

Given the current barrage of attention and research investments toward the production of next-generation fuels and chemicals, of which 2,3-butanediol (2,3-BD) produced by nonpathogenic Paenibacillus species is perhaps one of the most vigorously pursued, tools for engineering Paenibacillus species are intensely sought after. Exopolysaccharide (EPS) production during 2,3-BD fermentation constitutes a problem during downstream processing. Specifically, EPS negatively impacts 2,3-BD separation from the fermentation broth, thereby increasing the overall cost of 2,3-BD production. The results presented here demonstrate that inactivation of the levansucrase gene in P. polymyxa leads to diminished EPS accumulation. Additionally, a new method for an EPS assay and a simple protocol employing protoplasts for enhanced transformation of P. polymyxa were developed. Overall, although our study shows that levan is not the only EPS produced by P. polymyxa, it represents a significant first step toward developing cost-effective 2,3-BD fermentation devoid of EPS-associated complications during downstream processing.
KEYWORDS: 2,3-butanediol; levansucrase; homologous recombination; exopolysaccharide; polysaccharide polymerase; levan; acetoin; protoplast; cell efficiency
ABSTRACT
The formation of exopolysaccharides (EPSs) during 2,3-butanediol (2,3-BD) fermentation by Paenibacillus polymyxa increases medium viscosity, which in turn presents considerable technical and economic challenges to 2,3-BD downstream processing. To eliminate EPS production during 2,3-BD fermentation, we used homologous recombination to disable the EPS biosynthetic pathway in P. polymyxa. The gene which encodes levansucrase, the major enzyme responsible for EPS biosynthesis in P. polymyxa, was successfully disrupted. The P. polymyxa levansucrase null mutant produced 2.5 ± 0.1 and 1.2 ± 0.2 g/liter EPS on sucrose and glucose, respectively, whereas the wild type produced 21.7 ± 2.5 and 3.1 ± 0.0 g/liter EPS on the same substrates, respectively. These levels of EPS translate to 8.7- and 2.6-fold decreases in EPS formation by the levansucrase null mutant on sucrose and glucose, respectively, relative to that by the wild type, with no significant reduction in 2,3-BD production. Inactivation of EPS biosynthesis led to a considerable increase in growth. On glucose and sucrose, the cell biomass of the levansucrase null mutant (8.1 ± 0.8 and 6.5 ± 0.3 g/liter, respectively) increased 1.4-fold compared to that of the wild type (6.0 ± 0.1 and 4.6 ± 0.3 g/liter, respectively) grown on the same substrates. Evaluation of the genetic stability of the levansucrase null mutant showed that it remained genetically stable over fifty generations, with no observable decrease in growth or 2,3-BD formation, with or without antibiotic supplementation. Hence, the P. polymyxa levansucrase null mutant has potential for use as an industrial biocatalyst for a cost-effective large-scale 2,3-BD fermentation process devoid of EPS-related challenges.
IMPORTANCE Given the current barrage of attention and research investments toward the production of next-generation fuels and chemicals, of which 2,3-butanediol (2,3-BD) produced by nonpathogenic Paenibacillus species is perhaps one of the most vigorously pursued, tools for engineering Paenibacillus species are intensely sought after. Exopolysaccharide (EPS) production during 2,3-BD fermentation constitutes a problem during downstream processing. Specifically, EPS negatively impacts 2,3-BD separation from the fermentation broth, thereby increasing the overall cost of 2,3-BD production. The results presented here demonstrate that inactivation of the levansucrase gene in P. polymyxa leads to diminished EPS accumulation. Additionally, a new method for an EPS assay and a simple protocol employing protoplasts for enhanced transformation of P. polymyxa were developed. Overall, although our study shows that levan is not the only EPS produced by P. polymyxa, it represents a significant first step toward developing cost-effective 2,3-BD fermentation devoid of EPS-associated complications during downstream processing.
INTRODUCTION
The finite nature of fossil fuels, recurrent instability in oil price, and environmental concerns associated with crude oil consumption create an urgent need to develop sustainable alternatives to fossil fuels and their derivatives. An example is the industrial platform chemical 2,3-butanediol (2,3-BD), which is generated via cracking of petroleum-derived hydrocarbons (e.g., butenes) and has wide industrial applications. As a feedstock chemical, 2,3-BD can be used to produce 1,3-butadiene (1,3-BD), the monomer of synthetic rubber (1, 2), and methyl ethyl ketone (MEK), a fuel additive with higher heat of combustion than ethanol, and it has massive potential as a feedstock for a host of numerous pharmaceuticals, cosmetics, paints, and food preservatives (3, 4). It is also a solvent from which resins and lacquers are produced (1, 2).
Several microorganisms possess the metabolic machinery to convert carbohydrates to 2,3-BD. However, 2,3-BD is produced via a mixed acid fermentation pathway, where other products such as ethanol, acetoin, lactic, formic and acetic acids, and exopolysaccharides (EPSs) are cogenerated. Among these coproducts, EPS poses a significant challenge to the recovery and downstream processing of 2,3-BD postfermentation. Thus, we focused on genetic manipulation of Paenibacillus polymyxa, a nonpathogenic 2,3-BD producer, for reduced EPS production. Furthermore, P. polymyxa synthesizes levo-2,3-BD, the most desirable 2,3-BD isomer, because it can be easily dehydrated to 1,3-BD (5, 6). The other 2,3-BD isomers (meso- and dextro-2,3-BD) are the major fermentation products of the pathogenic 2,3-BD producers, such as Klebsiella spp., Enterobacter aerogenes, and Serratia marcescens (1, 2).
During 2,3-BD fermentation, P. polymyxa synthesizes the exopolysaccharide levan, a β-(2,6)-linked fructose polymer (7). Typically, P. polymyxa produces more than 50 g/liter EPS during fermentation (8). Consequently, EPS clogs bioreactor lines, impedes mixing of the fermentation broth, and, most importantly, complicates 2,3-BD downstream processing. Hence, extra purification steps would be required to remove EPS prior to 2,3-BD extraction at an industrial scale, adding to the overall production cost.
Levan is the most well-characterized EPS synthesized by P. polymyxa, and levansucrase plays a key role in levan production by P. polymyxa by serving as a conduit for the transfer of fructosyl residues to a growing levan chain (9). In its natural habitat, P. polymyxa produces EPS as a means of attaching itself to plant roots (10–12). Comparative analysis of nucleotide and protein sequences of P. polymyxa DSM 365 levansucrase relative to those from other P. polymyxa strains, whose genomes have been completely sequenced and annotated, was performed to ascertain the number of copies of levansucrase present in P. polymyxa DSM 365. Comparisons were conducted due to the absence of complete genome information on P. polymyxa DSM 365 (see Table S1 in the supplemental material). Our analyses revealed that P. polymyxa DSM 365 possesses a single copy of the levansucrase gene, with an open reading frame of 1,497 bp.
In this study, we targeted the P. polymyxa DSM 365 levansucrase gene for inactivation. Using double-crossover homologous recombination, we conducted pioneering work on the inactivation of the levansucrase gene of P. polymyxa DSM 365. Also, because acetic acid often accumulates in genetically modified solvent-producing Gram-positive bacteria and calcium supplementation of the growth medium mitigates acetic acid-induced stress (13), the culture medium in this study was supplemented with calcium chloride. While the P. polymyxa levansucrase null mutant developed in this study showed no activity for levansucrase, it produced EPS, albeit at miniscule levels, when grown on sucrose and on glucose. Our findings provide insights into the scope of the pathways for EPS formation in relation to substrate utilization during 2,3-BD fermentation by P. polymyxa. Furthermore, our results present a new context to consider toward the development of industrial strains for economical 2,3-BD production.
RESULTS
The levansucrase gene of P. polymyxa was successfully inactivated by double-crossover homologous recombination. An erythromycin resistance gene was inserted between upstream (210 bp) and downstream (213 bp) fragments of the levansucrase gene, creating a 1,224-bp levansucrase inactivation construct (Fig. 1). The inactivation construct included a stop codon downstream of the LevFragA sequence, which is immediately followed by a ribosome binding site (RBS) and a spacer sequence upstream of the erythromycin resistance gene. The construct also includes a transcription terminator sequence downstream of the erythromycin resistance gene that precedes the LevFragB sequence. The levansucrase inactivation construct is shown in Fig. 1. The stop codon, the RBS, the spacer sequence, and the transcription terminator sequences were organized in such a manner that only the erythromycin resistance gene was transcribed into mRNA. This approach eliminates any additional metabolic burden on P. polymyxa that may result from the inactivation construct. Following transformation of P. polymyxa, PCR screening of the resulting colonies using LevFragA_fwd/Erm_rev2, Erm_fwd/Erm_rev2, and Erm_fwd/LevFragB_rev primer pairs showed that the levansucrase null mutant possessed the LevFragA-ERM, ERM, and ERM-LevFragB sequences, corresponding to 1,008, 800, and 1,015 bp, respectively (Fig. 2), thus confirming successful double crossover.
FIG 1.

The levansucrase inactivation construct. (A) Construction of pGEM 7Zf-LevFragA-Erm-LevFragB from pGEM 7Zf(+). (B) Schematic representation of the double crossover leading to inactivation of the levansucrase gene using pGEM 7Zf-LevFragA-Erm-LevFragB. (C) Gel image showing levansucrase gene, LevFragA, LevFragB, ERM, LevFragA-ERM, and LevFragA-ERM-LevFragB gene fragments during the generation of the levansucrase inactivation construct.
FIG 2.

Inactivation of the levansucrase gene in P. polymyxa. (A) Screening for levansucrase null mutants by colony PCR. Lanes show bands corresponding to the LevFragA-ERM, ERM, and ERM-LevFragB fragments of the recombinant plasmid (or the plasmid control) from colonies 1 and 2. (B) Precipitated and floating EPS from cultures of the wild type and the levansucrase null mutant grown on sucrose medium.
Levansucrase inactivation disrupts EPS formation and enhances the growth of P. polymyxa.
Batch 2,3-BD fermentations were conducted with sucrose and glucose substrates to evaluate EPS formation by the levansucrase null mutant. The initial screening test showed that while inactivation of the levansucrase gene drastically reduced EPS production (Fig. 2B), the ability of the levansucrase null mutant to reassimilate acetic acid, a characteristic feature of P. polymyxa during fermentation (5), appeared to be disrupted (Fig. 3; see also Fig. S4 in the supplemental material). Therefore, to promote tolerance to and possibly enhance reassimilation of acetic acid, cultures were supplemented with CaCl2. In the shaken-flask fermentation, cultures were supplemented with 0, 0.2, and 0.4 g/liter CaCl2, whereas fermentations in 5-liter bioreactors were supplemented with 0 and 0.4 g/liter CaCl2 as control and treatment, respectively. With the levansucrase null mutant, supplementing the growth medium with CaCl2 did not lead to increased culture pH (Fig. 3B). Nonetheless, CaCl2 supplementation improved the fermentation characteristics of the levansucrase null mutant and the wild type (Fig. 3; see also Fig. S5 and S6).
FIG 3.

Fermentation profiles and levansucrase activity of the P. polymyxa levansucrase null mutant (Lev MT or LM) and the wild type (WT) grown on sucrose supplemented with 0, 0.2 and 0.4 g/liter CaCl2 (X represents no levansucrase activity).
With and without CaCl2 supplementation, EPS production in cultures of the levansucrase null mutant decreased at least 5.8- and 8.7-fold in the shaken flask and bioreactor, respectively, compared to that in the wild type in a sucrose-based medium (Table 1; Fig. 2B). Conversely, on sucrose, EPS production by the wild type increased at least 1.2-fold when 2,3-BD fermentation was scaled up to 2 liters in a 5-liter bioreactor (Tables 1 and 2). In addition, no measurable levansucrase activity was observed in the levansucrase null mutant, whereas more than 0.6 unit of levansucrase activity per mg of protein was detected in cultures of the wild type (Fig. 3F). The lack of levansucrase activity in the null mutant confirms successful inactivation of the levansucrase gene in P. polymyxa. However, despite the lack of levansucrase activity, the null mutant synthesized 2 to 3 g/liter EPS when grown on sucrose (Table 1). The presence of small amounts of EPS in cultures of the null mutant suggests that P. polymyxa produces EPS forms other than levan. Remarkably, EPS formation by the wild type and the levansucrase null mutant grown on glucose was significantly lower than that on sucrose. The wild type synthesized at least 4-fold more EPS on sucrose than on glucose. Conversely, levels of EPS production by the null mutant were similar on both sucrose and glucose substrates (Tables 1 and 3). Overall, EPS production by the levansucrase null mutant decreased at least 1.9-fold compared to that of the wild type when both strains were grown on glucose (Table 3; Fig. S4 to S6). Interestingly, levansucrase activity was not detected in either the wild type or the levansucrase null mutant grown on glucose.
TABLE 1.
Substrate consumed, growth, maximum products, 2,3-BD yield, and productivity during sucrose fermentation by the P. polymyxa DSM 365 wild type and levansucrase null mutanta
| Parameter | 0 g/liter CaCl2 |
0.2 g/liter CaCl2 |
0.4 g/liter CaCl2 |
|||
|---|---|---|---|---|---|---|
| WT | LM | WT | LM | WT | LM | |
| Sucrose consumed (g/liter) | 101.9 ± 0.3 A | 84.7 ± 3.0 B | 110.1 ± 0.5 A | 84.5 ± 1.2 A | 107.9 ± 0.4 A | 92.8 ± 1.3 B |
| Residual sucrose (g/liter) | 2.49 ± 0.0 A | 25.4 ± 3.4 B | NDb A | 25.6 ± 1.7 B | ND A | 17.3 ± 1.8 B |
| Residual glucose (g/liter) | 5.7 ± 0.1 A | ND B | ND A | ND A | 2.2 ± 0.0 A | ND B |
| Cell dry weight (g/liter) | 4.6 ± 0.3 A | 6.5 ± 0.3 B | 7.0 ± 0.7 A | 8.5 ± 0.7 B | 6.4 ± 0.6 A | 8.6 ± 0.6 B |
| 2,3-BD (g/liter) | 32.6 ± 0.7 A | 30.9 ± 2.3 A | 36.3 ± 2.0 A | 35.7 ± 1.7 A | 36.1 ± 2.5 A | 37.4 ± 0.9 A |
| 2,3-BD production cell efficiencyc | 10.2 ± 0.3 A | 7.3 ± 0.0 B | 5.2 ± 0.2 A | 5.0 ± 0.4 A | 11.2 ± 1.1 A | 5.1 ± 0.3 B |
| EPS (g/liter) | 17.4 ± 2.2 A | 3.0 ± 0.6 B | 18.4 ± 2.7 A | 2.9 ± 0.5 B | 17.6 ± 0.36 A | 2.9 ± 0.2 B |
| 2,3-BD yield (g/g) | 0.32 ± 0.01 A | 0.35 ± 0.02 A | 0.33 ± 0.02 A | 0.42 ± 0.03 B | 0.33 ± 0.02 A | 0.40 ± 0.02 A |
| 2,3-BD productivity (g/liter/h) | 0.68 ± 0.01 A | 0.62 ± 0.05 A | 1.01 ± 0.05 A | 1.04 ± 0.14 A | 0.75 ± 0.05 A | 0.78 ± 0.02 A |
| Acetoin (g/liter) | 2.6 ± 0.2 A | 4.1 ± 0.4 B | 21.6 ± 3.8 A | 5.0 ± 0.5 B | 1.3 ± 0.4 B | 6.0 ± 0.6 B |
| Ethanol (g/liter) | 5.7 ± 0.3 A | 6.8 ± 0.1 B | 5.8 ± 0.2 A | 5.1 ± 0.4 A | 5.8 ± 1.1 A | 5.5 ± 0.5 A |
| Acetic acid (g/liter) | 1.5 ± 0.3 A | 2.8 ± 0.1 B | 1.8 ± 0.1 A | 3.9 ± 1.2 B | 1.2 ± 0.3 A | 5.5 ± 0.2 B |
Fisher’s least significant difference (LSD) pairwise comparisons between wild type (WT) and levansucrase mutant (LM) were conducted. Values are means ± SD. Means with different uppercase letters across each row vary significantly at P < 0.05.
ND, not detected.
2,3-BD production cell efficiency was determined as the ratio of maximum 2,3-BD produced to the corresponding P. polymyxa cell growth.
TABLE 2.
Maximum substrate consumed, 2,3-BD and EPS formed, 2,3-BD yield, and productivity during fermentation of sucrose and glucose by the P. polymyxa wild type and levansucrase null mutant in a 5-liter bioreactora
| Parameter | 0 g/liter CaCl2 |
0.4 g/liter CaCl2 |
||
|---|---|---|---|---|
| WT | LM | WT | LM | |
| Sucrose based | ||||
| Sucrose consumed (g/liter) | 100.2 ± 1.4 A | 83.3 ± 7.8 A | 122.0 ± 2.6 A | 94.3 ± 2.8 B |
| 2,3-BD (g/liter) | 33.7 ± 0.7 A | 32.9 ± 0.5 A | 32.9 ± 0.5 A | 35.4 ± 1.2 A |
| EPS (g/liter) | 21.7 ± 2.5 A | 2.5 ± 0.1 B | 27.3 ± 0.4 A | 2.7 ± 0.0 B |
| 2,3-BD yield (g/g) | 0.34 ± 0.00 A | 0.39 ± 0.04 A | 0.27 ± 0.00 A | 0.37 ± 0.00 B |
| 2,3-BD productivity (g/liter/h) | 1.41 ± 0.03 A | 1.37 ± 0.02 A | 1.37 ± 0.02 A | 0.74 ± 0.03 B |
| Glucose based | ||||
| Glucose consumed (g/liter) | 100.00 ± 6.5 A | 93.4 ± 5.7 A | 101.8 ± 6.5 A | 100.6 ± 6.2 A |
| 2,3-BD (g/liter) | 34.5 ± 3.8 A | 39.5 ± 2.2 A | 39.8 ± 0.8 A | 42.2 ± 0.8 A |
| EPS (g/liter) | 3.1 ± 0.0 A | 1.2 ± 0.2 B | 2.1 ± 0.3 A | 1.3 ± 0.1 B |
| 2,3-BD yield (g/g) | 0.35 ± 0.06 A | 0.42 ± 0.05 A | 0.39 ± 0.02 A | 0.42 ± 0.03 A |
| 2,3-BD productivity (g/liter/h) | 0.72 ± 0.08 A | 0.66 ± 0.04 A | 0.66 ± 0.01 A | 0.88 ± 0.02 B |
Fisher’s LSD pairwise comparisons between wild type (WT) and levansucrase mutant (LM) were conducted. Values are means ± SD. Means with different uppercase letters across each row vary significantly at P < 0.05.
TABLE 3.
Substrate consumed, growth, maximum products, 2,3-BD yield, and productivity during glucose fermentation by the P. polymyxa DSM 365 wild type and levansucrase null mutanta
| Parameter | 0 g/liter CaCl2 |
0.2 g/liter CaCl2 |
0.4 g/liter CaCl2 |
|||
|---|---|---|---|---|---|---|
| WT | LM | WT | LM | WT | LM | |
| Glucose consumed (g/liter) | 85.6 ± 3.9 A | 87.8 ± 1.9 A | 96.1 ± 3.1 A | 105.0 ± 0.0 A | 94.7 ± 2.7 A | 106.7 ± 2.5 B |
| Residual glucose (g/liter) | 23.7 ± 3.2 A | 21.45 ± 1.2 A | 13.2 ± 2.3 A | 4.3 ± 0.7 B | 14.5 ± 2.0 A | 2.5 ± 1.8 B |
| Cell dry weight (g/liter) | 6.0 ± 0.1 A | 8.1 ± 0.8 B | 7.5 ± 0.1 A | 10.3 ± 0.8 B | 7.0 ± 0.2 A | 10.8 ± 0.4 B |
| 2,3-BD (g/liter) | 30.7 ± 1.2 A | 29.8 ± 2.0 A | 34.2 ± 2.7 A | 39.0 ± 1.3 A | 34.5 ± 3.4 A | 39.4 ± 4.4 A |
| 2,3-BD production cell efficiencyb | 5.7 ± 0.2 A | 4.2 ± 0.3 B | 5.2 ± 0.5 A | 3.9 ± 0.3 B | 5.8 ± 0.3 A | 4.1 ± 0.4 B |
| EPS (g/liter) | 5.5 ± 0.4 A | 2.27 ± 0.1 B | 4.2 ± 0.2 A | 2.4 ± 0.3 B | 5.4 ± 0.5 A | 2.8 ± 0.2 B |
| 2,3-BD yield (g/g) | 0.36 ± 0.01 A | 0.34 ± 0.02 A | 0.36 ± 0.02 A | 0.37 ± 0.01 A | 0.37 ± 0.04 A | 0.38 ± 0.04 A |
| 2,3-BD productivity (g/liter/h) | 0.51 ± 0.02 A | 0.57 ± 0.09 A | 0.71 ± 0.06 A | 1.62 ± 0.05 B | 0.72 ± 0.07 A | 1.64 ± 0.19 B |
| Acetoin (g/liter) | 3.3 ± 0.0 A | 2.4 ± 0.4 B | 10.1 ± 2.1 A | 2.8 ± 0.4 B | 8.8 ± 1.1 A | 2.6 ± 0.3 B |
| Ethanol (g/liter) | 5.8 ± 0.1 A | 7.1 ± 0.3 B | 5.6 ± 0.4 A | 8.7 ± 0.7 B | 5.4 ± 0.5 A | 9.4 ± 0.2 B |
| Acetic acid (g/liter) | 0.0 ± 0.0 A | 1.7 ± 0.0 B | 0.6 ± 0.0 A | 2.5 ± 0.9 B | 0.0 ± 0.0 B | 2.9 ± 0.1 B |
Fisher’s LSD pairwise comparisons between wild type (WT) and levansucrase mutant (LM) were conducted. Values are means ± SD. Means with different uppercase letters across each row vary significantly at P < 0.05.
2,3-BD production cell efficiency was determined as the ratio of maximum 2,3-BD produced to the corresponding P. polymyxa cell growth.
The growth of P. polymyxa increased considerably following levansucrase gene inactivation. In CaCl2-unsupplemented cultures, growth of the null mutant increased at least ∼1.4-fold with either sucrose or glucose compared to that of the wild type (Tables 1 and 3). However, addition of 0.2 and 0.4 g/liter CaCl2 to the sucrose- and glucose-based media increased the growth of the null mutant by 1.2- and 1.3-fold and ∼1.4- and 1.5-fold, respectively, compared to that of the wild type (Tables 1 and 3).
2,3-BD production and fermentation characteristics of the levansucrase null mutant.
(i) Without calcium chloride supplementation (shaken flasks). The overarching goal of this study was to eliminate EPS production as a means of improving the overall characteristics of 2,3-BD fermentation by P. polymyxa, particularly the downstream processes. However, toward possible deployment of the resulting strain in 2,3-BD fermentation, it was imperative to evaluate the 2,3-BD profile of the levansucrase null mutant. Thus, batch 2,3-BD fermentations were performed in shaken flasks and in 5-liter bioreactors, using sucrose and glucose as the substrates. Overall, inactivation of the levansucrase gene in P. polymyxa did not lead to significantly increased 2,3-BD production. In shaken flasks, despite the remarkable reduction in EPS production by the levansucrase null mutant, 2,3-BD concentration decreased ∼1.1-fold relative to that of the wild type when both strains were grown on sucrose not supplemented with CaCl2 (Fig. 3; Table 1). Notably, inactivation of the levansucrase gene in P. polymyxa appeared to disrupt sucrose uptake, because sucrose utilization by the null mutant decreased 1.2-fold relative to that by the wild type (Fig. 3; Table 3). Nonetheless, while 2,3-BD yield by the null mutant increased marginally (∼1.1-fold greater than the wild type) when both strains were grown on sucrose, the 2,3-BD production cell efficiency by the null mutant (product per cell) decreased significantly (Fig. 3; Table 1). When grown on CaCl2-unsupplemented glucose as the substrate, glucose utilization and 2,3-BD productivity of the levansucrase null mutant increased ∼1.03- and 1.12-fold, respectively, relative to that of the wild type (Fig. 3; Table 3). Conversely, in the same medium, 2,3-BD concentration and yield decreased marginally. In this medium, the 2,3-BD concentration and yield of the levansucrase null mutant were ∼1.03- and 1.06-fold lower, respectively, than for the wild type (Fig. 3; Table 3). Furthermore, the levansucrase null mutant was grown in wheat straw hydrolysates (WSH), a lignocellulosic biomass hydrolysate (LBH), and evaluated for 2,3-BD production. Interestingly, the levansucrase null mutant simultaneously utilized the sugars in WSH (glucose, xylose, and arabinose) and produced the same level of 2,3-BD as the glucose control when 60% WSH was used (14). Additionally, P. polymyxa showed high tolerance for lignocellulose-derived microbial inhibitory compounds (LDMICs), a characteristic that served P. polymyxa well in the conversion of WSH to 2,3-BD (14).
(ii) Supplementation of the growth medium with calcium chloride (shaken flasks). Following a previous study where we demonstrated that Ca2+ exerts positive effects on fermentative solvent production by recruiting assorted mechanisms (15), cultures of the P. polymyxa strains were supplemented with CaCl2. Generally, supplementation of the fermentation broth with 0.2 or 0.4 g/liter CaCl2 marginally enhanced 2,3-BD production, albeit to a slightly greater extent for the levansucrase null mutant (Fig. 3; Tables 1 and 3). In the presence of CaCl2, 2,3-BD production by the levansucrase null mutant and the wild type increased ∼1.2- and ∼1.1-fold, respectively, on sucrose and ∼1.3- and ∼1.1-fold, respectively, on glucose relative to that in the control culture with no CaCl2 supplementation (Tables 1 and 3). This likely stems from the increased sucrose and glucose utilization by the mutant and wild type following CaCl2 supplementation. Consequently, 2,3-BD yield and productivity by the levansucrase null mutant and wild type increased at least ∼1.1- and ∼1.3-fold on sucrose, respectively, whereas little to no improvement was observed with glucose (Tables 1 and 3).
(iii) 2,3-BD fermentation in a 5-liter bioreactor. Generally, 2,3-BD production improved more on glucose than on the sucrose when fermentation was scaled up from 30 ml to 2 liters in volume (Table 2). As observed with the shaken flasks, 2,3-BD production by the levansucrase null mutant decreased ∼1.02-fold relative to that by the wild type when both strains were grown on sucrose without CaCl2 supplementation. Conversely, 2,3-BD production by the levansucrase null mutant was ∼1.14-fold higher than the wild type when both strains were grown on glucose (Fig. 4 and Table 2). The slightly lower 2,3-BD production by the levansucrase null mutant on sucrose is ascribable to lower sucrose utilization (∼1.2-fold lower than the wild type) (Table 2). Nevertheless, the 2,3-BD yield of the levansucrase null mutant on sucrose was ∼1.15-fold greater than that of the wild type (Table 2). Furthermore, CaCl2 supplementation improved sucrose utilization by the levansucrase null mutant by 1.13-fold, resulting in a 1.08-fold increase in 2,3-BD concentration (Table 2). The 2,3-BD concentration and yield in CaCl2-supplemented cultures of the levansucrase null mutant grown on sucrose were ∼1.08- and ∼1.37-fold greater than those produced by the wild type (Table 2). The 2,3-BD concentration (Fig. 4) and yield during fermentation by the wild type increased by ∼1.14- and 1.06-fold, respectively, compared to those for the wild type grown in the control medium with no CaCl2 supplementation. Overall, the levansucrase null mutant converted sucrose to 2,3-BD with a diminished ability to produce EPS (Fig. 2B).
FIG 4.

Scale-up of 2,3-BD fermentation to a 2-liter working volume in a 5-liter bioreactor. Fermentation product profiles of the P. polymyxa levansucrase null mutant and the wild type grown on sucrose (A to C) and glucose (D to F) media supplemented with 0 and 0.4 g/liter CaCl2.
Expression levels of levansucrase and putative EPS genes in the wild type and the levansucrase null mutant.
Successful inactivation of the levansucrase gene in P. polymyxa was further confirmed by quantitative reverse transcriptase PCR (qRT-PCR), by measuring the mRNA transcript levels of the levansucrase and erythromycin resistance genes in the levansucrase null mutant and in the wild type. The mRNA samples obtained from cells grown on both sucrose- and glucose-based media were used. Levansucrase gene mRNA transcripts were not detected in the levansucrase null mutant grown on either sucrose or glucose (Fig. 5). Conversely, high levels of mRNA transcripts of the erythromycin resistance gene were detected in the levansucrase null mutant (Fig. 5). Thus, the levansucrase gene in the P. polymyxa was successfully inactivated.
FIG 5.
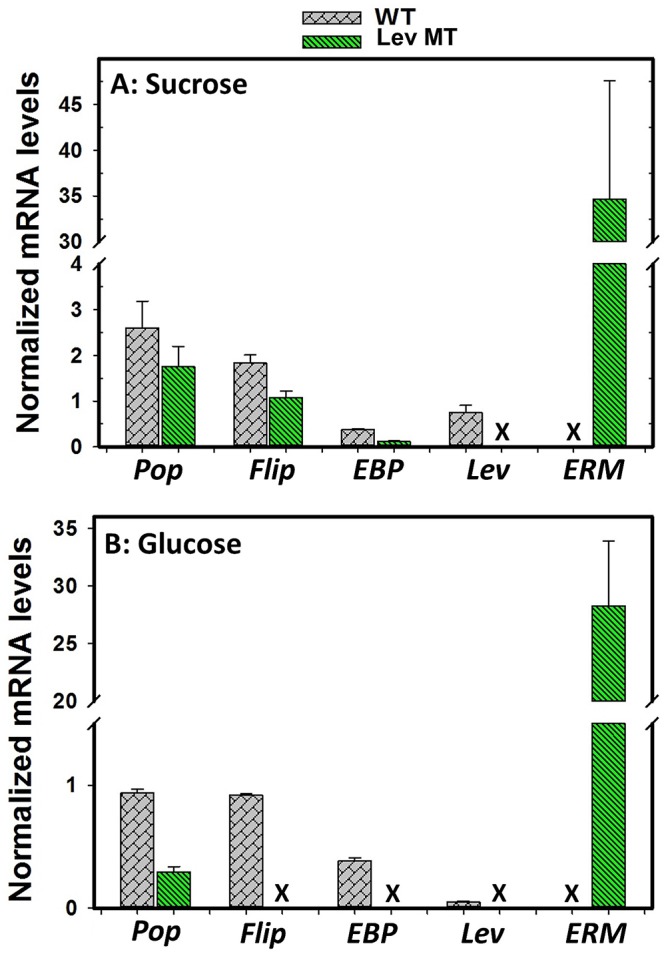
Comparisons of mRNA transcript levels of polysaccharide polymerase (Po), flippase (Fli or Flip), exopolysaccharide biosynthesis protein (EB), levansucrase (Lev), and erythromycin (ERM or ER) genes of P. polymyxa wild type (WT) and the levansucrase null mutant (Lev MT) grown on sucrose (A) and glucose (B) (X represents no mRNA detection).
Inactivation of levansucrase gene did not completely block EPS accumulation in P. polymyxa cultures. To delineate the likely source(s) of the observed EPSs in cultures of the levansucrase null mutant, qRT-PCR was used to probe the mRNA transcript levels of other putative exopolysaccharide biosynthesis genes in P. polymyxa, namely, polysaccharide polymerase (Pop), flippase (flip), and exopolysaccharide biosynthesis protein (EBP) genes. While Pop, flip, and EBP genes were expressed by the levansucrase null mutant grown on sucrose, it expressed only Pop when grown on glucose substrate (Fig. 5). The protein products of the Pop and EBP genes are putatively associated with biosynthesis and assembly of nonlevan exopolysaccharide chains (16). Different studies suggest that flippase might be membrane specific, where it assembles EPS repeating units and then translocates complete EPS across the membrane to the exterior environment (Fig. 6) (16, 17). The expression of Pop, flip, and EBP by the levansucrase null mutant suggests that P. polymyxa has the ability to synthesize other forms of EPS when grown on either sucrose or glucose.
FIG 6.

Schematic representation of likely pathways for EPS production by P. polymyxa. The diagram was conceived based on the results of our enzymatic assays, qRT-PCR data, and annotated metabolic network model of P. polymyxa on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. The red and black fonts or lines represent annotated and putative EPS pathways in P. polymyxa, respectively. 1, Levansucrase; 2, sugar PTS; 3, phosphoglucomutase; 4, UTP-glucose-1-phosphate uridylyltransferase; 5, glucose-1-phosphate thymidylyltransferase; 6, galactose-1-phosphate uridylyltransferase; 7, UDP-glucose-6-dehydrogenase; 8, dTDP-glucose-4,6-dehydratase; 9, dTDP-4-dehydrorhamnose-3,5-epimerase; 10, dTDP-4-dehydrorhamnose reductase; 11, polymerase synthase; 12, exopolysaccharide biosynthesis protein; 13, flippase. The dashed black arrows represent steps in EPS polymerization from nucleotide sugars. The genes that code for enzymes (numbers 11, 12, and 13) in blue font were analyzed by qRT-PCR.
Stability of the levansucrase null mutant.
Successful knockout of the levansucrase gene in P. polymyxa was confirmed by PCR, restriction digest analysis, levansucrase activity assay, qRT-PCR, antibiotic selection, plasmid curing, and genetic stability tests. As shown in Fig. S7 and S8, the maximum growth for each tested generation of the levansucrase null mutant under erythromycin pressure was to an optical density at 600 nm (OD600) of between 11.0 and 13.9, whereas without erythromycin, the growth ranged from an OD600 of 12.5 to 15.1. EPS concentrations at each tested tenth generation (G0 to G50) of the levansucrase null mutant, with and without erythromycin supplementation, were similar. The maximum 2,3-BD production by the null mutant grown under antibiotic pressure ranged from 35.3 to 39.4 g/liter, whereas without antibiotics, it ranged from 36.1 to 39.0 g/liter (see Table S2). Colony PCR and replica plating techniques were further employed to characterize the levansucrase null mutant for antibiotic resistance over multiple generations (G0 to G50). Notably, the levansucrase null mutant developed in this study retained resistance to erythromycin after 50 generations of growth, with or without erythromycin supplementation (see Fig. S9 and S10).
DISCUSSION
EPS production during 2,3-BD fermentation is undesirable for several reasons, most importantly because it increases the viscosity of the fermentation broth, which impairs mixing efficiency (8). Also, EPS accumulation negatively impacts 2,3-BD downstream processing by interfering with product purification and recovery, thereby increasing the overall cost of production. Therefore, this study was aimed at developing a mutant strain of P. polymyxa with diminished ability to synthesize EPS.
Inactivation of levansucrase gene disrupts EPS biosynthesis in P. polymyxa.
Knocking out the levansucrase gene in P. polymyxa resulted in a drastic reduction in EPS formation (Fig. 3C and 4C; see also Fig. S4C, S5C, and S6C in the supplemental material), indicating that levansucrase is indeed a key player in EPS biosynthesis in this organism. Additionally, the results presented here confirm that the open reading frame (ORF) targeted for knockout in the P. polymyxa DSM 365 shotgun sequence encodes levansucrase. The reduction in EPS biosynthesis by the levansucrase null mutant was more pronounced during growth on sucrose than on glucose. This is ascribable to the fact that EPS formation is more strongly favored by sucrose, because levansucrase hydrolyzes sucrose to release glucose and fructose (18) and then links most of the fructose molecules to form EPS. EPS production was almost completely abolished in the levansucrase null mutant, and levansucrase mRNA levels and activity were not detected in the levansucrase null mutant grown on either glucose or sucrose. Conversely, levansucrase mRNA levels and activity were detected in the wild type grown on sucrose (Fig. 3F). However, although low levels of levansucrase mRNA transcripts were detected in the wild type grown on glucose, there was no measurable levansucrase activity (Fig. 5), suggesting that sucrose likely plays a specific role in levansucrase gene expression. In fact, sucrose-mediated induction of levansucrase gene expression has been suggested by other authors (19, 20), although transcript levels were not quantified. Furthermore, levansucrase mRNA transcripts were observed in Zymomonas mobilis grown on both glucose and fructose (21), indicating that different mechanisms may govern levansucrase expression among species, which use EPS in functionally distinct ways that are adapted to their diverse ecological niches. Further studies are required to better delineate the role of sucrose in levansucrase gene expression in P. polymyxa.
Despite levansucrase gene inactivation, which was confirmed by PCR, restriction digest analysis, levansucrase activity assay, antibiotic sensitivity assay and replica plating, qRT-PCR, and plasmid curing, some EPSs were detected in cultures of both the wild type and the levansucrase null mutant grown on glucose, albeit in significantly small amounts in the levansucrase null mutant. Thus, P. polymyxa likely produces more than one type of extracellular polysaccharide, with sucrose favoring levan biosynthesis while glucose supports the production of other, uncharacterized polysaccharides (Fig. 6). These may include the EPS known to contain glucose, mannose, galactose, glucuronic acid, and pyruvate in the ratio of ∼10:7:4:4:1, with a repeating unit of at least three α-linked and four β-linked sugar residues (22). The formation of multiple EPSs is not unusual among EPS-producing microorganisms such as Bacillus spp., Z. mobilis, Leuconostoc mesenteroides, Agrobacterium radiobacter, Xanthomonas campestris, and Pseudomonas aeruginosa, as they produce alginate, xanthan, curdlan, or dextran from different sugars (23–29). Interestingly, the genes encoding the enzymes polysaccharide polymerase, flippase, and exopolysaccharide biosynthesis protein, which are putatively involved in the synthesis of other EPSs, were expressed in both the levansucrase null mutant and the wild type (Fig. 5 and 6). Physicochemical characterization of the small amounts of nonlevan EPS obtained in cultures of the levansucrase null mutant of P. polymyxa will shed greater light on the structural form of this EPS and the specific enzymes and genes responsible for its production.
Although EPS production was significantly lower in the levansucrase null mutant than in the wild type when both strains were grown on sucrose, this did not translate into significant increases in 2,3-BD production. More importantly, though, inactivation of EPS production did not impair 2,3-BD production. We infer, therefore, that EPS and 2,3-BD production are not metabolically linked in P. polymyxa—hence, disruption of the accumulation of one product does not affect the other. The usefulness of the levansucrase null mutant rests significantly on the capacity to retain 2,3-BD production following inactivation of EPS biosynthesis. Indeed, by abolishing levan accumulation in P. polymyxa, we have constructed a 2,3-BD-producing strain without the attendant burdens of levan on downstream processing. Notably, when grown on glucose, wild-type P. polymyxa produces significantly less EPS than wild-type cultures grown on sucrose. As a result, although inactivation of the levansucrase gene led to a considerable reduction in EPS accumulation, this reduction was far more significant in cultures of the null mutant grown on sucrose than in glucose-grown cultures, because lesser amounts of EPS are normally produced with glucose. The growth profile of the levansucrase null mutant suggests that levansucrase inactivation likely relieved growth-limiting machinery in the levansucrase null mutant, leading to the increase in growth observed for this strain. Unexpectedly, ethanol production increased in the levansucrase null mutant relative to that in the wild type only when both strains were grown on glucose. It is not clear why this occurred; thus, this warrants further investigation. Nevertheless, this result highlights ethanol biosynthesis as a veritable candidate for future inactivation in the levansucrase null mutant toward developing a 2,3-BD-overproducing strain.
Levansucrase inactivation in P. polymyxa interferes with acetic acid reassimilation.
Typically, 2,3-BD is produced via a mixed acid pathway, which results in the accumulation of acetic, formic, and lactic acids during fermentation. Acetic acid is then reassimilated with a concomitant increase in culture pH. However, elevated acetic acid accumulation was consistently observed for the null mutant under all culture conditions studied (Fig. 3B and E, S4B and E, S5E, and S6E), which may have contributed to the lower 2,3-BD production cell efficiency in the null mutant, suggesting that levansucrase may influence acetic acid reassimilation in some way. To our knowledge, there are no previous reports of possible links between EPS biosynthesis and acetic acid reassimilation in P. polymyxa or other 2,3-BD producers. Also, this indicates that EPS biosynthesis may play a broader role in the biology of P. polymyxa and perhaps other microorganisms that produce both 2,3-BD and EPS. Furthermore, it is likely that acetic acid accumulation in the levansucrase null mutant is a secondary or cascade effect stemming from downstream effectors of levansucrase not directly involved in EPS biosynthesis. Other solvent-producing, biphasic Gram-positive bacteria typically produce acids and then reabsorb them during solvent formation (13). Consequently, in solvent-producing Gram-positive bacteria, disruption of the native physiology tends to engender acid accumulation. Similar effects have been reported in Clostridium beijerinckii NCIMB 8052 following knockdown of acetoacetate decarboxylase (13) and in Clostridium acetobutylicum ATCC 842 (30).
Despite acetic acid accumulation and the attendant drop in culture pH, the levansucrase null mutant grew better than the wild type under all conditions tested, although growth was improved by CaCl2 in cultures of both strains grown on glucose or sucrose. The greater growth of the levansucrase null mutant cultures at relatively low pH suggests that levansucrase inactivation mitigates acid-mediated stress in P. polymyxa. Whereas this effect is not due to increased acetic acid reassimilation by the levansucrase null mutant (Fig. 3 and Tables 1 to 3) and seems more likely a mitigation of acetic acid-mediated stress, it warrants further investigation to delineate the underlying mechanism of action. In fact, supplementation of the growth medium with CaCl2 also did not lead to increased acetic acid reassimilation by the levansucrase null mutant (Fig. 3 and Tables 1 to 3). This suggests that the beneficial effect observed with CaCl2 (increased growth) was not a result of activation of enzymes involved in acetic acid reassimilation. On the contrary, CaCl2 appeared to confer resistance to acetic acid toxicity to the levansucrase null mutant of P. polymyxa. Notably, CaCl2 supplementation enhanced the growth of both the levansucrase null mutant and the wild type of P. polymyxa on both glucose and sucrose. Indeed, the underlying mechanism could be related to previous findings which demonstrated beneficial effects of calcium on fermenting microorganisms (13, 15). Calcium exerts global effects on cellular metabolism, sugar utilization, and stress mitigation in C. beijerinckii NCIMB 8052, including upregulation of heat shock and other proteins that participate in the repair of damaged or aberrant proteins, DNA synthesis, transcription, and repair of DNA damage (15). Additionally, calcium has been implicated in the stabilization of bacterial membranes, reducing the effect of membrane-damaging factors such as acids (30, 31). Although CaCl2 increased the growth of the levansucrase null mutant more than that of the wild type, the increased growth did not lead to a significant increase in 2,3-BD production by the levansucrase null mutant when compared to that of the wild type.
Stability of P. polymyxa levansucrase null mutant.
The stability of microbial strains intended for industrial bioprocesses is critical for uniform and consistent product accumulation. This is particularly important when genetically modified strains are used. The stability of the levansucrase null mutant over 50 generations (Fig. S7 and S8) indicates that it should be suited to an industrial production setting.
Concluding remarks.
The levansucrase gene was successfully inactivated in P. polymyxa via double-crossover homologous recombination, leading to a stable and faster-growing strain with significantly reduced EPS production. The ability of the levansucrase null mutant to retain the capacity for 2,3-BD biosynthesis on glucose makes it an attractive platform for generating a 2,3-BD-overproducing strain capable of fermenting lignocellulosic biomass hydrolysates. Although preliminary experiments indicate that the null mutant is capable of fermenting lignocellulosic biomass hydrolysates (wheat straw hydrolysate), further studies at a larger scale are needed to fully evaluate the utility of this strain for large-scale production of 2,3-BD from lignocellulosic biomass hydrolysates. This is because lignocellulosic biomass hydrolysates harbor a catalog of compounds, including furfural, 5-hydroxymethyl furfural, syringaldehyde, vanillin, ferulic acid, and vanillic acid, which are severely toxic to fermenting microorganisms. In fact, the inhibitory effects of these compounds largely account for the failure of several cellulosic ethanol scale-up efforts during the past decade. Whereas the levansucrase null mutant appeared to thrive in wheat straw hydrolysates, specific evaluation of its tolerance to the most abundant and toxic inhibitory compounds would be crucial toward efforts demonstrating the utility of this strain in large-scale production of 2,3-BD with lignocellulosic hydrolysates as the substrates. Unexpectedly, inactivation of EPS biosynthesis led to an increase in ethanol production. Hence, the levansucrase null mutant may be further optimized for possible large-scale production of 2,3-BD by inactivating ethanol biosynthesis. This would abolish undesirable diversion of carbons to a coproduct (ethanol) of lesser value and improve the 2,3-BD production cell efficiency. Lastly, supplementation of the growth medium with small amounts of CaCl2 mitigated growth-related metabolic disruptions that arose following metabolic engineering of P. polymyxa, a typical occurrence in solvent-producing Gram-positive bacteria.
MATERIALS AND METHODS
Microorganisms and culture conditions.
Paenibacillus polymyxa DSM 365 was procured from the German Collection of Microorganisms and Cell Culture, Braunschweig, Germany (DSMZ [Deutsche Sammlung von Mikroorganismen und Zellkulturen]). The lyophilized stock was reactivated by inoculating in Luria-Bertani (LB) broth, grown overnight (12 h), and then stored as glycerol stock (50% sterile glycerol) at −80°C. The microorganisms, vectors, and enzymes used in this study are shown in Table 4.
TABLE 4.
List of microorganisms, vectors, and enzymes used in this study and their respective characteristics and sources
| Strain, vector, or enzyme | Characteristic or genotype | Source |
|---|---|---|
| Strains | ||
| E. coli JM109 | endA1 recA1 gyrA96 relA1 | Promega Corporation |
| P. polymyxa DSM 365 | Wild type | DSMZ, Germany |
| P. polymyxa DSM 365 Lev null mutant | Δlev; Ermr | This study |
| Vectors | ||
| pMutin | Ermr | Bacillus Genetic Stock Center, OH |
| pGEM 7Zf(+) | f1 oriC lacZ; Ampr | Promega Corporation |
| Enzymes | ||
| GXL DNA polymerase | High fidelity, amplifies GC-rich templates | Takara Clontech |
| BamHI | New England BioLabs | |
| XhoI | New England BioLabs | |
| T4 DNA ligase | New England BioLabs |
Genomic DNA extraction and amplification of components of the levansucrase inactivation construct.
To extract genomic DNA (gDNA), P. polymyxa cells were grown in a previously described preculture medium (32) to an OD600 of 0.7. The cells were harvested by centrifugation at 10,000 × g and 4°C for 10 min and then suspended in Tris-HCl-EDTA (TE) buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). Zirconia/silica beads (0.1 mm; BioSpec Products, Inc., Bartlesville, OK) were added to the cells to a final concentration of 50% (wt/vol). The cells in the mixture were lysed using a TissueLyser LT (Qiagen, Hilden, Germany) at 50 oscillations/s for 2 min. The cell lysate was centrifuged at 10,000 × g and 4°C for 10 min, and the supernatant was transferred to a clean 2.0-ml Eppendorf tube. Phenol-chloroform genomic DNA (gDNA) extraction (33) was used to isolate P. polymyxa gDNA, which was then washed with 70% (vol/vol) ethanol. The gDNA was air dried at room temperature and reconstituted in 20 μl of nuclease-free water. The gDNA was stored at −20°C until use.
PCR primers targeting the levansucrase gene in P. polymyxa were designed using Eurofins genomics primer design tools (https://www.eurofinsgenomics.eu/en/ecom/tools/pcr-primer-design). The levansucrase gene was amplified with incorporation of XhoI and BamHI restriction sites at the appropriate locations. The design was such that the PCR primers would amplify short sequences (∼210 bp) up- and downstream of the levansucrase gene, designated LevFragA and LevFragB, respectively. Primers used to generate the constructs and their characteristics are shown in Table 5. First, the entire levansucrase gene was amplified from P. polymyxa genomic DNA using the LevFragA_fwd and LevFragB_rev primer pair. Then, LevFragA and LevFragB fragments were amplified from the gel-purified levansucrase gene by using the LevFragA_fwd/LevFragA_rev and LevFragB_fwd/LevFragB_rev primer pairs, respectively. An erythromycin resistance gene was amplified from the plasmid pMutin (BGSC, Columbus, OH) with an Erm_fwd and Erm_rev1 or Erm_rev2 primer pair, designed to incorporate ribosomal binding site (RBS), spacer, and transcription termination sequences. Erm_fwd and Erm_rev1 were first used to amplify the erythromycin resistance gene from pMutin. The gel-purified erythromycin resistance gene was reamplified using Erm_fwd and Erm_rev2, which ensured complete addition of the entire transcription termination sequence downstream of the erythromycin resistance gene sequence. Conventional PCR and splicing by overlap extension PCR (SOE-PCR) were carried out in a Bio-Rad iCycler thermal cycler (Bio-Rad, Hercules, CA) using PrimeStar GXL DNA polymerase (Clontech-TaKaRa, Mountain View, CA). A 50-μl reaction mix containing 5× PrimeStar GXL buffer (10 μl), deoxynucleoside triphosphates (dNTPs; 0.25 mM), primers (0.5 μM each), DNA template (∼5 ng/μl), and GXL DNA polymerase (1 μl) was used. The reaction conditions were (i) initial denaturation at 98°C for 2 min, (ii) 98°C for 20 s (1 cycle), (iii) hold at annealing temperature of primer pair for 30 s and 72°C for 1 min (35 cycles), (iv) final extension at 72°C for 10 min, and (v) hold at 4°C for 10 min (1 cycle). Nested PCR was employed for a one-step SOE-PCR with the following conditions: (i) initial denaturation at 98°C for 2 min, (ii) 98°C for 30 s, hold at annealing temperature of templates overlapping region for 30 s, and 72°C for 30 s (5 cycles), (iii) 98°C for 30 s, hold at annealing temperature of primer pair for 30 s, and 72°C for 30 s (30 cycles), (iv) final extension at 72°C for 5 min, and (v) hold at 4°C for 10 min. The annealing temperature of each primer pair was calculated from the primer with the lowest melting temperature.
TABLE 5.
List of primers used to generate the levansucrase inactivation construct and for qRT-PCR
| Primer name | Primer sequence (5′→3′)a |
|---|---|
| Levansucrase inactivation construct | |
| LevFragA_fwd | TGGGGATCCTTGAAGTTTAACAAATGGTTCAGTAAAGC |
| LevFragA_rev | CCTCCTAAACAGTTAGGACGGAACCTCATATTTCTCTTTGCC |
| LevFragB_fwd | ACTCTTATTTTTTTAATATTGTTTCATAGTGGCAATAACGTAGTCG |
| LevFragB_rev | GGGCTCGAGTTATTTCTTTCCATACTCATTTGGAG |
| Erm_fwd | TAACTGTTTAGGAGGACTGATAATATGAACAAAAATATAAAATATTCTCAAAAC |
| Erm_rev1 | TAAAAAAATAAGAGTTACCATTTATTACCTCCCGTTAAATAATAGATAAC |
| Erm_rev2 | GCCACTATGAAACAATATTAAAAAAATAAGAGTTACCATTTATTATTTCC |
| qRT-PCR | |
| Lev_Fwd | GTACAGCAAAGCGTCGGAAT |
| Lev_Rev | CCGGTTTCTGTTCCTGTGTT |
| Erythromycin_Fwd | GGTTGCTCTTGCACACTCAA |
| Erythromycin_Rev | CTGTGGTATGGCGGGTAAGT |
| Polysaccharide polymerase_Fwd | GCGTTCGTCGGTTTATCACT |
| Polysaccharide polymerase_Rev | GAATGCAGCCCTAGAACCTG |
| Flippase_Fwd | CGTTCCAAGCAGAAAGGAAG |
| Flippase_Rev | AGACAACAGCGAACCTGCTT |
| Exopolysaccharide biosynthesis protein_Fwd | GGTCACATTCTGGCCTGTCT |
| Exopolysaccharide biosynthesis protein_Rev | CTAAACAGCTTCGCCTTTGG |
| 16S rRNA_Fwd | GGCTTTCCAGCTACCTGTTG |
| 16S rRNA_Rev | ACGGCGTCTTCAAAGGAGTA |
Underlined sequences represent either restriction sites or RBS.
Subsequently, SOE-PCR was conducted to link LevFragA and ERM gene fragments to generate the LevFragA-ERM construct from LevFragA and ERM gene templates using the LevFragA_fwd and Erm_rev2 primer pair. The gel-purified LevFragA-ERM was used as a template alongside LevFragB to generate the inactivation construct, LevFragA-ERM-LevFragB, in another SOE-PCR using the LevFragA_fwd and LevFragB_rev primer pair.
Recombinant plasmid construction and transformation of competent E. coli JM109.
The LevFragA-ERM-LevFragB construct was ligated into pGEM 7Zf(+), a high-copy-number plasmid in Escherichia coli JM109 and nonreplicative in P. polymyxa. This plasmid possesses a filamentous phage f1 origin of replication recognized by E. coli but not by P. polymyxa. Consequently, pGEM 7Zf(+) is replicated into a circular single-stranded DNA (ssDNA) that enhances homologous recombination in vivo (34). The phage f1 origin of replication and the ability of pGEM 7Zf(+) to be replicated into stable circular DNA in E. coli are important for its application to gene inactivation via homologous recombination in P. polymyxa and other Gram-positive bacteria.
Next, pGEM 7Zf(+) and LevFragA-ERM-LevFragB were digested sequentially with XhoI and BamHI (New England BioLabs, Ipswich, MA) in a 50-μl reaction mixture consisting of CutSmart buffer (New England BioLabs, Ipswich, MA) and 0.02 μg/μl DNA. The mixture was incubated at 37°C for 1 h after addition of XhoI (1 μl), and for another 1 h following BamHI (1 μl) addition. The digested pGEM 7Zf(+) and LevFragA-ERM-LevFragB construct were independently gel purified and then ligated in a 20-μl reaction mixture to generate the pGEM 7Zf-LevFragA-ERM-LevFragB recombinant plasmid. The ligation reaction mixture consisted of 2 μl T4 DNA ligase buffer, 1 μl T4 DNA ligase (New England BioLabs, Ipswich, MA), and the gel-purified pGEM 7Zf(+) and LevFragA-ERM-LevFragB inserts in a 1:5 ratio, respectively. The reaction mixture was incubated overnight at 16°C followed by heating at 65°C for 10 min to quench the reaction.
The ligated pGEM 7Zf-LevFragA-ERM-LevFragB plasmid was chilled on ice for 20 min and then used to transform competent E. coli JM109 cells via heat shock at 42°C for 1 min, as previously described (35). The transformed E. coli JM109 cells were incubated at 37°C with shaking at 250 rpm for 1 h and then plated on LB agar supplemented with 50 μg/ml ampicillin, 20 mg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal), and 1 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG). The plates were incubated at 37°C overnight, after which white colonies were selected and screened for the presence of the correct insert via colony PCR and restriction digest analysis. Colonies with the correct insert were grown in LB broth supplemented with 50 μg/ml ampicillin for 12 h or to an OD600 of 0.9, and then the recombinant plasmid was isolated, purified, and stored at −20°C prior to use.
Electrotransformation of competent P. polymyxa protoplasts.
Following initial unsuccessful attempts to transform competent P. polymyxa cells with pGEM 7Zf-LevFragA-ERM-LevFragB by electroporation, competent P. polymyxa protoplasts were generated and used instead. To achieve this, the cell wall of P. polymyxa was removed according to a previously described method (36), with slight modification. Briefly, P. polymyxa cells were grown in tryptic soy broth (TSB) for 12 h until the OD600 reached 0.7. The cells were harvested and placed in 50-ml centrifuge tubes prechilled on ice for 20 min. The cells were washed twice with chilled 50 mM Tris-maleate buffer (pH 7.1) containing 2 mM dithiothreitol and then centrifuged at 1,000 × g and 4°C for 7 min. The cell pellets were harvested and resuspended in chilled (4°C) Tris-maleate buffer (pH 7.1) containing 0.6 M sucrose, 5 mM MgCl2, and 300 μg/ml lysozyme (Amresco, Solon, OH). The cell suspension was incubated in an ISOTEMP 220 water bath (Fisher Scientific, Pittsburg, PA) at 37°C for 60 min to generate P. polymyxa protoplasts. The protoplasts were harvested by centrifugation at 1,000 × g and 4°C for 7 min and were made competent by washing twice with 10% polyethylene glycol (PEG 8000). The protoplasts were then resuspended in 500 μl of 10% PEG 8000 to give a protoplast OD600 of 0.4. The competent P. polymyxa protoplasts were transformed with pGEM 7Zf-LevFragA-ERM-LevFragB via electroporation. Twenty microliters (100 μg DNA) of pGEM 7Zf-LevFragA-ERM-LevFragB was added to 100 μl of competent P. polymyxa protoplasts in a prechilled 0.2-cm electroporation cuvette, gently flicked with a finger to mix, and then placed on ice for 5 min. Electroporation was performed at 2.5 kV, 25 μF capacitance, and infinite resistance as previously described (37) in a Bio-Rad Gene Pulser Xcell electroporator (Bio-Rad, Hercules, CA). An electric pulse was delivered to the protoplasts once for between 2.5 and 4.1 ms. After electroporation, the protoplasts were placed on ice for 5 min, and then 500 μl of TSB was added and the mixture was incubated at 35°C for 6 h to allow for protoplast recovery. The recovered cells were plated on tryptic soy agar (TSA) supplemented with 35 μg/ml erythromycin and incubated at 35°C for 16 to 24 h. Colonies were picked, replated on fresh TSA plates supplemented with 35 μg/ml erythromycin, and then incubated at 35°C for 12 h. Fresh colonies were then selected, and the colony PCR technique was used to screen for the presence of the erythromycin resistance gene (38). Genomic DNA of colonies with the erythromycin resistance gene was extracted and used as the template to screen for the presence of LevFragA-ERM, ERM, and ERM-LevFragB fragments via PCR. The presence of all three fragments confirmed that the levansucrase gene was successfully inactivated via double-crossover homologous recombination. The resulting levansucrase null mutant was then plasmid cured using mitomycin C as previously described (38) to confirm the absence of pGEM 7Zf(+) in the cell.
Characterization of the P. polymyxa levansucrase null mutant.
The P. polymyxa levansucrase null mutant was characterized with respect to cell growth and EPS and 2,3-BD production. Batch fermentations were conducted in sucrose- and glucose-based media in 125-ml shaken flasks at 200 rpm and 35°C and then scaled up to 2 liters with an impeller speed of 300 rpm and 150 ml/min aeration rate in a 5-liter bioreactor at 35°C. For the fermentations, 50% glycerol stock (1 ml) of the P. polymyxa levansucrase null mutant was inoculated in 30 ml of preculture medium supplemented with 35 μg/ml erythromycin and incubated at 35°C and 200 rpm for 6 h until the OD600 reached 1.0 to 1.2. The actively growing P. polymyxa levansucrase null mutant (10% [vol/vol]) was inoculated in the fermentation medium containing 100 g/liter sucrose or glucose, supplemented with 35 μg/ml erythromycin. The preculture and fermentation medium components used in this study were as previously described (32). The fermentation medium was further supplemented with 0 to 0.4 g/liter CaCl2. The wild-type P. polymyxa culture was prepared as described for the levansucrase null mutant except without erythromycin supplementation. Batch shaken-flask 2,3-BD fermentations were conducted in loosely capped 125-ml Pyrex culture bottles with a 30-ml fermentation volume. All experiments were carried out in triplicate, and 2-ml samples were drawn at 0 h and then every 12 h until fermentation terminated at 72 h. Samples were analyzed for cell biomass, culture pH, and EPS, 2,3-BD, acetoin, acetic acid, and ethanol concentrations.
Quantitative reverse transcriptase PCR.
To further validate successful deletion of the levansucrase gene from the P. polymyxa genome, mRNA levels of the levansucrase gene were quantified by quantitative reverse transcriptase PCR (qRT-PCR) using cells grown on sucrose and glucose. In addition, qRT-PCR was used to confirm successful erythromycin gene integration in P. polymyxa. Specific primers (Table 5) for the disrupted region of levansucrase and erythromycin genes were used. Furthermore, owing to the presence of small amounts of EPS in cultures of the levansucrase null mutant (despite inactivation of the levansucrase gene), the mRNA levels of selected genes that putatively encode other EPS-producing enzymes, namely, flippase, polysaccharide polymerase, and exopolysaccharide biosynthesis protein, were quantified to ascertain their likely contributions to the remaining EPS. The specific primers used for qRT-PCR of these gene products are shown in Table 5. Total RNA was isolated from the wild type and the levansucrase null mutant after 12 h (point of maximum EPS accumulation) of growth using Tri reagent (Sigma, St. Louis, MO), according to the manufacturer’s protocol. The mRNA samples were used to encode cDNA, which was used for qRT-PCR as previously described (38). The mRNA expression levels of all the tested genes were normalized to that of P. polymyxa 16S rRNA (internal standard), and relative expression was determined using the threshold cycle (2-ΔΔCT) method (39).
Analytical methods.
Microbial cell growth was determined as cell dry weight by measuring the optical density (OD600) with a DU spectrophotometer (Beckman Coulter Inc., Brea, CA) and using a predetermined optical density (OD600) and cell dry weight standard curve. The standard curve was generated by growing P. polymyxa cells to exponential phase (10 h) in the preculture medium (32). The cells were harvested and washed twice with sterile distilled water, followed by centrifugation at 5,000 × g for 3 min. The cell pellet was reconstituted in sterile distilled water, and several dilutions (10% to 80%) of the cell suspension were obtained. The optical density (OD600) of each cell dilution was measured against that of sterile distilled water as a blank. Each diluted cell suspension was centrifuged in a preweighed Eppendorf tube at 10,000 × g for 10 min, and the aqueous phase was decanted. The cell pellets were then dried for 18 h in a TempCon oven (American Scientific Products, McGraw Park, IL) at 50°C. The dried cells were weighed, and the resulting weights were matched against the corresponding optical densities (OD600). A standard curve was generated from a plot of cell dry weight (g/liter) against absorbance at OD600.
Changes in pH were measured using an Accumet Basic pH meter (Fisher Scientific, Pittsburgh, PA). Concentrations of fermentation products—2,3-BD, acetoin, ethanol, and acetic acid—were quantified using a 7890A Agilent gas chromatograph (Agilent Technologies Inc., Wilmington, DE, USA) equipped with a flame ionization detector (FID) and a J&W 19091 N-213 capillary column (30 m [length] by 320 μm [internal diameter] by 0.5 μm [HP-Innowax film thickness]) as previously described (40).
The concentrations of sugars (sucrose, glucose, and fructose) were quantified by high-performance liquid chromatography (HPLC) using a Waters 2796 Bioseparations module equipped with an evaporative light scattering detector (ELSD; Waters, Milford, MA) and a 9-μm Aminex HPX-87P column (300 mm [length] by 7.8 mm [internal diameter]) connected in series to a 4.6-mm (internal diameter) by 3-cm (length) Aminex de-ashing guard column (Bio-Rad, Hercules, CA). The column temperature was maintained at 65°C. The mobile phase was HPLC-grade water (Waters Corporation, Milford, MA) maintained at a flow rate of 0.6 ml/min, as described previously (41).
The EPS produced during fermentation was quantified using a previously described procedure (42) with modification. Culture broth was centrifuged at 8,000 × g for 10 min to remove cell pellets, after which the EPS in the supernatant was precipitated with chilled 95% ethanol (10× the volume of the supernatant). The supernatant-ethanol mixture was kept overnight at 4°C and then centrifuged at 8,000 × g for 10 min. The resulting EPS pellet was dried in the oven at 60°C and then reconstituted in distilled water. The EPS-containing solution was vortexed vigorously to completely dissolve the EPS. The EPS was quantified by using the phenol-sulfuric acid method (43, 44). Briefly, 25 μl of 80% phenol was added to test tubes—one set containing 1 ml glucose or fructose standards (0 to 0.1 g/liter) and a second set containing 1 ml serially diluted EPS samples. The mixture was vortexed briefly, and 2.5 ml concentrated sulfuric acid (Fisher Scientific, Pittsburg, PA) was added to the mixture. The test tubes were then incubated at 25°C for 10 min. After incubation, the mixture was gently vortexed, and absorbance was measured at 490 nm against a reagent blank prepared similarly to the samples. A standard curve was generated by plotting the values of glucose concentration (x axis) against absorbance (OD490) (y axis), and EPS concentration was interpolated from the standard curve.
Levansucrase assay.
Levansucrase activity in the levansucrase null mutant and wild-type P. polymyxa was assayed using a previously described method with modification (45). Fermentation samples (from both the levansucrase null mutant and the wild type) were collected at the early and late stationary growth phases (24 and 36 h, respectively). The samples were centrifuged for 20 min at 8,600 × g and 4°C. The supernatant (200 μl) from each sample was divided into two portions (tubes A and B) containing 100 μl each. The supernatant in tube A was used to determine the levansucrase activity as depicted in Fig. S1 in the supplemental material. The reaction mixture for the levansucrase assay in tube A consisted of 400 μl of 1 M sucrose in 50 mM phosphate buffer (pH 6.0) and 100 μl of culture supernatant. The mixture was then incubated at 35°C for 1 h. Afterwards, the EPS produced during fermentation and during the levansucrase assay was precipitated with 95% ethanol (4°C) and was subsequently quantified and expressed as [EPS]A. Levansucrase activity was not determined in tube B. However, the EPS in tube B was precipitated with 95% ethanol, and the EPS was quantified as described above and was designated [EPS]B. The EPS produced during levansucrase activity was determined from the equation [EPS]L = [EPS]A − [EPS]B, where [EPS]L represents the concentration of EPS produced during the levansucrase assay. Protein concentrations in the supernatants were determined by the Bradford method (46). One unit of levansucrase activity was defined as the amount of protein (in milligrams) that catalyzed the formation of one micromole of EPS (levan) per minute at 35°C in a 1 M sucrose solution.
Determination of growth rate and generation time of the levansucrase null mutant.
To determine the stability of the levansucrase null mutant, the growth curve was first obtained. The levansucrase null mutant was grown in preculture medium, and the optical density (OD600) values of samples were measured at several time points until the cells reached the death phase of growth. The cell biomass concentrations were obtained from a predetermined correlation between optical density (OD600) and cell biomass. The cell biomass concentrations were plotted against time to generate the growth curve (see Fig. S2). Then, the cell biomass concentrations were plotted against time. The generation (doubling) time of the levansucrase mutant was determined from the exponential phase of the growth curve as 1.5 h (Fig. S2).
Assessment of stability of the P. polymyxa levansucrase null mutant.
The stability of the levansucrase null mutant was determined in the presence and absence of antibiotic (erythromycin) following growth for 50 generations. The levansucrase null mutant was grown in preculture medium supplemented with 35 μg/ml erythromycin until the OD600 reached 1.0 to 1.2. The actively growing cells (10% [vol/vol]) were transferred into fermentation medium containing 100 g/liter sucrose supplemented with 35 μg/ml erythromycin. This was designated G0 (generation zero). Several subcultures (every 3 h, two generations) were made from G0 until G50 (generation 50) was attained, and in each case, cultures were supplemented with 35 μg/ml erythromycin. Fermentations were then conducted using cells from G0, G10, G20, G30, G40, and G50 under antibiotic selective pressure. Samples (2 ml) were taken at 0 h and every 12 h until the fermentation ended and were then analyzed for cell growth and EPS and 2,3-BD production. The same experiment was conducted without antibiotic supplementation. Every tenth generation from zero to 50 (G0, G10, G20, G30, G40, and G50) was obtained as described above and then used to conduct fermentations. The antibiotic resistance profile of all the generations studied (with and without erythromycin supplementation) was assessed by PCR and replica plating onto erythromycin-containing plates. For each generation, samples of the P. polymyxa levansucrase null mutant cultures were taken during the stationary phase of growth (12 to 16 h), subcultured in fresh preculture medium, and then grown until another stationary growth phase (12 to 16 h) was attained. Subsequently, the cells were diluted to a concentration of 108 CFU/ml and then plated on TSA plates without antibiotic (erythromycin) supplementation. The plates were incubated at 35°C for 12 h. Colonies from each generation were selected and screened for the presence of the erythromycin resistance gene by PCR. Recombinant pGEM 7Zf(+) harboring the levansucrase inactivation construct was used as a PCR control for the erythromycin resistance gene. The presence of the erythromycin resistance gene in the colonies confirmed the stability of the P. polymyxa levansucrase null mutant over 50 generations. Colonies from the TSA plates (without erythromycin supplementation) were also transferred by replica plating to fresh TSA plates supplemented with erythromycin (35 μg/ml). The antibiotic-supplemented plates were incubated at 35°C for 12 h, and the resulting colonies were quantitatively compared to those on the plates without antibiotic supplementation. A schematic representation of the step-by-step procedure employed to evaluate the stability of the P. polymyxa levansucrase null mutant is shown in Fig. S3.
Statistical analysis.
The general linear model of Minitab 17 (Minitab Inc., State College, PA) was used for all statistical analyses. Analysis of variance (ANOVA) using Tukey’s method for pairwise comparisons was employed to compare differences between treatments. Differences in growth, sugar utilization, maximum product concentrations, and 2,3-BD yields and productivities were compared at the 95% confidence interval. 2,3-BD yield was expressed as the amount (in grams) of 2,3-BD produced from one gram of substrate (sucrose or glucose). 2,3-BD productivity was expressed as the amount (in grams per liter) of 2,3-BD produced per hour of fermentation. Additionally, 2,3-BD production cell efficiency was determined and expressed as the ratio of the maximum amount of 2,3-BD produced to the corresponding P. polymyxa cell growth.
Supplementary Material
ACKNOWLEDGMENTS
Salaries and research support were provided in part by state funds appropriated to the Ohio Agricultural Research and Development Center (OARDC), College of Food, Agricultural and Environmental Sciences (CFAES), and OARDC graduate and interdisciplinary grants and by United States Department of Agriculture–NIFA Hatch grants (project no. OHO01478 and OHO01333). Peloton Technologies LLC also provided financial support.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Celińska E, Grajek W. 2009. Biotechnology production of 2,3-butanediol–current state and prospects. Biotechnol Adv 27:715–725. doi: 10.1016/j.biotechadv.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Ji X, Huang H, Ouyang P. 2011. Microbial 2,3-butanediol production: a state-of-the-art review. Biotechnol Adv 29:351–364. doi: 10.1016/j.biotechadv.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 3.Syu MJ. 2001. Biological production of 2,3-butanediol. Appl Microbiol Biotechnol 55:10–18. doi: 10.1007/s002530000486. [DOI] [PubMed] [Google Scholar]
- 4.Garg S, Jain A. 1995. Fermentative production of 2,3-butanediol: a review. Bioresour Technol 51:103–109. doi: 10.1016/0960-8524(94)00136-O. [DOI] [Google Scholar]
- 5.Nakashimada Y, Marwoto B, Kashiwamura T, Kakizono T, Nishio N. 2000. Enhanced butanediol production by addition of acetic acid in Paenibacillus polymyxa. J Biosci Bioeng 90:661–664. doi: 10.1263/jbb.90.661. [DOI] [PubMed] [Google Scholar]
- 6.De Mas CD, Jansen NB, Tsao GT. 1988. Production of optically active 2,3-butanediol by Bacillus polymyxa. Biotechnol Bioeng 31:366–377. doi: 10.1002/bit.260310413. [DOI] [PubMed] [Google Scholar]
- 7.Donot F, Fontana A, Baccou JC, Schorr-Galindo S. 2012. Microbial exopolysaccharides: main examples of synthesis, excretion, genetics and extraction. Carbohydr Polym 87:951–962. doi: 10.1016/j.carbpol.2011.08.083. [DOI] [Google Scholar]
- 8.Häßler T, Schieder D, Pfaller R, Faulstich M, Sieber V. 2012. Enhanced fed-batch fermentation of 2,3-butanediol by Paenibacillus polymyxa DSM 365. Bioresour Technol 124:237–244. doi: 10.1016/j.biortech.2012.08.047. [DOI] [PubMed] [Google Scholar]
- 9.Liang TW, Wang SL. 2015. Recent advances in exopolysaccharides from Paenibacillus spp. production, isolation, structure, and bioactivities. Mar Drugs 13:1847–1863. doi: 10.3390/md13041847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lal S, Tabacchioni S. 2009. Ecology and biotechnological potential of Paenibacillus polymyxa: a minireview. Indian J Microbiol 49:2–10. doi: 10.1007/s12088-009-0008-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haggag WM. 2007. Colonization of exopolysaccharide producing Paenibacillus polymyxa on peanut roots for enhancing resistance against crown rot disease. Afr J Biotechnol 6:1568–1577. [Google Scholar]
- 12.Lebuhn M, Heulin T, Hartmann A. 2006. Production of auxin and other indolic and phenolic compounds by Paenibacillus polymyxa strains isolated from different proximity to plant roots. FEMS Microbiol Ecol 22:325–334. doi: 10.1111/j.1574-6941.1997.tb00384.x. [DOI] [Google Scholar]
- 13.Han B, Gopalan V, Ezeji TC. 2011. Acetone production in solventogenic Clostridium species: new insights from non-enzymatic decarboxylation of acetoacetate. Appl Microbiol Biotechnol 91:565–576. doi: 10.1007/s00253-011-3276-5. [DOI] [PubMed] [Google Scholar]
- 14.Okonkwo CC. 2017. Process development and metabolic engineering to enhance 2,3-butanediol production by Paenibacillus polymyxa DSM 365. PhD dissertation. The Ohio State University, Wooster, OH. [Google Scholar]
- 15.Han B, Ujor V, Lai LB, Gopalan V, Ezeji TC. 2013. Use of proteomic analysis to elucidate the role of calcium in acetone-butanol-ethanol fermentation by Clostridium beijerinckii NCIMB 8052. Appl Environ Microbiol 79:282–293. doi: 10.1128/AEM.02969-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rutering M, Cress BF, Schilling M, Ruhmann B, Koffas MAG, Sieber V, Schmid J. 2017. Tailor-made exopolysaccharides-CRISPR-Cas9 mediated genome editing in Paenibacillus polymyxa. Synth Biol 2:ysx007. doi: 10.1093/synbio/ysx007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yother J. 2011. Capsules of Streptococcus pneumoniae and other bacteria: paradigms for polysaccharide biosynthesis and regulation. Annu Rev Microbiol 65:563–581. doi: 10.1146/annurev.micro.62.081307.162944. [DOI] [PubMed] [Google Scholar]
- 18.Yanase H, Iwata M, Nakahigashi R, Kita K, Kato N, Tonomura K. 1992. Purification, crystallization, and properties of the extracellular levansucrase from Zymomonas mobilis. Biosci Biotechnol Biochem 56:1335–1337. doi: 10.1271/bbb.56.1335. [DOI] [Google Scholar]
- 19.Arrieta JG, Sotolongo M, Menéndez C, Alfonso D, Trujillo LE, Soto M, Ramírez R, Hernández L. 2004. A type II protein secretory pathway required for levansucrase secretion by Gluconacetobacter diazotrophicus. J Bacteriol 186:5031–5039. doi: 10.1128/JB.186.15.5031-5039.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bezzate S, Aymerich S, Chambert R, Czarnes S, Berge O, Heulin T. 2000. Disruption of the Paenibacilus polymyxa levansucrase gene impairs its ability to aggregate soil in the wheat rhizosphere. Environ Microbiol 2:333–342. doi: 10.1046/j.1462-2920.2000.00114.x. [DOI] [PubMed] [Google Scholar]
- 21.Gurunathan S, Gunasekaran P. 2004. Differential expression of Zymomonas mobilis sucrose genes (sacB and sacC) in Escherichia coli and sucrase mutants of Zymomonas mobilis. Braz Arch Biol Technol 47:329–338. doi: 10.1590/S1516-89132004000300001. [DOI] [Google Scholar]
- 22.Koepsell HJ, Tsuchiya HM, Hellman NN, Kazenko A, Hoffman CA, Sharpe ES, Jackson RW. 1953. Enzymatic synthesis of dextran acceptor specificity and chain initiation. J Biol Chem 200:793–801. [PubMed] [Google Scholar]
- 23.Esser K, Kadereit JW, Lüttge U, Runge M. 2012. Progress in botany 64: Genetics cell biology and physiology systematics and comparative morphology ecology and vegetation science. Springer Science & Business Media, Berlin, Germany. [Google Scholar]
- 24.Colvin KM, Irie Y, Tart CS, Urbano R, Whitney JC, Ryder C, Howell PL, Wozniak DJ, Parsek MR. 2012. The Pel and Psl polysaccharides provide Pseudomonas aeruginosa structural redundancy within the biofilm matrix. Environ Microbiol 14:1913–1928. doi: 10.1111/j.1462-2920.2011.02657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cooley BJ, Dellos-Nolan S, Dhamani N, Todd R, Waller W, Wozniak D, Gordon VD. 2016. Asymmetry and inequity in the inheritance of a bacterial adhesive. New J Phys 18:045019. doi: 10.1088/1367-2630/18/4/045019. [DOI] [Google Scholar]
- 26.Papagianni M, Psomas SK, Batsilas L, Paras SV, Kyriakidis DA, Liakopoulou-Kyriakides M. 2001. Xanthan production by Xanthomonas campestris in batch cultures. Process Biochem 37:73–80. doi: 10.1016/S0032-9592(01)00174-1. [DOI] [Google Scholar]
- 27.Saudagar PS, Singhal RS. 2004. Fermentative production of curdlan. Appl Biochem Biotechnol 118:21–31. doi: 10.1385/abab:118:1-3:021. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Z, Chen H. 2010. Fermentation performance and structure characteristics of xanthan produced by Xanthomonas campestris with a glucose/xylose mixture. Appl Biochem Biotechnol 160:1653–1663. doi: 10.1007/s12010-009-8668-y. [DOI] [PubMed] [Google Scholar]
- 29.Madden JK, Dea ICM, Steer DC. 1986. Structural and rheological properties of the extracellular polysaccharides from Bacillus polymyxa. Carbohydr Polym 6:51–73. doi: 10.1016/0144-8617(86)90012-3. [DOI] [Google Scholar]
- 30.Ren C, Gu Y, Hu S, Wu Y, Wang P, Yang Y, Yang C, Yang S, Jiang W. 2010. Identification and inactivation of pleiotropic regulator CcpA to eliminate glucose repression of xylose utilization in Clostridium acetobutylicum. Metab Eng 12:446–454. doi: 10.1016/j.ymben.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 31.Trček J, Mira NP, Jarboe LR. 2015. Adaptation and tolerance of bacteria against acetic acid. Appl Microbiol Biotechnol 99:6215–6229. doi: 10.1007/s00253-015-6762-3. [DOI] [PubMed] [Google Scholar]
- 32.Okonkwo CC, Ujor V, Mishra PK, Ezeji TC. 2017. Process development for enhanced 2,3-butanediol production by Paenibacillus polymyxa DSM 365. Fermentation 3:18. doi: 10.3390/fermentation3020018. [DOI] [Google Scholar]
- 33.Sambrook J, Russell DW. 2001. Molecular cloning, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 34.Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13amp18 and pUC19 vectors. Gene 33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 35.Görke B, Stülke J. 2008. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat Rev Microbiol 6:613–624. doi: 10.1038/nrmicro1932. [DOI] [PubMed] [Google Scholar]
- 36.Inukai M, Isono F, Takatsuki A. 1993. Selective inhibition of the bacterial translocase reaction in peptidoglycan synthesis by mureidomycins. Antimicrob Agents Chemother 37:980–983. doi: 10.1128/aac.37.5.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou Y, Johnson EA. 1993. Genetic transformation of Clostridium botulinum by electroporation. Biotechnol Lett 15:121–126. doi: 10.1007/BF00133010. [DOI] [Google Scholar]
- 38.Okonkwo CC, Ujor V, Ezeji TC. 2019. Chromosomal integration of aldo-keto-reductase and short-chain dehydrogenase/reductase genes in Clostridium beijerinckii NCMB 8052 enhanced tolerance to lignocellulose-derived microbial inhibitory compounds. Sci Rep 9:7634. doi: 10.1038/s41598-019-44061-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1110. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 40.Okonkwo CC, Ujor V, Ezeji TC. 2017. Investigation of the relationship between 2,3-butanediol toxicity and production during growth of Paenibacillus polymyxa. N Biotechnol 34:23–31. doi: 10.1016/j.nbt.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 41.Okonkwo CC, Azam MM, Ezeji TC, Qureshi N. 2016. Enhancing ethanol production from cellulosic sugars using Scheffersomyces (Pichia) stipitis. Bioprocess Biosyst Eng 39:1023–1032. doi: 10.1007/s00449-016-1580-2. [DOI] [PubMed] [Google Scholar]
- 42.Zhang J, Wang R, Jiang P, Liu Z. 2002. Production of an exopolysaccharide bioflocculant by Sorangium cellulosum. Lett Appl Microbiol 34:178–181. doi: 10.1046/j.1472-765x.2002.01068.x. [DOI] [PubMed] [Google Scholar]
- 43.Nielsen SS. 2010. Food analysis laboratory manual, p 47–53. Springer International, Cham, Switzerland. [Google Scholar]
- 44.Dubois M, Gilles JK, Hamilton PA, Rebers PA, Smith F. 1956. Colorimetric method for determination of sugars and related substances. Anal Chem 28:350–356. doi: 10.1021/ac60111a017. [DOI] [Google Scholar]
- 45.Euzenat O, Guibert A, Combes D. 1997. Production of fructo-oligosaccharides by levansucrase from Bacillus subtilis C4. Process Biochem 32:237–243. doi: 10.1016/S0032-9592(96)00058-1. [DOI] [Google Scholar]
- 46.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.