

Phagocytosis is the key mechanism for host control of Pseudomonas aeruginosa, a motile Gram-negative, opportunistic bacterial pathogen which frequently undergoes adaptation and selection for traits that are advantageous for survival. One such clinically relevant adaptation is the loss of bacterial motility, observed within chronic infections, that is associated with increased antibiotic tolerance and phagocytic resistance. Previous studies using phagocytes from a leukocyte adhesion deficiency type 1 (LAD-I) patient identified CD18 as a putative cell surface receptor for uptake of live P. aeruginosa.
KEYWORDS: Pseudomonas aeruginosa, phagocytosis, CD18, flagellar motility, N-glycans
ABSTRACT
Phagocytosis is the key mechanism for host control of Pseudomonas aeruginosa, a motile Gram-negative, opportunistic bacterial pathogen which frequently undergoes adaptation and selection for traits that are advantageous for survival. One such clinically relevant adaptation is the loss of bacterial motility, observed within chronic infections, that is associated with increased antibiotic tolerance and phagocytic resistance. Previous studies using phagocytes from a leukocyte adhesion deficiency type 1 (LAD-I) patient identified CD18 as a putative cell surface receptor for uptake of live P. aeruginosa. However, how bacterial motility alters direct engagement with CD18-containing integrins remains unknown. Here we demonstrate, with the use of motile and isogenic nonmotile deletion mutants of two independent strains of P. aeruginosa and with CRISPR-generated CD18-deficient cell lines in human monocytes and murine neutrophils, that CD18 expression facilitates the uptake of both motile and nonmotile P. aeruginosa. However, unexpectedly, mechanistic studies revealed that CD18 expression was dispensable for the initial attachment of the bacteria to the host cells, which was validated with ectopic expression of complement receptor 3 (CR3) by CHO cells. Our data support that surface N-linked glycan chains (N-glycans) likely facilitate the initial interaction of bacteria with monocytes and cooperate with CD18 integrins in trans to promote internalization of bacteria. Moreover, talin-1 and kindlin-3 proteins promote uptake, but not binding, of P. aeruginosa by murine neutrophils, which supports a role for CD18 integrin signaling in this process. These findings provide novel insights into the cellular determinants for phagocytic recognition and uptake of P. aeruginosa.
INTRODUCTION
Phagocytosis of bacteria by neutrophils and macrophages is the primary means of immune control and clearance of Pseudomonas aeruginosa, a motile opportunistic Gram-negative pathogen responsible for acute and chronic infections (1–3). Over the course of chronic infections, P. aeruginosa undergoes adaptation and selection of certain traits, such as the temporal transition from a motile to a nonmotile phenotype through the loss of flagellar swimming motility (4–8). This progressive loss of motility confers increased phagocytic resistance and antibiotic tolerance and promotes bacterial persistence (9–12). Therefore, there is ongoing interest in the cellular determinants for effective phagocytosis of P. aeruginosa with a particular focus on cell surface receptors that mediate the host-pathogen interactions. Several types of receptors have been implicated in innate immune host responses to P. aeruginosa. Notably, several of the Toll-like receptors (TLRs) are critical for inflammatory responses to P. aeruginosa but do not mediate the binding and uptake of the bacteria (13). To date, the only phagocytic cell surface receptor with genetic evidence to support that it is critical for uptake of P. aeruginosa is CD18, the common subunit of β2 integrins (14, 15). However, direct interactions between β2 integrins and P. aeruginosa, and how bacterial motility may modulate these interactions, have yet to be described.
β2 integrins are heterodimeric type I transmembrane proteins composed of a common β subunit (CD18) and an α subunit (CD11a through CD11d). Leukocyte adhesion deficiency type I (LAD-I) is a disease caused by mutations in the gene that encodes the CD18 protein (ITGB2), thus affecting β2 integrin function due to total absence or low surface expression of β2 integrins on leukocytes. Since β2 integrins are critical for the recruitment of neutrophils in response to bacterial infections, including those caused by P. aeruginosa, LAD-I patients suffer from recurrent and life-threatening bacterial infections (16–18). β2 integrins appear to have dual roles in mediating responsiveness to bacterial infections. First, they facilitate adhesion and migration of neutrophils to the sites of infections (19). Second, independent of migration, in vitro studies of phagocytic cells from a patient with LAD-I demonstrated that complement receptor 3 (CR3) (CD18/CD11b) facilitates nonopsonic phagocytosis of some P. aeruginosa strains; uptake of other strains was dependent on interactions with CD14 (a glycosylphosphatidylinositol [GPI]-anchored glycoprotein) (14, 20). In agreement with the initial findings, Wilson et al. subsequently showed that murine neutrophils deficient in CD18 expression were impaired in phagocytosis of heat-killed P. aeruginosa (15).
The initial step in recognition and phagocytic uptake of P. aeruginosa is cell surface interactions of the bacteria with phagocytes. We previously showed that nonmotile bacteria evade these interactions and therefore impair the ability of macrophages and neutrophils to bind and consequently phagocytose them (9, 10). Importantly, in the current work, we are guided by previous research on bacterial association with epithelial cells which determined that P. aeruginosa can bind directly to negatively charged N-linked glycan chains (N-glycans) on epithelial cell surfaces (21, 22). However, the relationship between N-linked glycosylation moieties and CD18 expression for host-bacterial (phagocyte-P. aeruginosa) interactions is not currently known.
Based on the aforementioned LAD data, the original premise of these studies was to determine how bacterial motility altered direct bacterial engagement with CD18-containing integrins. To do so, we employed CRISPR-generated CD18-deficient cell lines of human monocytes and murine neutrophils and, with the use of two independent strains of P. aeruginosa with genetically regulated motility, evaluated whether CD18-dependent uptake of P. aeruginosa was modulated by bacterial swimming motility. Quantitative phagocytosis assays revealed that the uptake of both motile and nonmotile P. aeruginosa was dependent on CD18 expression by human monocytes and murine neutrophils. However, surprisingly, mechanistic studies using complementary flow cytometry and cytotoxicity assays identified that CD18 expression on monocytes was dispensable for binding of bacteria by phagocytes, and this observation was validated with ectopic expression of CR3 by CHO cells. These disparate observations regarding the roles of CD18 for binding versus phagocytosis were subsequently reconciled by our data that surface N-glycans likely mediate the initial interaction of bacteria with monocytes and cooperate with CD18 (in trans) to promote uptake of bacteria. These data were validated by evidence that CD18 integrin activation on murine neutrophils is required for the uptake, but not binding, of P. aeruginosa in murine neutrophils. These findings expand our current understanding of the interactions of P. aeruginosa bacteria with phagocytic cells that functionally result in bacterial uptake and clearance.
RESULTS
CD18 expression is required for the uptake of motile and nonmotile P. aeruginosa by phagocytes.
Our lab’s previous findings showed that loss of flagellar swimming motility by P. aeruginosa, regardless of flagellar expression, confers in vitro and in vivo resistance (∼100-fold) to phagocytic clearance (9, 10). Based on previous findings (14, 15), we hypothesized that the expression of the β2 integrin, CD18, is necessary for binding and internalization of P. aeruginosa, with the role of CD18 expression on the uptake of nonmotile P. aeruginosa unknown. To evaluate the functional effect of the loss of CD18 expression on phagocytic uptake of P. aeruginosa, we performed quantitative gentamicin protection phagocytic assays with THP-1 human monocytic cell lines. Genetic deletion of CD18 was achieved through CRISPR-Cas9 targeting and verified by Western blotting for CD18 protein (Fig. 1, inset). Phagocytosis of swimming-competent PA14 wild type (WT) and PAO1 WT was diminished in THP-1 cells lacking CD18 expression (Fig. 1A and B). Interestingly, PA14 motABmotCD (have flagella but lack motility) and PAO1 fliC (flagellin structural gene) strains also exhibited decreases in their phagocytic uptake by CD18-deficient cells compared to wild-type cells (Fig. 1A and B). These findings were recapitulated by an independent CD18 knockout THP-1 cell line generated through CRISPR gene editing (data not shown). These data suggest that the effect of CD18 on phagocytosis may be broadly applicable to motile and nonmotile P. aeruginosa and that the effect is independent of flagellar expression.
FIG 1.

CD18-deficient phagocytes exhibit impaired phagocytosis of motile and nonmotile P. aeruginosa. THP-1 human monocytic cell line WT and CD18 KO were assayed for relative in vitro phagocytosis of PA14 WT or motABmotCD (A) and PAO1 WT or fliC bacteria (B) by gentamicin protection assay at an MOI of 10, as indicated. Murine neutrophils (WT or CD18 KO), differentiated from conditionally immortalized myeloid progenitors, were assayed for relative in vitro phagocytosis of PA14 WT or motABmotCD (C) and PAO1 WT or fliC bacteria (D) by gentamicin protection assay at an MOI of 10, as indicated. Phagocytic uptake levels were normalized as percentages of respective mean WT THP-1 or murine neutrophil phagocytosis. All data are analyzed using one-way ANOVA with Tukey’s post hoc analyses and are representative of at least two independent biological experiments (n ≥ 4). ***, P ≤ 0.0005 compared to WT cell type.
To determine whether the described findings are exclusive to human THP-1 monocytes or represent a widespread mechanism applicable to neutrophils, we tested the ability of CD18-deficient murine neutrophils (generated by CRISPR targeting) to phagocytose motile and nonmotile P. aeruginosa (15). Neutrophils were obtained by differentiating conditionally immortalized murine myeloid progenitors with granulocyte colony-stimulating factor and stem cell factor (23), and we confirmed that more than 80% of the cells expressed the murine neutrophil marker Ly6G (15). Murine neutrophils recapitulated the outcomes derived with the THP-1 cells, such that motile and nonmotile bacteria were internalized to a much lower extent by CD18-deficient cells (Fig. 1C and D). Importantly, these findings demonstrate that CD18 mechanistically contributes to the phagocytosis of both motile and nonmotile P. aeruginosa and has broad applicability to phagocytic cells across multiple cell types and species.
Cell surface association of P. aeruginosa with human monocytes is independent of CD18 expression.
Based on the observation that CD18 deficiency leads to decreased internalization of P. aeruginosa, we hypothesized that CD18 expression facilitates association with the bacteria and that, therefore, loss of CD18 would reduce the total association of bacteria by phagocytic cells. We quantitatively assessed the total cell surface association of the bacteria with human monocytes using complementary assays: (i) fluorescence-activated cell sorter (FACS) analyses, and (ii) bacterial type III secretion system (T3SS)-induced cytotoxicity, which is dependent upon interaction of the bacteria with the cell surface but not upon phagocytic uptake (24). We have previously demonstrated that nonmotile bacteria elicit less cytotoxicity by virtue of less cellular association, but both nonmotile and motile PA14 bacteria are equally capable of T3SS activity (9, 24). Consistent with previous data (9, 10), green fluorescent protein (GFP)-expressing PA14 WT and PAO1 bacteria exhibited significantly greater association with wild-type THP-1 cells than PA14 motABmotCD and PAO1 fliC (Fig. 2A and B). Intriguingly, infection of CD18-deficient cells resulted in similar bacterial association, as observed with CD18-expressing (wild-type) monocytes, and this outcome was observed with both motile and nonmotile P. aeruginosa (Fig. 2A and B). As an independent validation of the association data, wild-type and CD18-deficient monocytes infected with PA14 (WT) exhibited equivalent levels of cytotoxicity as measured by lactate dehydrogenase (LDH) release. This contrasted with the lower but comparable cytotoxicity in both cell lines infected with PA14 motABmotCD (Fig. 2C). These data suggest that cell surface association of bacteria to monocytes and the resulting cytotoxicity are independent of CD18 expression, supporting that initial bacterial interactions with the cells that result in phagocytosis are independent of CD18.
FIG 2.

Infection of CD18-deficient cells with bacteria elicits comparable bacterial association and cytotoxicity to WT cells. Flow cytometry to assay relative association of GFP-expressing PA14 WT or motABmotCD (A) or GFP-expressing PAO1 WT or fliC bacteria (B) with THP-1 WT or CD18 KO cells at 2 h postinfection (MOI of 25). The data are normalized to the mean fluorescence intensity (MFI) of uninfected THP-1 cells (set to 1,000). (C) Cytotoxicity of THP-1 WT or CD18 KO following infection with PA14 WT or motABmotCD (MOI of 25) was assayed using the LDH assay. Release of LDH into the culture supernatants was measured at 2 h postinfection. All data are analyzed using one-way ANOVA with Tukey’s post hoc analyses and are representative of at least two independent biological experiments (n ≥ 4). ***, P ≤ 0.0005; *, P ≤ 0.05; ns, not significant compared to WT cell type.
P. aeruginosa association to THP-1 cells is mediated by N-glycans.
In order to mechanistically elucidate the cell surface interaction between P. aeruginosa and THP-1 monocytes, we examined the relationship between N-glycosylation and CD18 for bacterial association to phagocytes. WT and CD18-deficient THP-1 cells were pretreated with tunicamycin, which blocks N-glycosylation of newly synthesized proteins prior to FACS analyses of bacterial association and quantitation of infection-elicited cytotoxicity. Inhibition of N-glycosylation with tunicamycin treatment induced a 50% reduction of N-glycans and reduced PA14 WT and PAO1 WT bacterial association with THP-1 WT cells, which is consistent with data derived from studies on epithelial cells (Fig. 3A and B) (21, 22). Importantly, we observed a similar decrease in bacterial association with CD18-deficient cells treated with tunicamycin (Fig. 3A and B). Taken together, these data demonstrate that bacterial binding of cell surface N-glycosylation is likely independent of the CD18 integrin and is acting in trans with the CD18 complex to mediate internalization.
FIG 3.

N-glycans mediate P. aeruginosa association to THP-1 cells, and CR3 overexpression on CHO cells does not promote bacterial association. Relative association of GFP-expressing PAO1 WT (A) or PA14 WT (B) (MOI of 25 at 2 h postinfection) to THP-1 WT or CD18 KO cells without, or after, treatment of cells with 1 μg/ml tunicamycin. The data are normalized to the mean fluorescence intensity (MFI) of uninfected THP-1 cells analyzed by flow cytometry (set to 1,000). (C) Flow cytometry of Chinese hamster ovary (CHO) and CHO-CR3 (CD11b/CD18) cells stained with anti-human CD11b antibody. CHO is represented in blue and CHO-CR3 in magenta. (D) Relative association of GFP-expressing PAO1 WT or PA14 WT bacteria with CHO and CHO-CR3 cells, quantitatively measured by FACS analyses. The data are normalized to the mean fluorescence intensity (MFI) of uninfected CHO cells (set to 1,000). All data are analyzed using one-way ANOVA with Tukey’s post hoc analyses and are representative of at least two independent biological experiments (n ≥ 4). ***, P ≤ 0.0005; **, P ≤ 0.005; *, P ≤ 0.05; ns, not significant compared to WT cell type.
CR3 expression on CHO cells is insufficient for bacterial association.
One possibility that was considered, with respect to our previous data, was that there were sufficient alternative receptor binding sites remaining on the CD18-deficient THP-1 cells such that the observed binding deficit upon loss of CD18 was masked. Therefore, we asked whether CR3 expression, including CD18, was sufficient to mediate bacterial binding. To do so, we tested whether ectopic expression of CR3 (CD11b/CD18) by CHO epithelial cells, which do not express any canonical phagocytic receptors, would be sufficient to promote the association of bacteria. We confirmed robust CR3 surface expression on CHO-CR3 cells by staining with anti-CD11b antibodies (Fig. 3C) and coincubated the cells with GFP-expressing bacteria prior to FACS analyses. The same cell lines had previously been used, with the same methodology (GFP-expressing bacteria), to demonstrate measurable CR3-dependent binding of Borrelia burgdorferi as well as zymosan (25, 26). Association of PAO1 WT and PA14 WT bacteria to cells with or without CR3 expression was equivalent (Fig. 3D). This supports that CR3 expression alone is insufficient to facilitate bacterial association. Overall, these data give us insight into productive cell surface interactions during P. aeruginosa infection of human monocytes, where bacterial ligands are initially recognized by and bind to N-glycans, which cooperate with CD18 integrin complexes to drive internalization.
Induction of inflammatory gene expression is regulated by P. aeruginosa infection but not by CD18 expression.
If CD18 expression controlled differential engagement of bacteria with phagocyte cell surfaces, concurrently, we would expect differential downstream inflammatory responses in CD18-deficient cells. However, based on our data on cell surface association (Fig. 2 and 3), we hypothesized that loss of CD18 expression would not significantly alter the transcriptional profiles in the context of infection. Therefore, we evaluated whether CD18 was required for controlling RNA expression of various downstream inflammatory genes in response to P. aeruginosa infection. We employed NanoString to find the top transcript differences between uninfected or infected (PA14 WT) THP-1 monocytes and CD18-deficient cells. As an important internal control, ITGB2, the gene that encodes the CD18 protein, was not expressed in the knockout cell lines (Fig. 4). Interestingly, we observed that anticipated inflammatory responses were dependent on P. aeruginosa infection but not on the expression of CD18. For instance, chemokine and cytokine receptor binding and activity were identified by gene ontology as some of the common molecular functions regulated by PA14 WT infection. In this regard, RNA expression of chemokines and cytokines such as interleukin-8 (IL-8), tumor necrosis factor (TNF), and NFKB2 was upregulated postinfection in wild-type monocytes (Fig. 4). In contrast, expression of receptors such as CCR2, HLA-B, and IL1RAP was downregulated postinfection (Fig. 4). However, for all of the aforementioned molecular pathways, similar transcription profiles were observed in CD18-expressing and -deficient cells following infection with PA14 WT (Fig. 4). Therefore, these results do not support that inflammatory responses dependent upon bacterial association with the cell are facilitated by CD18 expression.
FIG 4.
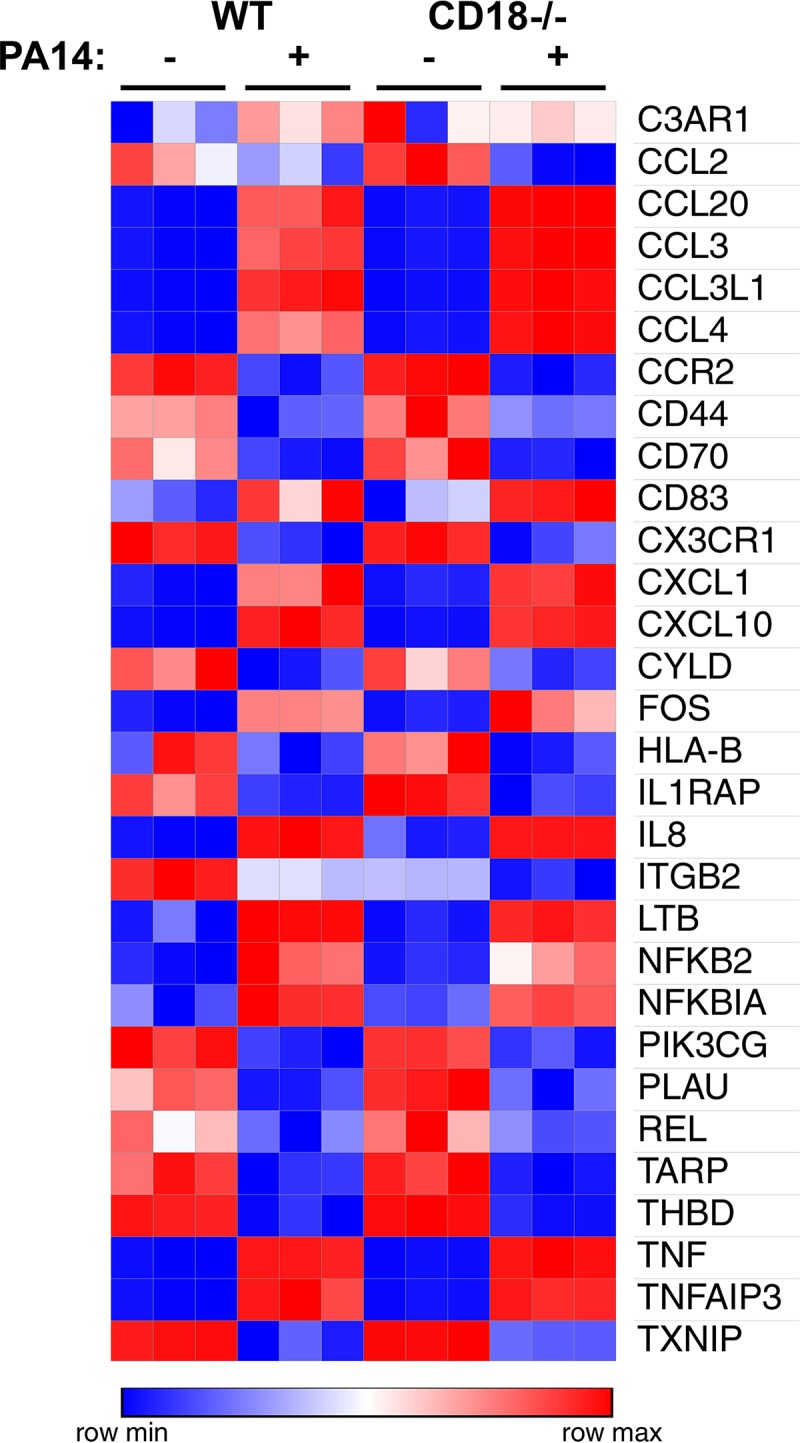
CD18 expression is dispensable for controlling mRNA levels of inflammatory genes in the context of P. aeruginosa infection. Top transcript differences between THP-1 WT and CD18 KO either uninfected or infected with PA14 WT bacteria (MOI of 25, set up in triplicates) using the PanCancer immune profiling panel for gene expression. The raw count expression data were analyzed using nSolver analysis software (NanoString). Heat map analysis shows the most highly expressed genes in red and the least highly expressed genes in blue, as shown in the legend.
CD18 integrin signaling is necessary for uptake and association of P. aeruginosa by murine neutrophils.
Next, we extended our studies with murine neutrophils to elucidate the role of the integrin-associated signaling complex for binding and uptake of P. aeruginosa. Talin and kindlin are intracellular proteins that play a major role in the regulation of integrin affinity. Integrin affinity is controlled by intracellular signal transduction (inside-out signaling) that triggers interaction between talin-1 and kindlin-3 proteins with distinct binding motifs in the short cytosolic tail of the β2 integrin (27). These interactions induce structural changes that eventually modulate the ligand-binding affinity of the headpiece (27). To test whether P. aeruginosa binding and uptake require signaling through talin-1 and kindlin-3, murine neutrophils deficient for those proteins were employed for FACS analyses and phagocytosis assays (15). Indeed, we observed a significant reduction in the total association (cell surface-bound and internalized bacteria) of GFP-expressing PA14 WT and PAO1 WT bacteria with murine neutrophils lacking talin-1, kindlin-3, and CD18 (Fig. 5A and B). Moreover, as expected, there was a significant decrease in the association of PA14 motABmotCD and PAO1 fliC compared to their respective motile wild-type strains (PA14 and PAO1 WT) in WT murine neutrophils. Rigorous uptake assays, quantitating only phagocytosed bacteria, recapitulated the outcomes of the total association experiments in that internalization of all strains tested required talin and kindlin activities (Fig. 5C and D). Thus, the total association deficit observed in the talin, kindlin, and CD18-deficient cell lines could either be due to deficiencies in both bacterial binding and uptake or is simply reflective of the phagocytic deficit. In order to resolve this, we also quantitatively measured the binding of bacteria (bound bacteria) to cells, which is the initial step of phagocytosis. To rigorously assess binding rather than total association, we treated cells with cytochalasin D (cytoD), an inhibitor of actin polymerization, prior to incubation with bacteria and the subsequent FACS analyses. CytoD blocks phagocytosis; hence, only cell surface-bound bacteria, in the absence of internalized bacteria, contribute to the observed outcome. For all the cellular genotypes treated with cytoD, we observed equivalent binding of GFP-expressing PA14 WT and PAO1 WT bacteria to neutrophils (Fig. 5E and F). Therefore, this suggests that the deficits in the total association of bacteria to kindlin, talin, and CD18-deficient cells were reflective of the phagocytic defect. Importantly, these data strongly support that the mechanism of internalization, but not binding, of P. aeruginosa by murine neutrophils requires CD18 integrin signaling through talin-1 and kindlin-3.
FIG 5.

CD18 integrin signaling is required for phagocytosis of P. aeruginosa by murine neutrophils. Flow cytometry to assay relative total association of GFP-expressing PA14 WT or motABmotCD (A) or GFP-expressing PAO1 WT or fliC bacteria (B) with murine neutrophils (WT, talin KO, kindlin KO, or CD18 KO) at 2 h postinfection (MOI of 25). Murine neutrophils (WT, talin KO, or kindlin KO) were assayed for relative in vitro phagocytosis of PA14 WT or motABmotCD (C) and PAO1 WT or fliC bacteria (D) by gentamicin protection assay at an MOI of 10, as indicated. Phagocytic uptake levels were normalized as percentages of respective mean WT phagocytosis. (E and F) Relative association of GFP-expressing PA14 WT or PAO1 WT (MOI of 25 at 45 min postinfection) to murine neutrophils (WT, talin KO, kindlin KO, or CD18 KO) without, or after, treatment of cells with 10 μM cytochalasin D (cytoD). The neutrophils were preincubated with cytoD for 30 min at 37°C prior to infection with bacteria. All flow cytometry data are normalized to the mean fluorescence intensity (MFI) of uninfected murine neutrophils (set to 1,000). All data are analyzed using one-way ANOVA with Tukey’s post hoc analyses and are representative of at least two independent biological experiments (n ≥ 4). ***, P ≤ 0.0005; **, P ≤ 0.005; *, P ≤ 0.05; ns, not significant.
Internalization of P. aeruginosa by murine neutrophils is mediated by CD18 but not CD11b alone.
Based on our observations that CD18 expression is required for uptake of P. aeruginosa by murine neutrophils, we wanted to study the effect of antibody blockade on bacterial internalization. Therefore, we pretreated wild-type and CD18-deficient murine neutrophils with GAME-46, a blocking antibody specific to CD18, and performed bacterial phagocytosis and binding/association assays. Functional blocking of CD18 in wild-type neutrophils resulted in diminished uptake of PA14 WT and PAO1 WT (Fig. 6A and B), which phenocopied CD18-deficient neutrophils. As a control, CD18-deficient neutrophils treated with the CD18-blocking antibody exhibited no effect on phagocytosis (Fig. 6A and B). Additionally, since CR3 (CD11b/CD18) is a possible candidate for phagocytosis of P. aeruginosa, among other bacteria (14, 28, 29), we decided to use a blocking antibody (M1/70) against CD11b, the alpha subunit which contains the ligand-binding domain. Unexpectedly, inhibition of CD11b did not abrogate uptake of PA14 WT or PAO1 WT P. aeruginosa, suggesting that CD11b interactions are insufficient for phagocytosis in murine neutrophils (Fig. 6C and D). Finally, we measured the binding and total association of bacteria to neutrophils in the presence or absence of the CD18-blocking antibody (GAME-46) and cytoD. Consistent with the phagocytosis phenotype, CD18 blocking decreased the total association of PAO1 WT bacteria with wild-type cells, whereas binding of bacteria was unaffected regardless of blocking CD18 (Fig. 6E). These data demonstrate that CD18 is required for phagocytosis of P. aeruginosa by murine neutrophils.
FIG 6.

Antibody blocking of CD18 but not CD11b is sufficient to reduce bacterial internalization of P. aeruginosa in murine neutrophils. Murine neutrophils (WT or CD18 KO) were assayed by gentamicin protection assay for relative in vitro phagocytosis of PA14 WT, PA14 motABmotCD (inset), or PAO1 WT (MOI of 10) in the presence of 10 μg/ml of GAME-46 CD18-blocking antibody (A and B) and M1/70 CD11b-blocking antibody (C and D) with the respective isotype controls (as indicated). The neutrophils were pretreated with the antibodies for 15 min on ice. Phagocytic uptake levels were normalized as percentages of respective mean WT phagocytosis. (E) Relative association of GFP-expressing PAO1 WT (MOI of 25 at 45 min postinfection) to murine neutrophils (WT or CD18 KO) in the absence or presence of 10 μg/ml GAME-46 and 10 μM cytoD. The neutrophils were preincubated with GAME-46 and cytoD for 30 min at 37°C prior to infection. The data are normalized to the mean fluorescence intensity (MFI) of uninfected murine neutrophils analyzed by flow cytometry (set to 1,000). All data are analyzed using one-way ANOVA with Tukey’s post hoc analyses and are representative of at least two independent biological experiments (n ≥ 4). ***, P ≤ 0.0005; *, P ≤ 0.05; ns, not significant.
DISCUSSION
P. aeruginosa is a motile bacterium that requires functional flagellar motility to initially establish an infection and colonize a host (30, 31). Indeed, P. aeruginosa mutants that lack flagella are often impaired in virulence in acute infection models (32). While flagellar motility is an early colonization factor, clinical isolates recovered from patients with chronic respiratory infections consistently demonstrate progressive loss of flagellar motility due to downregulation or mutation (7, 8). This observed alteration in bacterial phenotype is likely reflective of both the bacterial response to its environment and the selective pressure of phagocytes preferentially clearing motile bacteria. In this respect, phagocytes utilize swimming motility, a function of flagellar rotation, to recognize bacteria for internalization (9, 10). Specifically, previous work in our lab illustrated that bacterial flagellar motility facilitates engagement between the cell surface of the phagocyte and bacteria, which promotes subsequent uptake. The key mechanism of host control of P. aeruginosa is bacterial clearance by phagocytic cells (neutrophils and macrophages) (2). Therefore, we investigated the cellular determinants of phagocytic recognition of P. aeruginosa. In particular, we addressed the role of cell surface CD18-containing integrin complexes in mediating the differential interactions with motile and nonmotile P. aeruginosa.
CD18-containing integrins, including LFA-1 (CD11a/CD18), CR3 (CD11b/CD18), and CR4 (CD11c/CD18), are well characterized to mediate cellular activation, adhesion, and subsequent migration (16). CD18 complexes and CR3 in particular have also been recognized as important pattern recognition and phagocytic receptors for different pathogens. CR3 has been shown to mediate the internalization of bacteria in the presence (opsonic) or absence (nonopsonic) of complement-derived opsonins (iC3b) (29, 33, 34). Here, we report that CD18 expression on human monocytes and murine neutrophils is required for nonopsonic uptake of motile and nonmotile strains of P. aeruginosa. Nonopsonic phagocytosis is critical for host control during the early stages of infections and especially in sites with low levels of opsonins or opsonic receptors such as the lung (14, 35, 36). Based on previous data derived with the use of cells from a patient with the genetic condition LAD-I, which conveyed the initial indication that loss of CD18 expression reduces cellular uptake of P. aeruginosa, we anticipated that CD18 expression on phagocytes would be required for direct interactions with bacterial ligands. Unexpectedly, using complementary assays that evaluate bacterial interactions with phagocyte cell surfaces, our studies indicate that the CD18 receptors do not directly engage P. aeruginosa to facilitate uptake. With murine neutrophils, we observed a significant phagocytic deficit and decreased total cellular association of bacteria as a result of CD18 deficiency. However, we observed equivalent binding of bacteria to the CD18-deficient murine phagocytes. Likewise, with the THP-1 human monocytic cell line, we observed a significant phagocytic deficit but unaltered binding of bacteria by CD18-deficient cells. In validation of this observation, T3SS-induced cytotoxicity and downstream inflammatory responses, which depend on interactions of bacteria with the cell surface, were independent of CD18 expression. These data support that bacterial binding to phagocytes is likely not facilitated by CD18 and that the decrease in total association in murine neutrophils was reflective of the larger uptake deficit rather than a binding phenotype. We considered that phagocytes have many binding sites and putative receptors to which P. aeruginosa could bind, thus potentially masking a quantitative binding deficit in CD18 knockout cells. Therefore, we tested CHO cells that do not express any canonical phagocytic receptors in comparison to CHO-CR3 cells which ectopically express CR3. These assays directly address whether CR3 mediates bacterial association, and the outcomes further support that CR3 expression is dispensable for P. aeruginosa binding and cytotoxicity.
We were guided by previous reports with respect to what cell surface molecules mediate the initial interaction of P. aeruginosa with monocytes to subsequently promote CD18-dependent internalization. N-linked glycan chains (N-glycans) of glycoproteins, which are prominent cell surface-exposed structures (with long carbohydrate chains), are reported to be necessary for P. aeruginosa binding, entry, and elicited cytotoxicity at the apical surface of polarized epithelium (21, 22). Additionally, we previously demonstrated that exogenous addition of the polyphosphoinositides PIP3 and PIP2 induced cell surface binding and uptake of nonmotile P. aeruginosa (37). Therefore, we speculated that both motile and nonmotile P. aeruginosa may bind to negatively charged molecules on the cell surface, with N-glycans prioritized as logical candidate ligands. Our results illustrate that inhibition of N-glycosylation in THP-1 phagocytic cells that either express or do not express CD18 resulted in a reduction in bacterial association. Importantly, these data suggest that N-glycans are required and likely cooperate with CD18 integrins in trans to mediate uptake of P. aeruginosa; N-glycan-dependent binding in the CD18-deficient cells excludes that it is covalently associated glycans on CD18 that facilitate the interaction. In vitro binding assays in heterologous systems have revealed that P. aeruginosa preferentially binds to a mixture of complex N-glycans over single sugars, possibly recognizing specific ordered combinations of sugar sequences (21). In addition to N-glycoproteins, it is worth noting that heparan sulfate proteoglycans (HSPGs) were also shown to mediate P. aeruginosa binding and downstream events, including internalization and cell damage at the basolateral surface of the polarized epithelium (21, 22). Thus, future discovery of the identity of the glycoprotein receptors and specificity of the functional glycans on phagocytic cells that mediate initial productive interactions with P. aeruginosa will be very informative.
We observed that CD18 uses a canonical signaling pathway via the intracellular proteins talin-1 and kindlin-3 to promote uptake but not binding of P. aeruginosa by murine neutrophils. This further supports that the ability of integrins to signal and to activate are key steps for phagocytosis of P. aeruginosa bacteria. Antibody-blocking experiments in murine neutrophils revealed that CD18 is required, but the coreceptor CD11b, which contains the ligand-binding domain, is dispensable for the uptake of P. aeruginosa. This suggests a possible redundant function for CD11b in our system.
As previously noted, CD18-containing integrins function as phagocytic receptors for a variety of bacterial pathogens. Some examples of CR3-mediated phagocytosis of iC3b-opsonized bacteria include opsonized Staphylococcus aureus (33, 34, 38), Francisella tularensis (39), and Escherichia coli (38). However, CR3 has also been reported to facilitate the nonopsonic phagocytosis of various bacteria, including P. aeruginosa (14, 15, 40). Interestingly, van Bruggen et al. demonstrated that, with the use of LAD-I neutrophils and monoclonal antibodies against CD18 or CD11b in healthy human neutrophils, expression of CD18 and CD11b is necessary for the uptake of nonopsonized Salmonella enterica serovar Typhimurium but not for the opsonic uptake of the same bacterium (29). Thus, there may be value in revisiting previous reports that illustrate the interaction of CR3 with various bacterial species based on phagocytic assays to test whether glycosylation is required and possibly cooperates with CD18 integrins for bacterial internalization as observed with the studies herein of P. aeruginosa.
In summary, we have identified distinct roles for CD18 integrins expressed by phagocytes for the internalization of motile and nonmotile P. aeruginosa, but not the initial attachment of the bacteria to the host cells. We showed that N-glycans serve as cell surface ligands to facilitate integrin-mediated uptake of P. aeruginosa, with subsequent integrin signaling required for internalization of the bacteria. As discussed, this study provides a new understanding of the mechanisms by which integrins contribute to the binding and phagocytosis of P. aeruginosa, and these insights may have broad implications for host interactions with, and clearance of, other bacterial species.
MATERIALS AND METHODS
Bacteria.
P. aeruginosa strains of the PAO1 and PA14 backgrounds were kindly provided by D. Hogan and G. O’Toole (Dartmouth Medical School, Hanover, NH) and have been previously described and published (9, 41), and the PAO1 fliC and PA14 motABmotCD mutants have been complemented (41, 42). Bacterial strains expressing green fluorescent protein (GFP) were generated by transformation as previously described (10). Bacteria were cultured overnight at 37°C and subsequently subcultured to log-phase growth for 2 h in Luria-Bertani broth (LB), and the concentrations were determined by optical density at 600 nm.
Cells.
Guides targeting the ITGB2 gene in the THP-1 human monocytic cell line were selected using the Benchling software. The single guide RNA sequences targeting ITGB2 (5′ to 3′) are CACTGCTCGCCCTGGTGGGG and GCCGGGAATGCATCGAGTCG (data not shown). Guides were then cloned into the lentiCRISPR v2 lentiviral vector following the Zhang lab protocol (43). Lentivirus for each targeting construct was made by transfecting 293HEK cells in a 10-cm plate with 3 μg pMD2.G, 2 μg psPAX2, and 1 μg of targeting plasmid (plasmids were a gift from the Zhang lab). Lentiviral supernatant was collected at 72 h and used to infect THP-1 cells by spinfection at 2,500 rpm with 1:1,000 Polybrene. Selection was carried out over 2 weeks by exposing cells to 0.75 μg/ml of puromycin, and cells lacking surface expression of the CD18 protein were selected using the FACSAria III cell sorter.
Conditionally immortalized murine myeloid progenitors deficient in CD18, talin-1, or kindlin-3 expression were generated by CRISPR/Cas9 targeting of ITGB2, Tln1, or Fermt3 genes, respectively, and were validated by Western blotting or FACS analyses (15). Neutrophils were obtained by differentiating progenitors with 20 ng/ml granulocyte colony-stimulating factor and 20 ng/ml recombinant murine stem cell factor and removing 4-hydroxytamoxifen (23).
CHO and CHO-CR3 cells overexpressing human CR3 (CD11b/CD18) were kindly provided by R. Ingalls (Boston University) (26), validated herein for receptor cell surface expression by flow cytometry and previously validated for ligand-binding capability (25, 26).
Gentamicin protection assay.
Phagocytosis of live bacteria was performed as previously described (44). A total of 2.5 × 105 nonadherent phagocytic cells were incubated with the indicated bacterial genotype (multiplicity of infection [MOI] of 10) in serum-free Hanks balanced salt solution (HBSS; Corning Cellgro, Manassas, VA) for 45 min at 37°C, followed by incubation with 100 μg/ml gentamicin for 20 min at 37°C. The cells were then washed twice with HBSS and lysed with 500 μl 0.1% Triton X-100 solution in 1× phosphate-buffered saline (PBS; HyClone Laboratories, Logan, UT). Lysates were plated on LB plates and incubated overnight at 37°C. For phagocytosis assays, recovered CFU on LB plates are represented as the percentage of mean WT bacterial phagocytosis to quantitatively compare relative phagocytosis.
For antibody-blocking studies, 2.5 × 105 murine neutrophils were preincubated with 10 μg/ml of GAME-46 CD18-blocking antibody (catalog no. sc-19624; Santa Cruz Biotechnology), 10 μg/ml M1/70 CD11b-blocking antibody (catalog no. sc-23937; Santa Cruz Biotechnology), or the respective isotype controls for 15 min on ice before infection with specified bacterial strains (MOI of 10).
FACS-based bacterial association assay for fluorescent bacteria.
Phagocytes (human THP-1 cells or murine neutrophils) were incubated with GFP-expressing PA14 WT, PA14 motABmotCD, PAO1 WT, and PAO1 fliC bacterial strains (MOI of 25) for 45 min or 2 h at 37°C (as indicated) to measure total bacterial association (9, 10). THP-1 WT or CD18 knockout (KO) cells were incubated in the absence or presence of 1 μg/ml tunicamycin for 24 h at 37°C and 5% CO2 prior to infection with P. aeruginosa. For experiments assessing bacterial binding to phagocytes, murine neutrophils were preincubated with 10 μM cytochalasin D (cytoD; Enzo Life Sciences) for 30 min at 37°C before being coincubated with bacteria. As specified in the figure legend, neutrophils were pretreated with GAME-46 and cytoD for 30 min at 37°C prior to infection. FACS analyses were subsequently used to quantitatively measure the association of fluorescent bacteria with the bone marrow-derived dendritic cells (BMDCs), with nonassociated bacteria excluded by gating on scatter. In order to combine data derived from multiple independent experiments, the fluorescence values from each experiment were normalized to the mean fluorescence intensity (MFI) of uninfected BMDCs (set to 1,000).
LDH assay.
Human THP-1 cells (2.5 × 105 cells) were infected with subcultured bacteria at an MOI of 25 in a total volume of 300 μl HBSS for 2 h at 37°C. The CytoTox kit (Promega) was used according to the manufacturer’s protocol to measure lactate dehydrogenase (LDH) release from cell-free supernatants, representing cytotoxicity. Percent cytotoxicity is measured using the following formula: (A490 sample − A490 negative control)/(A490 positive control − A490 negative control) × 100, where A490 stands for absorbance at 490 nm, negative control is uninfected cells, and positive control is lysed cells (maximum LDH release).
NanoString analysis.
THP-1 WT and CD18 KO were either left uninfected or infected with PA14 WT bacteria (MOI of 25, set up in triplicates) for 3 h at 37°C. After RNA extraction, we compared the transcriptional profiles in the context of CD18 deficiency and P. aeruginosa infection using the PanCancer immune profiling panel for gene expression. The raw count expression data were analyzed using nSolver analysis software (NanoString). Briefly, raw counts were normalized to positive control probes, which recognize mRNAs spiked in with the samples at the time of analysis to control for sample application differences and the geometric mean of manufacturer-defined housekeeping genes, followed by log2 transformation. A cutoff was then chosen (with standard deviation based on comparison) for most differentially expressed genes among all four groups. Heat map analysis shows the most highly expressed genes in red and the least highly expressed genes in blue.
Statistical analyses.
Means ± standard deviations (SDs) derived from multiple independent experiments, with technical replicates, are shown for each graph. Sample sizes for each experiment are noted in the figure legends. As indicated, unpaired Student's t test with Welch’s correction or one-way analysis of variance (ANOVA) with Tukey’s post hoc analyses was performed using Prism 8.2.1 to determine statistical significance of the data. Statistical significance is represented in figures by asterisks.
ACKNOWLEDGMENTS
We thank Hannah Lust and Deborah Hogan (Geisel School of Medicine at Dartmouth) for help with NanoString analysis and George O’Toole and Deborah Hogan (Geisel School of Medicine at Dartmouth) for providing advice and reagents.
This work was supported by grants from the National Institutes of Health (NIH) (R21 AI121820, R03 AI135358, and R21 AI137656). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Andrews T, Sullivan KE. 2003. Infections in patients with inherited defects in phagocytic function. Clin Microbiol Rev 16:597–621. doi: 10.1128/cmr.16.4.597-621.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koh AY, Priebe GP, Ray C, Van Rooijen N, Pier GB. 2009. Inescapable need for neutrophils as mediators of cellular innate immunity to acute Pseudomonas aeruginosa pneumonia. Infect Immun 77:5300–5310. doi: 10.1128/IAI.00501-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kurahashi K, Sawa T, Ota M, Kajikawa O, Hong K, Martin TR, Wiener-Kronish JP. 2009. Depletion of phagocytes in the reticuloendothelial system causes increased inflammation and mortality in rabbits with Pseudomonas aeruginosa pneumonia. Am J Physiol Lung Cell Mol Physiol 296:L198–L209. doi: 10.1152/ajplung.90472.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hauser AR, Jain M, Bar-Meir M, McColley SA. 2011. Clinical significance of microbial infection and adaptation in cystic fibrosis. Clin Microbiol Rev 24:29–70. doi: 10.1128/CMR.00036-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winstanley C, O'Brien S, Brockhurst MA. 2016. Pseudomonas aeruginosa evolutionary adaptation and diversification in cystic fibrosis chronic lung infections. Trends Microbiol 24:327–337. doi: 10.1016/j.tim.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Folkesson A, Jelsbak L, Yang L, Johansen HK, Ciofu O, Hoiby N, Molin S. 2012. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat Rev Microbiol 10:841–851. doi: 10.1038/nrmicro2907. [DOI] [PubMed] [Google Scholar]
- 7.Luzar MA, Thomassen MJ, Montie TC. 1985. Flagella and motility alterations in Pseudomonas aeruginosa strains from patients with cystic fibrosis: relationship to patient clinical condition. Infect Immun 50:577–582. doi: 10.1128/IAI.50.2.577-582.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahenthiralingam E, Campbell ME, Speert DP. 1994. Nonmotility and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infect Immun 62:596–605. doi: 10.1128/IAI.62.2.596-605.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lovewell RR, Collins RM, Acker JL, O'Toole GA, Wargo MJ, Berwin B. 2011. Step-wise loss of bacterial flagellar torsion confers progressive phagocytic evasion. PLoS Pathog 7:e1002253. doi: 10.1371/journal.ppat.1002253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amiel E, Lovewell RR, O'Toole GA, Hogan DA, Berwin B. 2010. Pseudomonas aeruginosa evasion of phagocytosis is mediated by loss of swimming motility and is independent of flagellum expression. Infect Immun 78:2937–2945. doi: 10.1128/IAI.00144-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoiby N. 2011. Recent advances in the treatment of Pseudomonas aeruginosa infections in cystic fibrosis. BMC Med 9:32. doi: 10.1186/1741-7015-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mulcahy LR, Isabella VM, Lewis K. 2014. Pseudomonas aeruginosa biofilms in disease. Microb Ecol 68:1–12. doi: 10.1007/s00248-013-0297-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lovewell RR, Hayes SM, O'Toole GA, Berwin B. 2014. Pseudomonas aeruginosa flagellar motility activates the phagocyte PI3K/Akt pathway to induce phagocytic engulfment. Am J Physiol Lung Cell Mol Physiol 306:L698–L707. doi: 10.1152/ajplung.00319.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heale JP, Pollard AJ, Stokes RW, Simpson D, Tsang A, Massing B, Speert DP. 2001. Two distinct receptors mediate nonopsonic phagocytosis of different strains of Pseudomonas aeruginosa. J Infect Dis 183:1214–1220. doi: 10.1086/319685. [DOI] [PubMed] [Google Scholar]
- 15.Wilson ZS, Ahn LB, Serratelli WS, Belley MD, Lomas-Neira J, Sen M, Lefort CT. 2017. Activated beta2 integrins restrict neutrophil recruitment during murine acute pseudomonal pneumonia. Am J Respir Cell Mol Biol 56:620–627. doi: 10.1165/rcmb.2016-0215OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris ES, Weyrich AS, Zimmerman GA. 2013. Lessons from rare maladies: leukocyte adhesion deficiency syndromes. Curr Opin Hematol 20:16–25. doi: 10.1097/MOH.0b013e32835a0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizgerd JP, Horwitz BH, Quillen HC, Scott ML, Doerschuk CM. 1999. Effects of CD18 deficiency on the emigration of murine neutrophils during pneumonia. J Immunol 163:995–999. [PubMed] [Google Scholar]
- 18.Kumasaka T, Doyle NA, Quinlan WM, Graham L, Doerschuk CM. 1996. Role of CD 11/CD 18 in neutrophil emigration during acute and recurrent Pseudomonas aeruginosa-induced pneumonia in rabbits. Am J Pathol 148:1297–1305. [PMC free article] [PubMed] [Google Scholar]
- 19.Hanna S, Etzioni A. 2012. Leukocyte adhesion deficiencies. Ann N Y Acad Sci 1250:50–55. doi: 10.1111/j.1749-6632.2011.06389.x. [DOI] [PubMed] [Google Scholar]
- 20.Pollard AJ, Heale JP, Tsang A, Massing B, Speert DP. 2001. Nonopsonic phagocytosis of Pseudomonas aeruginosa: insights from an infant with leukocyte adhesion deficiency. Pediatr Infect Dis J 20:452–454. doi: 10.1097/00006454-200104000-00019. [DOI] [PubMed] [Google Scholar]
- 21.Bucior I, Mostov K, Engel JN. 2010. Pseudomonas aeruginosa-mediated damage requires distinct receptors at the apical and basolateral surfaces of the polarized epithelium. Infect Immun 78:939–953. doi: 10.1128/IAI.01215-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bucior I, Pielage JF, Engel JN. 2012. Pseudomonas aeruginosa pili and flagella mediate distinct binding and signaling events at the apical and basolateral surface of airway epithelium. PLoS Pathog 8:e1002616. doi: 10.1371/journal.ppat.1002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang GG, Calvo KR, Pasillas MP, Sykes DB, Hacker H, Kamps MP. 2006. Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat Methods 3:287–293. doi: 10.1038/nmeth865. [DOI] [PubMed] [Google Scholar]
- 24.Patankar YR, Lovewell RR, Poynter ME, Jyot J, Kazmierczak BI, Berwin B. 2013. Flagellar motility is a key determinant of the magnitude of the inflammasome response to Pseudomonas aeruginosa. Infect Immun 81:2043–2052. doi: 10.1128/IAI.00054-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hawley KL, Martin-Ruiz I, Iglesias-Pedraz JM, Berwin B, Anguita J. 2013. CD14 targets complement receptor 3 to lipid rafts during phagocytosis of Borrelia burgdorferi. Int J Biol Sci 9:803–810. doi: 10.7150/ijbs.7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hawley KL, Olson CM Jr., Iglesias-Pedraz JM, Navasa N, Cervantes JL, Caimano MJ, Izadi H, Ingalls RR, Pal U, Salazar JC, Radolf JD, Anguita J. 2012. CD14 cooperates with complement receptor 3 to mediate MyD88-independent phagocytosis of Borrelia burgdorferi. Proc Natl Acad Sci U S A 109:1228–1232. doi: 10.1073/pnas.1112078109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lefort CT, Ley K. 2012. Neutrophil arrest by LFA-1 activation. Front Immunol 3:157. doi: 10.3389/fimmu.2012.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Bruggen R, Drewniak A, Jansen M, van Houdt M, Roos D, Chapel H, Verhoeven AJ, Kuijpers TW. 2009. Complement receptor 3, not Dectin-1, is the major receptor on human neutrophils for beta-glucan-bearing particles. Mol Immunol 47:575–581. doi: 10.1016/j.molimm.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 29.van Bruggen R, Zweers D, van Diepen A, van Dissel JT, Roos D, Verhoeven AJ, Kuijpers TW. 2007. Complement receptor 3 and Toll-like receptor 4 act sequentially in uptake and intracellular killing of unopsonized Salmonella enterica serovar Typhimurium by human neutrophils. Infect Immun 75:2655–2660. doi: 10.1128/IAI.01111-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aggarwal NR, D'Alessio FR, Tsushima K, Files DC, Damarla M, Sidhaye VK, Fraig MM, Polotsky VY, King LS. 2010. Moderate oxygen augments lipopolysaccharide-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol 298:L371–L381. doi: 10.1152/ajplung.00308.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Toole GA, Kolter R. 1998. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol 30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- 32.Rossez Y, Wolfson EB, Holmes A, Gally DL, Holden NJ. 2015. Bacterial flagella: twist and stick, or dodge across the kingdoms. PLoS Pathog 11:e1004483. doi: 10.1371/journal.ppat.1004483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lukácsi S, Nagy-Baló Z, Erdei A, Sándor N, Bajtay Z. 2017. The role of CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in complement-mediated phagocytosis and podosome formation by human phagocytes. Immunol Lett 189:64–72. doi: 10.1016/j.imlet.2017.05.014. [DOI] [PubMed] [Google Scholar]
- 34.Sandor N, Kristof K, Parej K, Pap D, Erdei A, Bajtay Z. 2013. CR3 is the dominant phagocytotic complement receptor on human dendritic cells. Immunobiology 218:652–663. doi: 10.1016/j.imbio.2012.07.031. [DOI] [PubMed] [Google Scholar]
- 35.Lovewell RR, Patankar YR, Berwin B. 2014. Mechanisms of phagocytosis and host clearance of Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol 306:L591–L603. doi: 10.1152/ajplung.00335.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stokes RW, Thorson LM, Speert DP. 1998. Nonopsonic and opsonic association of Mycobacterium tuberculosis with resident alveolar macrophages is inefficient. J Immunol 160:5514–5521. [PubMed] [Google Scholar]
- 37.Demirdjian S, Hopkins D, Sanchez H, Libre M, Gerber SA, Berwin B. 2018. Phosphatidylinositol-(3,4,5)-trisphosphate induces phagocytosis of nonmotile Pseudomonas aeruginosa. Infect Immun 86:e00215-18. doi: 10.1128/IAI.00215-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Ziffle JA, Lowell CA. 2009. Neutrophil-specific deletion of Syk kinase results in reduced host defense to bacterial infection. Blood 114:4871–4882. doi: 10.1182/blood-2009-05-220806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dai S, Rajaram MV, Curry HM, Leander R, Schlesinger LS. 2013. Fine tuning inflammation at the front door: macrophage complement receptor 3-mediates phagocytosis and immune suppression for Francisella tularensis. PLoS Pathog 9:e1003114. doi: 10.1371/journal.ppat.1003114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones HE, Strid J, Osman M, Uronen-Hansson H, Dixon G, Klein N, Wong SY, Callard RE. 2008. The role of beta2 integrins and lipopolysaccharide-binding protein in the phagocytosis of dead Neisseria meningitidis. Cell Microbiol 10:1634–1645. doi: 10.1111/j.1462-5822.2008.01154.x. [DOI] [PubMed] [Google Scholar]
- 41.Toutain CM, Zegans ME, O'Toole GA. 2005. Evidence for two flagellar stators and their role in the motility of Pseudomonas aeruginosa. J Bacteriol 187:771–777. doi: 10.1128/JB.187.2.771-777.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang S, McCormack FX, Levesque RC, O'Toole GA, Lau GW. 2007. The flagellum of Pseudomonas aeruginosa is required for resistance to clearance by surfactant protein A. PLoS One 2:e564. doi: 10.1371/journal.pone.0000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanjana NE, Shalem O, Zhang F. 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Demirdjian S, Schutz K, Wargo MJ, Lam JS, Berwin B. 2017. The effect of loss of O-antigen ligase on phagocytic susceptibility of motile and non-motile Pseudomonas aeruginosa. Mol Immunol 92:106–115. doi: 10.1016/j.molimm.2017.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]