

Abstract
Heme-regulatory motifs (HRMs) are present in many proteins that are involved in diverse biological functions. The C-terminal tail region of human heme oxygenase-2 (HO2) contains two HRMs whose cysteine residues form a disulfide bond; when reduced, these cysteines are available to bind Fe3+-heme. Heme binding to the HRMs occurs independently of the HO2 catalytic active site in the core of the protein, where heme binds with high affinity and is degraded to biliverdin. Here, we describe the reversible, protein-mediated transfer of heme between the HRMs and the HO2 core. Using hydrogen-deuterium exchange (HDX)-MS to monitor the dynamics of HO2 with and without Fe3+-heme bound to the HRMs and to the core, we detected conformational changes in the catalytic core only in one state of the catalytic cycle—when Fe3+-heme is bound to the HRMs and the core is in the apo state. These conformational changes were consistent with transfer of heme between binding sites. Indeed, we observed that HRM-bound Fe3+-heme is transferred to the apo-core either upon independent expression of the core and of a construct spanning the HRM-containing tail or after a single turnover of heme at the core. Moreover, we observed transfer of heme from the core to the HRMs and equilibration of heme between the core and HRMs. We therefore propose an Fe3+-heme transfer model in which HRM-bound heme is readily transferred to the catalytic site for degradation to facilitate turnover but can also equilibrate between the sites to maintain heme homeostasis.
Keywords: heme oxygenase, hydrogen exchange mass spectrometry, enzyme mechanism, enzyme kinetics, protein dynamics, membrane protein, metalloprotein, conformational change, protein-protein interaction, conformational change, heme-regulatory motif, heme-binding site, intramolecular interaction, hydrogen-deuterium exchange mass spectrometry (HDX-MS)
Introduction
Heme degradation is a critical process in maintaining levels of this vital cofactor in living organisms. In humans, heme oxygenase (HO)3 is responsible for degrading heme to biliverdin through a pathway that requires cytochrome P450 reductase (CPR), NADPH, and O2. Curiously, however, humans and other amniotes express two isoforms of heme oxygenase, designated heme oxygenase-1 (HO1) and heme oxygenase-2 (HO2). HO1 and HO2 degrade heme with similar catalytic efficiencies (1) and have 55% sequence identity as well as a high degree of structural homology in the central core regions of the proteins in which heme binds at a conserved His residue (2, 3). The underlying reason for why both isoforms, given their similarities, are expressed is still not clear; however, distinguishing features of the two proteins continue to be identified that may provide insights.
Of particular interest in the quest to understand the unique characteristics of HO2 is the presence of two heme-regulatory motifs (HRMs), conserved sequences centered at a Cys-Pro dipeptide followed by a hydrophobic residue that can ligate Fe3+-heme via the Cys residue (4). The HRMs of HO2 are found in the region C-terminal to the catalytic core (approximately residues 249–288 in human HO2) that has little sequence homology with HO1, which lacks HRMs or even any Cys residues. Centered at Cys265-Pro266 and Cys282-Pro283 in human HO2, the two HRM sites bind Fe3+-heme independently of the core upon reduction of a disulfide bond between Cys265 and Cys282 (5, 6). These HRMs are conserved among all amniotes. Whereas Fe3+-heme coordinates to His45 in the core of the protein with high affinity (5, 6), the affinity of the HRMs for Fe3+-heme is weaker. The affinity of the Cys265 site (HRM1), which utilizes His256 as an additional axial heme ligand, is ∼2–6-fold weaker than the core, and the Cys282 site (HRM2) is yet another ∼10-fold weaker than HRM1 (5). The wide range of affinities at the three sites suggests that HO2 potentially senses and responds to Fe3+-heme levels over a wide range of concentrations, but the details of that response to heme binding at the HRMs has remained unclear.
HRM-mediated heme-dependent regulation occurs in several other proteins involved in heme metabolism. Despite the fact that HO1 does not contain any HRMs, its expression is regulated in an Fe3+-heme-dependent manner by the HRM-containing protein Bach-1 (7). Bach-1 acts as a repressor of HO1 in the absence of heme, but binding of Fe3+-heme to the HRMs of the protein prevents interactions with DNA, relieving repression (8). Additionally, Bach-1 is targeted for nuclear export, polyubquitination, and degradation upon heme binding to the HRMs (9, 10). Likewise, Fe3+-heme binding to an HRM of the enzyme that catalyzes the rate-limiting step of heme biosynthesis, aminolevulinic acid synthase (ALAS1), also targets this protein for degradation (11). Therefore, it appears that HRMs play an important role in regulating heme levels, and HO2 could logically participate in overall heme homeostasis through its HRMs, but nothing thus far has pointed to a clear rationale. HO2 expression has not been shown to be heme-dependent in the same way as HO1 (1), steady-state activity of HO2 is not altered in the absence of the HRMs (5, 6, 12, 13), and the Cys residues of the HRMs do not directly participate in the catalytic degradation of heme in HO2 (12).
The commonality between Bach-1 and ALAS1 is that both proteins are degraded upon Fe3+-heme binding to their respective HRMs. Indeed, other examples of HRM-containing proteins that are similarly targeted for degradation include p53 (14) and the circadian factors Period 2 (15) and Rev-erbβ (16), among others. However, as described in a recent review (17), no single function has been assigned to all HRMs, and not all HRMs have been assigned a function. Whereas it may then seem that comparing HO2 to other HRM-containing proteins offers no insights into HO2 function, a model proposed by Ikeda-Saito and co-workers (18) regarding Fe3+-heme binding to Bach-1 offered a different perspective on HRM function. In the model, Fe3+-heme binding to the HRMs of Bach-1 induces local conformational changes that alter interactions with its binding partners, DNA and Crm1. Crm1 is necessary for the heme-dependent nuclear export of Bach-1 (9).
We hypothesized that in HO2, similar heme-induced conformational changes affect intramolecular interactions between the HRM-containing tail and core regions of HO2. Furthermore, we hypothesized that these interactions promote transfer of Fe3+-heme from the tail to the core, effectively reloading the active site with heme at each catalytic turnover. These hypotheses are supported by our previous NMR characterization of HO2 (19), which suggested that the HRM-containing tail could dock onto the catalytic core when Fe3+-heme is limiting and the Cys in the HRMs are in the reduced, dithiol state (Fe3+core-HO2R; see Fig. 1). A subsequent study using hydrogen-deuterium exchange MS (HDX-MS) did not uncover any evidence of docking when both the core and the HRMs were fully occupied with Fe3+-heme (Fe3+core/HRM-HO2R) (20). To fully examine our hypothesis that the tail participates in heme transfer to the catalytic core, it was necessary to examine a construct designed to have a heme-loaded tail and an apo-core (Fe3+HRM-HO2R). However, due to the heme-binding affinities of the various sites on HO2 (with the core having the highest affinity), this state, where the HRMs are occupied with Fe3+-heme but the core is not, is not straightforward to study. Therefore, we devised strategies to obtain this form of the protein, allowing us to monitor changes in the protein that are specific to Fe3+-heme binding solely to the HRMs. Here, we demonstrate that heme-dependent interactions between the core and the HRM-containing region appear to facilitate Fe3+-heme transfer between the sites on HO2.
Figure 1.

A model of Fe3+-heme binding to HO2 and transfer of Fe3+-heme between binding sites. Upon reduction of a disulfide bond between Cys265 and Cys282, the HRMs as well as the core (His45) are available to bind heme, forming the Fe3+core/HRM-HO2R species. Heme is not shown to be bound to Cys282 in this model both for simplicity and because we expect this HRM to only bind heme under limiting circumstances due to the very weak affinity of the HRM. After turnover of heme to biliverdin (BV) in the presence of CPR, NADPH, and O2, HO2 is left in the Fe3+HRM-HO2R state: heme-loaded HRM and apo-core. Heme transfer from the HRM to the core results in the catalytically active Fe3+core-HO2R species. Our results also demonstrate that the heme transfer is reversible, allowing heme to equilibrate between the sites.
Results
Generation of an HO2 variant with an apo-core and fully heme-loaded HRMs
Due to the higher affinity of the core for heme than the HRMs (5), the form of the protein that has eluded us thus far is that in which heme is bound to the HRMs but not in the core (Fe3+HRM-HO2R). We therefore designed a variant of HO2 in which the heme-binding pocket could not accommodate heme, keeping the core in an “apo” state without perturbing heme binding to the HRMs. Previously, even with the substitution of His25, which directly coordinates to heme, or the substitution of Gly139, which coordinates to heme through an aquo ligand in the other axial position, heme was still able to insert itself into the HO1 heme-binding pocket (21–23). Given the conservation of these residues between HO1 and HO2, we replaced both His45 and Gly159 in HO2 (corresponding to His25 and Gly139 in HO1) with Trp in a soluble, truncated form of HO2 spanning residues 1–288 (HO2sol). The double variant (H45W/G159W), like WT (6), was isolated with a disulfide bond (0.84 ± 0.01 free thiols under denaturing conditions; HO2 contains three cysteines). Therefore, we reduced the H45W/G159W protein with tris(2-carboxyethyl)phosphine (TCEP) under anaerobic conditions, removed excess TCEP, and incubated the protein with 3 eq of heme (for three potential but only two hypothesized binding sites). After separation of protein from unbound heme, the spectrum of the protein was nearly identical to that of the heme-bound form of a truncated form of the protein, HO2tail (the “tail”), that spans residues 213–288, in which heme binds to both HRM1 and HRM2 (5) (Fig. 2). The results indicate that heme is not binding to the core. The spectrum of the heme-bound variant of HO2sol differs from the HO2tail spectrum only in the region around 280 nm, which can be accounted for by the difference in amino acid composition between the two constructs. HO2sol contains one Trp (plus an additional two in the H45W/G159W variant) and 12 Tyr, whereas HO2tail only contains two Tyr residues and no Trp.
Figure 2.
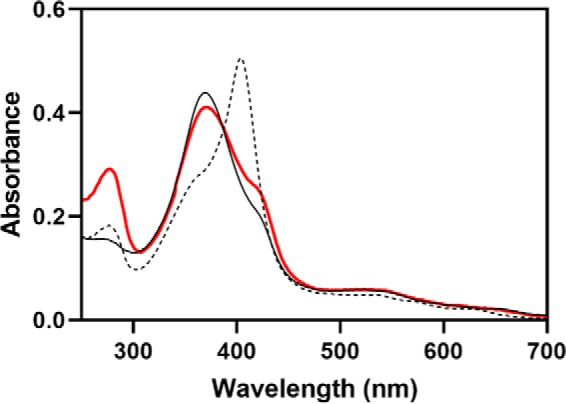
Characterization of the H45W/G159W variant of HO2. The absorbance spectrum of TCEP-reduced H45W/G159W HO2solR incubated with Fe3+-heme and separated from unbound heme is shown in red. The spectrum is of 5 μm protein in 50 mm Tris (pH 8.0), 50 mm KCl buffer at 20 °C. For comparison, the spectra of WT HO2tail, which binds heme at both the Cys265/His256 (HRM1) and Cys282 (HRM2) sites (solid black), and WT HO2solO, which binds heme at the core with His45 (dashed black), are shown.
Before testing the effect of heme binding to the HRMs on the structure and dynamics of H45W/G159W, we used CD spectroscopy and HDX-MS to ensure that the mutations at the catalytic site did not drastically perturb the overall HO2 structure. The similarities in the CD spectra of the WT and the H45W/G159W variant of a truncated form of HO2 spanning residues 1–248 (HO2core) suggested that the introduction of the two Trp residues in the catalytic site did not disturb the structure of the protein core appreciably (Fig. S1). Further, in comparing the H45W/G159W variant with WT HO2solO by HDX-MS, both lacking heme at the core and tail, there were not global or large-scale differences in deuteration, arguing against large perturbations due to the mutations. The only observed differences in exchange were specific to regions associated with heme binding (Fig. 3). These regions include the distal and proximal helices, which form the heme-binding pocket; the combined H45W and G159W substitutions resulted in protection against deuterium exchange relative to the WT protein. The distal helix, which has a high glycine content, is flexible in nature, allowing it to move in response to heme binding (3). Introduction of a Trp into the distal helix likely introduced more rigidity into this helix. There was slight deprotection in the residues C-terminal to the distal helix. Last, a significant level of protection in the area around residue 100 was observed in the H45W/G159W variant. We have previously noted protection from exchange in this region upon heme binding to the core of HO2, when protection is also observed in the distal helix (20), suggesting that the two regions interact in some way. Thus, overall, the H45W/G159W variant appears to have a similar global structure as WT HO2sol, with the substitutions having introduced some rigidity into the heme-binding pocket. These results also indicate that the H45W/G159W variant could serve as a mimic of the apo-WT protein in our investigation of the effect of heme binding at the HRM on the structure and dynamics of HO2 lacking heme at the core.
Figure 3.

HDX-MS comparisons of HO2sol. A, differences in deuterium incorporation between the apo states of the H45W/G159W variant and WT HO2sol. The differences in deuterium level are shown for labeling times 10 s, 1 min, 10 min, 1 h, and 4 h (left to right of each graph). In each panel, the peptic peptides where deuterium was measured are arranged N- to C-terminally (top to bottom), with approximate sequence numbering on the left side. Deuterium differences were calculated using the equation DH45W/G159W − DWT for each comparison and then colored according to the scale shown. A guide to the regions of each protein is shown at the far right. All data used to create this figure can be found in supporting Excel File S1. B, differences in deuterium incorporation between the heme-bound, TCEP-reduced state and the apo state of the H45W/G159W variant of HO2sol. Deuterium differences were calculated using the equation Dheme − Dapo for each comparison and then colored according to the scale shown C, mapping the data from the 10-s differences in B onto the structure for HO2 (Protein Data Bank code 2RGZ).
HDX-MS reveals that the HRM region interacts with the core of an HO2 variant in an Fe3+-heme–dependent manner
To probe the structural effects of heme binding to the HRMs when the core was unable to bind heme, we monitored HDX-MS of the H45W/G159W variant in two states: Fe3+HRM-HO2solR and apo-HO2solO. The dynamics of very specific regions were altered at early time points due to the presence of heme bound to the HRMs (Fig. 3). Upon the addition of heme to the HRMs of the H45W/G159W variant, the proximal helix experienced an increase in deuterium uptake, whereas a decrease was observed for the distal helix and the HRM-containing region (Fig. 3). Because we compared the H45W/G159W variant to itself (not to WT protein), changes in exchange were strictly due to the presence of heme, not the mutations themselves. Further, the changes observed with the H45W/G159W variant are quite distinct from those of previous observations in which heme was in the core (20). In those experiments, no difference in uptake was observed in WT Fe3+core/HRM-HO2solR relative to WT Fe3+core-HO2solO. Therefore, the protection observed here is not due to the C-terminal tail gaining any structure but, rather, is due to a direct interaction of the HRM-containing region with the core of the protein. Supporting this interpretation is the fact that all of the changes in dynamics are on the same time scale, suggesting that the events are related. Further, the time scale (10 s) on which the changes occur leads us to believe that this is a transient interaction between the two regions of the protein.
Fe3+-heme is transferred between the catalytic binding site in the core of HO2 and HRM1
The observed docking of the HRM region to the core when the HRMs, but not the core, were occupied with heme suggested to us that heme could be transferred between the binding sites. To test this hypothesis, we used two truncated forms of HO2 that have been characterized previously (5, 12, 20): HO2core, which spans residues 1–248 and includes the N terminus and the catalytic core of HO2, and HO2tail, which spans the HRM-containing region (residues 213–288). Here, we used a C282A variant of HO2tail; this variant reduces the number of heme-binding sites to one, simplifying the reaction. Because of the very weak affinity of the HRM centered at Cys282 (5), we expect this HRM to only bind heme under limiting circumstances; therefore, we focused on the HRM1 (Cys265/His256)-binding site, which has a more physiologically relevant heme-binding affinity. By essentially separating the protein into two parts, we were able to dictate which heme-binding sites were initially occupied. Further, because the heme-bound species of these two constructs have distinct spectral features (5), we were also able to monitor changes in binding site occupancy over time that would suggest transfer of heme between binding sites.
Upon mixing the heme-bound form of C282A HO2tail with the apo-form of HO2core, we observed changes consistent with heme leaving the Cys265 site and binding to the catalytic site. The initial increase in absorbance at 404 nm, indicative of heme binding to the catalytic core, was concurrent with an initial decrease in absorbance at 423 nm, indicative of heme leaving the HRM region (Fig. 4). After the initial changes, a slow, second phase in which the absorbance at 404 nm decreases was also observed, which will be discussed below. At a 1:1 molar ratio of HO2tail to HO2core, which would be the ratio in the full-length protein, the initial rates observed were 0.017 and 0.016 s−1 when monitoring at 404 and 423 nm, respectively. However, as the concentration of apo-HO2core was increased in the reaction, the rates remain unchanged.
Figure 4.

Fe3+-heme transfer or exchange between proteins. A, heme-bound C282A HO2tail (5 μm final concentration) was rapidly mixed with an equimolar concentration of apo-HO2core in a stopped-flow instrument within an anaerobic chamber. The data at 0.007 s (red), 6 s (green), 1000 s (blue), and several representative intermediate time points (black) are shown. All measurements were carried out at 20 °C using the 1-cm path length configuration in photodiode array mode with 2–4 replicates. B, the stopped-flow trace at 404 nm (blue) and 423 nm (red) from one of the replicates is shown. The data were fit to a double-exponential equation (black) using the Pro-data Viewer software provided by Applied Photophysics. C, the experiment described in A was repeated at varying concentrations of apo-HO2core, and the data at 404 nm were treated as described in B by fitting to a double-exponential equation. The rates were plotted as a function of the final concentration of apo-HO2core in the assay with k1 in blue on the left y axis and k2 in orange on the right y axis. Data represent the average ± S.D. (error bars) of 2–4 acquisitions. D–F, same as A–C except that heme-bound apo-HO2core was mixed with varying concentrations of C282A HO2tail. G–I, same as A except that heme-bound C282A HO2tail was mixed with varying concentrations of apo-H64Y/V68F-myoglobin (“green heme”), and the data at 600 nm were fit to a single-exponential equation.
The independence of the rate of transfer is instead reminiscent of the kinetics of heme exchange between holoproteins and apo-H64Y/V68F-myoglobin (“green heme”), which has become the standard assay for determining heme off-rates (24). Exchange between a heme-bound protein and another protein, which is in the apo-form, can be described by Reaction 1 (24),
where PH is the heme-bound protein, P is the apoprotein, H is free heme, Y is the second protein (usually apo-H64Y/V68F-myoglobin), and YH is the second protein in the heme-bound form. Assuming that the concentration of free heme is extremely low, Equation 1 is used to describe the observed rate constant (24).
| (Eq. 1) |
However, in the presence of excess Y, kobs = k−H, the heme off-rate for the protein that initially was in the heme-bound form, and the observed rate is independent of protein concentration. Indeed, when we used the same protein concentrations in the heme exchange reaction, but using apo-H64Y/V68F-myoglobin instead of apo-HO2core, the rate was independent of the concentration of the myoglobin (Fig. 4).
One key difference in the heme exchange reaction when using myoglobin rather than HO2core, however, is that the initial rate of heme transfer from C282A HO2tail to apo-HO2core (0.017 s−1) is faster than the rate of heme dissociation into solution (0.005 s−1) for heme-bound C282A HO2tail as determined by the apo-myoglobin assay despite the fact that the rate of association of heme from solution to HO2(1–248) (4.1 × 106 m−1 s−1) is similar to that of myoglobin (4.4 × 106 m−1 s−1) (25). The faster rate of heme transfer to the core than heme dissociation into solution thus yields some evidence of a protein-mediated heme transfer, as suggested by the HDX-MS results above, despite the fact that we have “unlinked” the two regions of the protein.
The other key difference in the heme exchange reactions relates to the second phase of the heme transfer reaction between heme-bound C282A HO2tail and apo-HO2core. As noted above, when monitoring the reaction at 404 nm (due to heme binding to the core), we observed a slow, second phase, which actually decreased in absorbance over time. A decrease in absorbance indicates loss of heme from the core, whereas absorbance for heme-bound H64Y/V68F-myoglobin remains steady over time. We hypothesize that the difference in the kinetic traces is due to the greater affinity of heme for H64Y/V68F-myoglobin relative to the HO2core, as reflected by their heme off-rates, 10−4–10−6 s−1 versus 10−2–10−3 s−1, respectively (25). Indeed, when we increased the heme affinity of HO2core by 100-fold to match that of myoglobin by substituting Gly163 with His (i.e. decreasing the off-rate to 10−4–10−6 s−1 (Fig. S2)), we no longer observed a second phase with decreasing absorbance (Fig. 5). In addition, the rate of heme transfer from C282A HO2tail to apo-G163H HO2core (0.022 s−1) is slightly faster than the rate of transfer to WT (0.017 s−1). This substitution creates a heme-binding pocket that can coordinate heme with bis-His ligation (26).
Figure 5.
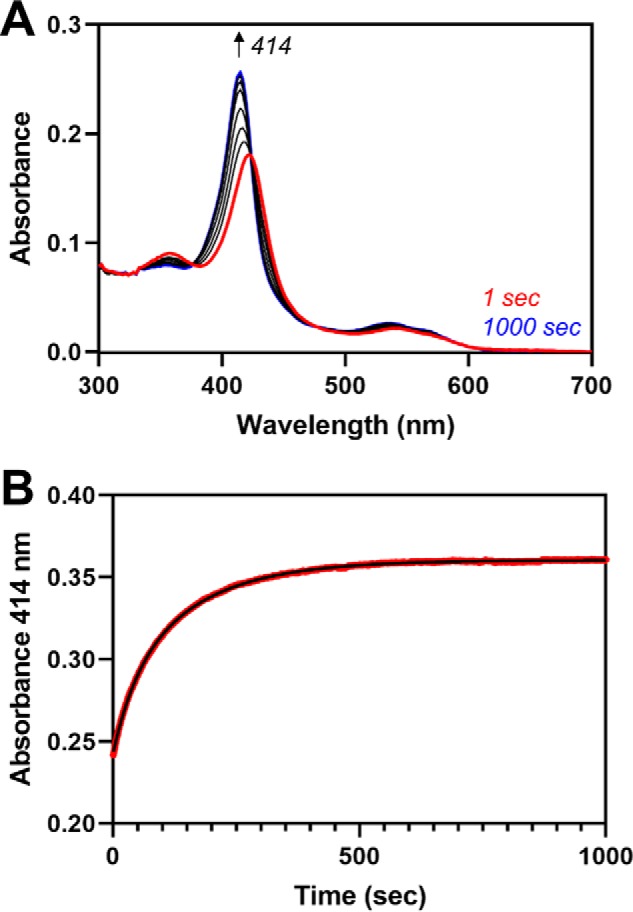
Fe3+-heme transfer between heme-bound C282A HO2tail and apo-G163H HO2core. A, heme-bound C282A HO2tail was mixed with an equimolar concentration of apo-G163H HO2core in a stopped-flow instrument within an anaerobic chamber as described in Fig. 4A. B, the absorbance change over time at 414 nm (red) was fit to a double-exponential equation (black).
A potential destination for heme released (or transferred) from HO2core in the presence of C282A HO2tail is back to the HRM-binding site. We have previously demonstrated that the heme off-rate for C282A HO2tail is slower than HO2core, resulting in weaker affinity for the HRM than the core (5). However, the heme association rates at the two sites are very similar (5). Therefore, we asked if heme could be transferred from the catalytic binding site to the HRMs of HO2. Indeed, when we mixed the apo-form of C282A HO2tail with the heme-bound form of HO2core, we observed changes consistent with heme leaving the catalytic site and binding to Cys265. The decrease in absorbance at 404 nm, indicative of heme leaving the catalytic core, was concurrent with an increase in absorbance at 423 nm, indicative of heme binding to HRM1 (Cys265/His256) (Fig. 4). After the initial changes, a slow, second phase in which the absorbance at 404 nm decreases was also observed. At a 1:1 molar ratio of HO2tail to HO2core, which would be the ratio in the full-length protein, the initial rate was 0.039 s−1 when monitoring at 404 nm, which is faster than heme transfer in the opposite direction, and the second, slow phase was 0.00013 s−1. Whereas the rate of the second phase was independent of the concentration, the rate of the first phase decreases with increasing apo-C282A HO2tail concentrations, suggestive of a protein-mediated transfer through conformational selection in which a ligand preferentially binds to a particular conformation of a protein (27) (see “Discussion”). Overall, the data indicate that heme is not transferred unidirectionally, but rather, heme is transferred between the sites.
Fe3+-heme equilibrates between binding sites on HO2
To complement the stopped-flow results just described, we performed assays to investigate the reaction on longer time scales. Specifically, we were interested in how heme equilibrates over longer time frames between the two sites. Previously, HO2core was used as the competitive heme-binding protein in an equilibrium-based competition assay (25). The assay is very similar to the assays performed in the stopped-flow experiments described above but monitors the end of the reaction rather than the initial phases. Simply put, a heme-bound protein (C282A HO2tail was used here) is incubated for long periods with varying concentrations of apo-HO2core, which acts as a competitor for heme. A plot of the fraction of heme bound to HO2core versus the concentration of added protein yields the Kd value.
Incubation of heme-bound C282A HO2tail with varying concentrations of the apo-form of HO2core demonstrated that heme equilibrates between the two sites (Fig. 6). A fit of the plot described above, using the Kd value of 3.6 nm for HO2core (25), yielded a Kd value of 54.9 nm for C282A HO2tail. The calculated Kd value is higher than previously calculated by measuring the rates of heme dissociation and association but lower than that determined by equilibrium heme titration (5). Whereas the difference in affinity between the core and C282A HO2tail was only 2–6-fold in the previous experiments, the competition assay here suggested a 15-fold difference in affinity. The most interesting aspect to the results, however, is that at near 1:1 protein levels (core to C282A HO2tail), the fraction of heme in the core began to level off at ∼60–70%.
Figure 6.
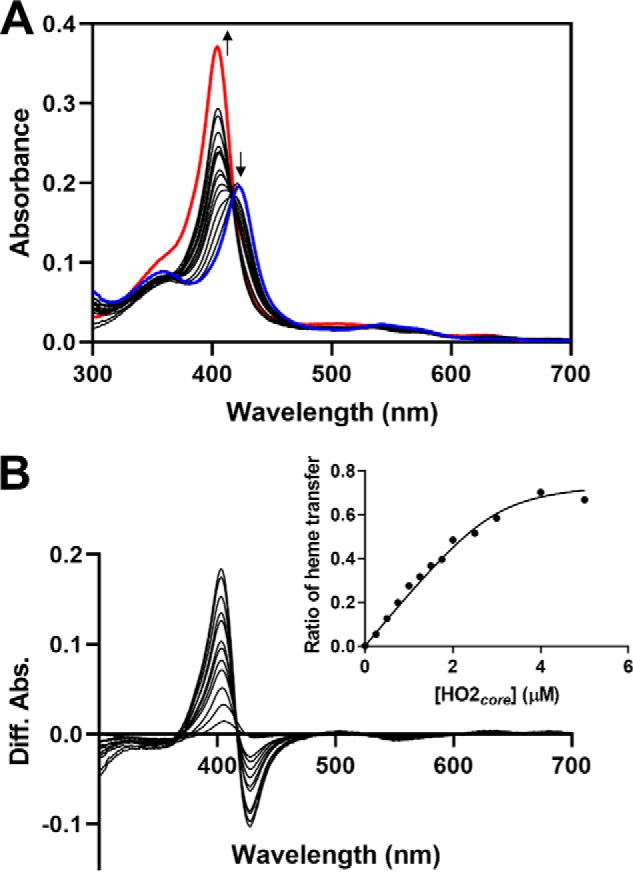
Fe3+-heme equilibration between C282A HO2tail and HO2core. Increasing concentrations of apo-HO2core were incubated with 3 μm heme-bound C282A HO2tail in an anaerobic chamber. A, after reaching equilibrium, UV-visible spectra were recorded. For comparison, the spectrum of heme-bound C282A HO2tail is shown in blue, and the spectrum of heme-bound HO2core is shown in red. B, difference spectra were generated from the data in A by subtracting the absorbance of heme-bound C282A HO2tail in the absence of added apo-HO2core (blue spectrum in A) from each spectrum. The ratio of heme transfer was calculated by normalizing the difference in absorbance at 404 nm (due to the formation of heme-bound HO2core) to the absorbance of fully heme-bound HO2core (in the absence of added C282A HO2tail). As shown in the inset, he ratio was plotted as a function of HO2core concentration and fit with the Morrison equation for tight-binding inhibitors.
We next asked whether heme also equilibrates between the binding sites in HO2sol, containing both the core and tail. Because we cannot vary the concentration of one site relative to the other as with the two separate proteins, we varied the concentration of heme that was incubated with the C282A variant of HO2solR. Difference spectra created by subtracting heme-bound HO2core from the spectra of C282A HO2sol at varying heme concentrations reveal that a fraction of heme binds at HRM1 even at low heme concentrations (∼0.5:1 heme/protein or ∼0.25:1 heme/total binding sites) under equilibrium conditions (Fig. 7). Further, to correlate the results of the assay using C282A HO2sol to the results of the competition assay using the two separate proteins above, we calculated how much heme was bound to the core of C282A HO2sol when incubated with equimolar heme (equivalent to 1:1 core/C282A HO2tail above). Consistent with the results above, ∼70% of the heme was bound to the core of the protein, suggesting that heme equilibrates between sites in the more full-length protein similarly to the two truncated proteins.
Figure 7.
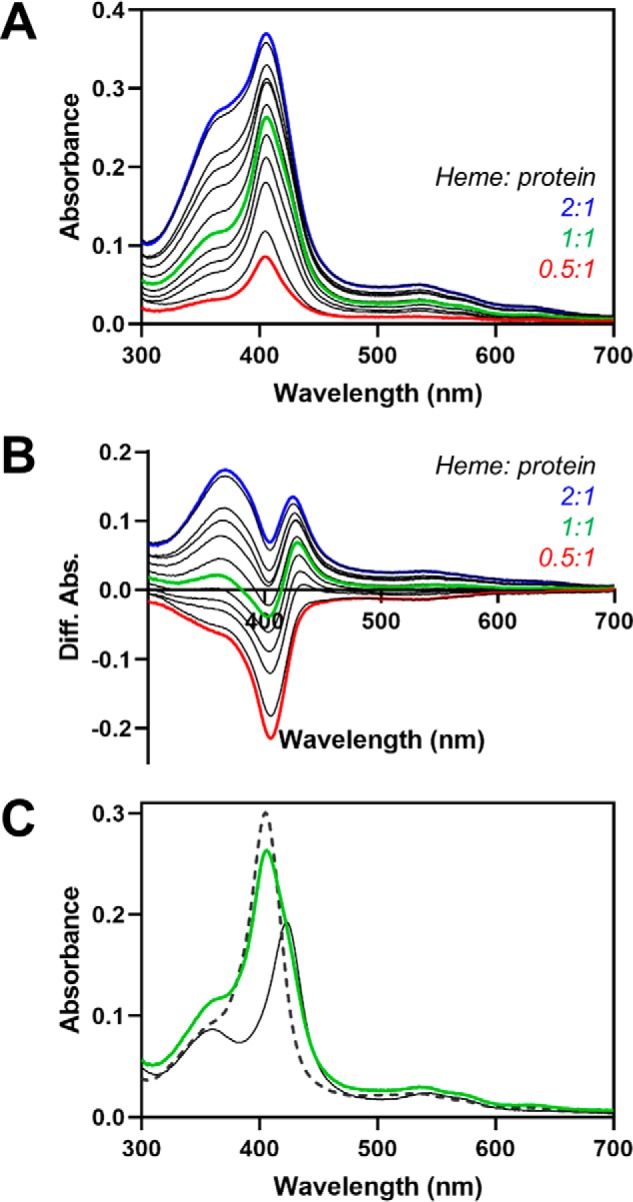
Fe3+-heme equilibration between HRM1 and the core in C282A HO2solR. Increasing concentrations of heme were incubated with C282A HO2solR in an anaerobic chamber. A, after reaching equilibrium, UV-visible spectra were recorded. B, difference spectra were generated by subtracting the absorbance of heme-bound C282A HO2tail (solid black spectrum in C). C, absorbance spectrum from the same assay in A in which the added heme concentration is equal to the protein concentration of C282A HO2solR (green). The spectra of heme-bound C282A HO2tail (solid black) and heme-bound HO2core (dashed black) are shown for reference and were used to calculate that ∼70% of the added heme is bound to the core.
CPR, but not biliverdin, inhibits transfer of Fe3+-heme from HRM1 to the core of HO2
To understand how heme transfer between the core and HRM-binding sites of HO2 relates to the catalytic activity of HO2, we asked how the equilibration of heme on HO2 is affected by CPR, the electron donor for HO, and biliverdin, the product of the HO reaction. CPR binds to HO2 through residues on the distal helix (specifically Lys169) as well as the helix immediately C-terminal to the distal helix (specifically Leu201) (28). A crystal structure of HO1 in complex with a tight-binding variant of CPR, ΔTGEE CPR (29), illustrates those interactions, which are conserved between HO2 and HO1 (30). Because our HDX-MS results indicate that the distal helix also interacts the HRM region of the protein, we predicted that CPR may compete with the HRMs for access to the heme binding site in the core of the protein. Indeed, as we increased the concentration of ΔTGEE CPR preincubated with apo-HO2core prior to mixing with the heme-bound form of C282A HO2tail, the transfer of heme to the core became slower (Fig. 8A).
Figure 8.
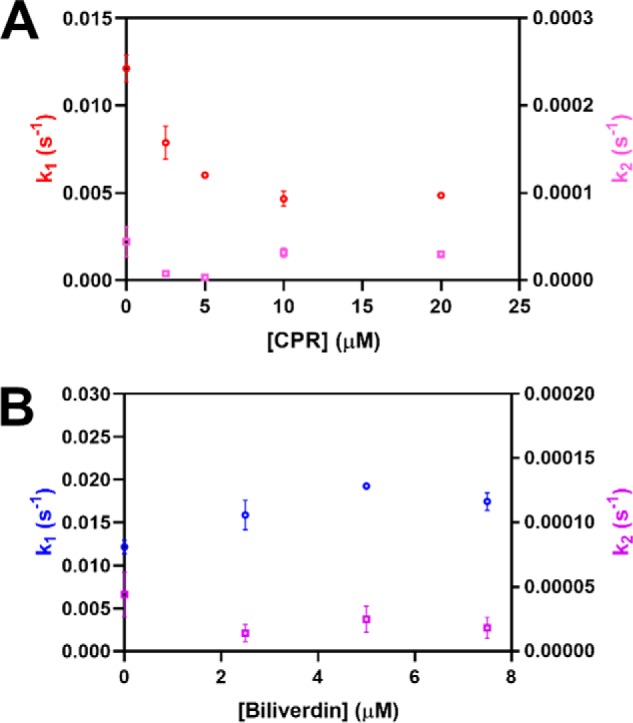
The effect of biliverdin or ΔTGEE CPR on Fe3+-heme transfer between heme-bound C282A HO2tail and apo-HO2core. Apo HO2core (10 μm) was incubated with varying concentrations of ΔTGEE CPR (A) or biliverdin (B) prior to being rapidly mixed with heme-bound C282A HO2tail (5 μm final concentration) in a stopped-flow instrument within an anaerobic chamber. All measurements were carried out at 20 °C using the 1-cm path length configuration in photodiode array mode in triplicate. The data at 404 nm were fit to a double-exponential equation using the Pro-data Viewer software provided by Applied Photophysics. The rates were plotted as a function of the final concentration of biliverdin or ΔTGEE CPR in the assay with k1 on the left y axis and k2 on the right y axis. Data represent the average ± S.D. of three acquisitions.
On the other hand, preincubation of apo-HO2core with biliverdin prior to mixing with heme-bound C282A HO2tail did not affect the rate of heme transfer to the core (Fig. 8B). Biliverdin, the product of the HO reaction, is converted to bilirubin by biliverdin reductase. In the absence of biliverdin reductase, release of biliverdin from HO1 was shown to be the rate-limiting step of heme turnover (31).
Fe3+-heme is transferred to the catalytic binding site of HO2 from HRM1 after turnover in the core
One of the key times, presumably, that heme would equilibrate between sites is after turnover, when the core is in the apo state and the HRMs contain heme. In an effort to mimic those conditions in vitro, we monitored the spectrum of HO2 to look for heme transfer after a single, fast turnover of heme in the core. Under single-turnover conditions, meaning limiting the concentration of NADPH, we could theoretically minimize the amount of heme that was transferred to the core from the HRMs and then turned over. Further, by ensuring the single turnover was fast by optimizing the concentration of CPR, we hypothesized that the heme transfer would occur after turnover was completed, thus allowing us a better opportunity to observe the transfer. As a control, we used Fe3+core-HO2solO, in which the HRMs are in a disulfide bond. Upon rapidly mixing NADPH with the protein that had been preincubated with WT CPR, we observed an initial decrease in absorbance at 404 nm due to heme turnover (as judged by the lack of spectral features consistent with heme bound to HO2) in the first 10 s, which fit to two phases (k1 = 2.25 s−1 and k2 = 0.40 s−1) (Fig. 9). The third phase of the reaction was a very slow (k3 = 0.00019 s−1) decrease in absorbance at 404 nm. Under those conditions, we felt confident we would be able to observe a heme transfer even if it occurred at a similar rate as the transfer of heme to HO2core from C282A HO2tail discussed above (0.017 s−1).
Figure 9.

Fe3+-heme transfer from HRM1 to the core after single turnover of heme in the core. A, heme-bound HO2solO (5 μm final concentration) and an equimolar concentration of CPR were rapidly mixed with a limiting amount of NADPH to initiate degradation of heme at the core. All measurements were carried out at 20 °C using the 1-cm path length configuration in photodiode array mode in triplicate. The stopped-flow trace at 404 nm (B, red) was fit to a triple-exponential equation (B, black) using the Pro-data Viewer software provided by Applied Photophysics. The inset is a semi-log plot of the data in B with the data shown in red and the fit in black. C and D, same as A and B except C282A Fe3+core/HRM-HO2solR was used in the assay in place of HO2solO, and data were fit to a quadruple-exponential equation. D–F, same as A and B except that an equimolar concentration of free heme was added to the heme-bound HO2solO and CPR solution before mixing with NADPH. Data were fit to a quadruple-exponential equation.
Heme transfer from HRM1 to the core was observed after single turnover of C282A Fe3+core/HRM-HO2solR, which has heme bound at both the core and the HRM-binding sites. Upon rapidly mixing NADPH with the protein that had been preincubated with WT CPR, we observed an initial decrease in absorbance at 404 nm due to heme turnover in the first 6 s, which fit to two phases (k1 = 2.30 s−1 and k2 = 0.15 s−1) (Fig. 9). However, unlike the control reaction with Fe3+core-HO2solO, there was an increase in absorbance at 404 nm after the initial decrease, suggesting reloading of heme to the catalytic core. Careful examination of the spectra revealed small shifts in the Soret band when comparing the initial spectrum, the spectrum at 6 s, and the final spectrum that is consistent with heme transfer. Furthermore, assuming that the only difference between the control and the HRM1-bound reaction was the heme transfer event, the difference spectra between the two reactions over the same time frame would reveal the heme transfer event. Indeed, the difference spectrum at 6 s was consistent with heme transfer and binding to the core. However, the Soret band did not fully reach 404 nm even by 1000 s. Despite the fact that we saw no effect in the HO2core/HO2tail assay, we considered the possibility that the product of the reaction, biliverdin limited complete transfer of heme to the core upon adding equimolar (to HO2) biliverdin reductase (Fig. S3). However, there was no change in the rates, nor did the final spectrum shift further toward 404 nm than in the absence of biliverdin reductase. Therefore, we hypothesize that after transfer, the heme equilibrates between the core and the tail, as described above for HO2core and HO2tail.
The absorbance increase at 404 nm due to heme transfer fit to two phases, k3 = 0.036 s−1 and k4 = 0.0023 s−1. When comparing the results of the single-turnover assay with C282A Fe3+core/HRM-HO2solR with those discussed earlier regarding heme transfer to HO2core from C282A HO2tail, we see that the initial rate of heme transfer is 2-fold faster for the more full-length protein than for the two truncated proteins (0.036 s−1 compared with 0.017 s−1). The rate of heme transfer from the HRMs to the core of C282A Fe3+core/HRM-HO2solR is comparable with the heme off-rates for C282A Fe3+core/HRM-HO2solR, as measured in an apo-H64Y/V68F-myoglobin assay (Fig. 11). The assay yields three rates (k1 = 0.095 s−1, k2 = 0.0075 s−1, and k3 = 0.00056 s−1); k2 is consistent with the off-rate observed for C282A HO2tail (Fig. 4) and is slower than the rate of heme transfer observed here. k1 and k3 can be assigned to heme dissociation from the core of the protein and thus would not be relevant in a comparison with the rates of heme transfer when the core is in the apo-form.
Figure 11.
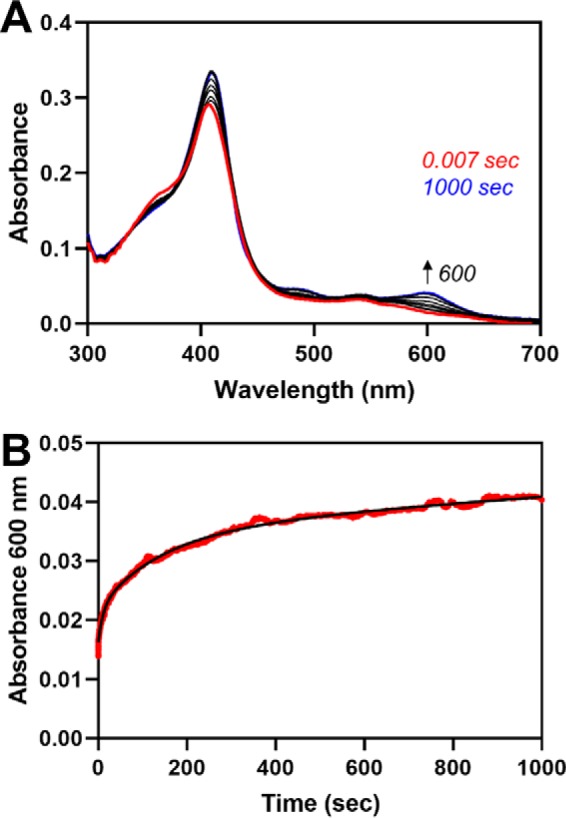
The Fe3+-heme off-rates for C282A Fe3+core/HRM-HO2solR, as measured in an apo-H64Y/V68F-myoglobin assay. A, C282A Fe3+core/HRM-HO2solR was mixed with a 10-fold excess of apo-H64Y/V68F-myoglobin in a stopped-flow instrument within an anaerobic chamber. B, the absorbance change over time at 600 nm (red) and the fit of the data to a triple-exponential equation (black).
Figure 10.
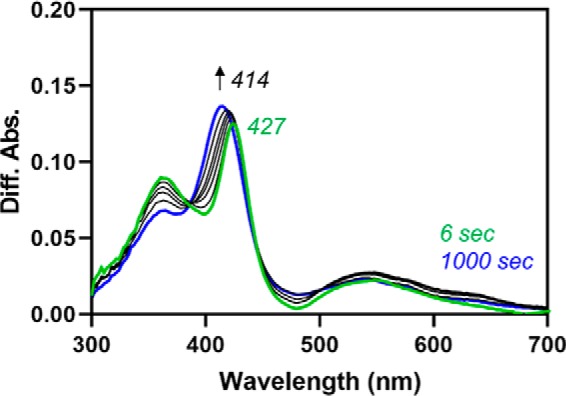
Fe3+-heme transfer from HRM1 to the core after single turnover of heme in the core as indicated by difference spectra. Data in Fig. 9A (heme-bound HO2solO/CPR mixed with NADPH) were subtracted from data in Fig. 9B (C282A Fe3+core/HRM-HO2solR/CPR mixed with NADPH) at the same time points to yield difference spectra. The difference spectra indicate the a shift in the absorbance maximum from 427 nm, which is near the absorbance maximum of heme-bound C282A HO2tail, to 414 nm, which is near the absorbance maximum of heme-bound HO2core.
Upon repeating the control reaction (WT Fe3+core-HO2solO preincubated with WT CPR and then rapidly mixed with NADPH) but adding an equivalent of heme to the solution, the rates of the rebinding event to the core (increase in absorbance at 404 nm after the initial decrease from turnover) were 0.11 s−1 (k3) and 0.016 s−1 (k4) (Fig. 9). The rate of heme acquisition from solution by the core (when the HRMs are in a disulfide bond) after single turnover (0.11 s−1) is faster than when heme is transferred from the HRMs (0.036 s−1), but the difference between the two rates is not so large that it would rule out heme transfer. Thus, we conclude that the results are consistent with the heme transfer model.
Discussion
One of the most intriguing features of HO2 is the presence of multiple heme-binding sites of varying affinity on a protein whose function is to degrade heme at just one of those sites. The presence of the additional heme binding sites, the redox-active HRMs, is the feature that distinguishes HO2 from HO1; furthermore, humans and all other amniotes express both HO isoforms. These structural and evolutionary characteristics suggest that the presence of HRMs in HO2 is crucial and could explain why amniotes express two HO isoforms; however, uncovering the functional significance of the HRMs had remained elusive. Upon considering a model proposed for another HRM-containing protein, Bach1, in which heme induces local conformational changes that alter interactions with its binding partners (18), we hypothesized that interactions between the core and the HRM-containing tail region of HO2 occur upon heme binding to the HRMs. Further, we predicted that, for HO2, the functional significance of the conformational change is to facilitate the transfer of heme between the catalytic core and the HRMs (Fig. 1).
Our characterization of Fe3+HRM-HO2R described here confirms the heme transfer model, allowing us to assign a novel function of the HRMs in HO2 as a reservoir that stores heme until the core is available for another round of catalysis. We observed heme transfer from HRM1 to the core when the two regions of the protein are expressed separately (heme-bound C282A HO2tail to apo-HO2core; 0.017 s−1). The rate of transfer is independent of protein concentration, suggesting that the core acts similarly to apo-H64Y/V68F-myoglobin (“green heme”) to accept heme from solution. Furthermore, the rate of heme transfer is enhanced when the two regions of the protein are expressed together (heme transfer after single turnover of heme bound to the core of HO2sol proceeds at a rate of 0.036 s−1), and the observed rate is faster than the rate of heme dissociation into solution (0.005 s−1) for C282A HO2tail as measured by the apo-H64Y/V68F-myoglobin assay, suggesting direct transfer of heme between the sites in HO2. Future studies will investigate mechanistic details of heme transfer, such as heme dissociation/association versus heme ligand exchange and/or conformational selection versus induced fit. We can, however, attribute the directionality of heme transfer (i.e. from HRM to the core) to a thermodynamic driving force, given that the core has an ∼10-fold higher affinity for heme than HRM1 (5). In addition, using HDX-MS, we observed heme-dependent conformational changes in a variant of HO2, H45W/G159W Fe3+HRM-HO2solR, when the core is in the apo state and the HRMs are occupied with heme. The results are consistent with the HRM region transiently docking on the core, with the HRMs specifically altering the dynamics of regions in the core—the proximal and distal helices—which could facilitate the heme transfer.
The model proposed here for heme transfer from the tail to the core of HO2 is consistent with that previously proposed for Bach1 in which heme binding to HRMs induces local conformational changes that alter interactions with binding partners. In the case of HO2, these are intramolecular interactions between two domains instead of separate proteins. It appears that the model may be applicable to other systems as well and explain why no single function has been assigned to all HRMs. Similar to HO2, the HRMs of most other systems are also located in unstructured regions of proteins (17). Indeed, the surface binding of heme has been shown to induce local structural definition around the HRM in a number of short peptides (32–35). Conformational changes beyond those observed in short peptides may also be introduced by heme ligation to HRMs in conjunction with histidine residues located outside the HRM region, as evidenced in HO2 (5), Rev-erbβ (36), and heme-regulated inhibitor (37, 38). Most importantly, this model suggests that heme binding to HRMs is mechanistically significant yet yields functionally different outcomes.
Not predicted by the model, however, are the intramolecular interactions detected in our previous NMR study of HO2, which suggested that the HRM-containing tail docks onto the heme-bound core in Fe3+core-HO2R (19). Consistent with the NMR data, we observed in this study the back-transfer of heme from the core to HRM1. We observed a slow back-transfer of heme from the core to HRM1 after it had been rapidly transferred from the heme-bound C282A HO2tail to apo-HO2core. Increasing the affinity of the core for heme by substitution of Gly163 with His eliminated the back-transfer, suggesting that equilibration of heme between the core and HRM1 is driven by thermodynamics, consistent with the Kd for the HRM1 being greater than that for the core. Indeed, our equilibrium assays indicate that the core is ∼60–70% occupied upon incubating heme-bound C282A HO2tail with the apo-form of HO2core in a 1:1 ratio or incubating C282A HO2solR in a 1:1 ratio with heme. Further, when the two regions of the protein were expressed separately, we observed heme transfer from the core to HRM1 at a rate (0.039 s−1) that is comparable with, and slightly faster than, transfer in the forward direction (from HRM1 to the core).
Whereas the rate of heme transfer from the tail to the core is independent of protein concentration, Fig. 4 reveals that the rate of heme transfer from the HO2 catalytic core to HRM1 decreases as a function of protein concentration. This profile is consistent with heme binding through a conformational selection model (27), in which a ligand preferentially binds to a particular conformation of a protein rather than binding to a protein to induce a conformation (induced fit). The core of HO2 has also previously been shown to display characteristics of conformational selection upon binding heme from solution, which distinguishes HO2 from other heme-binding proteins, including myoglobin and Rev-erbβ, which bind free heme in a two-step mechanism that is more consistent with induced fit (25). Given that we see heme-dependent conformational changes in both the Fe3+HRM-HO2solR and Fe3+core-HO2R states (19), conformational selection might be a strategy used by HO2 to facilitate heme transfer. Specifically, the HRMs in HO2 are located in an unstructured region of the protein (19, 20). Various studies indicate that intrinsically disordered proteins/regions of proteins, which play key roles in cellular regulation and signaling, bind specifically to other proteins and ligands while sampling many different conformations (39). In the case of HO2, it is only when the tail is interacting with the core (when just one of the two types of heme-binding sites is occupied), giving the protein a specific conformation, that heme transfer occurs. Thus, it remains unclear whether the conformational selection model describes the docking of the unstructured tail on the structured core or the binding of heme to HRM1. As mentioned above, future studies, which will aim to elucidate the mechanistic details of heme transfer (heme dissociation/association versus heme ligand exchange), will clarify why the kinetic profiles differ in the two directions of heme transfer.
We assign the HRMs as a dynamic reservoir for heme that can both transfer heme to and accept heme from the catalytic core of HO2. Consistent with our hypothesis, our characterization of heme transfer within HO2 is in contrast to the direct heme transfer from IsdA to IsdC within the Isd heme acquisition system of Staphylococcus aureus (40) or from Shp to HtsA within the heme acquisition machinery found in Streptococcus pyogenes (41). In these systems, whereas low affinities (Kd = 17–48 μm) for the holoprotein/apoprotein complex have been observed, heme transfer reactions are reported to have very fast rates (43–54 s−1) that are 40,000–70,000 times greater than that of heme dissociation. In another example, glyceraldehyde-3-phosphate dehydrogenase, which acts as a heme chaperone (42), binds to inducible nitric-oxide synthase with very high affinity (Kd = 0.6 nm) and enhances heme insertion into inducible nitric-oxide synthase relative to free heme (43). Rather, the HO2 model appears to be more mechanistically and functionally similar to the transfer of copper from CusF to CusB, components of the Escherichia coli efflux pump. In that system, the rate of complex formation between CusF and CusB is fast and transfer of copper is slow, proceeding at a rate of 0.015 s−1 (44). When copper is transferred from CusF to CusB, the efflux pump is activated; when copper is transferred back to CusF from CusB as metal concentrations decrease, the efflux pump is deactivated (45). Hence, transfer and back-transfer are key to regulating metal homeostasis.
In the instance of HO2, we also observe transfer in both directions and hypothesize here that both transfers are functionally significant, working together to regulate heme homeostasis. In the forward direction, meaning from the HRMs to the core, heme is transferred rapidly and directly to the core for degradation. While CPR is bound to HO2 during turnover, it appears that the tail is prevented from docking on the core. Instead, the HRMs would be available to bind heme, priming the system for the next round of turnover before the first round has even been completed when heme levels are elevated. As heme levels decrease, less heme would be available to bind to the HRMs, and less heme would be chaperoned to the HO2 core via the HRMs. In addition, the back-transfer of heme from the core to the tail may also indicate that heme can also be chaperoned back into the labile heme pool, equilibrating heme among the various locations. Of course, this simplified model does not account for how heme is chaperoned to the HRMs if the HRMs are indeed sensing the concentrations of labile heme. Nor does the model account for whether or not heme can be directly chaperoned to the core without the HRMs acting as an intermediary; this would also be relevant for acquisition of heme by HO1. Given the similarities between the core regions of HO2 and HO1, it is quite feasible that the same heme chaperone can deliver heme to both proteins, but the identity of such a chaperone has not been elucidated as of yet. These are questions under investigation in our laboratory. A chaperone that delivers heme directly to the core of HO2 would be particularly important when the protein is in the oxidized, disulfide-bond state. The HRMs of HO2 are 30–40% oxidized under normal growth conditions (46), making them unavailable to bind heme (5).
In conclusion, we have demonstrated that heme is transferred between binding sites on HO2 and that intramolecular interactions between the core and tail occur upon heme binding to the HRMs to facilitate heme transfer. We propose that the heme transfer from the HRMs to the core not only facilitates heme turnover in HO2 but also makes HO2 more responsive to changes in heme levels than previously thought by using a mechanism that is distinct from HO1, which has no HRMs, but perhaps consistent with that of other HRM-containing proteins by using heme binding to the HRMs to induce conformational changes in the protein. The results presented here suggest that future studies will cement a key role for HO2 in the larger context of heme trafficking and heme homeostasis.
Materials and methods
Protein expression and purification
HO2sol and HO2core were expressed from the pET28a vector in BL21(DE) cells (Life Technologies, Inc.) and purified by nickel-nitrilotriacetic acid–agarose affinity chromatography (Qiagen) as described previously (5, 19). In the case of HO2core, the N-terminal His6 tag was removed by treatment with thrombin prior to use (19), whereas the tag was not removed from HO2sol due to nonspecific cleavage of the protein upon thrombin treatment (20). The C282A variant of HO2tail was expressed from a modified version of pMSCG7 (generously provided by W. C. Brown of the High-throughput Protein Lab in the Center for Structural Biology at the University of Michigan), purified by nickel-nitrilotriacetic acid–agarose affinity chromatography (Qiagen), and treated with tobacco etch virus protease to remove the N-terminal affinity tag, as described previously (5). The G163H variant of HO2core was constructed using the Q5 site-directed mutagenesis kit (New England Biolabs) to introduce the mutation, and the H45W/G159W variant of HO2core was constructed using the QuikChange site-directed mutagenesis protocol (Stratagene). Variants were expressed and purified as described for WT HO2core above. The C282A, H45W, G159W, and H45W/G159W variants of HO2sol were introduced into a pMSCG10 vector harboring HO2sol described previously (20) using the QuikChange site-directed mutagenesis protocol (Stratagene). Variants were expressed and purified as described for WT HO2sol above, except that the affinity tag was removed by treatment with tobacco etch virus protease.
Residues 236–239 of an N-terminally truncated form of CPR (the first 66 were deleted) were deleted using the QuikChange site-directed mutagenesis protocol (Stratagene) in two successive reactions to create the ΔTGEE CPR variant. The WT and the ΔTGEE variant of N-terminally truncated CPR were purified as described previously (28). Biliverdin was purified as described previously (28). H64Y/V68F-myoglobin (green heme) was purified and converted to the apo-form as described previously (25) using an expression vector generously supplied by J. Olson (Rice University, Houston, TX).
Free thiol determination
Free thiol concentrations were measured by a modified 5,5′-dithiobis-(2-nitrobenzoic acid) assay described previously (25) in which 50 μl of 5 mm 5,5′-dithiobis-(2-nitrobenzoic acid) was added to 950 μl of 6 m guanidine hydrochloride in 100 mm Tris, pH 8.0. 100 μl of sample was added, and the mixture was incubated at room temperature for 15 min before spectra were recorded. DTT was used to standardize the assay.
Preparation and analysis of Fe3+-heme–bound HO2
Heme-bound samples were prepared as described previously in 50 mm Tris, pH 8.0, buffer with 50 mm KCl (5). As with other reduced, dithiol samples, the C282A variants of HO2sol and HO2tail were treated with TCEP in an anaerobic chamber and separated from the TCEP prior to incubation with heme. Cys265 has previously been shown to be in the sulfenic acid form upon replacement of Cys282 with Ala (6). Absorbance spectra of heme-bound proteins were recorded in 50 mm Tris, pH 8.0, buffer with 50 mm KCl at 20 °C on a Shimadzu UV-2600 UV-visible spectrophotometer.
CD spectroscopy
CD spectra of the protein samples were acquired in phosphate buffer (50 mm potassium phosphate, pH 8) at a concentration of 5 μm on a J-1500 CD Spectrometer (JASCO). Each spectrum was acquired with five scans in a cuvette with a path length of 0.1 cm.
HDX-MS
All HDX-MS experiments were performed in an identical manner regardless of the presence of heme or mutation. Deuterium labeling was carried out in triplicate. Labeling was initiated by diluting 3 μl of HO2 (33.3 μm; 50 mm Tris and 50 mm KCl at pH 8.0) 15-fold in deuterated buffer at room temperature. The labeling reactions were quenched by decreasing the temperature to 0 °C and decreasing the pH to 2.5 by adding 30 μl of quench buffer (150 mm NaH2PO4, 100 mm TCEP, pH 2.2) at five time points (10 s, 1 min, 10 min, 1 h, and 4 h). Upon quenching, the samples were injected immediately into a Waters nanoACQUITY UPLC equipped with HDX technology. The samples were digested online using a pepsin column at 15 °C. Peptides were then trapped on a VanGuard BEH C18 1.7-μm guard column. Following trapping, peptides were eluted from the guard column and separated using a 5–85% water/acetonitrile gradient (0.1% formic acid) over 10 min at a flow rate of 60 μl/min using a Waters ACQUITY UPLC HSS T3 1.8-μm 1.0 × 50-mm analytical column. Mass spectral analysis was performed on a Waters Synapt G2-Si with a standard electrospray ionization source in positive resolution mode. Peptides were identified from triplicate HDMSE analyses, and data were analyzed using PLGS 3.0.1 (Waters Corp.). Peptide masses were identified using a minimum of 250 ion counts for the low-energy threshold and a 50 ion count threshold for their fragment ions. The peptides identified in PLGS were then analyzed in DynamX 3.0 (Waters Corp.) implementing a minimum products per amino acid cut-off of 0.2, at least two consecutive product ions, and a maximum MH+ error of 10 ppm. A database containing only the sequence from human HO2 (Uniprot P-30519) was used with no cleavage specificity and no post-translational modifications considered. Those peptides meeting the filtering criteria were further processed by DynamX to create deuterium uptake graphs. Relative deuterium incorporation was calculated by subtracting the centroid of the isotopic distribution of the undeuterated control from the centroid of the isotopic distribution from a given labeling experiment.
Fe3+-heme transfer assays
The heme-bound form of the indicated variant and construct of HO2 (final concentration 5 μm) was rapidly mixed with apo-HO2 at the indicated concentrations in an Applied Photophysics stopped-flow spectrophotometer with the sample handling unit housed in an anaerobic chamber. In some assays, the apo-form of HO2 was premixed with varying concentrations of either ΔTGEE CPR or biliverdin prior to the assay. All measurements were carried out at 20 °C in 50 mm Tris, pH 8.0 buffer, with 50 mm KCl using the 1-cm path length configuration in photodiode array mode. When C282A HO2tail was used in the apo-form, the protein was reduced with TCEP in the anaerobic chamber as described above prior to use in the assay. Stopped-flow traces at the indicated wavelengths were fit with a double-exponential equation using the Pro-data Viewer software provided by Applied Photophysics.
Fe3+-heme off-rate measurements
Heme off-rates were measured as described previously on a Shimadzu UV-2600 UV-visible spectrophotometer to monitor the change in absorbance at 600 nm due to heme binding to apo-H64Y/V68F-myoglobin (green heme) (5). In other experiments, heme-bound HO2 was rapidly mixed apo-H64Y/V68F-myoglobin at the indicated protein concentrations in an Applied Photophysics stopped-flow spectrophotometer with the sample handling unit housed in an anaerobic chamber. All measurements were carried out at 20 °C in 50 mm Tris, pH 8.0, buffer with 50 mm KCl using the 1-cm path length configuration in photodiode array mode. Stopped-flow traces at the indicated wavelengths were fit using the Pro-data Viewer software provided by Applied Photophysics.
Fe3+-heme competition assays
The HO2core competition assay was performed as described previously (25) with minor modifications. In 50 mm Tris, pH 8.0, buffer with 50 mm KCl, varying concentrations of apo-HO2core were mixed with the preformed complex of 3 μm TCEP-reduced C282A HO2tail and 2.5 μm heme in an anaerobic chamber. Samples were incubated at ambient temperature for 48 h before they were removed from the anaerobic chamber, and spectra were recorded on a Shimadzu UV-2600 UV-visible spectrophotometer at 20 °C. Spectra were treated and fit to the Morrison equation for tight-binding inhibitors as described previously (25).
The HO2sol heme competition assay was performed similarly except with varying concentrations of heme. The C282A variant of HO2sol was reduced with TCEP as described above in an anaerobic chamber. After the removal of TCEP, the protein was mixed with varying concentrations of heme in 50 mm Tris, pH 8.0, buffer with 50 mm KCl. Samples were incubated at ambient temperature for 48 h before they were removed from the anaerobic chamber, and spectra were recorded on a Shimadzu UV-2600 UV-visible spectrophotometer at 20 °C.
Single-turnover assays
Heme degradation by HO2 requires O2, but the Applied Photophysics stopped-flow spectrophotometer with the sample handling unit is housed in an anaerobic chamber. Therefore, immediately prior to the assay, the instrument was extensively rinsed with aerobic buffer. Then solutions for syringe A (10 μm heme-bound HO2solO or C282A HO2solR (prepared as described above) and 10 μm CPR in aerobic buffer (50 mm Tris, pH 8.0, buffer with 50 mm KCl); in some instances, the mixture for syringe A also included 10 μm CPR, 10 μm heme, or 10 μm biliverdin) and for syringe B (35 μm NADPH in the same buffer) were prepared, immediately brought into the anaerobic chamber in sealed tubes, and rapidly loaded into the instrument. All measurements were carried out at 20 °C using the 1-cm path length configuration in photodiode array mode. Stopped-flow traces at the indicated wavelengths were fit using the Pro-data Viewer software provided by Applied Photophysics.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (47) partner repository with the data set identifier PXD017538.
Author contributions
A. S. F., B. A. K., J. R. E., and S. W. R. conceptualization; A. S. F., A. L. G., and T. E. W. data curation; A. S. F., B. A. K., and T. E. W. software; A. S. F., B. A. K., T. E. W., J. R. E., and S. W. R. formal analysis; A. S. F., J. R. E., and S. W. R. supervision; A. S. F., B. A. K., J. R. E., and S. W. R. validation; A. S. F., A. L. G., L. L., T. E. W., M. C. B., J. R. E., and S. W. R. investigation; A. S. F. and B. A. K. visualization; A. S. F., A. L. G., T. E. W., M. C. B., and J. R. E. methodology; A. S. F. writing-original draft; A. S. F., A. L. G., B. A. K., L. L., M. C. B., J. R. E., and S. W. R. writing-review and editing; B. A. K., J. R. E., and S. W. R. resources; J. R. E. and S. W. R. funding acquisition; J. R. E. and S. W. R. project administration.
Supplementary Material
This work was supported by National Institutes of Health Grant R01-GM123513 (to S. W. R.) and a research collaboration with the Waters Corporation (J. R. E.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains supporting Excel File S1 and Figs. S1–S3.
- HO
- heme oxygenase
- CPR
- cytochrome P450 reductase
- Fe3+core-HO2
- heme oxygenase-2 with Fe3+-heme bound to the core at His45
- Fe3+core/HRM-HO2R
- heme oxygenase-2 in which the core and the HRMs are fully occupied with Fe3+-heme
- Fe3+HRM-HO2R
- heme oxygenase-2 with heme-loaded HRMs and an apo-core
- HDX
- hydrogen-deuterium exchange
- HO1
- heme oxygenase-1
- HO2
- heme oxygenase-2
- HO2core
- heme oxygenase-2 construct spanning amino acids 1–248
- HO2sol
- soluble heme oxygenase-2 construct spanning amino acids 1–288
- HO2tail
- heme oxygenase-2 construct spanning amino acids 213–288
- HO2R
- heme oxygenase-2 in which the disulfide bond between Cys265 and Cys282 is reduced
- HRM
- heme-regulatory motif
- TCEP
- tris(2-carboxyethyl)phosphine
- ALAS1
- aminolevulinic acid synthase.
References
- 1. Maines M. D., Trakshel G. M., and Kutty R. K. (1986) Characterization of two constitutive forms of rat-liver microsomal heme oxygenase: only one molecular-species of the enzyme is inducible. J. Biol. Chem. 261, 411–419 [PubMed] [Google Scholar]
- 2. Schuller D. J., Wilks A., Ortiz de Montellano P. R., and Poulos T. L. (1999) Crystal structure of human heme oxygenase-1. Nat. Struct. Biol. 6, 860–867 10.1038/12319 [DOI] [PubMed] [Google Scholar]
- 3. Bianchetti C. M., Yi L., Ragsdale S. W., and Phillips G. N. Jr. (2007) Comparison of apo- and heme-bound crystal structures of a truncated human heme oxygenase-2. J. Biol. Chem. 282, 37624–37631 10.1074/jbc.M707396200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang L., and Guarente L. (1995) Heme binds to a short sequence that serves a regulatory function in diverse proteins. EMBO J. 14, 313–320 10.1002/j.1460-2075.1995.tb07005.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fleischhacker A. S., Sharma A., Choi M., Spencer A. M., Bagai I., Hoffman B. M., and Ragsdale S. W. (2015) The C-terminal heme regulatory motifs of heme oxygenase-2 are redox-regulated heme binding sites. Biochemistry 54, 2709–2718 10.1021/acs.biochem.5b00266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yi L., and Ragsdale S. W. (2007) Evidence that the heme regulatory motifs in heme oxygenase-2 serve as a thiol/disulfide redox switch regulating heme binding. J. Biol. Chem. 282, 21056–21067 10.1074/jbc.M700664200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sun J., Hoshino H., Takaku K., Nakajima O., Muto A., Suzuki H., Tashiro S., Takahashi S., Shibahara S., Alam J., Taketo M. M., Yamamoto M., and Igarashi K. (2002) Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 21, 5216–5224 10.1093/emboj/cdf516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ogawa K., Sun J., Taketani S., Nakajima O., Nishitani C., Sassa S., Hayashi N., Yamamoto M., Shibahara S., Fujita H., and Igarashi K. (2001) Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 20, 2835–2843 10.1093/emboj/20.11.2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suzuki H., Tashiro S., Hira S., Sun J., Yamazaki C., Zenke Y., Ikeda-Saito M., Yoshida M., and Igarashi K. (2004) Heme regulates gene expression by triggering Crm1-dependent nuclear export of Bach1. EMBO J. 23, 2544–2553 10.1038/sj.emboj.7600248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zenke-Kawasaki Y., Dohi Y., Katoh Y., Ikura T., Ikura M., Asahara T., Tokunaga F., Iwai K., and Igarashi K. (2007) Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol. Cell. Biol. 27, 6962–6971 10.1128/MCB.02415-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kubota Y., Nomura K., Katoh Y., Yamashita R., Kaneko K., and Furuyama K. (2016) Novel mechanisms for heme-dependent degradation of ALAS1 protein as a component of negative feedback regulation of heme biosynthesis. J. Biol. Chem. 291, 20516–20529 10.1074/jbc.M116.719161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Davydov R., Fleischhacker A. S., Bagai I., Hoffman B. M., and Ragsdale S. W. (2016) Comparison of the mechanisms of heme hydroxylation by heme oxygenases-1 and-2: kinetic and cryoreduction studies. Biochemistry 55, 62–68 10.1021/acs.biochem.5b00943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McCoubrey W. K. Jr., Huang T. J., and Maines M. D. (1997) Heme oxygenase-2 is a hemoprotein and binds heme through heme regulatory motifs that are not involved in heme catalysis. J. Biol. Chem. 272, 12568–12574 10.1074/jbc.272.19.12568 [DOI] [PubMed] [Google Scholar]
- 14. Shen J., Sheng X., Chang Z., Wu Q., Wang S., Xuan Z., Li D., Wu Y., Shang Y., Kong X., Yu L., Li L., Ruan K., Hu H., Huang Y., et al. (2014) Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep. 7, 180–193 10.1016/j.celrep.2014.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang J., Kim K. D., Lucas A., Drahos K. E., Santos C. S., Mury S. P., Capelluto D. G. S., and Finkielstein C. V. (2008) A novel heme-regulatory motif mediates heme-dependent degradation of the circadian factor Period 2. Mol. Cell. Biol. 28, 4697–4711 10.1128/MCB.00236-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carter E. L., Gupta N., and Ragsdale S. W. (2016) High affinity heme binding to a heme regulatory motif on the nuclear receptor Rev-erbβ leads to its degradation and indirectly regulates its interaction with nuclear receptor corepressor. J. Biol. Chem. 291, 2196–2222 10.1074/jbc.M115.670281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fleischhacker A. S., Carter E. L., and Ragsdale S. W. (2018) Redox regulation of heme oxygenase-2 and the transcription factor, Rev-Erb, through heme regulatory motifs. Antioxid. Redox Signal. 29, 1841–1857 10.1089/ars.2017.7368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hira S., Tomita T., Matsui T., Igarashi K., and Ikeda-Saito M. (2007) Bach1, a heme-dependent transcription factor, reveals presence of multiple heme binding sites with distinct coordination structure. IUBMB Life 59, 542–551 10.1080/15216540701225941 [DOI] [PubMed] [Google Scholar]
- 19. Bagai I., Sarangi R., Fleischhacker A. S., Sharma A., Hoffman B. M., Zuiderweg E. R. P., and Ragsdale S. W. (2015) Spectroscopic studies reveal that the heme regulatory motifs of heme oxygenase-2 are dynamically disordered and exhibit redox-dependent interaction with heme. Biochemistry 54, 2693–2708 10.1021/bi501489r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kochert B. A., Fleischhacker A. S., Wales T. E., Becker D. F., Engen J. R., and Ragsdale S. W. (2019) Dynamic and structural differences between heme oxygenase-1 and -2 are due to differences in their C-terminal regions. J. Biol. Chem. 294, 8259–8272 10.1074/jbc.RA119.008592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun J., Loehr T. M., Wilks A., and Ortiz de Montellano P. R. (1994) Identification of histidine-25 as the heme ligand in human liver heme oxygenase. Biochemistry 33, 13734–13740 10.1021/bi00250a026 [DOI] [PubMed] [Google Scholar]
- 22. Liu Y., Moënne-Loccoz P., Hildebrand D. P., Wilks A., Loehr T. M., Mauk A. G., and Ortiz de Montellano P. R. (1999) Replacement of the proximal histidine iron ligand by a cysteine or tyrosine converts heme oxygenase to an oxidase. Biochemistry 38, 3733–3743 10.1021/bi982707s [DOI] [PubMed] [Google Scholar]
- 23. Liu Y., Koenigs Lightning L., Huang H., Moënne-Loccoz P., Schuller D. J., Poulos T. L., Loehr T. M., and Ortiz de Montellano P. R. (2000) Replacement of the distal glycine 139 transforms human heme oxygenase-1 into a peroxidase. J. Biol. Chem. 275, 34501–34507 10.1074/jbc.M004245200 [DOI] [PubMed] [Google Scholar]
- 24. Hargrove M. S., Singleton E. W., Quillin M. L., Ortiz L. A., Phillips G. N. Jr., Olson J. S., and Mathews A. J. (1994) His64(E7) → Tyr apomyoglobin as a reagent for measuring rates of hemin dissociation. J. Biol. Chem. 269, 4207–4214 [DOI] [PubMed] [Google Scholar]
- 25. Carter E. L., Ramirez Y., and Ragsdale S. W. (2017) The heme-regulatory motif of nuclear receptor Rev-erb β is a key mediator of heme and redox signaling in circadian rhythm maintenance and metabolism. J. Biol. Chem. 292, 11280–11299 10.1074/jbc.M117.783118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lad L., Koshkin A., de Montellano P. R. O., and Poulos T. L. (2005) Crystal structures of the G139A, G139-NO and G143H mutants of human heme oxygenase-1: a finely tuned hydrogen-bonding network controls oxygenase versus peroxidase activity. J. Biol. Inorg. Chem. 10, 138–146 10.1007/s00775-004-0620-6 [DOI] [PubMed] [Google Scholar]
- 27. Vogt A. D., and Di Cera E. (2013) Conformational selection is a dominant mechanism of ligand binding. Biochemistry 52, 5723–5729 10.1021/bi400929b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Spencer A. L. M., Bagai I., Becker D. F., Zuiderweg E. R. P., and Ragsdale S. W. (2014) Protein/protein interactions in the mammalian heme degradation pathway: heme oxygenase-2, cytochrome P450 reductase, and biliverdin reductase. J. Biol. Chem. 289, 29836–29858 10.1074/jbc.M114.582783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hamdane D., Xia C., Im S. C., Zhang H., Kim J. J. P., and Waskell L. (2009) Structure and function of an NADPH-cytochrome P450 oxidoreductase in an open conformation capable of reducing cytochrome P450. J. Biol. Chem. 284, 11374–11384 10.1074/jbc.M807868200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sugishima M., Sato H., Higashimoto Y., Harada J., Wada K., Fukuyama K., and Noguchi M. (2014) Structural basis for the electron transfer from an open form of NADPH-cytochrome P450 oxidoreductase to heme oxygenase. Proc. Natl. Acad. Sci. U.S.A. 111, 2524–2529 10.1073/pnas.1322034111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu Y., and Ortiz de Montellano P. R. (2000) Reaction intermediates and single turnover rate constants for the oxidation of heme by human heme oxygenase-1. J. Biol. Chem. 275, 5297–5307 10.1074/jbc.275.8.5297 [DOI] [PubMed] [Google Scholar]
- 32. Brewitz H. H., Kühl T., Goradia N., Galler K., Popp J., Neugebauer U., Ohlenschläger O., and Imhof D. (2015) Role of the chemical environment beyond the coordination site: structural insight into Fe-III protoporphyrin binding to cysteine-based heme-regulatory protein motifs. Chembiochem 16, 2216–2224 10.1002/cbic.201500331 [DOI] [PubMed] [Google Scholar]
- 33. Kühl T., Sahoo N., Nikolajski M., Schlott B., Heinemann S. H., and Imhof D. (2011) Determination of hemin-binding characteristics of proteins by a combinatorial peptide library approach. Chembiochem 12, 2846–2855 10.1002/cbic.201100556 [DOI] [PubMed] [Google Scholar]
- 34. Kühl T., Wissbrock A., Goradia N., Sahoo N., Galler K., Neugebauer U., Popp J., Heinemann S. H., Ohlenschläger O., and Imhof D. (2013) Analysis of Fe(III) heme binding to cysteine-containing heme-regulatory motifs in proteins. ACS Chem. Biol. 8, 1785–1793 10.1021/cb400317x [DOI] [PubMed] [Google Scholar]
- 35. Schubert E., Florin N., Duthie F., Henning Brewitz H., Kühl T., Imhof D., Hagelueken G., and Schiemann O. (2015) Spectroscopic studies on peptides and proteins with cysteine-containing heme regulatory motifs (HRM). J. Inorg. Biochem. 148, 49–56 10.1016/j.jinorgbio.2015.05.008 [DOI] [PubMed] [Google Scholar]
- 36. Pardee K. I., Xu X., Reinking J., Schuetz A., Dong A., Liu S., Zhang R., Tiefenbach J., Lajoie G., Plotnikov A. N., Botchkarev A., Krause H. M., and Edwards A. (2009) The structural basis of gas-responsive transcription by the human nuclear hormone receptor REV-ERBβ. PLoS Biol. 7, e43 10.1371/journal.pbio.1000043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Igarashi J., Sato A., Kitagawa T., Yoshimura T., Yamauchi S., Sagami I., and Shimizu T. (2004) Activation of heme-regulated eukaryotic initiation factor 2 α kinase by nitric oxide is induced by the formation of a five-coordinate NO-heme complex: optical absorption, electron spin resonance, and resonance raman spectral studies. J. Biol. Chem. 279, 15752–15762 10.1074/jbc.M310273200 [DOI] [PubMed] [Google Scholar]
- 38. Miksanova M., Igarashi J., Minami M., Sagami I., Yamauchi S., Kurokawa H., and Shimizu T. (2006) Characterization of heme-regulated eIF2 α kinase: roles of the N-terminal domain in the oligomeric state, heme binding, catalysis, and inhibition. Biochemistry 45, 9894–9905 10.1021/bi060556k [DOI] [PubMed] [Google Scholar]
- 39. Wright P. E., and Dyson H. J. (2015) Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 16, 18–29 10.1038/nrm3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu M., Tanaka W. N., Zhu H., Xie G., Dooley D. M., and Lei B. (2008) Direct hemin transfer from IsdA to IsdC in the iron-regulated surface determinant (Isd) heme acquisition system of Staphylococcus aureus. J. Biol. Chem. 283, 6668–6676 10.1074/jbc.M708372200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nygaard T. K., Blouin G. C., Liu M., Fukumura M., Olson J. S., Fabian M., Dooley D. M., and Lei B. (2006) The mechanism of direct heme transfer from the streptococcal cell surface protein shp to HtsA of the HtsABC transporter. J. Biol. Chem. 281, 20761–20771 10.1074/jbc.M601832200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sweeny E. A., Singh A. B., Chakravarti R., Martinez-Guzman O., Saini A., Haque M. M., Garee G., Dans P. D., Hannibal L., Reddi A. R., and Stuehr D. J. (2018) Glyceraldehyde 3-phosphate dehydrogenase is a chaperone that allocates labile heme in cells. J. Biol. Chem. 293, 14557–14568 10.1074/jbc.RA118.004169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chakravarti R., Aulak K. S., Fox P. L., and Stuehr D. J. (2010) GAPDH regulates cellular heme insertion into inducible nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 107, 18004–18009 10.1073/pnas.1008133107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chacón K. N., Perkins J., Mathe Z., Alwan K., Ho E. N., Ucisik M. N., Merz K. M., and Blackburn N. J. (2018) Trapping intermediates in metal transfer reactions of the CusCBAF export pump of Escherichia coli. Commun. Biol. 1, 192 10.1038/s42003-018-0181-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chacón K. N., Mealman T. D., McEvoy M. M., and Blackburn N. J. (2014) Tracking metal ions through a Cu/Ag efflux pump assigns the functional roles of the periplasmic proteins. Proc. Natl. Acad. Sci. U.S.A. 111, 15373–15378 10.1073/pnas.1411475111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yi L., Jenkins P. M., Leichert L. I., Jakob U., Martens J. R., and Ragsdale S. W. (2009) Heme regulatory motifs in heme oxygenase-2 form a thiol/disulfide redox switch that responds to the cellular redox state. J. Biol. Chem. 284, 20556–20561 10.1074/jbc.M109.015651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Pérez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S., et al. (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 10.1093/nar/gky1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (47) partner repository with the data set identifier PXD017538.