

Abstract
P-glycoprotein (Pgp), a member of the ATP-binding cassette family, is one of the major causes of multidrug resistance in tumors. Current clinical treatments to overcome MDR involve the co-delivery of a Pgp inhibitor and a chemotherapeutic. A concern for this treatment that has led to varied clinical trial success is the associated systemic toxicities involving endogenous Pgp. Local drug delivery systems, such as in situ forming implants (ISFIs), alleviate this problem by delivering a high concentration of the drug directly to the target site without the associated systemic toxicities. ISFIs are polymeric drug solutions that undergo a phase transition upon injection into an aqueous environment to form a solid drug eluting depot allowing for a high initial intratumoral drug concentration. In this study, we have developed an ISFI capable of overcoming the Pgp resistance by co-delivering a chemotherapeutic, Doxorubicin (Dox), with a Pgp inhibitor, either Pluronic P85 or Valspodar (Val). Studies investigated in vitro cytotoxicity of Dox when combined with either Pgp inhibitor, effect of the inhibitors on release of Dox from implants in PBS, in vivo Dox distribution and retention in a subcutaneous flank colorectal murine tumor, and therapeutic response characterized by tumor growth curves and histopathology. Dox + Val showed a 4-fold reduction in the 50% lethal dose (LD50) after 48 hours. Concurrent delivery of Dox and Val showed the greatest difference at 16 days post injection for both Dox penetration and retention. This treatment group had a 5-fold maximum Dox penetration compared to Dox alone ISFIs (0.53 ± 0.22 cm vs 0.11 ± 0.11 cm, respectively, from the center of the ISFI). Additionally, there was a 3-fold increase in normalized total intratumoral Dox intensity with the Dox + Val ISFIs compared to Dox alone ISFIs (0.54 ± 0.11 vs 0.18 ± 0.09, respectively). Dox + Val ISFIs showed a 2-fold reduction in tumor growth and a 27.69% increase in necrosis 20 days post-injection compared to Dox alone ISFIs. These findings demonstrate that co-delivery of Dox and Val via ISFI can avoid systemic toxicity issues seen with clinical Pgp inhibitors.
Subject terms: Cancer imaging, Chemotherapy
Introduction
Annually, approximately 650,000 cancer patients receive neoadjuvant, standard or adjuvant chemotherapy1. Systemic chemotherapy is severely hampered by its dose-limiting toxicity, inefficient transport, and limited retention in tumors leading to sub-lethal doses2. These sub-lethal concentrations of the drug can cause mutations in cancer cells leading to multidrug-resistant (MDR) tumors. MDR is described as the ability of cancer cells to be resistant to a multitude of drugs that are structurally and functionally different3. Numerous mechanisms are involved in the development of MDR4. One of the most studied mechanisms of MDR is the upregulation of transmembrane ATP-binding cassette (ABC) transporters, such as P-glycoprotein (Pgp). They function through ATP hydrolysis at the intracellular domain causing a mechanical conformation of the Pgp thus ejecting the drug into the extracellular space3,5,6.
Numerous approaches have been developed in order to overcome Pgp mediated MDR in several types of cancers6–10. Current clinical trials attempt to systemically deliver a Pgp inhibitor, either synthetic or natural compounds, which modulate the expression and functionality of Pgp5,11–13. Pluronic P85 (P85), a tri-block synthetic copolymer, has been extensively studied and has been shown to disrupt ATP production in MDR cells, leading to the reduced functionality of Pgp13–16. Site-specific inhibitors, such as Valspodar (Val), competitively bind with the drug binding domain in Pgp and directly inhibit the conformation change required to eject the drug16–18. Val has made it to Phase III clinical trials for patients with acute myeloid leukemia but was terminated early due to excessive mortality19 and interactions with cytochrome P450 3A4, a liver enzyme, causing poor drug metabolism leading to toxic drug plasma concentrations20–22.
Lack of clinical success, mainly due to systemic toxicity, has created a need for better delivery of these Pgp inhibitor. Local drug delivery systems (DDS) have been used in preclinical research for glioblastomas, hepatocellular carcinoma, lung, and abdominal cancers, as an alternative method to deliver high concentrations of drug to the target site without associated systemic toxicities11,23–27. However, local DDS struggle with intratumoral drug retention within the target site due to several factors, such as the convective flow from the interstitial fluid/blood vessels, excretion due to Pgp, intracellular drug metabolism, and many more28–30. Here, we have developed an in situ forming implant (ISFI)31 capable of locally delivering a Pgp inhibitor and chemotherapeutic, through a minimally invasive injection procedure using a small-gauge needle. Our delivery system was tested in a murine colorectal cancer (CRC) model. Lack of clinical success are attributed to MDR which occurs in 90% of patients with metastatic CRC32–34. This approach can concurrently address the systemic toxicity issues and improve local drug retention within the tumor over time. Upon injection into an aqueous environment (e.g. a tumor), the ISFI will phase invert from a liquid solution into a solid depot, co-releasing a chemotherapeutic, Doxorubicin (Dox), and a Pgp inhibitor, P85 or Val. In this study, we have evaluated the ability of both Pgp inhibitors to improve the Dox penetration and retention intratumorally and enhance the therapeutic efficacy.
Methods and Methods
Materials
Poly(DL-lactic-co-glycolic) (PLGA, acid-capped, 75:25, MW 28.8 kDa, inherent viscosity 0.28 dL/g) was obtained from Evonik Corp (Parsippany, NJ). N-methyl-2-pyrrolidinone (NMP) and Valspodar were obtained from Sigma–Aldrich (St. Louis, Missouri). Dox HCl was obtained from LC Laboratories (Woburn, MA). Pluronic P85 were obtained from BASF (Ludwigshafen, Germany). RPMI-1640, fetal bovine serum, and penicillin-streptomycin were obtained from ThermoFisher Scientific (Waltham, MA). WST-1 was obtained from Roche Applied Sciences (Penzberg, Germany). All items were used as received.
Tumor cells
Human colorectal carcinoma cells, HCT-15, were chosen due to documented overexpression of Pgp35, and were obtained from American Type Culture Collection (Rockville, MD). HCT-15 cells were maintained in RPMI-1640 media supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin in an atmosphere of 5% CO2 at 37 °C.
Cytotoxicity of co-incubation of Dox and Pgp inhibitor
To determine inherent toxicity of each Pgp inhibitor, HCT-15 cells were seeded in a 96 well plates at 5000 cells/well in 200 µL of FBS supplemented media and allowed to reattach overnight. After attachement, the media was replaced with 200 µL of varying Pgp inhibitor concentrations (0 to 100 µg/mL for Val and 0 to 1000 µg/mL for P85 in FBS supplemented media) for 24 and 48 hours. After the exposure time, cells were washed in 1X PBS twice and viability was determined by incubating the cells in 100 µL of WST-1 (1:10 dilution of stock WST-1 in no FBS supplemented RPMI 1640) for 3 hours.
To determine chemosensitization effects, HCT-15 cells were seeded in a 96 well plates at 5000 cells/well in 200 µL FBS supplemented media and allowed to reattach overnight. After attachment, the media was replaced with 200 µL of varying concentration of Dox (0 to 1000 µg/mL) and the highest non-lethal concentration of the Pgp inhibitor seen for 24 and 48 hrs (1 µg/mL for Val and 0.1 µg/mL for P85). After the exposure time, cell viability was determined by washing two times in 1X PBS and incubating cells in 100 µL of WST-1 for 3 hours (1:10 dilution of stock WST-1 in no FBS supplemented RPMI 1640).
Cell viability was calculated by comparing the absorbance of the treatment group to the no treatment group using a plate reader at an absorbance of 450 nm (Tecan Ltd, Infinite 200 series) and displayed as the 50% lethal dose (LD50), the amount of Dox required to reduce cell viability to 50%. The resistance reversion index (RRI) was calculated with the following formula:
where a larger index value correlates to a better sensitization. Nonlinear regression curve fits ([inhibitor] vs response (three parameters)) were applied to each cell viability curve in Prism 8. These curves reported LD50 values for each treatment group which was used to determine the RRI values. Goodness of fit (R2) values are reported in each figure.
ISFI solution preparation
ISFI solutions consisted of PLGA co-dissolved with Dox and a Pgp inhibitor in NMP. Mass ratios of PLGA to NMP to Dox were 39:60:1. P85 and Valspodar were incorporated at 5 wt.% and 1 wt.%, respectively. The addition of the Pgp inhibitor reduced the overall wt.% of the NMP. Dox mass ratio was determined by delivering a clinical dosage to the tumor36. Val mass ratio was determined by delivering a third of current phase II and III trials37,38. Pluronic mass ratio was determined by delivering similar quantities to the phase II study using Pluronic39. The in vivo Pgp inhibitor concentration was also equivalent to the concentration used in the in vitro cytotoxicity assay. The components of the ISFI solution were added together and allowed to mix overnight inside an incubator shaker at 37 °C. ISFI solutions were used within 24 h of mixing.
ISFI Dox release
To measure the rate of Dox release, 50 μL of each ISFI were injected into 10 mL of 1X PBS in a glass scintillation vial, capped and placed inside an incubator shaker at 37 °C at 60 rpm. At predetermined time points (t = 2, 4, 6 h and 1, 2, 3, 5, 7, 10 and 14 days), PBS bath-side solution was collected to measure release. Within the first day, 1 mL of PBS was collected and replaced with 1 mL of fresh PBS. After the first day, 1 mL of PBS was collected, then the rest of the solution was removed and replaced with a fresh 10 mL of PBS. This was done to ensure infinite sink conditions, where the bath-side concentration would be 10 times less than the maximum solubility of Dox in PBS. Cumulative release of drug was determined by measuring the fluorescence intensity of the samples collected and compared to a standard curve using a plate reader at an excitation/emission wavelength of 495/595 nm (Tecan Ltd, Infinite 200 series). The release was normalized to the theoretical 1 wt.% Dox loading prior to injection.
Subcutaneous HCT-15 mouse tumor model
Studies involving 4-week-old BALB/c nude mice (n=5/group) were received by the Athymic Animal & Xenograft Core and approved by the Case Western Reserve University Institutional Animal Care and Use Committee. All animal experiments were performed in accordance with relevant guidelines and regulations of Case Western Reserve University Institutional Animal Care and Use Committee. Prior to the commencement of experiments the mice were maintained in an aseptic environment for 7 days to acclimate to the new environment. All food, water, and bedding were sterilized before use. HCT-15 cells (5 × 106 cells in 0.1 mL of 1X PBS) were subcutaneously injected into the dorsal side of each mouse. All procedures were performed in a laminar flow hood using aseptic techniques. Tumor growth was monitored daily until the beginning of the study and then on 2, 4, 8, 12, 16, and 20 days after the start of the study.
Intratumoral Dox distribution and tumor growth study
Subcutaneous tumors were grown for 10 days then injected with 40 μl of ISFI solution through a 21-gauge needle (Fig. 1A). Mass of the ISFI syringe was measured before and after injection to ensure equivalent ISFI injections. Dox distribution was evaluated 0, 2, 4, 8, 12, 16, and 20 days post-injection with a fluorescent optical imaging system (CRI Maestro, Caliper Life Sciences) with a blue excitation (445–490 nm)/green emission (long pass 580 nm) filter. Total Dox intensity, radial distribution, and max penetration were analyzed using a custom-made MATLAB (Mathworks) script followed by image analysis in ImageJ. Here, intratumoral ISFI images were imported into MATLAB where the image background fluorescence determined from mice with no ISFI injections was removed (Fig. 1B). In ImageJ, a line intensity profile plugin with a line width of 300 was used on the image to measure the signal intensity across the tumor. All points in this profile were added to determine the total Dox intensity in the tumor and normalized to the total intensity post injection40,41. Radial distributions were plotted from these intensity profiles where maximum intratumoral Dox penetration of each formulation was determined. Nonlinear regression lines were fitted to each radial distribution using Prism 8. Penetration values were collected above 0.1 normalized intensity to ensure a sufficient signal to noise ratio. It is important to note that Dox distributes radially from the ISFI. Optical images will combine fluorescent intensities above and below the ISFI into a single pixel value as seen in Fig. 1C. Tumor size was monitored every other day. Tumor dimensions were measured using a caliper. and tumor volume was calculated using the following formula:
where V = tumor volume, L = length tumor and W = width of tumor42.
Figure 1.

(A) Intratumoral ISFI injection into a subcutaneous murine tumor. (B) Intratumoral Dox loaded ISFI (red) fluorescent image after masking the background mouse signal. (C) Schematic of Dox distribution above and below ISFI being collected into a single pixel value after image acquisition.
Histopathology analysis
After resection, tumors were fixed with paraformaldehyde and frozen in an optical cutting temperature (OCT) compound. Samples were sliced at 7 μm and stained with hematoxylin and eosin. Histological slices were imaged with a microscope slide scanner (Olympus vs120). The necrotic area was calculated using ImageJ by drawing a region of interest around the necrosis area seen in the H&E stain. The necrosis percentage was determined by dividing the necrotic area by the area of the slice.
Statistical analysis
Statistical analysis was performed using either a student t-test or ANOVA, assuming unequal variances between data sets in Prism 8. Significant differences among means of groups were evaluated using a Tukey multiple comparison test. All data are reported as mean ± standard deviation.
Results
Cytotoxicity of co-incubation of Dox and Pgp inhibitor
Inherent toxicity curves of HCT-15 cells incubated with either P85 or Val for 24 and 48 hr are shown in Fig. 2A. Nonlethal concentrations (~100% cell viability) determined for Val and P85 for both 24 and 48 hours were 1 μg/mL and 0.1 μg/mL, respectively. These concentrations were then used for the following chemosensitization study. After a 24-hour incubation, Dox + Val showed a 1.33 fold decrease and Dox + P85 showed a 0.57 fold decrease in LD50 compared to Dox alone. After a 48-hour incubation, Dox + Val showed a 4 fold decrease and Dox + P85 showed a 1.38 fold decrease in LD50 compared to Dox alone. Resistance reversion index (RRI) values are shown in Table 1.
Figure 2.

(A) Inherent cytotoxicity of Val and P85 incubated at 24 and 48 hrs. (B) Chemosensitization cytotoxicity curves of co-incubation varying Dox concentration and non-lethal Pgp inhibitor concentration. Co-incubation of Dox and Val after 48 hours showed a 4-fold reduction in the LD50. N = 5 for all samples.
Table 1.
LD50 and resistance reversion index (RRI) values for each treatment.
| Treatment | 24 hr LD50 (μg/mL) | 24 hr RRI | 48 hr LD50 (μg/mL) | 48 hr RRI |
|---|---|---|---|---|
| Dox | 3.47 | 1 | 0.68 | 1 |
| Dox + P85 | 6.05 | 0.57 | 0.49 | 1.38 |
| Dox + Val | 2.61 | 1.33 | 0.17 | 4 |
ISFI Dox release
Effects on Dox release from the ISFI with the addition of a inhibitor were evaluated in a PBS dissolution study over the course of 20 days as shown in Fig. 3. Dox + Val ISFIs showed a significant (p < 0.05) reduction in the release of Dox compared to Dox alone and Dox + P85 ISFIs up to 6 hours post-injection. Dox, Dox + P85, and Dox + Val ISFIs showed a 29.5 ± 5.6%, 35.8 ± 1.0%, 27.3 ± 1.6%, respectively, release of Dox over the course of 1 day. Over the next 20 days, no significant differences were seen in the release of Dox with the incorporation of either Pgp inhibitor. The average release after 21 days post-injection for Dox, Dox + P85, and Dox + Val ISFIs were 59.61 ± 4.93%, 66.86 ± 2.44%, and 61.75 ± 9.89%, respectively.
Figure 3.
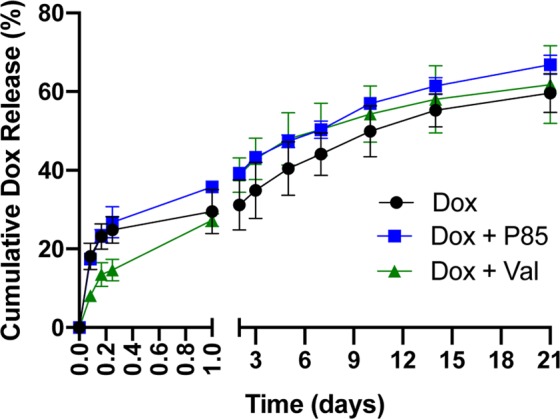
Effects of incorporating a Pgp inhibitor on the release of Dox from the ISFI. No significant differences were seen with the addition of a Pgp inhibitor after 1 day. N = 3 for all samples.
Intratumoral Dox distribution
To evaluate the effects of the Pgp inhibitor on Dox distribution within a tumor, 2D fluorescence images of the tumors were collected in mice over 20 days following ISFI injection; representative images are shown in Fig. 4. Total fluorescent intensity per area at each time point is plotted in Fig. 5. Tumors treated with Dox + Val ISFIs showed a significant increase in drug retention compared to tumors treated with Dox alone throughout the 20-day study. The greatest difference can be seen at 16 days with 3-fold increase in normalized intratumoral Dox intensity with in the Dox + Val ISFIs compared to Dox alone ISFIs (0.54 ± 0.11 vs 0.18 ± 0.09, respectively). Radial fluorescence intensity was measured from the center of the ISFI over time and shown in Fig. 6. Post injection, an equivalent release of Dox is seen between all treatment groups. At 2, 12, and 16 days, the Dox + Val ISFIs showed a significant increase in the retention of Dox at distances further from the ISFI center compared to Dox alone. Additional radial distribution data is provided in Supplemental Fig. 2. Figure 7 shows that Dox + Val ISFIs had a significant maximum intratumoral Dox penetration on days 2, 12, and 16 days post injection. The greatest difference was seen at 16 days; Dox + Val ISFIs showed a nearly 5-fold increase in maximum Dox penetration compared to Dox alone ISFIs (0.53 ± 0.22 vs 0.11 ± 0.11 cm, respectively, from the center of the ISFI).
Figure 4.

Representative optical fluorescent images of intratumorally injected Dox-loaded ISFIs. Dashed white lines indicate tumor boundaries in each case. Retention of Dox intensity can be seen up to 20 days post-injection for Dox + Val ISFIs. Scale bar = 1 cm. N = 5 for all samples. Fluorescent intensity are in arbitrary units.
Figure 5.
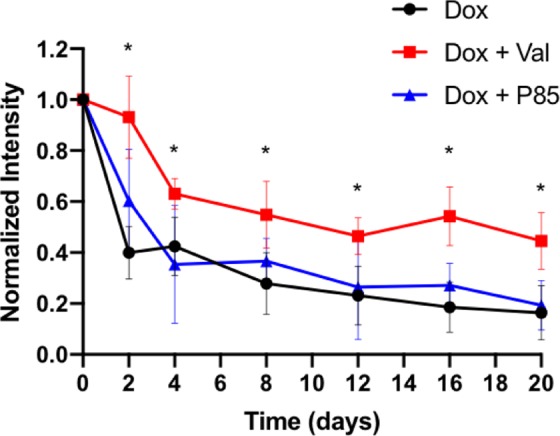
Normalized intratumoral Dox fluorescent intensity over time. Dox + Val ISFIs showed a significant increase in the normalized Dox intensity over time suggesting greater intratumoral Dox retention. *p < 0.05. N = 5 for all samples.
Figure 6.

(A) Radial distributions of Dox intensities from the center of the intratumoral ISFI over time. Nonlinear regression lines were fitted to each curve and shown as a dashed line. Dox + Val ISFIs showed a consistent increase in radial intensity over time compared to the other groups. Dashed gray lines represent edge of the ISFI. Black lines are placed at 0.1 normalized intensity to ensure sufficient SNR. Maximum Dox penetration values were determined from this 0.1 normalized intensity with respect to distance and shown in Fig. 7. N = 5 for all samples.
Figure 7.
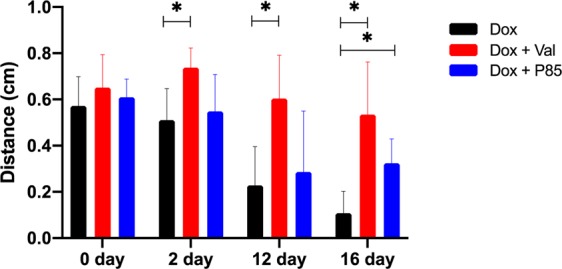
Maximum intratumoral Dox penetration from the center of the ISFI. Dox + Val ISFIs showed a significantly higher Dox retention at further distances from the center of the ISFI over time. *p < 0.05. N = 5 for all samples.
Subcutaneous tumor growth study
Therapeutic efficacy of concurrently delivering a Dox and a Pgp inhibitor from an ISFI was evaluated over 20 days Tumor growth studies (Fig. 8) showed a significant inhibition of tumor growth when co-delivering Dox with Val compared to all other groups. At the end of the 20-day study, tumors treated with Dox + Val ISFIs showed a normalized tumor volume of 2.00 ± 1.06. This was a 2-fold reduction compared to Dox alone and Dox + P85 ISFIs (4.36 ± 1.20 and 4.71 ± 2.07, respectively).
Figure 8.

Intratumoral ISFI treatment. (A) Tumor volumes measured over time, and (B) average tumor volumes for each treatment group at the end of the 20-day study. Dox + Val ISFIs showed a significant inhibition of tumor growth after 20 days. *p < 0.05. N = 4 for NT and N = 5 for all other samples. (NT: no treatment).
Histological evaluation
In order to assess the degree of necrosis, H&E staining was performed for each treatment group. Representative H&E slices are shown in Fig. 9A–D and calculated necrosis percentage is seen in Fig. 9E. As expected, the NT group showed the lowest necrosis percentage at 15.53 ± 6.83%. Interestingly, tumors treated with Dox alone showed greater necrosis compared to tumors treated with Dox + P85 ISFIs (46.13 ± 13.55% vs 32.07 ± 14.72%, respectively). Treatment with Dox + Val ISFIs showed the greatest amount of necrosis (73.82 ± 13.73%).
Figure 9.

Representative H&E histological images of (A) No-treatment (B) Dox alone (C) Dox + P85 and (D) Dox + Val ISFIs. (E) Necrosis percentage of all tumor slices. Dox + Val ISFIs showed the greatest necrosis after 20 days. N = 4 for NT and N=5 for all other samples. (NT: no treatment). Necrotic area is shown in a dashed black line in (B).
Discussion
A single non-lethal exposure to chemotherapeutics can lead to MDR, where one of the best-understood mechanisms is the upregulation of Pgp drug efflux transporters that presents itself on the tumor cell membrane43. Pgp effluxes chemotherapeutics from the cytoplasm causing sublethal concentrations within the tumor cell12,44,45. Current treatments in clinical trials include delivering competitive inhibitors, such as Val37,38, concurrently with the chemotherapeutic. However, these treatments suffer from disruptions to drug metabolism, toxic drug plasma concentrations, and interactions with endogenous Pgp37,38,46. Local drug delivery systems, such as ISFIs, can deliver high concentrations of the drug directly to the tumor site, thus avoiding systemic toxicities seen in clinical Pgp inhibitors47. In this study we locally co-delivered Dox and a Pgp inhibitor that led to a greater retention of the Dox within the tumor mass and enhanced the therapeutic outcomes.
Chemosensitization effects were investigated by co-incubating Dox and a Pgp inhibitor with HCT-15 cells, which have been shown to overexpress Pgp48. The co-incubation of Dox with either Val or P85 showed an enhanced reduction in the LD50 as seen with a high RRI value. Pluronic P85, a tri-block copolymer composed of polyethylene oxide (PEO) and polypropylene oxide (PPO) in the ratio of PEO26-PPO40-PEO26, has been studied extensively. It has been demonstrated to sensitize the cell through disruption in mitochondrial ATP activity by inhibition of the electron transport chain13,15. This decrease in ATP production reduces the efficacy of Pgp, an ATP-dependent efflux transporter, allowing for the enhanced cellular accumulation of Dox enhancing the cytotoxicity. Val has been characterized as a competitive substrate inhibitor of Pgp, where it complexes with the drug binding domains of Pgp inhibiting the mechanical conformational change required to eject the drug6,16,20,45. This inhibition allows for increased intracellular concentration of Dox leading to greater cytotoxicity.
Dox release studies showed that the addition of Val to the implants resulted in a significantly smaller burst release of Dox within 6 hours, which may be due to Val’s high lipophilicity altering the solvent/non-solvent exchange kinetics49. These reports are consistent with other studies, where the addition of lipophilic molecules into ISFIs, such as glycerol monostearate, ethyl heptanoate, stearic acid, ethyl heptanoate, methyl heptanoate, and ethyl nonoate, reduce solvent excretion therefore leading to a reduction in burst release50–53. Dox + P85 ISFIs showed no effect on the burst release, which was equivalent to a previous study54. During the diffusion phase where the ISFI is completely solidified, the Dox + P85 and Dox+ Val ISFIs showed no significant differences compared to Dox ISFI. These studies show that although Val has an effect on the burst release of ISFI, the overall release (21 days) of Dox was equivalent between all groups.
Optical fluorescent images of the intratumorally injected Dox + Val ISFIs showed a greater retention of Dox throughout the 20-day study which suggests a greater concentration of Dox intratumorally. Radial distribution profiles of Dox + Val ISFIs showed a greater penetration of Dox from the center of the ISFI over the course of the study. These profiles were used to determine the maximum Dox penetration, where Dox + Val ISFIs showed a significant increase in maximum Dox penetration over time. This may be due to the lower molecular weight of Val (1214 kDa) compared to P85 (4600 kDa), which would allow greater diffusion of Val into more distal tumor cells to elicit a greater retention of the distally penetrated Dox55,56.
Tumor volume measurements and histopathology analysis were performed to evaluate the therapeutic efficacy of a single treatment of co-delivering Dox with a Pgp inhibitor. As expected, all treatment groups showed a significant reduction in tumor growth over 20 days compared to the no-treatment group. Tumors treated with Dox + Val ISFIs showed the greatest inhibition of tumor growth, where a two-fold decrease was seen compared to tumors treated with either Dox alone or Dox + P85 ISFIs. This was validated with histological analysis showing a greater necrosis percentage for the Dox + Val ISFIs. The improvement in therapeutic efficacy of the ISFIs loaded with a Pgp inhibitor correlates well with the optical intratumoral Dox images. A greater maximum penetration and total intratumoral retention of Dox seen with the Dox + Val ISFIs suggests there is a larger area of treatment leading to a better therapeutic outcome.
A limitation to the current work is that the treatment only consisted of a single injection of ISFI at the beginning of the study. As shown with the release curves, approximately 50% of the Dox is released within 10 days and another 10% in the following 10 days22,31,57–59. Allowing the study to progress further than 20 days and using multiple injections of the ISFI will likely elicit stronger therapeutic effects. This approach as well as incorporation of varying doses of Pgp inhibitors into the ISFIs and examining their distribution within tumor tissue will be the subject of future studies.
Conclusions
MDR tumors that express Pgp have poor treatment responses due to required elevated drug concentrations to reach therapeutic levels. Current clinical trials using Pgp inhibitors suffer due to systemic toxicity. ISFIs provide an excellent alternative to deliver elevated concentrations of both the chemotherapeutic and Pgp inhibitor bypassing the associated systemic toxicities seen in clinical trials. In this study, Dox + Val ISFIs were able to demonstrate enhanced distribution and retention of the drug leading to a reduced tumor growth and increased necrosis compared to Dox alone ISFIs. This concurrent local delivery of a chemotherapeutic and Pgp inhibitor serves as a promising approach to treat MDR tumors without the toxicities seen with the systemic chemotherapy approach.
Supplementary information
Acknowledgements
This work was supported by the National Institute of Biomedical Imaging and Bioengineering and National Cancer Institute of the National Institutes of Health under award number R01EB016960 (to AAE). CH was supported by T32-EB007509, and F31CA200373. Views and opinions of and endorsements by the author(s) do not reflect those of the National Institutes of Health.
Author contributions
Dr. Selva Jeganathan collected and analyzed all data and wrote the manuscript. Ms. Emily Budziszewski helped with all in vivo studies, including animal monitoring, injections, and acquistion of fluorescent images. Dr. Christopher Hernandez had significant intellectual input into the design and analysis of all data collected. Mr. Anshul Dhingra contributed to the collection and analysis of in vitro cytotoxicity data. Dr. Agata Exner contributed to the oversight of the entire project along with significant intellectual input into the data analysis and discussion of the manuscript. All authors reviewed the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-63636-x.
References
- 1.Halpern MT, Yabroff KR. Prevalence of Outpatient Cancer Treatment in the United States: Estimates from the Medical Panel Expenditures Survey (MEPS) Cancer Invest. 2008;26:647–651. doi: 10.1080/07357900801905519. [DOI] [PubMed] [Google Scholar]
- 2.Dai Q, et al. Quantifying the Ligand-Coated Nanoparticle Delivery to Cancer Cells in Solid Tumors. ACS Nano. 2018;12:8423–8435. doi: 10.1021/acsnano.8b03900. [DOI] [PubMed] [Google Scholar]
- 3.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP–dependent transporters. Nat. Rev. Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 4.Wu Q, Yang Z, Nie Y, Shi Y, Fan D. Multi-drug resistance in cancer chemotherapeutics: Mechanisms and lab approaches. Cancer Lett. 2014;347:159–166. doi: 10.1016/j.canlet.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 5.Kathawala RJ, Gupta P, Ashby CR, Chen Z-S. The modulation of ABC transporter-mediated multidrug resistance in cancer: A review of the past decade. Drug Resist. Updat. 2015;18:1–17. doi: 10.1016/j.drup.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Ozben T. Mechanisms and strategies to overcome multiple drug resistance in cancer. FEBS Lett. 2006;580:2903–2909. doi: 10.1016/j.febslet.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 7.Coley HM. Mechanisms and strategies to overcome chemotherapy resistance in metastatic breast cancer. Cancer Treat. Rev. 2008;34:378–390. doi: 10.1016/j.ctrv.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 8.Markman JL, Rekechenetskiy A, Holler E, Ljubimova JY. Nanomedicine therapeutic approaches to overcome cancer drug resistance. Adv. Drug Deliv. Rev. 2013;65:1866–1879. doi: 10.1016/j.addr.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee G, Joung J-Y, Cho J-H, Son C-G, Lee N. Overcoming P-Glycoprotein-Mediated Multidrug Resistance in Colorectal Cancer: Potential Reversal Agents among Herbal. Medicines. Evidence-Based Complement. Altern. Med. 2018;2018:1–9. doi: 10.1155/2018/3412074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Z, et al. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett. 2016;370:153–164. doi: 10.1016/j.canlet.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 11.Chung M-F, Liu H-Y, Lin K-J, Chia W-T, Sung H-W. A pH-Responsive Carrier System that Generates NO Bubbles to Trigger Drug Release and Reverse P-Glycoprotein-Mediated Multidrug Resistance. Angew. Chemie Int. Ed. 2015;54:9890–9893. doi: 10.1002/anie.201504444. [DOI] [PubMed] [Google Scholar]
- 12.Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 13.Kabanov AV, Batrakova EV, Alakhov VY. Pluronic® block copolymers as novel polymer therapeutics for drug and gene delivery. J. Control. Release. 2002;82:189–212. doi: 10.1016/S0168-3659(02)00009-3. [DOI] [PubMed] [Google Scholar]
- 14.Alakhova DY, Zhao Y, Li S, Kabanov AV. Effect of Doxorubicin/Pluronic SP1049C on Tumorigenicity, Aggressiveness, DNA Methylation and Stem Cell Markers in Murine Leukemia. Plos One. 2013;8:e72238. doi: 10.1371/journal.pone.0072238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Batrakova EV, et al. Effects of pluronic and doxorubicin on drug uptake, cellular metabolism, apoptosis and tumor inhibition in animal models of MDR cancers. J. Control. Release. 2010;143:290–301. doi: 10.1016/j.jconrel.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robey RW, et al. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer. 2018;18:452–464. doi: 10.1038/s41568-018-0005-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tidefelt U, et al. P-Glycoprotein inhibitor valspodar (PSC 833) increases the intracellular concentrations of daunorubicin in vivo in patients with P-glycoprotein-positive acute myeloid leukemia. J. Clin. Oncol. 2000;18:1837–44. doi: 10.1200/JCO.2000.18.9.1837. [DOI] [PubMed] [Google Scholar]
- 18.Atadja P, Watanabe T, Xu H, Cohen D. PSC-833, a frontier in modulation of P-glycoprotein mediated multidrug resistance. Cancer Metastasis Rev. 1998;17:163–168. doi: 10.1023/A:1006046201497. [DOI] [PubMed] [Google Scholar]
- 19.Advani R, et al. Treatment of refractory and relapsed acute myelogenous leukemia with combination chemotherapy plus the multidrug resistance modulator PSC 833 (Valspodar) Blood. 1999;93:787–95. doi: 10.1182/blood.V93.3.787. [DOI] [PubMed] [Google Scholar]
- 20.Advani R, et al. Treatment of refractory and relapsed acute myelogenous leukemia with combination chemotherapy plus the multidrug resistance modulator PSC 833 (Valspodar) Blood. 1999;93:787–95. doi: 10.1182/blood.V93.3.787. [DOI] [PubMed] [Google Scholar]
- 21.Palmeira A, Sousa E, Vasconcelos MH, Pinto MM. Three Decades of P-gp Inhibitors: Skimming Through Several Generations and Scaffolds. Curr. Med. Chem. 2012;19:1946–2025. doi: 10.2174/092986712800167392. [DOI] [PubMed] [Google Scholar]
- 22.Solorio L, Wu H, Hernandez C, Gangolli M, Exner AA. Ultrasound-guided intratumoral delivery of doxorubicin from in situ forming implants in a hepatocellular carcinoma model. Ther. Deliv. 2016;7:201–212. doi: 10.4155/tde-2015-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krukiewicz K, Zak JK. Biomaterial-based regional chemotherapy: Local anticancer drug delivery to enhance chemotherapy and minimize its side-effects. Mater. Sci. Eng. C. 2016;62:927–942. doi: 10.1016/j.msec.2016.01.063. [DOI] [PubMed] [Google Scholar]
- 24.Miller MA, et al. Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nat. Commun. 2015;6:8692. doi: 10.1038/ncomms9692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bastiancich C, Danhier P, Préat V, Danhier F. Anticancer drug-loaded hydrogels as drug delivery systems for the local treatment of glioblastoma. J. Control. Release. 2016;243:29–42. doi: 10.1016/j.jconrel.2016.09.034. [DOI] [PubMed] [Google Scholar]
- 26.Belz JE, et al. Sustained Release Talazoparib Implants for Localized Treatment of BRCA1-deficient Breast Cancer. Theranostics. 2017;7:4340–4349. doi: 10.7150/thno.18563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramazani F, et al. Locoregional cancer therapy using polymer-based drug depots. Drug Discov. Today. 2016;21:640–647. doi: 10.1016/j.drudis.2016.02.014. [DOI] [PubMed] [Google Scholar]
- 28.Weinberg BD, Patel RB, Exner AA, Saidel GM, Gao J. Modeling doxorubicin transport to improve intratumoral drug delivery to RF ablated tumors. J. Control. Release. 2007;124:11–19. doi: 10.1016/j.jconrel.2007.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weinberg BD, Ai H, Blanco E, Anderson JM, Gao J. Antitumor efficacy and local distribution of doxorubicin via intratumoral delivery from polymer millirods. J. Biomed. Mater. Res. Part A. 2007;81A:161–170. doi: 10.1002/jbm.a.30914. [DOI] [PubMed] [Google Scholar]
- 30.Weiser JR, Saltzman WM. Controlled release for local delivery of drugs: barriers and models. J. Control. Release. 2014;190:664–673. doi: 10.1016/j.jconrel.2014.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mach J, et al. Noninvasive characterization of in situ forming implants using diagnostic ultrasound. J. Control. Release. 2010;143:183–190. doi: 10.1016/j.jconrel.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA. Cancer J. Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 33.Hammond WA, Swaika A, Mody K. Pharmacologic resistance in colorectal cancer: a review. Ther. Adv. Med. Oncol. 2016;8:57–84. doi: 10.1177/1758834015614530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Longley D, Johnston P. Molecular mechanisms of drug resistance. J. Pathol. 2005;205:275–292. doi: 10.1002/path.1706. [DOI] [PubMed] [Google Scholar]
- 35.Lee, J. S. et al. Rhodamine efflux patterns predict P-glycoprotein substrates in the National Cancer Institute drug screen. Mol. Pharmacol. 46 (1994). [PubMed]
- 36.Yang F, Teves SS, Kemp CJ, Henikoff S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim. Biophys. Acta - Rev. Cancer. 2014;1845:84–89. doi: 10.1016/j.bbcan.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fracasso PM, et al. Phase II study of paclitaxel and valspodar (PSC 833) in refractory ovarian carcinoma: a gynecologic oncology group study. J. Clin. Oncol. 2001;19:2975–82. doi: 10.1200/JCO.2001.19.12.2975. [DOI] [PubMed] [Google Scholar]
- 38.Lissoni, A. A., Manikhas, G. M. & Fracasso, P. M. Phase III Study of Valspodar (PSC 833) Combined With Paclitaxel and Carboplatin Compared With Paclitaxel and Carboplatin Alone in Patients With Stage IV or Suboptimally Debulked. Artic. J. Clin. Oncol., 10.1200/JCO.2007.14.9807 (2008). [DOI] [PubMed]
- 39.Valle JW, et al. A phase 2 study of SP1049C, doxorubicin in P-glycoprotein-targeting pluronics, in patients with advanced adenocarcinoma of the esophagus and gastroesophageal junction. Invest. New Drugs. 2011;29:1029–1037. doi: 10.1007/s10637-010-9399-1. [DOI] [PubMed] [Google Scholar]
- 40.Bielecki, P. et al. Enhancing fluorescein distribution from in situ forming PLGA implants using therapeutic ultrasound. In IEEE International Ultrasonics Symposium, IUS, 10.1109/ULTSYM.2017.8091587 (2017).
- 41.Jeganathan, S. et al. Ultrasound-Enhanced Distribution and Treatment Efficacy of Dox-Loaded Intratumoral In Situ Forming Implants in Murine HCT-15 Tumors. in 2018 IEEE International Ultrasonics Symposium (IUS) 1–4 (IEEE, 2018), 10.1109/ULTSYM.2018.8580108
- 42.Faustino-Rocha A, et al. Estimation of rat mammary tumor volume using caliper and ultrasonography measurements. Lab Anim. (NY) 2013;42:217–224. doi: 10.1038/laban.254. [DOI] [PubMed] [Google Scholar]
- 43.Gottesman, M. M. & Pastan, I. BIOCHEMISTRY OF MULTIDRUG RESISTANCE MEDIATED BY THE MULTIDRUG TRANSPORTER l * **. [DOI] [PubMed]
- 44.Wong HL, et al. A mechanistic study of enhanced doxorubicin uptake and retention in multidrug resistant breast cancer cells using a polymer-lipid hybrid nanoparticle system. J. Pharmacol. Exp. Ther. 2006;317:1372–81. doi: 10.1124/jpet.106.101154. [DOI] [PubMed] [Google Scholar]
- 45.Shen F, et al. Dynamic assessment of mitoxantrone resistance and modulation of multidrug resistance by valspodar (PSC833) in multidrug resistance human cancer cells. J. Pharmacol. Exp. Ther. 2009;330:423–9. doi: 10.1124/jpet.109.153551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loor F. Valspodar: current status and perspectives. Expert Opin. Investig. Drugs. 1999;8:807–835. doi: 10.1517/13543784.8.6.807. [DOI] [PubMed] [Google Scholar]
- 47.Solorio L, et al. Noninvasive characterization of the effect of varying PLGA molecular weight blends on in situ forming implant behavior using ultrasound imaging. Theranostics. 2012;2:1064–77. doi: 10.7150/thno.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alvarez M, et al. Generation of a drug resistance profile by quantitation of mdr-1/P-glycoprotein in the cell lines of the National Cancer Institute Anticancer Drug Screen. J. Clin. Invest. 1995;95:2205–14. doi: 10.1172/JCI117910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karlgren M, et al. Classification of Inhibitors of Hepatic Organic Anion Transporting Polypeptides (OATPs): Influence of Protein Expression on Drug–Drug Interactions. J. Med. Chem. 2012;55:4740–4763. doi: 10.1021/jm300212s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Solorio Luis, Sundarapandiyan Divya, Olear Alex, Exner AgataA. The Effect of Additives on the Behavior of Phase Sensitive in Situ Forming Implants. J. Pharm. Sci. 2015;104:3471–3480. doi: 10.1002/jps.24558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bakhshi R, Vasheghani-Farahani E, Mobedi H, Jamshidi A, Khakpour M. The effect of additives on naltrexone hydrochloride release and solvent removal rate from an injectablein situ forming PLGA implant. Polym. Adv. Technol. 2006;17:354–359. doi: 10.1002/pat.717. [DOI] [Google Scholar]
- 52.Zare M, et al. Effect of additives on release profile of leuprolide acetate in anin situ forming controlled-release system:In vitro study. J. Appl. Polym. Sci. 2008;107:3781–3787. doi: 10.1002/app.27520. [DOI] [Google Scholar]
- 53.Mashak A, Mobedi H, Ziaee F, Nekoomanesh M. The effect of aliphatic esters on the formation and degradation behavior of PLGA-based in situ forming system. Polym. Bull. 2011;66:1063–1073. doi: 10.1007/s00289-010-0386-7. [DOI] [Google Scholar]
- 54.Patel RB, Carlson AN, Solorio L, Exner AA. Characterization of formulation parameters affecting low molecular weight drug release from in situ forming drug delivery systems. J. Biomed. Mater. Res. Part A. 2010;9999A:NA–NA. doi: 10.1002/jbm.a.32724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kozlov MY, Melik-Nubarov NS, Batrakova EV, Kabanov AV. Relationship between Pluronic Block Copolymer Structure, Critical Micellization Concentration and Partitioning Coefficients of Low Molecular Mass Solutes. Macromolecules. 2000;33:3305–3313. doi: 10.1021/ma991634x. [DOI] [Google Scholar]
- 56.Karlgren M, et al. Classification of inhibitors of hepatic organic anion transporting polypeptides (OATPs): influence of protein expression on drug-drug interactions. J. Med. Chem. 2012;55:4740–63. doi: 10.1021/jm300212s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hernandez, C. et al. Macroporous acrylamide phantoms improve prediction of in vivo performance of in situ forming implants. J. Control. Release243 (2016). [DOI] [PMC free article] [PubMed]
- 58.Patel RB, Solorio L, Wu H, Krupka T, Exner AA. Effect of injection site on in situ implant formation and drug release in vivo. J. Control. Release. 2010;147:350–358. doi: 10.1016/j.jconrel.2010.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.solorio L, Sundarapandiyan D, Olear A, Exner AA. The Effect of Additives on the Behavior of Phase Sensitive In Situ Forming Implants. J. Pharm. Sci. 2015;104:3471–3480. doi: 10.1002/jps.24558. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.