once randomised always analysed'. We considered and reported if the principle 'last observation carried forward' was used in the included RCTs.
Data synthesis
As we were interested in the effects of each drug, we reported the results for each comparison separately and did not perform a meta‐analysis. We did not plan any network meta‐analysis for indirect comparisons of multiple interventions, which would also have been hampered by the use of different measures to assess outcomes in the included RCTs.
'Summary of findings' tables
We summarised the main findings of the review using 'Summary of findings' tables according to methods and recommendations described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We included the following outcomes.
Disability due to FAP progression
Severity of peripheral neuropathy
Quality of life
Number of participants who died during the trial
-
Adverse events, encompassing:
number of dropouts due to adverse events
number of participants experiencing at least one severe adverse event
Two review authors (FM and ST) worked independently on 'Summary of findings' assessments. Where there were multiple outcome measures among those predetermined for a specific outcome in the protocol of this review, we reported in the 'Summary of findings' tables the one with higher clinical relevance and inter‐rater reproducibility. For instance, among outcome measures assessing the severity of peripheral neuropathy in Berk 2013, we chose to report scores combining clinical and neurophysiological assessments, rather than purely clinical scores. Furthermore, 'Summary of findings' tables report outcome measures at the longest time interval available among those predetermined in the protocol of this review, as this represents the most relevant time interval for people affected by a chronic progressive disease. We used the five GRADEpro GDT considerations (study limitations, consistency of effect, imprecision, indirectness, and publication bias) to assess the certainty of the body of evidence for each outcome. We downgraded the certainty of studies from high according to whether these considerations are present to a serious degree. We used footnotes to aid the reader's understanding of our judgements where necessary.
Reaching conclusions
We based our conclusions only on findings from the quantitative or narrative synthesis of included studies for this review. We avoided making recommendations for practice. Our implications for research suggest priorities for future research and outline what the remaining uncertainties are for the topic.
We conducted the review according to the published protocol and reported any deviations from it in the Differences between protocol and review section of this review. Additional methods for use in future updates, if more trials are available and meta‐analysis becomes possible, are shown in Appendix 6.
Results
Description of studies
See: 'Characteristics of included studies' section; 'Characteristics of excluded studies' section.
Results of the search
We found a total of 719 references from the database search. After deduplication, we screened the resulting 477 references at title and abstract stage. We identified no additional papers by reviewing reference lists of included studies, narrative reviews, and major textbooks on peripheral neuropathies, by conducting a citation search via the Web of Science on 18 November 2019, or through email correspondence with known experts in the field between 2017 and 2018. We selected 147 references for full‐text screening. None of these papers required translation. We also found public synopses of the four identified randomised clinical trials (RCTs) on ClinicalTrials.gov (www.clinicaltrials.gov), and of two identified RCTs on ClinicalTrialsRegister.eu (www.clinicaltrialsregister.eu) when we conducted complementary searches on clinical trials registers and pharmaceutical company websites. See Figure 1 for a PRISMA flow diagram showing the study selection process for this review. We did not identify any ongoing RCTs that fulfilled our predetermined inclusion criteria.
1.
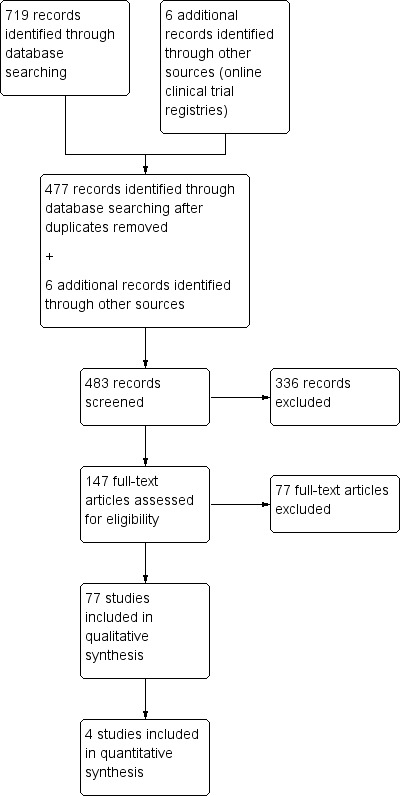
PRISMA chart for searched to included studies for FAPs
Included studies
See: 'Characteristics of included studies' section.
We included four studies, with a total of 655 participants. These studies provided data for four different comparisons.
Tafamidis versus placebo (128 participants; Coelho 2012)
Diflunisal versus placebo (130 participants; Berk 2013)
Patisiran versus placebo (225 participants; Adams 2018)
Inotersen versus placebo (172 participants; Benson 2018)
All studies were randomised, double‐blind, multicentre, international trials conducted in outpatient settings. Coelho 2012 evaluated the efficacy and safety of the transthyretin (TTR) kinetic stabiliser tafamidis 20 mg once daily compared to placebo during 18 months of treatment in adults with early‐stage TTR‐related familial amyloid polyneuropathy (FAP) due to Val30Met mutation in the TTR gene. Berk 2013 investigated the efficacy and safety of the TTR kinetic stabiliser diflunisal 250 mg twice daily compared to placebo during 24 months of treatment in adults with TTR‐FAP due to Val30Met and non‐Val30Met mutations in the TTR gene at any stage. Adams 2018 explored the efficacy and safety of the TTR small interfering RNA patisiran 0.3 mg/kg once every three weeks compared to placebo after 18 months of treatment in adults with TTR‐FAP due to Val30Met and non‐Val30Met mutations in the TTR gene at any stage, who might previously have received TTR kinetic stabilisers. Benson 2018 investigated the efficacy and safety of the TTR antisense oligonucleotide inotersen 300 mg once weekly compared to placebo after 65 weeks of treatment in adults with TTR‐FAP due to Val30Met and non‐Val30Met mutations in the TTR gene at early stages, who might previously have been treated with TTR kinetic stabilisers. Coelho 2012, Adams 2018, and Benson 2018 were funded by the manufacturer of the pharmacological agent under investigation.
Excluded studies
We excluded 77 full‐text articles that had been considered as possibly eligible for this review at the title and abstract screening stage. In particular, we excluded open‐label extensions of RCTs previously completed.
We listed 11 studies of the articles reviewed in full text as Excluded studies. These were:
seven non‐randomised studies, of which four were open‐label extension studies involving included interventions (Adams 2015; Coelho 2013; Plante‐Bordeneuve 2018; Waddington‐Cruz 2016), and three were other non‐randomised studies (Barroso 2017; Russo 2013; Suhr 2015);
one open‐label study of an investigational drug (Gillmore 2015);
an abstract reporting on drug development (Sah 2011);
a paper comparing tamafidis results from a RCT with those of an open‐label study (Gundapaneni 2018);
a RCT of tafamidis focusing on cardiomyopathy, not neuropathy (Maurer 2018).
See: the 'Characteristics of excluded studies' section.
Risk of bias in included studies
We presented the 'Risk of bias' assessment of each included study in the 'Characteristics of included studies' tables. Figure 2 summarises the review authors’ 'Risk of bias' judgments for all included studies.
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
All studies reported methods of randomisation and allocation concealment and we rated them at low risk of bias (Coelho 2012; Berk 2013; Adams 2018; Benson 2018).
Blinding
Participants and investigators were blinded to the interventions in Coelho 2012, Berk 2013, Adams 2018, and Benson 2018, which we rated at low risk of bias. As for the blinding of outcome assessment, we rated three studies at unclear risk of bias (Coelho 2012; Adams 2018; Benson 2018), as the statistical analysis of results was conducted by personnel of the sponsor, whereas we rated Berk 2013 at low risk of bias.
Incomplete outcome data
In Coelho 2012, after randomisation of 128 participants, 1/65 (1.5%) in the tafamidis group and 2/63 (3.2%) in the placebo group were excluded from the intention‐to‐treat (ITT) population due to absence of post‐baseline evaluations or genetic testing not confirming TTR‐FAP. Coelho 2012 then reported a loss to follow‐up of 34/125 (27.2%) participants, with an ITT analysis; we therefore rated this study at high risk of bias. Berk 2013 reported 67/130 (51.5%) dropouts and mentioned an ITT analysis which included all randomised participants who initiated treatment, thus being judged at high risk of bias. After reporting that 40/225 (17.8%) participants discontinued the trial regimen and 32/225 (14.2%) were withdrawn from the trial, Adams 2018 defined a modified ITT population as all participants who underwent randomisation and received at least one dose of patisiran or placebo; thus, we rated this study at low risk of bias. In Benson 2018, one participant in the inotersen group underwent randomisation in error and did not begin the trial regimen. Although Benson 2018 mentioned that the efficacy analyses included all randomly‐assigned participants who received at least one dose of a trial regimen and who had at least one post‐baseline efficacy assessment for the mNIS+7 Ionis version or the Norfolk Quality of Life‐Diabetic Neuropathy Questionnaire (Norfolk QOL‐DN) score, there was no clear mention of ITT analysis. We therefore judged this study at unclear risk of bias.
Selective reporting
All included studies reported all prespecified outcomes in their published report papers (Coelho 2012; Berk 2013; Adams 2018; Benson 2018), or public synopses on online clinical trial registries, or both. Therefore, we rated these studies at low risk of bias.
Other potential sources of bias
We rated three studies at unclear risk of other potential sources of bias because of some involvement of the sponsor in the design and conduct of the study (Coelho 2012; Adams 2018; Benson 2018). We rated Berk 2013 at low risk of bias.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4
Summary of findings for the main comparison. Tafamidis compared to placebo for people with transthyretin‐familial amyloid polyneuropathy (TTR‐FAP).
| Tafamidis compared to placebo for people with transthyretin‐familial amyloid polyneuropathy (TTR‐FAP) | ||||||
| Patient or population: people with early‐stage TTR‐FAP Setting: outpatients Intervention: tafamidis Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with tafamidis | |||||
|
Disability due to FAP progression Not measured |
‐ | ‐ | ‐ | ‐ | ‐ | Disability due to FAP progression was not measured |
|
Severity of peripheral neuropathy (expressed as the change in NIS‐LL) Scale from: 0 (normal) to 88 (total impairment) Follow‐up: 18 monthsa |
The mean baseline NIS‐LL score in the placebo group was 11.44 points. The mean change from baseline in the placebo group was an increase (worsening) of 5.40 points | The mean change in NIS‐LL score in the tafamidis group was 3.21 points lower (better) than in the placebo group (5.63 points lower to 0.79 points lower) | ‐ | 125 (1 RCT)b | ⊕⊕⊝⊝ Lowc | MCID is 2 NIS‐LL points according to a Peripheral Nerve Society consensus group (Dyck 1997). Tafamidis probably slightly reduces the number of people with progression of peripheral neuropathy compared to placebo. |
| Quality of life (expressed as the change in 35‐Item Norfolk QOL‐DN total score) Scale from: ‐2 (best possible quality of life) to 138 (worst possible quality of life) Follow‐up: 18 monthsa | The mean baseline Norfolk QOL score in the placebo group was 30.80 points. The mean change from baseline in the placebo group was an increase (worsening) of 6.90 points | The mean change in Norfolk QOL score in the tafamidis group was 4.50 points lower (better) than in the placebo group (11.27 points lower to 2.27 points higher) | ‐ | 125 (1 RCT)b | ⊕⊝⊝⊝ Very lowd | MCID was not provided nor reported in the literature. Quality of life may decline slightly less with tafamidis than placebo. |
| Number of participants who died during the trial Follow‐up: 18 monthsa | Study population | RR 0.65 (0.11 to 3.74) | 128 (1 RCT)e | ⊕⊝⊝⊝ Very lowf | It is uncertain whether tafamidis had an effect on the number of participants who died during the trial compared to placebo. | |
| 5 per 100 | 3 per 100 (1 to 18) | |||||
| Number of dropouts due to adverse events Follow‐up: 18 monthsa | Study population | RR 1.29 (0.30 to 5.54) | 128 (1 RCT)e | ⊕⊝⊝⊝ Very lowf | It is uncertain whether tafamidis had an effect on the number of dropouts due to adverse events compared to placebo. | |
| 5 per 100 | 6 per 100 (1 to 26) | |||||
| Number of participants experiencing at least one severe adverse event Follow‐up: 18 monthsa | Study population | RR 1.16 (0.37 to 3.62) | 128 (1 RCT)e | ⊕⊝⊝⊝ Very lowf | It is uncertain whether tafamidis had an effect on the number of participants experiencing at least one severe adverse event compared to placebo. | |
| 8 per 100 | 9 per 100 (3 to 29) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; FAP: familial amyloid polyneuropathy; MCID: minimum clinically important difference; NIS‐LL: Neuropathy Impairment Score of the lower limbs; QOL‐DN: Quality of Life‐Diabetic Neuropathy Questionnaire; RCT: randomised clinical trial; RR: risk ratio; TTR: transthyretin. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
aEnd of the study. bIntention‐to‐treat population. cWe downgraded the certainty of evidence by one level for risk of bias (high risk of attrition bias in the single RCT included in this comparison) and by one level for serious imprecision (CI encompassed a clinically important effect and little or no effect). dWe downgraded the certainty of evidence by one level for risk of bias (high risk of attrition bias in the single RCT included in this comparison), by one level for serious imprecision (CI is wide and includes the possibility of both harms and benefits) and by one level for serious indirectness (mechanisms by which this intervention would affect QOL are not direct). eRandomised population (safety population). fWe downgraded the certainty of evidence by one level for risk of bias (high risk of attrition bias in the single RCT included in this comparison), by two levels for very serious imprecision (small event numbers; and CI includes the possibility of both harms and benefits) and by one level for serious indirectness (events collected by a non‐systematic assessment).
Summary of findings 2. Diflunisal compared to placebo for people with transthyretin‐familial amyloid polyneuropathy (TTR‐FAP).
| Diflunisal compared to placebo for people with transthyretin‐familial amyloid polyneuropathy (TTR‐FAP) | ||||||
| Patient or population: people with TTR‐FAP Setting: outpatient Intervention: diflunisal Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with diflunisal | |||||
|
Disability due to FAP progression (expressed as the change in Kumamoto Score) Scale from: 0 (normal) to 102 (worst disease severity). Follow‐up: 24 monthsa |
The mean baseline Kumamoto Score in the placebo group was 16.7 points. The mean change from baseline in the placebo group was an increase (worsening) of 8.0 points. |
The mean change in Kumamoto Score in the diflunisal group was 4.90 points lower (better) than in the placebo group (7.89 points lower to 1.91 points lower). | ‐ | 130 (1 RCT) | ⊕⊕⊝⊝ Lowb | MCID not provided nor reported in the literature. Diflunisal probably slightly decreases disability due to FAP progression compared to placebo. |
|
Severity of peripheral neuropathy (expressed as the change in NIS+7) Scale from: 0 (normal) to 270 (total impairment) Follow‐up: 24 monthsa |
The mean baseline NIS+7 score in the placebo group was 59.0 points. The mean change from baseline in the placebo group was an increase (worsening) of 26.3 points. |
The mean change in NIS+7 score in the diflunisal group was 18.10 points lower (better) than in the placebo group (26.03 points lower to 10.17 points lower). | ‐ | 130 (1 RCT) | ⊕⊕⊝⊝ Lowb | MCID detectable by neuromuscular experts is 2 NIS+7 points according to the international Peripheral Nerve Society (PNS 1995). Diflunisal probably slightly reduces the worsening of peripheral neuropathy compared to placebo. |
|
Quality of life (expressed as the change in SF‐36 physical component score) Scale from: 0 (worst possible quality of life) to 100 (best possible quality of life) Follow‐up: 24 monthsa |
The mean baseline SF‐36 QOL score (physical component) in the placebo group was 34.8 points. The mean change from baseline in the placebo group was a decrease (worsening) of 4.9 points. |
The mean change in SF‐36 QOL score (physical component) in the diflunisal group was 6.10 points higher (better) than in the placebo group (2.56 points higher to 9.64 points higher). | ‐ | 130 (1 RCT) | ⊕⊝⊝⊝ Very lowc |
MCID proposed in the SF‐36v2 2011 is 2 points on the physical component summary. QOL (physical status) may decline slightly less with diflunisal than placebo. |
|
Quality of life (expressed as the change in SF‐36 mental component score) Scale from: 0 (worst possible quality of life) to 100 (best possible quality of life). Follow‐up: 24 monthsa |
The mean baseline SF‐36 QOL score (mental component) in the placebo group was 46.5 points. The mean change from baseline in the placebo group was a decrease (worsening) of 0.9 points. |
The mean change in SF‐36 QOL score (mental component) in the diflunisal group was 4.40 points higher (better) than in the placebo group (0.19 points lower to 8.99 points higher). | ‐ | 130 (1 RCT) | ⊕⊝⊝⊝ Very lowd | MCID proposed in the SF‐36v2 2011 is 3 points on the mental component summary. Quality of life (mental status) may decline slightly less with diflunisal than placebo. |
| Number of participants who died during the trial Follow‐up: 24 monthsa | Study population | RR 0.46 (0.15 to 1.41) | 130 (1 RCT) | ⊕⊝⊝⊝ Very lowe | There is evidence that there may be fewer deaths among people treated with diflunisal | |
| 14 per 100 | 6 per 100 (2 to 19) | |||||
| Number of dropouts due to adverse events Follow‐up: 24 monthsa | Study population | RR 2.06 (0.39 to 10.87) | 130 (1 RCT) | ⊕⊝⊝⊝ Very lowe | Diflunisal may increase the number of dropouts due to adverse events. | |
| 6 per 100 | 11 per 100 (3 to 36) | |||||
| Number of participants experiencing at least one severe adverse event Follow‐up: 24 monthsa | Study population | RR 0.77 (0.18 to 3.32) | 130 (1 RCT) | ⊕⊝⊝⊝ Very lowe | Diflunisal may have no clear effect on the number of participants experiencing at least one severe adverse event. | |
| 6 per 100 | 5 per 100 (1 to 20) | |||||
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; FAP: familial amyloid polyneuropathy; MCID: minimum clinically important difference; NIS+7: Neuropathy Impairment Score plus 7 nerves test; QOL: quality of life; RCT: randomised clinical trial; RR: risk ratio; SD: standard deviation; SF‐36: 36‐Item Short‐Form Health Survey; TTR: transthyretin. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
aEnd of the study. bWe downgraded the certainty of evidence by one level for risk of bias (high risk of attrition bias in the single RCT included in this comparison) and by one level for serious imprecision. cWe downgraded the certainty of evidence by one level for risk of bias (high risk of attrition bias in the single RCT included in this comparison), by one level for serious imprecision and by one level for serious indirectness (data regarding QOL provided for SF‐36 physical and mental components separately). dWe downgraded the certainty of evidence by one level for risk of bias (high risk of attrition bias in the single RCT included in this comparison), by one level for serious imprecision (CI includes the possibility of both harms and benefits) and by one level for serious indirectness (data regarding QOL provided for SF‐36 physical and mental components separately and mechanisms by which this intervention would affect QOL are not direct). eWe downgraded the certainty of evidence by one level for risk of bias (high risk of attrition bias in the single RCT included in this comparison) and by two levels for very serious imprecision (small event numbers and CI includes the possibility of both harms and benefits).
Summary of findings 3. Patisiran compared to placebo for people with transthyretin‐familial amyloid polyneuropathy (TTR‐FAP).
| Patisiran compared to placebo for people with transthyretin‐familial amyloid polyneuropathy (TTR‐FAP) | ||||||
| Patient or population: people with TTR‐FAP Setting: outpatient Intervention: patisiran Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with patisiran | |||||
|
Disability due to FAP progression (expressed as the change in R‐ODS) Scale from: 0 (worst disability) to 48 (normal) Follow‐up: 18 monthsa |
The mean baseline R‐ODS score in the placebo group was 29.8 points. The mean change from baseline in the placebo group was a decrease (worsening) of 8.9 points.b |
The mean change in R‐ODS score in the patisiran group was 8.90 points higher (better) than in the placebo group (7.00 points higher to 10.80 points higher).b | ‐ | 225 (1 RCT) | ⊕⊕⊕⊝ Moderatec | MCID not provided nor reported in the literature. Patisiran probably decreases disability due to FAP progression compared to placebo. |
|
Severity of peripheral neuropathy
(expressed as the change in mNIS+7ALN) Scale from: 0 (normal) to 304 (total impairment) Follow‐up: 18 monthsa |
The mean baseline mNIS+7ALN score in the placebo group was 74.6 points. The mean change from baseline in the placebo group was an increase (worsening) of 27.96 points.b |
The mean change in mNIS+7ALN score in the patisiran group was 33.99 points lower (better) than in the placebo group (39.86 points lower to 28.13 points lower).b | ‐ | 225 (1 RCT) | ⊕⊕⊕⊝ Moderatec | MCID not available for this modified NIS+7 scores. Patisiran probably slightly reduces the worsening of peripheral neuropathy compared to placebo. |
|
Quality of life
(expressed as the change in 35‐Item Norfolk QOL‐DN total score) Scale from: ‐4 (best possible quality of life) to 136 (worst possible quality of life) Follow‐up: 18 monthsa |
The mean baseline Norfolk QOL score in the placebo group was 55.5 points. The mean change from baseline in the placebo group was an increase (worsening) of 14.4 pointsb | The mean change in Norfolk QOL score in the patisiran group was 21.10 points lower (better) than in the placebo group (27.20 points lower to 15.00 points lower).b | ‐ | 225 (1 RCT) | ⊕⊕⊝⊝ Lowd | MCID not provided nor reported in the literature. Quality of life may decline slightly less with patisiran than placebo. |
| Number of participants who died during the trial Follow‐up: 18 monthsa | Study population | RR 0.61 (0.21 to 1.74) | 225 (1 RCT) | ⊕⊕⊝⊝ Lowe | There may be no clear difference between the patisiran group and the placebo group in the number of participants who died during the trial. | |
| 8 per 100 | 5 per 100 (2 to 14) | |||||
| Number of dropouts due to adverse events Follow‐up: 18 monthsa | Study population | RR 0.33 (0.13 to 0.82) | 225 (1 RCT) | ⊕⊕⊝⊝ Lowf | There may be little or no difference between the patisiran group and the placebo group in the number of dropouts due to adverse events. | |
| 13 per 100 | 5 per 100 (2 to 12) | |||||
| Number of participants experiencing at least one severe adverse event Follow‐up: 18 monthsa | Study population | RR 0.91 (0.64 to 1.28) | 225 (1 RCT) | ⊕⊕⊝⊝ Lowe | There may be little or no difference between the patisiran group and the placebo group in the number of participants with at least one severe adverse event. | |
| 40 per 100 | 37 per 100 (26 to 52) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval;FAP: familial amyloid polyneuropathy;MCID: minimum clinically important difference; mNIS+7ALN: modified Neuropathy Impairment Score plus 7 nerves test (Alnylam);QOL‐DN: Quality of Life‐Diabetic Neuropathy Questionnaire; RCT: randomised clinical trial;ROD‐S: Rasch‐built Overall Disability Scale; RR: risk ratio; TTR: transthyretin. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
aEnd of the study. bMeans change from baseline and mean differences between groups refer to least‐squares means and least‐squares mean differences, respectively. cWe downgraded the certainty of evidence by one level for serious imprecision. dWe downgraded the certainty of evidence by one level for serious imprecision and by one level for serious indirectness (mechanisms by which this intervention would affect QOL are not direct). eWe downgraded the certainty of evidence by two levels for very serious imprecision (small event numbers and CI includes the possibility of both harms and benefits). fWe downgraded the certainty of evidence by two levels for very serious imprecision (small event numbers and wide CI).
Summary of findings 4. Inotersen compared to placebo for people with transthyretin‐familial amyloid polyneuropathy (TTR‐FAP).
| Inotersen compared to placebo for people with transthyretin‐familial amyloid polyneuropathy (TTR‐FAP) | ||||||
| Patient or population: people with TTR‐FAP Setting: outpatient Intervention: inotersen Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with inotersen | |||||
|
Disability due to FAP progression Not measured |
‐ | ‐ | ‐ | ‐ | ‐ | Disability due to FAP progression was not measured. |
|
Severity of peripheral neuropathy
(expressed as the change in mNIS+7Ionis) Scale from: ‐22.3 (normal) to 346.3 (total impairment) Follow‐up: 66 weeksa |
The mean baseline mNIS+7Ionis score in the placebo group was 74.8 points. The mean change from baseline in the placebo group was an increase (worsening) of 23.89 pointsb | The mean change in mNIS+7Ionis score in the inotersen group was 19.73 points lower (better) than in the placebo group (26.50 lower to 12.96 lower)b | ‐ | 172 (1 RCT) | ⊕⊕⊕⊝ Moderatec | The MCID detectable is 2 points. Inostersen probably reduces the worsening of peripheral neuropathy compared to placebo. |
|
Quality of life (expressed as the change in 35‐Item Norfolk QOL‐DN total score) Scale from: ‐4 (best possible quality of life) to 136 (worst possible quality of life) Follow‐up: 66 weeksa |
The mean baseline Norfolk QOL score in the placebo group was 48.7 points. The mean change from baseline in the placebo group was an increase (worsening) of 10.77 pointsb | The mean change in Norfolk QOL score in the inotersen group was 10.85 points lower (better) than in the placebo group (17.25 lower to 4.45 lower)b | ‐ | 172 (1 RCT) | ⊕⊕⊝⊝ Lowd | MICD not provided nor reported in the literature. Inotersen may have little effect on the change in QOL. |
| Number of participants who died during the trial Follow‐up: 66 weeksa | Study population | RR 5.94 (0.33 to 105.60) | 172 (1 RCT) | ⊕⊕⊝⊝ Lowe | The number of participants who died during the trial may be greater in the inotersen group than in the placebo group. | |
| 0/60 deaths | 5/112 deaths | |||||
| Number of dropouts due to adverse events Follow‐up: 66 weeksa | Study population | RR 8.57 (1.16 to 63.07) | 172 (1 RCT) | ⊕⊕⊝⊝ Lowf | The number of dropouts due to adverse events may be greater in the inotersen group than the placebo group. | |
| 2 per 100 | 14 per 100 (2 to 100) | |||||
| Number of participants experiencing at least one severe adverse event Follow‐up: 66 weeksa | Study population | RR 1.48 (0.85 to 2.57) | 172 (1 RCT) | ⊕⊕⊝⊝ Lowe | The number of participants experiencing at least one severe adverse event may be greater in the inotersen group than in the placebo group. | |
| 22 per 100 | 32 per 100 (18 to 56) | |||||
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; FAP: familial amyloid polyneuropathy; MCID: minimum clinically important difference; mNIS+7Ionis: modified Neuropathy Impairment Score plus 7 nerves test (Ionis version); QOL‐DN: Quality of Life‐Diabetic Neuropathy Questionnaire; RCT: randomised clinical trial; RR: risk ratio; TTR: transthyretin. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
aEnd of the study. bMean change from baseline and mean differences between groups refer to least‐squares means and least‐squares mean differences, respectively. cWe downgraded the certainty of evidence by one level for serious imprecision. dWe downgraded the certainty of evidence by one level for serious imprecision and by one level for serious indirectness (mechanisms by which this intervention would affect QOL are not direct). eWe downgraded the certainty of evidence by two levels for very serious imprecision (small event numbers and CI includes the possibility of both harms and benefits). fWe downgraded the certainty of evidence by two levels for very serious imprecision (small event numbers and wide CI).
See: Table 1; Table 2; Table 3; Table 4.
Among the included studies, one study investigated tafamidis versus placebo (Coelho 2012), one study compared diflunisal with placebo (Berk 2013), one study explored patisiran versus placebo (Adams 2018), and one study examined inotersen versus placebo (Benson 2018). As all included studies investigated a different pharmacological agent compared to placebo, we did not perform a standard pairwise meta‐analysis. We had not planned any network meta‐analysis for indirect comparisons of multiple interventions for this review. The number of studies and available data are currently too limited for formal indirect comparisons to be made between drugs. In addition, they would have been difficult to perform since different measures to assess outcomes were used in the included randomised clinical trials (RCTs).
Tafamidis versus placebo in TTR‐FAP
See: Table 1.
One study, including 128 randomised participants (intention‐to‐treat (ITT) population, 125 participants) contributed data to the comparison of tafamidis versus placebo (Coelho 2012). This study followed the principle of 'last observation carried forward'. We obtained data for Coelho 2012, both from published report papers and the online trial registries ClinicalTrials.gov (www.clinicaltrials.gov) and ClinicalTrialsRegister.eu (www.clinicaltrialsregister.eu). The predetermined outcomes at time points selected for this review were available at 12 months and at the end of the follow‐up period (18 months) for efficacy outcomes and at the end of the follow‐up period (18 months) for acceptability and tolerability outcomes. As results for continuous variables were available as means ± standard deviations (SDs) for changes within groups and as least‐square mean differences (MDs) ± standard errors (SEs) for differences between groups, we calculated MDs and 95% CIs between groups in Review Manager 5 (Review Manager 2014), using available data for changes within groups.
Primary outcome measure: disability due to FAP progression
Coelho 2012 did not report our predetermined primary outcome measure.
Secondary outcome measures
Severity of peripheral neuropathy
In Coelho 2012 (ITT population, N = 125), the severity of peripheral neuropathy, expressed as the change from baseline in the Neuropathy Impairment Score ‐ Lower Limb (NIS‐LL) at 12 months and 18 months was a secondary endpoint. The NIS‐LL ranges from 0 to 88, with higher scores indicating greater deficits (minimum clinically important difference (MCID): 2 points; Table 5).
At 12 months, the mean (standard deviation (SD)) increase from baseline in the NIS‐LL was 1.005 ± 3.964 points in the tafamidis group and 4.835 ± 7.697 points in the placebo group. Therefore, tafamidis may lead to slightly less worsening of peripheral neuropathy compared to placebo (MD ‐3.83 points, 95% confidence interval (CI) ‐5.99 to ‐1.67; P < 0.001; Analysis 1.1), but absolute values of change are small in both groups. At 18 months, the mean (SD) increase from baseline in the NIS‐LL was 2.193 ± 4.372 points in the tafamidis group and 5.402 ± 8.661 points in the placebo group, once again suggesting that there may be slightly less worsening of peripheral neuropathy with tafamidis than with placebo (MD ‐3.21 points, 95% CI ‐5.63 to ‐0.79 points; P = 0.009; low‐certainty evidence; Analysis 1.1).
1.1. Analysis.
Comparison 1 Tamafidis versus placebo in TTR‐FAP, Outcome 1 Severity of peripheral neuropathy (expressed as the change from baseline in NIS‐LL).
We downgraded the certainty of evidence once for risk of bias as the evidence is from a single study judged at high risk of attrition bias due to its dropout rate, and once for serious imprecision as the evidence is from a single study involving 128 participants and CIs encompass a clinically important effect and little or no effect. The absolute values of change only just exceed the MCID at 12 months and may exceed it at 18 months.
Change in modified body mass index (mBMI)
Coelho 2012 (ITT population, N = 125) investigated the change from baseline in the mBMI at 12 months and 18 months as a secondary endpoint.
At 12 months, the mean (SD) change from baseline in the mBMI was an increase of 19.4 ± 71.8 kg/m2 g/L in the tafamidis group and a decrease of 30.8 ± 74.9 kg/m2 g/L in the placebo group (MD 50.20 kg/m2 g/L, 95% CI 24.46 to 75.94; P < 0.001; Analysis 1.2). At 18 months, the mean (SD) change from baseline in the mBMI was an increase of 37.9 ± 73.7 kg/m2 g/L in the tafamidis group and a decrease of 32.7 ± 88.6 kg/m2 g/L in the placebo group (MD 70.60 kg/m2 g/L, 95% CI 41.96 to 99.24; P < 0.001; Analysis 1.2). Therefore, at both time points, tafamidis may improve nutritional status compared to placebo.
1.2. Analysis.
Comparison 1 Tamafidis versus placebo in TTR‐FAP, Outcome 2 Change in mBMI.
We downgraded the certainty of evidence by one level for risk of bias as the evidence is from a single study judged at high risk of attrition bias due to its dropout rate, and by one level for serious imprecision as the evidence is from a single study involving 128 participants and CIs are wide. Furthermore BMI is an indirect outcome measure potentially influenced by many other aspects of life other than the drug itself.
Quality of life
In Coelho 2012 (ITT population, N = 125), quality of life, expressed as the change from baseline in the 35‐item Norfolk Quality of Life‐Diabetic Neuropathy (QOL‐DN) Questionnaire total score over 12 months was a secondary endpoint; the change over 18 months was a co‐primary endpoint. The Norfolk QOL‐DN total score ranges from ‐2 to 138, with lower scores indicating better quality of life (MCID not provided nor reported in the literature; Table 5).
At 12 months, the mean (SD) increase from baseline in the Norfolk QOL score was 1.10 ± 14.70 points in the tafamidis group and 4.60 ± 19.00 points in the placebo group. At 18 months, the mean (SD) increase from baseline in the Norfolk QOL score was 2.40 ± 14.60 points in the tafamidis group and 6.90 ± 22.90 points in the placebo group. It was therefore, uncertain whether tafamidis leads to any change from baseline of quality of life after 12 months of treatment (MD ‐3.50 points, 95% CI ‐9.48 to 2.48; P = 0.25; Analysis 1.3) and 18 months of treatment (MD ‐4.50 points, 95% CI ‐11.27 to 2.27; P = 0.19; Analysis 1.3) because the certainty of evidence is very low.
1.3. Analysis.
Comparison 1 Tamafidis versus placebo in TTR‐FAP, Outcome 3 Quality of life (expressed as the change from baseline in Norfolk QOL‐DN total score).
We downgraded the certainty of evidence by one level for serious risk of bias (as the evidence is from a single study with high risk of attrition bias due to its dropout rate), by one level for serious imprecision (as the evidence is from a single study involving 128 participants and CIs are wide and include the possibility of both harms and benefits), and by one level for serious indirectness (as mechanisms by which this intervention would affect quality of life are not direct).
Severity of depression
Coelho 2012, did not assess depression using our predetermined outcome measures.
Number of participants who died during the trial
According to the published report by Coelho and colleagues (Coelho 2012), 5 (3.9%) of the 128 randomised participants died during the trial, including 2/65 (3.1%) participants in the tafamidis group and 3/63 (4.8%) participants in the placebo group, with no clear difference between groups (RR 0.65, 95% CI 0.11 to 3.74; P = 0.63; very low‐certainty evidence; Analysis 1.4). In the tafamidis group, one person died from cardiac tamponade after insertion of a pacemaker, and the cause was unknown in another. In the placebo arm, one participant died from sepsis, one from hepatic failure, and one from an unknown cause. The trialists stated that all deaths were a consequence of complications following liver transplantation and none happened during the study or before liver transplantation.
1.4. Analysis.
Comparison 1 Tamafidis versus placebo in TTR‐FAP, Outcome 4 Number of participants who died during the trial (published paper ‐ see text).
There are some discrepancies in mortality results between the published report and results available on ClinicalTrialsRegister.eu (www.clinicaltrialsregister.eu). According to the latter, 1/65 (1.5%) participants in the tafamidis group and 3/63 (4.8%) participants in the placebo group died during the trial (RR 0.32, 95% CI 0.03 to 3.02; P = 0.32), with little or no between‐group difference. We received no response to our request for clarification.
Adverse events
i) Number of participants experiencing at least one adverse event
In Coelho 2012 (N = 128), 60/65 (92.3%) tafamidis‐treated participants and 61/63 (96.8%) placebo‐treated participants experienced at least one adverse event during the trial, with little or no difference between groups (RR 0.95, 95% CI 0.88 to 1.04; P = 0.26; Analysis 1.5). In the same population, 39/65 (60.0%) tafamidis‐treated participants and 43/63 (68.2%) placebo group participants reported having experienced at least one adverse event related, or possibly related, to the study medication; there was no clear between‐group difference (RR 0.88, 95% CI 0.68 to 1.14; P = 0.33).
1.5. Analysis.
Comparison 1 Tamafidis versus placebo in TTR‐FAP, Outcome 5 Number of participants experiencing at least one adverse event.
ii) Number of participants experiencing mild adverse events
In Coelho 2012 (N = 128), there was no clear between‐group difference in the number of participants experiencing mild adverse events: 57/65 (87.7%) in the tafamidis group and 56/63 (88.9%) in the placebo group (RR 0.99, 95% CI 0.87 to 1.12; P = 0.83; Analysis 1.6).
1.6. Analysis.
Comparison 1 Tamafidis versus placebo in TTR‐FAP, Outcome 6 Number of participants experiencing mild adverse events.
iii) Number of dropouts due to adverse events
Coelho 2012 (N = 128) reported that 4/65 (6.1%) tafamidis‐treated participants and 3/63 (4.8%) placebo‐treated participants discontinued study medication due to a treatment‐emergent adverse event (RR 1.29, 95% CI 0.30 to 5.54; P = 0.73; very low‐certainty evidence; Analysis 1.7), which indicated little or no between‐group difference. These adverse events were diarrhoea, nausea, urticaria, and pregnancy (with normal outcome) in the tafamidis group, and paraesthesia associated with fatigue, nausea with unintentional weight loss, and worsening cardiac amyloidosis in the placebo group.
1.7. Analysis.
Comparison 1 Tamafidis versus placebo in TTR‐FAP, Outcome 7 Number of dropouts due to adverse events.
iv) Number of participants experiencing at least one severe adverse event
In Coelho 2012 (N = 128), 6/65 (9.2%) tafamidis‐treated participants and 5/63 (7.9%) placebo‐treated participants experienced at least one serious treatment‐emergent adverse event (RR 1.16, 95% CI 0.37 to 3.62; P = 0.79; very low‐certainty evidence; Analysis 1.8), indicating no clear difference between groups. The serious adverse events reported in the tafamidis group were urinary tract infections (2 participants), conduction disorder (1 participant), localised infection (1 participant), pneumonia (1 participant), viral infection (1 participant), and urticaria (1 participant).
1.8. Analysis.
Comparison 1 Tamafidis versus placebo in TTR‐FAP, Outcome 8 Number of participants experiencing at least one severe adverse event.
For all outcomes related to mortality and adverse events, we downgraded the certainty of evidence by one level for serious risk of bias (evidence is from a single study judged at high risk of attrition bias due to dropout rate), by two levels for very serious imprecision (there are small event numbers, and CIs include the possibility of both harms and benefits), and by one level for serious indirectness (events were collected by a non‐systematic assessment).
Diflunisal versus placebo in TTR‐FAP
See: Table 2.
One study, including 130 participants (ITT population) contributed data to the comparison of diflunisal versus placebo (Berk 2013). This study followed the principle of 'last observation carried forward'. We obtained data for Berk 2013 from both published papers and ClinicalTrials.gov (www.clinicaltrials.gov). The outcomes preselected for this review were available at 12 months and at the end of the follow‐up period (24 months). Means with 95% CIs for change within groups and MDs with 95% CIs and P values for differences between groups were available for continuous data; therefore, we did not impute missing SDs or SEs.
Primary outcome measure: disability due to FAP progression
Berk 2013 (N = 130) reported disability due to FAP progression as the change from baseline in the Kumamoto Score (KS) at 12 months and 24 months, as a secondary outcome. The KS ranges from 0 to 102, with higher scores indicating greater disease severity (MCID not provided nor reported in the literature; Table 5).
At 12 months, the mean increase from baseline in the KS was 1.9 points (95% CI 0.1 to 3.7) in the diflunisal group and 4.1 points (95% CI 2.0 to 6.2) in the placebo group, with a MD between groups of ‐2.20 points (95% CI ‐4.91 to 0.51; P = 0.115; Analysis 2.1) corresponding to little or no between‐group difference. At 24 months, the mean increase from baseline in the KS was 3.1 points (95% CI 1.1 to 5.1) in the diflunisal group and 8.0 points (95% CI 5.7 to 10.3) in the placebo group. Therefore, diflunisal may slightly reduce worsening of disability due to FAP progression compared to placebo (MD ‐4.90 points, 95% CI ‐7.89 to ‐1.90; P = 0.002; low‐certainty evidence; Analysis 2.1).
2.1. Analysis.
Comparison 2 Diflunisal versus placebo in TTR‐FAP, Outcome 1 Disability due to FAP progression (expressed as the change from baseline in KS).
We downgraded the certainty of evidence by one level for serious risk of bias (evidence is from a single study judged at high risk of attrition bias due to dropout rate) and by one level for serious imprecision (evidence is from a single study involving 130 participants).
Secondary outcome measures
Severity of peripheral neuropathy
In Berk 2013 (N = 130), the primary endpoint was severity of peripheral neuropathy expressed as the change from baseline in the Neuropathy Impairment Score plus 7 nerves test (NIS+7) at 12 months and 24 months. The NIS+7 ranges from 0 to 270, with higher scores indicating greater deficits (MCID: 2 points; Table 5).
At 12 months, the mean increase from baseline in the NIS+7 was 6.2 points (95% CI 2.8 to 9.6) in the diflunisal group and 12.5 points (95% CI 8.6 to 16.4) in the placebo group, with a MD between groups of ‐6.30 points (95% CI ‐11.38 to ‐1.22; P = 0.017; Analysis 2.2), which may slightly favour diflunisal. At 24 months, the mean increase from baseline in the NIS+7 was 8.2 points (95% CI 2.9 to 13.5) in the diflunisal group and 26.3 points (95% CI 20.2 to 32.4) in the placebo group, with a MD between groups of ‐18.10 points (95% CI ‐26.03 to ‐10.17; low‐certainty evidence) favouring diflunisal (P < 0.001; Analysis 2.2).
2.2. Analysis.
Comparison 2 Diflunisal versus placebo in TTR‐FAP, Outcome 2 Severity of peripheral neuropathy (expressed as the change from baseline in the NIS+7).
We downgraded the certainty of evidence by one level for serious risk of bias, as the evidence is from a single study judged at high risk of attrition bias due to the dropout rate, and by one level for serious imprecision, as the evidence is from a single study involving 130 participants.
Berk 2013 (N = 130) also investigated, as secondary endpoints, the severity of peripheral neuropathy with the NIS and the NIS‐LL. The NIS ranges from 0 to 244, with higher scores indicating greater deficits (MCID: 2 points; Table 5) and the NIS‐LL ranges from 0 to 88, with higher scores indicating greater deficits (MCID: 2 points; Table 5).
At 12 months, the mean increase from baseline in the NIS was 4.10 points (95% CI 1.30 to 6.90) in the diflunisal group and 10.10 points (95% CI 6.9 to 13.3) in the placebo group, with a MD between groups of ‐6.00 points (95% CI ‐10.17 to ‐1.83; P = 0.006; Analysis 2.3) favouring diflunisal. At 24 months, the mean increase from baseline in the NIS was 6.40 points (95% CI 1.6 to 11.2) in the diflunisal group and 23.20 points (95% CI 17.9 to 28.5) in the placebo group, with a MD between groups of ‐16.80 points (95% CI ‐23.81 to ‐9.78; P < 0.001; Analysis 2.3) in favour of diflunisal.
2.3. Analysis.
Comparison 2 Diflunisal versus placebo in TTR‐FAP, Outcome 3 Severity of peripheral neuropathy (expressed as the change from baseline in the NIS).
Regarding the NIS‐LL, the mean increase from baseline at 12 months was 3.2 points (95% CI 1.2 to 5.2) in the diflunisal group and 6.0 points (95% CI 3.8 to 8.2) in the placebo group, with a MD between groups of ‐2.80 points (95% CI ‐5.72 to 0.12; P = 0.063; Analysis 2.4), corresponding to little or no between‐group difference. At 24 months, the mean increase from baseline in the NIS‐LL was 3.80 points (95% CI 1.0 to 6.6) in the diflunisal group and 12.10 points (95% CI 8.9 to 15.3) in the placebo group, with a MD between groups of ‐8.30 points (95% CI ‐12.47 to ‐4.13) in favour of diflunisal (P < 0.001; Analysis 2.4).
2.4. Analysis.
Comparison 2 Diflunisal versus placebo in TTR‐FAP, Outcome 4 Severity of peripheral neuropathy (expressed as the change from baseline in the NIS‐LL).
Change in modified body mass index (mBMI)
Berk 2013 (N = 130) investigated the change from baseline in the mBMI at 12 months and 24 months as secondary endpoints.
At 12 months, the mean decrease from baseline in the mBMI was 18.7 kg/m2 g/L (95% CI ‐51.5 to 14.1) in the diflunisal group and 38.5 kg/m2 g/L (95% CI ‐74.90 to ‐2.10) in the placebo group. At 24 months, the mean decrease from baseline in the mBMI was 33.7 kg/m2 g/L (95% CI ‐69.20 to 1.80) in the diflunisal group and 67.9 kg/m2 g/L (95% CI ‐108.10 to ‐27.70) in the placebo group. There was therefore, no clear between‐group difference in the worsening of nutritional status after 12 months (MD 19.80 kg/m2 g/L, 95% CI ‐28.27 to 67.87; P = 0.422; Analysis 2.5) and 24 months (MD 34.20 kg/m2 g/L, 95% CI ‐18.42 to 86.82; P = 0.21; Analysis 2.5).
2.5. Analysis.
Comparison 2 Diflunisal versus placebo in TTR‐FAP, Outcome 5 Change in mBMI.
There was serious risk of bias, as the single study has a high risk of attrition bias due to the dropout rate, and very serious imprecision as the evidence is from a single study involving 130 participants and CIs are wide and encompass both a clinically positive and a largely negative effect. Furthermore, this endpoint is indirect (see above).
Quality of life
In Berk 2013 (N = 130), the change from baseline in the quality of life measured with the 36‐Item Short‐Form Health Survey (SF‐36) physical component score and SF‐36 mental component score were secondary endpoints. Both scales range from 0 to 100, with lower scores indicating worse status (physical component score MCID: 2 points; mental component score MCID: 3 points; Table 5.
At 12 months, the mean change from baseline in the SF‐36 physical component score was an increase of 0.7 points (95% CI ‐1.1 to 2.5) in the diflunisal group and a decrease of 1.9 (95% CI ‐3.9 to 0.1) in the placebo group, with a MD between groups of 2.60 (95% CI ‐0.04 to 5.24; P = 0.06; Analysis 2.6), corresponding to an uncertain between‐group difference. At 24 months, the mean change from baseline in the SF‐36 physical component score was an increase of 1.2 points (95% CI ‐1.2 to 3.6) in the diflunisal group and a decrease of 4.9 (95% CI ‐7.6 to ‐2.2) in the placebo group. The MD between groups was 6.10 points (95% CI 2.56 to 9.64; P = 0.001; very low‐certainty evidence; Analysis 2.6), so it was uncertain whether diflunisal improves quality of life.
2.6. Analysis.
Comparison 2 Diflunisal versus placebo in TTR‐FAP, Outcome 6 Quality of life (expressed as the change from baseline in SF‐36 physical score).
At 12 months, the mean increase from baseline in the SF‐36 mental component score was 2.5 points (95% CI 0.0 to 5.0) in the diflunisal group and 0.8 points (95% CI ‐2.0 to 3.6) in the placebo group. At 24 months, the mean change from baseline in the SF‐36 mental component score was an increase of 3.5 points (95% CI 0.4 to 6.6) in the diflunisal group and a decrease of 0.9 points (95% CI ‐4.4 to 2.6) in the placebo group. Therefore, it was uncertain if diflunisal leads to a change in the SF‐36 mental component score at 12 months (MD 1.70 points, 95% CI ‐1.98 to 5.38; P = 0.37; Analysis 2.7) 24 months (MD 4.40 points, 95% CI ‐0.19 to 8.99; P = 0.063; Analysis 2.7) because the certainty of the evidence is very low.
2.7. Analysis.
Comparison 2 Diflunisal versus placebo in TTR‐FAP, Outcome 7 Quality of life (change in SF‐36 mental score).
We downgraded the certainty of evidence by one level for serious risk of bias (high risk of attrition bias due to dropout rate), by one level for serious imprecision (CIs include the possibility of both harms and benefits), and by one level for serious indirectness (as mechanisms by which this intervention would affect quality of life are not direct).
Severity of depression
Berk 2013 did not assess depression using our predetermined outcome measures.
Number of participants who died during the trial
According to Berk 2013, 13 deaths occurred in the randomised population (N = 130) during the 24‐month study period, including 4 (6.25%) deaths among 64 participants in the diflunisal group and 9 (13.64%) deaths among 66 participants in the placebo group (RR 0.46, 95% CI 0.15 to 1.41; P = 0.17; very low‐certainty evidence; Analysis 2.8). Therefore, it is uncertain whether diflunisal decreases mortality. Causes of deaths were not reported. The trialists stated that 12/13 (92.3%) deaths occurred after discontinuation of the study medication.
2.8. Analysis.
Comparison 2 Diflunisal versus placebo in TTR‐FAP, Outcome 8 Number of participants who died during the trial.
We graded the certainty of evidence as very low, downgrading by one level for serious risk of bias (single study at high risk of attrition bias due to dropout rate), two levels for very serious imprecision (small event numbers; and CI includes the possibility of both harms and benefits).
Adverse events
i) Number of participants experiencing at least one adverse event
No data for this outcome were reported in Berk 2013.
ii) Number of participants experiencing mild adverse events
In Berk 2013 (N = 130), it is uncertain whether there were between‐group differences in the number of participants experiencing mild adverse events, with 29/64 (45.3%) in the diflunisal group and 27/66 (40.9%) in the placebo group (RR 1.11, 95% CI 0.75 to 1.65; P = 0.61; Analysis 2.9).
2.9. Analysis.
Comparison 2 Diflunisal versus placebo in TTR‐FAP, Outcome 9 Number of participants experiencing mild adverse events.
iii) Number of dropouts due to adverse events
In Berk 2013 (N = 130), 4/64 (6.2%) diflunisal‐treated participants and 2/66 (3.0%) placebo‐treated participants discontinued study medication due to treatment‐related adverse events, with a RR of 2.06 (95% CI 0.39 to 10.87; P = 0.39; very low‐certainty evidence; Analysis 2.10). Therefore, it is uncertain whether diflunisal has an effect on treatment discontinuation caused by adverse events. The adverse events that led to discontinuation of the trial regimen were gastrointestinal bleeding, congestive cardiac failure, glaucoma, and nausea in the diflunisal group, and headache and renal failure in the placebo group.
2.10. Analysis.
Comparison 2 Diflunisal versus placebo in TTR‐FAP, Outcome 10 Number of dropouts due to adverse events.
iv) Number of participants experiencing at least one severe adverse event
In Berk 2013 (N = 130), 3/64 (4.7%) participants in the diflunisal arm and 4/66 (6.1%) participants in the placebo arm discontinued study medication due to treatment‐related adverse events, thus being uncertain whether diflunisal has an effect on severe adverse events (RR 0.77, 95% CI 0.18 to 3.32; P = 0.73; very low‐certainty evidence; Analysis 2.11).
2.11. Analysis.
Comparison 2 Diflunisal versus placebo in TTR‐FAP, Outcome 11 Number of participants experiencing at least one severe adverse event.
For mortality and available adverse event outcomes, we downgraded the certainty of evidence by one level for serious risk of bias (evidence is from a single study judged at high risk of attrition bias due to the dropout rate) and by two levels for very serious imprecision (single study involving 130 participants; small event numbers; and CIs include the possibility of both harms and benefits).
Patisiran versus placebo in TTR‐FAP
See: Table 3.
One study (ITT population 225 participants) compared patisiran versus placebo (Adams 2018). We obtained data for Adams 2018 from both published papers and the online trial registries ClinicalTrials.gov (www.clinicaltrials.gov) and ClinicalTrialsRegister.eu (www.clinicaltrialsregister.eu). The outcomes prespecified for this review were only available at the end of the follow‐up period (18 months). Results for continuous variables were provided as least‐square means and SEs for change within groups and least‐square MDs with 95% CIs and P values for differences between groups, and we did not impute any missing mean, SD, MD, or 95% CI.
Primary outcome measure: disability due to FAP progression
Adams 2018 (N = 225) did not report any of the primary outcome measures described in the protocol of this review. However, it explored disability due to FAP progression, expressed as the change from baseline in the Rasch‐built Overall Disability Scale (R‐ODS) at 18 months, as a secondary outcome. The R‐ODS ranges from 0 to 48, with lower scores indicating greater disability (MCID not provided; Table 5).
At 18 months, the least‐squares mean (SE) change from baseline in the R‐ODS was 0.0 points (0.59) in the patisiran group and ‐8.9 points (0.88) in the placebo group. Therefore, patisiran probably decreases disability due to FAP progression compared to placebo (least‐squares MD 8.90 points, 95% CI 7.00 to 10.80; P < 0.001; moderate‐certainty evidence; Analysis 3.1).
3.1. Analysis.
Comparison 3 Patisiran versus placebo in TTR‐FAP, Outcome 1 Disability due to FAP progression (expressed as the change from baseline in the R‐ODS), at 18 months.
We downgraded the certainty of evidence by one level for serious imprecision, as evidence is from a single study including 225 participants.
Secondary outcome measures
Severity of peripheral neuropathy
In Adams 2018 (N = 225), the severity of peripheral neuropathy, expressed as the change from baseline in the modified Neuropathy Impairment Score plus 7 nerves test (mNIS+7ALN) was the primary efficacy endpoint. The scale ranges from 0 to 304, with higher scores indicating greater deficits (MCID not provided; Table 5) .
At 18 months, the least‐squares mean (SE) change from baseline in the mNIS+7ALN was a decrease of 6.03 points (1.74) in the patisiran group and an increase of 27.96 points (2.60) in the placebo group. The least‐squares MD between groups was ‐33.99 points (95% CI ‐39.86 to ‐28.12; P < 0.001; moderate‐certainty evidence; Analysis 3.2). This indicates that peripheral neuropathy probably worsened slightly less with patisiran than placebo.
3.2. Analysis.
Comparison 3 Patisiran versus placebo in TTR‐FAP, Outcome 2 Severity of peripheral neuropathy (expressed as the change from baseline in the mNIS+7Alnylam), at 18 months.
We downgraded the certainty of evidence by one level for serious imprecision, as evidence is from a single study including 225 participants.
Change in modified body mass index (mBMI)
Adams 2018 (N = 225) investigated the change from baseline in the mBMI at 18 months as a secondary endpoint.
At 18 months, the least‐squares mean (SE) decrease from baseline in the mBMI was 3.7 kg/m2 g/L (9.57) in the patisiran group and 119.4 kg/m2 g/L (14.51) in the placebo group, with a least‐squares MD between groups of 115.70 kg/m2 g/L (95% CI 82.40 to 149.00; P < 0.001; Analysis 3.3), indicating that nutritional status probably worsened less in patisiran‐treated participants than the placebo group.
3.3. Analysis.
Comparison 3 Patisiran versus placebo in TTR‐FAP, Outcome 3 Change in mBMI, at 18 months.
We downgraded the certainty of evidence by one level for serious imprecision, as the evidence is from a single study involving 225 participants, and CIs are wide.
Quality of life
The quality of life, expressed as the change from baseline in the Norfolk QOL‐DN total score was a secondary endpoint in Adams 2018 (N = 225). The scale ranges from ‐4 to 136, with lower scores indicating better quality of life (MCID not provided nor reported in the literature; Table 5).
At 18 months, the least‐squares mean (SE) change from baseline in the Norfolk QOL‐DN total score was a decrease of 6.7 points (1.77) in the patisiran group and an increase of 14.4 (2.73) in the placebo group (least‐squares MD ‐21.10 points, 95% CI ‐27.20 to ‐15.00; P < 0.001; low‐certainty evidence; Analysis 3.4). This indicates that quality of life may decline slightly less with patisiran than placebo.
3.4. Analysis.
Comparison 3 Patisiran versus placebo in TTR‐FAP, Outcome 4 Quality of life (change in Norfolk QOL‐DN total score), at 18 months.
We downgraded the certainty of evidence by one level for serious imprecision (sample size is below 400) and by one level for serious indirectness (mechanisms by which this intervention would affect quality of life are not direct).
Severity of depression
Adams 2018 did not assess depression using our predetermined outcome measures.
Number of participants who died during the trial
Adams 2018 reported 13 (5.8%) deaths in the randomised population out of 225 participants during the 18‐month study period, including seven (4.7%) deaths in the patisiran group (148 participants) and six (7.8%) deaths in the placebo group (77 participants) (RR 0.61, 95% CI 0.21 to 1.74; P = 0.35; low‐certainty evidence; Analysis 3.5). There may therefore, be little or no difference in mortality in participants treated with patisiran compared to those who received placebo. In the patisiran‐treated population, two deaths occurred as a consequence of cardiac arrest, two due to sudden cardiac death (one of which after more than 30 days off treatment), two due to cardiac failure, and one due to pulseless electrical activity. In the placebo group, causes of deaths were subarachnoid haemorrhage, staphylococcal sepsis, anaemia associated with gastrointestinal haemorrhage, acute kidney failure associated with urinary tract infection and bacteraemia, metastatic colorectal cancer, and ischaemic stroke. The investigators considered all deaths unlikely or not related to the intervention drug.
3.5. Analysis.
Comparison 3 Patisiran versus placebo in TTR‐FAP, Outcome 5 Number of participants who died during the trial.
Adverse events
i) Number of participants experiencing at least one adverse event
In Adams 2018 (N = 225), 143/148 (96.6%) patisiran‐treated participants and 75/77 (97.4%) placebo group participants experienced at least one adverse event (RR 0.99, 95% CI 0.95 to 1.04; P = 0.74; Analysis 3.6), with no clear between‐group difference.
3.6. Analysis.
Comparison 3 Patisiran versus placebo in TTR‐FAP, Outcome 6 Number of participants experiencing at least one adverse event.
ii) Number of participants experiencing mild adverse events
There was not enough detail to impute the prevalence of mild adverse events among all adverse events reported.
iii) Number of dropouts due to adverse events
In Adams 2018 (N = 225), 7/148 (4.7%) patisiran‐treated participants and 11/77 (14.3%) placebo‐treated participants were reported to have discontinued study medication due to adverse events (RR 0.33, 95% CI 0.13 to 0.82; P = 0.017; low‐certainty evidence; Analysis 3.7). The most frequent adverse events that led to discontinuation of the trial regimen were cardiac failure in the patisiran group (2/148 participants, 1.3%) and acute kidney injury in the placebo group (3/77 participants, 3.9%). For the same RCT, the online trial registries ClinicalTrials.gov (www.clinicaltrials.gov) and ClinicalTrialsRegister.eu (www.clinicaltrialsregister.eu) reported that 8/148 (5.4%) patisiran‐treated participants and 10/77 (13.0%) placebo‐treated participants discontinued study medication due to adverse events (RR 0.42, 95% CI 0.17 to 1.01; P = 0.05). Although we were unable to clarify this discrepancy, this study provides evidence that there may be no clear difference in dropouts due to adverse events with patisiran compared to placebo.
3.7. Analysis.
Comparison 3 Patisiran versus placebo in TTR‐FAP, Outcome 7 Number of participant dropouts due to adverse events (data from published paper ‐ see text).
iv) Number of participants experiencing at least one severe adverse event
Adams 2018 (N = 225) reported that 54/148 (36.5%) patisiran‐treated participants and 31/77 (40.3%) placebo group participants experienced at least one serious adverse event during the study (RR 0.91, 95% CI 0.64 to 1.28; P = 0.58; low‐certainty evidence; Analysis 3.8), thus suggesting that patisiran may lead to little or no difference in the incidence of severe adverse events. The most frequent severe adverse events reported in the patisiran group (N = 148) were diarrhoea (8 participants, 5.4%), cardiac failure (6 participants, 4.0%), pneumonia (3 participants, 2.0%), orthostatic hypotension (3 participants, 2.0%), and complete atrioventricular block (3 participants, 2.0%).
3.8. Analysis.
Comparison 3 Patisiran versus placebo in TTR‐FAP, Outcome 8 Number of participants experiencing at least one severe adverse event.
For all outcomes related to mortality and adverse events, we downgraded the certainty of evidence by two levels for very serious imprecision, as evidence is provided by a single study involving 225 participants, there are small event numbers, and CIs include the possibility of both harms and benefits.
Inotersen versus placebo in TTR‐FAP
See: Table 4.
One study, including 172 participants (ITT population) contributed data to the comparison of inotersen versus placebo (Benson 2018). We obtained information on results of Benson 2018 from both published papers and the online trial registry ClinicalTrials.gov (www.clinicaltrials.gov). The predetermined outcomes at time points selected for this review were available at the end of the follow‐up period (65 to 66 weeks) only. Results for continuous variables were provided both as means ± standard deviations (SDs) and least‐square means and SEs for change within groups, and as least‐square MDs and 95% CIs for differences between groups. We therefore calculated MDs and 95% CIs between groups in Review Manager 5 (Review Manager 2014), entering values of means ± SDs available for changes within groups.
Primary outcome measure: disability due to FAP progression
Benson 2018 did not report our predetermined primary outcome measure.
Secondary outcome measures
Severity of peripheral neuropathy
In Benson 2018 (N = 172), the severity of peripheral neuropathy, expressed as the change from baseline in the modified Neuropathy Impairment Score plus 7 nerves test (Ionis version) (mNIS+7Ionis) was one of the primary efficacy endpoints. The mNIS+7Ionis ranges from ‐22.3 to 346.3, with higher scores indicating greater deficits (MCID not provided; Table 5).
At week 66, the mean increase (SD) from baseline in the mNIS+7Ionis score was 4.16 ± 15.672 points in the inotersen group and 23.89 ± 24.19 points in the placebo group. The MD between groups was ‐19.73 (95% CI ‐26.5 to ‐12.96; P < 0.001; moderate‐certainty evidence; Analysis 4.1). This indicated that peripheral neuropathy worsened slightly less with inotersen than with placebo.
4.1. Analysis.
Comparison 4 Inotersen versus placebo in TTR‐FAP, Outcome 1 Severity of peripheral neuropathy (expressed as the change from baseline in the mNIS+7Ionis), at 66 weeks.
We downgraded the certainty of evidence by one level for serious imprecision, as evidence is from a single study involving 172 participants.
Benson 2018 (N = 172) also investigated the severity of peripheral neuropathy with other outcome measures, whose ranges did not correspond to the scales predetermined in the protocol of this review for this outcome.
Change in modified body mass index (mBMI)
Benson 2018 (N = 172) provided data on the change from baseline in the mBMI at week 65 as a secondary endpoint.
At week 65, the mean (SD) decrease from baseline in the mBMI was 73.32 ± 96.31 kg/m2 g/L in the inotersen group and 85.21 ± 91.26 kg/m2 g/L in the placebo group, thus revealing no clear between‐group difference in effect on nutritional status (MD 11.89 kg/m2 g/L, 95% CI ‐17.29 to 41.07; P = 0.43; Analysis 4.2).
4.2. Analysis.
Comparison 4 Inotersen versus placebo in TTR‐FAP, Outcome 2 Change in mBMI from baseline to 65 weeks.
We downgraded the certainty of evidence by one level for serious imprecision, as evidence is from a single study involving 172 participants.
Quality of life
In Benson 2018 (N = 172), the quality of life, expressed as the change from baseline in the Norfolk QOL‐DN total score was a primary efficacy endpoint. This scale has a range of ‐4 to 136, with lower scores indicating better quality of life (MCID not provided; Table 5).
At week 66, the mean (SD) change from baseline in the Norfolk QOL‐DN total score was a decrease of 0.08 ± 18.97 points in the inotersen group and an increase of 10.77 ± 21.13 points in the placebo group, with a MD between groups of ‐10.85 points (95% CI ‐17.25 to ‐4.45; low‐certainty evidence) favouring inotersen (P < 0.001; Analysis 4.3).
4.3. Analysis.
Comparison 4 Inotersen versus placebo in TTR‐FAP, Outcome 3 Quality of life (change in Norfolk QOL‐DN total score), at 66 weeks.
We downgraded the certainty of evidence by one level for serious imprecision (sample size is below 400) and by one level for serious indirectness (mechanisms by which this intervention would affect quality of life are not direct).
Benson 2018 (N =172) also reported the change from baseline in the SF‐36 physical component score as a measure of quality of life and a tertiary outcome. The SF‐36 physical component scale ranges from 0 to 100, with lower scores indicating worse status (MCID not provided; Table 5). At week 66, the least‐squares mean decrease (SE) from baseline in the SF‐36 physical component score was 0.05 points (0.80) in the inotersen group and 3.65 points (1.01) in the placebo group, with a least‐squares MD of ‐3.60 points (95% CI ‐6.13 to ‐1.07; P = 0.005; Analysis 4.4) favouring inotersen.
4.4. Analysis.
Comparison 4 Inotersen versus placebo in TTR‐FAP, Outcome 4 Quality of life (change in SF‐36 physical component), at 66 weeks.
Severity of depression
Benson 2018 did not assess depression using our predetermined outcome measures.
Number of participants who died during the trial
In Benson 2018, five deaths occurred in the ITT population (172 participants) during the trial, all in the inotersen group (5/112, 4.5%), with a RR of 5.94 (95% CI 0.33 to 105.60; P = 0.22; low‐certainty evidence; Analysis 4.5). Among these, one participant died as a consequence of intracranial haemorrhage in association with very severe thrombocytopenia (platelet count < 25,000 per cubic millilitre), possibly related to the interventional drug, whereas four deaths were consistent with progression or complication of TTR‐FAP (2 deaths due to cachexia, 1 due to intestinal perforation, and 1 due to congestive heart failure).
4.5. Analysis.
Comparison 4 Inotersen versus placebo in TTR‐FAP, Outcome 5 Number of participants who died during the trial.
Adverse events
i) Number of participants experiencing at least one adverse event
In Benson 2018 (N = 172), 111/112 (99.1%) inotersen‐treated participants and 60/60 (100%) placebo‐treated participants experienced at least one adverse event (RR 0.99, 95% CI 0.96 to 1.03; P = 0.75; Analysis 4.6). In the same population, 87/112 (77.7%) tafamidis‐treated participants and 23/60 (38.3%) placebo‐treated participants reported having experienced at least one adverse event that was considered by the investigators to be related, or possibly related, to the study medication, with a RR of 2.03 (95% CI 1.45 to 2.84; P < 0.001; Analysis 4.7).
4.6. Analysis.
Comparison 4 Inotersen versus placebo in TTR‐FAP, Outcome 6 Number of participants experiencing at least one adverse event.
4.7. Analysis.
Comparison 4 Inotersen versus placebo in TTR‐FAP, Outcome 7 Number of participants experiencing at least one adverse event related to the study medication.
ii) Number of participants experiencing mild adverse events
Adams 2018 (N = 172) reported that 110/112 (98.2%) participants in the inotersen group and 60/60 (100.0%) participants in the placebo group experienced mild adverse events (RR 0.99, 95% CI 0.95 to 1.02; P = 0.44; Analysis 4.8), thus showing little or no difference between groups.
4.8. Analysis.
Comparison 4 Inotersen versus placebo in TTR‐FAP, Outcome 8 Number of participants experiencing mild adverse events.
iii) Number of dropouts due to adverse events
In Benson 2018 (N = 172), 16/112 (14.3%) inotersen‐treated participants and 1/60 (1.7%) placebo‐treated participants discontinued study medication due to treatment‐emergent adverse events, with a RR of 8.57 (95% CI 1.16 to 63.07; P = 0.035; low‐certainty evidence; Analysis 4.9), suggesting that inotersen may increase the number of dropouts due to adverse events. These were thrombocytopenia, abdominal distension, intestinal perforation, nausea, vomiting, pyrexia, hypersensitivity, cachexia, arthralgia, myalgia, chorea, dementia, embolic stroke, intracranial haemorrhage, myelopathy, myoclonus, acute kidney injury, glomerulonephritis, tubulointerstitial nephritis, pruritus, reticular erythematous mucinosis, and deep vein thrombosis in the inotersen group, and pain, increased weight, and arthralgia in the placebo arm.
4.9. Analysis.
Comparison 4 Inotersen versus placebo in TTR‐FAP, Outcome 9 Number of dropouts due to adverse events.
iv) Number of participants experiencing at least one severe adverse event
In Benson 2018 (N = 172), 36/112 (32.1%) inotersen‐treated participants and 13/60 (21.7%) placebo‐treated participants experienced at least one severe adverse event (RR 1.48, 95% CI 0.85 to 2.57; P = 0.16; low‐certainty evidence; Analysis 4.10). In the same population, 8/112 (7.1%) inotersen‐treated participants and 1/60 (1.7%) placebo‐treated participants experienced at least one severe adverse event that was considered by the investigators to be related or possibly related to study medication, with a RR of 4.29 (95% CI 0.55 to 33.46; P = 0.16; Analysis 4.11). The most frequent severe adverse events in the inotersen group (N = 112) were glomerulonephritis (3 participants, 2.7%), which caused permanent haemodialysis in one participant (0.9%), and very marked thrombocytopenia (3 participants, 2.7%), which was responsible for an intracranial haemorrhage, leading to death in one participant.
4.10. Analysis.
Comparison 4 Inotersen versus placebo in TTR‐FAP, Outcome 10 Number of participants experiencing at least one severe adverse event.
4.11. Analysis.
Comparison 4 Inotersen versus placebo in TTR‐FAP, Outcome 11 Number of participants experiencing at least one severe adverse event related to the study medication.
Discussion
Summary of main results
Four randomised controlled trials (RCTs) with a total of 655 randomised participants with transthyretin (TTR)‐related familial amyloid polyneuropathy (FAP) were eligible for inclusion in this review. The four comparisons in the included studies were: 1) tafamidis versus placebo (1 study, 128 participants), 2) diflunisal versus placebo (1 study, 130 participants), 3) patisiran versus placebo (1 study, 225 participants), and 4) inotersen versus placebo (1 study, 172 participants). We did not find any study that looked at pharmacological treatment for people with apolipoprotein AI‐FAP, gelsolin‐FAP, or beta‐2‐microglobulin‐FAP.
Two studies explored disability due to FAP progression using our prespecified measures (Berk 2013; Adams 2018). Berk 2013 suggested that diflunisal might reduce progression of disability (Kumamoto Score) compared to placebo, whereas Adams 2018 reported that patisiran probably slightly decreased disability due to FAP progression compared to placebo (Rasch‐built Overall Disability Scale).
All included RCTs investigated the change from baseline in severity of peripheral neuropathy, through different measures of nerve impairment (Coelho 2012; Berk 2013; Adams 2018; Benson 2018). The results of Coelho 2012 suggested that tafamidis might reduce progression of peripheral neuropathy compared to placebo, documented by the Neuropathy Impairment Score in the Lower Limbs (NIS‐LL). Evidence from Berk 2013 indicated that diflunisal might reduce worsening of peripheral neuropathy compared to placebo, measured on the NIS plus 7 nerve tests (NIS+7). According to Adams 2018, patisiran reduced progression of peripheral neuropathy compared to placebo, measured on the modified NIS+7 Alnylam version. Inotersen reduced worsening of peripheral neuropathy compared to placebo, measured on the mNIS+7 Ionis version (Benson 2018).
All the included studies assessed the change from baseline in quality of life on different scales (Coelho 2012; Berk 2013; Adams 2018; Benson 2018). Quality of life was an indirect measure of the drug function and could be influenced by many factors. There was little or no difference in the change of quality of life with tafamidis in comparison to placebo (according to the Norfolk QOL‐DN total score; Coelho 2012) and no clear difference between diflunisal and placebo (as measured by the 36‐Item Short‐Form Health Survey (SF‐36) physical component and mental component scores; Berk 2013). In Adams 2018 and Benson 2018, patisiran (measured on the Norfolk QOL‐DN total score) and inotersen favoured quality of life compared to placebo, respectively.
For tafamidis (Coelho 2012) and diflunisal (Berk 2013), no clear differences compared to placebo were found in the numbers of participants who died, who dropped out due to adverse events, and who experienced at least one severe adverse event.
According to Adams 2018, there may be little or no difference in mortality, dropouts due to adverse events, or number of people experiencing severe adverse events between participants who received patisiran compared to those who received placebo. In Benson 2018, data indicated that there may be a higher number of deaths and of participants with severe adverse events in the inotersen group than in the placebo group, and revealed that there may be more dropouts due to adverse events with inotersen compared to placebo. In particular, three cases of glomerulonephritis and three cases of thrombocytopenic intracranial haemorrhage were reported.
Overall completeness and applicability of evidence
This review primarily assessed whether any disease‐modifying pharmacological treatment reduced disability due to FAP progression in people with TTR‐FAP. It also evaluated other parameters, such as severity of peripheral neuropathy, modified body mass index (mBMI), quality of life, severity of depression, and safety (number of deaths during the trial, number of participants with at least one adverse event, number of participants with mild adverse events, dropouts due to adverse events, and number of participants with at least one severe adverse event).
Coelho 2012 examined tafamidis versus placebo only for the treatment of early‐stage TTR‐FAP due to Val30Met mutation in the TTR gene. Berk 2013, Adams 2018, and Benson 2018 investigated, respectively, diflunisal, patisiran, and inotersen versus placebo in people with TTR‐FAP at any stage due to either Val30Met or non‐Val30Met mutation in the TTR gene.
Only two of the included studies provided data for the primary outcome of this review using our predetermined outcome measures (Berk 2013; Adams 2018). All included studies evaluated the severity of peripheral neuropathy and the change from baseline in the quality of life using different outcome measures or slightly different score range for the same outcome measure (Coelho 2012; Berk 2013; Adams 2018; Benson 2018). All included studies explored the change from baseline in the mBMI as a measure of the nutritional status/global wasting (Coelho 2012; Berk 2013; Adams 2018; Benson 2018). None of the included studies reported a specific evaluation of depression. All included studies provide evidence for safety and tolerability (mortality and adverse events) (Coelho 2012; Berk 2013; Adams 2018; Benson 2018).
We reported results for people with TTR‐FAP only, as we found no evidence on pharmacological treatment of ApoAI‐FAP, Gel‐FAP, and B2M‐FAP in the current literature.
The number of studies and available data are currently too limited for formal indirect comparisons to be made between drugs, which would also have been hampered by the use of different outcome measures in the included studies.
Certainty of the evidence
Data for disability due to FAP progression were available only for diflunisal and patisiran (Berk 2013; Adams 2018). Evidence for the effects of diflusinal and patisiran versus placebo on disability due to FAP progression was of low and moderate certainty, respectively (Berk 2013; Adams 2018). We downgraded the certainty of evidence for both comparisons by one level due to serious imprecision as data were from single studies with small sample sizes (Berk 2013; Adams 2018). In addition, we downgraded the certainty of evidence for the comparison between diflunisal and placebo by one level due to serious risk of bias since data were from a single study which we judged to be at high risk of attrition bias due to its dropout rate (Berk 2013).
All included RCTs investigated the effects of a drug (tamafidis, diflunisal, patisiran or inotersen) on the change in the severity of peripheral neuropathy compared to placebo (Coelho 2012; Berk 2013; Adams 2018; Benson 2018). We downgraded the certainty of evidence for this outcome to low for tafamidis and diflunisal (serious risk of bias in the single RCTs included in these comparisons and serious imprecision; Coelho 2012; Berk 2013). We downgraded the certainty of evidence for this outcome to moderate for patisiran and inotersen as data were from single studies with small sample sizes (Adams 2018; Benson 2018).
Data on quality of life were available for all four drugs in comparison with placebo (Coelho 2012; Berk 2013; Adams 2018; Benson 2018). The certainty of evidence for this outcome was very low for tafamidis and diflunisal as we downgraded the certainty of evidence by one level for risk of bias, by one level for serious imprecision, and by one level for serious indirectness (Coelho 2012; Berk 2013). We downgraded the certainty of evidence twice for patisiran and inotersen: once for serious imprecision due to small sample size and once for serious indirectness, because the interventions would affect quality of life only by indirect mechanisms (Adams 2018; Benson 2018).
Data for mortality, dropouts, and adverse events were available for all four treatments (Coelho 2012; Berk 2013; Adams 2018; Benson 2018). We downgraded the certainty of evidence for tafamidis and diflunisal to very low for mortality, dropouts due to adverse events and severe adverse events due to serious risk of bias and very serious imprecision (Coelho 2012; Berk 2013). For patisiran and inotersen, we downgraded the certainty of evidence for these outcomes by two levels for very serious imprecision due to small sample sizes, small event numbers, and wide CIs, which in some cases include the possibility of both harms and benefits, which made findings of low certainty (Adams 2018; Benson 2018).
Potential biases in the review process
We are confident that we have identified all relevant studies in this review as this is a small, albeit growing field and we supplemented our search strategy with checking references, searching online clinical trial registries, and contacting experts in the field. We followed standard methodological procedures to reduce bias in the review process and we adhered to the inclusion and exclusion criteria prespecified in the protocol in order to limit subjectivity (New Reference). In addition, the fact that there was almost complete consensus between review authors responsible for study selection suggests that the risk of selection bias in this part of the review process was probably low. We made efforts to trace study authors and additional data from manufacturers in order to clarify minor discrepancies in data obtained from published report papers and online clinical trial registries, but we were unable to do so. If we can source supplementary data, we will consider them in future updates. It should be noted that reviews that include a small number of trials, such as ours, have limitations in relation to events that occur infrequently, such as adverse events. We also consider a limitation of this review that we did not plan a network meta‐analysis to compare different pharmacological agents; however this would not have been possible with currently available trials.
Agreements and disagreements with other studies or reviews
To our knowledge, this is the first systematic review on pharmacological treatments for FAPs.
Open‐label extensions of previously completed RCTs and minor evidence for pharmacological agents not investigated in RCTs
We briefly summarise below evidence from open‐label extensions of the RCTs included in this review and minor evidence regarding pharmacological agents not previously investigated in RCTs.
Tafamidis
In a 12‐month open‐label extension study of tafamidis (NCT00791492; Coelho 2013), 86 people with TTR‐FAP who completed the pivotal study of tafamidis were enrolled (Coelho 2012), of whom 85 either continued on tafamidis 20 mg once daily (44 participants) or were switched from placebo to tafamidis 20 mg once daily (41 participants). This study did not assess disability due to FAP progression using our predetermined outcome measures. At 12 months, the mean (standard deviation (SD)) increase from baseline in the NIS‐LL (range: 0 to 88, with higher scores indicating greater deficits; minimum clinically important difference (MCID): 2 points; Table 5) was 1.36 ± 4.77 points in participants who continued on tafamidis and 1.6 ± 8.2 points in participants switched from placebo to tafamidis. At the same time point, the mean (SD) change from baseline in the mBMI was a decrease of 0.12 ± 1.16 kg/m2 g/L in the tafamidis‐tafamidis group and an increase of 0.73 ± 1.45 kg/m2 g/L in the placebo‐tafamidis group. After 12 months, the mean (SD) change from baseline in the Norfolk‐QOL total score (range: ‐2 to 138, with lower scores indicating better quality of life; MCID not provided; Table 5) was an increase of 2.25 ± 8.91 points in participants who continued on tafamidis and a decrease of 2.33 ± 15.66 points in participants switched from placebo to tafamidis. No participants died or discontinued treatment due to an adverse event during this study. In Coelho 2013, 5/44 (11.4%) participants in the tafamidis‐tafamidis group and 4/41 (9.8%) participants in the placebo‐tafamidis group experienced one severe adverse event, whereas 24/44 (54.5%) participants who continued on tafamidis and 24/41 (58.5%) participants who were switched from placebo to tafamidis had at least one mild adverse event.
An open‐label, multicentre, international study investigated tafamidis 20 mg once daily for 12 months in people with non‐Val30Met TTR‐FAP (NCT00630864; Merlini 2013). This study did not assess disability due to FAP progression using our predetermined outcome measures. At 12 months, the mean (SD) increase from baseline in the NIS‐LL (range: 0 to 88, with higher scores indicating greater deficits; MCID: 2 points; Table 5) was 2.7 ± 6.21 points. At the same time point, the mean (SD) increase from baseline in the mBMI was 16.6 ± 89.33 kg/m2 g/L. After 12 months, the mean (SD) change from baseline in the Norfolk‐QOL total score (range: ‐2 to 138, with lower scores indicating better quality of life; MCID not provided; Table 5) was an increase of 0.1 ± 18.01 points. No participant died during this study. In Merlini 2013, 17/21 (80.9%) participants experienced at least one adverse event, 13/21 (61.9%) participants had a mild adverse event, 1/21 (4.8%) discontinued due to an adverse event, and 8/21 (38.1%) participants were reported to have severe adverse events.
An ongoing, not recruiting, open‐label extension study of tafamidis was designed to obtain additional, long‐term efficacy and safety data on tafamidis 20 mg once daily over a 10‐year period and was initiated in 2009 (NCT00925002; Waddington‐Cruz 2016; Barroso 2017). It enrolled 93 people with TTR‐FAP who had completed the 12‐month open‐label extension study of tafamidis (NCT00791492; Coelho 2013) or the open‐label study to evaluate TTR stabilisation, safety, tolerability, and efficacy of tafamidis in participants with non‐Val30Met TTR‐FAP (NCT00630864; Merlini 2013).
A prospective non‐RCT of tafamidis was conducted by the French Network for FAP and enrolled 37 participants with Val30Met TTR‐FAP both in early and advanced stages between December 2009 and July 2011. In the 12‐month follow‐up period, 24% of participants deteriorated with respect to disability, measured on the Polyneuropathy Disability Score (Table 5), and 31% of participants deteriorated with respect to the severity of neuropathy, measured on the NIS‐LL (Table 5). Adverse events caused 7/37 (18.9%) participants to withdraw from the study. A total of 19 adverse events were reported, including febrile urinary tract infections and cases of severe diarrhoea (Lozeron 2013).
A phase 3, open‐label study evaluated the safety, tolerability, and efficacy of tafamidis 20 mg once daily for up to 78 weeks in 10 Japanese people with Val30Met or non‐Val30Met TTR‐FAP (NCT01435655). This study did not assess disability due to FAP progression according to our predetermined outcome measures. At week 78, the mean (SD) increase from baseline in the NIS‐LL (which was herein reported with a different range: 0 to 44, with higher scores indicating greater deficits) was 3.3 ± 4.74 points, whereas the mean (SD) increase from baseline in the mBMI was 53.65 ± 81.39 kg/m2 g/L. After 78 weeks, the mean (SD) increase from baseline in the Norfolk‐QOL total score (range: ‐4 to 136, with lower scores indicating better quality of life; MCID not provided; Table 5) was 10.8 ± 13.69. During this study, 2/10 (20%) participants died, 10/10 (100%) experienced adverse events, and 7/10 (70%) had severe adverse events.
Patisiran
A phase 2, open‐label extension study evaluated long‐term safety, tolerability, and efficacy of patisiran in people with TTR‐FAP (NCT01961921; Adams 2015), who completed the pivotal RCT of patisiran (Adams 2018). This study was initiated in 2013 and enrolled 27 participants, who received patisiran 0.3 mg/kg intravenously every three weeks for up to two years. At month 24, the median decrease from baseline in the Rasch‐built Overall Disability Scale (which was herein reported with a different range: 0 to 100, with lower scores indicating greater disability) was 1.0 point (full range ‐14.0 to 8.0). At the same time point, the median decrease from baseline in the mNIS+7Alnylam (range: 0 to 304, with higher scores indicating greater deficits; MCID not provided; Table 5) was 7.0 points (full range ‐34.63 to 15.38), and the median decrease from baseline in the mBMI was 39.85 kg/m2 g/L (full range ‐368.8 to 258.9). During this study, 2/27 (7.4%) participants died, 26/27 (96.3%) experienced at least one adverse event, and 7/27 (25.9%) had a severe adverse event.
Inotersen
An ongoing, phase 3, open‐label extension study was designed to evaluate long‐term efficacy and safety of inotersen in people with TTR‐FAP who were enrolled and completed the pivotal RCT of inotersen (Benson 2018), and started in 2014 (NCT02175004; Plante‐Bordeneuve 2018). Participants received inotersen 300 mg weekly subcutaneously up to five years. Definitive results have not been released to date.
Other evidence
Pre‐clinical studies demonstrated that both the tetracycline antibiotic doxycycline and a combination of doxycycline and the antiapoptotic agent tauroursodeoxycholic acid (TUDCA) inhibited amyloid formation and promoted the disruption of amyloid deposits in murine FAP models (Cardoso 2006; Cardoso 2010). Based on these data, in 2012 the European Medicinal Agency granted doxycycline orphan drug designation for TTR‐FAP. A phase 2 open‐label study was designed to evaluate the efficacy, tolerability, safety, and pharmacokinetics of orally administered doxycycline (100 mg, twice daily) and TUDCA (250 mg, 3 times daily) for one year (Obici 2012). Results provided evidence that the investigational molecules stabilised the disease for at least one year in the majority of participants, with a favourable toxicity profile (Obici 2012).
SOM0226/CRX1008 (tolcapone) is a repositioned molecule which is being evaluated for its recently recognised activity in TTR stabilisation and fibril disruption (Sant'Anna 2016). A proof‐of‐concept open‐label, phase 2a clinical trial was conducted in six healthy volunteers and 15 people with TTR‐FAP and asymptomatic carriers (Reig 2015). This study demonstrated that a single oral dose of 200 mg or three doses of 100 mg SOM0226 stabilised plasmatic TTR in all participants studied, and did not report any adverse events related to the investigational compound, thus supporting further clinical development of SOM0226 for the treatment of TTR‐FAP.
Epigallocatechin‐3‐gallate (EGCG), the most abundant polyphenol in green tea, has been observed to reduce extracellular TTR deposition in the peripheral nervous system of a FAP transgenic mouse model (Ferreira 2012). Subsequently, a single‐centre observational study reported no progression in left ventricular wall thickness and mass on echocardiography in a small cohort of people with hereditary and non‐hereditary forms of cardiac TTR amyloidosis, including eight people with hereditary TTR cardiomyopathy, after daily consumption of EGCG in the form of green tea or green tea extract for 12 months (Kristen 2012). However, no studies have investigated the potential effect of EGCG on FAP progression.
Authors' conclusions
Implications for practice.
To date, evidence on the pharmacological treatment of familial amyloid polyneuropathies (FAPs) from randomised clinical trials (RCTs) is limited to transthyretin (TTR)‐FAP, and no RCT directly compares disease‐modifying pharmacological treatments for TTR‐FAP. The four RCTs included in this review investigate disease‐modifying drugs for TTR‐FAP versus placebo and are individually either too small, short, or both, for the review authors to draw confident, high‐certainty conclusions about the efficacy, acceptability, and tolerability of the pharmacological agents under investigation, especially in the long term. The review authors underline that the severity of the disease, challenges of new drug development and RCT management, and lack of alternative treatments, necessitate flexibility in clinical practice. Results indicate that patisiran and inotersen are probably beneficial in TTR‐FAP, and tafamidis and diflunisal may be beneficial in TTR‐FAP. The lower certainty of evidence for tafamidis and diflunisal is mainly due to the high dropout rate in their pivotal RCTs. As for safety data, the occurrence of severe adverse events, including glomerulonephritis and thrombocytopenic intracranial haemorrhage with inotersen highlights the need for a close monitoring of renal profile and platelet count in people on this medication. A full assessment of people with TTR‐FAP, including cardiological and ophthalmological assessment is recommended. As direct comparative studies for TTR‐FAP will be hampered by sample size and costs required to demonstrate superiority of one drug over another, long‐term non‐randomised open‐label studies to monitor the efficacy and safety of these drugs are needed.
Implications for research.
Various approaches have been taken in the research and development of disease‐modifying treatments for TTR‐FAP over the past decades, and a number of pharmacological agents are currently available. Drug development for TTR‐FAP has encountered a number of challenges typically associated with rare genetic conditions, as well as others specific for this complex multisystem disease.
First, the number of people available to participate in clinical trials is limited. The pivotal trials enrolled a small number of participants and often showed a high dropout rate. For example, the trial investigating tafamidis presented a higher than anticipated liver transplantation dropout rate, which reduces certainty in the findings (Coelho 2012).
Second, the small participant populations may be highly heterogeneous, both genotypically and phenotypically. Nuances in clinical manifestations and progression of TTR‐FAP are not completely understood due to the lack of natural history studies, especially in nonendemic areas and for uncommon mutations, such as many non‐Val30Met variants. Moreover, genetic, epigenetic, and environmental factors that may influence phenotypic expression should be clarified. These aspects are critical in determining delayed diagnosis and participant enrolment in clinical trials. Therefore, efforts should be undertaken in order to create observational registries worldwide to improve understanding of the disease. In addition, results from RCTs should be critically analysed within homogeneous participant subgroups, in terms of profile of organ involvement and genotype.
Third, the detection of appropriate clinically meaningful outcomes and relevant duration of follow‐up to measure efficacy and safety are mandatory, and require understanding of their rate of occurrence and variability, both of which contribute to difficulties in powering a study. The follow‐up periods of available RCTs were too short for evaluation of long‐term safety and efficacy in relation to the natural progression of the disease. On the other hand, the combination of double‐blind trials and open‐label extension studies resembles the design of a delayed‐start trial, and is aimed at distinguishing between long‐term effects on disease progression and effects on symptoms. The design of these studies (in which only the initial phase is randomised, double‐blind, and placebo‐controlled) precludes direct assessment of the extent to which investigational drugs preserve neurologic function and quality of life in the long term, compared with placebo. However, it may represent a good compromise between the need to promptly provide a possibly beneficial treatment to people with a severe progressive disorder and the challenges of drug development. A consensus on the best surrogate endpoints or biomarkers to consider in clinical trials may help future direct or indirect comparisons between different drugs.
Fourth, as TTR‐related amyloidosis is a multisystem disorder, evaluation of treatment efficacy in future clinical trials for FAP should be extended to other involved organs, including the heart and eyes. Available pharmacological treatments for TTR‐FAP are not curative at the moment; at most, they may change severe phenotypes into milder ones. As a chronic and progressive condition, TTR‐FAP requires ongoing treatment and management of complications of long‐standing disease. In particular, monitoring long‐term complications, such as cardiomyopathy, ophthalmic manifestations and leptomeningeal involvement is necessary. The optimal duration of treatment is uncertain and the lack of long‐term data from RCTs hampers considerations of quality of life.
Finally, as for other orphan drugs, development of pharmacological treatments for TTR‐FAP may be influenced by potential conflict of interest of patient advocacy organisations, industry sponsorship, and media coverage, which may overemphasise benefits and harms.
Future trials should use sensitive and validated disability and clinical scores that are likely to extract meaningful effects and allow direct or indirect comparisons of results. Cost‐effectiveness measurements should also be considered in future trials, as the treatments that have been used and those that are likely to be used in the future are expensive. Trial endpoints should also be planned according to the chronicity of the disorder and meaningful in patient terms, particularly overall disability.
Acknowledgements
The Methods section includes sections of standard text provided by Cochrane Neuromuscular. The search strategy was developed by the Information Specialist of Cochrane Neuromuscular (Angela Gunn), in collaboration with the review authors. The review authors would like to thank Marianna Purgato for her helpful comments on the protocol for this review. We are grateful to Violaine Planté‐Bordeneuve, Isabel Conceicao, and Marcia Waddingon‐Cruz, who provided information via email correspondence during the search stage for this review. The authors would also like to acknowledge and thank the Cochrane Neuromuscular editorial base for their input, with a special mention for the Managing Editor (Ruth Brassington) for her invaluable support. Finally, the authors are most grateful to the peer reviewers Sarah Nevitt, University of Liverpool, UK; Colin Chalk, McGill University, Montreal, Canada; Alexander Rossor, UCL Institute of Neurology and the National Hospital for Neurology and Neurosurgery, London, UK; and Carlos Carlos Heras‐Palou, and UK ATTR Amyloidosis Patients’ Association, London.
This project was supported by the National Institute for Health Research (NIHR) via Cochrane Infrastructure funding to Cochrane Neuromuscular. The views and opinions expressed herein are those of the review authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, National Health Service, or the Department of Health. Cochrane Neuromuscular is also supported by the Queen Square Centre for Neuromuscular Diseases.
Appendices
Appendix 1. Cochrane Neuromuscular Specialised Register (CRS Web) search strategy
Search run on 18 November 2019
#1 MESH DESCRIPTOR Amyloid Neuropathies, Familial WITH QUALIFIER DT AND INREGISTER #2 MESH DESCRIPTOR Amyloid Neuropathies WITH QUALIFIER DT AND INREGISTER #3 MESH DESCRIPTOR Amyloidosis WITH QUALIFIER DT AND INREGISTER #4 MESH DESCRIPTOR peripheral nervous system diseases WITH QUALIFIER DT AND INREGISTER #5 #3 AND #4 AND INREGISTER #6 #1 OR #2 OR #5 AND INREGISTER #7 ("familial amyloid*" NEAR2 (neuropath* or polyneuropath*)):ti,ab AND INREGISTER #8 ("hereditary amyloid*" NEAR2 (neuropath* or polyneuropath*)):ti,ab AND INREGISTER #9 amyloidosis NEAR2 (neuropath* or polyneuropath*) AND INREGISTER #10 (transthyretin NEAR3 amyloidosis):ti,ab AND INREGISTER #11 (transthyretin TTR):ti,ab AND INREGISTER #12 "ttr fap":ti,ab AND INREGISTER #13 ("fap type 1" or "fap type I" or "fap type 2" or "fap type II" or "fap type 3" or "fap type III" or "fap type 4" or "fap type IV"):TI,AB AND INREGISTER #14 (Apolipoprotein NEAR5 FAP):TI,AB AND INREGISTER #15 (Apolipoprotein NEAR familial):TI,AB AND INREGISTER #16 (Apolipoprotein NEAR hereditary):TI,AB AND INREGISTER #17 (gelsolin NEAR hereditary):TI,AB AND INREGISTER #18 "hereditary amyloidosis":ti,ab AND INREGISTER #19 (("Van Allen type" or "Iowa type" or "Finnish type" or "Meretoja type") and amyloid*):ti,ab AND INREGISTER #20 "hereditary transthyretin‐mediated (hATTR) amyloidosis" AND INREGISTER #21 hereditary NEAR3 amyloidosis AND INREGISTER #22 #7 or #8 or #9 or #10 or #11 or #12 or #13 or #14 or #15 or #16 or #17 or #18 or #19 or #20 or #21 AND INREGISTER #23 MESH DESCRIPTOR Oligonucleotides, Antisense EXPLODE ALL AND INREGISTER #24 MESH DESCRIPTOR RNA antisense EXPLODE ALL AND INREGISTER #25 ((tafamidis or meglumine or FX1006A or diflunisal or dolobid or dolobis or dolocid or MK647)):AB,EH,EMT,KW,KY,MH,TI AND INREGISTER #26 ((doxycycline or "tauroursodeoxycholic acid" or TUDCA or "RNA‐targeted therap*" or "RNA interfer*" or "antisense oligonucleotide*" or "ISIS TTR*" or "ISIS 420915" or "ALN‐TTR0*")):AB,EH,EMT,KW,KY,MH,TI AND INREGISTER #27 drug:ti,ab AND INREGISTER #28 prealbumin AND INREGISTER #29 #23 OR #24 OR #25 OR #26 OR #27 OR #28 AND INREGISTER #30 #22 AND #29 AND INREGISTER #31 #6 OR #30 AND INREGISTER
Appendix 2. Cochrane Central Register of Controlled Trials (CENTRAL) (CRS Web) search strategy
Search run on 18 November 2019
#1 MESH DESCRIPTOR Amyloid Neuropathies, Familial WITH QUALIFIER DT AND CENTRAL:TARGET #2 MESH DESCRIPTOR Amyloid Neuropathies WITH QUALIFIER DT AND CENTRAL:TARGET #3 MESH DESCRIPTOR Amyloidosis WITH QUALIFIER DT AND CENTRAL:TARGET #4 MESH DESCRIPTOR peripheral nervous system diseases WITH QUALIFIER DT AND CENTRAL:TARGET #5 #3 AND #4 AND CENTRAL:TARGET #6 #1 OR #2 OR #5 AND CENTRAL:TARGET #7 ("familial amyloid*" NEAR2 (neuropath* or polyneuropath*)):ti,ab AND CENTRAL:TARGET #8 ("hereditary amyloid*" NEAR2 (neuropath* or polyneuropath*)):ti,ab AND CENTRAL:TARGET #9 amyloidosis NEAR2 (neuropath* or polyneuropath*) AND CENTRAL:TARGET #10 (transthyretin NEAR3 amyloidosis):ti,ab AND CENTRAL:TARGET #11 (transthyretin TTR):ti,ab AND CENTRAL:TARGET #12 "ttr fap":ti,ab AND CENTRAL:TARGET #13 ("fap type 1" or "fap type I" or "fap type 2" or "fap type II" or "fap type 3" or "fap type III" or "fap type 4" or "fap type IV"):TI,AB AND CENTRAL:TARGET #14 (Apolipoprotein NEAR5 FAP):TI,AB AND CENTRAL:TARGET #15 (Apolipoprotein NEAR familial):TI,AB AND CENTRAL:TARGET #16 (Apolipoprotein NEAR hereditary):TI,AB AND CENTRAL:TARGET #17 (gelsolin NEAR hereditary):TI,AB AND CENTRAL:TARGET #18 "hereditary amyloidosis":ti,ab AND CENTRAL:TARGET #19 (("Van Allen type" or "Iowa type" or "Finnish type" or "Meretoja type") and amyloid*):ti,ab AND CENTRAL:TARGET #20 "hereditary transthyretin‐mediated (hATTR) amyloidosis" AND CENTRAL:TARGET #21 hereditary NEAR3 amyloidosis AND CENTRAL:TARGET #22 #7 or #8 or #9 or #10 or #11 or #12 or #13 or #14 or #15 or #16 or #17 or #18 or #19 or #20 or #21 AND CENTRAL:TARGET #23 MESH DESCRIPTOR Oligonucleotides, Antisense EXPLODE ALL AND CENTRAL:TARGET #24 MESH DESCRIPTOR RNA antisense EXPLODE ALL AND CENTRAL:TARGET #25 ((tafamidis or meglumine or FX1006A or diflunisal or dolobid or dolobis or dolocid or MK647)):AB,EH,EMT,KW,KY,MH,TI AND CENTRAL:TARGET #26 ((doxycycline or "tauroursodeoxycholic acid" or TUDCA or "RNA‐targeted therap*" or "RNA interfer*" or "antisense oligonucleotide*" or "ISIS TTR*" or "ISIS 420915" or "ALN‐TTR0*")):AB,EH,EMT,KW,KY,MH,TI AND CENTRAL:TARGET #27 drug:ti,ab AND CENTRAL:TARGET #28 prealbumin AND CENTRAL:TARGET #29 #23 OR #24 OR #25 OR #26 OR #27 OR #28 AND CENTRAL:TARGET #30 #22 AND #29 AND CENTRAL:TARGET #31 #6 OR #30 AND CENTRAL:TARGET
Appendix 3. MEDLINE (OvidSP) search strategy
Database: Ovid MEDLINE(R) and Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations, Daily and Versions(R) <1946 to November 15, 2019> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 randomized controlled trial.pt. (494660) 2 controlled clinical trial.pt. (93427) 3 randomi#ed.ti,ab. (594923) 4 placebo.ab. (203151) 5 drug therapy.fs. (2158069) 6 randomly.ab. (322080) 7 trial.ab. (484880) 8 groups.ab. (1978072) 9 or/1‐8 (4598231) 10 exp animals/ not humans.sh. (4643868) 11 9 not 10 (3985510) 12 Amyloid Neuropathies, Familial/dt (116) 13 Amyloid Neuropathies/dt [Drug Therapy] (30) 14 amyloidosis/dt (1307) 15 peripheral nervous system diseases/ (22438) 16 14 and 15 (13) 17 14 and polyneuropathies/dt (2) 18 or/12‐13,16‐17 (159) 19 (familial amyloid adj2 (neuropath* or polyneuropath*)).mp. (929) 20 (transthyretin adj3 amyloidosis).mp. (1133) 21 transthyretin TTR fap.mp. (3) 22 (fap type 1 or fap type I or fap type 2 or fap type II or fap type 3 or fap type III or fap type 4 or fap type IV).mp. (58) 23 (Apolipoprotein adj5 FAP).mp. (3) 24 (apolipoprotein adj5 hereditary amyloidosis).mp. (6) 25 (gelsolin adj5 hereditary amyloidosis).mp. (6) 26 related hereditary amyloidosis.mp. (18) 27 ((Van Allen type or Iowa type or Finnish type or Meretoja type) and amyloid).mp. (85) 28 or/19‐27 (1985) 29 exp Oligonucleotides, Antisense/ (15570) 30 exp RNA, Antisense/ (135729) 31 (tafamidis or meglumine or FX1006A or diflunisal or dolobid or dolobis or dolocid or MK647 or prealbumin).mp. (15939) 32 (doxycycline or tauroursodeoxycholic acid or TUDCA or RNA‐targeted therap* or RNA interfer* or antisense oligonucleotide* or ISIS TTR* or ISIS 420915 or ALN‐TTR0*).mp. (93041) 33 drug*.tw. (1539196) 34 or/29‐33 (1746627) 35 18 or (28 and 34) (1099) 36 11 and 35 (248) 37 remove duplicates from 36 (244)
Appendix 4. Embase (OvidSP) search strategy
Database: Embase <1974 to 2019 November 15> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 crossover‐procedure.sh. (61385) 2 double‐blind procedure.sh. (167720) 3 single‐blind procedure.sh. (37256) 4 randomized controlled trial.sh. (580660) 5 (random* or crossover* or cross over* or placebo* or (doubl* adj blind*) or allocat*).tw,ot. (1712290) 6 trial.ti. (286320) 7 controlled clinical trial/ (462825) 8 or/1‐7 (2032915) 9 exp animal/ or exp invertebrate/ or animal.hw. or non human/ or nonhuman/ (26631387) 10 human/ or human cell/ or human tissue/ or normal human/ (20361884) 11 9 not 10 (6335996) 12 8 not 11 (1807373) 13 limit 12 to (conference abstracts or embase) (1525052) 14 familial amyloid polyneuropathy/ (1914) 15 amyloid neuropathy/ (1343) 16 amyloidosis/ (26422) 17 peripheral neuropathy/ or neuropathy/ (85297) 18 16 and 17 (982) 19 (familial amyloid adj2 (neuropath$ or polyneuropath*)).mp. (2415) 20 (transthyretin adj3 amyloidosis).mp. (1823) 21 transthyretin TTR fap.mp. (8) 22 transthyretin familial polyneuropathy.mp. (1) 23 Hereditary ATTR hATTR amyloidosis.mp. (13) 24 "ATTR amyloidosis"/ (839) 25 (Hereditary adj3 amyloidosis).mp. (1212) 26 (fap type 1 or fap type I or fap type 2 or fap type II or fap type 3 or fap type III or fap type 4 or fap type IV).mp. (63) 27 (apolipoprotein adj5 hereditary amyloidosis).mp. (6) 28 (Apolipoprotein adj5 FAP).mp. (3) 29 (apolipoprotein adj5 hereditary amyloidosis).mp. (6) 30 (gelsolin adj5 hereditary amyloidosis).mp. (13) 31 related hereditary amyloidosis.mp. (38) 32 ((Van Allen type or Iowa type or Finnish type or Meretoja type) and amyloid).mp. (96) 33 or/14‐15,18‐32 (6405) 34 antisense oligodeoxynucleotide/ (5384) 35 complementary RNA/ (8547) 36 (tafamidis or meglumine or FX1006A or diflunisal or dolobid or dolobis or dolocid or MK647 or prealbumin).mp. (36444) 37 (doxycycline or tauroursodeoxycholic acid or TUDCA or RNA‐targeted therap* or RNA interfer* or antisense oligonucleotide* or ISIS TTR* or ISIS 420915 or ALN‐TTR0*).mp. (142561) 38 drug*.tw. (2128226) 39 or/34‐38 (2293581) 40 13 and 33 and 39 (226) 41 remove duplicates from 40 (220)
Appendix 5. Online registers for clinical trials strategy
Search run on 18 November 2019.
Online registers: World Health Organization International Clinical Trials Registry Platform (ICTRP)(www.who.int/ictrp), ClinicalTrials.gov (www.clinicaltrials.gov), and ClinicalTrialsRegister.eu (www.clinicaltrialsregister.eu) Search Strategy: simple search of different combination of the following terms: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
1 "familial amyloid polyneuropathy" 2 "amyloid neuropathy" 3 "amyloidosis" 4 "transthyretin" 5 "apolipoprotein" 6 "gelsolin" 7 "beta‐2‐microglobulin"
Appendix 6. Methods specified in the protocol
Our protocol described the following methods for data presentation and analysis.
Measures of treatment effect
Dichotomous data
We will report dichotomous data as risk ratios (RRs), or risk differences (RDs), with 95% confidence intervals (CIs).
Continuous data
For continuous data, if meta‐analysis is possible in future, we will calculate mean differences (MDs) or, for outcomes that are conceptually the same but measured in different ways, standardised mean differences (SMDs) and 95% CIs. If some scales increase with disease severity whilst others decrease, we will multiply the mean values from one set of studies by –1 to ensure that all the scales point in the same direction (Higgins 2011).
Unit of analysis issues
For trials with a cross‐over design, only results from the first randomisation period will be considered. Where a trial involves more than two treatment arms, especially two appropriate dose groups of the same drug, the different dose arms will be pooled and considered to be one. In case we identify cluster placebo‐controlled randomised trials, we plan to use the intracluster correlation coefficient (ICC), where provided, to adjust for cluster effects.
Assessment of heterogeneity
In accordance with theCochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), we will quantify heterogeneity using the I² statistic. Higgins 2011 recommends overlapping intervals for I² interpretation as follows:
0% to 40%: might not be important;
30% to 60%: may represent moderate heterogeneity;
50% to 90%: may represent substantial heterogeneity; and
75% to 100%: considerable heterogeneity.
Heterogeneity classification based on I² will be looked at in conjunction with a visual inspection of the forest plots. We will also use the Chi² test and its P value to determine the direction and magnitude of the treatment effects.
Assessment of reporting biases
We will create and examine funnel plots to explore possible small study biases only if there are more than 10 trials in a single analysis.
Data synthesis
We will use a random‐effects model to calculate treatment effects. We choose the random‐effects model as it takes into account differences between studies even when there is no evidence of statistical heterogeneity and gives a more conservative estimate than the fixed‐effect model. We note that the random‐effects model gives added weight to small studies, which can either increase or decrease the effect size. We will apply a fixed‐effect model, on primary outcomes only, to see whether it markedly changes the effect size.
Subgroup analysis and investigation of heterogeneity
If a study of doubtful eligibility for the systematic review, appears to be an outlier, or has missing data that are impossible to retrieve, we will perform analyses with and without inclusion of the trial and will compare these results with each other. We plan to consider the following diagnostic subgroups separately:
participants with autonomic involvement and participants without autonomic involvement;
participants with neuropathic pain and participants without neuropathic pain;
participants with cardiac involvement and participants without cardiac involvement; and
participants with renal involvement and participants without renal involvement.
If groups within any of the subgroups are found to be significantly different from one another, we will run metaregression for exploratory analyses of additive or multiplicative influences of the variables in question.
We will use the following outcomes in subgroup analyses:
change in disability;
death;
change in quality of life.
We will use the formal test for subgroup interactions in Review Manager 5 (Review Manager 2014).
Sensitivity analysis
We plan to conduct the following sensitivity analyses. We will examine whether the results change and check for the robustness of the observed findings by:
excluding trials at high risk of bias (i.e. trials with inadequate allocation concealment and blinding, with incomplete data reporting and/or with high probability of selective outcome reporting);
excluding trials with dropout rates greater than 20%;
excluding studies funded by the pharmaceutical company marketing each available pharmacological agent;
excluding unpublished studies (if there are any).
Data and analyses
Comparison 1. Tamafidis versus placebo in TTR‐FAP.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Severity of peripheral neuropathy (expressed as the change from baseline in NIS‐LL) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1 12 months | 1 | 125 | Mean Difference (IV, Fixed, 95% CI) | ‐3.83 [‐5.99, ‐1.67] |
| 1.2 18 months | 1 | 125 | Mean Difference (IV, Fixed, 95% CI) | ‐3.21 [‐5.63, ‐0.79] |
| 2 Change in mBMI | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 12 months | 1 | 125 | Mean Difference (IV, Fixed, 95% CI) | 50.2 [24.46, 75.94] |
| 2.2 18 months | 1 | 125 | Mean Difference (IV, Fixed, 95% CI) | 70.6 [41.96, 99.24] |
| 3 Quality of life (expressed as the change from baseline in Norfolk QOL‐DN total score) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 12 months | 1 | 125 | Mean Difference (IV, Fixed, 95% CI) | ‐3.50 [‐9.48, 2.48] |
| 3.2 18 months | 1 | 125 | Mean Difference (IV, Fixed, 95% CI) | ‐4.5 [‐11.27, 2.27] |
| 4 Number of participants who died during the trial (published paper ‐ see text) | 1 | 128 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.11, 3.74] |
| 5 Number of participants experiencing at least one adverse event | 1 | 128 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.88, 1.04] |
| 6 Number of participants experiencing mild adverse events | 1 | 128 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.87, 1.12] |
| 7 Number of dropouts due to adverse events | 1 | 128 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.29 [0.30, 5.54] |
| 8 Number of participants experiencing at least one severe adverse event | 1 | 128 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.37, 3.62] |
Comparison 2. Diflunisal versus placebo in TTR‐FAP.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Disability due to FAP progression (expressed as the change from baseline in KS) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1 12 months | 1 | 130 | Mean Difference (IV, Fixed, 95% CI) | ‐2.20 [‐4.91, 0.51] |
| 1.2 24 months | 1 | 130 | Mean Difference (IV, Fixed, 95% CI) | ‐4.9 [‐7.89, ‐1.91] |
| 2 Severity of peripheral neuropathy (expressed as the change from baseline in the NIS+7) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 12 months | 1 | 130 | Mean Difference (IV, Fixed, 95% CI) | ‐6.3 [‐11.38, ‐1.22] |
| 2.2 24 months | 1 | 130 | Mean Difference (IV, Fixed, 95% CI) | ‐18.1 [‐26.03, ‐10.17] |
| 3 Severity of peripheral neuropathy (expressed as the change from baseline in the NIS) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 12 months | 1 | 130 | Mean Difference (IV, Fixed, 95% CI) | ‐6.0 [‐10.17, ‐1.83] |
| 3.2 24 months | 1 | 130 | Mean Difference (IV, Fixed, 95% CI) | ‐16.80 [‐23.82, ‐9.78] |
| 4 Severity of peripheral neuropathy (expressed as the change from baseline in the NIS‐LL) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 12 months | 1 | 130 | Mean Difference (IV, Fixed, 95% CI) | ‐2.8 [‐5.72, 0.12] |
| 4.2 24 months | 1 | 130 | Mean Difference (IV, Fixed, 95% CI) | ‐8.3 [‐12.47, ‐4.13] |
| 5 Change in mBMI | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 12 months | 1 | 130 | Mean Difference (IV, Fixed, 95% CI) | 19.8 [‐28.27, 67.87] |
| 5.2 24 months | 1 | 130 | Mean Difference (IV, Fixed, 95% CI) | 34.2 [‐18.42, 86.82] |
| 6 Quality of life (expressed as the change from baseline in SF‐36 physical score) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.1 12 months | 1 | 130 | Mean Difference (IV, Fixed, 95% CI) | 2.60 [‐0.04, 5.24] |
| 6.2 24 months | 1 | 130 | Mean Difference (IV, Fixed, 95% CI) | 6.10 [2.56, 9.64] |
| 7 Quality of life (change in SF‐36 mental score) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 7.1 12 months | 1 | 130 | Mean Difference (IV, Fixed, 95% CI) | 1.7 [‐1.98, 5.38] |
| 7.2 24 months | 1 | 130 | Mean Difference (IV, Fixed, 95% CI) | 4.4 [‐0.19, 8.99] |
| 8 Number of participants who died during the trial | 1 | 130 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.15, 1.41] |
| 9 Number of participants experiencing mild adverse events | 1 | 130 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.75, 1.65] |
| 10 Number of dropouts due to adverse events | 1 | 130 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.06 [0.39, 10.87] |
| 11 Number of participants experiencing at least one severe adverse event | 1 | 130 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.18, 3.32] |
Comparison 3. Patisiran versus placebo in TTR‐FAP.
Comparison 4. Inotersen versus placebo in TTR‐FAP.
Characteristics of studies
Characteristics of included studies [ordered by year of study]
Coelho 2012.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group | |
| Participants | Outpatients Setting: 8 centres in Argentina, Brazil, France, Germany, Portugal (2 centres), Spain, and Sweden Number: 128 randomised participants. ITT population (all randomised participants who received at least 1 dose of study medication and who had at least 1 post‐baseline assessment for both co‐primary endpoints or who discontinued due to LT): 125 participants (64 in the tafamidis group, 61 in the placebo group) Mean (SD) age (ITT population): 39.8 ± 12.7 years in the tafamidis group, 38.4 ± 12.9 years in the placebo group. Age was > 65 years in 5/64 (7.8%) of participants in the tafamidis group and in 3/61 (4.9%) in the placebo group Gender (ITT population): males were 32/64 (50.0%) in the tafamidis group and 26/61 (42.6%) in the placebo group Race (ITT population): White were 56/64 (87.5%) in the tafamidis group and 54/61 (88.5%) in the placebo group Median (IQR) disease duration (ITT population): 28.0 months (IQR 13.8 to 41.7) in the tafamidis group, 21.0 months (IQR 13.5 to 72.2) in the placebo group Key inclusion criteria: age 18 to 75 years inclusive; TTR‐FAP with documented Val30Met mutation in the TTR gene and biopsy‐confirmed amyloid deposits; peripheral or autonomic neuropathy with a Karnofsky ≥ 50 Key exclusion criteria: presence of primary amyloidosis, other causes of sensorimotor neuropathy, absence of a recordable sensory threshold for vibration perception in both feet, liver function test abnormalities, prior LT, renal insufficiency (creatinine clearance < 30 mL/min), New York Heart Association classification ≥ 3, any comorbidity anticipated to limit survival to < 18 months, chronic use of non‐protocol‐approved nonsteroidal anti‐inflammatory drugs |
|
| Interventions |
Oral self‐administration Duration of treatment period: 18 months |
|
| Outcomes | Co‐primary endpoints (time point: 18 months):
Secondary endpoints:
|
|
| Funding | Support from:
|
|
| Conflicts of interest among main investigators | T Coelho's institution received support from FoldRx Pharmaceuticals, which was acquired by Pfizer Inc. in October 2010. T Coelho received support from Pfizer Inc. M Waddington‐Cruz, O Suhr, and IM Conceicao received support from FoldRx Pharmaceuticals, which was acquired by Pfizer Inc. in October 2010, and from Pfizer Inc. V Planté‐Bordeneuve, P Lozeron, HHJ Schmidt, and P Trigo received support from FoldRx Pharmaceuticals, which was acquired by Pfizer Inc. in October 2010. JM Campistol received support from FoldRx Pharmaceuticals, which was acquired by Pfizer Inc. in October 2010, and from Wyeth (Pfizer Inc.). JW Kelly was founder, shareholder and option holder, and paid consultant of FoldRx Pharmaceuticals, which was acquired by Pfizer Inc. in October 2010. J Chan, R Labaudiniere, J Packman, A Wilson, and DR Grogan were employees of FoldRx Pharmaceuticals, which was acquired by Pfizer Inc. in October 2010, during the conduct of the study. |
|
| Notes | ClinicalTrials.gov Identifier: NCT00409175 Clinicaltrialsregister.eu Identifier: 2006‐002792‐41 Information obtained from both published report papers and online clinical trial registries Maximum duration of study: 20 months (screening period: 1 month, treatment period: 18 months, final telephone contact: 1 month after the last dose of study medication) Dates: 2007 to 2009 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomized by a central computerized telerandomization system, in a 1:1 ratio". Comment: probably done |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients were randomized by a central computerized telerandomization system, in a 1:1 ratio, to self‐administer [...] tafamidis [...] or matching placebo. [...]". Comment: probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double‐blind trial". "Patients were randomized by a central computerized telerandomization system, in a 1:1 ratio, to self‐administer [...] tafamidis [...] or matching placebo. [...] The active drug was provided in soft‐gelatin capsules [...]. The packaging, appearance, and constitution of the placebo capsules were identical to those of the active‐drug capsules except for the absence of tafamidis". Quote (from the final public disclosure synopsis provided by Pfizer Inc.): "Blinded safety summaries were provided [...] 24 months after enrollment commenced. [...] An interim efficacy and safety analysis was conducted [...]. The interim analysis was unblinded, but the blind remained unbroken for subjects and Investigators, and the Sponsor was not aware of the results of the interim analysis". Quote (from ClinicalTrials.gov): "Masking: Quadruple (Participant, Care Provider, Investigator, Outcomes Assessor)". Comment: probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double‐blind trial"; "Patients returned to the clinical sites during the double‐blind treatment period". Quote (from the final public disclosure synopsis provided by Pfizer Inc.): "Blinded safety summaries were provided [...] 24 months after enrollment commenced. [...] An interim efficacy and safety analysis was conducted [...]. The interim analysis was unblinded, but the blind remained unbroken for subjects and Investigators, and the Sponsor was not aware of the results of the interim analysis". Quote (from ClinicalTrials.gov): "Masking: Quadruple (Participant, Care Provider, Investigator, Outcomes Assessor)". Comment: probably done, but the statistical analysis was conducted by an employee of the sponsor. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "The primary efficacy analyses were performed on the intent‐to‐treat (ITT) population (all randomized patients who received at least 1 dose of study medication and who had 1 postbaseline assessment for both coprimary endpoints or who discontinued due to liver transplantation). For patients with postbaseline assessments, the last‐observation‐carried‐forward method was used to impute missing data at month 18"; "Analyses of the coprimary endpoints were performed in an efficacy‐evaluable (EE) population consisting of ITT patients who completed the study per protocol. This EE population was prespecified as it was anticipated that the majority of patients enrolled would be on the liver transplant list and that many would undergo liver transplantation during the study
if a donor organ became available"; "Analyses of the secondary endpoints were conducted in the ITT population using a repeated measures analysis of variance model [...] Only observed values were used". Comment: there was a high dropout rate in this RCT. As the original protocol of this RCT was not accessible, we could not verify whether populations on which analyses of the outcomes have been performed were prespecified. Some continuous outcome data of interest for this review were only descriptively presented and no measures of variability were published so that they cannot be entered in a meta‐analysis. For dichotomous outcomes most data were obtained from unpublished data. |
| Selective reporting (reporting bias) | Low risk | Comment: results for all predetermined outcomes were obtained by either published reports, online clinical trial registries, or both. |
| Other bias | Unclear risk | Comment: this study was funded by FoldFox Pharmaceuticals, which was acquired by Pfizer Inc. in October 2010. Most Investigators received funding from the Sponsor or were employees of the Sponsor during the conduct of this trial. |
Berk 2013.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group | |
| Participants | Outpatients Setting: 8 centres in England, Italy, Japan (2 centres), Sweden, and the USA (3 centres) Number: 130 randomised participants (64 in the diflunisal group, 66 in the placebo group) Mean (SD) age: 60.3 ± 11.7 years in the diflunisal group, 59.2 ± 12.2 years in the placebo group Gender: males were 43/64 (67.2%) in the diflunisal group and 44/66 (66.7%) in the placebo group Race: Asian 8/64 (12.5%) in the diflunisal group and 6/66 (9.1%) in the placebo group; Black 1/64 (1.6%) in the diflunisal group and 5/66 (7.6%) in the placebo group; White 52/64 (81.3%) in the diflunisal group and 50/66 (75.8%) in the placebo group; Other 0/64 (0.0%) in the diflunisal group and 1/66 (1.5%) in the placebo group; Multiracial 3/64 (4.7%) in the diflunisal group and 4/66 (6.1%) in the placebo group TTR genotype: Val30Met 36/64 (56.3%) in the diflunisal group and 35/66 (53.0%) in the placebo group; no‐Val30Met 28/64 (43.8%) in the diflunisal group and 31/66 (47.0%) in the placebo group Key inclusion criteria: age 18 to 75 years; biopsy‐confirmed amyloid deposition by Congo Red staining; mutant TTR genopositivity by DNA sequence analysis; signs of sensorimotor or autonomic neuropathy clinically detectable by a trained neurologist; routinely spent more than 50% of waking hours out of bed or chair (Eastern Co‐operative Oncology Group performance status < 3). Key exclusion criteria: alternative causes of sensorimotor polyneuropathy; limited survival prognosis (< 2 years); prior LT; severe congestive heart failure (New York Heart Association class IV) or renal insufficiency (estimated creatinine clearance < 30 mL/min); ongoing anticoagulation |
|
| Interventions |
Duration of treatment period: 24 months |
|
| Outcomes | Primary outcome:
Secondary outcomes:
|
|
| Funding | Grants from the National Institute of Neurological Diseases and Stroke (grant R01‐NS051306), the Orphan Products Division of the US Food and Drug Administration (grant FD‐R‐002532), the Young Family Amyloid Research Fund, and the National Center for Advancing Translational Sciences, National Institutes of Health (grant UL1‐TR000157). Merck Sharp and Dohme Inc supplied study drug (diflunisal) |
|
| Conflicts of interest among main investigators | "Dr. Ole Suhr has received support from Pfizer for activities as Chairman of The Transthyretin Amyloidosis Outcome Survey (THAOS), ISIS, and Alnylam Pharmaceuticals. Dr. Giampaolo Merlini has received honoraria from Pfizer. Dr. Jeffery Kelly reports financial holdings in FoldRx Pharmaceuticals, Inc. Drs. Berk, Obici, Zeldenrust, Litchy, and Dyck have received honoraria from Alnylam, ISIS, and Pfizer Pharmaceuticals." | |
| Notes | ClinicalTrials.gov Identifier: NCT00294671 EudraCT number: 2006‐001066‐16 Information obtained from both published report papers and one online clinical trial registry Duration: 24 months (treatment period) Dates: 2006 to 2012 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomly assigned in a 1:1 manner"; "Randomization was performed in permuted blocks of 2 to 4 stratified for mutant TTR (non‐V30M versus V30M) and study site"; "Study drug was prepackaged according to a computer‐generated randomization scheme". Comment: probably done |
| Allocation concealment (selection bias) | Low risk | Quotes: "randomization was performed in permuted blocks of 2 to 4 stratified for mutant TTR (non‐Val30Met versus Val30Met) and study site"; "Study drug was prepackaged according to a computer‐generated randomization scheme and dispensed by independent investigational pharmacists using sequential study IDs". Comment: probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quotes: "Study drug was [...] dispensed by independent investigational pharmacists using sequential study IDs"; "patients, investigators, study coordinators, and investigational pharmacists were unaware of treatment assignments"; "the randomization code was not broken at any time during the study". Comment: probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Study drug was [...] dispensed by independent investigational pharmacists using sequential study IDs"; "patients, investigators, study coordinators, and investigational pharmacists were unaware of treatment assignments"; "the randomization code was not broken at any time during the study". Comment: probably done |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "analysis of missingness completely at random for the primary and secondary outcomes using the permutation test indicated dependence of dropout on the outcome values". Comment: 24 months (primary outcome): 24/64 missing from intervention group and 38/66 missing from placebo group; 67 participants discontinued study treatment before completing the 2‐year protocol (27 from the diflunisal group and 40 from the placebo group). 5 participants in the placebo group and 3 in the diflunisal group discontinued study drug and acquired diflunisal outside the study but completed 2‐year NIS+7 testing. There was a high dropout rate in this RCT. |
| Selective reporting (reporting bias) | Low risk | Comment: outcome measures declared in the 'methods' section are reported by either published reports, online clinical trial registries, or both. |
| Other bias | Low risk | Comment: we did not observe any other potential risk of bias. Merck Sharp and Dohme Inc. was only stated to support this trial by providing study drug (diflunisal) and had no role in the design and conduct of the study. |
Benson 2018.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group | |
| Participants | Outpatients Setting: 24 centres, in Argentina, Brazil (3 centres), France (2 centres), Germany, Italy (2 centres), New Zealand, Portugal (2 centres), Spain (2 centres), UK, and USA (9 centres) Number: 173 randomised participants. Efficacy analysis population: 172 participants (112 in the inotersen group, 77 in the placebo group) Mean (SD) age: 59.0 ± 12.5 years in the inotersen group, 59.5 ± 14.0 years in the placebo group Gender: males were 77/112 (69%) in the inotersen group and 41/60 (68%) in the placebo group Race: Asian 1/112 (< 1%) in the inotersen group and 3/60 (5%) in the placebo group; Black 3/112 (3%) in the inotersen group and 1/60 (2%) in the placebo group; White 105/112 (94%) in the inotersen group and 53/60 (88%) in the placebo group; Other or multiracial 3/112 (3%) in the inotersen group and 3/60 (5%) in the placebo group TTR genotype: Val30Met 56/112 (50%) in the inotersen group and 33/60 (55%) in the placebo group; non‐Val30Met 56/112 (50%) in the inotersen group and 27/60 (45%) in the placebo group Key inclusion criteria: age 18 to 82 years, stage 1 or stage 2 TTR‐FAP, NIS of 10 to 130, TTR mutation, documented amyloid deposit determined on biopsy Key exclusion criteria: clinically significant abnormalities in screening laboratory values, Karnofsky performance status of 50 or less, other causes of polyneuropathies besides TTR‐FAP, previous LT, heart failure of New York Heart Association class III or higher |
|
| Interventions |
Subcutaneous administration; participants received three subcutaneous injections on alternate days during the first week to reach near steady‐state drug levels, followed by one subcutaneous injection once weekly for the next 64 weeks Participants also received vitamin A supplements (approximately 3000 IU daily) |
|
| Outcomes | Primary outcomes (time point: week 66):
Secondary outcomes (time point: week 66):
Other assessments (safety evaluation):
|
|
| Funding | Funded by Ionis Pharmaceuticals, Inc | |
| Conflicts of interest among main investigators | Merrill D Benson reported other support from Ionis Pharmaceuticals during the conduct of the study, as well as personal fees and non‐financial support from Ionis Pharmaceuticals outside the submitted work. Márcia Waddington‐Cruz reported grants and personal fees from Ionis Pharmaceuticals during the conduct of the study. John L Berk reported personal fees and non‐financial support from Alnylam Pharmaceuticals outside the submitted work. Michael Polydefkis reported grants and non‐financial support from Ionis Pharmaceuticals during the conduct of the study. Peter J Dyck reported other support from Ionis Pharmaceuticals, Inc. during the conduct of the study. Annabel K Wang reported personal fees and other support from Ionis Pharmaceuticals, Inc. during the conduct of the study. Violaine Planté‐Bordeneuve reported personal fees and non‐financial support from Ionis Pharmaceuticals during the conduct of the study. Fabio A Barroso reported grants, personal fees and non‐financial support from Ionis during the conduct of the study. Thomas H Brannagan reported grants and personal fees from Ionis during the conduct of the study. William J Litchy reported other support from Ionis Pharmaceuticals, Inc. during the conduct of the study. David Adams reported other support from Ionis during the conduct of the study. Amil M Shah reported grants from Ionis Pharmaceuticals during the conduct of the study. Scott D Solomon reported grants from Ionis during the conduct of the study. Brett P Monia reported other support from Ionis during the conduct of the study. Steven G Hughes reports personal fees and other support from Ionis Pharmaceuticals, Inc. outside the submitted work. T Jesse Kwoh reported personal fees and other support from Ionis Pharmaceuticals, Inc. outside the submitted work. Bradley W McEvoy personal fees and other support from Ionis Pharmaceuticals, Inc. outside the submitted work. Shiangtung W Jung reported personal fees and other support from Ionis Pharmaceuticals, Inc. outside the submitted work. Brenda F Baker reported personal fees and other support from Ionis Pharmaceuticals, Inc. outside the submitted work. Elizabeth J Ackermann reported personal fees and other support from Ionis Pharmaceuticals, Inc. outside the submitted work. Morie A Gertz reported personal fees from Ionis. Teresa Coelho reported non‐financial support and other from Ionis during the conduct of the study. |
|
| Notes | ClinicalTrials.gov Identifier: NCT01737398 Clinicaltrialsregister.eu: 2012‐001831‐30 Information obtained from both published report papers and one online clinical trial registry Maximum duration of study: 66 weeks: 65 weeks (treatment period), 1 week (post‐intervention evaluation) Dates: 2013 to 2017 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (form report): "Patients were randomly assigned, in a 2:1 ratio", Quote (from the final protocol): "Using an Interactive Voice/Web‐Response System (IXRS), eligible patients will be randomized 2:1 to receive ISIS 420915 or placebo, respectively"; "A permuted block schedule will be used". Comment: probably done |
| Allocation concealment (selection bias) | Low risk | Quote (from the final protocol): "The Study Drug is contained in stoppered glass vials and will be provided to the Study Center by the Sponsor. The Sponsor will provide the Investigator with packaged Study Drug labeled in accordance with specific country regulatory requirements". Comment: probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote (from the final protocol): "The Sponsor, participants, monitors, and Study Center personnel will be blinded throughout the study until all participants have completed the treatment period and the EOT [end of treatment] efficacy assessments, and the database has been locked"; "In order to ensure maintenance of the study blind, TTR, RBP4, and retinol values will not be available to the Sponsor, monitors, Investigators, Study Center Personnel, or the participants." Comment: probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote (from report): "the sponsor (Ionis Pharmaceuticals) was responsible for data analysis". Quote (from the final protocol): "The Sponsor, participants, monitors, and Study Center personnel will be blinded throughout the study until all participants have completed the treatment period and the EOT efficacy assessments, and the database has been locked"; "In order to ensure maintenance of the study blind, TTR, RBP4, and retinol values will not be available to the Sponsor, monitors, Investigators, Study Center Personnel, or the participants". Comment: probably done, but data analysis was conducted by the sponsor |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: efficacy analyses included all randomly assigned participants who received at least one dose of a trial regimen and who had at least one post‐baseline efficacy assessment for the mNIS+7 Ionis version or the Norfolk QOL‐DN score. There was no clear mention of ITT analysis. |
| Selective reporting (reporting bias) | Low risk | Comment: results for all predetermined outcomes were obtained by either published report papers, online clinical trial registries, or both. |
| Other bias | Unclear risk | This study was fully supported by Ionis Pharmaceuticals. |
Adams 2018.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group | |
| Participants | Outpatients Setting: 50 centres, in Argentina, Australia, Bulgaria, Brasil (3 centres), Canada, Cyprus, France (5 centres), Germany (3 centres), Italy (3 centres), Japan (3 centres), Korea (2 centres), Malaysia, Mexico, Netherlands, Portugal (2 centres), Spain (4 centres), Sweden, Taiwan (2 centres), Turkey, UK (2 centres), USA (11 centres) Number: 225 randomised participants (148 in the patisiran group, 77 in the placebo group) Median age (range): 62 years (24 to 83) in the patisiran group, 63 years (34 to 80) in the placebo group Gender: males were 109/148 (74%) in the patisiran group and 58/77 (75%) in the placebo group Race: Asian 27/148 (18%) in the patisiran group and 25/77 (32%) in the placebo group; Black 4/148 (3%) in the patisiran group and 1/77 (1%) in the placebo group; White 113/148 (76%) in the patisiran group and 50/77 (65%) in the placebo group; Other 1/148 (< 1%) in the patisiran group and 0/77 (0%) in the placebo group; Multiracial 2/148 (1%) in the patisiran group and 0/77 (0%) in the placebo group; missing data 1/148 (< 1%) in the patisiran group and 1/77 (1%) in the placebo group TTR genotype: Val30Met 56/148 (38%) in the patisiran group and 40/77 (52%) in the placebo group; non‐Val30Met 92/148 (62%) in the patisiran group and 37/77 (48%) in the placebo group Key inclusion criteria: age 18 to 85 years; a documented pathogenic variant in the TTR gene; a diagnosis of hereditary transthyretin amyloidosis with peripheral neuropathy, with a NIS of 5 to 130 and a PDS of IIIb or lower; adequate liver and renal function. Key exclusion criteria: prior LT or plan to undergo LT during the study period; known human immunodeficiency virus infection; history of malignancy within 2 years, except for basal or squamous cell carcinoma of the skin or carcinoma in situ of the cervix that has been successfully treated; recently received an investigational agent or device; currently taking diflunisal, tafamidis, doxycycline, or tauroursodeoxycholic acid |
|
| Interventions |
Intravenous administration Duration of treatment period: 18 months |
|
| Outcomes | Primary outcome:
Secondary outcomes:
|
|
| Funding | Support from: Alnylam Pharmaceuticals | |
| Conflicts of interest among main investigators | David Adams reported grants from Alnylam during the conduct of the study. Alejandra Gonzalez‐Duarte reported personal fees from Alnylam Pharmaceuticals outside the submitted work. William D O’Riordan reported other support from Alnylam Pharmaceuticals during the conduct of the study. Arnt V Kristen reported personal fees and other support from Alnylam Pharmaceuticals during the conduct of the study. Teresa Coelho reported personal fees and non‐financial support from Alnylam Pharmaceuticals. John L Berk reported personal fees from Alnylam Pharmaceuticals. Michelle M. Mezei reported personal fees from Alnylam during the conduct of the study. Juan Buades reported personal fees and non‐financial support from Alnylam Pharmaceuticals during the conduct of the study. Thomas H Brannagan reported grants from Alnylam during the conduct of the study, as well as personal fees from Alnylam. Yesim Parman reported other support from Alnylam Pharmaceuticals during the conduct of the study. Scott D Solomon reported grants and personal fees from Alnylam during the conduct of the study. Michael Polydefkis reported grants and personal fees from Alnyam Pharmaceuticals during the conduct of the study. Peter J Dyck reported other support from Alnylam, Inc. during the conduct of the study. Pritesh J Gandhi reported personal fees and other support from Alnylam Pharmaceuticals outside the submitted work. Jihong Chen reported personal fees from Alnylam Pharmaceuticals outside the submitted work. Andrew L Strahs reported personal fees from Alnylam Pharmaceuticals outside the submitted work. Saraswathy V Nochur reported personal fees and other support from Alnylam Pharmaceuticals outside the submitted work. Marianne T Sweetser was an employee of Alnylam Pharmaceuticals during the conduct of the study. Pushkal P Garg reported personal fees and other support from Alnylam Pharmaceuticals outside the submitted work. Akshay K Vaishnaw reported personal fees from Alnylam Pharmaceuticals outside the submitted work. Ole B Suhr, MD, PhD reports personal fees and non‐financial support from Alnylam Pharamceuticals during the conduct of the study. |
|
| Notes | ClinicalTrials.gov Identifier: NCT01960348 Clinicaltrialsregister.eu Identifier: 2013‐002987‐17 Information obtained from both published report papers and online clinical trial registries Maximum duration of study: 18 months Dates: 2013 to 2017 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (from report): "patients were randomly assigned in a 2:1 ratio". Quote (from final protocol): "Patients will be randomized via an interactive response system (IRS)"; "Upon signing the informed consent form, the patient will be assigned a screening number by the IRS. The Investigator or his/her delegate will contact the IRS (via phone or web) after confirming that the patient fulfils all the inclusion criteria and none of the exclusion criteria. The patient will be randomized via the IRS, assigned a subject number and a study treatment". Comment: probably done |
| Allocation concealment (selection bias) | Low risk | Quote (from final protocol): "To maintain the blind, all IV infusion bags and lines will have amber‐colored covers added prior to leaving the pharmacy". Comment: probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote (from final protocol): "Only the pharmacist and designated site personnel who dispense or administer study drug will be unblinded to the study treatment. All other site personnel will be blinded to the treatment. Study personnel performing assessments related to the efficacy endpoints will be different from the Investigator and other personnel managing the patient, all of whom will also remain blinded to any clinical laboratory results that could potentially unblind them (e.g., TTR levels, vitamin A levels, thyroid function tests)." Comment: probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote (from final protocol): "An Interim Analysis Committee (IAC), which will include at least 1 statistician (blinded) comprised of 2 statisticians (1 blinded and 1 unblinded) independent of the conduct of the study, will be responsible for the implementation of the interim analysis and for the calculations and recommendations surrounding whether an adjustment to the sample size is warranted, and if so, the appropriate adjustment, based on the study’s primary endpoint data from the interim analysis. The Sponsor, the CROs, and all other parties conducting the study will remain blinded to all interim analyses until study completion. The IAC will follow the procedure outlined in the committee’s charter". Comment: probably done, but it not clear if the sponsor was involved in data analysis. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote (from report): "The modified intention‐to‐treat (ITT) population included all the patients who underwent randomization and received at least one dose of patisiran or placebo". Comment: there are clear data on dropouts. |
| Selective reporting (reporting bias) | Low risk | Comment: results for all predetermined outcomes were obtained by either published report papers, online clinical trial registries, or both. |
| Other bias | Unclear risk | This study was fully supported by Alnylam Pharmaceuticals. |
BMI: body mass index; EE: efficacy‐evaluable; EOT: end of treatment; ITT: intention‐to‐treat; IQR: interquartile range; KS: Kumamoto Score; LT: liver transplantation; mBMI: modified body mass index; mNIS: modified Neuropathy Impairment Score; N: number of participants; NIS: Neuropathy Impairment Score; NIS+7: Neuropathy Impairment Score plus 7 nerve tests; NIS‐LL: Neuropathy Impairment Score in the lower limbs; Norfolk QOL‐DN: Norfolk Quality of Life–Diabetic Neuropathy Questionnaire; PDS: Polyneuropathy Disability Score; R‐ODS: Rasch‐built Overall Disability Scale; RBP4: retinol binding protein 4; RCT: randomised clinical trial; SD: standard deviation; SF‐36: Short Form 36 Health Survey Questionnaire; TTR: transthyretin; TTR‐FAP: transthyretin‐familial amyloid polyneuropathy.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Adams 2015 | A report on a phase 2 open‐label extension study of patisiran. Not randomised |
| Barroso 2017 | A paper reporting interim results of an open‐label, single‐treatment study investigating safety and efficacy of tafamidis in TTR‐FAP. Not randomised |
| Coelho 2013 | A 12‐month, open‐label, multicentre, international, single‐arm trial (ClinicalTrial.gov, NCT00791492) evaluating the safety and efficacy of tafamidis in people with Val30Met TTR‐FAP who completed the pivotal trial of tafamidis continues to receive tafamidis or are switched from placebo 20 mg once daily for 12 months. Not randomised |
| Gillmore 2015 | A report on a phase 2, open‐label extension study of revusiran, an investigational RNAi therapeutic for the treatment of people with transthyretin cardiac amyloidosis. We excluded this study as it is not randomised and does not focus on FAP (neuropathy). |
| Gundapaneni 2018 | A paper reporting a post hoc analysis comparing data from the pivotal trial of tafamidis (NCT00409175; Coelho 2012) and an open‐label study to evaluate TTR stabilisation, safety, tolerability, and efficacy of tafamidis in people with non‐Val30Met TTR‐FAP (NCT00630864; Merlini 2013). Not randomised |
| Maurer 2018 | A paper reporting on a multicentre, international, randomised double‐blind, placebo‐controlled, phase 3 trial of tafamidis in people with transthyretin amyloid cardiomyopathy. We excluded this study as it does not focus on FAP (neuropathy). |
| Plante‐Bordeneuve 2018 | A report mentioning an ongoing, phase 3, open‐label extension study (ClinicalTrial.gov, NCT02175004) designed to evaluate long‐term efficacy and safety of inotersen in people with TTR‐FAP who were enrolled and completed the pivotal RCT of inotersen up to 5 years. Not randomised |
| Russo 2013 | A conference abstract presenting a small study investigating the efficacy of tafamidis in 7 people with TTR‐FAP. Not randomised |
| Sah 2011 | A conference abstract which presents an update on the development of ALN‐TTR01 as a therapeutic option for the treatment of TTR‐related amyloidosis as well as of a second‐generation RNAi therapeutic targeting TTR. Not a trial |
| Suhr 2015 | A paper reporting a multicentre, international, open‐label, multiple‐dose escalation phase 2 study (Clinicaltrials.gov, NCT01617967) which evaluates the safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple doses of patisiran (ALN‐TTR02) in people with FAP. Not randomised |
| Waddington‐Cruz 2016 | A paper reporting interim results from an open‐label extension study (ClinicalTrials.gov, NCT00925002) which evaluates the long‐term safety and efficacy of tafamidis in people with Val30Met TTR‐FAP over a 10‐year period. Not randomised |
FAP: familial amyloid polyneuropathy; RCT: randomised controlled trial; RNAi: ribonucleic acid inhibitor; TTR: transthyretin; TTR‐FAP: transthyretin‐familial amyloid polyneuropathy.
Differences between protocol and review
We changed the name of the condition in the review title to familial amyloid polyneuropathy, which is more commonly used in the literature.
We extended the 'Background' section in order to provide more information, which is useful to understand other parts of the review, and we added references to the most recent literature on the topic.
We highlighted that Isis Pharmaceuticals, Inc. announced that the company has changed its name to Ionis Pharmaceuticals, Inc. during the preparation of the protocol for this review. Therefore, we reviewed the new pharmaceutical company website www.ionispharma.com (see: 'Searching other resources' paragraph).
We included a description of quasi‐randomisation in the 'Types of studies' section, for explanatory purposes.
We included B2M gene mutations under 'Types of participants' for completeness, as B2M‐FAP has emerged as an extremely rare form of FAP in recent years.
We included the clarification that data from open‐label extension periods of RCTs are reported in the 'Discussion' section but not in the 'Effects of interventions' section, as our inclusion criteria specified RCTs and quasi‐RCTs.
We slightly reformulated the headings of outcomes in the 'Methods' and 'Results' sections as well as in the 'Summary of findings' tables in order that they are the same in different comparisons.
We included the Rasch‐built Overall Disability Scale (R‐ODS) as measure of disability due to FAP progression in the 'Types of outcome measures' and in Table 5 and added a relevant reference.
We included the modified NIS+7Alnylam and NIS+7Ionis as measures of severity of peripheral neuropathy in the 'Types of outcome measures' and in Table 5 and added relevant references because these measures have replaced the NIS+7 (already included in our outcome measures) in recent clinical trials.
We included reasons for the choice of outcome measures and time intervals reported in the 'Summary of findings' tables in the 'Summary of findings tables' paragraph of the 'Methods' section.
We moved methods not applicable in the current review to Appendix 6.
Contributions of authors
FM is the contact person with the editorial base.
Three review authors (FM, GZ, and ST) selected studies, extracted data, and performed data analysis.
FM, GMF, and ST wrote the first draft of the review.
All review authors (FM, GMF, LS, FioM, GZ, TC, and ST) commented on the first draft of the review and approved the final manuscript.
FM and ST are the guarantors of the final review.
Declarations of interest
FM: was sponsored by Pfizer Inc. to attend the ARiA (Advances and Research in TTR Amyloidosis) VI Symposium in Wien in 2017.
GMF: Gian Maria Fabrizi was sponsored by: Akcea Therapeutics to attend the 2019 American Academy of Neurology Conference in Philadelphia and by Pfizer Inc. to attend the 2018 ARia (Advances and Research in TTR Amyloidosis) VII symposium, in Frankfurt. GMF received fees for Board membership from Akcea Therapeutics (Akcea Scientific Advisory Board, Milan January 29 2019) and Alnylam Pharmaceuticals (Alnylam Scientific Advisory Board, Milan December 11 2019).
LS: none known.
FioM: none known.
GZ: none known.
TC: received fees from Pfizer Inc. for her participation as member of the Advisory Board "Amiloidosi ereditaria TTR correlata" (Milan, 2016).
ST: none known; ST is a specialist in neurology and manages people with peripheral neuropathies, including familial amyloid polyneuropathy.
The review is not compliant with Cochrane Commercial Sponsorship policy; the update will have a majority of authors and lead author free of conflicts.
New
References
References to studies included in this review
Adams 2018 {published data only (unpublished sought but not used)}
- Adams D, Gonzalez‐Duarte A, O'Riordan W, Yang CC, Yamashita T, Kristen A, et al. Evaluation of quality of life and disability in patients with hereditary transthyretin‐mediated (HATTR) amyloidosis with polyneuropathy following treatment with patisiran, an investigational RNAI therapeutic:results from the phase 3 APOLLO study. Neurology 2018;90(15 Suppl 1):S31.003. [EMBASE: 622309208] [Google Scholar]
- Adams D, Gonzalez‐Duarte A, O'Riordan W, Yang CC, Yamashita T, Kristen A, et al. Patisiran, an investigational RNAi therapeutic for patients with hereditary transthyretin‐mediated (hATTR) amyloidosis with polyneuropathy: results from the phase 3 APOLLO study. Neurology 2018;90(15 Suppl 1):CT.001. [EMBASE: 622307825] [Google Scholar]
- Adams D, Gonzalez‐Duarte A, O'Riordan W, Yang CC, Yamashita T, Kristen A, et al. Patisiran, an investigational RNAi therapeutic for patients with hereditary transthyretin‐mediated (hATTR) amyloidosis with polyneuropathy: results from the phase 3 APOLLO study. Orphanet Journal of Rare Diseases 2017;12(Suppl 1):165. [EMBASE: 619389882] [Google Scholar]
- Adams D, Gonzalez‐Duarte A, O'Riordan WD, Yang CC, Ueda M, Kristen AV, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. New England Journal of Medicine 2018;379(1):11‐21. [PUBMED: 29972753 ] [DOI] [PubMed] [Google Scholar]
- Adams D, Pritesh JG. Outcomes of patients with hereditary transthyretin‐mediated amyloidosis with early onset V30M versus all other mutations in APOLLO, a phase 3 study of patisiran. Revue Neurologique 2019;175(Suppl 1):S129. [Google Scholar]
- Adams D, Suhr OB, Dyck PJ, Litchy WJ, Leahy RG, Chen J, et al. Trial design and rationale for APOLLO, a phase 3, placebo‐controlled study of patisiran in patients with hereditary ATTR amyloidosis with polyneuropathy. BMC Neurology 2017;17(1):181. [PUBMED: 28893208] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajroud‐Driss S, Adams D, Coelho T, Polydefkis M, Gonzalez‐Duarte A, Quan D, et al. Impact of patisiran on overall health status in HATTR amyloidosis: Results from the APOLLO trial. Neurology. Conference: 71st Annual Meeting of the American Academy of Neurology, AAN 2019;92(15 Suppl 1):2019. [Google Scholar]
- Ajroud‐Driss S, Adams D, Coelho T, Polydefkis M, Gonzalez‐Duarte A, Quan D, et al. Impact of patisiran on overall health status in hATTR amyloidosis: results from the APOLLO trial. Journal of the Peripheral Nervous System 2018;23(4):272‐3. [Google Scholar]
- Berk JL, Conceicao I, Waddington‐Cruz M, Polydefkis M, Coelho T, Partisano AM, et al. Evaluating the efficacy of patisiran in patients with hATTR amyloidosis in the phase 2 OLE trial and the phase 3 trial. Annals of Neurology. Conference: 143rd annual meeting American Neurological Association, ANA 2018;84(Suppl 22):S180. [Google Scholar]
- Coelho T, Adams D, Gonzalez‐Duarte A, O'Riordan W, Yang CC, Yamashita T, et al. Impact of prior TTR stabilizer use in patients with hereditary transthyretin‐mediated amyloidosis in the APOLLO phase‐3 study of patisiran. European Journal of Neurology 2018;25(Suppl 2):17‐8. [EMBASE: 623298109] [Google Scholar]
- Coelho T, Adams D, Gonzalez‐Duarte A, O'Riordan W, Yang CC, Yamashita T, et al. Outcomes of patients with hereditary transthyretin‐mediated amyloidosis with early onset V30M versus all other mutations in APOLLO, a phase 3 study of patisiran. European Journal of Neurology 2018;25(Suppl 2):353‐4. [EMBASE: 623298597 ] [Google Scholar]
- Coelho T, Adams D, González‐Duarte A, O'Riordan W, Yang CC, Polydefkis M, et al. Transthyretin reduction with patisiran in the APOLLO phase 3 study. Journal of Neuromuscular Diseases 2018;5(Suppl 1):S261‐2. [Google Scholar]
- Gonzalez‐Duarte A, Adams D, Mauermann M, Coelho T, Yang CC, Polydefkis M, et al. Impact of patisiran on autonomic neuropathy in hereditary transthyretin‐mediated (hATTR) amyloidosis patients. Clinical Autonomic Research 2018;28(5):466. [Google Scholar]
- Gonzalez‐Duarte A, Adams D, O'Riordan W, Yang CC, Yamashita T, Kristen A, et al. Changes in neuropathy stage in patients with hATTR amyloidosis following patisiran treatment: analysis from APOLLO. Journal of the Peripheral Nervous System 2018;23(4):400. [Google Scholar]
- Judge DP, Gonzalez‐Duarte A, Dispenzieri A, Lin H, Merkel M, Wang Y, et al. Impact of patisiran, an RNAi therapeutic, on orthostatic intolerance in patients with hereditary transthyretin‐mediated amyloidosis. Journal of Cardiac Failure 2019;25(8 Suppl):S78. [Google Scholar]
- Mauermann M, Adams D, Gonzalez‐Duarte A, Coelho T, Yang CC, Polydefkis M, et al. Impact of patisiran on autonomic neuropathy in hereditary transthyretin‐mediated amyloidosis patients. Journal of Neuromuscular Diseases 2018;5(Suppl 1):S220‐1. [Google Scholar]
- Merlini G, Solomon S, Adams D, Coelho T, Damy T, Mauer M, et al. Impact of patisiran on Norfolk QOL‐DN in patients with hereditary transthyretin‐mediated amyloidosis: results from the cardiac subpopulation in the phase 3 APOLLO study. European Journal of Heart Failure 2018;20(Suppl 1):383. [EMBASE: 622650519 ] [Google Scholar]
- Obici L, Coelho T, Adams D, Gonzalez‐Duarte A, O'Riordan W, Yang CC, et al. Impact of patisiran on Norfolk Quality of Life Questionnaire Diabetic Neuropathy (QOL‐DN) in patients with hereditary transthyretin‐mediated amyloidosis: results from the phase‐3 APOLLO study. European Journal of Neurology 2018;25(Suppl 2):459. [EMBASE: 623297736] [Google Scholar]
- Obici L, Coelho T, Adams D, Gonzalez‐Duarte A, O'Riordan W, Yang CC, et al. Impact of patisiran, an investigational RNAi therapeutic, on nutritional status in patients with hereditary transthyretin‐mediated amyloidosis. European Journal of Neurology 2018;25(Suppl 2):17. [EMBASE: 623298051] [Google Scholar]
- Partisano A, Schmidt H, O'Riordan W, Yamashita T, Berk JL, Tournev I, et al. Reasons for discontinuation of TTR stabilizers and/or TTR fibril disrupter: an analysis of baseline demographics of patients with hereditary ATTR (hATTR) amyloidosis in the phase 3 APOLLO study. Orphanet Journal of Rare Diseases 2017;12(Suppl 1):165. [EMBASE: 619390286] [Google Scholar]
- Polydefkis M, Adams D, Coelho T, Kristen A, Gonzalez‐Duarte A, Berk J, et al. Infusion related reactions in patients with hATTR amyloidosis treated with patisiran. Journal of the Peripheral Nervous System 2018;23(4):352. [Google Scholar]
- Quan D, Adams D, González‐Duarte A, Polydefis M, Kristen A, Tournev I, et al. Evaluating the impact of patisiran on disability using the Rasch‐built overall disability scale (R‐ODS) in patients with hereditary transthyretin‐mediated (HATTR) amyloidosis in the APOLLO study. Muscle & Nerve 2018;58(Suppl 2):S51. [Google Scholar]
- Schmidt H, O'Riordan W, Yamashita T, Berk JL, Tournev I, Mezei M, et al. Reasons for discontinuation of TTR stabilizers and/or TTR fibril disrupter: an analysis of baseline demographics of patients with hATTR amyloidosis in the phase 3 APOLLO study. Annals of Neurology 2017;82(Suppl 21):S100‐1. [EMBASE: 619084531 ] [Google Scholar]
- Slama MS, Solomon S, Adams D, Coelho T, Damy T, Merlini G, et al. Analysis of NT‐proBNP baseline levels in APOLLO as a predictor of survival in hereditary transthyretin‐mediated (hATTR) amyloidosis. European Journal of Heart Failure 2018;20(Suppl 1):576. [EMBASE: 622651967] [Google Scholar]
- Solomon SD, Adams D, Kristen A, Grogan M, González‐Duarte A, Maurer MS, et al. Effects of patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin‐mediated amyloidosis. Analysis of the APOLLO study. Circulation 2019;139(4):431‐43. [DOI] [PubMed] [Google Scholar]
- Zhang X, Goel V, Attarwala H, Sweetser MT, Clausen VA, Robbie GJ. Patisiran pharmacokinetics, pharmacodynamics, and exposure‐response analyses in the phase 3 APOLLO trial in patients with hereditary transthyretin‐mediated (hATTR) amyloidosis. Journal of Clinical Pharmacology 2019;19:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Sweetser MT, Robbie GJ. Results from APOLLO phase III study of patisiran, the first approved RNAi therapeutic, in hereditary ATTR amyloidosis patients with polyneuropathy. Clinical Pharmacology and Therapeutics 2019;105(Suppl 1):S31. [Abstract: E‐009] [Google Scholar]
Benson 2018 {published data only}
- Anonymous. Improvement in quality of life in patients with hereditary transthyretin amyloidosis with polyneuropathy and cardiomyopathy treated with inotersen in the phase 3 study NEURO‐TTR. Heart & Lung 2019;48(5):469‐71. [Google Scholar]
- Benson M, Kincaid J, Ackermann E, Monia B. A phase 3 study to evaluate ISIS‐TTRRX in patients with transthyretin familial amyloid polyneuropathy (TTRFAP): study design and baseline demographics. Neurology 2015;84(14 Suppl):S50.006. [EMBASE: 71922192 ] [Google Scholar]
- Benson M, Waddington‐Cruz M, Wang A, Polydefkis M, Plante‐Bordeneuve V, Berk J, et al. Safety and efficacy of inotersen in patients with hereditary transthyretin amyloidosis with polyneuropathy (hATTR‐PN). Orphanet Journal of Rare Diseases 2017;12(Suppl 1):165‐6. [EMBASE: 619389834] [Google Scholar]
- Benson MD, Waddington‐Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. New England Journal of Medicine 2018;379(1):22‐31. [PUBMED: 29972757] [DOI] [PubMed] [Google Scholar]
- Coelho T, Wang A, Waddington‐Cruz M, Polydefkis M, Dyck P, Scheinberg M, et al. Inotersen improves quality of life and neuropathy in patients with hereditary transthyretin (hATTR) amyloidosis with polyneuropathy: results of the phase 3 study NEURO‐TTR. European Journal of Neurology 2018;25(Suppl 2):376. [EMBASE: 623298254] [Google Scholar]
- Conceicao I, Berk J, Wang A, Coelho T, Waddington‐Cruz M, Polydefkis M, et al. Baseline characteristics of patients with hereditary transthyretin (hATTR) amyloidosis with polyneuropathy enrolled in the phase 3 study NEURO‐TTR demonstrate significant disease burden. European Journal of Neurology 2018;25(Suppl 2):375. [EMBASE: 623298181] [Google Scholar]
- Dyck PJ, Coelho T, Cruz MW, Brannagan T, Khella S, Karam C, et al. Responsiveness of neuropathy symptom and change (NSC) score components in inotersen treatment of hereditary transthyretin amyloidosis polyneuropathy. Neurology 2019;92(15 Suppl):P3.9‐060. [Google Scholar]
- Dyck PJ, Coelho T, Waddington Cruz M, Brannagan T, Khella S, Karam C, et al. Responsiveness of Neuropathy Symptom and Change (NSC) score components in inotersen treatment of hereditary transthyretin amyloidosis polyneuropathy. European Journal of Neurology 2019;26(Suppl 1):715. [Google Scholar]
- Dyck PJ, Kincaid JC, Wiesman JF, Polydefkis M, Litchy WJ, Mauermann ML, et al. Responsiveness of mNIS+7 components in inotersen treatment of hereditary transthyretin amyloidosis polyneuropathy. European Journal of Neurology 2019;26(Suppl 1):714. [Google Scholar]
- Gertz M, Wang A, Cruz MW, Polydefis M, Dyck P, Scheinberg M, et al. Safety and efficacy of inotersen in patients with hereditary transthyretin amyloidosis with polyneuropathy (NEURO‐TTR). Muscle & Nerve 2018;58(Suppl 2):S55. [Google Scholar]
- Gertz M, Wang AK, Coelho T, Cruz MW, Polydefkis MJ, Dyck PJ, et al. Safety and efficacy of inotersen in patients with hereditary transthyretin amyloidosis with polyneuropathy (NEURO‐TTR). Neurology 2018;90(15 Suppl 1). [Abstract: N2.001] [Google Scholar]
- Gertz M, Wang AK, Coelho T, Waddington‐Cruz M, Polydefkis MJ, Dyck PJ, et al. Safety and efficacy of inotersen in patients with hereditary transthyretin amyloidosis with polyneuropathy (NEURO‐TTR). Neurology 2018;90(15 Suppl 1):N2.001. [EMBASE: 622308128] [Google Scholar]
- Maurer MS, Heitner S, Drachman B, Whelan C, Guthrie S, Tai L, et al. Inotersen improves quality of life in patients with hereditary transthyretin amyloidosis with polyneuropathy and cardiomyopathy: results of the phase 3 study NEURO‐TTR. Journal of the American College of Cardiology 2018;71(11 Suppl 1):1217M‐03. [EMBASE: 621784581] [Google Scholar]
- Polydefkis M, Cruz MW, Dyck P, Scheinberg M, Berk J, Barroso F, et al. Inotersen improves Norfolk quality of life‐diabetic neuropathy (Norfolk QOL‐DN) measures in patients with hereditary transthyretin (HATTR) amyloidosis in the phase 3 study NEURO‐TTR. Muscle & Nerve 2018;58(Suppl 2):S54. [Google Scholar]
- Vita G, Polydefkis M, Coelho T, Waddington‐Cruz M, Dyck P, Scheinberg M, et al. Inotersen improved Norfolk quality of life‐diabetic neuropathy (Norfolk QOL‐DN) measures in patients with hereditary transthyretin (hATTR) amyloidosis treated in the phase‐3 study NEURO‐TTR. European Journal of Neurology 2018;25(Suppl 2):568‐9. [EMBASE: 623298443] [Google Scholar]
- Waddington‐Cruz M, Ackermann EJ, Polydefkis M, Heitner SB, Dyck PJ, Barroso FA, et al. Hereditary transthyretin amyloidosis: baseline characteristics of patients in the NEURO‐TTR trial. Amyloid 2018;25(3):180‐8. [DOI] [PubMed] [Google Scholar]
- Wang AK, Coelho T, Waddington‐Cruz M, Polydefkis MJ, Dyck PJ, Scheinberg M, et al. Safety and efficacy of inotersen in patients with hereditary transthyretin amyloidosis with polyneuropathy (neuro‐TTR). Annals of Neurology 2017;82(Suppl 21):S109. [EMBASE: 619084794] [Google Scholar]
- Yarlas A, Merlini G, White MK, Kessler AS, Lovley A, Guthrie SD, et al. Impact of inotersen on functioning and activities of daily living for patients with hereditary TTR Amyloidosis: results from a double‐blind placebo‐controlled trial. Blood 2018;132(Suppl 1):4812. [Google Scholar]
- Yarlas A, Merlini G, White MK, Kessler AS, Lovley A, Guthrie SD, et al. Impact of inotersen on functioning and activities of daily living for patients with hereditary TTR amyloidosis: results from a double‐blind placebo‐controlled trial. Blood 2018;132(Suppl 1):4812. [Google Scholar]
- Yarlas A, Merlini G, White MK, Kessler AS, Lovley A, Guthrie SD, et al. Impact of inotersen on subgroups of patients with hereditary TTR amyloidosis: results from a double‐blind placebo‐controlled trial. Blood 2018;132(Suppl 1):4803. [Google Scholar]
Berk 2013 {published data only}
- Berk JL, Dyck PJ, Obici L, Zeldenrust SR, Sekijima Y, Yamashita T, et al. The diflunisal trial: update on study drug tolerance and disease progression. Amyloid 2010;17(Suppl 1):78. [EMBASE: 70725032] [DOI] [PubMed] [Google Scholar]
- Berk JL, Dyck PJ, Obici L, Zeldenrust SR, Sekijima Y, Yamashita T, et al. The diflunisal trial: update on study drug tolerance and disease progression. Amyloid 2011;18(Suppl 1):196‐7. [PUBMED: 21838485] [DOI] [PubMed] [Google Scholar]
- Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 2013;310(24):2658–67. [PUBMED: 24368466] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk JL, Suhr OB, Sekijima Y, Yamashita T, Heneghan M, Zeldenrust SR, et al. The Diflunisal Trial: study accrual and drug tolerance. Amyloid 2012;19(Suppl 1):37‐8. [PUBMED: 22551208] [DOI] [PubMed] [Google Scholar]
Coelho 2012 {published data only (unpublished sought but not used)}
- Coelho T. Tafamidis: a novel and effective oral treatment for familial amyloid neuropathies. European Journal of Neurology 2012;19(Suppl 1):8. [EMBASE: 70938581] [Google Scholar]
- Coelho T, Maia L, Martins Da Silva A, Waddingtion‐Cruz M, Plante‐Bordeneuve V, Lozeron P, et al. Tafamidis (Fx‐1006A): A first‐in‐class disease‐modifying therapy for transthyretin familial amyloid. Amyloid 2010;17(Suppl 1):75‐6. [EMBASE: 70725028] [Google Scholar]
- Coelho T, Maia L, Martins Da Silva A, Waddingtion‐Cruz M, Plante‐Bordeneuve V, Lozeron P, et al. Tafamidis and nerve fiber function in transthyretin‐type familial amyloid polyneuropathy. Clinical Neurophysiology 2012;123(6):e56. [EMBASE: 70824746] [Google Scholar]
- Coelho T, Maia L, Martins Da Silva A, Waddingtion‐Cruz M, Plante‐Bordeneuve V, Suhr OB, et al. A landmark clinical trial of a novel small molecule transthyretin stabilizer, Fx‐1006A, in patients with TTR amyloid polyneuropathy: a phase II/III randomized, double‐blind, placebo‐controlled study. Journal of the Peripheral Nervous System 2009;14(Suppl 2):35‐6. [EMBASE: 70008473] [Google Scholar]
- Coelho T, Maia LF, Martins da Silva A, Waddington‐Cruz M, Planté‐Bordeneuve V, Lozeron P, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2012;79(8):785‐92. [PUBMED: 22843282] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keohane D, Schwartz J, Gundapaneni B, Stewart M, Amass L. Tafamidis delays disease progression in patients with early stage transthyretin familial amyloid polyneuropathy: additional supportive analyses from the pivotal trial. Amyloid 2017;24(1):30‐6. [PUBMED: 28393570] [DOI] [PubMed] [Google Scholar]
- Keohane D, Schwartz J, Gundapaneni B, Stewart M, Amass L. Tafamidis reduces disease progression in patients with transthyretin familial amyloid polyneuropathy: Supportive post‐hoc analyses of a pivotal trial. European Journal of Neurology 2015;22(Suppl 1):413. [EMBASE: 71933792] [Google Scholar]
- Keohane D, Schwartz J, Gundapaneni B, Stewart M, Amass L. Tafamidis reduces disease progression in patients with transthyretin familial amyloid polyneuropathy: supportive post‐hoc analyses of a pivotal trial. Orphanet Journal of Rare Diseases 2015;10(Suppl 1):P11. [EMBASE: 613537301] [Google Scholar]
- Suhr OB, Conceicao I, Karayal O, Ericzon BG. Nutritional status and autonomic function in clinical trials of tafamidis for transthyretin familial polyneuropathy. European Journal of Neurology 2012;19(Suppl 1):775. [EMBASE: 70939980] [Google Scholar]
- Suhr OB, Conceicao IM, Karayal ON, Mandel FS, Huertas PE, Ericzon B. Post hoc analysis of nutritional status in patients with transthyretin familial amyloid polyneuropathy: impact of tafamidis. Neurology and Therapy 2014;3(2):101‐12. [PUBMED: 26000226] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Adams 2015 {published data only}
- Adams D, Coelho T, Conceicao I, Waddington Cruz M, Schmidt H, Buades J, et al. Phase 2 open‐label extension (OLE) study of patisiran for the treatment of hereditary ATTR (hATTR) amyloidosis: 24‐month safety and efficacy in subgroup of patients with cardiac involvement. European Journal of Heart Failure 2017;19(Suppl S1):19. [Google Scholar]
- Adams D, Coelho T, Conceicao I, Waddington‐Cruz M, Schmidt H, Buades J, et al. Phase 2 open‐label extension (OLE) study of patisiran with or without a TTR stabilizer for the treatment of hereditary ATTR (hATTR) amyloidosis with polyneuropathy. European Journal of Neurology 2017;24(Suppl 1):31‐2. [Google Scholar]
- Adams D, Coelho T, Conceicao I, Waddington‐Cruz M, Schmidt H, Buades J, et al. Phase 2 open‐label extension (OLE) study of patisiran, an investigational RNA interference (RNAi) therapeutic for the treatment of hereditary attr amyloidosis with polyneuropathy. Value in Health 2017;20(5):A211‐2. [Google Scholar]
- Adams D, Suhr O, Conceicao I, Waddington‐Cruz M, Schmidt H, Buades J, et al. Phase 2 open‐label extension (OLE) study of patisiran, an investigational siRNA agent for familial amyloidotic polyneuropathy (FAP). Orphanet Journal of Rare Diseases 2015;10:1‐2. [Google Scholar]
Barroso 2017 {published data only}
- Barroso FA, Judge DP, Ebede B, Li H, Stewart M, Amass L, et al. Long‐term safety and efficacy of tafamidis for the treatment of hereditary transthyretin amyloid polyneuropathy: results up to 6 years. Amyloid 2017;24(3):194‐204. [DOI] [PubMed] [Google Scholar]
Coelho 2013 {published data only}
- Coelho T, Maia LF, Silva AM, Waddington‐Cruz M, Planté‐Bordeneuve V, Suhr OB, et al. Long‐term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. Journal of Neurology 2013;260(11):2802‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gillmore 2015 {published data only}
- Gillmore JD, Falk RH, Maurer MS, Hanna M, Karsten V, Vest J, et al. Phase 2, open‐label extension (OLE) study of revusiran, an investigational RNAi therapeutic for the treatment of patients with transthyretin cardiac amyloidosis. Orphanet Journal of Rare Diseases 2015;10:1‐2. [Google Scholar]
Gundapaneni 2018 {published data only}
- Gundapaneni B, Sultan MB, Keohane DJ, Schwartz J. Tafamidis delays disease progression comparably across Val30Met and non‐Val30Met genotypes in transthyretin familial amyloid polyneuropathy. Journal of the Peripheral Nervous System 2017;22(3):294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundapaneni BK, Sultan MB, Keohane DJ, Schwartz JH. Tafamidis delays disease progression comparably across Val30Met and non‐Val30Met genotypes in transthyretin familial amyloid polyneuropathy. Orphanet Journal of Rare Diseases 2017;12(Suppl 1):165. [Google Scholar]
- Gundapaneni BK, Sultan MB, Keohane DJ, Schwartz JH. Tafamidis delays neurological progression comparably across Val30Met and non‐Val30Met genotypes in transthyretin familial amyloid polyneuropathy. European Journal of Neurology 2018;25(3):464‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Maurer 2018 {published data only}
- Maurer MS, Elliott P, Merlini G, Shah SJ, Waddington‐Cruz M, Flynn A, et al. Design and rationale of the phase 3 ATTR‐ACT clinical trial (Tafamidis in Transthyretin Cardiomyopathy Clinical Trial). Circulation: Heart Failure 2017;10(6):e003815. [DOI] [PubMed] [Google Scholar]
- Maurer MS, Schwartz JH, Gundapaneni B, Perry M, Merlini G, Waddington‐Cruz M, et al. Treatment for patients with transthyretin amyloid cardiomyopathy. New England Journal of Medicine 2018;379(11):1007‐16. [DOI] [PubMed] [Google Scholar]
Plante‐Bordeneuve 2018 {published data only}
- Dasgupta NR, Falk RH, Drachman BM, Maurer MS, Whelan CJ, Brannagan T, et al. Safety of inotersen treatment in patients with transthyretin amyloid cardiomyopathy. Journal of the American College of Cardiology 2019;73(9 Suppl 1):909. [Google Scholar]
- Plante‐Bordeneuve V, Brannagan T, Wang A, Coelho T, Waddington‐Cruz M, Polydefkis M, et al. Long‐term efficacy and safety of Inotersen in patients with hereditary transthyretin (hATTR) amyloidosis treated in the open‐label extension of the phase‐3 study NEURO‐TTR. European Journal of Neurology 2018;25(Suppl 2):30. [Google Scholar]
Russo 2013 {published data only}
- Russo M, Obici L, Calabrese D, Piscosquito G, Luigetti M, Manganelli F, et al. Neurological data collection protocol on tafamidis meglumine treatment for familial amyloid polyneuropathy: Preliminary data on patients enrolled by the Italian network. Journal of the Peripheral Nervous System 2013;18:S99. [Google Scholar]
- Russo M, Stancanelli C, Gentile L, Toscano A, Vita G, Mazzeo A. Safety and tolerability of orally administered tafamidis meglumine in TTR FAP: Preliminary data at 3‐month follow‐up. Journal of the Peripheral Nervous System 2012;17:S48‐9. [Google Scholar]
Sah 2011 {published data only}
- Sah DWY, Chen Q, Costelha S, Butler J, Fishman S, Rossomando A, et al. ALN‐TTR, an RNAI therapeutic for the treatment of transthyretin amyloidosis. Nucleic Acid Therapeutics 2011;21(5):A55‐6. [Google Scholar]
Suhr 2015 {published data only}
- Suhr OB, Coelho T, Buades J, Pouget J, Conceicao I, Berk J, et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi‐dose study. Orphanet Journal of Rare Diseases 2015;10:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Waddington‐Cruz 2016 {published data only}
- Waddington‐Cruz M, Amass L, Keohane D, Schwartz J, Gundapaneni B, Li H. Early intervention with tafamidis provides long‐term benefit in delaying neurological progression in patients with transthyretin familial amyloid polyneuropathy. Orphanet Journal of Rare Diseases 2015;10(Suppl 1):P6. [Google Scholar]
- Waddington‐Cruz M, Amass L, Keohane D, Schwartz J, Li H, Gundapaneni B. Early intervention with tafamidis provides long‐term (5.5‐year) delay of neurologic progression in transthyretin hereditary amyloid polyneuropathy. Amyloid 2016;23(3):178‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington‐Cruz M, Amass L, Keohane DJ, Schwartz J, Li H, Gundapaneni B. Early intervention with tafamidis provides long‐term benefit in delaying neurological progression in patients with transthyretin familial amyloid polyneuropathy. Journal of the Peripheral Nervous System 2015;20(2):244. [Google Scholar]
Additional references
Abbott 2002
- Abbott CA, Carrington AL, Ashe H, Bath S, Every LC, Griffiths J, et al. The North‐West Diabetes Foot Care Study: incidence of, and risk factors for, new diabetic foot ulceration in a community‐based patient cohort. Diabetic Medicine 2002;19(5):377‐84. [DOI] [PubMed] [Google Scholar]
Adams 2013
- Adams D. Recent advances in the treatment of familial amyloid polyneuropathy. Therapeutic Advances in Neurological Disorders 2013;6(2):129‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Adams 2014
- Adams A, Théaudin M, Cauquil C, Algalarrondo V, Slama M. FAP neuropathy and emerging treatments. Current Neurology and Neuroscience Reports 2014;14(3):435‐46. [DOI] [PubMed] [Google Scholar]
Andrade 1952
- Andrade C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain 1952;75(3):408–27. [DOI] [PubMed] [Google Scholar]
Beck 1988
- Beck AT, Steer RA, Garbin MG. Psychometric properties of the Beck Depression Inventory: twenty‐five years of evaluation. Clinical Psychology Review 1988;8(1):77‐100. [Google Scholar]
Bril 1999
- Bril V. NIS‐LL: the primary measurement scale for clinical trial endpoints in diabetic peripheral neuropathy. European Neurology 1999;41(Suppl 1):8‐13. [DOI] [PubMed] [Google Scholar]
Cardoso 2006
- Cardoso I, Saraiva MJ. Doxycycline disrupts transthyretin amyloid: evidence from studies in a FAP transgenic mice model. FASEB Journal 2006;20(2):234‐9. [DOI] [PubMed] [Google Scholar]
Cardoso 2010
- Cardoso I, Martins D, Ribeiro T, Merlini G, Saraiva MJ. Synergy of combined Doxycycline/TUDCA treatment in lowering Transthyretin deposition and associated biomarkers: studies in FAP mouse models. Journal of Translational Medicine 2010;8:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Castro 2016
- Castro J, Miranda B, Castro I, Carvalho M, Conceição I. The diagnostic accuracy of Sudoscan in transthyretin familial amyloid polyneuropathy. Clinical Neurophysiology 2016;127(5):2222‐7. [DOI] [PubMed] [Google Scholar]
Chiang 2015
- Chiang MC, Tseng MT, Pan CL, Chao CC, Hsieh ST. Progress in the treatment of small fiber peripheral neuropathy. Expert Review of Neurotherapeutics 2015;15(3):305‐13. [DOI] [PubMed] [Google Scholar]
Coelho 2017
- Coelho T, Vinik A, Vinik EJ, Tripp T, Packman J, Grogan DR. Clinical measures in transthyretin familial amyloid polyneuropathy. Muscle & Nerve 2017;55(3):323‐32. [DOI] [PubMed] [Google Scholar]
Coutinho 1980
- Coutinho P, Martins da Silva A, Lopes Lima J, Resende Barbosa A. Forty years of experience with type I amyloid neuropathy. Review of 483 cases. In: Glenner G, Costa P, Freitas A editor(s). Amyloid and Amyloidosis. Amsterdam: Execerpta Medica, 1980:88‐98. [Google Scholar]
Covidence [Computer program]
- Veritas Health Innovation. Covidence. Melbourne, Australia: Veritas Health Innovation, Version accessed 10 January 2017.
Denier 2007
- Denier C, Ducot B, Husson H, Lozeron P, Adams D, Meyer L, et al. A brief compound test for assessment of autonomic and sensory‐motor dysfunction in familial amyloid polyneuropathy. Journal of Neurology 2007;254(12):1684–8. [DOI] [PubMed] [Google Scholar]
Dubrey 2015
- Dubrey S, Ackermann E, Gillmore J. The transthyretin amyloidoses: advances in therapy. Postgraduate Medical Journal 2015;91(1078):439‐48. [DOI] [PubMed] [Google Scholar]
Dyck 1995
- Dyck PJ, Litchy WJ, Lehman KA, Hokanson JL, Low PA, O'Brien PC. Variables influencing neuropathic endpoints: the Rochester Diabetic Neuropathy Study of Healthy Subjects. Neurology 1995;45(6):1115‐21. [DOI] [PubMed] [Google Scholar]
Dyck 1997
- Dyck PJ, Davies JL, Litchy WJ, O’Brien PC. Longitudinal assessment of diabetic polyneuropathy using a composite score in the Rochester Diabetic Neuropathy Study cohort. Neurology 1997;49(1):229‐39. [DOI] [PubMed] [Google Scholar]
Dyck 2017
- Dyck PJ, Kincaid JC, Dyck PJ, Chaudhry V, Goyal NA, Alves C, et al. Assessing mNIS+7Ionis and international neurologists' proficiency in a familial amyloidotic polyneuropathy trial. Muscle & Nerve 2017;56(5):901‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ferreira 2012
- Ferreira N, Saraiva MJ, Almeida MR. Epigallocatechin‐3‐gallate as a potential therapeutic drug for TTR‐related amyloidosis: "in vivo" evidence from FAP mice models. PLoS One 2012;7(1):e29933. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hamilton 1960
- Hamilton M. A rating scale for depression. Journal of Neurology, Neurosurgery & Psychiatry 1960;23:56‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Hund 2012
- Hund E. Familial amyloidotic polyneuropathy: current and emerging treatment options for transthyretin‐mediated amyloidosis. The Application of Clinical Genetics 2012;5:37‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kempler 2011
- Kempler P, Amarenco G, Freeman R, Frontoni S, Horowitz M, Stevens M, et al. Management strategies for gastrointestinal, erectile, bladder, and sudomotor dysfunction in patients with diabetes. Diabetes/Metabolism Research and Reviews 2011;27(7):665‐77. [DOI] [PubMed] [Google Scholar]
Kollmer 2015
- Kollmer J, Hund E, Hornung B, Hegenbart U, Schönland SO, Kimmich C, et al. In vivo detection of nerve injury in familial amyloid polyneuropathy by magnetic resonance neurography. Brain 2015;138(Pt 3):549‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kollmer 2017
- Kollmer J, Sahm F, Hegenbart U, Purrucker JC, Kimmich C, Schönland SO, et al. Sural nerve injury in familial amyloid polyneuropathy: MR neurography vs clinicopathologic tools. Neurology 2017;89(5):475‐84. [DOI] [PubMed] [Google Scholar]
Kristen 2012
- Kristen AV, Lehrke S, Buss S, Mereles D, Steen H, Ehlermann P, et al. Green tea halts progression of cardiac transthyretin amyloidosis: an observational report. Clinical Research in Cardiology 2012;101(10):805‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lacomblez 1989
- Lacomblez L, Bouche P, Bensimon G, Meininger V. A double‐blind, placebo‐controlled trial of high doses of gangliosides in amyotrophic lateral sclerosis. Neurology 1989;39(12):1635–7. [DOI] [PubMed] [Google Scholar]
Lozeron 2013
- Lozeron P, Théaudin M, Mincheva Z, Ducot B, Lacroix C, Adams D, French Network for FAP (CORNAMYL). Effect on disability and safety of Tafamidis in late onset of Met30 transthyretin familial amyloid polyneuropathy. European Journal of Neurology 2013;20(12):1539‐45. [DOI] [PubMed] [Google Scholar]
Lunn 2016
- Lunn M, Hanna M, Howard R, Parton M, Rahman S, Reilly M, et al. Nerve and muscle disease. In: Clarke C, Howard R, Rossor M, Shorvon S editor(s). Neurology: a Queen Square Textbook. 2nd Edition. Oxford, UK: Wiley Blackwell, 2016:391‐473. [Google Scholar]
Mariani 2015
- Mariani LL, Lozeron P, Théaudin M, Mincheva Z, Signate A, Ducot B, et al. Genotype‐phenotype correlation and course of transthyretin familial amyloid polyneuropathies in France. Annals of Neurology 2015;78(6):901‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Masuda 2017
- Masuda T, Ueda M, Suenaga G, Misumi Y, Tasaki M, Izaki A, et al. Early skin denervation in hereditary and iatrogenic transthyretin amyloid neuropathy. Neurology 2017;88(23):2192‐7. [DOI] [PubMed] [Google Scholar]
Maule 2007
- Maule S, Papotti G, Naso D, Magnino C, Testa E, Veglio F. Orthostatic hypotension: evaluation and treatment. Cardiovascular & Hematological Disorders Drug Targets 2007;7(1):63‐70. [DOI] [PubMed] [Google Scholar]
Merlini 2013
- Merlini G, Planté‐Bordeneuve V, Judge DP, Schmidt H, Obici L, Perlini S, et al. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non‐Val30Met transthyretin amyloidosis. Journal of Cardiovascular Translational Research 2013;6(6):1011‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group. Preferred Reporting Items for Systematic Reviews and Meta‐Analyses: The PRISMA Statement. PLoS Medicine 2009;6(7):e1000097. [DOI: 10.1371/journal.pmed1000097] [DOI] [PMC free article] [PubMed] [Google Scholar]
Murphy 2011
- Murphy SM, Herrmann DN, McDermott MP, Scherer SS, Shy ME, Reilly MM, et al. Reliability of the CMT neuropathy score (second version) in Charcot‐Marie‐Tooth disease. Journal of the Peripheral Nervous System 2011;16(3):191‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nasr 2017
- Nasr SH, Dasari S, Mills JR, Theis JD, Zimmermann MT, Fonseca R, et al. Hereditary lysozyme amyloidosis variant p.Leu102Ser associates with unique phenotype. Journal of the American Society of Nephrology 2017;28(2):431‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Obici 2012
- Obici L, Cortese A, Lozza A, Lucchetti J, Gobbi M, Palladini G, et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid 2012;19(Suppl 1):34‐6. [DOI] [PubMed] [Google Scholar]
Planté‐Bordeneuve 2011
- Planté‐Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurology 2011;10(12):1086‐97. [DOI] [PubMed] [Google Scholar]
Planté‐Bordeneuve 2018
- Planté‐Bordeneuve V. Transthyretin familial amyloid polyneuropathy: an update. Journal of Neurology 2018;265(4):976‐83. [DOI] [PubMed] [Google Scholar]
PNS 1995
- Peripheral Nerve Society. Diabetic polyneuropathy in controlled clinical trials: consensus report of the Peripheral Nerve Society. Annals of Neurology 1995;38(3):478–82. [DOI: 10.1002/ana.410380323; PUBMED: 7668839] [DOI] [PubMed] [Google Scholar]
Rabin 2001
- Rabin R, Charro F. EQ‐5D: a measure of health status from the EuroQolGroup. Annals of Medicine 2001;33(5):337‐43. [DOI] [PubMed] [Google Scholar]
Rapezzi 2010
- Rapezzi C, Quarta CC, Riva L, Longhi S, Gallelli I, Lorenzini M, et al. Transthyretin‐related amyloidoses and the heart: a clinical overview. Nature Reviews. Cardiology 2010;7(7):398‐408. [DOI] [PubMed] [Google Scholar]
Rapezzi 2013
- Rapezzi C, Quarta CC, Obici L, Perfetto F, Longhi S, Salvi F, et al. Disease profile and differential diagnosis of hereditary transthyretin‐related amyloidosis with exclusively cardiac phenotype: an Italian perspective. European Heart Journal 2013;34(7):520‐8. [DOI] [PubMed] [Google Scholar]
Reig 2015
- Reig N, Ventura S, Salvadó M, Gámez J, Insa R. SOM0226, a repositioned compound for the treatment of TTR amyloidosis. Orphanet Journal of Rare Disease 2015;10(Suppl 1):P9. [Google Scholar]
Review Manager 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Rowczenio 2014
- Rowczenio DM, Noor I, Gillmore JD, Lachmann HJ, Whelan C, Hawkins PN, et al. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Human Mutation 2014;35(9):E2403‐12. [10.1002/humu.22619] [DOI] [PubMed] [Google Scholar]
Rowczenio 2015
- Rowczenio D, Wechalekar A. Mutations in transthyretin gene. amyloidosismutations.com/mut‐attr.php 2015.
Sales‐Luís 1990
- Sales‐Luís ML, Galvão M, Sousa G, Alves MM, Serrão R. Plasma exchanges on: the treatment of familial amyloidotic polyneuropathy, Portuguese type. Familial amyloidotic polyneuropathy and other transthyretin related disorders. Arquivos de Medicina 1990;3:361‐9. [Google Scholar]
Sant'Anna 2016
- Sant'Anna R, Gallego P, Robinson LZ, Pereira‐Henriques A, Ferreira N, Pinheiro F, et al. Repositioning tolcapone as a potent inhibitor of transthyretin amyloidogenesis and associated cellular toxicity. Nature Communications 2016;7:10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
SF‐36v2 2011
- User's manual for the SF‐36v2 survey. 3rd ed. Lincoln (RD), Quality Metric Incorporated 2011.
Shy 2005
- Shy ME, Blake J, Krajewski K, Fuerst DR, Laura M, Hahn AF, et al. Reliability and validity of the CMT neuropathy score as a measure of disability. Neurology 2005;64(7):1209‐14. [DOI] [PubMed] [Google Scholar]
Sipe 2014
- Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJ, et al. Nomenclature 2014: amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid 2014;21(4):221‐4. [DOI] [PubMed] [Google Scholar]
Steen 1983
- Steen L, Ek B. Familial amyloidosis with polyneuropathy. A long‐term follow‐up of 21 patients with special reference to gastrointestinal symptoms. Acta Medica Scandinavica 1983;214:387–97. [PubMed] [Google Scholar]
Suanprasert 2014
- Suanprasert N, Berk JL, Benson MD, Dyck PJ, Klein CJ, Gollob JA, et al. Retrospective study of a TTR FAP cohort to modify NIS+7 for therapeutic trials. Journal of the Neurological Sciences 2014;344(1‐2):121‐8. [DOI] [PubMed] [Google Scholar]
Suhr 1984
- Suhr O, Danielsson A, Holmgren G, Steen L. Malnutrition and gastrointestinal dysfunction as prognostic factors for survival in familial amyloidotic polyneuropathy. Journal of Internal Medicine 1994;235(5):479‐85. [DOI] [PubMed] [Google Scholar]
Tashima 1999
- Tashima K, Ando Y, Terazaki H, Yoshimatsu S, Suhr OB, Obayashi K, et al. Outcome of liver transplantation for transthyretin amyloidosis: follow‐up of Japanese familial amyloidotic polyneuropathy patients. Journal of the Neurological Sciences 1999;171(1):19‐23. [DOI] [PubMed] [Google Scholar]
Valleix 2012
- Valleix S, Gillmore JD, Bridoux F, Mangione PP, Dogan A, Nedelec B, et al. Hereditary Systemic Amyloidosis Due to Asp76Asn Variant β2‐Microglobulin. New England Journal of Medicine 2012;366(24):2276‐83. [DOI] [PubMed] [Google Scholar]
van Nes 2011
- Nes SI, Vanhoutte EK, Doorn PA, Hermans M, Bakkers M, Kuitwaard K, et al. Rasch‐built Overall Disability Scale (R‐ODS) for immune‐mediated peripheral neuropathies. Neurology 2011;76(4):337‐45. [DOI] [PubMed] [Google Scholar]
Vinik 2005
- Vinik EJ, Hayes RP, Oglesby A, Bastyr E, Barlow P, Ford‐Molvik SL, et al. The development and validation of the Norfolk QOL‐DN, a new measure of patients’ perception of the effects of diabetes and diabetic neuropathy. Diabetes Technology & Therapeutics 2005;7(3):497‐508. [DOI] [PubMed] [Google Scholar]
Ware 1992
- Ware JE Jr, Sherbourne CD. The MOS 36‐item short‐form health survey (SF‐36). I. Conceptual framework and item selection. Medical Care 1992;30(6):473–83. [PubMed] [Google Scholar]
Yamamoto 2007
- Yamamoto S, Wilczek H, Nowak G, Larsson M, Oksanen A, Iwata T, et al. Liver transplantation for familial amyloidotic polyneuropathy (FAP): a single‐center experience over 16 years. American Journal of Transplantation 2007;7(11):2597–604. [DOI] [PubMed] [Google Scholar]
Yates 1980
- Yates JW, Chalmer B, McKegney FP. Evaluation of patients with advanced cancer using the Karnofsky performance status. Cancer 1980;45:2220–4. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Magrinelli 2016
- Magrinelli F, Fabrizi GM, Santoro L, Manganelli F, Cavallaro T, Zanette G, et al. Pharmacological treatment for familial amyloid neuropathy. Cochrane Database of Systematic Reviews 2016, Issue 10. [DOI: 10.1002/14651858.CD012395] [DOI] [PMC free article] [PubMed] [Google Scholar]