

Abstract
Background
Thrombophilia is becoming a more frequently reported disorder these years. Hereditary protein S deficiency is one of the anticoagulant deficiencies that eventually results in thrombophilia.
Case presentation
A 24‐year‐old male patient was suffering from unexplained thrombosis for the second time with a family history of deep venous thrombosis. Screening tests for anticoagulant proteins found the activity of protein S markedly lowered (5.0%). The patient was discharged after anticoagulation treatment. Four years later, the review still showed the activity of protein S in his plasma decreased (16.0%). Molecular genetic analysis revealed him homozygous for a missense mutation, c.664G>A, in the exon7 of PROS1. The mutation discovered here is the first mutation affecting the codon 222 of PROS1. This mutation results in the replacement of the glycine at the codon 222 of protein S with arginine, leading to a reduction of protein S function.
Conclusions
The finding of this mutation may help with the understanding of the mechanism of protein S deficiency, especially in the Chinese population.
Keywords: mutation, PROS1, protein S deficiency, pulmonary embolism, thrombophilia
1. INTRODUCTION
Thrombophilia is a hereditary or acquired abnormality of the hemostatic mechanism that increases the risk of thrombosis.1 Hereditary thrombophilia mainly includes disorders or deficiencies of anticoagulant proteins, plasminogens, coagulation factors, etc Protein S (PS, natural anticoagulant) is synthesized in the liver and found in vascular endothelial cells and platelet alpha granules. As a multifunctional vitamin K‐dependent single‐strand glycoprotein, it is involved in coagulation, inflammation, angiogenesis, malignant tumors, etc Being natural anticoagulation, it is located mostly in vascular endothelial cells and blood platelets.2
2. CASE REPORT
A 24‐year‐old male patient presented to the emergency department with a complaint of right‐sided chest pain for 2 days and hemoptysis for 1 day. Chest CT scan showed bilateral lung pneumonia, pulmonary fibrosis, and right‐sided pleural effusions. The patient's previous medical history showed a deep venous thrombosis (DVT) in the left lower extremity 9 years ago, and this patient was treated with low‐molecular‐weight heparin (LMWH) and warfarin. In family history, one of the patient's siblings suffered from DVT during pregnancy. Physical examination showed an increased respiratory rate (30/min) and low‐grade fever (T 37.8°C). ABG: PO2 78.3 mm Hg (1 mm Hg = 0.133 kPa), PCO2 30.6 mm Hg, and P(A‐a)O2 20.5 mm Hg, indicating respiratory alkalosis and low‐grade hypoxemia.
Ten days later, this patient experienced swelling in the right leg. Laboratory tests reported a markedly high level of D‐Dimer (10 000 μg/L). Ultrasonography analysis of lower limb vessels found multiple acute thromboses in the right leg, old thrombi in the left leg, and blood clotting in the middle‐lower part of inferior vena cava (Figure 1A). CTPA showed pulmonary embolism (PE) at the lower level of the basic lobe of the right lung (Figure 1B). However, the myocardial enzyme, troponin, and BNP were normal. Screening test analysis revealed that anticardiolipin antibody, anti‐beta 2 glycoprotein I antibodies, anti‐extracted nuclear antigens autoantibody, lupus anticoagulant, rheumatic diseases, antineutrophil cytoplasmic antibody, and HBV were all negative. Hereditary thrombophilia was then considered for the diagnosis. Further screening analysis of anticoagulant proteins demonstrated a markedly low level of protein S (PS) activity (5%, reference value: 55%‐130%) while those of antithrombin Ⅲ (AT‐Ⅲ) and protein C (PC) were normal. The patient was incidentally diagnosed with protein S deficiency combined with deep venous thrombosis and pulmonary embolism. After anticoagulation treatment, the symptoms were relieved and the level of D‐Dimer recovered to normal value. Four years later, in August 2018, this patient revisited the hospital without any discomfort. Physical examination found varicose veins in the lower extremities. Ultrasonography analysis found old thrombi in the left common iliac vein (Figure 1C), which was not reported in the previous treatment. Further analysis showed that the activity of PS was 16.0%, which was still significantly lower than the normal values.
Figure 1.
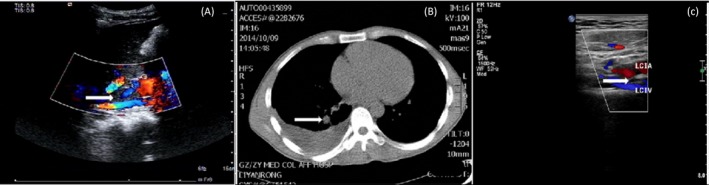
A, Ultrasonography of inferior vena cava. The arrow points to the thrombi in the middle‐lower part of inferior vena cava. B, CTPA of the patient. The arrow points to the location of a pulmonary embolism at the lower level of the basic lobe of the right lung. C, The arrow points to the old thrombi in the left common iliac vein
The patient agreed for molecular genetic analysis. Genomic DNA was extracted from peripheral blood leukocytes for high‐throughput genetic sequencing analysis. With the patient's permission and the approval of ethics committee, 2 mL of peripheral venous blood samples was collected from both the patient and the patient's daughter into Ethylene Diamine Tetraacetic Acid (EDTA) anticoagulant tube. Genomic DNA was extracted from leukocyte using QIAamp DNAout kit according to the manufacturer's instructions. The DNA was fragmented, purified and amplified before captured, and purified twice using a customized panel probe (Illumina Inc). Afterward, the samples were amplified and purified again to achieve the ultimate cDNA library. The related exons were then sequenced on a NextSeq 500 sequencer (Illumina Inc). All data were compared with the reference sequences (UCSC hg19) by the BWA algorithm.3 The data were then annotated.4 With the clinical data of the patient and bioinformatics software (PolyPhen2, LRT, Mutation Taster, etc), we were able to analyze the mutation of PROS1 of this patient. As a result, the patient was homozygous for a missense mutation (c.664G>A) at position 664 in exon7 of PROS1, leading to a replacement of the glycine at the codon 222 of PS with arginine (Figure 2). The patient's 3‐year‐old daughter showed heterozygous (c.664G/A) for this mutation (Figure 2). The daughter was reported to have a decreased PS activity (24.5%). The mutation discovered here is the first reported mutation affecting the codon 222 of PROS1, which might be the contributing factor to the protein S deficiency in the patient and the patient's daughter.
Figure 2.
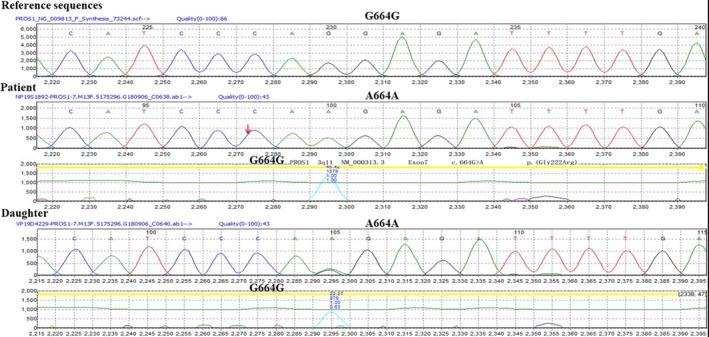
Gene sequencing of the patient and the patient's daughter. A homozygous mutation was found in the patient (A664A), and a homozygous mutation (G664A) was observed in the patient's daughter
In the first generation, the father had a heterozygous protein S gene mutation and protein S deficiency; the mother was dead. In the second generation, the son was a proband with protein S gene mutation in the new homozygote and protein S deficiency, which are currently reported in the article. In the second generation, the oldest sister did not do genetic testing or protein detection; the second oldest sister had heterozygous protein S gene mutation and protein S deficiency. In the third generation, each son from each older sister did not have gene/protein detection; the daughter from the proband had new heterozygous protein S gene mutation and protein S deficiency. Therefore, it can be concluded from the family survey that this newly discovered protein S gene mutation should be an inherited genetic mutation.
3. DISCUSSION
Protein S exists in the circulation in two forms: a complex form bound to complement protein C4b‐binding protein by the SHBG‐like domain and a free form. Only free protein S (FPS) participates in the coagulation procedure as an inhibitor of coagulation. FPS acts as a cofactor to activated protein C in the protein C anticoagulant pathway, assisting in the inhibition of Factor Xa and the inactivation of Factors Va and VIIa. Protein S deficiency (PSD) could be described in three types. All three types of PSD have the manifestation of decreased PS function; therefore, a continuously significant decrease in PS activity could be regarded as a reminder of PSD.5, 6 Herein, we report a novel mutation Gly222Arg in chromosome 3q11(PROS1) which leads to protein S deficiency in a patient with multiple venous thrombosis and pulmonary embolism.
Different from Europe, in which activated protein C resistance, coagulation Factor V Leiden, and prothrombin G20210A mutation are the most common types in thrombophilia etiology; PSD and other anticoagulant protein deficiencies appear to be the major cause of thrombophilia in Asia. PSD is an autosomal dominant disorder according to pedigree analysis.7 The condition of this patient's daughter is consistent with this conclusion. PS in the circulation is encoded by PROS1 gene on 3q11.2 while PROS2 on 3q11.1 has other functions.
Although PS type does not affect the treatment of a patient, previous findings have established that PS deficiency is critical in patients of Caucasian ethnicity.8, 9 While a missense mutation, PS Tokushima (p.Lys196Glu), is the most common mutation leading to PSD in Japan, the PS mutation in China is highly different from those in other countries without a confirmed prevalent mutation.8 In this case, we discovered a novel missense mutation (c.664G>A) in the exon 7 of PROS1, resulting in the replacement of the glycine at the codon 222 of PS with arginine, which might cause a reduction of PS function.
Xu J, Peng G, Ouyang Y. A novel mutation Gly222Arg in PROS1 causing protein S deficiency in a patient with pulmonary embolism. J Clin Lab Anal. 2020;34:e23111 10.1002/jcla.23111
REFERENCES
- 1. Walker ID, Davidson JF, Colvin BT, et al. Guidelines on the investigation and management of thrombophilia. The British Committee for Standards in Haematology[J]. J Clin Pathol. 1990;43:703‐709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Suleiman L, Négrier C, Boukerche H. Protein S: A multifunctional anticoagulant vitamin K‐dependent protein at the crossroads of coagulation, inflammation, angiogenesis, and cancer[J]. Crit Rev Oncol Hematol. 2013;88:637‐654. [DOI] [PubMed] [Google Scholar]
- 3. Li H, Durbin R. Fast and accurate long‐read alignment with Burrows‐Wheeler transform. [J]Bioinformatics. 2010;26:589‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang LU, Zhang J, Yang J, Ying D, Lau YL, Yang W. PriVar: a toolkit for prioritizing SNVs and indels from next‐generation sequencing data[J]. Bioinformatics. 2012;29(1):124‐125. [DOI] [PubMed] [Google Scholar]
- 5. Stavenuiter F, Davis NF, Duan E, Gale AJ, Heeb MJ. Platelet protein S directly inhibits procoagulant activity on platelets and microparticles[J]. Thromb Haemost. 2013;109:229‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Labrouche S, Reboul MP, Guérin V, Vergnes C, Freyburger G. Protein C and protein S assessment in hospital laboratories. Blood Coagul Fibrinolysis. 2003;14(6):531‐538. [DOI] [PubMed] [Google Scholar]
- 7. Hamasakin N. Unmasking Asian thrombophilia: is APC dysfunction the real culprit? J Thromb Haemost. 2012;10:2016‐2018. [DOI] [PubMed] [Google Scholar]
- 8. Wypasek E, Corral J, Alhenc‐Gelas M, et al. Genetic characterization of antithrombin, protein C, and protein S deficiencies in Polish patients. Pol Arch Intern Med. 2017;127:512‐523. [DOI] [PubMed] [Google Scholar]
- 9. Wypasek E, Potaczek DP, Alhenc‐Gelas M, Undas A. PROS1 mutations associated with protein S deficiency in Polish patients with residual vein obstruction on rivaroxaban therapy. Thromb Res. 2014;134(1):199‐201. [DOI] [PubMed] [Google Scholar]