

Abstract
Family-based penetrance is frequently cited as a major challenge for translating penetrance estimates from familial populations to asymptomatic populations. A systematic review was performed to assess the literature evidencing penetrance estimates in patients without a family history of disease, following the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) framework. Initially 1592 papers were identified, which were filtered to a final nine, through application of inclusion and exclusion criteria. Fundamental differences in the identified papers prevented combination of papers using meta-analysis, so thematic analysis to produce a narrative synthesis was performed. Key themes included disease risk modifiers, evidence, study limitations and bias. A methodological appraisal too was used to assess quality of included studies. It is evident from the findings that the evidence base for penetrance estimates in individuals without a family history of disease is limited. Future work is needed to refine design of penetrance studies and the impact of incorrect estimates.
Subject terms: Genetic testing, Genetic testing
Introduction
Traditionally, genetics has used a phenotype-driven approach to identify cases with an underlying genetic pathology and likely causative variants [1]. An example of this is for developmental delay (DD), where trio analysis of affected individuals, typically children, is used to identify likely causative variants [2, 3]. Some variants are known to have variable penetrance, which can be caused by many different mechanism, leading to reduced or increased risk [4].
One example of a disorder which causes DD is Rett syndrome, which is an X-linked disorder caused by MECP2 variants, predominantly affecting girls [5]. Mosaicism can alter the phenotype of this disease though skewed inactivation of the X-chromosomes (XCI), which was first identified in twins with differing phenotypes [6]. In 2007, a woman was identified, through her affected son, to be asymptomatic for Rett syndrome with a C-terminal truncation variant in MECP2, which usually produces a classical phenotype. This was the first case of non-penetrance and was predicted to be linked to highly skewed XCI. It also suggested that screening mothers of Rett syndrome patients is indicated, as this could affect reproductive decisions [7].
Genetic testing for inherited disease is frequently offered to family members of affected individuals. For example, BRCA1 and BRCA2 testing is offered to individuals with a known history of familial breast and ovarian cancer (FBOC). Screening has the advantage of informing life-time risk, and whether preventive action is advisable [8]. However, individuals with a family history of FBOC, who are negative for BRCA1 and BRCA2 variants established to be associated with an increased risk of breast and ovarian cancer, may still have a higher cancer risk relative to the wider population, indicating further risk factors are involved [9, 10].
Calculating prevalence and penetrance of variants requires population sampling and is more accurate the larger sample populations and more homogeneous cohorts [11]. Technological developments have reduced the costs and time associated with genome-wide sequencing [12] and NGS has been essential to projects such as ExAC, 1000 Genomes, and the 100,000 Genomes Project [13–15]. ExAC, a whole exome sequencing (WES) cohort of more than 60,000 people, harboured ~54 variants reported as disease causing and 41 of these were too common in the population to be classified as pathogenic [15]. ExAC has also been used successfully to confirm pathogenicity of variants validating clinical decisions [16].
A qualitative study of expert opinion on reporting of secondary findings, highlighted the challenges facing reporting without having complete penetrance studies linking genotype-to-phenotype in unaffected populations. There is concern over the extent to which family-based penetrance impacts pathogenicity through unknown risk modifiers [17]. Genetic profiling is used to inform clinical and reproductive decisions, so individuals may feel they should take action, especially if they have seen the effect of the disease and wish to prevent disease risk [18]. If there is uncertainty around penetrance between populations, there is a risk of misinformation which could lead to unnecessary intervention. If the mechanisms behind risk modifiers were identified, this information would allow genomic counsellors and clinicians to more accurately estimate risk informing clinical action.
This systematic review addressed the question: What is the evidence base for variant penetrance estimates in individuals with no family history of the condition?
Methods
Study design
A systematic search was performed and reported following the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidance [19, 20]. This process allowed for an explicit and reproducible strategy for selection of literature. The methodological search was performed through Ovid, a journal search engine, facilitating searches in Medline, EMBASE and Global Health. Ovid also allowed for search terms to be exploded, to incorporate all known similar terms within literature to be incorporated into one search. The search was performed in September 2019.
Search strategy
A broad search strategy being deemed to be most appropriate to give the best chance of capturing all available evidence pertaining to the research question. Terms used included “incidental findings,” “genetic testing,” “genetic counselling,” “penetrance” and “genetic disease.” The complete bank of terms, including how the terms were exploded and combined using Boolean operators is documented in Appendix 1 as a full electronic search strategy for one of the databases. Free searching was used in addition to add unknown terms when exploding, such as secondary findings (SF). Reference searching of final papers was performed to identify further relevant papers.
Inclusion and exclusion criteria
Included papers had to have been published in English between January 2009 and September 2019. Key inclusion criteria were that the papers had to be primary literature or a systematic review of primary literature (see Table 1). There also had to be some reference to the use of a population identified through means other than family history. This could for example be the use of a cohort database, such as 1000 genomes, or through a population screen. Exclusion criteria were clearly defined. All articles unrelated to genetics were excluded, for example relating to medical imaging. Genetic related review papers, articles without abstracts, or articles linked to ethical, legal and social implications were also excluded. This review excluded papers related to epigenetic or mitochondrial disease and papers aiming to inform clinical management of a disease or case studies pertaining to individuals affected by disease. Papers were excluded for having a familial focus when estimating penetrance or similar risk estimates.
Table 1.
Inclusion and exclusion criteria for papers to be included within the current review.
| Include | Exclude |
|---|---|
|
Articles where: • The study is primary literature OR a systematic review of primary literature • The population is not obtained through means of family history only • The written language is English • The paper was published between January 2008 and November 2018 |
Studies that: • Are unrelated to genetics • Are review papers • Discuss ethical, legal and social implications • Study epigenetic or mitochondrial disease • Define clinical management of disease • Are case studies • Use family history based penetrance only |
Search outcomes
Figure 1 is a PRISMA flow-chart showing the stages involved in the search. This search identified 1592 papers and reduced to 1356 once duplicates were removed. At each stage of analysis, two independent researchers screened papers and met to discuss and reach consensus on which papers to progress to the next stage. Disagreement over paper inclusion was resolved through recruitment of a third researcher. These papers were initially title searched to remove all papers deemed to be clearly irrelevant to the research question. This left 175 articles had their abstracts screened. Of these, 25 met the criteria for a full-text search. Ten studies met the inclusion criteria to be included in the final analysis. All final papers were identified by the Medline search, but only two were identified by searching Global Health alone. Reference searching and discussion with an experienced genomics researcher identified a further eight papers, of which two met the inclusion criteria and were retained.
Fig. 1. PRISMA flow-chart showing the stages of the systematic review.
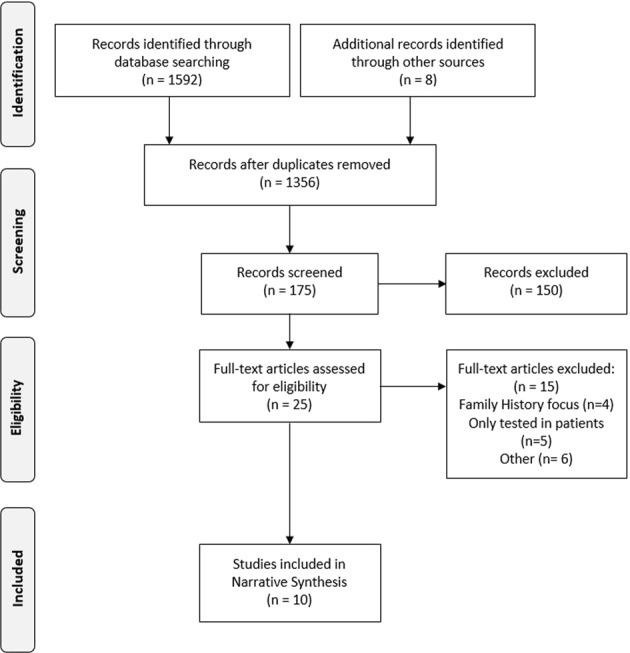
PRISMA flow diagram of study selection. The screening process identified ten studies from an initial pool of 1592 as being relevant to the current review and having satisfied the inclusion criteria. This PRISMA flow diagram demonstrates the process of identifying and screening relevant studies.
Quality assessment
Quality assessment of identified literature was performed by two independent researchers using a validated tool for qualitative and quantitative study appraisal [21]. This analysis generated a comparable score to assess the methodological strength of identified literature. Quality assessment scores were not used to exclude papers due to the paucity of literature in this area, however all included papers scored highly regardless.
Analysis of final literature
Due to the disparity of aims and methodologies of the identified papers, it was not feasible to do a meta-analysis of results. Therefore, a narrative synthesis was performed according to the Cochrane guidelines [22]. Thematic analysis was used to generate themes from the included studies [23]. This technique has been adopted for use in systematic reviews and is effective for analysis of small samples of quantitative literature [18, 24]. Identified themes were confirmed between researchers to minimise bias and ensure fair representation of identified papers within this review. The Cochrane Risk of Bias tool was referred to, to ensure assessment of the robustness of evidence in identified papers and to reconcile limitations within this review [25].
Results
Ten papers met the inclusion criteria for this search, with core features described in Table 2 [26–35]. These trials used either observational [27, 28] or case-control design which focused on specific diseases to identify the cohort of interest. The diseases these papers focused on were haemochromatosis [31], Huntington’s disease [34], familial and sporadic breast and ovarian cancer (FBOC and SPBCOC, respectively) [26], cystic fibrosis [32], retinitis pigmentosa [33], diabetes [29], Turner syndrome [35], 47,XXX syndrome [35] and prion disease [30].
Table 2.
Description of key characteristics of final papers from the literature search.
| Reference | Title | Aim | Number of participants | Key findings | Quality assessment |
|---|---|---|---|---|---|
| Cardenosa et al. [26] | Low penetrance alleles as risk modifiers in familial and sporadic breast cancer | Investigate the relevance of HUS1, CASP8, and FGFR2 polymorphisms and miRNA-146A genes, as risk modifiers in hereditary breast or ovarian cancer or sporadic breast or ovarian cancer patients. →Spanish cohort | 340 familial cases, 198 sporadic cases and 189 control cases |
BRCA1/2 sequencing only in FBCOC group. BRCA1/2 FBCOC group risks were associated with FGFR2, and miRNA-146A. Non-BRCA1/2 FBCOC risks were associated with HUS1 and FGFR2. HUS1 led to a younger age of onset for cancer. SPBCOC group had no significant associations. CASP8 had no significant association |
81% |
| Cassa et al. [27] | Large number of genetic variants considered to be pathogenic are common in asymptomatic individuals | Assess the frequency of HGMD variants classified as pathogenic in an asymptomatic population. →International cohort | 1092 |
3744 HGMD variants found in cohort with MAF ≥ 0.01. The average individual had on 294 variants in homozygous form and 942 variants in heterozygous form previously identified pathogenic variant 903 identified variants had supporting biological evidence. PolyPhen2 software predictions: As MAF increase, likely pathogenicity decreased. 58% of disease variants were predicted as pathogenic. |
100% |
| Chen et al. [28] | Analysis of 589, 306 genomes identifies individuals resilient to severe mendelian childhood diseases. | Analysis and screening of genotype data (SNP array, WES, WGS studies) to identify asymptomatic individuals for severe childhood-onset disease. →global cohort |
n = 589,306 Genotyped: 518,721 WES: 67,061 WGS: 3052 |
Screen identified 13 individuals harbouring pathogenic variants for eight Mendelian childhood diseases. Use of confirmation sequencing in five individuals. No follow-up of candidates possible. Creation of a more detailed disease gene panel and a pipeline/filtering process to identify individuals of interest. |
100% |
| Laver et al. [29] | The Common p. R114W HNF4A mutation causes a distinct clinical subtype of monogenic diabetes. | Evaluate if p. R114W is pathogenic and to define clinical subtype. |
n = 30/2289 MODY cohort with p.R114W with 51 family members n = 100 HNF4A probands with 224 family members n = 9188 type 2 Diabetes patients n = 12,890 healthy controls |
p.R114W is enriched in MODY cohort compared with control population (OR = 49.4, p = 3.7 × 10−37). p.R114W is enriched in MODY cohort compared with type 2 diabetes patients (OR = 15.6, p = 2 × 10−21). Inheritance pattern consistent with reduced penetrance compared to other HNF4A variants (46% p.R114W don’t have diabetes by 36 vs. 29% for other HNF4A). Median age of diagnosis 34 vs. 24 for p.R114W compared with other HNF4A variants. Observed that birthweight not increased as with other HNF4A variants and lower response to sulphonylureas. Clinical subtype defined as low penetrance, normal birthweight and reduced response to sulphonylureas. |
|
| Minikel et al. [15] | Quantifying prion disease using large population cohorts | Re-evaluate the penetrance of reported pathogenic variants in PRNP. |
Prion cases: 16,025 (no genotyping) ExAC: 60,706 (WES) 23&me: 531,575 (genotyped) |
Variants are more prevalent within ExAC (30-fold) and 23&me (ninefold) than predicted based upon disease incidence, genetic role in disease and life expectancy. Variants with high levels of evidence not present in cohorts. Three variants had evidence to suggest they were not benign but were too prevalent to follow mendelian disease. Penetrance evaluated at between 0.1 and 10%. Heterozygous loss of PrP function seems tolerated. |
100% |
| Pedersen and Milman [31] | Genetic screening for HFE haemochromatosis in 6020 Danish men: penetrance of C282Y, H63D and S65C variants | Assess penetrance of HFE variants within a Danish cohort C282Y, H63D, and S65C on the phenotypic expression of iron status markers. |
6020 genotyped 1452 Phenotyped: Control group (CG): 331 |
Allele frequency of variants and penetrance estimates for C282Y/C282Y (84%), C282Y/H63D (18%) and H63D/H63D (13.2) homozygotes, and C282Y/wildtype (8%). No follow-up on C282Y homozygote with non-penetrance. |
100% |
| Robinet et al. [32] | The very low penetrance of cystic fibrosis for the R117H mutations: a reappraisal for genetic counselling and newborn screening | Delineate the overall disease phenotype associated with R117H for cystic fibrosis (CF) and evaluate penetrance of the [R117H] + [F508del] genotype and identify is IVS8 poly (T) influences phenotype variance. →French cohort | 278 cases, 5245 healthy adults, 3,527,352 newborns screened |
Penetrance estimation for phenotypes. 0.03% penetrance for classical CF with [R117H] + [F508del] phenotype. Population prevalence of genotype in France was equivalent to 3650 individuals. Identified 25.7% of predicted newborns with [R117H] + [F508del] vs. 83.8% of predicted homozygous F508del newborns. Very low prevalence of T5 in French population so could not assess if it has a role in pathogenicity. |
100% |
| Rose et al. [33] | Transcriptional regulation of PRPF31 gene expression by MSR1 repeat elements causes incomplete penetrance in retinitis pigmentosa | Assess the role of copy number variance of MSR1 element in PRPF31 and the associated risk with Retinitis Pigmentosa. →International cohort |
624 CG: 354 |
Symptomatic daughter had 3× copies of MSR1 and asymptomatic father had hemizygous 4× copies of MRS1 In vitro evidence that 3× MSR1 induces haploinsufficiency for PRPF31 and other genes indicating it acts as a generic risk modifier. All individuals with retinitis pigmentosa had 3/3 MSR1 genotype. Proportion of Asian-origin families who were asymptomatic was significantly lower, and the rate of 4× MSR1 was significantly lower in Asian populations. MSR1 was shown to be a generic modulator of activity on other gene function and activity appeared to be location and not orientation specific. Having four MSR1 copies seemed to play a protective role for this disease and it was hypothesised that MRS1 copy number may play a role in other diseases. |
100% |
| Sequeiros et al. [34] | Large normal and reduced penetrance alleles in Huntington disease: instability in families and frequency at the laboratory, at the clinic and in the population | Confirm frequencies and study variation in size of transmission of large normal and reduced penetrant alleles. →Portuguese cohort |
Consultands = 146 Diagnostic lab = 1214 CG: 886 |
Large normal alleles had similar allele frequency (and genotype frequency) for all populations: Consultands = 3.8% (6.8), diagnostic lab = 3.5 (6.8), CG = 3% (6%) 13 cases of parent-offspring transmission, the ten large normal alleles all stably transmitted, and three reduced penetrance alleles showed instability (either anticipation or contraction). Instability in sibling groups indicated to increase with parental age, with one set of four siblings showing anticipation leading to change from 38 in oldest sibling to 41 in youngest. |
100% |
| Tuke et al. [35] | Mosaic Turner syndrome shows reduced penetrance in adult population study. | Identify from large cohort how many have an X chromosome imbalance. Characterise the phenotypic characteristics of women with X chromosome aneuploidy. |
n = 244,848 n = 326 with X chromosome imbalance |
Identified a greater number of mosaic 45,X/46,XX within cohort than expected (76/100,000). 47,XXX had expected clinical phenotypes. Non-mosaic 45,X individuals consistent with clinical characteristics of Turner syndrome (short stature and primary amenorrhoea). 55% known diagnosis of Turner syndrome. Partial X chromosome deletion and isochromosome caused short stature phenotype. Mosaic 45,X/46,XX were one average 3.5 cm shorter but with weak correlation (r = 0.23). No difference in mean age of menopause or number of pregnancies between mosaic 45,X/46,XX and 46XX women and no increased incidence of pregnancy loss. Hypertension and blood pressure not raised in 45,X/46,XX women and not more likely to have had a cardiac operation or be on blood pressure medication than 46,XX women. Study concluded that most women with mosaic 45,X/46,XX will not require clinical follow-up. |
100% |
All included studies scored full marks in the quality appraisal, with one exception. Cardenosa et al.’s study design was not entirely appropriate to the study aim, because only selected populations underwent BRCA1/2 sequencing, preventing comprehensive comparison. In addition, the sporadic population was disproportionately small relative to the familial population resulting in underpowering and researchers did not fully discuss how this limitation could impact the generalisability of their findings.
Summary of thematic analysis findings
Seven themes were identified from the included studies: variant classification, clinical implications, evidence, bias, study design, study limitations and disease risk modifiers. Categories and themes identified can be seen in Fig. 2. In the interest of brevity, only the themes most relevant to the aims of this review are discussed below.
Fig. 2. A schematic depicting the themes and categories generated from the included studies.

The 48 categories generated by the thematic analysis are represented in this schematic as small boxes. The seven themes are represented by large boxes. The colours indicate the overarching theme that the category belongs to.
Disease risk modifiers
Most evidence for variations in penetrance comes from incomplete penetrance within families. Rose et al. identified the link between three or four copies of MRS1 in penetrance of retinitis pigmentosa [33]. Cassa et al. indicated that either erroneous findings or risk modifiers must occur at higher frequencies than previously believed, due to identifying a high proportion of variants annotated as pathogenic from the Human Gene Mutation Database (HGMD) in the 1000 genome cohort [27]. Chen et al. identified 13 individuals harbouring variants for severe childhood-onset disorders without apparent phenotype [28]. This suggests that risk modifiers exist even for variants which are classified as 100% penetrant, which has wide implications for the rare disease community.
Population studies were used to identify if there is variation in prevalence or penetrance of a disease.
Rose et al. compared European and Asian-origin populations, because Asian populations have higher penetrance for retinitis pigmentosa, and identified that Asian populations have a lower prevalence of the four copies of MSR1 genotype [33]. The approach to calculating prevalence varied between studies, with Thauvin-Robinet et al. using partners of CF patients, whereas Pedersen and Milman used a country-wide sampling approach [31, 36]. Thauvin-Robinet et al. used their approach to calculate the number of newborns with the [R117H] + [F508del] genotype, which they compared with the true number identified [31]. They also aimed to see if the IVS8 poly(T) played a role in modifying phenotype. However, T5, which is believed to be more pathogenic, had a very low prevalence in the French cohort. In populations with higher prevalence of T5, it has been associated with higher penetrance [37]. Pedersen and Milman used a genotype-driven approach to population sampling to calculate prevalence within the population [31]. They phenotyped a proportion of this population to calculate penetrance of these variants, which was a novel approach to penetrance estimation for haemochromatosis [38].
Tuke et al. identified mosaic X chromosome 45,X/46,XX in a higher than expected number of patients within women in the UK Biobank cohort (76/100,000) [35]. Turner syndrome is caused by 45,X genotype with key phenotypes related to short stature, cardiac disorders and reproduction. Comparison between these mosaic individuals and normal 46,XX women identified no significant increase in cardiac or reproductive incidents. Clinically this has significant implications as women identified as 45,X/46,XX appear to require no clinical follow-up.
Biological mechanisms for how a variant causes disease were presented either by building upon previous work, which has indicated a mechanism [26], or by studying the mechanism as part of the studies design [33]. Cardenosa et al. use a combination of biological evidence and case studies to select variants to study [26]. Rose et al. performed the only study in this review which aimed to identify the biological mechanisms underlying the risk variance [33]. In their study, they used a combination of in vivo and in vitro evidence to show that 3× MSR1 CNV induces haploinsufficiency through inhibition of transcription and that 4× MSR1 CNV may act as a rescue mechanism. It is likely that MSR1 has pleiotropic effects, as it modified the activity of other genes when inserted into the promoter region.
Evidence
Rose et al. were the only study to actively explain how the underlying biology affects disease risk [33]. Evidence quality is also dependent upon appropriate study design. Chen et al. included several aspects in their design to manage potential limitations [28]. They chose to only look at severe childhood-onset Mendelian diseases to increase certainty in findings. When they attempted to reduce stringency, the number of individuals identified and the subsequent complexity of verifying variants increased. A multistage filtering process was used, including assessing data quality to exclude candidates, to maximise the validity of the final candidates. They identified 13 individuals that are asymptomatic; however, only five could have confirmation sequencing performed which is part of the final stage of their process, so only these candidates definitively have been shown to harbour these variants.
Thauvin-Robinet et al. wanted to delineate the phenotype of R117H to calculate the prevalence and penetrance of [R117H] + [F508del] for causing CF [32]. They used a CF phenotype test and then genotyped for CF variants in phenotype-positive newborns, only identifying ~25% of predicted newborns. However, certain newborns may not present with the screened phenotype, so it could be a poor indicator for that genotype. Pedersen and Milman, by comparison, used a genotype-driven approach to identify prevalence and phenotyped afterwards to calculate penetrance [31, 38].
Laver et al. questioned the pathogenicity of R114W within HNF4A, due to being more common in the population relative to the expected population occurrence [29]. In addition, this variant did not exhibit the same phenotypes as other HNF4A MODY variants. They were able to identify that this variant is enriched in the MODY cohort and is not linked to type 2 diabetes. They were also able to demonstrate that relative to other HNF4A variants, R114W has a later median disease onset of approximately 10 years. This was consistent with the hypothesis that R114W has a reduced penetrance.
Sequeiros et al. used a combination of data samples to provide evidence that large normal alleles are common in the population, as an indicator that they are likely stably transmitted [34]. In addition, new intermediate alleles, identified through sperm analysis, are more unstable than those of identical size in the general population, although samples were only taken from four men [39].
Cardenosa et al. had the lowest score using the appraisal tool, largely due to uncertainty over the way they conducted their sequencing. They chose to only sequence BRCA1/2 in the FBOC group, and not the control group or the SPBCOC group. While BRCA1/2 are rarely found in sporadic cancer, only genotyping could confirm that BRCA1/2 is not present in the sporadic or control group [40]. Genotyping of BRCA1/2 in the control group would be important to indicate if BRCA1/2 variants occurred in the population. The researchers also grouped BRCA1/2 into one group with both breast and ovarian cancer, which may be confounding as BRCA1 and BRCA2 are separate genes and are associated with different cancer risks [41].
Cassa et al. designed their study to provide evidence of reduced or non-penetrance of variants listed by HGMD as pathogenic, within the 1000 Genomes cohort [27]. They identified 6917 variants of which 3744 had a minor allele frequency of ≥0.01. Some disease-associated variants have known varied penetrance, but this is insufficient to explain the volume of identified variants. There was a high number of disease variants (n = 583) identified in this cohort with a MAF ≥ 0.01. This study provided evidence of erroneous findings, and likely unknown risk modifiers within the cohort. They based their rational on the fact that many variants are identified in symptomatic populations in studies with small cohorts. There are often limited validation studies and unmatched control populations. Their study indicated that the probability of observing a variant may not be equivalent to the risk of developing the disease. In a previous study by the same researchers, 10.6% of HGMD variants had sufficient clinical relevance and scientific validity to be shared with research participants [42].
Minikel et al. were limited in the evidence which they could gather due to lack of standardisation of genotyping across the countries involved in case collection [30].
Bias and study limitations
Several of these studies worked to demonstrate the effect of bias and minimise its impact. Key stated limitations within this review were missing diagnosis [32] and follow-up [28, 31, 32]. Missing description of limitations was identified within Cardenosa et al. [26].
Chen et al. were unable to follow-up candidates due to limited consent. This is especially a challenge within the 23&me cohort and other direct-to-consumer genetic testing (DTC-GT) companies, where inadequate consent prevents re-contact. Chen et al. attempted to expand their analysis in candidates from the UK10K, a WES cohort [28]. However, only five of the identified variants were annotated and none indicated a putative role. WGS would probably be required to identify putative variants for further study.
Chen et al. attempted to collect the largest possible sample of individuals to optimise the chance of identifying candidates of interest, including a combination of genotyped, whole exome and whole genome cohorts. SNP arrays made up 88% of samples, with 68% being from 23&me alone [28]. SNP arrays had the lowest coverage (377 loci vs. 699 for WES) and were responsible for the low coverage of loci (36.5%). Chen et al. do highlight that the exact composition of commercial pan-ethnic screening panels cannot be scrutinised [28]. They attempt to overcome issues around incomplete screening panels by creating their own expanded allele panel, which is more comprehensive than other commercial panels.
Study design can also act to limit the scope of a study, as can be seen in the decision to only BRCA1/2 genotype FBOC limited the findings of Cardenosa et al. [26]. It is suspected that these individuals had already been BRCA genotyped and the researchers did not have the resources to BRCA1/2 genotype the rest of the cohort. As this may have confounded the result, the design limits the scope of the findings.
Cassa et al. based their rationale upon the hypothesis that some variants which are classified as pathogenic are erroneous findings, and some of the other studies identified provided evidence which further supported these claims [27]. Rose et al. identified a mechanism which underlies variance between populations, which Cassa et al. hypothesised would happen when asymptomatic individuals were studied further [27, 33]. Thauvin-Robinet et al. confirmed that R117H is more commonly associated with asymptomatic patients than affected patients, making it potentially an erroneous finding if it were used in clinical decisions, such as prenatal testing [32].
While potential factors which may have biased the papers have been discussed in detail, they are likely to only have a moderate to low risk of bias due to their efforts to consider and manage limitations and bias within their design [25].
Discussion
This systematic review has summarised the sources of data for genetic variant penetrance estimates in a population setting. It has shown that the evidence base in this context is extremely limited. Limitations of study design, strength of evidence and disease risk modifiers may account for discrepancies between estimates generated form the traditional phenotype first, family history based approach and observed prevalence in the wide population or within genotype first cohorts.
Disease risk modifiers are central to understanding why disease phenotype varies between individuals. The included studies addressed indications of varied penetrance, support for reduced risk variants and biological mechanisms underlying variants. Data from one of the studies [35] suggests that mosaicism may be a powerful modulator of disease pathogenicity. The included studies demonstrate evidence to suggest that disease risk modifiers are relatively common in disease cohorts. This evidence is most robust when biological evidence is coupled with population data and so it can be suggested that genomic data repositories be combined wherever possible to increase the strength of such studies. This information has the potential to impact genomic counselling and treatment considerations.
Observational studies can be used to reveal weaknesses within current research. This is particularly important where the discovery cohort is significantly different to the population in which the testing will occur. Large discrepancies between predicted penetrance and observed disease should cause cautious consideration of the original estimates and prompt discussions about how to counsel and manage patients. Standardisation of case selection across countries would improve penetrance studies because it would create greater certainty of the levels of ascertainment, especially regarding lower penetrance variants. In addition, it would allow for better ascertainment of founder variants in specific regions which guide clinical management of affected families.
Bias and limitations are common issues within research, although their origin differs. Bias is the systematic misrepresentation of findings, due to omission of outcomes from their derivation, whereas limitations are factors that a researcher cannot control, which restrict methodology and conclusions. Limitations have the power to bias findings if they are unreported. A number of the included studies contained significant limitations. One of these being the issue of correctly phenotyping patients. Missing diagnosis can occur when phenotypes are mild preventing sufficient recognition to prompt genetic testing. Certain phenotypes are ambiguous and common, so an individual could easily dismiss these symptoms. More newborns possessed the [R117H] + [F508del], who were not identified by phenotyping; however, without genotyping, they could not be identified to calculate true penetrance. To overcome this limitation, all newborns would need genotyping and clinical follow-up to calculate true penetrance [32]. When assessing individuals who are suspected to be asymptomatic, follow-up is essential to clarifying if they are truly non-penetrant or reduced penetrance. This could prove particularly difficult on a population level, where DTC-GT data has been used and inadequate consent prevents re-contact. This is just one issue which needs to be addressed within the wider discussion of consent for DTC-GT [43, 44].
Another issue of note to DTC-GT and GWAS alike is the use of SNP arrays. SNP arrays are designed primarily for use in genome-wide association (GWA) studies, which are based upon the premise that common variation contributing to disease are present in frequencies >1% in the population and these studies are dependent upon large samples to create statistical power [45, 46]. This means that they are less effective at picking up rare variants, especially between ethnic populations, where variant prevalence differs and the composition of the arrays may be designed for a specific population 46. Up to 40% of variants identified by DTC-GT companies are believed to be false positives and some variants which these companies classify as pathogenic are classified as benign by software [47]. Recent work has further highlighted how poor these arrays perform for rare pathogenic variants, with over 80% of those identified being false positives [48]. This has profound implications for downstream clinical follow-up.
The close to significant findings of FGFR2 for SPBCOC in the Cardenosa study, indicates either too small a sample size or that BRCA1/2 genotyping may have confounded results, especially as this variant has been significant for this group in other studies [49, 50]. A recent GWA study again linked FGFR2 to breast cancer risk, although a different variant within this gene [51]. The Cardenosa study also does not characterise the spread of age of onset among the cohort, despite penetrance being known to vary in BRCA1/2 FBOC cohorts [52]. The non-BRCA1/2 FBOC group had a higher rate of breast cancer than the BRCA1/2 FBOC group, so this information could have implications for further study, although this is not commented upon in the discussion [26]. The grouping of BRCA1 and BRCA2 into the same cohort also warrants explanation as they confer different cancer risks [53].
Clinical implications
The major clinical implications of these studies relate to genomic counselling; either to guide genetic testing [32–34], inform risk reporting of findings [28, 31], or to guide changes to improve research and clinical interactions [27, 28]. These studies all recognised a need for further research to expand the applicability of findings to other populations, to identify mechanisms underlying findings and to validate findings. Therefore, the clinical implications of these papers can be considered as a need to ensure sufficient evidence before reporting findings and that complete consent must be taken to ensure follow-up is possible.
Generalisability: secondary findings and the 100,000 Genome Project
The clinical implications of these papers and themes expand beyond the individual diseases into a wider discussion of how incidental findings or SF should be included in clinical practice. The American College of Medical Genetics and Genomics (ACMG) created a list of genes, which they recommended should be sequenced alongside any genomic test. These genes were selected for their potential actionability [54, 55]. The resultant debate has led to some very different opinions regarding if SF should be reported, and if they are, what criteria make them actionable.
ACMG listed RET variants causing medullary thyroid carcinoma in childhood as being clinically actionable [56]. However, there have been cases of reduced penetrance, including a woman with a high-risk variant who developed thyroid cancer at 87 [57]. Management of this variant would normally involve thyroid removal, which is a major operation with a long-term impact on quality of life. If there are risk modifiers involved, these need to be identified to improve clinical decisions and minimise the risk of unnecessary medical intervention. This is the foundation of much of the reticence around incorporating SF into clinical settings [17].
In the UK, the 100,000 Genome Project has raised a lot of questions about how SF (termed additional findings in the project) should be managed [58–60], especially with uncertainty around family-based penetrance [17].
There have been efforts to create quantitative measures of actionability to facilitate incorporation into clinical reporting [61, 62]. Much of the study into the evidence base of reporting has been inconclusive, because many variants lack validation [17, 27, 42, 63]. The impact of disease risk modifiers is a complicating factor in the reporting of SF, with this review identifying several studies suggesting risk modification is common [27, 28] and that there are identifiable mechanisms underlying variance in penetrance [26, 31–33]. The role of ascertainment bias was explicitly addressed in Laver et al. when trying to evaluate risk as a key aspect of clinical management of patients [30]. The authors discuss the impact of ascertainment bias and ways to minimise this when evaluating whether variants are reduced penetrance or likely benign.
Cardenosa et al.’s design came closest to calculating family-based penetrance by using control vs. sporadic vs. familial populations. The familial group was additionally characterised by BRCA1/2 sequencing, which made comparison between these populations difficult and somewhat reduced the validity of the data to address this specific question [26]. However, this study did not aim to calculate penetrance for these variants, rather to assess the strength of association, whereas Pedersen and Milman used population screening to calculate penetrance for HFE variants using a genotype-to phenotype approach [31].
The most direct approach to characterising penetrance differences requires comparison between control, familial and sporadic populations. Large-scale projects such as UK Biobank and the 100,000 Genome Project will provide an invaluable resource for this. The large populations allow for comparison, with knowledge of phenotypes, and if any candidates of interest are identified, the whole genome can be interrogated to identify putative risk modifiers. The participants have indicated if they are available for follow-up, and thus can be further characterised to assess if the variants they have are benign or exhibit reduced penetrance. Tissue samples are also stored to allow for confirmation sequencing of putative variants or individuals can be re-contacted to collect samples [64]. The longitudinal study of participants will support age-stratified penetrance estimates, which is especially relevant to cancer risk.
Alternatively, the approach taken by Pedersen and Milman is effective for calculating penetrance [31]. They were able to genotype a large number of individuals for a specific disease and then follow-up a proportion for phenotyping. This approach would only be able to identify family-based penetrance, if combined with clinical data to identify individuals with a known family history for the disease. Penetrance could then be calculated separately between the familial and the sporadic groups to identify any statistically significant differences between the groups. Statistical significance would be an indicator for risk modifiers and these individuals could be further genotyped to identify putative variants. This approach would favour diseases which are easily phenotyped and relatively common in the population, as haemochromatosis is in the Danish population.
Limitations of this review
The biggest limitation of this review is that the small number of papers identified and the diversity of aims and methodologies which prevented meta-analysis of the data and direct testing of whether penetrance estimates are significantly different in cohorts where patients lack a positive family history. This suggests that the evidence base as a whole is poor and that extensive further research is needed in this area, however anecdotally it appears where such comparisons have been carried out, variants have higher penetrance in cohorts with a positive family history.
Conclusion
It remains unclear how much evidence there is to suggest that family-based penetrance impacts variability in disease risk. That being said, there is some indication that mechanisms exist which could explain variability between sporadic and familial groups and between ethnic populations. Concerns over how best to manage SF, and pressure to actively report findings, make this a pressing issue, which needs to be studied further. Further research is warranted encompassing familial, sporadic and control groups for a given condition or population sampling combined with clinical and phenotypic data to assess variant penetrance more extensively.
Further research into the ethical consequences of reporting SF without specific penetrance estimates in the relevant population is needed before they should be integrated into clinical practice.
Supplementary information
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41431-019-0556-5) contains supplementary material, which is available to authorised users.
References
- 1.Beskow LM, Linney KN, Radtke RA, Heinzen EL, Goldstein DB. Ethical challenges in genotype-driven research recruitment. Genome Res. 2010;20:705–9. doi: 10.1101/gr.104455.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, Van Kogelenberg M, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 2015;385:1305–14. doi: 10.1016/S0140-6736(14)61705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.LELM Vissers, KJM VanNimwegen, Schieving JH, Kamsteeg EJ, Kleefstra T, Yntema HG, et al. A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genet Med. 2017;19:1055–63. doi: 10.1038/gim.2017.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H. Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet. 2013;132:1077–130. doi: 10.1007/s00439-013-1331-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–8. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 6.Miyake K, Yang C, Minakuchi Y, Ohori K, Soutome M, Hirasawa T, et al. Comparison of genomic and epigenomic expression in monozygotic twins discordant for Rett syndrome. PLoS ONE. 2013;8:e66729. [DOI] [PMC free article] [PubMed]
- 7.Dayer AG, Bottani A, Bouchardy I, Fluss J, Antonarakis SE, Haenggeli C-A, et al. MECP2 mutant allele in a boy with Rett syndrome and his unaffected heterozygous mother. Brain Dev. 2007;29:47–50. doi: 10.1016/j.braindev.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 8.Rousset-Jablonski C, Gompel A, C. R-J. Screening for familial cancer risk: focus on breast cancer. Maturitas. 2017;105:69–77. doi: 10.1016/j.maturitas.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 9.Goldgar D, Venne V. BRCA phenocopies or ascertainment bias? J Med Genet. 2007;44:86. [PMC free article] [PubMed] [Google Scholar]
- 10.Smith A, Moran A, Boyd MC, Bulman M, Shenton A, Smith L, et al. Phenocopies in BRCA1 and BRCA2 families: evidence for modifier genes and implications for screening. J Med Genet. 2007;44:10–5. doi: 10.1136/jmg.2006.043091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Colonna V, Pistis G, Bomba L, Mona S, Matullo G, Boano R, et al. Small effective population size and genetic homogeneity in the Val Borbera isolate. Eur J Hum Genet. 2013;21:89–94. doi: 10.1038/ejhg.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borry P, Bentzen HB, Budin-Ljøsne I, Cornel MC, Howard HC, Feeney O, et al. The challenges of the expanded availability of genomic information: an agenda-setting paper. J Community Genet. 2017;9:103–16. doi: 10.1007/s12687-017-0331-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marx V. The DNA of a nation. Nature. 2015;524:503–5. doi: 10.1038/524503a. [DOI] [PubMed] [Google Scholar]
- 14.Shendure J. Human genomics: a deep dive into genetic variation. Nature. 2016;536:277–8. doi: 10.1038/536277a. [DOI] [PubMed] [Google Scholar]
- 15.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobayashi Y, Yang S, Nykamp K, Garcia J, Lincoln SE, Topper SE. Pathogenic variant burden in the ExAC database: an empirical approach to evaluating population data for clinical variant interpretation. Genome Med. 2017;9:1–14. doi: 10.1186/s13073-017-0403-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ormondroyd E, Mackley MP, Blair E, Craft J, Knight JC, Taylor JC, et al. “Not pathogenic until proven otherwise”: perspectives of UK clinical genomics professionals toward secondary findings in context of a Genomic Medicine Multidisciplinary Team and the 100,000 Genomes Project. Genet Med. 2018;20:320–8. [DOI] [PMC free article] [PubMed]
- 18.Godino L, Turchetti D, Jackson L, Hennessy C, Skirton H. Impact of presymptomatic genetic testing on young adults: a systematic review. Eur J Hum Genet. 2016;24:496–503. doi: 10.1038/ejhg.2015.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akobeng AK. Understanding systematic reviews and meta-analysis. Arch Dis Child. 2005;90:845–8. doi: 10.1136/adc.2004.058230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moher D, Liberati A, Tetzlaff J AD. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6:7. [DOI] [PMC free article] [PubMed]
- 21.Kmet LM, Lee RC, Cook LS. Standard quality assessment criteria for evaluating primary research papers from a variety of fields. Edmonton: Alberta Heritage Foundation for Medical Research (AHFMR). AHFMR - HTA Initiative #13. 2004.
- 22.Ryan R. Cochrane consumers and communication review group: data synthesis and analysis. Cochrane Consum Commun Rev Gr. 2013;2013:1–5. [Google Scholar]
- 23.Braun V, Clarke V. Using thematic analysis in psychology. Qual Res Psychol. 2006;3:77–101. [Google Scholar]
- 24.Thomas J, Harden A. Methods for the thematic synthesis of qualitative research in systematic reviews. BMC Med Res Methodol. 2008;8:1–10. doi: 10.1186/1471-2288-8-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Higgins J, Altman D, Gøtzsche P, Jüni P, Moher D, Oxman AD, et al. The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. Br Med J. 2011;343:889–93. doi: 10.1136/bmj.d5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Esteban Cardenosa E, de Juan Jimenez I, Palanca Suela S, Chirivella Gonzalez I, Segura Huerta A, Santaballa Beltran A, et al. Low penetrance alleles as risk modifiers in familial and sporadic breast cancer. Fam Cancer. 2012;11:629–36. doi: 10.1007/s10689-012-9563-1. [DOI] [PubMed] [Google Scholar]
- 27.Cassa CA, Tong MY, Jordan DM. Large numbers of genetic variants considered to be pathogenic are common in asymptomatic individuals. Hum Mutat. 2013;34:1216–20. doi: 10.1002/humu.22375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen R, Shi L, Hakenberg JJ, Naughton B, Sklar P, Zhang J, et al. Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat Biotechnol. 2016;34:531–8. doi: 10.1038/nbt.3514. [DOI] [PubMed] [Google Scholar]
- 29.Laver TW, Colclough K, Shepherd M, Patel K, Houghton JAL, Dusatkova P, et al. The common p.R114W HNF4A mutation causes a distinct clinical subtype of monogenic diabetes. Diabetes. 2016;65:3212–7. doi: 10.2337/db16-0628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minkel EV, Vallabh S, Lek M, Estrada K, Samocha K. Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med. 2016;8:322ra9. doi: 10.1126/scitranslmed.aad5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pedersen P, Milman N. Genetic screening for HFE hemochromatosis in 6,020 Danish men: penetrance of C282Y, H63D, and S65C variants. Ann Hematol. 2009;88:775–84. doi: 10.1007/s00277-008-0679-1. [DOI] [PubMed] [Google Scholar]
- 32.Thauvin-Robinet C, Munck A, Huet F, Genin E, Bellis G, Gautier E, et al. The very low penetrance of cystic fibrosis for the R117H mutation: a reappraisal for genetic counselling and newborn screening. J Med Genet. 2009;46:752–8. doi: 10.1136/jmg.2009.067215. [DOI] [PubMed] [Google Scholar]
- 33.Rose AM, Shah AZ, Venturini G, Krishna A, Chakravarti A, Rivolta C, et al. Transcriptional regulation of PRPF31 gene expression by MSR1 repeat elements causes incomplete penetrance in retinitis pigmentosa. Sci Rep. 2016;6:19450. doi: 10.1038/srep19450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sequeiros J, Ramos EM, Cerqueira J, Costa MC, Sousa A, Pinto-Basto J, et al. Large normal and reduced penetrance alleles in Huntington disease: instability in families and frequency at the laboratory, at the clinic and in the population. Clin Genet. 2010;78:381–7. doi: 10.1111/j.1399-0004.2010.01388.x. [DOI] [PubMed] [Google Scholar]
- 35.Tuke MA, Ruth KS, Wood AR, Beaumont RN, Tyrrell J, Jones SE, et al. Mosaic Turner syndrome shows reduced penetrance in an adult population study. Genet Med. 2018. 10.1038/s41436-018-0271-6. [DOI] [PMC free article] [PubMed]
- 36.Lefebvre M, Sanlaville D, Marle N, Thauvin-Robinet C, Gautier E, Chehadeh SE, et al. Genetic counselling difficulties and ethical implications of incidental findings from array-CGH: a 7-year national survey. Clin Genet. 2016;89:630–5. doi: 10.1111/cge.12696. [DOI] [PubMed] [Google Scholar]
- 37.Massie RJH, Poplawski N, Wilcken B, Goldblatt J, Byrnes C, Robertson C. Intron-8 polythymidine sequence in Australasian individuals with CF mutations R117H and R117C. Eur Respir J. 2001;17:1195–200. doi: 10.1183/09031936.01.00057001. [DOI] [PubMed] [Google Scholar]
- 38.Rossi E, Olynyk JK, Jeffrey GP. Clinical penetrance of C282Y homozygous HFE hemochromatosis. Expert Rev Hematol. 2004;25:183–90. [PMC free article] [PubMed] [Google Scholar]
- 39.Hendricks AE, Latourelle JC, Lunetta KL, Cupples LA, Wheeler V, MacDonald ME, et al. Estimating the probability of de novo HD cases from transmissions of expanded penetrant CAG alleles in the Huntington disease gene from male carriers of high normal alleles (27-35 CAG) Am J Med Genet A. 2009;149A:1375–81. doi: 10.1002/ajmg.a.32901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rohini R, Chun J, Powell SN. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat Rev Cancer. 2012;12:68–78. doi: 10.1038/nrc3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mersch J, Jackson M, Park M, Nebgen D, Peterson SK, Singletary C, et al. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. 2015;121:269–75. doi: 10.1002/cncr.29041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cassa CA, Savage SK, Taylor PL, Green RC, McGuire AL, Mandl KD. Disclosing pathogenic genetic variants to research participants: quantifying an emerging ethical responsibility. Genome Res. 2012;22:421–8. doi: 10.1101/gr.127845.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spector-Bagdady K. Reconceptualising consent for direct-to-consumer health services. Am J Law Med. 2015;41:568–616. doi: 10.1177/0098858815622191. [DOI] [PubMed] [Google Scholar]
- 44.Phillips AM. Reading the fine print when buying your genetic self online: direct-to-consumer genetic testing terms and conditions. N Genet Soc. 2017;36:273–95. [Google Scholar]
- 45.Cantor RM, Lange K, Sinsheimer JS. Prioritizing GWAS results: a review of statistical methods and recommendations for their application. Am J Hum Genet. 2010;86:6–22. doi: 10.1016/j.ajhg.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feng T, Zhu X. Statistical. Hum Genet. 2012;850:453–64. [Google Scholar]
- 47.Tandy-Connor S, Guiltinan J, Krempely K, Reineke P, Gutierrez S, LaDuca H, et al. False positive results reported by direct-to-consumer genetic tests highlight the importance of clinical confirmation testing for appropriate patient care. Genet Med. 2018;00:1–7. doi: 10.1038/gim.2018.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weedon M, Jackson L, Harrison J, Ruth K, Tyrrell J, Hattersley A, et al. Very rare pathogenic genetic variants detected by SNP-chips are usually false positives: implications for direct-to-consumer genetic testing. 2019. bioRxiv, 696799, 10.1101/696799.
- 49.Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007;39:870–4. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Easton DF, Pooley KA, Dunning AM, Pharoah PDP, Thompson D, Ballinger DG, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–93. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baxter JS, Leavy OC, Dryden NH, Maguire S, Johnson N, Fedele V, et al. Capture Hi-C identifies putative target genes at 33 breast cancer risk loci. Nat Commun. 2018;9:1028. doi: 10.1038/s41467-018-03411-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Begg CB, Haile RW, Borg A, Malone KE, Concannon P, Thomas DC, et al. Variation of Breast Cancer Risk Among BRCA1/2 Carriers. JAMA. 2008;299:194–201. doi: 10.1001/jama.2007.55-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007;25:1329–33. doi: 10.1200/JCO.2006.09.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–74. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–55. doi: 10.1038/gim.2016.190. [DOI] [PubMed] [Google Scholar]
- 56.ACMG. Incidental findings in clinical genomics: a clarification. Genet Med. 2013;15:664–6. doi: 10.1038/gim.2013.82. [DOI] [PubMed] [Google Scholar]
- 57.Jaggard MKJ, MacRae C, Ifeacho S, Robinson S, Tolley NS. New presentation of familial medullary thyroid carcinoma in 87-year-old patient with high-risk RET proto-oncogene codon 620 mutation. J Laryngol Otol. 2009;123:796–800. doi: 10.1017/S0022215108003472. [DOI] [PubMed] [Google Scholar]
- 58.Wright C, Middleton A, Burton H, Cunningham F, Humphries SE, Hurst J, et al. Policy challenges of clinical genome sequencing. BMJ. 2013;347:f6845. doi: 10.1136/bmj.f6845. [DOI] [PubMed] [Google Scholar]
- 59.Hallowell N, Hall A, Alberg C, Zimmern R. Revealing the results of whole-genome sequencing and whole-exome sequencing in research and clinical investigations: some ethical issues. J Med Ethics. 2015;41:317–21. doi: 10.1136/medethics-2013-101996. [DOI] [PubMed] [Google Scholar]
- 60.Knoppers BM, Zawati MH, Senecal K. Return of genetic testing results in the era of whole-genome sequencing. Nat Rev Genet. 2015;16:553–9. doi: 10.1038/nrg3960. [DOI] [PubMed] [Google Scholar]
- 61.Berg JS, Foreman AKM, O’Daniel JM, Booker JK, Boshe L, Carey T, et al. A semiquantitative metric for evaluating clinical actionability of incidental or secondary findings from genome-scale sequencing. Genet Med. 2016;18:467–75. doi: 10.1038/gim.2015.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hunter JE, Irving SA, Biesecker LG, Buchanan A, Jensen B, Lee K, et al. A standardized, evidence-based protocol to assess clinical actionability of genetic disorders associated with genomic variation. Genet Med. 2016;18:1258–68. doi: 10.1038/gim.2016.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jackson L, Goldsmith L, O’Connor A, Skirton H. Incidental findings in genetic research and clinical diagnostic tests: a systematic review. Am J Med Genet A. 2012;158A:3159–67. doi: 10.1002/ajmg.a.35615. [DOI] [PubMed] [Google Scholar]
- 64.Benjamin C, Boudioni M, Ward H, Marston E, Lindenmeyer A, Bangee M. NHS Genomic Medicine Centres: National Service Evaluation of the Consent Process and Participant Materials used in the 100,000 Genomes Project. Genomics England. 2016:1.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.