

The translocated actin recruiting phosphoprotein (Tarp) is a multidomain type III secreted effector used by Chlamydia trachomatis. In aggregate, existing data suggest a role of this effector in initiating new infections. As new genetic tools began to emerge to study chlamydial genes in vivo, we speculated as to what degree Tarp function contributes to Chlamydia’s ability to parasitize mammalian host cells. To address this question, we generated a complete tarP deletion mutant using the fluorescence-reported allelic exchange mutagenesis (FRAEM) technique and complemented the mutant in trans with wild-type tarP or mutant tarP alleles engineered to harbor in-frame domain deletions.
KEYWORDS: Chlamydia trachomatis, Tarp, effector, actin cytoskeleton, actin, cytoskeleton, effector functions
ABSTRACT
The translocated actin recruiting phosphoprotein (Tarp) is a multidomain type III secreted effector used by Chlamydia trachomatis. In aggregate, existing data suggest a role of this effector in initiating new infections. As new genetic tools began to emerge to study chlamydial genes in vivo, we speculated as to what degree Tarp function contributes to Chlamydia’s ability to parasitize mammalian host cells. To address this question, we generated a complete tarP deletion mutant using the fluorescence-reported allelic exchange mutagenesis (FRAEM) technique and complemented the mutant in trans with wild-type tarP or mutant tarP alleles engineered to harbor in-frame domain deletions. We provide evidence for the significant role of Tarp in C. trachomatis invasion of host cells. Complementation studies indicate that the C-terminal filamentous actin (F-actin)-binding domains are responsible for Tarp-mediated invasion efficiency. Wild-type C. trachomatis entry into HeLa cells resulted in host cell shape changes, whereas the tarP mutant did not. Finally, using a novel cis complementation approach, C. trachomatis lacking tarP demonstrated significant attenuation in a murine genital tract infection model. Together, these data provide definitive genetic evidence for the critical role of the Tarp F-actin-binding domains in host cell invasion and for the Tarp effector as a bona fide C. trachomatis virulence factor.
INTRODUCTION
Similar to the requirement of viruses, the sexually transmitted bacterium Chlamydia trachomatis (serovars D to K, L1, L2, and L3) must enter a suitable host cell in order to grow and cause disease (1). Consequently, interruption of chlamydial invasion of tissues is an attractive target for novel approaches to reduce the burden of urethritis and cervicitis and potential sequelae of epididymo-orchitis, proctitis, pelvic inflammatory disease, tubal infertility, ectopic pregnancy, and miscarriage (2). Much of the success of C. trachomatis as an intracellular pathogen stems from chlamydial proteins evolved to hijack host cellular processes (3). Some of these early effector proteins translocate into the mammalian cell via a type III secretion apparatus where they promote invasion and establishment of the parasitophorous vacuole (4). Specifically, one early effector called translocated actin recruiting phosphoprotein (Tarp) is hypothesized to manipulate the host cytoskeleton (both directly and indirectly) to create favorable cell surface changes for bacterial invasion of mammalian cells and tissues (5). A complete understanding of how Tarp alters the cell surface to promote invasion may lead to strategies to prevent C. trachomatis infections.
Tarp associates directly with both globular actin (G-actin) and filamentous actin (F-actin) via small alpha helical domains contained within the C-terminal region of the protein (6–8). Tarp binding to actin in vitro promotes actin nucleation and actin bundling, suggesting that Tarp directs cytoskeletal changes in the host cell during entry (6, 8). Furthermore, Tarp likely contributes to cytoskeletal changes indirectly following Tarp phosphorylation by host cell kinases. Tyrosine residues within the phosphorylation domain are phosphorylated by members of the Src family kinases (SFKs) and by other tyrosine kinases, Syk and Abl/Arg kinases (9–11). Phosphorylated Tarp recruits the guanine nucleotide exchange factors Sos1 and Vav2, and disruption of these signaling proteins with small interfering RNA (siRNA) interferes with chlamydial entry presumably by failing to activate the host cell Arp2/3 complex (12).
Phosphorylated Tarp may also play a role in altering host cell signaling shortly after invasion to favor parasitization. Tarp associates with Src-homology domain 2 (SH2)-containing protein phosphoinositide 3-kinase (PI3K) and Src homology 2 domain-containing transforming protein (SHC) to modify host cell susceptibility to cell death (12, 13).
Tarp’s multifaceted ability to interact with the host cell has been partially characterized, and multiple protein domains have been found to be required for in vitro activities such as actin binding, actin nucleation, actin bundling, oligomerization, and phosphorylation (6–8). The contribution of Tarp and the protein domain activities of Tarp have not been genetically assessed in vivo. In this work, we generated a complete C. trachomatis tarP mutant using fluorescence-reported allelic exchange mutagenesis (FRAEM). Additionally, the mutant was complemented in trans with wild-type (WT) and mutant tarP alleles engineered to harbor in-frame domain deletions designed to inactivate each specific function. We report that Tarp is critical for C. trachomatis invasion of HeLa cells, and this activity is dependent on the C-terminal F-actin-binding domains of the protein. C. trachomatis entry was also found to induce host cell shape changes. Although Tarp was not essential for propagation of C. trachomatis in culture, the tarP mutant was severely attenuated in a mouse genital tract infection model. These findings strongly support a role for Tarp in Chlamydia trachomatis pathogenesis and, for the first time, define Tarp as a bona fide virulence factor.
RESULTS
Generation of a complete C. trachomatis tarP deletion mutant using FRAEM technology.
The Tarp effector is encoded by an apparent monocistronic gene and is hypothesized to play a critical role in C. trachomatis entry of mammalian host cells. In order to definitively examine the role of Tarp in a C. trachomatis infection model, we employed fluorescence-reported allelic exchange mutagenesis (FRAEM) technology to replace the tarP gene with genes encoding both green fluorescent protein (GFP) and β-lactamase as previously described (14) (Fig. 1A). A mutant clone was isolated via sequential limiting dilution, and the absence of tarP was confirmed by quantitative real-time PCR (qPCR) (Fig. 1B) and Western blotting analysis (Fig. 1C). The presence of the chlamydial plasmid pL2 and absence of shuttle vector pSUmC-encoded mCherry were also confirmed via qPCR (Fig. 1B). Whole-genome sequencing analysis confirmed that allelic exchange was limited to the tarP locus, yet the clone did harbor two nucleotide changes compared to the parent WT strain. The first was an A→G transition within an intergenic region upstream of ctl0611 while the second represented a missense mutation resulting in a Y→M change at residue 302 of the 30S ribosomal protein S1. Interestingly, the tarP mutant generated inclusion sizes and progeny inclusion-forming units (IFUs) (Fig. 1D and E) similar to wild-type C. trachomatis over one developmental cycle, suggesting Tarp is not required for chlamydial development in HeLa cells. Furthermore, the fact that a tarP mutant was successfully generated using FRAEM also indicates that tarP is not an essential gene for propagation in tissue culture.
FIG 1.

Generation of C. trachomatis ΔtarP. (A) Schematic of the tarP locus in C. trachomatis L2. FRAEM was used to delete the entire tarP (ctl0716) sequence and insert a cassette containing gfp and bla. (B) The loss of tarP and presence of gfp were confirmed in the resulting strain via qPCR. Curing of pSUmC was confirmed using primers specific for mcherry, and retention of endogenous pL2 was verified using primers specific for plasmid-encoded pgp7-8. (C) Chemiluminescence immunoblot of EB material from wild-type (WT) or mutant Chlamydia (tarp) with TarP-specific antibodies. MOMP was probed as a loading control. Cultures of HeLa cells were infected with equal IFU of WT or tarP chlamydiae. At 0, 12, 24, 36, and 48 h postinfection, one replicate was processed for enumeration of progeny EBs on fresh HeLa monolayers (D) while the other was fixed and stained for measurement of inclusion areas (24 h is shown) (E).
Tarp production is restored in the tarP mutant carrying the chlamydial shuttle vector engineered to express wild-type or mutant tarP alleles.
In order to restore the Tarp effector to the tarP mutant, we employed a chlamydial shuttle vector engineered to express wild-type or mutant tarP alleles (15). These plasmids have been successfully used to express dominant negative Tarp effectors in wild-type Chlamydia trachomatis L2 (15). Mutant tarP alleles included in-frame deletions of the phosphorylation domain, the proline-rich domain, and the actin-binding domain as well as a truncation which resulted in deletion of F-actin-binding domains 1 and 2 (Fig. 2A). Transformation of the tarP mutant was accomplished by coinfection of HeLa cells with the tarP mutant clone lacking the endogenous plasmid and wild-type C. trachomatis transformants harboring each of the shuttle vectors described above. GFP-positive, drug-resistant inclusions were isolated by limiting dilution and expanded under antibiotic selection for several passages, density gradient purified, and tested for the presence of Tarp by Western blotting analysis. All of the coinfections successfully led to transfer of the desired shuttle vector into the tarP mutant resulting in production of wild-type or domain deletion mutant Tarp effectors (Fig. 2B).
FIG 2.
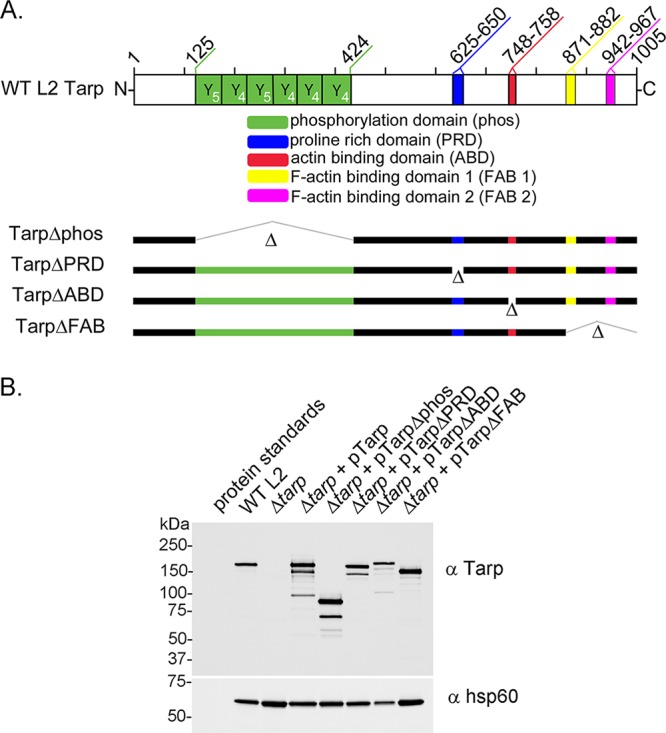
Schematic representation of Tarp and the complementation clones expressed in the tarP mutant. (A) Chlamydia trachomatis (L2) Tarp harbors an N-terminal tyrosine-rich repeat region, which is also referred to as the phosphorylation domain (phos; YYY, green boxes), a proline-rich domain (PRD; blue box), a single G-actin-binding domain (ABD; red box), and two F-actin-binding domains (yellow box [FAB 1] and pink box [FAB 2]). In-frame deletions engineered to remove specific protein domains for complementation are indicated with hatch marks and the corresponding Δ designation (Δphos, ΔPRD, ΔABD, ΔFAB 1, and ΔFAB 2). Numbers along the top of the schematic indicate amino acid positions encoded within the C. trachomatis L2 tarP gene. (B) Protein extracts from density gradient purified wild-type C. trachomatis L2 (WT L2), tarP deletion mutant (Δtarp), and mutants complemented from plasmid-based expression employing the Tarp promoter with full-length tarP (Δtarp + pTarp), tarP lacking the phosphorylation domain (Δtarp + pTarpΔphos), tarP lacking the proline-rich domain (Δtarp + pTarpΔPRD), tarP lacking the actin-binding domain (Δtarp + pTarpΔABD), or tarP lacking the F-actin-binding domains (Δtarp + pTarpΔFAB). EBs were resolved by SDS-PAGE and visualized by immunoblotting analysis with Tarp (α Tarp) and C. trachomatis heat shock protein 60 (α Hsp60)-specific antibodies. The molecular masses of protein standards are shown.
Complementation of the Chlamydia trachomatis tarP mutant restores Tarp secretion and phosphorylation.
In order to examine Tarp phosphorylation of the mutant and complemented clones, a secretion assay was performed as previously described (5). It has been shown that phosphotyrosine-containing proteins are not observed in purified elementary bodies (EBs). Interestingly, however, Tarp phosphorylation by mammalian tyrosine kinases occurs shortly after Tarp translocation into the host cell during invasion (5). This modification is readily detected by Western blotting analysis of protein lysates generated from HeLa-infected cells with a phosphotyrosine-specific antibody and, therefore, serves as a marker for Tarp delivery into the host cell cytosol. Accordingly, Tarp phosphorylation was detected for wild-type C. trachomatis and the tarP mutant clones complemented with full-length Tarp, Tarp lacking the proline-rich domain, Tarp lacking the G-actin-binding domain, or Tarp lacking the F-actin-binding domains (Fig. 3A). Interestingly, the extent of phosphotyrosine observed in the complement clones harboring Tarp lacking the G-actin-binding domain or the F-actin-binding domains was reduced compared to that of the complement clones harboring full-length Tarp or Tarp lacking the proline-rich domain. Tarp phosphorylation was not detected for the tarP mutant or the mutant complemented with Tarp lacking the tyrosine-rich phosphorylation domain (Fig. 3A). The lack of phosphorylation of the mutant complemented with Tarp harboring the domain deletion of the phosphorylation domain was predicted given that the tyrosine amino acids expected to be phosphorylated are missing. Although it remained a possibility in the absence of phosphorylation, domain translocation of the TarpΔphos protein into the host cell does not occur. In order to test this possibility, we examined whether TarpΔphos could be detected in the host cell soluble fraction following subcellular fractionation of infected HeLa cells as previously described (15). Protein pellet and supernatant samples sequentially obtained from 800, 10,000, and 100,000 × g centrifugal spins indicated that TarpΔphos was detectable in the soluble fraction (100,000 × g supernatant) as was the soluble control, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Fig. 3B). Chlamydial EBs were restricted to the 10,000 × g pellet as demonstrated by tracking the nonsecreted chlamydial antigen major outer membrane protein (MOMP) (Fig. 3B) and other undefined EB antigens detected with polyclonal anti-EB serum (data not shown). Wild-type C. trachomatis served as a positive control and wild-type Tarp was observed in the soluble fraction as previously reported (Fig. 3B) (15). These data taken together indicate that all of the Tarp proteins from the tarP mutant complement clones were secreted into the mammalian host cell during invasion.
FIG 3.
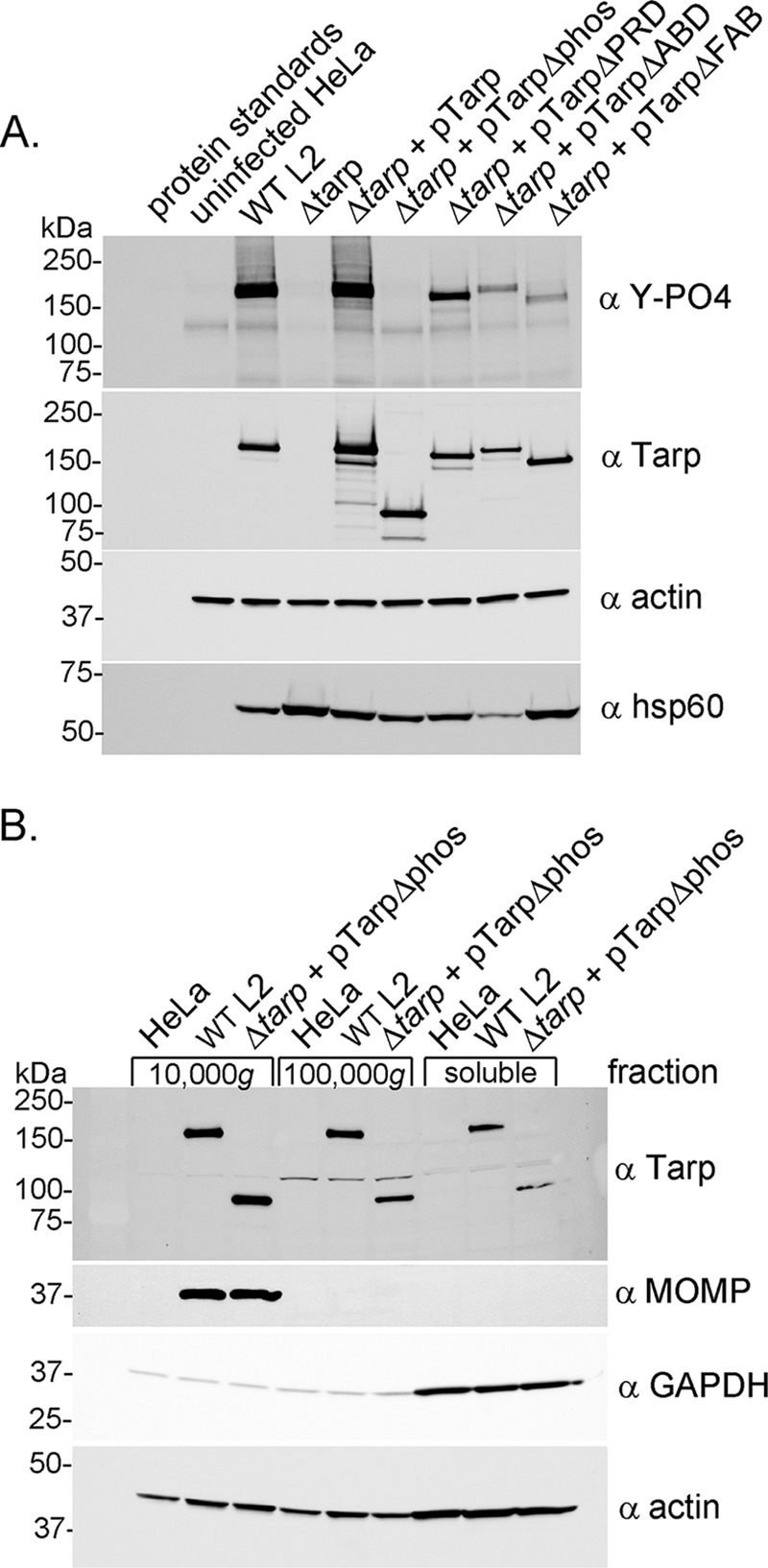
Tarp secretion and phosphorylation by Chlamydia trachomatis L2 tarP mutant and complemented clones. (A) Protein lysates were generated from HeLa cells infected with wild-type C. trachomatis L2 (WT L2), a tarP deletion mutant (Δtarp), and mutants complemented with full-length tarP (Δtarp + pTarp), tarP lacking the phosphorylation domain (Δtarp + pTarpΔphos), tarP lacking the proline-rich domain (Δtarp + pTarpΔPRD), tarP lacking the actin-binding domain (Δtarp + pTarpΔABD), or tarP lacking the F-actin-binding domains (Δtarp + pTarpΔFAB). Protein samples underwent immunoblotting analysis with phosphotyrosine (α Y-PO4), Tarp (α Tarp), actin (α actin), and C. trachomatis heat shock protein 60 (α Hsp60)-specific antibodies. A protein lysate generated from uninfected HeLa cells (uninfected HeLa) served as a negative control. (B) Subcellular fractionation of C. trachomatis-infected cells by differential centrifugation out to 100,000 × g yields a soluble Tarp fraction that is distinct from intact elementary bodies. Total lysates derived from HeLa cells alone or HeLa cells infected with wild-type C. trachomatis L2 (WT L2) or the tarP mutant complemented with pTarpΔphos (Δtarp + pTarpΔphos) underwent subcellular fractionation by centrifugation. Fractions were resolved by SDS-PAGE and transferred to nitrocellulose for immunoblotting analysis with antibodies specific for phosphotyrosine (α Y-PO4); Tarp (α Tarp); C. trachomatis major outer membrane protein (α MOMP); glyceraldehyde-3-phosphate dehydrogenase, a soluble protein marker (α GAPDH); and actin, a protein expected to be present in all fractions (α actin).
Chlamydia trachomatis Tarp is important for invasion of host cells, and the Tarp F-actin-binding domains are required for this activity.
To test the invasion potential of the tarP mutant and complement clones, we performed invasion assays to quantitate the number of elementary bodies that enter a host cell in a 1-h time period. Strikingly, the tarP mutant demonstrated a significant reduction in host cell invasion compared to that of wild-type C. trachomatis and tarP mutant complemented with wild-type tarP (Fig. 4). Interestingly, the tarP mutants harboring Tarp lacking the phosphorylation domain (TarpΔphos) and Tarp lacking the F-actin-binding domains (TarpΔFAB) both demonstrated a significant reduction in EB invasion compared to that of the wild type. An invasion phenotype was not observed for the tarP mutant carrying Tarp lacking the proline-rich domain (TarpΔPRD) or the G-actin-binding domain (TarpΔABD).
FIG 4.
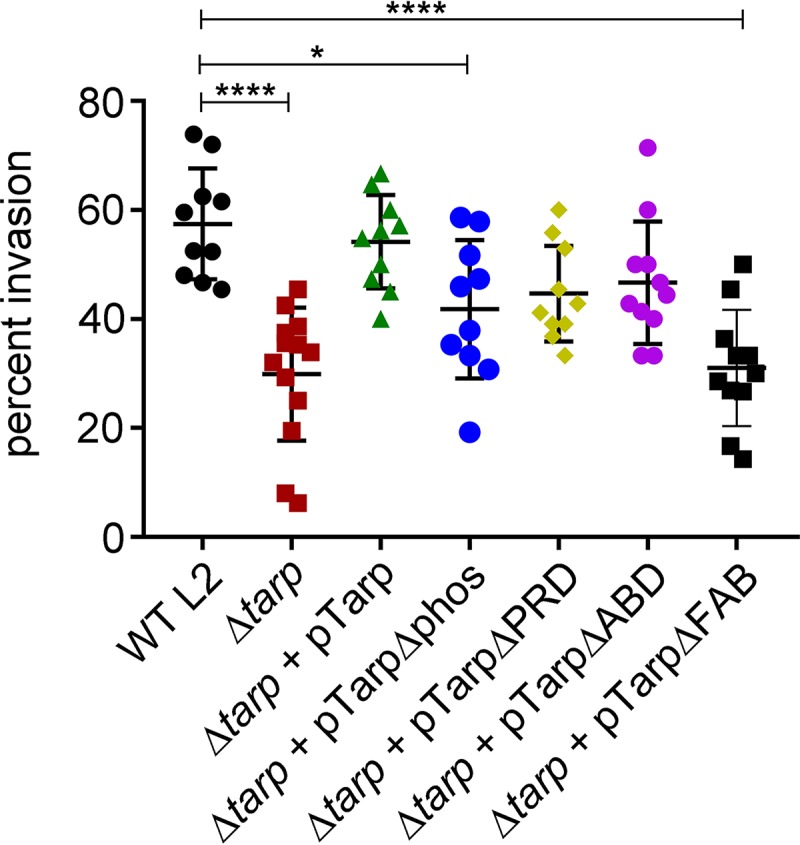
The C. trachomatis tarP mutant is deficient in chlamydial entry (and the C-terminal F-actin-binding sites play a key role in invasion). Wild-type Chlamydia trachomatis (WT L2, black circles), C. trachomatis tarP mutant (Δtarp, red squares), and C. trachomatis tarP mutant complemented with pTarp (Δtarp + pTarp, green triangles), pTarpΔphos (Δtarp +pTarpΔphos, blue circles), pTarpΔPRD (Δtarp + pTarpΔPRD, yellow diamonds), pTarpΔABD (Δtarp + pTarpΔABD, purple circles), or pTarpΔFAB (Δtarp + pTarpΔFAB, black squares) were examined for chlamydial invasion of HeLa 229 cells after 1 h. The graph presented is from one representative experiment of three. Data sets were compared with one-way ANOVA and Tukey’s multiple comparison test of the mean. *, P < 0.05; ****, P < 0.0001.
Chlamydia trachomatis mediates host cell shape changes during entry.
The Tarp effector has been biochemically shown to increase the rate of actin polymerization, bind to actin filaments, and promote the formation of actin bundles (6, 8). We hypothesized that these effector-induced cytoskeletal changes might manifest into morphological host cell changes during the early stages of infection. In support of this, hypertrophic microvilli have been observed on the cell surface of chlamydia-infected cells when observed by scanning electron microscopy (16). We wondered whether C. trachomatis was capable of inducing even more dramatic cell shape changes and whether the Tarp effector might contribute to this change. In order to examine cell shape changes triggered by a chlamydial infection, HeLa cells were infected with wild-type C. trachomatis, the tarP mutant, or the tarP mutant complemented with wild-type or mutant tarP alleles. Infected cells were examined with a Zeiss 710 confocal microscope, and the maximum cell height, length, and width were determined by z-stack analysis and ImageJ. HeLa cells infected with wild-type C. trachomatis were found to be significantly thicker than uninfected cells (Fig. 5A). The tarP mutant yielded a cell height similar to uninfected control cells while the tarP mutant complemented with wild-type tarP produced a cell height similar to that of the wild type (Fig. 5A). Further, the C. trachomatis-induced cell shape change was abrogated with the actin-destabilizing drug cytochalasin D (Fig. 5A), which indicates that this cell shape change is due to cytoskeletal changes of the host cell. In contrast to differences observed in cell height, the maximum length and width measured for each infected population of cells were heterogeneous and not significantly different.
FIG 5.
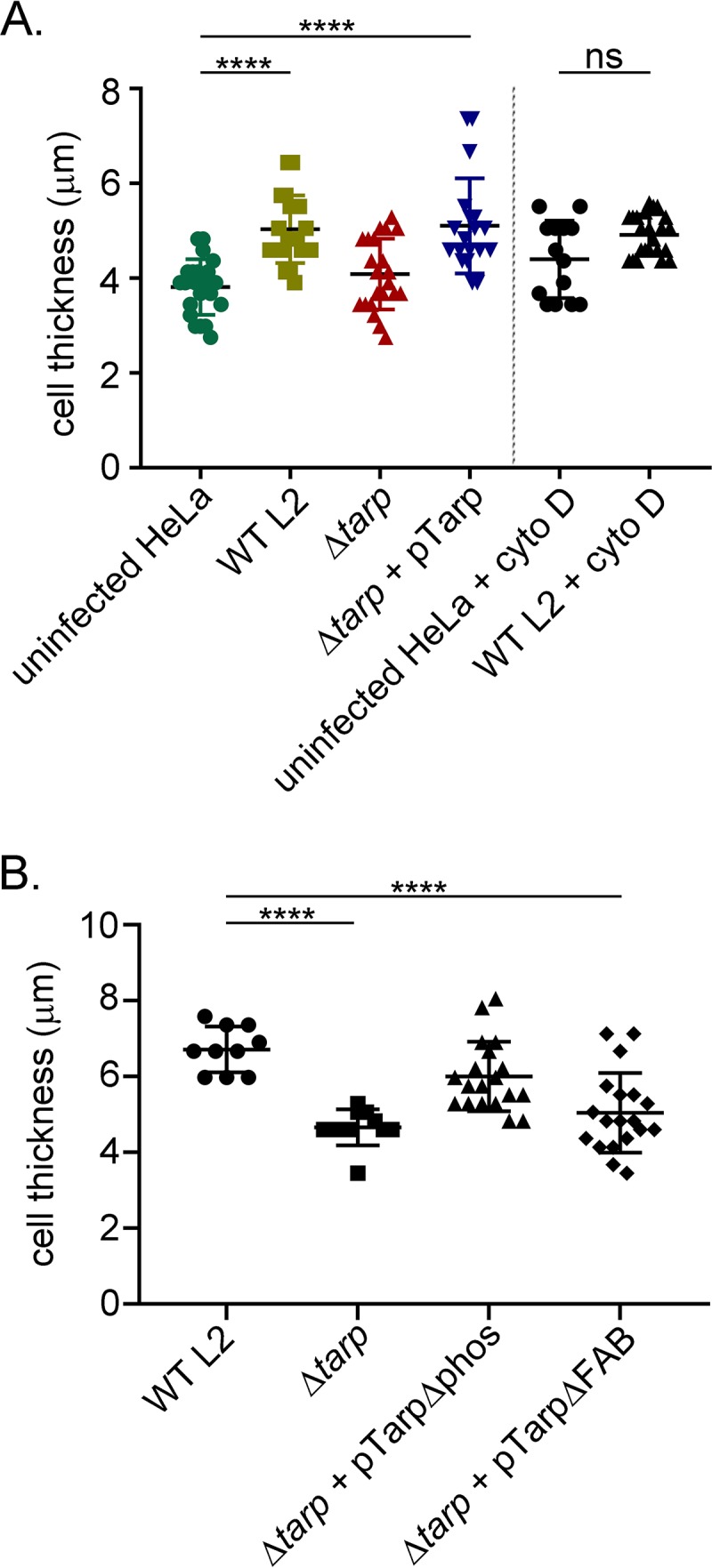
Chlamydia trachomatis invasion of HeLa cells alters the host cell shape. (A) Wild-type Chlamydia trachomatis L2 (WT L2, light green squares), tarP mutant (ΔtarP, red triangles), or tarP mutant complemented with pTarp (Δtarp + pTarp, blue upside down triangles) underwent chlamydial invasion of HeLa 229 cells for 30 min. Bacteria were visualized by staining with anti-MOMP primary antibody followed by goat anti-mouse Alexa Fluor 488 secondary antibody. The actin cytoskeleton was stained with Alexa Fluor 568 Phalloidin. The cell thickness was compared to uninfected HeLa cells (uninfected HeLa, green circles) by z-stack analysis on a Zeiss 710 inverted confocal microscope. Samples were compared using one-way ANOVA and Tukey’s multiple comparison test. ****, P < 0.0001. The graph presented is from one representative experiment of three performed. Host cells pretreated with the actin-destabilizing drug cytochalasin D (uninfected HeLa + cyto D, black circles) or drug-treated cells infected with wild-type C. trachomatis (WT L2 + cyto D, black triangles) did not undergo changes to cell shape. (B) Similarly, HeLa 229 cells were infected with wild-type Chlamydia trachomatis L2 (WT L2, black circles), tarP mutant (Δtarp, black squares), or tarP mutant complemented with pTarpΔphos (Δtarp + pTarpΔphos, black triangles) or pTarpΔFAB (Δtarp + pTarpΔFAB, black diamonds), and the cell thickness was quantified as described in panel A.
The two mutant-complemented clones engineered to express TarpΔphos and TarpΔFAB, which demonstrated retarded entry kinetics (Fig. 4), were tested for their ability to increase the infected HeLa cell height. Interestingly, only the mutant expressing Tarp missing the F-actin-binding domains failed to increase the cell height (Fig. 5B), suggesting that this domain is responsible for this activity. Together, these data support the hypothesis that Tarp is driving cytoskeletal changes, which result in alterations in cell shape. However, these findings do not rule out the possibility that the observed cell thickness differences result from a difference in EB internalization, which is significantly reduced in the absence of Tarp (Fig. 4) as well as in the presence of cytochalasin D (16).
Attenuation of C. trachomatis tarP in a murine infection model.
Finally, we extended characterization of the tarP strain to the murine infection model in order to test the overall requirement of Tarp during in vivo infection. Initial experiments using the intravaginal infection route in susceptible C3H/HeJ mice indicated that Tarp was essential for infection, yet we were unable to demonstrate complementation using the tarP clone expressing the wild-type tarP allele in trans on the chlamydial shuttle vector (not shown). Inclusions from swabs of tarP complement-infected mice were stained with Tarp-specific antibodies, indicating that lack of complementation was not due to plasmid loss (data not shown). Although no growth defect was detected in tissue culture for the tarP mutant, in the absence of genetic complementation, we could not rule out the possibility that point mutations present in the chromosome conferred the observed attenuation in mice. It was also possible that nonphysiologic levels of Tarp abundance during ectopic expression resulted in disruption of the host-pathogen balance. To test this possibility, we adapted the FRAEM technology to restore wild-type tarP in cis to the null clone using allelic replacement. This was accomplished by transforming the tarP mutant clone with pSUmC containing tarP and 2.5 kb of 5′ and 3′ flanking DNA for homologous recombination. Since tarP is monocistronic (Fig. 6A), we were also able to include aadA downstream of tarP to convey antibiotic selection. Transformants were passaged until nonfluorescent, spectinomycin-resistant, and penicillin G-sensitive inclusions were detected. A clone was isolated, and the presence of tarP and endogenous pL2 was confirmed via qPCR (not shown). As expected, allelic replacement resulted in essentially WT levels of Tarp being restored in HeLa-infected cultures (Fig. 6B and C). This clone was then tested in the intravaginal murine infection model. Mice were infected with equal IFUs of WT, tarP, or cis-tarP Chlamydia, and shed IFUs were enumerated over time. The absence of tarP correlated with early clearance of bacteria compared to WT and cis-tarP strains (Fig. 6D). Enumeration of chlamydiae revealed a ca. 2-log decrease in average shed mutant IFUs at day 3 postinfection (Fig. 6E), indicating a defect in establishing infection for the mutant clone. The tarP-deficient chlamydiae were no longer detectible by day 15 postinfection, whereas infections with both WT and cis-tarP strains persisted until day 31. Two-way analysis of variance (ANOVA) indicated a significant difference in shedding for the null mutant compared to that for the WT, whereas no significant difference was indicated between WT and cis-tarP strains. We conclude that Tarp is an important virulence protein required to establish and maintain infectivity in vivo.
FIG 6.

Attenuation of C. trachomatis tarP in a murine model can be reversed by cis complementation with WT tarP. (A) Schematic of allelic replacement strategy to restore tarP to the mutant strain. FRAEM was used to replace the gfp-bla cassette in the ΔtarP mutant with the tarP gene and a downstream aadA selection marker. (B) Representative fluorescence-based Western blot of tarP levels in 24-h culture material derived from equal infections with wild-type (WT), mutant (tarp), and cis-complemented (cis-tarp) Chlamydia. Hsp60 was probed as a loading control. (C) Signal intensity of Tarp-specific signal was normalized to Hsp60, and values are plotted as a function WT Tarp signal. Error bars represent one standard deviation from blots derived from 3 separate experiments. Groups of 5 female C3H/HeJ mice were infected intravaginally with equal input IFUs, and shed IFUs were enumerated every 4 days from day 3 to 31 postinfection. Shed bacteria were enumerated by passage in HeLa cells and are represented as number of mice actively shedding detectible chlamydiae (D) or numbers of detected inclusions (E). Data are represented with standard deviation, and two-way repeated measures (RM) ANOVA was performed to establish statistical significance (*, P < 0.0001) compared to the wild type.
DISCUSSION
Tarp is one of a handful of known prepackaged chlamydial effector proteins secreted from the EB into the host cell during pathogen entry and is likely involved in the molecular events that give rise to EB invasion of human tissues and the start of a chlamydial infection. Biochemically, Tarp promotes actin polymerization and actin bundle formation (6, 8). These reactions occur in vitro in the absence of other host cell proteins, and it is hypothesized that Tarp catalyzes these reactions near the inner membrane of the host cell cytoplasm juxtaposed to the attached EB on the host cell surface (5). The current models suggest that altered cytoskeletal changes promote EB penetration across the host cell surface, but a more detailed molecular mechanism of EB entry has not been described. As genetic tools have emerged to characterize chlamydial genes, the Tarp gene has remained a gene of importance. Interestingly, strategies to generate a tarP mutant using ethyl methanesulfonate (EMS) or TargeTron have not resulted in viable clones, leading to the suggestion that tarP may be an essential gene. However, we report herein the successful generation of a tarP mutant by fluorescence-reported allelic exchange mutagenesis (FRAEM). At this time, it is not clear why one deletion strategy is more efficacious than another but deletion of tarP with FRAEM suggests that multiple approaches to delete a gene of interest in C. trachomatis might be prudent. Successful generation and isolation of the tarP mutant via FRAEM indicate that Tarp is a nonessential gene for the overall growth of C. trachomatis in tissue culture cells. In fact, when progeny counts were compared to wild-type C. trachomatis after one developmental cycle, no difference in IFUs was observed. In stark contrast to this phenotype, Tarp was absolutely essential in the comparatively complex milieu of an in vivo infection. The impaired ability to establish an infection was apparent by the significant decrease in IFUs shed by intravaginally infected mice as early as day 3. We believe the early clearance seen by day 11 is most likely manifested by deficiencies in cellular infection since C. trachomatis L2 infections are confined to the lower genital tract (17) and do not require adaptive immunity for resolution (18). In addition, lack of another invasion-related effector, tmeA, also results in early clearance of chlamydiae (19). It is unclear why our initial experiments using trans expression of Tarp failed to complement this phenotype, yet we speculate that nonphysiological levels of Tarp could disrupt the delicate host-pathogen balance in favor of the host. Indeed, early studies of type III effectors in other bacterial systems indicated a need for physiological levels of expression to achieve complementation in animal infection models (20, 21). Regardless, our newly demonstrated ability to use allelic replacement for cis-complementation should provide an effective means of complementation for future studies.
Upon a more focused in vitro analysis of EB entry examined 1 h postinfection, the tarP mutant demonstrated a significant reduction in invasion compared to that of wild-type C. trachomatis. The invasion phenotype was complemented by reintroduction of the tarP gene on a chlamydial shuttle vector. We hypothesize that the absence of Tarp results in retarded invasion kinetics, but eventually sufficient numbers of EBs gain entry to produce infectious progeny (titers) similar to those of wild-type Chlamydia trachomatis. Interestingly, an invasion defect resulting in normal titers is similarly detected for Chlamydia trachomatis mutants lacking the early effector TmeA (19).
The generation of a tarP mutant also enabled us to analyze the function of mutant tarP alleles expressed from the chlamydial shuttle vector. We have previously described mutant Tarp effectors expressed from the shuttle vector in wild-type C. trachomatis, but these clones maintained the endogenous wild-type tarP gene and, therefore, the phenotypes observed were the result of EBs producing both wild-type and mutant Tarp proteins (15). Herein, production of the mutant Tarp proteins in the absence of wild-type Tarp allowed us to examine Tarp translocation and phosphorylation as well as assess the contribution of various Tarp protein domains to invasion. We found that while all domain deletion mutant Tarp proteins were secreted, only those Tarp proteins which harbored the tyrosine-rich repeat phosphorylation domain were phosphorylated. The extent of Tarp phosphorylation for those mutants that were missing either the G-actin-binding domain or F-actin-binding domains appeared to be lower than those of the wild type and mutant complemented with wild-type tarP. We speculate that the Tarp proteins that are deficient in binding to the actin cytoskeleton are less likely to encounter host tyrosine kinases, some of which are associated with actin filaments themselves (22). We believe it is less likely that the domain deletions may result in changes in the protein folding restricting access to the phosphorylation sites, as we have not observed alterations in the phosphorylation of recombinant Tarp proteins harboring the same mutations. Furthermore, nuclear magnetic resonance (NMR) analysis of the actin-binding domain of Tarp, which is described as intrinsically disordered, is stabilized following association with host cell actin (23). Therefore, failure to properly engage the host cytoskeleton is likely to prevent the stabilization of Tarp, which may be needed for optimal kinase engagement.
Analysis of EB entry of the tarP mutant complement clones engineered to express mutant tarP alleles revealed that the absence of the C-terminal domain of the Tarp protein, which contains the F-actin-binding and actin-bundling sites, resulted in attenuation of EB invasion of host cells to the same extent as the tarP mutant alone. Our results suggest that actin bundling is more important than actin nucleation during chlamydial entry. It is possible that the different mutant tarP alleles used in our studies may not adequately disrupt actin nucleation, as two separate domains (G-actin-binding domain and phosphorylation domain) may both contribute to actin nucleation in different ways (6, 24). We have hypothesized that Tarp-mediated host actin bundles are unique. We recently discovered that actin bundles generated by Tarp are more flexible than α actinin or fascin-generated actin bundles, and this characteristic may promote C. trachomatis entry (25). Like Tarp, the Salmonella SipC effector is able to promote actin nucleation and actin bundle formation via two different protein domains. Furthermore, analysis of SipC deletion mutants revealed that Salmonella entry of host cells was inhibited to the greatest extent when the SipC actin bundling function was removed (26). Even though Tarp and SipC lack primary amino acid sequence similarity, it is interesting that the two unrelated pathogens, Chlamydia and Salmonella, employ effectors with the same overall function, and in both cases, the actin bundling activities are critical for pathogen invasion of host cells.
Chlamydia trachomatis invasion of host cells requires cytoskeletal rearrangements, as cytochalasin D-treated cells are refractory to infection (16). In untreated cells, EB entry of tissue culture cells triggers the formation of numerous microvilli, a structure which requires orchestrated actin polymerization and bundle formation (16). Examination of the host cell by confocal microscopy revealed that chlamydia-infected cells were thicker than uninfected controls, requiring a greater number of z-stacks to image cells. Interestingly, the tarP mutant did not induce a cell shape change at the 15-, 30-, and 60-min time points examined, as the cell height measured for the mutant was the same as the height for the uninfected controls. To confirm that the cell shape change was due to cytoskeletal changes to the host cell and not to unforeseen osmotic or other physical pressures, we examined wild-type infected cells with cytochalasin D treatment and, as predicted, did not observe a cell shape change. We believe that the cell shape change is determined by the high infectious dose and that Tarp promotes the efficient entry of EBs by driving cytoskeletal changes in the host cell. We propose that Tarp triggers the formation of new actin filaments directly by binding to G-actin and indirectly via the activation of the Arp2/3 complex following Tarp phosphorylation. The newly generated actin filaments are then placed into Tarp-mediated actin bundles, which are uncharacteristically flexible. However, in the absence of Tarp, EBs infect tissue culture cells, albeit using a less efficient Tarp-independent entry mechanism. The altered entry mechanism and kinetics do not require the dramatic cytoskeletal changes induced by C. trachomatis, which harbors wild-type Tarp. It is intriguing to speculate that a high C. trachomatis infectious dose in vivo might lead to cell shape changes, which may allow other genital mucosal pathogens, such as HIV or human papillomavirus (HPV), more favorable host receptor engagement. Epidemiological data and in vitro experiments indicate that C. trachomatis infection predisposes patients and cells to other sexually transmitted infections (STIs), such as HIV and HPV, and many models suggest that this association is driven by the inflammatory response (27, 28). Our data, however, suggest that the connection between C. trachomatis and other STIs could also be driven by physical changes to the infected host cells within tissues.
Taken together, our analysis of the tarP mutant and complemented clones reveals a role for Tarp in the efficient entry of host cells mediated by Tarp’s F-actin-binding and -bundling domain. Furthermore, the tarP mutant is attenuated in a murine infection model, allowing us to define Tarp as a bona fide virulence factor.
MATERIALS AND METHODS
Organisms and cell culture.
C. trachomatis L2 (LGV 434) was propagated in HeLa 229 cells (ATCC CCL-2.1) or McCoy B cells (ATCC CRL-1696) and purified by diatrizoate meglumine and diatrizoate sodium density gradient centrifugation (29). All tissue culture cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% l-glutamine unless otherwise stated.
SDS-PAGE and immunoblotting.
Proteins were separated on SDS 4 to 12% polyacrylamide gels (Thermo Fisher Scientific, Waltham, MA) and transferred to 0.45-μm nitrocellulose immobilization membrane (Schleicher & Schuell, Keene, NH). Immunoblotting employed peroxidase-conjugated secondary antibodies (Chemicon International, Temecula, CA) and SuperSignal West Pico chemiluminescent substrate (Pierce). The anti-actin C4 monoclonal antibody was purchased from Chemicon International. The anti-actin polyclonal antibody was purchased from Cytoskeleton, Inc. The anti-phosphotyrosine 4G10 monoclonal antibody was purchased from Upstate (Millipore). The antichlamydial EB polyclonal antibody, the MOMP monoclonal antibody, and the GAPDH monoclonal antibody were all purchased from Pierce. The antichlamydial Hsp60 A57-B9 monoclonal antibody was purchased from Thermo Fisher Scientific. Polyclonal rabbit antibodies directed toward Chlamydia trachomatis L2 LGV 434 Tarp (CT456) and MOMP (CT861) have been previously described (5, 30).
FRAEM-related constructs and C. trachomatis transformations.
The tarP-specific suicide plasmid was generated using described molecular techniques (31). Briefly, 3.8 kb of 5′ and 3.9 kb of 3′ flanking DNA was PCR amplified from C. trachomatis L2 genomic DNA and mobilized into pUC18A using primer combinations 5’tarp@pUC18F (5′-GGTCTGACGCTCAGTGGAACG AGCAAGCGCTACCGTGAAAGG-3′) + 5’tarp@pUC18R (5′-GGGTTCCGCGCACATTTCCAACTACAAATTAAATAAAAACAACAGCCGATTTAATTAGATTTTAAAAAGTTGT-3′) and 3’tarp@pUC18F (5′-CACATGGCATGGATGAACTATACAAGTAA AAAGCAAAGGAGAACAGACGAGCAGAA-3′) + 3’tarp@pUC18R (5′-CTCCCGGCATCCGCTTACAGCCATGCGAGTGCCCGGAAAAA-3′), respectively. A gfp-blaM cassette was inserted into the tarP locus using insertion PCR and primers Bla-gfp@tarpF (5′-ACAACTTTTTAAAATCTAATTAAATCGGCTGTTGTTTTTATTTAATTTGTAGTTGGAAATGTGCGCGGAACCC-3′) and Bla-gfp@tarpR (5′-TTCTGCTCGTCTGTTCTCCTTTGCTTTTTACTTGTATAGTTCATCCATGCCATGTG-3′). The resulting DNA was mobilized into pSUmC to generate pSU-Δtarp using primers HomRR@pSUmC-F (5′-CTGCAGGTACCGGTCGACCATTCGGTCTGACGCTCAGTGGAACG-3′) and HomRR@pSUmC-R (5′-GATCTTTCTACGGGGTCTGACGCTC CTGGCGTTACCCAACTTAATCGC-3′). The cis complement construct, pSU-cistarp, was created via Gibson Assembly of three PCR-amplified fragments, using NEB HiFi DNA assembly master mix. The first fragment (11.4 kb) was derived from the divergent PCR of the template pSUmC-aadA-GFP-SbfI using primers SpecTarpR2 (5′-GGGCCTGCAGGTTACCAATGCTTAATCAGTGAGGCACC-3′) and aadASbfITarp-F (5′-GACCTGCAGGGCGTCAGACC-3′) to delete gfp as well as add an SbfI restriction site downstream of aadA. The second fragment (5.4 kb) contained the tarP open reading frame as well as the 2.5-kb region upstream DNA and was amplified from Chlamydia trachomatis L2, using primers Tarp5armUpF (5′-TCCTCCGTCACTGCAGGTACCGGGACTTAGAATCTAAAAACCTTGTCACGCTTTCTG-3′) and TarpUpR (5′-GGCATGATGATGAATGGTCGATTATCCTACGGTATCAATCAGTGAGCTTAGC-3′). The final 2.5-kb fragment was also amplified from Chlamydia trachomatis L2 using primers TarpdownF (5′-ATTAAGCATTGGTAACCTGCAAAAGCAAAGGAGAACAGACGAGCAGAAC-3′) and TarpdownR2 (5′-TTCTACGGGGTCTGACGCAGTTAAAGGAATTGGAACAGCAGGCGG-3′) and includes the 2.5-kb region downstream of tarP. All three PCR fragments were digested with DpnI and purified via phenol chloroform extraction and ethanol precipitation. The 11.4-kb fragment was then digested with both SalI-HF and SbfI-HF and again purified via phenol chloroform extraction and ethanol precipitation. All fragments were joined through a Gibson assembly reaction via NEB HiFi DNA assembly master mix in accordance with the manufacturer’s protocol. One microliter of the reaction mixture was used to transform NEB 10-beta electrocompetent Escherichia coli (New England Biolabs). All constructs were verified by direct DNA sequencing prior to mobilization into Chlamydia.
FRAEM was accomplished essentially as described (31) using plasmid DNA isolated from the E. coli dam dcm mutant (NEB). The tarP deletion clone was created by transforming C. trachomatis L2 with pSU-Δtarp and selection with penicillin G. Allelic replacement was accomplished by transforming C. trachomatis ΔtarP with pSU-cistarp and selecting with spectinomycin. Clonal isolates were generated by sequential limiting dilution in 384 plates, and genome integrity was confirmed via whole-genome sequencing. A consequence of the FRAEM technique in the generation of C. trachomatis mutants is the potential loss of the endogenous plasmid. We took advantage of the creation of the plasmid minus the tarP mutant and used it to transform our complementation plasmids restoring both the Tarp gene and endogenous C. trachomatis plasmid genes.
Invasion assay and indirect immunofluorescence microscopy.
Intrinsically fluorescent EBs were purified from cell cultures supplemented with CellTracker red CMTPX dye as previously described (24). Briefly, CMPTX-labeled C. trachomatis EBs (multiplicity of infection [MOI], ∼50) were added to HeLa 229 cells prepared in 24-well plates with coverslips. The cultures were then incubated at 37°C for 1 h. The cultures were fixed with 4% paraformaldehyde at room temperature for 15 min and rinsed with phosphate-buffered saline (PBS). The cells were not permeabilized. Extracellular EBs were labeled for 1 h with a monoclonal antibody specific for chlamydial major outer membrane protein (MOMP). After four washes in PBS, secondary antibody conjugated to Alexa 488 was added for 1 h. Coverslips were rinsed and mounted in ProLong Gold antifade reagent (Invitrogen). Cells were examined with a Zeiss Axio Observer A1 microscope. The number of green (external) and red (total) EBs was determined for each host cell. These data were then used to determine the percentage of internalized EBs. Twenty fields of view were taken from each cover slip, and these percentages were then averaged together to give a final invasion rate.
Subcellular fractionation and protein extraction.
Chlamydia trachomatis-infected cells underwent subcellular fractionation as previously described (15, 32). Briefly, C. trachomatis-infected HeLa 229 cells or host cells alone were removed from flasks and suspended in 100 mM KCl, 10 mM HEPES (pH 7.7), 2 mM MgCl2, and 2 mM ATP (buffer A) and disrupted by sonication using a Misonix S-4000 ultrasonic liquid processor disintegrator equipped with a microtip (Misonix Incorporated, Farmingdale, NY). All cell lysates underwent subcellular fractionation by sequential centrifugation in which supernatants and pellets were separated. Lysates were initially subject to an 800 × g spin for 15 min at 4°C. The 800 × g supernatants were then subjected to a 10,000 × g spin for 30 min at 4°C. The remaining 10,000 × g supernatant underwent a 100,000 × g spin for 1 h at 4°C. Protein sample buffer was added to all pellets, and supernatants and proteins were resolved by SDS-PAGE and transferred to nitrocellulose membranes for immunoblotting with antibodies specific for Tarp, actin, GAPDH, and MOMP.
Chlamydia trachomatis development.
HeLa 229 cells were seeded into 6-well plates (2 × 105 cells/well) 24 h prior to infection. Individual wells were infected with wild-type Chlamydia trachomatis L2 (LGV 434) or the Chlamydia trachomatis tarP mutant. All host cells and bacteria were collected from select wells at 0, 12, 24, 36, and 48 h. Cell lysates were then frozen at –80°C until samples from all time points had been collected. Cell lysates were thawed on ice and diluted and then placed onto HeLa cells grown on 16-mm circular coverslips contained within 24-well plates. After a 40-h incubation, infected cells were then immunostained and observed under a fluorescence microscope for inclusion formation. Twenty fields of view were taken from each cover slip (the experiment was performed in triplicate), and cover slip counts were averaged. Averages were plotted using GraphPad Prism software.
Shape change of individual cells.
HeLa cells were infected with C. trachomatis L2 EBs (MOI, 1,000) and incubated at 37°C for 30 min. The infected cells were then fixed with 4% paraformaldehyde and permeabilized by PBS + 0.1% Triton-X. After the infected cells were washed with PBS three times, they were stained with mouse anti-MOMP primary antibody and goat anti-mouse Alexa Fluor 488 secondary antibody. The actin cytoskeletons of the cells were stained with Alexa Fluor 568 Phalloidin as per manufacturer’s protocol. Cells were then mounted on poly-l-lysine-coated microscope slides with ProLong Gold antifade solution (Invitrogen, Karlsruhe, Germany), and the images were acquired in a Zeiss 710 inverted confocal microscope. The z-stack images were acquired from the coverslip to the top of the cell at a step size of 230 nm using a 63×/1.4 oil objective. The images were analyzed by ZEN 2.3 SP1 imaging software. To obtain the estimated cellular thickness, the total number of steps with visible Phalloidin fluorescence was multiplied by 230 nm. Images were also analyzed by ImageJ to obtain the maximum length and width of each cell. Samples were compared using one-way ANOVA and Tukey’s multiple comparison test. ****, P < 0.0001.
Murine infectivity studies.
Groups of 5 female C3H/HeJ (Jackson Laboratory) 6- to 8-week-old mice were intravaginally infected as described (33). Mice were pretreated with 2.5 mg medroxyprogesterone 5 days prior to infection and then intravaginally infected with 5 × 105 IFU of each chlamydial strain. Mice were swabbed for shed bacteria beginning on day 3 and then every 4 days until no chlamydiae were detected for all WT-infected mice. Recovered IFUs were enumerated on fresh HeLa cells as described above. All manipulations were reviewed and approved by the University of Kentucky Institutional Animal Care and Use Committee.
ACKNOWLEDGMENTS
We thank members of the Mollie W. Jewett laboratory for helpful discussions and review of this manuscript as well as acknowledge the technical assistance of Caryl-Lynn Stone and Robert Hayman.
This work is supported by the NIAID NIH grants AI065530 and AI124649 awarded to K.A.F. and the NIAID NIH grant AI139242 awarded to T.J.J.
We declare that this research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Moulder JW, Hatch TP, Kuo CC, Schachter J, Storz J. 1984. Chlamydia, p 729–739. In Krieg NR. (ed), Bergey’s manual of systematic bacteriology. Williams & Wilkins, Baltimore, MD. [Google Scholar]
- 2.Pizarro-Cerdá J, Cossart P. 2006. Bacterial adhesion and entry into host cells. Cell 124:715–727. doi: 10.1016/j.cell.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 3.Mueller KE, Plano GV, Fields KA. 2014. New frontiers in type III secretion biology: the Chlamydia perspective. Infect Immun 82:2–9. doi: 10.1128/IAI.00917-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferrell JC, Fields KA. 2016. A working model for the type III secretion mechanism in Chlamydia. Microbes Infect 18:84–92. doi: 10.1016/j.micinf.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clifton DR, Fields KA, Grieshaber SS, Dooley CA, Fischer ER, Mead DJ, Carabeo RA, Hackstadt T. 2004. A chlamydial type III translocated protein is tyrosine-phosphorylated at the site of entry and associated with recruitment of actin. Proc Natl Acad Sci U S A 101:10166–10171. doi: 10.1073/pnas.0402829101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jewett TJ, Fischer ER, Mead DJ, Hackstadt T. 2006. Chlamydial TARP is a bacterial nucleator of actin. Proc Natl Acad Sci U S A 103:15599–15604. doi: 10.1073/pnas.0603044103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jewett TJ, Miller NJ, Dooley CA, Hackstadt T. 2010. The conserved Tarp actin binding domain is important for chlamydial invasion. PLoS Pathog 6:e1000997. doi: 10.1371/journal.ppat.1000997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiwani S, Alvarado S, Ohr RJ, Romero A, Nguyen B, Jewett TJ. 2013. Chlamydia trachomatis Tarp harbors distinct G and F actin binding domains that bundle actin filaments. J Bacteriol 195:708–716. doi: 10.1128/JB.01768-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elwell CA, Ceesay A, Kim JH, Kalman D, Engel JN. 2008. RNA interference screen identifies Abl kinase and PDGFR signaling in Chlamydia trachomatis entry. PLoS Pathog 4:e1000021. doi: 10.1371/journal.ppat.1000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jewett TJ, Dooley CA, Mead DJ, Hackstadt T. 2008. Chlamydia trachomatis tarp is phosphorylated by src family tyrosine kinases. Biochem Biophys Res Commun 371:339–344. doi: 10.1016/j.bbrc.2008.04.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mehlitz A, Banhart S, Hess S, Selbach M, Meyer TF. 2008. Complex kinase requirements for Chlamydia trachomatis Tarp phosphorylation. FEMS Microbiol Lett 289:233–240. doi: 10.1111/j.1574-6968.2008.01390.x. [DOI] [PubMed] [Google Scholar]
- 12.Lane BJ, Mutchler C, Al Khodor S, Grieshaber SS, Carabeo RA. 2008. Chlamydial entry involves TARP binding of guanine nucleotide exchange factors. PLoS Pathog 4:e1000014. doi: 10.1371/journal.ppat.1000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mehlitz A, Banhart S, Maurer AP, Kaushansky A, Gordus AG, Zielecki J, Macbeath G, Meyer TF. 2010. Tarp regulates early Chlamydia-induced host cell survival through interactions with the human adaptor protein SHC1. J Cell Biol 190:143–157. doi: 10.1083/jcb.200909095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mueller KE, Wolf K, Fields KA. 2016. Gene deletion by fluorescence-reported allelic exchange mutagenesis in Chlamydia trachomatis. mBio 7:e01817-15. doi: 10.1128/mBio.01817-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parrett CJ, Lenoci RV, Nguyen B, Russell L, Jewett TJ. 2016. Targeted disruption of Chlamydia trachomatis invasion by in trans expression of dominant negative tarp effectors. Front Cell Infect Microbiol 6:84. doi: 10.3389/fcimb.2016.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carabeo RA, Grieshaber SS, Fischer E, Hackstadt T. 2002. Chlamydia trachomatis induces remodeling of the actin cytoskeleton during attachment and entry into HeLa cells. Infect Immun 70:3793–3803. doi: 10.1128/iai.70.7.3793-3803.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Connell CM, Ferone ME. 2016. Chlamydia trachomatis genital infections. Microb Cell 3:390–403. doi: 10.15698/mic2016.09.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sturdevant GL, Caldwell HD. 2014. Innate immunity is sufficient for the clearance of Chlamydia trachomatis from the female mouse genital tract. Pathog Dis 72:70–73. doi: 10.1111/2049-632X.12164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKuen MJ, Mueller KE, Bae YS, Fields KA. 2017. Fluorescence-reported allelic exchange mutagenesis reveals a role for Chlamydia trachomatis TmeA in invasion that is independent of host AHNAK. Infect Immun 85:e00640-17. doi: 10.1128/IAI.00640-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trulzsch K, Sporleder T, Igwe EI, Russmann H, Heesemann J. 2004. Contribution of the major secreted yops of Yersinia enterocolitica O:8 to pathogenicity in the mouse infection model. Infect Immun 72:5227–5234. doi: 10.1128/IAI.72.9.5227-5234.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Black DS, Bliska JB. 2000. The RhoGAP activity of the Yersinia pseudotuberculosis cytotoxin YopE is required for antiphagocytic function and virulence. Mol Microbiol 37:515–527. doi: 10.1046/j.1365-2958.2000.02021.x. [DOI] [PubMed] [Google Scholar]
- 22.Van Etten RA, Jackson PK, Baltimore D, Sanders MC, Matsudaira PT, Janmey PA. 1994. The COOH terminus of the c-Abl tyrosine kinase contains distinct F- and G-actin binding domains with bundling activity. J Cell Biol 124:325–340. doi: 10.1083/jcb.124.3.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tolchard J, Walpole SJ, Miles AJ, Maytum R, Eaglen LA, Hackstadt T, Wallace BA, Blumenschein TMA. 2018. The intrinsically disordered Tarp protein from chlamydia binds actin with a partially preformed helix. Sci Rep 8:1960. doi: 10.1038/s41598-018-20290-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carabeo RA, Dooley CA, Grieshaber SS, Hackstadt T. 2007. Rac interacts with Abi-1 and WAVE2 to promote an Arp2/3-dependent actin recruitment during chlamydial invasion. Cell Microbiol 9:2278–2288. doi: 10.1111/j.1462-5822.2007.00958.x. [DOI] [PubMed] [Google Scholar]
- 25.Ghosh S, Park J, Thomas M, Cruz E, Cardona O, Kang H, Jewett T. 2018. Biophysical characterization of actin bundles generated by the Chlamydia trachomatis Tarp effector. Biochem Biophys Res Commun 500:423–428. doi: 10.1016/j.bbrc.2018.04.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Myeni SK, Zhou D. 2010. The C terminus of SipC binds and bundles F-actin to promote Salmonella invasion. J Biol Chem 285:13357–13363. doi: 10.1074/jbc.M109.094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buckner LR, Amedee AM, Albritton HL, Kozlowski PA, Lacour N, McGowin CL, Schust DJ, Quayle AJ. 2016. Chlamydia trachomatis infection of endocervical epithelial cells enhances early HIV transmission events. PLoS One 11:e0146663. doi: 10.1371/journal.pone.0146663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schust DJ, Ibana JA, Buckner LR, Ficarra M, Sugimoto J, Amedee AM, Quayle AJ. 2012. Potential mechanisms for increased HIV-1 transmission across the endocervical epithelium during C. trachomatis infection. Curr HIV Res 10:218–227. doi: 10.2174/157016212800618093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scidmore MA. 2005. Cultivation and laboratory maintenance of Chlamydia trachomatis. Curr Protoc Microbiol 11:11A.1. doi: 10.1002/9780471729259.mc11a01s00. [DOI] [PubMed] [Google Scholar]
- 30.Hower S, Wolf K, Fields KA. 2009. Evidence that CT694 is a novel Chlamydia trachomatis T3S substrate capable of functioning during invasion or early cycle development. Mol Microbiol 72:1423–1437. doi: 10.1111/j.1365-2958.2009.06732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mueller KE, Wolf K, Fields KA. 2017. Chlamydia trachomatis transformation and allelic exchange mutagenesis. Curr Protoc Microbiol 45:11A.3.1–11A.3.15. doi: 10.1002/cpmc.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cox B, Emili A. 2006. Tissue subcellular fractionation and protein extraction for use in mass-spectrometry-based proteomics. Nat Protoc 1:1872–1878. doi: 10.1038/nprot.2006.273. [DOI] [PubMed] [Google Scholar]
- 33.Darville T, Andrews CW Jr, Laffoon KK, Shymasani W, Kishen LR, Rank RG. 1997. Mouse strain-dependent variation in the course and outcome of chlamydial genital tract infection is associated with differences in host response. Infect Immun 65:3065–3073. doi: 10.1128/IAI.65.8.3065-3073.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]