

Abstract
Pyruvate kinase muscle isoform 2 (PKM2) is a key glycolytic enzyme involved in ATP generation and critical for cancer metabolism. PKM2 is expressed in many human cancers and is regulated by complex mechanisms that promote tumor growth and proliferation. Therefore, it is considered an attractive therapeutic target for modulating tumor metabolism. Various stimuli allosterically regulate PKM2 by cycling it between highly active and less active states. Several small molecules activate PKM2 by binding to its intersubunit interface. Serine and cysteine serve as an activator and inhibitor of PKM2, respectively, by binding to its amino acid (AA)-binding pocket, which therefore represents a potential druggable site. Despite binding similarly to PKM2, how cysteine and serine differentially regulate this enzyme remains elusive. Using kinetic analyses, fluorescence binding, X-ray crystallography, and gel filtration experiments with asparagine, aspartate, and valine as PKM2 ligands, we examined whether the differences in the side-chain polarity of these AAs trigger distinct allosteric responses in PKM2. We found that Asn (polar) and Asp (charged) activate PKM2 and that Val (hydrophobic) inhibits it. The results also indicate that both Asn and Asp can restore the activity of Val-inhibited PKM2. AA-bound crystal structures of PKM2 displayed distinctive interactions within the binding pocket, causing unique allosteric effects in the enzyme. These structure-function analyses of AA-mediated PKM2 regulation shed light on the chemical requirements in the development of mechanism-based small-molecule modulators targeting the AA-binding pocket of PKM2 and provide broader insights into the regulatory mechanisms of complex allosteric enzymes.
Keywords: pyruvate kinase, crystal structure, enzyme kinetics, allosteric regulation, glycolysis, amino acids, asparagine, aspartate, pyruvate kinase muscle isoform 2 (PKM2), valine, valine, fluorescence binding, cancer metabolism, oligomerization
Introduction
Glycolysis is an important metabolic pathway utilized by cancer cells for tumor growth and proliferation (1). Pyruvate kinase muscle isoform 2 (PKM2)2 is an essential enzyme involved in regulating glycolysis in cancer cells. It catalyzes the terminal step of glycolysis, by transferring a phosphoryl group from phosphoenolpyruvate (PEP) to ADP, generating pyruvate and ATP (2, 3). PKM2 is mainly expressed in embryonic cells, normal proliferating cells, and cancer cells (4). In contrast to the other PKM isoform, PKM1, which is expressed in skeletal muscle, heart, and brain and exists in a constitutively active tetrameric state, PKM2 can exist in different oligomeric states: an active tetramer and a less active dimer/monomer.
The activity and oligomeric state of PKM2 are regulated by various synthetic small molecules (such as TEPP-46, DASA-58 (1), 1-(sulfonyl)-5-(arylsulfonyl)indoline (5), and shikonin (1, 6)), metabolites (such as fructose 1,6-bisphosphate (FBP) (7), succinyl-5-aminoimidazole-4-carboxamide-1-ribose 5′-phosphate (SAICAR) (8–10)), and amino acids (AAs) (serine (11), cysteine (12, 13), alanine (14–16), phenylalanine (17), and tryptophan (16)). Depending upon tumors' requirement for energy or metabolic intermediates, the activity of PKM2 is either up- or down-regulated by cycling between the two oligomeric states (3). Due to the unique ability of cancer cells to perform aerobic glycolysis (18) and the preferential expression of PKM2 in tumors (4), the enzyme has been an attractive therapeutic target for regulating tumor metabolism (19).
Previous studies with small molecules have shown that proper regulation of glycolytic flux is vital for cell proliferation and tumor growth (1, 5, 20, 21). In particular, PKM2 inhibitors, such as peptide aptamers, bind to tetrameric PKM2 and permanently fixate it to the dimeric form, resulting in reduced glycolysis and decelerated cellular proliferation (20, 21). Although dimeric PKM2 is required for the accumulation of glycolytic intermediates used in the synthesis of cellular building blocks (3), a lack of ATP due to PKM2 inhibition can lead to low cell division rates (20). Conversely, several PKM2 activators, such as TEPP-46, DASA-58, and 1-(sulfonyl)-5-(arylsulfonyl)indoline derivative bind dimeric PKM2 at the dimer-dimer interface and convert it to the active tetrameric form (1, 5). It has been demonstrated that activation of PKM2 down-regulates cell proliferation by generating more ATP and fewer metabolic intermediates (1, 5). Likewise, AAs such as Ser (11) and Cys (13) activate or inhibit PKM2 by binding to the AA-binding pocket of the enzyme and stabilize or destabilize the tetrameric state, respectively. Although several PKM2 activators have been designed to bind to the dimer-dimer interface of PKM2, the AA-binding pocket is an attractive site that can be targeted for developing mechanism-based activators or inhibitors for regulating tumor metabolism.
An open question, which is mainly due to the lack of detailed structure-function studies with small molecule probes containing varying functional groups, is how the AA-binding site of PKM2 communicates with the active site, making Cys an inhibitor and Ser an activator. On the basis of ligand binding and kinetic studies of PKM1 with various AAs, it has been proposed that the main-chain carboxylate and amine functionalities of the AAs are primarily responsible for binding to PKM1, and groups beyond the β-carbon are responsible for eliciting the allosteric signal (22). From the crystal structures of PKM2-Ser and PKM2-Cys, the differential regulatory roles can be posited for the distinct chemical properties of the side chains. In the present study, we hypothesized that the polarity of AA side chains is likely responsible for altering the dynamics of the largely polar and hydrophilic binding pocket (13), consequently resulting in a distinct long-range allosteric effect. Because of the importance of PKM2 in cancer metabolism, understanding how the difference in functional groups of amino acids regulate PKM2 activity and oligomeric state will aid in our understanding of desired parameters required for developing small-molecule chemical probes for modulating PKM2 function.
To gain insight into how side-chain polarity of AAs influence PKM2, Asn, Asp, and Val were examined. Whereas the polar Asn and the charged Asp activate PKM2, hydrophobic Val is an inhibitor. We observe that Asp and Asn stabilize PKM2 in a tetrameric state, whereas Val inhibits PKM2 by shifting the tetramer to a mixture of tetramer and dimer/monomer equilibrium. Notably, both Asn and Asp restore Val-mediated inhibition. Competition assays show that Val can displace Ser from PKM2, leading to reduced catalytic activity. On the basis of the current findings, including the crystal structures of PKM2 in complex with AAs (PKM2-Asn, PKM2-Asp, and PKM2-Val), it can be inferred that the mechanism of PKM2 activation by AAs is mediated via their side chains, whereas the main chains of Val as well as Cys are responsible for inhibiting PKM2.
Results
Asn and Asp allosterically activate PKM2 and Val is an inhibitor
To examine whether polar, charged AA ligands activate PKM2 and a hydrophobic AA, such as Val, acts as an inhibitor, activity studies were performed using 20 nm PKM2. The concentrations of corresponding AA and PEP were varied in the presence of a fixed concentration of ADP (0.8 mm). The extent by which PKM2 activity is altered depends on the AA concentrations used (Fig. S1, A–C). Both Asn and Asp increased the catalytic efficiency of PKM2 by ∼3-fold (8.2 ± 0.8 × 103 mm−1 min−1 without Asn versus 22 ± 3.1 × 103 mm−1 min−1 with 450 μm Asn and 10.5 ± 2.4 × 103 mm−1 min−1 without Asp versus 24.6 ± 1.6 × 103 mm−1 min−1 with 450 μm Asp) (Fig. 1A, Fig. S1 (A and B) and Table S1). The increase in catalytic efficiencies of PKM2 in the presence of Asn and Asp led to a concomitant decrease in the Km for PEP (Table S1). At 1.4 mm PEP, Asn and Asp activated PKM2 with a half-maximal effective concentration (EC50) of 38.2 ± 1.3 and 112 ± 33 μm, respectively, suggesting that Asn might be a slightly better activator (Fig. S2A).
Figure 1.

Effect of Asn, Asp, and Val on PKM2 activity and activation of Val inhibited PKM2 by various effector molecules. The activity assays with Asn, Asp, and Val were performed with 20 nm PKM2 with the PEP concentration varying between 0.1 and 12 mm. For the reactivation assays, the enzyme concentration was 12.5 nm, and the PEP concentration range was 0.1–20 mm. The ADP concentration was kept fixed at 0.8 mm for all experiments. A, varying concentrations of AAs were incubated with PKM2, and kinetic data at 450 μm (Asp and Asn) and 4 mm (Val) concentration were chosen to calculate the catalytic efficiency. The kcat/Km increases from 8.2 ± 0.8 × 103 to 22 ± 3.1 × 103 mm−1 min−1 in the presence of Asn and from 10.5 ± 2.4 × 103 to 24.6 ± 1.6 × 103 mm−1 min−1 with Asp. Val decreases the catalytic efficiency from 8.6 ± 1.4 × 103 to 1.8 ± 0.2 × 103 mm−1 min−1. B–E, PKM2 was inhibited with Val (1 or 5 mm), and the activity was resuscitated by adding an increasing amount of Asn, Asp, Ser, and FBP. Kinetic parameters are listed in Table S2. F, displacement of Ser activated PKM2 with increasing concentrations of Val. The catalytic efficiency decreases from 110 ± 17 × 103 mm−1 min−1 in the presence of 1 mm Ser to 44 ± 4.6 × 103 mm−1 min−1 in the presence of 1 mm Ser and 2 mm Val. Kinetic parameters are listed in Table S3.
Activity assays with increasing concentrations of Val decreased the catalytic efficiency of PKM2 by ∼5-fold (8.6 ± 1.4 × 103 mm−1 min−1 without Val versus 1.8 ± 0.2 × 103 mm−1 min−1 with 4 mm Val) (Fig. 1A and Fig. S1C), with a simultaneous increase in the Km for PEP (Table S1). Val inhibited PKM2 with an IC50 of 671 ± 12 μm at 4 mm PEP (Fig. S2A, inset).
Elimination of Val-mediated PKM2 inhibition by different allosteric effectors and displacement of bound Ser by Val
The intracellular concentration of amino acids is known to significantly increase in tumors compared with normal cells (23, 24). To understand whether physiological concentrations of AA activators (Asn, Asp, or Ser) can eliminate Val-mediated PKM2 inhibition, activity assays were conducted with increasing concentrations of the activators at a fixed concentration of Val (1 mm). As the cellular concentrations of Asn, Asp, Ser, and Val are ∼0.5–2 mm (23, 25), the concentrations of these AAs were varied within this range in the activity assays. The catalytic efficiency of Val inhibited PKM2 (4.9 ± 0.7 × 103 mm−1 min−1) increased in the presence of 1 mm Asn (12.3 ± 1.4 × 103 mm−1 min−1), Asp (11.3 ± 1.4 × 103 mm−1 min−1), and Ser (50.5 ± 8.1 × 103 mm−1 min−1), indicating that these activators can stimulate PKM2 (Fig. 1 (B–D), Fig. S1 (D–F), and Table S2). These results demonstrate for the first time that PKM2 inhibition by Val can be reversed in the presence of physiological concentrations of Asn, Asp, and Ser.
FBP, a known allosteric activator of PKM2 (7), eliminates the inhibitory effects of Cys (13) and Phe (26). To validate whether FBP can stimulate PKM2 in the presence of Val, activity assays were conducted using varying concentrations of FBP, in the presence of excess Val (5 mm). Indeed, within cellular concentrations (∼20–40 μm (26)) used in the assays, FBP increased the catalytic efficiency of Val inhibited PKM2 (0.9 ± 0.05 × 103 mm−1 min−1, with 5 mm Val and without FBP) in a concentration-dependent manner, with 0.4 μm FBP causing a complete reactivation of PKM2 (17.2 ± 2.9 × 103 mm−1 min−1 with 0.4 μm FBP and 5 mm Val versus 10.7 ± 1.5 × 103 mm−1 min−1 without FBP/Val) with a concomitant decrease in the Km for PEP (Fig. 1E, Fig. S1G, and Table S2).
As Ser is one of the AA activators that has been studied in greater detail, to examine whether an inhibitor such as Val can displace Ser from the PKM2-Ser complex, competitive assays were performed by first activating PKM2 with Ser (1 mm). Subsequently, Ser was displaced with increasing concentration of Val, in the range of 0.5–2 mm (physiological concentration of Val in malignant cells is 0.9 ± 0.5 mm (23)). 2 mm Val decreased the catalytic efficiency of Ser-activated PKM2 by ∼2.5-fold (110 ± 17 × 103 mm−1 min−1 (no Val) and 44 ± 4.6 × 103 mm−1 min−1 (with 2 mm Val)), indicating that Val is able to influence PKM2 activity by displacing Ser (Fig. 1F, Fig. S1H, and Table S3).
Asn, Asp, and Val allosterically regulate PKM2 activity by influencing PEP binding
Allosteric regulators of PKM2 have been shown to influence the activity by altering its binding affinity for substrate PEP (1, 11, 13, 17, 27). To understand how the presence of Asn, Asp, or Val alters substrate-binding affinity of PKM2, ligand-binding studies were carried out by monitoring changes in the intrinsic tryptophan fluorescence (Fig. 2 and Table 1). None of the AAs influence the ADP-binding affinity of PKM2 (Kd of 0.2–0.3 mm, with and without AAs) (Fig. 2A). However, the presence of 1 mm Asn or Asp increases the PEP-binding affinity of PKM2 by ∼30-fold, with Kd decreasing from 10 ± 2 μm (absence of AA) to ∼0.3 μm (Fig. 2B, inset). In contrast, the presence of 1 mm Val reduces the PEP-binding affinity of PKM2 (Kd of 81 ± 21 μm) (Fig. 2B). These results indicate that Asn and Asp increase PKM2 activity by enhancing the binary complex formation with substrate PEP, whereas Val-mediated inhibition occurs by reducing PEP binding. These results are consistent with previous observations where various small molecules, such as TEPP-46 (1), DASA-58 (1), and AAs (Cys (13), Ser (11), and Phe, Ala, and Trp (16)) modulate PKM2 activity by altering its affinity for PEP without perturbing the ADP-binding affinity.
Figure 2.

The presence of Asn, Asp, and Val alters the binding of PKM2 for substrate PEP, but not ADP. A, binding of ADP in the absence (black) and presence of 1 mm Asn (magenta), Asp (salmon), and Val (green), showing that the AAs do not influence the ADP-binding affinity of PKM2. B, PEP-binding affinity of PKM2 in the absence (black) and presence of 1 mm Asn (magenta), Asp (salmon), or Val (green). Kd values were as follows: 10 ± 2 μm (no AA ligand), 0.27 ± 0.0 μm (Asn), 0.29 ± 0.01 μm (Asp), and 81 ± 21 μm (Val). Error bars, S.E.
Table 1.
AAs alter the PEP binding affinity of PKM2 and vice versa
The concentration of PKM2 was kept constant at 1.6 μm for all of the studies. In the top two rows, the binding affinities of PKM2 for PEP and ADP were calculated in the absence and presence of AAs with 5 mm MgCl2 in the buffer. In the bottom three rows, binding affinities of PKM2 for the AAs in the absence and presence of PEP (2 mm) + MgCl2 (1 mm) and MgCl2 (1 mm) are shown (related to Figs. 2 and 3).
| Titrant | Ligand (concentration in mm) | Kd |
|---|---|---|
| μm | ||
| PEP | 10 ± 2 | |
| Asn (1) | 0.27 ± 0.0 | |
| Asp (1) | 0.29 ± 0.01 | |
| Val (1) | 81 ± 21 | |
| Val (10) | 505 ± 26 | |
| ADP | 237 ± 54 | |
| Asn (1) | 345 ± 33 | |
| Asp (1) | 585 ± 188 | |
| Val (1) | 363 ± 57 | |
| Asn | 11 ± 2 | |
| PEP (2) + MgCl2 (1) | 2 ± 0 | |
| MgCl2 (1) | 27 ± 5 | |
| Asp | 35 ± 7 | |
| PEP (2) + MgCl2 (1) | 8 ± 1 | |
| MgCl2 (1) | 95 ± 16 | |
| Val | 757 ± 120 | |
| PEP (2) + MgCl2 (1) | 4296 ± 604 | |
| MgCl2 (1) | 2170 ± 317 |
To gain insight into the binding affinities of PKM2 for the AA ligands, the Kd values of PKM2 for Asn, Asp, and Val were determined to be 11 ± 2, 35 ± 7, and ∼0.7 ± 0.1 mm, respectively (Fig. 3 and Table 1).
Figure 3.

Effect of PEP and MgCl2 on the binding of Asn, Asp, and Val to PKM2. The studies were conducted at a fixed concentration of MgCl2 (1 mm) and PEP (2 mm) while the concentration of the AA was varied. Shown is binding of PKM2 to Asn in the absence (●) and presence of MgCl2 (▴)/MgCl2 + PEP (■) (A), Asp in the absence (●) and presence of MgCl2 (▴)/MgCl2 + PEP (■) (B), and Val in the absence (●) and presence of MgCl2 (▴)/MgCl2 + PEP (■) (C). The dissociation constants (Kd) of all AAs to PKM2 are listed in Table 1. Error bars, S.E.
Functionally relevant bidirectional allosteric coupling in the presence of Asn, Asp, and Val
As described in the previous section, Asn/Asp and Val used as ligands increased and decreased the PEP-binding affinity of PKM2, respectively (Table 1 and Fig. 2B). To investigate whether the presence of PEP as a ligand influences the binding affinities of PKM2 for Asn, Asp, or Val, binding studies were conducted using AAs as titrants (Fig. 3 and Table 1). The affinity of PKM2 for Asn and Asp increased in the presence of PEP + MgCl2 (Kd values of 2 and 8 μm for Asn and Asp, respectively) (Fig. 3 (A and B) and Table 1). In contrast, the affinity of PKM2 for Val decreased by ∼6-fold in the presence of PEP (Kd of ∼0.7 mm without PEP + MgCl2 versus ∼4 mm with PEP + MgCl2) (Fig. 3C and Table 1). These results are consistent with the occurrence of a functionally relevant bidirectional coupling between the AA-binding site and the active site of PKM2, also observed before with Cys/Ser (13).
In a previous study, it was demonstrated that phosphoglycolate, a PEP analog, activated yeast pyruvate kinase by promoting tetrameric conformation (28). Thus, based on the binding studies, one can speculate that the presence of PEP reinforces the tetrameric state of PKM2, which facilitates Asn/Asp binding, whereas PEP has the reverse effect on Val binding to PKM2. The binding studies also show that the presence of MgCl2 decreased the affinities of PKM2 for AAs by ∼3-fold (Asn: Kd of 11 μm without MgCl2 versus 27 μm with MgCl2; Asp: Kd of 35 μm without MgCl2 versus 95 μm with MgCl2; Val: Kd of ∼0.7 mm without MgCl2 versus ∼2.2 mm with MgCl2) (Fig. 3 and Table 1). These results indicate that the magnesium ion and active-site residues involved in magnesium binding are likely responsible for allosteric communication between the active site and the AA-binding site.
Asn, Asp, and Val modulate PKM2 activity by influencing the oligomeric state
To understand how the oligomeric state of PKM2 is influenced by the AAs, gel filtration studies were carried out by incubating PKM2 (0.5 mg/ml or 8.3 μm) with an excess of each AA. Indeed, the presence of Val converted the native tetrameric state of PKM2 (208 kDa) to a mixture of tetramer (215 kDa) and dimer/monomer equilibrium (99 kDa), similar to that observed previously with Cys (13) (Fig. 4 (A and D) and Table 2). As the protein that elutes at ∼13.7 ml corresponds to a calculated molecular weight (MW) of 99 kDa, which is between a complete dimer (120 kDa) and a monomer (60 kDa), that fraction exists in a dimer/monomer equilibrium. It is known that the oligomeric state of PKM2 is concentration-dependent, with higher concentrations favoring more tetramer (29, 30). To examine how Asn, Asp, and Val influence PKM2 at lower concentrations, gel filtration studies were carried out with 0.1 mg/ml (1.7 μm) and 0.3 mg/ml (5 μm) PKM2 (Fig. 4D and Fig. S3). As expected, a dimer/monomer equilibrium is evident in the absence of any AA at 0.1 mg/ml PKM2 as well as in the presence of Asn or Asp (Fig. 4D and Table S4). With Val, the dimer/monomer peak is much more prominent at 0.1 (Fig. 4D) and 0.3 mg/ml (Fig. S3 and Table S4) compared with that with 0.5 mg/ml PKM2 (Fig. 4, A–C).
Figure 4.

Gel filtration chromatograms depicting changes in the oligomeric state of PKM2 in the presence of allosteric regulators, Asn, Asp, Ser, and Val. The experiments were conducted at fixed concentrations of corresponding AA ligand (10 mm) and enzyme (0.5 and 0.1 mg/ml). PEP, MgCl2, and FBP concentrations were kept at 5, 10, and 1 mm, respectively. Shown is an overlay of PKM2 with Val (green), Asn (magenta), Asp (orange), and without amino acid (black) (A and D); Val (green), Val + MgCl2 (brown), Val + MgCl2 + PEP (red), and Val + FBP (pale blue) (B); and Val (green), Val + Asn (magenta), Val + Asp (orange), and Val + Ser (blue) (C). In A and D, concentrations of PKM2 were at 0.5 and 0.1 mg/ml, respectively. In B and C, MgCl2, MgCl2 + PEP, FBP, Ser, Asn, and Asp shift the oligomeric state of Val-inhibited PKM2 from a mixture of tetramer and dimer/monomer equilibrium (green trace) to a near tetramer. The standards (158 and 44 kDa) and theoretical molecular weight of dimer (120 kDa) and monomer (60 kDa) are marked as ticks on the upper axis. As the protein that elutes at ∼13.7 ml corresponds to a calculated molecular weight of 99 kDa, that fraction exists in a dimer/monomer equilibrium.
Table 2.
Gel filtration elution volumes, experimental, and theoretical MW for PKM2 in the presence and absence of Asn, Asp, Val, Val + MgCl2, Val + MgCl2 + PEP, Val + FBP, Asn + Val, Asp + Val, and Ser + Val
All AA concentration was kept fixed at 10 mm. Concentrations of FBP, PEP, and MgCl2 were 1, 5, and 10 mm, respectively.
| Allosteric regulator | Theoretical MW |
Elution volume |
Experimental MWa |
||||
|---|---|---|---|---|---|---|---|
| Tetramer | Dimer | Monomer | Tetramer | Dimer/monomer | Tetramer | dimer/monomer | |
| kDa | ml | kDa | |||||
| 240 | 12.28 | 208 | |||||
| Asn | 240 | 12.26 | 211 | ||||
| Asp | 240 | 12.16 | 223 | ||||
| Val | 240 | 120 | 60 | 12.22 | 13.61 | 215 | 99 |
| Val + MgCl2 | 240 | 12.11 | 229 | ||||
| Val + MgCl2 + PEP | 240 | 12.08 | 233 | ||||
| Val + FBP | 240 | 12.11 | 229 | ||||
| Val + Asn | 240 | 120 | 60 | 12.41 | 13.81 | 224 | 100 |
| Val + Asp | 240 | 120 | 60 | 12.4 | 13.84 | 225 | 98 |
| Val + Ser | 240 | 12.47 | 216 | ||||
a Experimental MWs were calculated from elution volumes using gel filtration calibration curves.
As described in the previous section, the presence of MgCl2 and/or PEP reduced the Val-binding affinity of PKM2. To understand how the substrate and/or Mg2+ ion influence the oligomeric state of PKM2 in the presence of Val, gel filtration studies were performed with PKM2 + MgCl2 and PKM2 + PEP + MgCl2 with Val present. With either MgCl2 or PEP and MgCl2, Val-inhibited PKM2 shifts to a tetrameric conformation (229/233 kDa) from a mixture of tetramer and dimer/monomer equilibrium (Fig. 4B and Table 2). Likewise, the addition of FBP to PKM2-Val-inhibited complex, leads to a change in the oligomeric state to the native tetrameric form (229 kDa) (Fig. 4B and Table 2). As expected, incubation of PKM2 with Asp or Asn did not alter the tetramer (Fig. 4 (A and D) and Table 2).
To understand whether the activation of Val-inhibited PKM2 by Asn/Asp/Ser occurs through oligomeric state changes, size-exclusion chromatography was redone by incubating PKM2 with Val and each of the activator AAs. Asn, Asp, and Ser altered the oligomeric state of Val-inhibited PKM2 from a mixture of tetramer and dimer/monomer equilibrium to nearly a tetramer (Fig. 4C and Table 2). Thus, the size-exclusion chromatography results indicate that Val inhibits PKM2 by shifting the tetrameric form to a mixture of tetramer and dimer/monomer equilibrium states, whereas Asp and Asn activate PKM2 by reinforcing the tetrameric conformation in a manner similar to Ser.
Crystal structures of PKM2-Asn, PKM2-Asp, and PKM2-Val complexes
Overall structures of PKM2-Asn, PKM2-Asp, and PKM2-Val
To gain insight into the structural basis of allosteric activation and inhibition mechanism by the AAs, crystal structures of PKM2-Asn (PDB: 6V74), PKM2-Asp (PDB: 6V75), and PKM2-Val (PDB: 6V76) were determined in the P1211 space group at 2.32, 2.85, and 2.75 Å, respectively. The structures of all three AA-bound complexes of PKM2 contain four molecules in the asymmetric unit. An alignment of all three structures of the complexes with PKM2-FBP (PDB: 1T5A) showed that the overall structures are similar, and the tetramers exist in the relaxed state (R-state), with little to no difference in the relative orientation of the subunits (Figs. S4A and S5A). For all three structures, each monomer typically consists of one molecule of FBP, oxalate, and Mg2+ ion and the corresponding AA ligand (Fig. S4B).
Each PKM2 monomer has four domains, namely, the N- (AAs 1–44), A- (AAs 44–116 and 219–389), B- (AAs 117–218), and C- (AAs 390–531) domains (7). Structural alignment of a monomer (excluding the B-domain) of PKM2-Asn, PKM2-Asp, or PKM2-Val over 361–398 Cα atoms with the monomer of PKM2-FBP (PDB: 1T5A) resulted in root mean square deviation (RMSD) values of 0.37, 0.59, and 0.40 Å, respectively, indicating that the monomeric structures are fairly similar (Fig. S4B). The B-domain in all three complexes was highly mobile as observed previously for other PKM2 structures (7, 31). Although FBP was not added during the crystallization process, it is known to co-purify with PKM2 (9) and consequently was observed in each of the chains in all three structures of the AA-bound complexes (Fig. S4B). In the active sites of all chains in PKM2-Asn and PKM2-Asp, Mg2+ was present, but in PKM2-Val, it was only found in chains B and C. Likewise, oxalate was found in all chains of PKM2-Asn. In contrast, oxalate was only bound to the active sites of chains B and C of PKM2-Val and in chain C of PKM2-Asp (Fig. S4B). The overall architecture of the active site remains unchanged in all the three AA-bound complexes of PKM2.
Amino acid–binding pocket of PKM2-Asn, PKM2-Asp, and PKM2-Val complexes
In all three complexes of PKM2 with bound Asn, Asp, and Val, the corresponding ligands were located in the known AA-binding pocket of PKM2 (Figs. 5 and 6 and Fig. S6). The AA ligands were positioned in the respective electron density following an occupancy test as detailed under “Experimental procedures.” The AA ligands are held to the binding pocket through various hydrogen bond interactions. In particular, the main-chain amino groups form hydrogen bonds with the imidazole of His-464. Also, the amino group of Val interacts with the backbone carbonyl of Ile-469, similar to that observed previously for other AA-bound structures (11, 13, 16). One of the oxygen atoms in the main-chain carboxylate group of the bound AAs interacts with the side chain of Arg-106, which is located in the AA-binding pocket. As seen in PKM2-Cys (PDB: 6NU1) (13) and other AA-bound structures (11, 17), the side chain of Arg-106 flips inward to interact with the carboxylate group of the bound AA. However, in the absence of any AA, such as PKM2-FBP (PDB: 1T5A) the side chain of Arg-106 is flipped outward away from the AA-binding site (7) (Fig. 5 and Fig. S6). The second oxygen atom in the carboxylate group of bound AAs forms an H-bond with the amine group of Asn-70 in PKM2-Asn, PKM2-Asp, and PKM2-Val complexes (Fig. 5 and Fig. S6).
Figure 5.
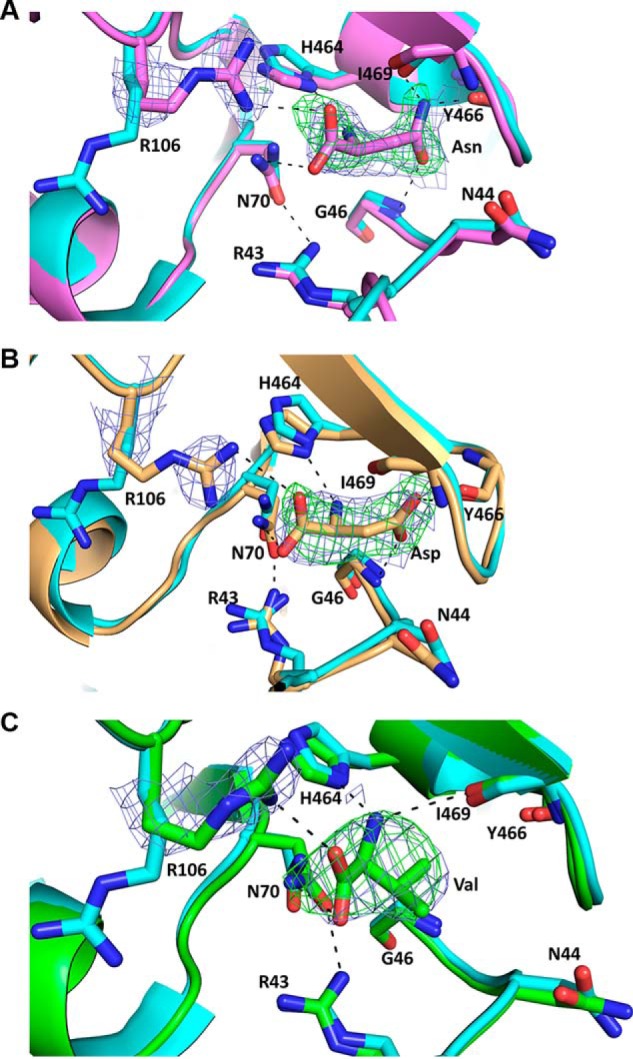
Crystal structures of the AA-binding pocket of PKM2 with bound Asn (violet) in chain C (A), Asp (light orange) in chain B (B), and Val (green) in chain C (C). PKM2-AA complex superimposed on PKM2-FBP (cyan, PDB: 1T5A). Dashed lines represent hydrogen bond interactions between the AA and residues involved in binding. Composite omit 2Fo − Fc maps (blue mesh) were generated for Arg-106 and all of the ligands and are contoured at 1σ. For all of the structures, the Fo − Fc omit maps (green mesh) for the AA ligands are contoured at 3σ. Red, oxygen; blue, nitrogen; protein backbone color, carbon.
Figure 6.
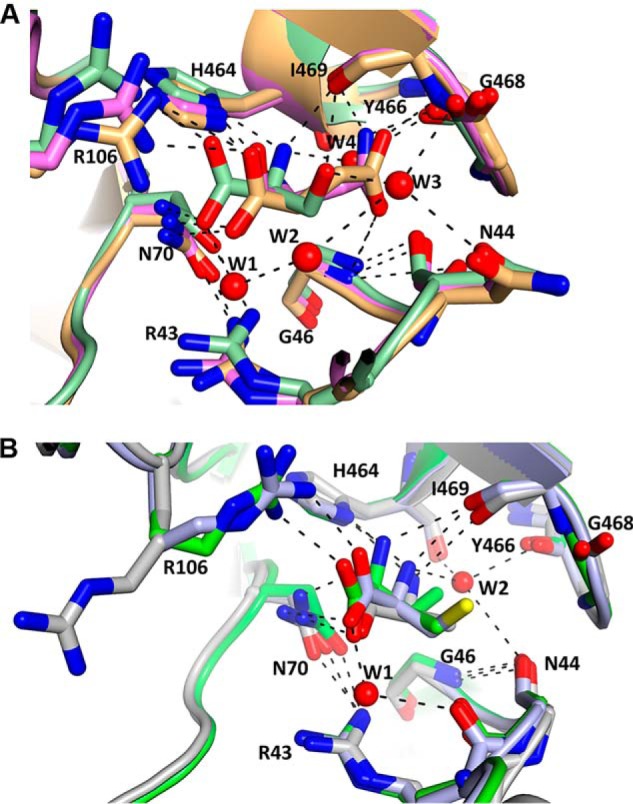
Superimposition of the AA-binding site of PKM2 with activators and inhibitors bound. A, PKM2-Asn (PDB: 6V74, chain C, violet), PKM2-Asp (PDB: 6V75, chain B, light orange), and PKM2-Ser (PDB: 4B2D, chain A, pale green). B, PKM2-Val (PDB: 6V76, chain C, green), PKM2-Ala (PDB: 6GG3, chain A, gray), and PKM2-Cys (PDB: 6NU1, chain C, light blue). Spheres, water molecules present in PKM2-Ser and PKM2-Cys. Dashed lines, H-bonds. The side chains of Asn, Asp, and Ser can form H-bonds with neighboring residues in the binding pocket, whereas these interactions are absent in Val, Cys, and Ala. Red, oxygen; blue, nitrogen; protein backbone color, carbon.
In PKM2-Asn and PKM2-Asp, the AA side chains form various H-bond interactions with the residues in the binding pocket, similar to that observed in PKM2-Ser (Figs. 5 (A and B) and 6A and Fig. S6 (A and B)). Whereas Ser forms water-mediated contacts (PDB: 4B2D), the binding pocket in the presence of Asn or Asp lacks water molecules, and thus the AAs form direct contacts with the binding pocket residues. In particular, the amine group in the side chain of Asn in PKM2-Asn forms two H-bond interactions with the backbone carbonyl atoms of Tyr-466 (2.26 Å) and Ile-469 (3.07 Å) (Fig. 5A and Fig. S6A). Also, the side-chain carbonyl (C=O) of Asn forms an H-bond to the -NH group of Gly-46 (2.70 Å) (Fig. 5A and Fig. S6A). In PKM2-Asp, the side-chain carboxylate groups of Asp form H-bond interactions with the backbone amides of Ile-469 (2.58 Å) and Gly-46 (3.11 Å) (Fig. 5B and Fig. S6B). The involvement of Gly-46 has not been observed in other AA-bound structures of PKM2. As expected, the methyl groups in the side chain of Val in the PKM2-Val complex do not interact with residues in the binding pocket (Fig. 5C and Fig. S6C). The crystal structures display that the AA activators form additional interactions via their side chains, whereas such interactions are not possible with the side chain of inhibitory AAs.
Discussion
PKM2 is an attractive therapeutic target for regulating glycolytic flux in cancer cells, with drugs like TLN-232 currently in phase II clinical trials (32). Although several small molecule activators of PKM2, such as TEPP-46, DASA-58, and others, have been shown to bind to the dimer-dimer interface of PKM2, to our knowledge, currently no small-molecule drugs are available targeting the AA-binding pocket. Interestingly, several AAs, such as Cys (13), Ser (11), Phe (16), Ala (16), and Trp (16), have been observed to allosterically regulate PKM2 activity by binding to the AA-binding pocket. However, due to lack of sufficient structure-function studies, we do not have a clear understanding of the functional group requirements and chemical nature of small molecules important for regulating PKM2. In particular, it is puzzling why Cys acts as an inhibitor of PKM2, whereas Ser is an activator despite both being similar in size and binding in a similar orientation within the enzyme.
We hypothesized that the difference in polarity between the side chains of Ser and Cys is responsible for their differential regulatory effect. In the present study, we investigated the mechanism by which polar, charged, and hydrophobic AAs, such as Asn, Asp, and Val, modulate PKM2 activity. The activity assays clearly illustrate that polar (Asn) and charged (Asp) AAs activate PKM2, whereas the hydrophobic Val inhibits PKM2 in a concentration-dependent manner. These results agree with the fact that the relatively polar Ser serves as an activator and Cys is an inhibitor of PKM2. The catalytic efficiencies of PKM2, in the presence of Asn (62 ± 5 × 103 mm−1 min−1), Asp (46 ± 5 × 103 mm−1 min−1), and Ser (70 ± 1 × 103 mm−1 min−1) are quite similar, indicating that at saturating concentrations (3 mm), comparable levels of PKM2 activation are achieved with all three activators (Fig. S2B and Table S5).
Our competition assays demonstrate that Val-inhibited PKM2 can be reactivated by Asn/Asp at physiological concentrations of the AAs (Fig. 1 (B and C) and Table S2). On the basis of these findings, one can speculate how cancer cells might be modulating PKM2 activity in the glycolytic pathway using Asn, Asp, and Val. It is known that a high rate of glycolysis in cancer cells results in the accumulation of pyruvate (3), which in turn produces Val. An increase in Val levels in cancer cells (34) can inhibit PKM2, and the activity can be restored by physiologically available concentrations of Asn and Asp (∼1 mm (23, 25)). Inhibition of PKM2 leads to a buildup of glycolytic intermediates necessary for the biosynthesis of nucleotides, amino acids (such as Ser (11)), and lipids (3), thus providing nutrients required for cell division.
It is also known that Val gets catabolized to fuel the tricarboxylic acid cycle (35). Therefore, one can speculate that while the Val pool decreases, there is an increase in the levels of Asp and Asn, which are produced from the tricarboxylic acid cycle (36, 37). It is possible that both Asn and Asp, along with Ser, can activate PKM2 in the presence of Val in cells and can contribute toward cell proliferation. Indeed, previous studies have shown that Asn (38), Asp (39), and Ser (40) are critical for tumor growth. Competition assays also show that Val can displace Ser from PKM2 and cause a decrease in the catalytic activity of PKM2 (Fig. 1F and Table S3). We can speculate that when PKM2 is activated by Ser, there is a lack of glycolytic intermediates, but Val synthesized from excess pyruvate can replace Ser and decrease PKM2 activity. This decrease in glycolytic flux initiates the process of accumulating metabolic intermediates, and the cycle starts anew.
The EC50 of Asn (38.2 ± 1.3 μm) was calculated to be ∼3-fold lower than that of Asp (112 ± 1.3 μm). This difference is in agreement with their relative binding affinities for PKM2, as Asn binds more tightly compared with Asp (Kd of 11 ± 2 μm (Asn) versus 35 ± 7 μm (Asp)) (Fig. S2A and Table 1). The EC50 and binding affinity values are within the physiological limit of the AAs found in normal and tumor cells, indicating that they can activate PKM2 intracellularly (23, 25). The crystal structures of PKM2-Asn and PKM2-Asp provide a structural basis for the lower binding affinity of Asp compared with that of Asn (Figs. 5 (A and B) and 6A and Fig. S6 (A and B)). Asn forms six interactions with surrounding residues in the binding pocket, whereas Asp can form only five H-bonds. The carboxylate in the side chain of Asp does not interact with the carbonyl of Tyr-466 in the binding pocket, unlike Asn, where the amide nitrogen group in the side chain is involved in an additional interaction with Tyr-466. Thus, the enhanced interaction of Asn within the binding pocket via H-bonds compared with that of Asp is responsible for the increased affinity of Asn for PKM2. The crystal structures of PKM2-Asn and PKM2-Asp complexes show that these two AAs stabilize PKM2 in a tetrameric R-state, as seen previously with PKM2-Ser structure (16) (Fig. S5A). Thus, based on activity assays, crystal structures, and gel filtration studies, it can be inferred that the increase in activity of PKM2 in the presence of Asn or Asp is due to the stabilization in an active, tetrameric R-state (Fig. S5A).
In contrast to the activators, Val binds poorly to PKM2 (Kd of ∼0.8 mm) compared with Cys (5.5 μm) (13, 23), although the Kd and IC50 values (∼0.7 mm) are close to the physiological concentrations of these AAs found in normal and cancer cells (23, 25). A close comparison of the crystal structures of PKM2-Val and PKM2-Cys provides a rationale for the difference in their binding affinities. In PKM2-Val structure, Val forms direct H-bonds with Arg-106, Asn-70, Ile-469, and His-464, which is similar to other inhibitory AA-bound PKM2 structures (13, 16). However, Val is tethered to the AA-binding pocket through four H-bond interactions with the neighboring residues, whereas Cys, being weakly polar, forms two additional water-mediated H-bonds with residues in the binding pocket (Fig. 6B). The nonpolar nature of Val excludes water molecules from the binding pocket, and the additional H-bonds might be responsible for the better binding affinity of Cys compared with Val. The exclusion of water molecules from the binding pocket in the presence of Val is similar to the crystal structures of PKM2 with nonpolar and/or bulky AAs, such as Ala (PDB: 6GG3), Phe (PDB: 6GG4), and Trp (PDB: 6GG5), where there is a dearth of water molecules in the AA-binding pocket (16).
In the presence of Val, PKM2 exists as a mixture of tetramer and dimer/monomer equilibrium in solution (Fig. 4 and Table 2). It is the dimer/monomer fraction that contributes toward the reduced activity of PKM2. Thus, the mechanism of inhibition of PKM2 by either Val (or Cys) is different from the inhibition mechanism by Phe, Ala, or Trp (16), where the enzyme exists in a tense state (T-state) while remaining as a tetramer (Fig. S5B). We believe that the dimer/monomer fraction of PKM2 observed with Val bound to the AA-binding pocket is responsible for the reduction of PEP binding in the active site of PKM2. The presence of Val reduced the binding affinity of PKM2 for PEP by ∼8-fold (Kd = 10 μm without Val versus 81 μm with Val) (Table 1). Indeed, previous studies have shown that dimeric PKM2 exhibits reduced PEP-binding affinity (41). In contrast, the presence of Asn or Asp increased the PEP-binding affinity of PKM2 (Table 1). These results illustrate the existence of allosteric signal communication from the AA-binding site to the active site of PKM2. On the basis of ligand-binding studies, one can speculate that PEP and magnesium ion along with active-site residues (Glu-272 and Asp-296) associated with magnesium binding are potentially involved in allosteric communication with the AA-binding site. The presence of PEP and/or Mg2+ has been shown to significantly influence the binding affinities of Asn, Asp, and Val for PKM2. In particular, Val binds weakly to PKM2 (Kd ∼0.8 mm), and introducing Mg2+ to the Val-bound complex of PKM2 further decreased the affinity for Val (Kd ∼2 mm), consequently reverting to its native tetrameric state from a mixture of tetramer and dimer/monomer equilibrium (Fig. 4B and Tables 1 and 2). Furthermore, PKM2 remains in the tetrameric state when PEP and Mg2+ are added to the Val bound complex, and this is because PEP further lowers the binding affinity of Val for PKM2 (∼4 mm) (Fig. 4B and Tables 1 and 2). Taken together, our findings show the occurrence of functionally relevant bidirectional communication between the active site and AA-binding site of PKM2 in the presence of Asn, Asp, and Val.
The Val-bound complex of PKM2 is tetrameric in crystallo, which is due to the high concentration of enzyme employed for crystallization conditions (10 mg/ml), as discussed in our previous reports (13, 30). The PKM2-Val complex exists in the R-state similar to PKM2-Cys structure, whereas other inhibitor-bound structures of PKM2, such as PKM2-Phe, PKM2-Ala, and PKM2-Trp, existed in the T-state (Fig. S5B) (16). We believe that the presence of FBP predisposes PKM2-Val and PKM2-Cys to a R-state, whereas FBP is absent in the structures of PKM2 in complex with Phe, Trp, and Ala. Furthermore, FBP was able to activate PKM2 in the presence of Val (Figs. 1E and 4B). Although FBP copurifies with PKM2 (9), we believe that a major fraction of the total enzyme lacks FBP. Consequently, the addition of FBP activates Val-inhibited PKM2. Taken together, the structures of PKM2 with inhibitory AAs (Val, Cys (13), Ala, Phe, and Trp (16)) along with the gel filtration data with Val confirm that FBP overrides the inhibitory effect by driving the enzyme from a mixture of tetramer and dimer/monomer equilibrium to a tetrameric R-state.
A close comparison of the AA-binding pockets of PKM2-Asn/Asp/Ser (activators) and PKM2-Val/Cys/Phe/Trp/Ala (inhibitors) (13, 16) displayed some striking features for PKM2-Asn and PKM2-Asp complexes, distinct from other reported AA-bound structures. First, the backbone -NH2 group of either Asp or Asn does not interact with the backbone carbonyl oxygen of Ile-469 (Fig. 6A). The lack of this H-bond interaction can be attributed to the difference in the relative orientation of Asp/Asn in the binding pocket of PKM2 compared with other AAs. Second, in PKM2-Asn or PKM2-Asp, the relatively long polar side chains point toward Gly-46, Tyr-466, and Ile-469 and are anchored to the AA-binding pocket via direct H-bond interactions. In contrast, these contacts are unattainable with the relatively short side chain of Ser (11) or less polar Cys (13) and/or bulky hydrophobic AAs (16), when occupied in the same AA-binding pocket. Instead, the side chain of Ser is involved in water (W3)-mediated interactions with the backbone carbonyl of Gly-468 and Asn-44 (Fig. 6A) (PDB: 4B2D). Although water molecules were found in the binding pocket of PKM2-Cys, the weakly polar side chain prevents any interactions that are formed by the polar and charged AAs (Fig. 6, A and B). Third, in contrast to PKM2-Ser/Cys, no water molecules were found in the AA-binding pocket of Asp- or Asn-bound complexes. The larger space occupied by Asp/Asn relative to Ser/Cys excludes any water molecules from the binding pocket. A previous all-atom molecular dynamics simulation demonstrated that Ile-469, Asn-44, and Gly-468 are part of a community network and are involved in the allosteric signal communication of PKM2 by Ser (42). Interestingly, in Asn/Asp bound structures of PKM2, Ile-469, Gly-46, and Asn-44, along with Asn-70, form a network and interact with the bound AA ligand, and one can posit that these residues are potentially involved in allosteric signal transmission. As noted previously, Asn-70 resides at one end of a β-strand that connects the AA-binding site to the active site (Fig. 5A of Ref. 13). Thus, based on the PKM2-Asn and PKM2-Asp structures in the current study along with PKM2-Cys and PKM2-Ser, one can envision potential residues involved in long-range allosteric regulation of PKM2 by AAs.
Conclusions
The results described here demonstrate that Asn and Asp activate PKM2, whereas Val is an inhibitor. From the present findings, it can be inferred that small molecules with polar functional groups that bind in an orientation similar to that of Ser/Asn/Asp will activate PKM2. In contrast, chemical probes with predominantly nonpolar functional groups will inhibit PKM2. We present here a mechanistic proposal as to how Val, Asn, and Asp can work in tandem to assist in cancer cell proliferation by regulating PKM2. This work will add to our understanding of the dynamic regulation mechanism of PKM2. Future studies can be directed toward developing polar/nonpolar small molecules to target the AA-binding pocket of PKM2 and modulate its function. In addition, cell-based assays with PKM2 can validate the findings with Asn, Asp, and Val. Finally, further research with PKM2 variants might provide insight into residues potentially involved in allosteric signal communication between the active site and the AA-binding site of PKM2.
Experimental procedures
Expression and purification of human PKM2
The human PKM2 gene in the pET28a vector was a generous gift from M. G. Vander Heiden. The plasmid was transformed into Escherichia coli BL21(DE3)pLysS cells (Life Technologies, Inc.). Single colonies were used to initiate starter cultures in Luria–Bertani medium. The overnight-grown starter culture was used to inoculate 2 liters of Terrific Broth medium. Following inoculation, the cells were grown at a temperature of 37 °C/180 rpm until the optical density at 600 nm reached 2.0. The cells were induced with 0.2 mm isopropyl β-d-1-thiogalactopyranoside and were grown further for 22 h at 22 °C. Cells were harvested at 4000 rpm at 4 °C for 30 min. 50–60 g of cell pellet was resuspended in 60–80 ml of 20 mm HEPES, pH 7.5, 150 mm NaCl, 5 mm imidazole, and 5% glycerol (buffer A). The cell suspension was lysed by sonication (8 s on, 20 s off, 14-min total time), and the cell lysate was centrifuged for 30 min at 30,000 rpm. The supernatant containing His6-PKM2 was purified using an immobilized nickel-affinity resin pre-equilibrated with buffer A. The column loaded with the protein was washed with buffer A followed by buffer A containing 50 mm imidazole. The protein was eluted with buffer A containing 450 and 1000 mm imidazole, and the eluted fractions were analyzed by SDS-PAGE for purity. Pure fractions were dialyzed overnight in 50 mm Tris-HCl, pH 7.5, 150 mm KCl, 0.5 mm tris(3-hydroxypropyl) phosphine, 5% glycerol, and 0.5 mm EDTA (storage buffer). For separating any aggregated protein molecules, the dialyzed protein was passed through a HiLoad Superdex 200 16/600 (GE Healthcare) gel filtration column pre-equilibrated with storage buffer. Protein concentrations were measured using a NanoDrop 2000c spectrophotometer (Thermo Scientific, Waltham, MA) at 280 nm, using the MW of 59.96 kDa (corresponding to PKM2 monomer) and molar extinction coefficient (ϵ280 nm) of 29.91 m−1 mm−1. The readout was in absorbance and protein concentration in mg/ml. The path length of the cuvette was set at 1 mm. Typically, 100-μl aliquots of 31 mg/ml protein concentration were made and stored at −80 °C until use. The protein concentrations used in all experiments described here refer to the monomer.
Pyruvate kinase activity assays
PKM2 activity was measured by the PKM2-lactate dehydrogenase (LDH) coupled assay in the presence and absence of different AAs using an Epoch microplate spectrophotometer (BioTek, Winooski, VT), as described previously (13). Briefly, 20 nm PKM2 was dissolved in 20 mm Tris-HCl, pH 7.5, 150 mm KCl, 5 mm MgCl2, 4 units/ml LDH, and 0.5 mm NADH. Varying concentrations (0–8 mm) of Val/Asn/Asp were added to the enzyme solution and incubated on ice for 15 min while keeping the ADP concentration fixed at 0.8 mm. Reactions (100 μl) were initiated by increasing the PEP concentration from 0.1 to 12 mm. The initial velocities were calculated in GraphPad Prism and plotted against PEP concentration. The resulting curves were fit using the Michaelis–Menten equation (Equation 1),
| (Eq. 1) |
where Y is the initial velocity, Vmax is the maximum velocity, X is the substrate (PEP) concentration, and Km is the Michaelis-Menten constant for PEP. For reactivation assays with different AAs and FBP, 12.5 nm PKM2 in the activity assay buffer was incubated on ice for 15 min along with 0.5 mm NADH, 4 units/ml LDH, 1 mm Val (with Asn/Asp/Ser), or 5 mm Val (with FBP). Varying concentrations of FBP (0–0.4 μm) or activator AAs (0–2 mm) were used, and the reactions were initiated with increasing concentrations of the PEP followed by the measurement of initial velocity as described above. For the displacement assay of PKM2 with Val in the presence of Ser, the enzyme was incubated with 1 mm Ser followed by inhibition with 0–2 mm Val with other experimental conditions being the same as described above. The PEP concentration range used for this experiment was 0.1–2.0 mm. All experiments of PKM2 with Asp were conducted at pH 7.5 or higher and consequently the AA is in the deprotonated state.
To compare kinetic parameters with Ser-stimulated turnover, the catalytic efficiency in the presence of 3 mm Ser was determined to be 70 ± 1 × 103 mm−1 min−1 (Table S5). This value is in close agreement with the calculated catalytic efficiencies of ∼32 ± 0.9 × 103 mm−1 min−1 with ∼3 mm Ser (16) and ∼88 ± 20 × 103 mm−1 min−1 with 200 mm Ser (26). Furthermore, to validate quality of the protein to that of previous studies, the catalytic efficiency of PKM2 was determined to be ∼8 ± 0.8 × 103 mm−1 min−1 to ∼17 ± 2.4 × 103 mm−1 min−1 (Tables S1 and S2). The values agree well with calculated catalytic efficiency of PKM2 from two previous studies, which were ∼14 ± 0.7 × 103 mm−1 min−1 (16) and ∼17 ± 2 × 103 mm−1 min−1 (26).
Ligand-binding assays
All binding studies were performed with 1.6 μm PKM2 in 20 mm Tris-HCl (pH 7.5) and 150 mm KCl using a Cary Eclipse fluorescence spectrophotometer (Agilent Technologies, Santa Clara, CA). The binding of ADP and PEP in the presence and absence of Val, Asn, or Asp was assessed by quenching of the intrinsic Trp fluorescence of PKM2. Briefly, PKM2 was dissolved in the buffer containing 5 mm MgCl2 and incubated for 15 min in the absence and presence of either 1 mm Asn/Asp or 1 and 10 mm Val. Titration experiments were performed with varying concentrations of PEP or ADP while measuring the decrease in Trp fluorescence.
To determine how PEP and Mg2+ influence the binding affinity of the AAs with PKM2, fluorescence data were collected by varying the concentration of the AAs in the absence and presence of MgCl2 and PEP + MgCl2, respectively. The wavelengths used for excitation and emission of PKM2 were 295 and 340 nm with slit widths of 5 and 10 nm, respectively. The fluorescence intensity was converted into fractional saturation and fitted to the one-site specific binding equation (Equation 2) or the Hill equation (Equation 3) using GraphPad Prism (San Diego, CA),
| (Eq. 2) |
| (Eq. 3) |
where Bmax is the maximum binding, X is titrant concentration, Y is fractional saturation, Khalf is the equilibrium constant at which the titrant occupies half of the binding sites, Kd is the dissociation constant, and h is the Hill coefficient. As the initial Kd was lower than the PKM2 concentration used, data for PEP-binding experiments in the presence of Asn and Asp were fitted using a ligand-binding model (Equation 4) as reported previously (43, 44) with Sigma Plot 12.5 (Systat Software Inc., Point Richmond, CA),
| (Eq. 4) |
where f is fluorescence signal resulting from PEP binding to PKM2, fm is the maximum fluorescence intensity, f0 is the signal from PKM2 in buffer solution, P and x are total protein and added PEP concentration, respectively, Kd is the dissociation constant, and n is the number of binding sites.
Gel filtration analysis
Solutions of PKM2 (0.1, 0.3, and 0.5 mg/ml) in storage buffer containing 10 mm tris(3-hydroxypropyl) phosphine (to control the redox state of the enzyme) in the absence and presence of Asn, Asp, and Val were injected separately onto a Superdex 200 10/300 GL gel filtration column (24 ml; GE Healthcare) via a 0.2-ml loop at a rate of 0.5 ml/min using an AKTA Pure FPLC system (GE Healthcare). The concentrations of all the AAs used in gel filtration chromatographic experiments were kept constant at 10 mm. To understand the effect of different PKM2 regulators on Val bound PKM2, in independent experiments, PKM2 was incubated for 15 min with Val and MgCl2/MgCl2 + PEP/FBP/Asn/Asp/Ser, and 0.2 ml of each solution was injected into the column. MgCl2, PEP, and FBP concentrations were kept constant at 10, 5, and 1 mm, respectively. The column was calibrated using gel filtration MW standards (Bio-Rad) containing vitamin B12 (1.35 kDa), horse myoglobin (17 kDa), chicken ovalbumin (44 kDa), bovine γ-globulin (158 kDa), and bovine thyroglobulin (670 kDa). The eluted protein was detected by monitoring UV absorbance at 280 nm. The intensity data were normalized by setting the highest intensity value to 100%, and the normalized values were plotted against elution volume in GraphPad Prism.
Crystallization of PKM2 with amino acids
To crystallize PKM2 with the AAs under investigation, 10 mg/ml PKM2 in storage buffer was mixed with 2 mm oxalate, 5 mm MgCl2, and 20 mm Asn/100 mm Asp/280 mm Val, and the solution was incubated for 30 min at 4 °C. 1 μl each of the protein and precipitant solutions were set up for crystallization by the sitting-drop vapor diffusion method at room temperature. Crystals of PKM2 with Asn (PKM2-Asn) and Asp (PKM2-Asp) were obtained in the precipitant solution containing 0.2 m NaBr, 0.1 m Bistris propane, pH 8.5. The PEG 3350 concentration in the precipitant solution was 20% and 18% for Asn- and Asp-bound crystals, respectively. Crystals of PKM2 with Val (PKM2-Val) were obtained in a precipitant solution consisting of 0.2 m NaBr, 0.1 m Bistris propane, pH 8.5, and 24% PEG 3350. Diamond-shaped crystals were obtained in a week for PKM2-Val and PKM2-Asp, whereas the crystals were rod-shaped for PKM2-Asn. The crystals were looped and washed in a precipitant solution containing mother liquor and 25% (v/v) glycerol. Crystals of PKM2-Val and PKM2-Asp were additionally soaked in mother liquor containing 40 and 20 mm concentrations of the respective AAs. All crystals were cryocooled by flash-freezing in liquid nitrogen.
Data collection and structure determination
X-ray diffraction data for all of the PKM2-AA crystals were collected at 100 K at beamline 4.2.2 at the Advanced Light Source (Berkeley, CA) using an RDI CMOS_8 M detector. For the PKM2-Asn crystal, the detector distance was kept at 350 mm with a 0.5-s exposure time and 0.1° oscillation. The data for PKM2-Val were collected using a 1-s exposure time, an oscillation of 0.1°, and a 350-mm detector distance, whereas for the PKM2-Asp crystal, the exposure time and oscillation were kept unchanged, but the detector was moved to 340 mm. The images were processed using the XDS package (45) and SCALA (CCP4) (46). Molecular replacement for all of the structures was performed with PHASER using chain C of PKM2-Cys structure (PDB: 6NU1) as the search model. Iterative rounds of refinement and model building were carried out using PHENIX (47) and COOT (48), respectively.
For all three AA-bound structures of PKM2, the His tag at the N terminus and the first 13 residues are disordered in all the chains and were not modeled. In PKM2-Asn, the B-domain was poorly modeled in all chains except chain B. In particular, residues 126–131 in chain A, residues 125–130 and 183–197 in chain C, and 121–203 in chain D of PKM2-Asn were untraced due to the absence of electron density in that region. In PKM2-Asp and PKM2-Val, the B-domain could only be modeled in chain A and it was either partially modeled or left unmodeled in chains B, C, and D. Specifically, in chain D of the PKM2-Asp structure, the B-domain comprising residues 121–214 was left unmodeled due to poor electron density along with residues 126–129 and 191–192 of chain B. For PKM2-Val structure, B-domain residues 125–129 and 150–151 of chain B, and 121–206 of chain D were missing due to poor quality of electron density.
Refinement strategies that were employed in the initial rounds of refinement for all three AA-bound PKM2 structures include rigid body (excluding PKM2-Asn), restrained coordinate refinement, individual isotropic ADP, simulated annealing (SA), occupancy, and target weights. SA was turned off in the later stages of refinement, and water molecules were added into clear densities. Likewise, at later stages, after Ramachandran outliers were fixed, only individual isotropic ADP refinement was used.
Ligands were added into distinct positive electron density. In PKM2-Asn structure, one molecule of Asn was added only to chain C. Due to weak density, Asn was not modeled in the AA-binding pocket of chains A, B, and D. Likewise, a molecule of Asp was added to chains B and C of PKM2-Asp. The AA-binding pocket in chain A was occupied by two water molecules, and no ligand was modeled in the binding pocket of chain D due to lack of electron density. In PKM2-Val, two molecules of Val were added in the AA-binding pocket of chains B and C, whereas it was left empty in chains A and D due to poor electron density. The AA ligands were positioned by generating omit maps, wherein either the ligand (for the SA omit map) or the ligand and surrounding bulk solvent within a 5-Å radius (for the polder omit map) was omitted (49). To confirm the position of ligands, occupancy of each of the AAs was set at 0.6, and six rounds of refinement were done. Post-refinement, the resulting occupancy of the AAs was observed to be ∼0.8–0.9. The SA omit map for the AAs of the new model (with occupancy at 0.8) did not show any significant difference from that of the models where the occupancy was set to 1. Therefore, the structures with ligand occupancy at 1 were retained as the final model. To ensure that the electron density of the ligands does not belong to glycerol (present in the cryoprotectant solutions), it was placed in the SA omit map density of the ligands in Asn/Asp/Val bound to PKM2 (PKM2-Asn/Asp/Val). For PKM2-Asp/Asn, the density is significantly elongated to be a good fit for glycerol. For PKM2-Val, glycerol was observed to fit in the density of the ligand. To ensure that the SA omit map density is not of glycerol, we compared PKM2-Val with a previously reported structure of PKM2 (PDB: 3SRD), containing glycerol in the AA–binding pocket. In the reported structure (PDB: 3SRD), Arg-106, which binds to the AA ligand, is flipped outward from the binding pocket. However, in all three structures (PKM2-Asn/Asp/Val), Arg-106 flips toward the binding pocket and anchors one of the carboxylate oxygens. Thus, we can conclude that the SA omit map density for Asp/Asn/Val belongs to the AAs and not glycerol. (Fig. 5). In PKM2-Asn and PKM2-Asp structures, a glycerol molecule is located near the AA-binding pocket, and this site was shown to be occupied by water molecules in both PKM2-Ser (PDB: 4B2D) and PKM2-Cys (PDB: 6NU1). The PKM2-Val structure does not contain either water or glycerol molecule at that site.
The final models were verified by generating composite omit maps. The structures were validated with the wwPDB validation server (50). All figures were prepared using PyMOL (33). Coordinates for all three structures were deposited in the Protein Data Bank (PDB entries 6V74, 6V75, and 6V76). Detailed information about data processing, refinement, and structure validation is provided in Table 3.
Table 3.
X-ray data collection and refinement statistics
Values in parentheses are for the highest-resolution shell. Rmerge = ∑∑j|Ihj − 〈Ih〉|/∑∑jIhj Rmeas = ∑√(n/(n − 1)) ∑j|Ihj − 〈Ih〉|/∑∑jIhj multiplicity-weighted Rmerge. Rwork = Σh{|Fo| − |Fc|}/∑h|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively. Rfree was calculated as R but for an independent 5% test set of reflections excluded from the modeling and refinement process. asu, asymmetric unit.
| Parameters | Values |
||
|---|---|---|---|
| PKM2-Asn | PKM2-Asp | PKM2-Val | |
| PDB code | 6V74 | 6V75 | 6V76 |
| Data collection | |||
| Space group | P1211 | P1211 | P1211 |
| Unit cell dimensions | |||
| a, b, c (Å) | 80.99, 154.58, 92.15 | 82.52, 157.96, 93.03 | 81.04, 154.98, 91.96 |
| α, β, γ (degrees) | 90, 102.54, 90 | 90, 101.08, 90 | 90, 102.36, 90 |
| Beamline | ALS 4.2.2 | ALS 4.2.2 | ALS 4.2.2 |
| Wavelength (Å) | 1.000030 | 1.000030 | 1.000030 |
| Oscillation range (degrees) | 180 | 180 | 180 |
| Resolution range (Å) | 77.75–2.32 (2.45–2.32) | 72.07–2.85 (3.00–2.85) | 77.72–2.75 (2.90–2.75) |
| Observation reflections | 632,664 (72,775) | 197,364 (26,617) | 416,317 (59,761) |
| Unique reflections | 92,793 (12,554) | 54,344 (7939) | 57,593 (8407) |
| Redundancy | 6.8 (5.8) | 3.6 (3.4) | 7.2 (7.1) |
| CC½ | 0.999 (0.847) | 0.998 (0.716) | 0.998 (0.878) |
| Completeness (%) | 97.2 (90.2) | 99.6 (99.9) | 100.0 (100.0) |
| I/σ(I) | 19.9 (2.4) | 14.8 (1.7) | 17.3 (3.2) |
| Rmerge (%) | 5.7 (58.4) | 6.0 (59.6) | 8.3 (56.4) |
| Rmeas (%) | 6.7 (70.3) | 8.3 (82) | 9.8 (66.4) |
| Wilson B-factor (Å2) | 44.3 | 69.0 | 52.2 |
| Refinement | |||
| Rwork (%) | 23.3 (32.8) | 24.6 (36.6) | 22.3 (28.6) |
| Rfree (%) | 27.5 (38.6) | 28.4 (42.3) | 26.5 (36.4) |
| Resolution range (Å) | 58.6–2.32 | 55.5–2.85 | 61.4–2.75 |
| No. of reflections | 92675 | 54194 | 57548 |
| No. of molecules/asu | 4 | 4 | 4 |
| No. of atoms | |||
| Protein | 14046 | 13426 | 13828 |
| Oxalate | 24 | 6 | 12 |
| Magnesium | 4 | 4 | 2 |
| Polyethylene glycol | – | 21 | – |
| Fructose 1,6-biphosphate | 80 | 80 | 80 |
| Chlorine | 2 | 1 | 2 |
| Glycerol | 102 | 71 | 72 |
| Potassium | 4 | 6 | 1 |
| Tris-hydroxymethyl-methyl-ammonium | 8 | ||
| Asn | 9 | ||
| Asp | 18 | ||
| Val | 16 | ||
| Water | 156 | 58 | 64 |
| Average B-factor (Å2) | |||
| Protein | 49.9 | 67.3 | 54.1 |
| Oxalate | 50.1 | 60.9 | 50.9 |
| Magnesium | 53.2 | 58.9 | 49.8 |
| Polyethylene glycol | 73.4 | ||
| Fructose 1,6-bisphosphate | 47.5 | 69.0 | 54.5 |
| Chlorine | 47.3 | 47.8 | 46.7 |
| Glycreol | 55.9 | 71.2 | 63.3 |
| Potassium | 63.3 | 77.4 | 50.5 |
| Tris-hydroxymethyl-methyl-ammonium | 62.5 | ||
| Asn | 52.6 | ||
| Asp | 61.9 | ||
| Val | 49.4 | ||
| Water | 46.5 | 60.5 | 48.4 |
| RMSD bond lengths (Å) | 0.006 | 0.006 | 0.003 |
| RMSD bond angles (degrees) | 0.66 | 0.76 | 0.60 |
| Ramachandran favored (%) | 96.1 | 92.6 | 94.8 |
| Ramachandran allowed (%) | 3.25 | 6.85 | 4.5 |
| Ramachandran outliers (%) | 0.62 | 0.56 | 0.66 |
Data availability
Atomic coordinates and structure factors have been deposited in the Protein Data Bank as entries 6V74, 6V75, and 6V76. All other data are contained within the paper. The amino acid sequence of this protein can be accessed through the UniProt KB Protein Database under UniProt no. P14618.
Author contributions
S. N. and M. D. data curation; S. N. and M. D. formal analysis; S. N. and M. D. validation; S. N. and M. D. investigation; S. N. and M. D. visualization; S. N. and M. D. methodology; S. N. writing-original draft; S. N. and M. D. writing-review and editing; M. D. conceptualization; M. D. resources; M. D. software; M. D. supervision; M. D. funding acquisition; M. D. project administration.
Supplementary Material
Acknowledgments
X-ray diffraction data were collected at beamline 4.2.2 of the Molecular Biology Consortium at the Advanced Light Source (ALS). We thank Dr. Jay Nix for help with the data collection. ALS is a Department of Energy Office of Science User Facility under contract DE-AC02-05CH11231. We acknowledge use of resources at the Carver College of Medicine's Protein Crystallography Facility at the University of Iowa. We thank Dr. Lokesh Gakhar for helping with X-ray data collection and data processing. We thank the reviewers for the insightful comments.
The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1–S5 and Figs. S1–S6.
- PKM2
- pyruvate kinase muscle isoform 2
- PEP
- phosphoenolpyruvate
- FBP
- fructose 1,6-bisphosphate
- SAICAR
- succinyl-5-aminoimidazole-4-carboxamide-1-ribose 5′-phosphate
- AA
- amino acid
- RMSD
- root mean square deviation
- MW
- molecular weight
- SA
- simulated annealing
- RMSD
- root mean square deviation
- R-state
- relaxed state
- T-state
- tense state
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- PDB
- Protein Data Bank
- LDH
- lactate dehydrogenase.
References
- 1. Anastasiou D., Yu Y., Israelsen W. J., Jiang J. K., Boxer M. B., Hong B. S., Tempel W., Dimov S., Shen M., Jha A., Yang H., Mattaini K. R., Metallo C. M., Fiske B. P., Courtney K. D., et al. (2012) Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol. 8, 839–847 10.1038/nchembio.1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ashizawa K., McPhie P., Lin K. H., and Cheng S. Y. (1991) An in vitro novel mechanism of regulating the activity of pyruvate kinase M2 by thyroid hormone and fructose 1,6-bisphosphate. Biochemistry 30, 7105–7111 10.1021/bi00243a010 [DOI] [PubMed] [Google Scholar]
- 3. Wu S., and Le H. (2013) Dual roles of PKM2 in cancer metabolism. Acta Biochim. Biophys. Sin. 45, 27–35 10.1093/abbs/gms106 [DOI] [PubMed] [Google Scholar]
- 4. Munday K. A., Giles I. G., and Poat P. C. (1980) Review of the comparative biochemistry of pyruvate-kinase. Comp. Biochem. Physiol. B 67, 403–411 10.1016/0305-0491(80)90328-4 [DOI] [PubMed] [Google Scholar]
- 5. Yacovan A., Ozeri R., Kehat T., Mirilashvili S., Sherman D., Aizikovich A., Shitrit A., Ben-Zeev E., Schutz N., Bohana-Kashtan O., Konson A., Behar V., and Becker O. M. (2012) 1-(Sulfonyl)-5-(arylsulfonyl)indoline as activators of the tumor cell specific M2 isoform of pyruvate kinase. Bioorg. Med. Chem. Lett. 22, 6460–6468 10.1016/j.bmcl.2012.08.054 [DOI] [PubMed] [Google Scholar]
- 6. Chen J., Xie J., Jiang Z., Wang B., Wang Y., and Hu X. (2011) Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 30, 4297–4306 10.1038/onc.2011.137 [DOI] [PubMed] [Google Scholar]
- 7. Dombrauckas J. D., Santarsiero B. D., and Mesecar A. D. (2005) Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry 44, 9417–9429 10.1021/bi0474923 [DOI] [PubMed] [Google Scholar]
- 8. Yan M., Chakravarthy S., Tokuda J. M., Pollack L., Bowman G. D., and Lee Y. S. (2016) Succinyl-5-aminoimidazole-4-carboxamide-1-ribose 5′-phosphate (SAICAR) activates pyruvate kinase isoform M2 (PKM2) in its dimeric form. Biochemistry 55, 4731–4736 10.1021/acs.biochem.6b00658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Keller K. E., Tan I. S., and Lee Y. S. (2012) SAICAR stimulates pyruvate kinase isoform M2 and promotes cancer cell survival in glucose-limited conditions. Science 338, 1069–1072 10.1126/science.1224409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Keller K. E., Doctor Z. M., Dwyer Z. W., and Lee Y. S. (2014) SAICAR induces protein kinase activity of PKM2 that is necessary for sustained proliferative signaling of cancer cells. Mol. Cell 53, 700–709 10.1016/j.molcel.2014.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chaneton B., Hillmann P., Zheng L., Martin A. C. L., Maddocks O. D. K., Chokkathukalam A., Coyle J. E., Jankevics A., Holding F. P., Vousden K. H., Frezza C., O'Reilly M., and Gottlieb E. (2012) Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 491, 458–462 10.1038/nature11540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ikeda Y., and Noguchi T. (1998) Allosteric regulation of pyruvate kinase M2 isozyme involves a cysteine residue in the intersubunit contact. J. Biol. Chem. 273, 12227–12233 10.1074/jbc.273.20.12227 [DOI] [PubMed] [Google Scholar]
- 13. Srivastava D., Nandi S., and Dey M. (2019) Mechanistic and structural insights into cysteine-mediated inhibition of pyruvate kinase muscle isoform 2. Biochemistry 58, 3669–3682 10.1021/acs.biochem.9b00349 [DOI] [PubMed] [Google Scholar]
- 14. Imamura K., Taniuchi K., and Tanaka T. (1972) Multimolecular forms of pyruvate kinase: II. Purification of M2-type pyruvate kinase from Yoshida ascites hepatoma 130 cells and comparative studies on the enzymological and immunological properties of the three types of pyruvate kinases, L, M1, and M2. J. Biochem. 72, 1001–1015 10.1093/oxfordjournals.jbchem.a129962 [DOI] [PubMed] [Google Scholar]
- 15. Schulz J., Sparmann G., and Hofmann E. (1975) Alanine-mediated reversible inactivation of tumour pyruvate kinase caused by a tetramer-dimer transition. FEBS Lett. 50, 346–350 10.1016/0014-5793(75)80524-2 [DOI] [PubMed] [Google Scholar]
- 16. Yuan M., McNae I. W., Chen Y., Blackburn E. A., Wear M. A., Michels P. A. M., Fothergill-Gilmore L. A., Hupp T., and Walkinshaw M. D. (2018) An allostatic mechanism for M2 pyruvate kinase as an amino-acid sensor. Biochem. J. 475, 1821–1837 10.1042/BCJ20180171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morgan H. P., O'Reilly F. J., Wear M. A., O'Neill J. R., Fothergill-Gilmore L. A., Hupp T., and Walkinshaw M. D. (2013) M2 pyruvate kinase provides a mechanism for nutrient sensing and regulation of cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 110, 5881–5886 10.1073/pnas.1217157110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Warburg O. (1956) On the origin of cancer cells. Science 123, 309–314 10.1126/science.123.3191.309 [DOI] [PubMed] [Google Scholar]
- 19. Tennant D. A., Durán R. V., and Gottlieb E. (2010) Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer 10, 267–277 10.1038/nrc2817 [DOI] [PubMed] [Google Scholar]
- 20. Spoden G. A., Rostek U., Lechner S., Mitterberger M., Mazurek S., and Zwerschke W. (2009) Pyruvate kinase isoenzyme M2 is a glycolytic sensor differentially regulating cell proliferation, cell size and apoptotic cell death dependent on glucose supply. Exp. Cell Res. 315, 2765–2774 10.1016/j.yexcr.2009.06.024 [DOI] [PubMed] [Google Scholar]
- 21. Spoden G. A., Mazurek S., Morandell D., Bacher N., Ausserlechner M. J., Jansen-Dürr P., Eigenbrodt E., and Zwerschke W. (2008) Isotype-specific inhibitors of the glycolytic key regulator pyruvate kinase subtype M2 moderately decelerate tumor cell proliferation. Int. J. Cancer 123, 312–321 10.1002/ijc.23512 [DOI] [PubMed] [Google Scholar]
- 22. Williams R., Holyoak T., McDonald G., Gui C., and Fenton A. W. (2006) Differentiating a ligand's chemical requirements for allosteric interactions from those for protein binding: phenylalanine inhibition of pyruvate kinase. Biochemistry 45, 5421–5429 10.1021/bi0524262 [DOI] [PubMed] [Google Scholar]
- 23. Brodzki A., Tatara M. R., Pasternak K., Rozanska D., and Szponder T. (2005) Free amino acids in skin neoplastic tissues and serum in dogs. Bull. Vet. Inst. Pulawy 49, 231–235 [Google Scholar]
- 24. Lukey M. J., Katt W. P., and Cerione R. A. (2017) Targeting amino acid metabolism for cancer therapy. Drug Discov. Today. 22, 796–804 10.1016/j.drudis.2016.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Canepa A., Filho J. C., Gutierrez A., Carrea A., Forsberg A. M., Nilsson E., Verrina E., Perfumo F., and Bergström J. (2002) Free amino acids in plasma, red blood cells, polymorphonuclear leukocytes, and muscle in normal and uraemic children. Nephrol. Dial. Transplant. 17, 413–421 10.1093/ndt/17.3.413 [DOI] [PubMed] [Google Scholar]
- 26. Macpherson J. A., Theisen A., Masino L., Fets L., Driscoll P. C., Encheva V., Snijders A. P., Martin S. R., Kleinjung J., Barran P. E., Fraternali F., and Anastasiou D. (2019) Functional cross-talk between allosteric effects of activating and inhibiting ligands underlies PKM2 regulation. Elife 8, e45068 10.7554/eLife.45068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kung C., Hixon J., Choe S., Marks K., Gross S., Murphy E., DeLaBarre B., Cianchetta G., Sethumadhavan S., Wang X., Yan S., Gao Y., Fang C., Wei W., Jiang F., et al. (2012) Small molecule activation of PKM2 in cancer cells induces serine auxotrophy. Chem. Biol. 19, 1187–1198 10.1016/j.chembiol.2012.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jurica M. S., Mesecar A., Heath P. J., Shi W., Nowak T., and Stoddard B. L. (1998) The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure 6, 195–210 10.1016/S0969-2126(98)00021-5 [DOI] [PubMed] [Google Scholar]
- 29. Wang P., Sun C., Zhu T., and Xu Y. (2015) Structural insight into mechanisms for dynamic regulation of PKM2. Protein Cell 6, 275–287 10.1007/s13238-015-0132-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Srivastava D., Razzaghi M., Henzl M. T., and Dey M. (2017) Structural investigation of a dimeric variant of pyruvate kinase muscle isoform 2. Biochemistry 56, 6517–6520 10.1021/acs.biochem.7b01013 [DOI] [PubMed] [Google Scholar]
- 31. Kalaiarasan P., Kumar B., Chopra R., Gupta V., Subbarao N., and Bamezai R. N. (2015) In silico screening, genotyping, molecular dynamics simulation and activity studies of SNPs in pyruvate kinase M2. PLoS ONE 10, e0120469 10.1371/journal.pone.0120469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sborov D. W., Haverkos B. M., and Harris P. J. (2015) Investigational cancer drugs targeting cell metabolism in clinical development. Expert Opin. Investig. Drugs 24, 79–94 10.1517/13543784.2015.960077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. DeLano W. L. (2012) The PyMOL Molecular Graphics System, version 1.5.0.1, Schroedinger, LLC, New York [Google Scholar]
- 34. Hattori A., Tsunoda M., Konuma T., Kobayashi M., Nagy T., Glushka J., Tayyari F., McSkimming D., Kannan N., Tojo A., Edison A. S., and Ito T. (2017) Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 545, 500–504 10.1038/nature22314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shan Y., Gao Y., Jin W., Fan M., Wang Y., Gu Y., Shan C., Sun L., Li X., Yu B., Luo Q., and Xu Q. (2019) Targeting HIBCH to reprogram valine metabolism for the treatment of colorectal cancer. Cell Death Dis. 10, 618 10.1038/s41419-019-1832-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alkan H. F., and Bogner-Strauss J. G. (2019) Maintaining cytosolic aspartate levels is a major function of the TCA cycle in proliferating cells. Mol. Cell. Oncol. 6, e1536843 10.1080/23723556.2018.1536843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang J., Fan J., Venneti S., Cross J. R., Takagi T., Bhinder B., Djaballah H., Kanai M., Cheng E. H., Judkins A. R., Pawel B., Baggs J., Cherry S., Rabinowitz J. D., and Thompson C. B. (2014) Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol. Cell 56, 205–218 10.1016/j.molcel.2014.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Krall A. S., Xu S., Graeber T. G., Braas D., and Christofk H. R. (2016) Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 7, 11457 10.1038/ncomms11457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sullivan L. B., Luengo A., Danai L. V., Bush L. N., Diehl F. F., Hosios A. M., Lau A. N., Elmiligy S., Malstrom S., Lewis C. A., and Vander Heiden M. G. (2018) Aspartate is an endogenous metabolic limitation for tumour growth. Nat. Cell Biol. 20, 782–788 10.1038/s41556-018-0125-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mattaini K. R., Sullivan M. R., and Vander Heiden M. G. (2016) The importance of serine metabolism in cancer. J. Cell Biol. 214, 249–257 10.1083/jcb.201604085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Eigenbrodt E., Reinacher M., Scheefers-Borchel U., Scheefers H., and Friis R. (1992) Double role for pyruvate kinase type M2 in the expansion of phosphometabolite pools found in tumor cells. Crit. Rev. Oncog. 3, 91–115 [PubMed] [Google Scholar]
- 42. Yang J., Liu H., Liu X., Gu C., Luo R., and Chen H. F. (2016) Synergistic allosteric mechanism of fructose-1,6-bisphosphate and serine for pyruvate kinase M2 via dynamics fluctuation network analysis. J. Chem. Inf. Model. 56, 1184–1192 10.1021/acs.jcim.6b00115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schnicker N. J., and Dey M. (2016) Bacillus anthracis prolyl 4-hydroxylase modifies collagen-like substrates in asymmetric patterns. J. Biol. Chem. 291, 13360–13374 10.1074/jbc.M116.725432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brummett A. E., Schnicker N. J., Crider A., Todd J. D., and Dey M. (2015) Biochemical, kinetic, and spectroscopic characterization of Ruegeria pomeroyi DddW–a mononuclear iron-dependent DMSP lyase. PLoS ONE 10, e0127288 10.1371/journal.pone.0127288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liebschner D., Afonine P. V., Moriarty N. W., Poon B. K., Sobolev O. V., Terwilliger T. C., and Adams P. D. (2017) Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr. D Biol. Crystallgr. 73, 148–157 10.1107/S2059798316018210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Berman H., Henrick K., and Nakamura H. (2003) Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 10, 980 10.1038/nsb1203-980 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors have been deposited in the Protein Data Bank as entries 6V74, 6V75, and 6V76. All other data are contained within the paper. The amino acid sequence of this protein can be accessed through the UniProt KB Protein Database under UniProt no. P14618.