

Key Readings Index
The Normal Cell, 3
Causes of Cell Injury, 8
Reversible Cell Injury, 11
Acute Cell Swelling, 11
Irreversible Cell Injury and Cell Death, 13
Cell Death by Oncosis (Oncotic Necrosis), 14
Coagulative Necrosis, 17
Caseous Necrosis, 18
Liquefactive Necrosis, 19
Gangrenous Necrosis, 19
Cell Death by Apoptosis, 21
Chronic Cell Injury and Cell Adaptations, 22
Atrophy, 23
Hypertrophy, 24
Hyperplasia, 25
Metaplasia, 25
Dysplasia, 25
Intracellular Accumulations, 25
Extracellular Accumulations, 30
Pathologic Calcification, 33
Pigments, 35
Cell Cycle, 41
Cellular Aging, 42
Genetic Basis of Disease, 43
Summary, 43
E-Glossary 1-1 Glossary of Abbreviations and Terms
AA—Amyloid A protein
AIF—Apoptosis-inducing factor
AL—Amyloid protein composed of immunoglobulin light chains
Apaf-1—Apoptosis activating factor 1
ATG—Autophagy-related gene products
ATP—Adenosine triphosphate
Bak—Bcl-2 antagonist/killer, a proapoptotic protein
Bax—Bcl-2 associated X protein, a proapoptotic protein
Bcl-2—B lymphocyte lymphoma 2 family of regulatory proteins
Bid—BH3-interacting domain death agonist
BMP3—Bone morphogenetic protein 3
C5—Complement component 5
C5b—Complement fragment 5b
C6—Complement component 6
C7—Complement component 7
C8—Complement component 8
C9—Complement component 9
cAMP—Cyclic adenosine monophosphate
CD3—Cluster of differentiation (classification determinant) protein 3
CD59—Cluster of differentiation glycoprotein 59
CDK—Cyclin-dependent kinase
cGMP—Cyclic guanosine monophosphate
CHS—Chédiak-Higashi syndrome
CNS—Central nervous system
CYP—Member of the cytochrome P450 family
DD—Death domain
DDR—DNA damage response
DISC—Death-inducing signaling complex
DNA—Deoxyribonucleic acid
DOPA—Dihydroxyphenylalanine
DR—Death receptor
ECM—Extracellular matrix
ER—Endoplasmic reticulum
FAD—Flavin adenine dinucleotide
FADD—Fas-associated death domain
FasL—Fas ligand
FGF4—Fibroblast growth factor 4
FLIP—(FADD-like interleukin 1 β-converting enzyme)-inhibitory protein, an antiapoptotic protein
FOXO—Forkhead box protein O
H&E—Hematoxylin and eosin
IGF-1—Insulin-like growth factor-1
IL-1—Interleukin 1
IL-6—Interleukin 6
IL-10—Interleukin 10
LC—Light chain
LYST—Lysosomal trafficking regulator gene
MAC—Membrane attack complex
MAPK—Mitogen-activated protein kinase
MDR1—Multidrug resistance 1 gene
MLKL—Mixed lineage kinase domain-like
MOMP—Mitochondrial outer membrane permeabilization
MPT—Mitochondrial permeability transition
mRNA—Messenger ribonucleic acid
mtDNA—Mitochondrial DNA
mTOR—Mammalian target of rapamycin
NAD—Nicotinamide adenine dinucleotide
NADPH—Nicotinamide adenine dinucleotide phosphate
NFκB—Nuclear factor κ B
NK—Natural killer
NO—Nitric oxide
NOR—Nucleolar organizing region
p53—Protein 53, product of the tumor protein TP53 gene
PAS—Periodic acid–Schiff
PCR—Polymerase chain reaction
PFK1—Phosphofructokinase 1
PPARγ—Peroxisome proliferator-activated receptor γ
PTH—Parathyroid hormone
PUMA—p53-upregulated modulator of apoptosis
rER—Rough endoplasmic reticulum
RIPK—Receptor-interacting protein-serine/threonine kinase
RNA—Ribonucleic acid
ROS—Reactive oxygen species
rRNA—Ribosomal ribonucleic acid
SASP—Senescence-associated secretory phenotype
sER—Smooth endoplasmic reticulum
SMAC—Second mitochondrial activator of caspases
SNARE—Soluble NSF (N-ethylmaleimide–sensitive fusion protein) attachment protein receptor
SOD—Superoxide dismutase
spp.—Species (plural)
TCA cycle—Tricarboxylic acid cycle, also known as citric acid cycle or Krebs cycle
TERC—Telomerase RNA subunit template component
TERT—Telomerase reverse transcriptase
TGF-α—Transforming growth factor-α
TGF-β—Transforming growth factor-β
TNF-α—Tumor necrosis factor-α
TNFR—Tumor necrosis factor receptor
TRADD—TNF receptor-associated death domain
TRAILR—TNF-related apoptosis-inducing ligand receptor
tRNA—Transfer ribonucleic acid
UBL—Ubiquitin-like
ULK1—UNC-51–like autophagy-activating kinase 1
VEGF—Vascular endothelial growth factor
VMP1—Vacuolar membrane protein 1
VPS34—Vacuolar protein sorting 34
Introduction
The goals of this chapter are to explain and illustrate the structure and function of cells and how they are interconnected with mechanisms of and responses to cell and tissue injury, such as adaptation, degeneration, and death. This information will serve as the underpinnings for materials presented in the remaining chapters covering general pathology and for comprehending materials presented on disease mechanisms and pathogeneses in subsequent chapters that cover pathology of organ systems.
Pathology is the study of disease from all perspectives. This pathology textbook begins with a 6-chapter general pathology section followed by 15 chapters of pathology of organ systems (systemic pathology). Although this layout parallels the instruction of pathology in many veterinary schools, the division into general pathology and systemic pathology is somewhat artificial. General pathology is the study of the reaction of cells or tissues to injury with a focus on the mechanisms of that response. In the first six chapters of this book, the response to injury is classified as cellular adaptations (degenerative, regenerative, or restorative), vascular disorders, inflammation, or neoplasia, with an additional chapter on the mechanisms of infectious diseases and one on disorders of immunity. These categorizations simplify the teaching and learning of general pathology. However, in the living body, cell injury provokes a variety of vascular, inflammatory, and immune-mediated responses in addition to disturbances of growth. These reactions not only extend beyond the injured cell to the organ or organismal level but also can occur simultaneously or in rapid succession. This first chapter is focused on the cellular responses to injury, not only on the degeneration that can progress to cell death but also on the adaptations of surviving cells. In subsequent general pathology chapters of Section I, more emphasis will be placed on the interaction among cells of different types, as well as the interaction of cells with their stroma, with other organ systems, and with circulating cells and molecules.
Systemic pathology is the study of systemic disease (i.e., disease that affects the system, meaning the entire organism). It is not a separate discipline from general pathology, but a different approach to the study of disease, in which the principles of general pathology are applied at the level of the tissue or organ or even the entire body. As for general pathology, the learning process is simplified by categorization, so Section II of this book is arranged in chapters based on a particular organ system. Again, this subdivision is arbitrary, and the student must bear in mind that disease seldom, if ever, affects only one organ or tissue. It also helps to remember that most organs or tissues respond in a similar way to a particular type of injury, hence the value in mastering the concepts of general pathology before the organ system approach. There is no optimum arrangement of the organ system chapters, so pathology of organ systems can be taught in different sequences in different curricula.
Pathologists are specialists in the discipline of pathology. Although general pathology and systemic pathology are educationally useful divisions of the discipline, pathologists are seldom categorized as general pathologists or systemic pathologists but, instead, are often classified as specialists in a particular organ system. For example, a dermatopathologist specializes in skin diseases; a neuropathologist, in diseases of the nervous system. In North America, pathologists are certified as anatomic pathologists, interested especially in the morphologic changes of gross (macroscopic) pathology and histopathology (microscopic pathology of tissues), or as clinical pathologists, who work more with microscopic and biochemical evaluations of blood, urine, and other bodily fluids or with cytologic samples, in which individual cells are studied rather than the intact tissue. Although there is overlap between anatomic and clinical pathology, the focus of this book is anatomic pathology; clinical pathology is taught separately in most veterinary curricula. After certification, many anatomic pathologists specialize further in practice. Diagnostic pathologists are involved in autopsy (syn: necropsy; postmortem gross and histologic examination along with correlation of ancillary test results) and histologic examination of surgical biopsy specimens. Some diagnostic pathologists limit their practice to surgical (biopsy) pathology. Toxicologic and other experimental pathologists study the tissue, cellular, and molecular mechanisms of disease in a research setting.
In the practice of pathology the goal is to answer a question or solve a problem. The question depends on the nature of the investigation. In diagnostic pathology an autopsy (syn: necropsy) may be performed to determine the cause of death in an individual or in a group of animals or to explain decreased production in a herd, flock, kennel, or cattery. In forensic pathology the purpose of an autopsy is to determine the nature of death from a legal perspective. Surgical pathology (histologic examination of surgically excised tissue specimens) not only facilitates diagnosis and prognosis for a living animal but also can be the basis for therapy. Experimental pathologists contribute from the design to the end point of an investigation with the goal of correlating morphologic changes with clinical, functional, and biochemical parameters to elucidate the mechanisms of disease.
Most veterinary students will practice internal medicine or surgery, rather than pathology, yet pathology is an integral part of veterinary education and practice. Pathology is the link between basic sciences, such as anatomy and physiology, and clinical sciences and is the foundation for a lifetime of learning, diagnosing, and understanding disease in living and dead animals. The practicing veterinarian and the pathologist form a team at the forefront of animal and public health.
Basic Terminology
The language of pathology shares many words with other biomedical disciplines but also has its share of unique terms. First, the student should remember that pathology is the study of disease and that the word “pathology” is not equivalent to “lesion(s).” A lesion is a structural abnormality in a tissue or organ. All too often, we hear or read a phrase such as “No pathology is observed in the liver.” This would be more correctly written as “No lesions are observed in the liver.”
Traditionally, in veterinary medicine, the word necropsy has been applied to a postmortem examination of an animal to distinguish it from autopsy (postmortem examination of a human being). The word necropsy (literally, having a look at something dead) also served to distinguish the examination of a dead creature from biopsy, a look at a sample from a living animal (human or nonhuman). In the interest of one health, and in recognition that it is unnecessary to distinguish a postmortem examination of a human from that of a nonhuman animal, the word autopsy (literally, having a look with one's own eyes) has been applied to the postmortem examination of any creature. One could argue, because pathologists use microscopy in addition to their own eyes, that the word autopsy could just as well be applied to other diagnostic imaging modalities (e.g., radiography or computed tomography). However, by convention, the term has been relegated to the discipline of pathology, and the autopsy or biopsy examination generally implies a search for lesions.
The student will find that much of the vocabulary of pathology is common to that of other courses. However, certain words mean one thing in one discipline and something quite different in another. Indeed, the precise meaning—and precision is the object in writing or speaking about pathology—of a word depends very much on the context. For example, the word malacia, which literally means softening, implies a lack of mineralization when applied to bones, but when applied to the brain, it means necrosis. That brings us to an important word that pathologists use a lot and that figures prominently in this chapter—necrosis. It means death or, more precisely, the structural changes that follow death, not of the entire organism, but of cells within an organ or tissue. Do not worry, there will be more on necrosis in subsequent sections!
The Normal Cell
Knowledge of anatomy and of normal anatomic variations is prerequisite to lesion recognition and interpretation. Structure is covered briefly at the beginning of each of the organ system pathology chapters in Section II. The anatomic focus in this chapter is on the cell.
Components of Normal Cells and Their Vulnerabilities
A clear understanding of normal cell structure and function is essential to the study of cellular responses to injury. The cell can be visualized simplistically as a membrane-enclosed structure, subdivided into smaller functional units (organelles) by these membranes (Fig. 1-1 ). This interconnecting system of membrane-bound compartments is termed the cytocavitary network. The function of individual organelles depends in great part on the biochemistry of their membrane and intracellular matrix (i.e., gel component of the cytoplasm that supports the functions of the organelle). Cell membranes and organelles are targets for injury by microbes and various genetic, metabolic, and toxic diseases that are addressed in greater detail in the pathology of organ systems chapters.
Figure 1-1.
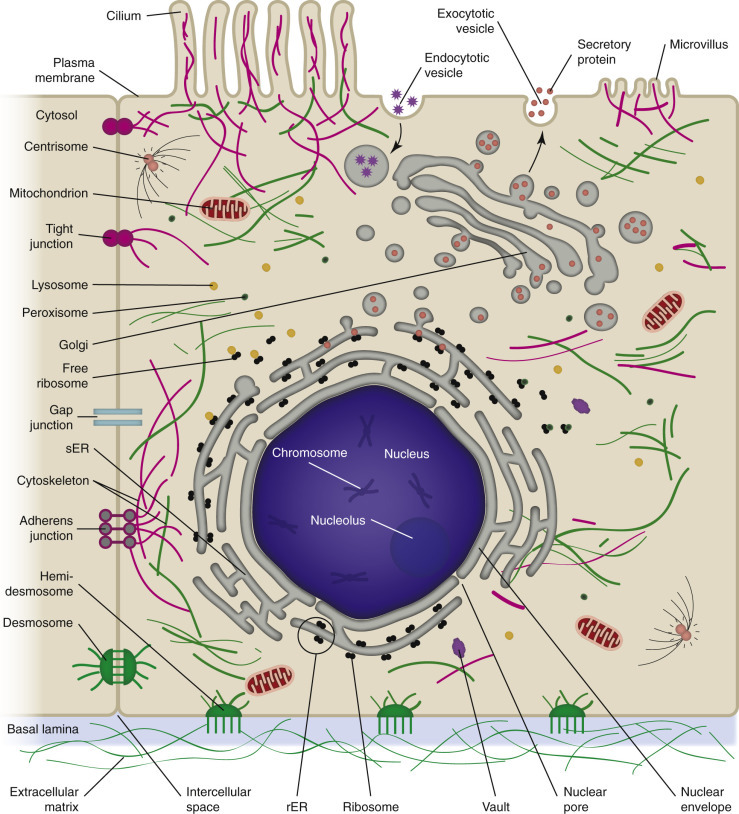
Cell Structure and the Organization of Organelles, Cytoskeleton, and Membrane Enhancements.
rER, Rough endoplasmic reticulum; sER, smooth endoplasmic reticulum.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Cell Membranes (Cytocavitary System)
Cell membranes are fluidic phospholipid bilayers that enclose cells and their organelles (Fig. 1-2 ). The two main functions of these membranes are (l) to serve as selective barriers (i.e., barrier systems [see Chapter 4]) and (2) to form a structural base for the membrane-associated proteins (enzymes and receptors) that determine cell function. The term fluidic indicates that proteins and lipids in the membrane are not immovable but can travel as part of the cytocavitary system (Fig. 1-3 ) throughout the physical extent of the cell. As an example of this process of “fluidic” movement, transmembrane proteins used as cell surface receptors are synthesized and assembled in the rough endoplasmic reticulum (rER), inserted into membranes in the Golgi complex, and moved (fluidic) to the cell's surface at the plasma membrane via the cytocavitary system (see Fig. 1-3).
Figure 1-2.
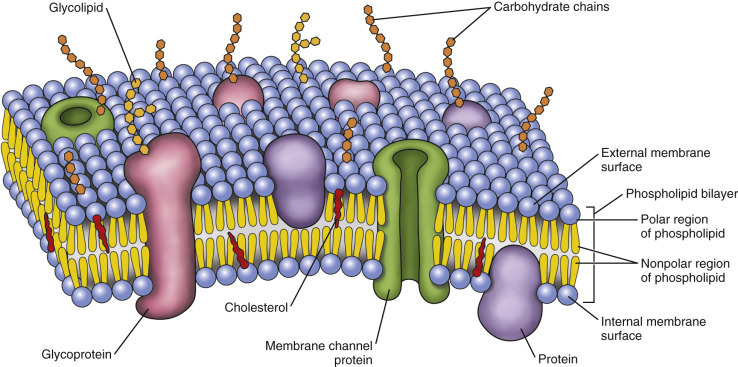
Fluid Mosaic Model of Cell Membrane Structure.
The lipid bilayer provides the basic structure and serves as a relatively impermeable barrier to most water-soluble molecules.
Figure 1-3.
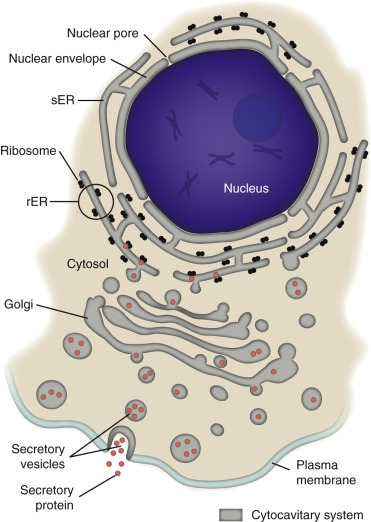
Cytocavitary System.
The rough endoplasmic reticulum (rER) and Golgi complex function in synthesis of proteins and glycoproteins used in and secreted from cells. Transcription, translation, assembly, modification, and packaging of these molecules occur in an orderly sequence from the nucleus to the plasma membrane as shown. Smooth endoplasmic reticulum (sER) is involved in the synthesis of lipids, steroids, and carbohydrates and in the metabolism of exogenous substances.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
The plasma membrane encloses the entire cell and thus is its first contact with harmful substances, agents, and infectious microbes. Microvilli and cilia (see Fig. 1-1) are specialized areas of the plasma membrane that are often altered in disease. Plasma membranes separate the interior of the cell from the external environment, neighboring cells, or the extracellular matrix (ECM). Surface proteins, such as fibronectin, play a role in cell-to-cell and cell-to-ECM interactions. Transmembrane proteins embedded in the phospholipid bilayer serve in a variety of essential structural, transport, and enzymatic functions (Fig. 1-4 ). Ligand-receptor interactions play key roles in these functions. Ligands are signaling molecules (also known as first messengers) (i.e., autocrine, paracrine, and endocrine signals [see Fig. 12-1]) that bind to receptors in the plasma membrane (cell surface receptors), cytoplasm (cytoplasmic receptors), or nucleus (nuclear receptors). Ligands may be cell associated, such as those on the surface of infectious microbes (see Fig. 4-31), or extracellular, such as hormones, growth factors, cytokines, cell recognition molecules, and neurotransmitters.
Figure 1-4.
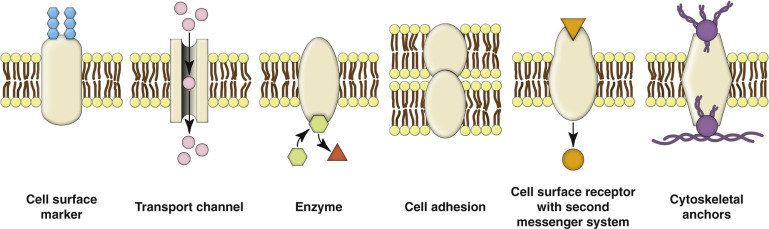
Functions of Transmembrane Proteins.
Transmembrane proteins that span the phospholipid bilayer of cell membranes serve a variety of structural, transport, signaling, and enzymatic functions.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Cytoplasmic and nuclear receptors, through control of gene expression, regulate cellular development, homeostasis, metabolism, and aging. Ligands that bind these receptors include lipophilic substances, such as steroid hormones, vitamins, and xenobiotic endocrine disruptors that cross plasma and nuclear membranes by passive diffusion.
Cell surface receptors are central to the pathogenesis of many disorders discussed throughout this book. As an extension of a transmembrane protein, cell surface receptors receive and interpret extracellular signals (i.e., ligands) from the environment. When a ligand binds to an appropriate surface receptor, conformational changes in the transmembrane protein result in a process called signal transduction (signaling molecule → specific receptor protein on the plasma membrane → second messenger transmits the signal into the cell → physiologic response) and the activation (i.e., second messenger system [see later discussion]) or inhibition of the receptor's biochemical pathway. There are hundreds of different types of glycoprotein and lipoprotein transmembrane receptors; each type is linked to a specific intracellular biochemical pathway, and individual cells contain many of these receptors based on their function as determined by their genome. Transmembrane receptors are often used by infectious microbes to invade cells or use cell systems during their life cycles, thus initiating a process that can injure the host cell. These receptors and their roles in the mechanisms of infectious disease are discussed in detail in Chapter 4.
A unique transmembrane protein receptor is involved in the notch-signaling pathway. Ligand activation of notch signaling results in the formation of a cytoplasmic second messenger that enters the nucleus and modifies gene expression during embryonic development and homeostasis. During development, notch signaling allows specific types of cells and tissues to develop, organize, and grow. If a specific cell type expresses a trait essential for the development of a specific tissue type, ligands are released from the “essential” cell that bind notch receptors on adjacent cells. Signal transduction and second messenger systems are activated, leading to the inhibition of division and development of affected “bystander” cells. This outcome allows specific types of cells to increase in number during development, while inhibiting other less essential cell types. Notch-signaling pathways are involved in the development of neural tissues, blood vessels, heart, pancreas, mammary gland, T lymphocytes, hematopoietic lineages, and other cell types. Notch-signaling pathways also play a role in mature animals. They appear to determine, for example, whether enteric stem cells differentiate into villous enterocytes with secretory or absorptive functions. Diseases that kill or injure enteric crypt stem cells (e.g., parvovirus) or villous enterocytes (e.g., coronaviruses) probably disrupt notch-signaling pathways, leading to a lack of secretory or absorptive enterocytes during healing with failure to return to “normal” function (see Chapter 7).
Second Messenger Systems.
Cells are in continuous contact with a wide variety of extracellular molecules (see first messengers earlier). Examples of first messenger molecules include microbial ligands (see also Chapter 4), hormones, growth factors, neurotransmitters, and xenobiotics. First messenger interactions typically involve the binding of a ligand to its transmembrane protein receptor, which activates a second messenger system (E-Fig. 1-1). Examples of second messenger molecules include Ca2+, cyclic adenosine monophosphate (cAMP), cyclic guanosine monophosphate (cGMP), inositol triphosphate, diacylglycerol, arachidonic acid, and nitric oxide (NO). The second messenger initiates an intracellular signal transduction cascade that stimulates or alters a metabolic pathway. Thus second messenger systems translate “first messages” from the plasma membrane into specific actions within the cell and its organelles to maintain homeostasis or defend against infection or other injury.
E-Figure 1-1.
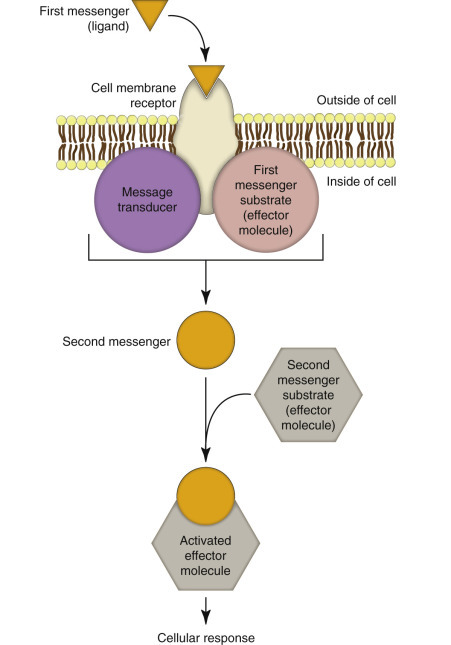
Messenger Systems in Cells.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Cytosol versus Cytoplasm
Whereas the term cytoplasm refers to the light microscopically visible portion of the cell that is inside the plasma membrane and outside the nuclear envelope (see the next section), the term cytosol specifies the cytoplasmic matrix (i.e., the gel portion of the cytoplasm that surrounds organelles). The cytosol contains water, dissolved ions, and macromolecules, such as proteins.
Nucleus
Animals are made of eukaryotic cells, meaning cells that have a nucleus, which, except in mammalian erythrocytes, is retained throughout the life of the cell. The nucleus (see Fig. 1-1) is readily visible by light microscopy because it contains chromatin (DNA complexed with histones), which is well stained by hematoxylin. Uncoiled chromatin is called euchromatin and is dispersed throughout the nucleus and actively involved in production of messenger RNA (mRNA). Tightly coiled chromatin is called heterochromatin and is clumped around the inner nuclear membrane and is inactive (see also E-Fig. 1-22). The nucleus is surrounded by an inner and an outer nuclear membrane that together form the nuclear envelope. The inner and outer nuclear membranes merge at the nuclear pore complexes, which allow bidirectional trafficking between the nucleus and the cytosol. The inner nuclear membrane is more “nuclear” in its biochemistry and serves to segregate and maintain the unique biochemistry of the nucleus, whereas the outer nuclear membrane has features more like those of the endoplasmic reticulum (ER), with which it is continuous. This differentiation and arrangement is essential for translation of genetic material (DNA and RNA) into gene products (proteins).
E-Figure 1-22.
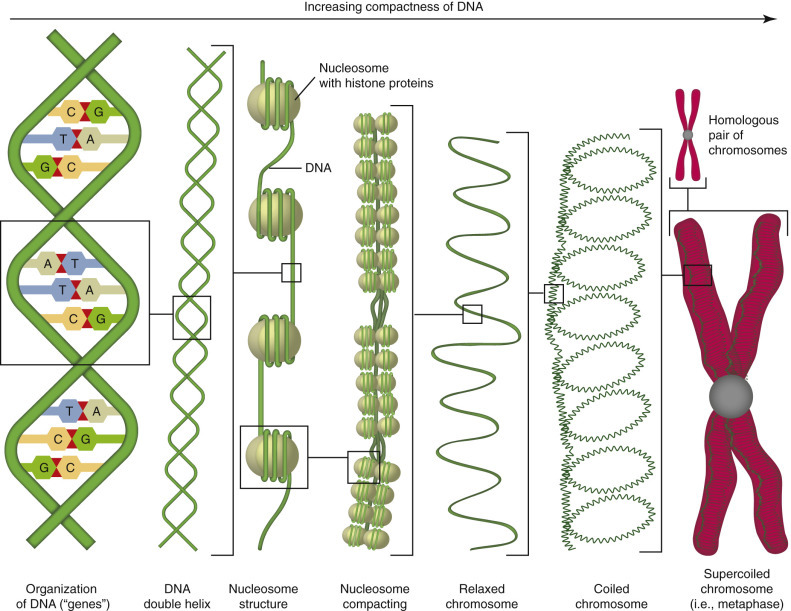
Organization of DNA.
DNA has an antiparallel configuration with one strand arranged 5′ to 3′ in one direction, and the other strand in the opposite direction. A purine is bound to a pyrimidine by hydrogen bonds: A : T and G : C. The double helix is the result of bonds in the phosphate backbone. DNA is organized around histone proteins into nucleosomes, which are compacted and progressively coiled into relaxed chromosomes, coiled chromosomes, and finally supercoiled chromosomes. DNA is inactive in this coiled form and must be uncoiled for transcription and translation.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Nucleolus.
The nucleolus (see Fig. 1-1) is a non–membrane-bound structure within the nucleus that forms around chromosomal loci of the ribosomal RNA (rRNA) genes known as nucleolar organizing regions (NORs). The nucleolus is the site of transcription and processing of rRNA and of assembly of preribosomal subunits. Thus it consists of ribosomal DNA, RNA, and ribosomal proteins, including RNA polymerases, imported from the cytosol. At the light microscopic level, the nucleolus can be inconspicuous in inactive cells or quite prominent in cells with high protein production.
Rough Endoplasmic Reticulum
The ER is a membrane-bound network of flattened saclike cisternae (see Figs. 1-1 and 1-3). The membrane of the rER is continuous with the outer nuclear membrane, so the luminal contents of the rER and of the nuclear envelope communicate. rER is so named because attached ribosomes impart a rough appearance (at the ultrastructural level) to its membrane as opposed to the appearance of the smooth ER (sER), which lacks surface ribosomes. The main function of rER is protein synthesis. Translation of mRNA with assembly of amino acids into peptides begins on ribosomes that are free in the cytosol. When the developing peptide is detected by a signal recognition particle, translation pauses until the ribosomal peptide–mRNA complex is attached to the outer surface of the rER. Protein formation continues in the membrane or lumen of the rER until a signal peptidase removes the signal peptide, at which time the newly formed protein can be transported to the cellular or extracellular site where it is needed or to the Golgi complex for further processing (see Fig. 1-3). Transmission electron microscopy is generally required to visualize the rER; however, cells that produce abundant protein and thus have abundant rER tend to have more basophilic cytoplasm because of the ample nucleic acid (RNA) in ribosomes.
Ribosomes.
Ribosomes facilitate the synthesis of proteins in cells (i.e., translation) (see Figs. 1-1 and 1-3). Their function is to “translate” information encoded in mRNA into polypeptide chains of amino acids that make up proteins. There are two types of ribosomes, free and fixed (also known as membrane bound). They are identical in structure but differ in locations within the cell. Free ribosomes are located in the cytosol and are able to move throughout the cell, whereas fixed ribosomes are attached to the rER. Free ribosomes synthesize proteins that are released into the cytosol and used within the cell. Fixed ribosomes synthesize proteins that are (1) inserted into the cell membrane (transmembrane proteins) at the rER and subsequently moved (fluid mosaic membrane model) to their final destinations usually within the plasma membrane or (2) placed in membrane-bound vesicles and moved through the Golgi complex (see next paragraph) to the plasma membrane and released via exocytosis into the extracellular environment.
Golgi Complex
The Golgi complex, also commonly called the Golgi apparatus, is a series of flattened membrane-bound sacs with its inner face (cis or entry face) near the rER in a paranuclear position (see Fig. 1-3). Proteins made in the rER are delivered to the entry face of the Golgi complex by transport vesicles. As the proteins traverse the Golgi complex, they are processed (e.g., carbohydrate moieties added through glycosylation) and packaged into secretory vesicles to be released from the outer (trans) face of the Golgi complex into the cytosol, either for use by the cell that produced them, as in the case of lysosomal enzymes, or (more commonly) for delivery to the plasma membrane for export. Transmission electron microscopy is usually required to visualize the Golgi complex. However, an active Golgi complex, such as that needed for processing and packaging of immunoglobulin molecules, is large enough to impart a paranuclear eosinophilic pallor to plasma cells in a hematoxylin and eosin (H&E)–stained histologic section.
Smooth Endoplasmic Reticulum
sER is a membrane-bound network of tubules (see Figs. 1-1 and 1-3) without surface ribosomes. sER is not involved in protein synthesis. Its main function is the synthesis of lipids, steroids, and carbohydrates, as well as the metabolism of exogenous substances, such as drugs or toxins. Cells, such as hepatocytes, that are important for synthesis of lipids and metabolism of drugs or toxins have abundant sER, as do cells that produce steroid hormones, such as adrenocortical cells and certain testicular or ovarian cells. Cells with abundant sER have pale eosinophilic, finely vacuolated cytoplasm.
Mitochondria
Mitochondria are dynamic organelles that can change shape, undergo fission and fusion, and move about within the cell. They can be large enough (up to 1 µm) to resolve with the light microscope, especially in muscle from athletic animals such as racehorses. Because most cellular processes require “energy,” a major mitochondrial function is the generation of energy as adenosine triphosphate (ATP) through oxidative phosphorylation. Mitochondria are also involved in programmed cell death (e.g., apoptosis), signaling, cell differentiation, and cell growth. Mitochondria contain their own genome (see later section on the Genetic Basis of Disease), which consists mainly of circular DNA that encodes transfer and rRNAs as well as some mitochondrial proteins. However, most of the genes that encode mitochondrial proteins are located in the nucleus of the cell. Mitochondria have a biochemically distinct inner and outer membrane. The inner membrane is folded into cristae that project into the central matrix of the mitochondrion (see Figs. 1-1 and 1-5 ). Some mitochondrial structural proteins and enzymes are made on free ribosomes and then imported from the cytosol to the appropriate mitochondrial compartment (outer membrane, intermembrane space, inner membrane, or matrix). Mitochondria also establish close contact, perhaps via tethering proteins, with the ER.
Figure 1-5.
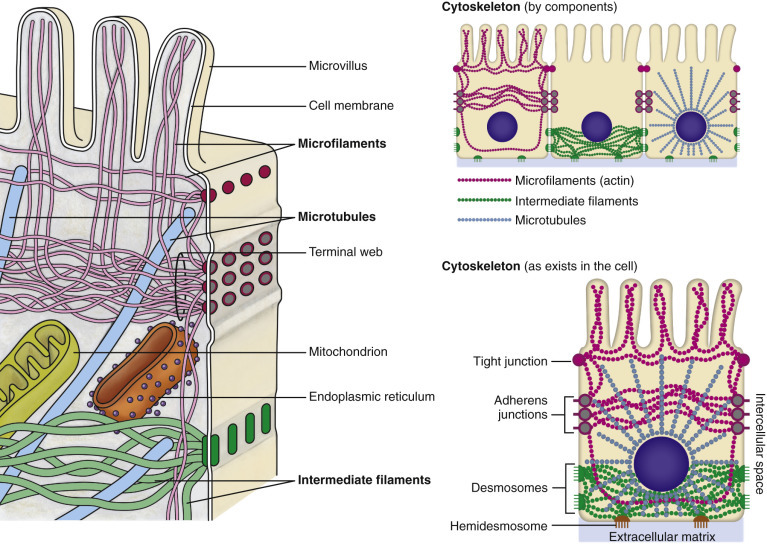
Cytoskeleton.
The complexity and interrelations of microfilaments, intermediate filaments, and microtubules with the plasma membrane and other organelles are depicted.
Oxidative Phosphorylation
Mitochondria produce energy in their matrix by converting pyruvate (produced from glucose or other carbohydrates by glycolysis in the cytosol) and amino or fatty acids to acetyl coenzyme A. As acetyl coenzyme A is oxidized to CO2 in the citric acid cycle (also known as the tricarboxylic acid or Krebs cycle), the coenzymes oxidized nicotinamide adenine dinucleotide (NAD+) and flavin adenine dinucleotide (FAD) are reduced to NADH and FADH2, respectively. These reduced coenzymes provide electrons for stepwise transfer (in the inner mitochondrial membrane) to molecular oxygen. The energy released from this electron transport generates a proton gradient across the inner mitochondrial membrane that drives oxidative phosphorylation with the production of ATP from adenosine diphosphate (ADP) and inorganic phosphate.
Vaults
Vaults are rather recently discovered barrel-shaped organelles (see Fig. 1-1) that are thought to function in transporting large molecules (e.g., mRNA or proteins) between the nucleus and other intracellular locations. Their octagonal profile may facilitate docking at nuclear pores.
Lysosomes and Peroxisomes
Lysosomes are membrane-bound vesicles (see Fig. 1-1; also see E-Fig. 1-27, A) that contain enzymes (acid hydrolases) that can digest most chemical compounds (nucleic acids, carbohydrates, proteins, or lipids) endogenous to the cell or extracellular substances taken up by endocytosis or phagocytosis. Enzymes contained in lysosomes are synthesized by the rER (i.e., fixed ribosomes), processed and packaged in the Golgi complex, and released in vesicles from the outer surface of the Golgi complex into the cytosol.
E-Figure 1-27.
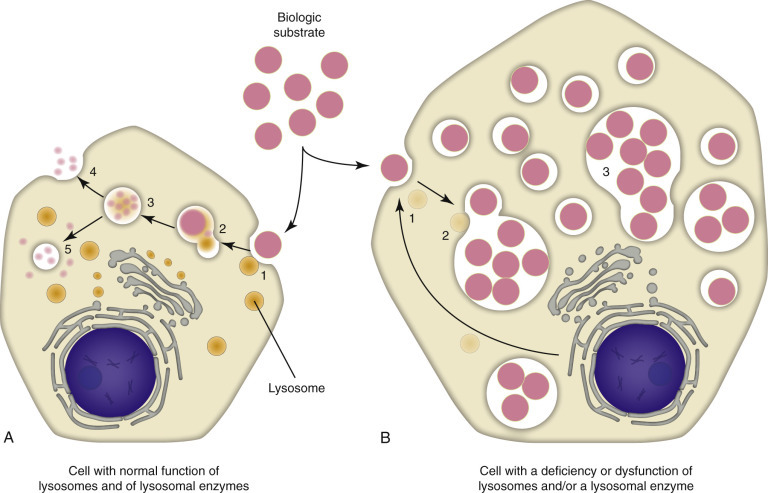
Mechanisms and Morphology of Lysosomal Storage Diseases.
A, Normal cell. A biologic substrate such as glycogen is taken into the cell via endocytosis (1), undergoes endosome-lysosome fusion (2), and the substrate is enzymatically-processed (3) by a series of lysosomal enzymes to one or more water-soluble end products that are released from the cell (4) or recycled within the cell (5). B, Defective cell. If the cell has acquired a deficiency (1) or dysfunction of lysosomes and/or lysosomal enzymes (2), the catabolism of the biologic substrate is defective and insoluble intermediates accumulate in the lysosomes (3). Note the accumulation of unprocessed substrate in the defective cell and its larger size when compared with the normal cell.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Peroxisomes (see Fig. 1-1) are membrane-bound vesicles that are specialized for the β-oxidation of fatty acids and degradation by catalase of the hydrogen peroxide produced. They may be distinguished from lysosomes by an electron-dense core. Peroxisomes can import large protein complexes; their function depends on communication with the Golgi complex, mitochondria, and the cytosol. Peroxisomes are generated de novo by budding from the ER but are also capable of replication through fission. Enzymes contained in peroxisomes are synthesized on free ribosomes in the cytosol, then transported into peroxisomes.
The Cytoskeleton: Microfilaments, Intermediate Filaments, and Microtubules
The cytoskeleton (Fig. 1-5) is a structural network that regulates the shape and movement of the cell and its organelles, cell division, and biochemical pathways. It consists of three integrated components: actin microfilaments (6 to 7 nm in diameter), intermediate filaments (approximately 10 nm in diameter) of different types depending on the cell type, and microtubules (approximately 25 nm in diameter). The function of most organelles requires their interaction with the cytoskeleton.
The following are general concepts: (1) microfilaments facilitate cell motility (e.g., ameboid movement [chemotaxis], cilia, pseudopodia); (2) intermediate filaments facilitate the physical strength and shape of cells and tissues, often via junctional complexes; and (3) microtubules move organelles and vesicles within the cytosol of a cell and chromosomes via mitotic spindles during cell division.
Cellular Inclusions
Cellular inclusions are composed of molecules, such as glycogen, proteins, nucleic acids, lipids, hemosiderin, and calcium, that accumulate as metabolic by-products, breakdown products of macromolecular complexes, or as a result of cell injury. Certain infectious microbes, especially viruses, can also produce intranuclear or cytoplasmic inclusions (see Figure 1-11, Figure 1-32, and 9-83). Cellular inclusions are “free” within the cytosol (i.e., not membrane bound).
Figure 1-11.
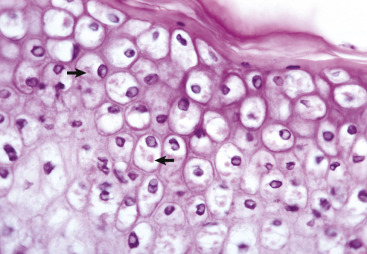
Ballooning Degeneration, Papular Stomatitis, Oral Mucosa, Ox.
Cells infected by certain poxviruses (e.g., papular stomatitis virus) cannot regulate their volume and undergo hydropic degeneration at certain stages of the infection. These cells may become so distended (ballooning degeneration) that they eventually rupture. Note cytoplasmic viral inclusion bodies (arrows). H&E stain.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Figure 1-32.
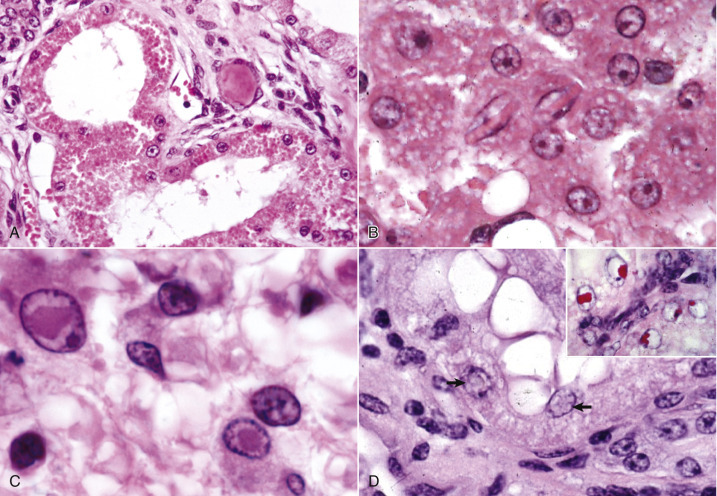
Cell Droplets and Inclusion Bodies.
A, Protein resorption droplets, kidney, dog. The cytoplasm of proximal tubular epithelial cells is filled with eosinophilic droplets—protein that has been resorbed by the cells from the glomerular filtrate. H&E stain. B, Crystalloids, hepatocytes, dog. Note the elongated eosinophilic crystalline inclusions in the nucleus of two hepatocytes. C, Viral inclusion bodies, canine distemper, brain, dog. Note the intranuclear eosinophilic inclusion bodies in astrocytes. H&E stain. D, Lead inclusion bodies, kidney, dog. The intranuclear inclusions (arrows) in renal tubular epithelial cells are difficult to see with an H&E stain. Inset, The lead inclusion bodies are acid-fast (red) and easily observed with Ziehl-Neelsen stain.
(A and C courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee. B courtesy Dr. D.D. Harrington, College of Veterinary Medicine, Purdue University; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia. D courtesy Dr. W. Crowell, College of Veterinary Medicine, The University of Georgia; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia. Inset courtesy Dr. W. Crowell, College of Veterinary Medicine, The University of Georgia; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia.)
Intercellular Junctions and the Extracellular Matrix
The cell connects and communicates with neighboring cells of the same type via intercellular junctions (Fig. 1-6 ). Certain cell types (e.g., basilar epithelial cells) also attach to a basal lamina and its contiguous connective tissue via hemidesmosomes, literally half a desmosome, in the ECM. These cell types interact with the ECM via integrin-mediated adhesions between ECM ligands, such as fibronectin or various collagens, and the cell's actin cytoskeleton. The ECM (see Chapter 3) is produced by fibroblasts and a variety of other supportive mesenchymal cells and includes such components as collagens and proteoglycans of basement membranes and the interstitium. Connections with neighboring cells and with the ECM are essential for normal cellular structure and function, including proliferation, migration, and signaling.
Figure 1-6.
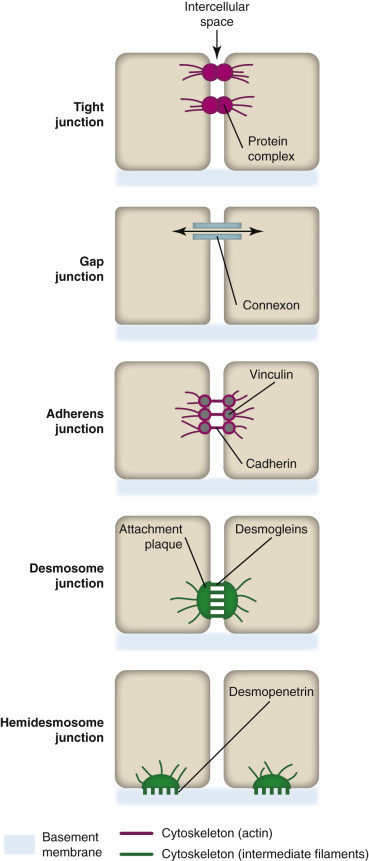
Intercellular Junctions and Hemidesmosomes.
A variety of intercellular junctions connect certain cell types (e.g., epithelial cells) to each other and facilitate intercellular communication. Some types of cell (e.g., basilar epithelial cells) are connected to a basement membrane by hemidesmosomes.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Causes of Cell Injury
Injury to tissues and organs begins at the cellular level. Rudolf Virchow (1821-1902), known as the father of cellular pathology, based his study of diseased cells on the observation of structural alterations (morphologic lesions). However, Virchow also realized that biochemical changes in the cell, which preceded the appearance of lesions, more completely explained the functional disturbances in diseased cells and, in some cases, were the only detectable changes. Thus the pathologist must always correlate lesions with their biochemical bases and remember that a cell can be damaged functionally (biochemically) yet have no apparent morphologic alterations.
Simplistically, cell injury disrupts cellular homeostasis. Cells are injured by numerous and diverse causes (etiologic agents) from intrinsic and extrinsic sources; however, all of these causes, and they number in the thousands, activate one or more of four final common biochemical mechanisms leading to cell injury (Essential Concept 1-1 ). These fundamental underlying biochemical mechanisms of cell injury are (1) ATP depletion, (2) permeabilization of cell membranes, (3) disruption of biochemical pathways, and (4) damage to DNA. These four mechanisms will be discussed in greater detail in later sections of this chapter.
Essential Concept 1-1. Mechanisms of Cell Injury.
The fundamental pathogenesis of cell injury is a perturbation of homeostasis. Cell injury is initiated at the molecular level, and, although the specific causes are diverse and numerous, the basic mechanisms can be categorized as follows:
-
1.
Adenosine triphosphate (ATP) depletion
-
2.
Permeabilization of cell membranes
-
3.
Disruption of biochemical pathways, especially those of protein synthesis
-
4.
DNA damage
Although certain injurious agents can cause ATP depletion, membrane damage, pathway disruption, or DNA damage in isolation, more often there is interplay among these basic mechanisms. Anything that decreases the supply of oxygen and other nutrients to the cell or that damages mitochondria directly halts oxidative phosphorylation, leading to rapid depletion of ATP, even in those cells that can switch to anaerobic glycolysis. The ATP depletion results in additional cell damage by causing failure of energy-dependent enzymes, in particular the cell membrane adenosinetriphosphatase ion pumps that control cell volume and electrolyte balance. Mitochondria are the major site of ATP generation and are also one of the most vulnerable organelles of the cell. Importantly, mitochondrial injury results not only in ATP depletion but also in increased permeability of mitochondrial membranes with resultant loss of calcium homeostasis and activation of enzymes, such as phospholipases, proteases, and endonucleases, hence inflicting damage on mitochondrial and other cell membranes, structural and enzymatic proteins, and nucleic acids.
Cells have a limited repertoire of responses to injury, depending on the cell type and the nature of the injury. These responses can be categorized as (1) adaptation, (2) degeneration, or (3) death. A cell may adapt to a stimulus or sublethal injury positively, with increased efficiency or productivity, or undergo degeneration with diminished functional capacity. The response to injury can be reversible, with eventual restoration (i.e., healing) of normal or near-normal cellular structure and function, or irreversible with progression from degeneration to death of the cell (Fig. 1-7 ). Irreparable DNA damage can result in permanent growth arrest (senescence), cell death, or malignant transformation. Not surprisingly, mitochondria, which are perhaps the organelles most susceptible to injury, are also thought to direct many of the processes of cellular adaptation, degeneration, and death through apoptosis or programmed necrosis (Fig. 1-8 ).
Figure 1-7.
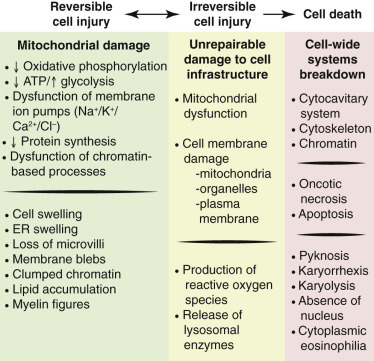
Postulated Sequence of Events in Reversible and Irreversible Ischemic Cell Injury.
Although reduced oxidative phosphorylation and adenosine triphosphate (ATP) concentration have a central role, ischemia can damage membranes directly. ER, Endoplasmic reticulum.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Figure 1-8.
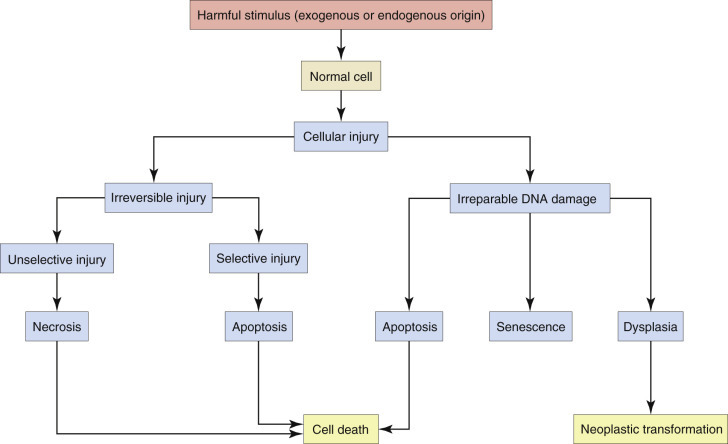
Stages in the Cellular Response to Irreversible Injury or Irreparable DNA Damage.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
The more common causes (etiologic agents) of cellular injury are grouped, discussed, and illustrated in the following sections.
Oxygen Deficiency
Hypoxia, a reduction in oxygen supply, is one of the most common and most important causes of injury; indeed, it is often the ultimate cause of cell injury. Hypoxia can result from inadequate oxygenation of blood as a result of cardiac or respiratory failure, reduction of vascular perfusion (ischemia), reduced O2 transport by erythrocytes (as in anemia or carbon monoxide [CO] toxicosis), or inhibition of respiratory enzymes of the cell (e.g., cyanide toxicosis).
Physical Agents
Physical agents of cell injury include mechanical trauma, temperature extremes, radiation, and electric shock. Trauma can damage cells directly (e.g., crushing or tearing), or indirectly by disruption of the blood supply to these cells and tissues. Low-intensity heat can damage blood vessels, accelerate certain cellular reactions, or halt those reactions with temperature-sensitive enzymes. Extreme heat denatures enzymes and other proteins. Cold causes vasoconstriction, limiting the blood supply to cells and tissues; extreme cold literally freezes cells with formation of ice crystals within the cytosol that disrupt cell membranes. Ionizing and ultraviolet radiation are the most important types of radiation causing cellular injury. Ionizing radiation, with its frequencies above the ultraviolet range, ionizes atoms or molecules, which then cause direct cell membrane or organelle damage or the production of free radicals that react with other cellular components, especially DNA. Ionizing radiation injury is a localized side effect of radiation therapy for cancer. Ultraviolet (frequencies just above that of visible light) radiation injury develops from exposure of sparsely haired and lightly pigmented skin (or other minimally pigmented tissues, such as the conjunctiva) to sunlight. Ultraviolet radiation can disrupt cellular bonds with the formation of reactive oxygen species (ROS). It also damages DNA, mainly through the formation of pyrimidine dimers. Electrical currents generate heat as they pass through tissues (e.g., skin, with high resistance), which can result in burns. Once the current enters the body, it is conducted through tissues of least resistance, especially the nervous system, where disruption of impulses in brainstem respiratory centers, the cardiac conduction system, or neuromuscular junctions results in indirect injury to cells and tissues.
Infectious Microbes
Infectious microbes (see also Chapter 4) differ from other injurious agents in that they can replicate once they gain access to cells or tissues. Infectious microbes range from protein molecules without nucleic acids (e.g., prions) through microbes (e.g., viruses and bacteria) to macroscopic parasites and injure cells in diverse ways. Viruses tend to subvert the host cell's DNA synthesis in the production of their own gene products; many bacteria produce toxins. Injury is exacerbated in many infectious diseases by the inflammatory (see Chapters 3 and 4) and immune (see Chapter 5) responses against the infectious microbe.
Nutritional Imbalances
Nutritional deficiencies, excesses, and imbalances all predispose the cell to injury. Animals can adapt to short-term dietary deficiencies in protein or calories through glycolysis, lipolysis, and catabolism of muscle protein; however, long-term starvation leads to atrophy of cells and tissues. In contrast, caloric excess can overload cells with glycogen and lipids and lead to obesity with metabolic disturbances that predispose the obese animal to a variety of diseases. Certain dietary deficiencies or imbalances of essential amino acids, fatty acids, vitamins, or minerals can lead to muscle wasting, decreased stature, increased susceptibility to infection, metabolic disturbances, and a host of other diseases, depending on which elements are missing from or disproportionate in the diet.
Genetic Derangement
Selective breeding of domestic animals for a particular conformational or dispositional phenotype has resulted in decreased genetic diversity in purebred animals and increased prevalence of inherited diseases (see subsequent section on the Genetic Basis of Disease and pertinent chapters in Section II, Pathology of Organ Systems), as well as a familial predilection for disease conditions with more complex inheritance, such as metabolic abnormalities, neoplasia, autoimmune diseases, and increased susceptibility to infection. Since the sequencing of the genomes of domestic animals, the genetic basis has been discovered for more and more of these phenotypes and associated familial diseases. For example, a single insulin-like growth factor-1 (IGF-1) haplotype is common to toy and miniature dog breeds, but generally absent in giant breeds; a fibroblast growth factor 4 (FGF4) retrogene is associated with chondrodysplastic conformation. Some conformational phenotypes are strongly linked to pathologic conditions (e.g., a missense mutation in bone morphogenetic protein 3 [BMP3] is linked to the extreme brachycephalic phenotype of Cavalier King Charles spaniels and Brussels griffons). Interestingly, bone morphogenetic protein genes also determine patterning in the developing brain and spinal cord, so the brachycephalic conformation in these breeds is associated with Chiari-like malformation of the cerebellum and syringomyelia of the cervical spinal cord.
Workload Imbalance
Cells can compensate for increased workload with an increase in size (hypertrophy [e.g., muscle]) or, if capable, in number (hyperplasia [e.g., adrenal cortex]). Cells that cannot meet an increased demand may undergo degeneration or death. Conversely, cells that are no longer necessary or that no longer receive the stimulus of physical exercise, innervation, hormones, or growth factors tend to shrink as in the disuse atrophy or denervation atrophy in skeletal muscles or the physiologic atrophy of the mammary gland after weaning of the offspring. Excessive cells, for example, neurons in the developing brain, are also removed by programmed cell death (apoptosis).
Chemicals, Drugs, and Toxins
Chemicals, including drugs and toxins, can alter cellular homeostasis. The therapeutic effect of pharmaceutical agents (drugs) is achieved by perturbing the homeostasis of selected populations of cells, ideally within tolerable limits. Chemicals are considered toxins if they alter homeostasis in a harmful way (outside of tolerable limits) with no beneficial pharmaceutical effect. Of course, many chemicals are beneficial or therapeutic at certain doses and harmful at higher doses. Chemicals affect cells by binding receptors, inhibiting or inducing enzymes or otherwise altering metabolic pathways, producing free radicals, increasing membrane permeability, or damaging chromosomes or structural components of the cell. The susceptibility of a cell to chemical injury depends on such factors as its mitotic rate and its ability to bind, take up, concentrate, or metabolize the chemical.
Immunologic Dysfunction
Immunologic dysfunction can result in cell injury either through a failure to respond effectively (immunodeficiency) to infectious microbes (see Chapter 4) or other harmful foreign antigens or through an excessive response (allergic or hypersensitivity reaction) to a foreign antigen or an inappropriate reaction to self-antigens (autoimmune disease). See Chapter 5 for more complete information on immunodeficiencies, hypersensitivity reactions, and autoimmune diseases.
Aging
Cells and tissues age because of accumulated damage to their proteins, lipids, and nucleic acids. Much of the damage of aging is attributed to ROS, DNA mutations, and cellular senescence (see the subsequent section on Cellular Aging). Cumulative damage to DNA predisposes aged animals to the development of neoplasia. In cells that can replicate, the telomeres at the ends of chromosomes are shortened with each successive division, eventually causing the cell to stop dividing. Not surprisingly, many cancer cells have active telomerase to maintain the length of their telomeres. In cells with little regenerative capacity, such as neurons, accumulation of lipofuscin and other metabolic products contributes to their degeneration and loss, leading to cerebrocortical atrophy in the aging brain. However, many of the common “aging lesions” in geriatric animals (e.g., nodular hyperplasia in the liver [see Fig. 8-65], pancreas [see Fig. 8-91], or spleen [see Fig. 13-90; see E-Figs. 13-9, 13-14, and 13-15E-Figs. 13-9E-Figs. 13-14E-Figs. 13-15] of dogs; cholesterol granulomas in the choroid plexus of horses [see Fig. 14-87]; siderofibrotic plaques in the canine spleen [see Figs. 13-71 and 13-72; see E-Figs. 13-9 and 13-10]; even thyroid C-cell adenomas in horses [see Fig. 12-30]) are generally disregarded as incidental findings (i.e., not the cause of death) at autopsy.
Reversible Cell Injury
The initial response of the cell to perturbation of homeostasis is acute cell swelling. If the injury is not too severe or too prolonged, the cell can recover and return to normal structure and function. Therefore acute cell swelling is, up to a point, a reversible change (Essential Concept 1-2 ).
Essential Concept 1-2. Reversible Cell Injury.
Cell injury is classified as reversible if the injured cell can regain homeostasis and return to a morphologically (and functionally) normal state. Acute cell swelling is the classic morphologic change in reversible injury; however, it is also the typical early change of irreversible cell injury. Irrespective of the nature of the initial injury, hypoxia is often the ultimate cause of acute cell swelling because it results in adenosine triphosphate depletion. The hypoxic cell then swells because of loss of volume control when membrane adenosine triphosphatase ionic pumps fail. Acute cell swelling is also a response to direct cell membrane damage from lipid peroxidation (by reactive oxygen species), binding of certain toxins, damage to ion channels, or insertion of transmembrane pore-forming complexes. Because acute cell swelling is a common early response to both reversible and irreversible injury, it is well to think of this morphologic change as a marker of potentially reversible cell injury. Cells, depending on their reparative or regenerative capacities, may recover from potentially irreversible cell injury; however, if the injury is severe or sustained, acute cell swelling becomes the initial step in the process of cell death. If the injury is not so severe as to be lethal, then the cell may not succumb but (again depending on the nature of the injury and of the cell) is unlikely to recover completely or to return to its “normal” structural and functional state.
Acute Cell Swelling
Cell swelling, a fundamental and common expression of cell injury (Fig. 1-9 ), is also known as hydropic degeneration because it is the influx of water along with sodium ions when the sodium-potassium ion pumps fail that causes the swelling. If not stopped, acute cell swelling will cause lysis and death of the cell. The term hydropic degeneration is commonly used when the change occurs in certain types of cells, such as hepatocytes or renal tubular epithelial cells. In other tissues (e.g., keratinocytes in the epidermis), cell swelling from influx of water is called ballooning degeneration. In the central nervous system (CNS), cell swelling of glial cells, especially prominent in astrocytes, is termed cytotoxic edema. In any tissue, acute cell swelling is a degenerative change in which the cellular enlargement is the result of increased water volume. Acute cell swelling therefore is quite different from hypertrophy, in which the enlargement of cells is caused by an adaptive increase in number and/or size of organelles.
Figure 1-9.
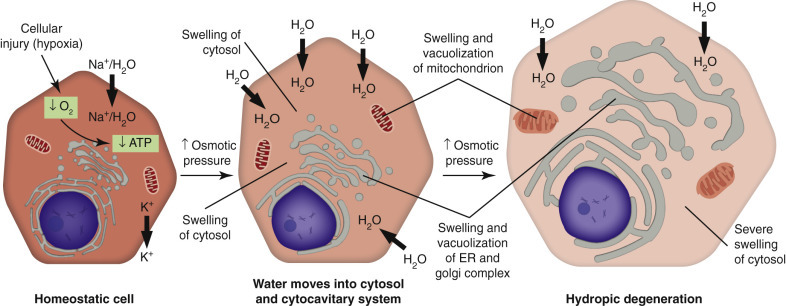
The Process of Acute Cell Swelling (Hydropic Degeneration).
ATP, Adenosine triphosphate; ER, endoplasmic reticulum.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Mechanisms of Acute Cell Swelling
In normal cells, sodium-potassium adenosine triphosphatases (Na+/K+-ATPases) function as ionic pumps, specifically, as active transporters of cations across cell membranes (see Fig. 1-9). For each ATP molecule hydrolyzed, the ionic pump exports (i.e., outside the cell) three Na+ ions and imports (i.e., inside the cell) two K+ ions. The resultant electrochemical gradient generates energy that is especially important in establishing and maintaining the membrane potential of neurons and of cardiac and skeletal muscle cells and pH homeostasis within the cytosol of the cell. Because water diffuses passively along the osmotic gradient, the ATPase pump also controls cell volume. The best-studied models of acute cell swelling are (1) hypoxia-induced failure of ATP synthesis (and hence the ATPase pumps) and (2) carbon tetrachloride (CCl4)–induced membrane damage. Notably, the cardiac glycosides, digitalis and ouabain, specifically inhibit Na+/K+-ATPase pumps.
Acute Cell Swelling Resulting from Hypoxic Injury
Hypoxia is the end result of decreased oxygen concentration at any point in its passage from air into the respiratory tract through hemoglobin uptake and transport by the vasculature to cells, where it drives mitochondrial oxidative phosphorylation. Ischemia is a local decrease in blood supply to tissue with resultant decreased delivery of oxygen (hypoxia), glucose, and other nutrients to the cell, as well as decreased removal of metabolic wastes. Because any injury to the respiratory or cardiovascular system can lead to hypoxia, it is commonly the ultimate cause of acute cell swelling. When cellular oxygen is depleted, oxidative phosphorylation stops, and the cell must switch to anaerobic metabolism (i.e., glycolysis) or die. As production of ATP declines, the resultant drop stimulates hexokinases, phosphofructokinase 1 (PFK1), and other enzymes of glycolysis. PFK1 catalyzes the phosphorylation of fructose 6-phosphate to fructose 1,6-bisphosphate, another integral step in glycolysis. The end products of glycolysis are ATP and pyruvate and heat. This anaerobic generation of ATP (though less efficient than oxidative phosphorylation) contributes to short-term survival of the cell. In addition, pyruvate produced by glycolysis can enter the tricarboxylic acid (TCA) cycle. However, certain specialized cells (e.g., neurons) cannot generate ATP anaerobically and therefore need a continuous supply of oxygen and glucose. This dependency makes neurons one of the cells that are most susceptible to a deficiency or lack of oxygen.
The early events in acute cell swelling (see Figs. 1-7 and 1-9) caused by hypoxia or ischemia are potentially reversible if the injury is mild or of short duration. With the depletion of cellular oxygen, oxidative phosphorylation stops. The resultant deficiency of ATP causes failure of the Na+/K+-ATPase pumps with influx of Na+, Ca2+, and water into the cytosol, and loss of K+ and Mg2+ from the cytosol. The electrolyte imbalance and influx of water expand the cytosol and swell mitochondria and the cytocavitary network. Ultrastructurally, chromatin is clumped, the cytosol is electron lucent, ribosomes detach from rER, and the ER becomes vesiculated. Damaged membranes coil into whorls (also known as “myelin figures”). Cytoskeletal damage causes the plasma membrane to lose microvilli or other specialized structures and to undergo blebbing (the formation of multiple irregular bulges). With light microscopy the acutely swollen cell has an expanded and rounded profile with pale eosinophilic or vacuolated cytoplasm. The cytoplasmic pallor and vacuolation is the result of dispersion of organelles and dilution of cytosolic proteins by the influx of water. The ATP deficiency also prompts a switch to anaerobic metabolism with production of ATP (and pyruvate) through glycolysis. Glycolysis depletes cellular glycogen, leads to an accumulation of lactate with decreased intracellular pH, and produces heat, which if excessive may also injure the cell.
Acute Cell Swelling Resulting from Specific Types of Cell Membrane Injury
Cell membranes can also be selectively injured by chemical modification of their phospholipids by free radicals (i.e., lipid peroxidation), by covalent binding of toxins to macromolecules, by interference with ion channels, and by insertion of transmembrane complexes. CCl4 is an example of cell membrane injury caused by chemical modifications (see the following section). Cell membranes can also be injured directly by defensive molecules of the immune system and by bacterial cytotoxins (see later).
Carbon Tetrachloride and Cell Membrane Injury
As an example, CCl4 injures hepatocytes in diverse and complicated ways that have not been fully elucidated, but CCl4 toxicosis provides a useful model to study cell membrane injury. CCl4 is activated by cytochrome P450 (CYP; mainly CYP2) mixed-function oxidases in hepatocellular sER to form the trichloromethyl radical (CCl3 •), which binds cellular macromolecules and disrupts biochemical processes, particularly lipid metabolism. CCl3 • also interacts with oxygen to form the highly reactive trichloromethylperoxy radical (CCl3OO•), which initiates a lipid peroxidation chain reaction, especially in the phospholipids of cell membranes (not only the plasma membrane, but also those of the ER, mitochondria, and Golgi complex [cytocavitary network]). Reactive metabolites of CCl4 also cause hypomethylation of rRNA and the release of growth factors and cytokines such as tumor necrosis factor-α (TNF-α), NO, transforming growth factor-α (TGF-α), transforming growth factor-β (TGF-β), interleukin 1 (IL-1), interleukin 6 (IL-6), and interleukin 10 (IL-10) from Kupffer cells in hepatic sinusoids. Hepatic stellate cells also respond to growth factors and cytokines by ceasing their lipid storage functions, assuming a myofibroblastic phenotype, and producing type I collagen, thus following hepatic injury with hepatic fibrosis.
Notably, CCl4 toxicosis develops along two major pathways; both begin with free radical formation, and both injure the hepatocyte in what is initially a reversible process. The first process, haloalkylation (covalent binding of CCl3 • to macromolecules), disrupts biochemical processes, especially triglyceride secretion into plasma, oxidative phosphorylation, and calcium transport, before any structural damage is evident. The other pathway, lipid peroxidation induced mainly by CCl3OO•, is a secondary (indirect) chain reaction that takes time to develop. It is mainly responsible for the cell membrane injury of CCl4 toxicosis, but the effects of both pathways are thought to act in combination to cause cell death. Depending on the dose and duration of exposure and other factors, CCl4 toxicosis causes a variety of lesions via membrane injury, including steatosis (fatty liver), apoptosis and regeneration of hepatocytes, and hepatic necrosis with fibrosis. Hepatocellular membrane injury typically commences in centrilobular hepatocytes (see Fig. 8-15), where mixed-function oxidase activity in the sER is highest, but with higher doses of CCl4 occurs throughout the lobule, leading to massive hepatic necrosis.
Molecules of the Immune System and Cell Membrane Injury.
Cell membranes can also be injured directly by the membrane attack complex (MAC) of the complement pathway, by bacterial cytolysins, and by molecules from natural killer (NK) cells (see Chapters 3, 4, and 5Chapter 3Chapter 4Chapter 5). The MAC, bacterial cytolysins, and NK cells exert their effect in part by forming a pore or channel that disrupts the lipid bilayers of the plasma membrane. The MAC is assembled from terminal components of the complement pathway, which are abundant in blood. Assembly of the MAC begins with enzymatic cleavage of complement fragment 5b (C5b) from complement component 5 (C5). Complement component 6 (C6) binds a labile site on C5b to produce a stable intermediate. Subsequent binding of complement component 7 (C7) renders the MAC precursor lipophilic. With binding of the α, β, and γ subunits of complement component 8 (C8), the MAC precursor penetrates a nearby cell membrane lipid bilayer. Binding and oligomerization of complement component 9 (C9) then completes formation of the MAC, which creates a lytic pore that is part of the innate immune response to bacteria. Cluster of differentiation 59 (CD59), a glycoprotein receptor on the surface of leukocytes, epithelial cells, and endothelial cells (and overexpressed on some cancer cells), blocks penetration of cell membranes by the C5b-8 precursor and blocks incorporation of C9 into the MAC, thereby protecting host cells against cell membrane injury.
Morphologic Changes: Their Detection and Evaluation
Although the modern pathologist relies on many nonmorphologic techniques (e.g., isolation and identification of infectious agents, quantification of chemical deficiencies or excesses, detection of genetic abnormalities) to determine the cause of disease, pathology is based on morphologic changes, specifically on the gross and microscopic structural abnormalities (lesions) that develop in diseased cells, tissues, and organs. The pathologist discovers a lesion because of its macroscopic or histologic differences from normal cells or tissue, and either recognizes that lesion or, for unfamiliar lesions, categorizes it as a degenerative change, vascular disturbance, inflammation, or disturbance of growth. Gross diagnosis, based on recognition of lesions at physical examination, surgery, or autopsy (syn: necropsy), is definitive for some conditions (e.g., bone fractures and other traumatic injuries). The skilled and experienced pathologist can even diagnose certain infectious, nutritional, or neoplastic diseases with accuracy at the macroscopic level. However, most gross examinations are followed by histologic examination for the final morphologic diagnosis.
For histologic examination, selected tissues are routinely fixed in a 10% phosphate buffered formalin. These fixatives include (1) 10% buffered neutral formalin (10% BNF), which in practice has a pH of 6.8 and is not iso-osmolar, or (2) Carson’s fixative, which is iso-osmolar and has a pH of 7.2. Carson’s fixative is also suitable as an emergency fixative for electron microscopy. In general, representative specimens from most organ systems should be fixed in formalin at autopsy, even if histologic examination is anticipated for only a few tissues or organs. Because formalin penetrates tissue from the cut surface at approximately 1 mm/day, most parenchymal organs are sliced at 4 to 5 mm thick before immersion in the fixative. Exceptions are the brain and spinal cord, which are often fixed intact (after opening or removing the dura mater) or in much thicker slices because these tissues are easily damaged by slicing. Hollow organs, such as intestine, can be also be fixed in longer segments, provided that the lumen is flushed with the fixative.
After an appropriate interval for fixation, depending on the nature of the tissue and thickness of the slice among other factors, the fixed specimen is trimmed into tissue cassettes for histologic processing. Routine histologic processing, which is automated in most laboratories, entails dehydration through increasing concentrations of ethanol, followed by xylene, which is miscible with the paraffin wax in which the tissues will be embedded. Tissues are often processed as early as 24 hours after immersion in formalin to expedite diagnosis, though ideal fixation periods tend to vary between 2 and 7 days. Tissues should seldom be held in formalin longer than a couple of weeks because the cross-linking of aldehydes—formalin is a formaldehyde solution—can interfere with histochemical or immunohistochemical techniques. However, once the tissue is embedded, the paraffin blocks can be stored for decades with little or no apparent effect on immunohistochemistry.
For routine histologic examination, paraffin sections are cut at approximately 5 µm thickness and stained with H&E to achieve differential staining of nuclei, cytoplasm, and extracellular structures. Simplistically, hematoxylin stains nuclei and certain cytoplasmic structures, such as ribosomes, that contain nucleic acids, whereas eosin stains proteins, both cytoplasmic and extracellular (e.g., collagen). Other histochemical techniques are used to identify substances (e.g., mucins, glycogen, amyloid) that are either not differentially stained by H&E or are stained only weakly. In immunohistochemistry, an immune reaction (binding of antibody to an antigen) is combined with histochemistry for more specific identification of microbes in the case of infectious disease or of cellular antigens in neoplastic or noninfectious diseases. Immunohistochemistry has the added value (over many immunologic or molecular techniques) of colocalizing the antigen of interest with a particular cell type or even with a particular cellular compartment. However, molecular techniques, such as polymerase chain reaction (PCR), can be adapted to histologic sections with in situ hybridization.
Morphology of Acute Cell Swelling
Gross Appearance.
Acute cell swelling increases the volume and weight of parenchymal organs and imparts pallor to them. It is important to distinguish hydropic degeneration from more positive adaptations, such as hypertrophy or hyperplasia, which, if extensive, also increase the size of an organ. Liver and kidney (especially the renal cortex) are two organs in which the lesions of acute cell swelling can be striking (see Chapters 8 and 11). An affected liver weighs more than normal, appears pale and swollen with rounded edges, and has an accentuated lobular pattern (Fig. 1-10, A ). In the CNS the cell swelling of cytotoxic edema has little effect on the color of neuroparenchyma but does increase the weight and volume of the affected tissue. Even a slight increase in volume of the brain has catastrophic consequences because there is little space in the cranium to accommodate swelling (see Chapter 14).
Figure 1-10.
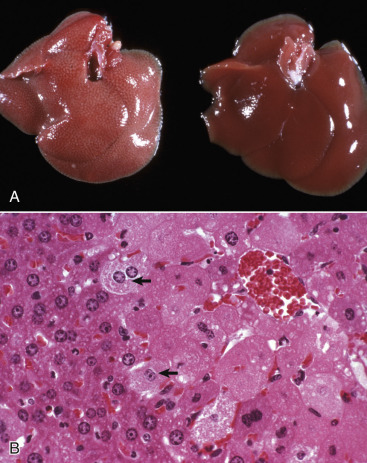
Acute Cell Swelling, Liver, Mouse.
A, Hepatic swelling in a mouse exposed to chloroform 24 hours previously. The accentuated lobular pattern and slight pallor in the liver on the left are the result of acute cell swelling (hydropic degeneration) and necrosis of centrilobular hepatocytes. The right liver is normal. B, Liver from a mouse with chloroform toxicosis. Although many hepatocytes in the centrilobular areas (at right) are necrotic, several cells at the interface of normal and necrotic (arrows) are still undergoing acute cell swelling (hydropic degeneration). H&E stain.
(Courtesy Dr. L.H. Arp.)
Microscopic Appearance.
The influx of water in hydropic degeneration dilutes the cytosol, separates its organelles, and distends the cell, giving affected cells a swollen, pale, and finely vacuolated appearance. In renal proximal tubules, swollen epithelial cells impinge on the tubular lumen. In the liver, swollen hepatocytes and endothelial cells compress hepatic sinusoids.
Hydropic degeneration and cloudy swelling are terms for the microscopic appearance of acute cell swelling (see Fig. 1-10, B). In addition to endothelial cells, hepatocytes, and renal tubular epithelial cells, other epithelial cells, neurons, and glial cells are particularly prone to acute cell swelling. The clear cytoplasmic vacuoles in affected cells are mainly water-distended mitochondria or cisternae of the Golgi complex or ER; therefore these vacuoles are not labeled by histochemical techniques to detect fat or glycogen (two other causes of cytoplasmic vacuolation). Ballooning degeneration is an extreme variant of hydropic degeneration that is typically seen in keratinocytes of stratified squamous epithelium of the skin. Poxviruses are a classic cause of ballooning degeneration of keratinocytes of epidermal or mucosal (e.g., esophagus) stratified squamous epithelium (Fig. 1-11 ).
Ultrastructural Appearance.
Ultrastructurally, the acutely swollen epithelial cell loses plasma membrane structures, such as cilia and microvilli, and develops cytoplasmic “blebs” at apical cell surfaces. The cytosol is electron lucent, mitochondria are swollen, and cisternae of the ER and Golgi complex are dilated. The cytocavitary network fragments into vesicles. Proteins and Ca2+ precipitate in the cytosol and in organelles, especially mitochondria. Acute cell swelling in the CNS has other distinctive features (see Chapter 14).
Significance and Fate of Acute Cell Swelling
If the injury is brief and mild, many cells can recover and regain normal or near-normal structure and function. Recovered cells can phagocytize their own damaged organelles (autophagy); these autophagosomes may ultimately appear as lipofuscin granules, indicative of previous injury. However, even with reversible injury, impaired regulation of water and electrolyte balance across cell membranes is generally accompanied by disruption of other cellular processes. The ultimate effect on the animal depends on the number of cells affected, reparative and regenerative abilities of the cell, and the importance of the disrupted biochemical processes, such as ATP synthesis. With severe, lengthy, or repetitive injury, acute cell swelling can progress beyond the “point of no return” and become an early stage in the process of cell death. In summary, the acute cell swelling of hydropic degeneration reflects potentially reversible, sublethal cell injury. However, unless the injury to essential cells in vital organs (e.g., brain, heart, lung, liver, or kidney) is stopped quickly, it can progress to cell and tissue death, loss of essential physiologic functions, and possibly death of the animal (Fig. 1-12 ).
Figure 1-12.
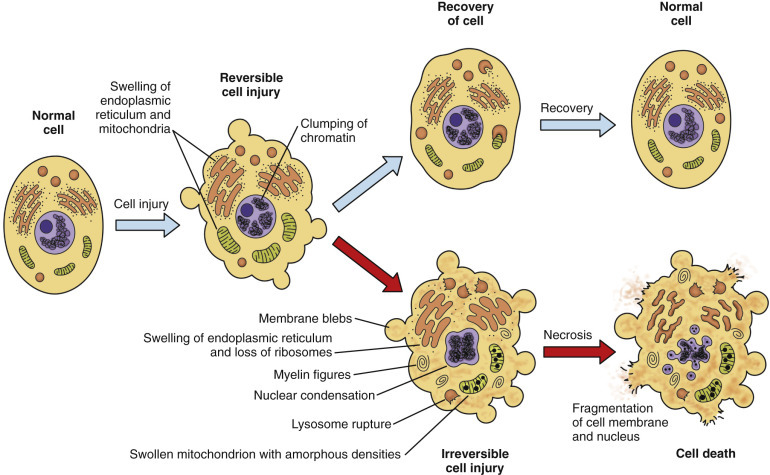
Normal Cell and the Changes in Reversible and Irreversible Cell Injury.
Reversible injury is characterized by generalized swelling of the cell, its organelles (especially mitochondria), and the cytocavitary network. Other changes include blebbing of the plasma membrane, detachment of ribosomes from ER, and clumping of nuclear chromatin. Irreversible injury is characterized by increased cell swelling, disruption of lysosomes, formation of amorphous densities in mitochondria, membrane disruption in the cytocavitary network, and severe nuclear changes. Irreversible nuclear changes include pyknosis (severe condensation of chromatin), followed by karyorrhexis (nuclear fragmentation) and karyolysis (nuclear dissolution). Laminated structures (myelin figures) derived from injured cell membranes can appear during reversible injury, but become more pronounced in irreversibly injured cells.
Irreversible Cell Injury and Cell Death
Major mechanisms of acute cell swelling, as discussed and illustrated earlier, are (1) hypoxia, (including ischemia) and (2) membrane injury caused by lipid peroxidation or the formation of lytic pores through insertion of a MAC via the complement pathway or by bacterial cytolysins. The cellular response to injury depends on (1) the type of cell injured and its susceptibility and/or resistance to hypoxia and direct membrane injury and (2) the nature, severity, and duration of the injury. As examples, neurons, cardiac myocytes, endothelium, and epithelium of the proximal tubule of the kidney are cells that are extremely susceptible to hypoxia, whereas fibroblasts, adipocytes, and other mesenchymal structural cells are less susceptible.
The response to injury can be degenerative, adaptive, or completely reversible with restoration of normal structure and function for the affected cell; however, with more severe or persistent injury, acute cell swelling can progress to irreversible cell injury and cell death. The cellular alterations that differentiate reversible cell injury from irreversible cell injury have been and are being studied extensively.
Cell Death
The death of cells is an essential “value-added” part of embryonic development and maturation of the fetus and of homeostasis within populations of adult somatic cells. In these physiologic examples of cell death, cells that are no longer needed are removed during development or remodeling of tissues. However, cell death is also a point-of-no-return response to severe injury, and it is this pathologic form of cell death that is the topic of this section. Cell death typically assumes one of two morphologic forms (Fig. 1-13 ): necrosis or apoptosis. The term necrosis has evolved to mean death by swelling of the cell (oncosis) with eventual rupture of cell membranes. Necrotic cell death typically involves groups or zones of cells and elicits an inflammatory reaction because of the release of cell contents into the ECM. Apoptosis, in contrast, is directed by cellular signaling cascades and typically affects individual cells. Apoptosis is a process of condensation and shrinkage of the cell and its organelles with eventual fragmentation of the cell. Importantly, apoptotic cell fragments remain membrane bound; thus no cellular components that could induce inflammation are released. Autophagy is a third possible mechanism of cell death, but it is more commonly a means of cell survival. (See subsequent section on Autophagy under Chronic Cell Injury and Cell Adaptation.)
Figure 1-13.
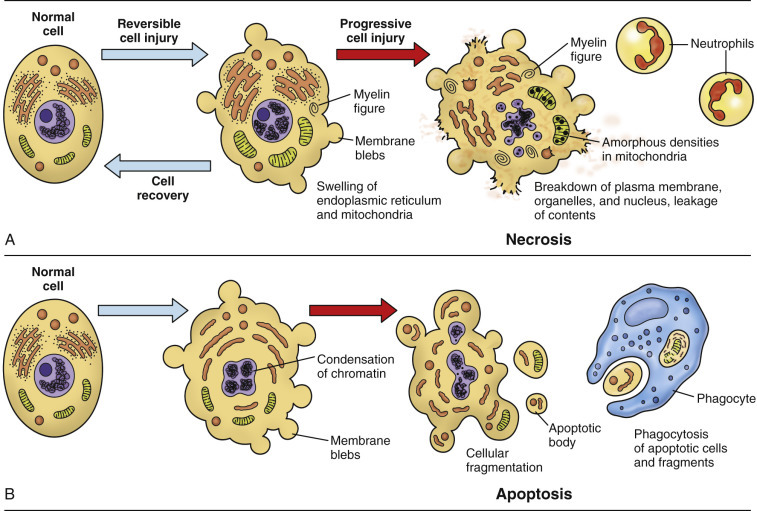
The Sequential Ultrastructural Changes of Necrosis and Apoptosis.
A, In necrosis, leakage of cell contents through the ruptured plasma membrane into the extracellular matrix elicits inflammation. B, In apoptosis, cellular fragments are extruded as plasma membrane-bound apoptotic bodies that are recognized by phagocytes but do not cause inflammation.
Whereas apoptosis has long been recognized as a regulated or programmed process, not only responsible for physiologic removal of surplus cells but also occurring as a reaction to certain injuries, necrosis was once considered an entirely accidental and random response to injury. However, with the discovery that inhibition of apoptosis could shift cells from apoptotic death to a regulated process of oncotic death, the idea arose that necrosis could, at least in certain situations, be regulated by cellular signaling pathways.
Cell Death by Oncosis (Oncotic Necrosis)
Oncotic cell death results from irreversible cell injury that, for example, is caused by hypoxia, ischemia, or direct damage to cell membranes (Essential Concept 1-3 ). Ischemia causes particularly extensive cell injury because the decreased perfusion results in not only an oxygen deficit (hypoxia) but also a deficiency of glucose and other nutrients, plus an accumulation of toxic metabolic by-products. Cell swelling, resulting from loss of volume control (see later), is the fundamental mechanism of oncotic necrosis and distinguishes it from apoptosis. Just as in reversible acute cell swelling, the initial O2 deficit in irreversible acute cell swelling causes an uncoupling of oxidative phosphorylation and a switch to anaerobic glycolysis with accumulation of lactic acid and a resulting decrease in pH of the cytosol. The Na+/H+ exchanger exports the excess H+ in exchange for Na+. However, because glycolysis is less efficient in ATP production than oxidative phosphorylation, the decreased ATP concentration leads to failure of ionic ATPase pumps and a loss of volume control (i.e., failure of Na+/K+-ATPase pumps with influx of Na+, Ca2+, and water). In addition, the normal function of enzymes, contractile proteins, membrane pumps, and other protein-based mechanisms in the cell occurs in a very narrow pH range around 7.0. With glycolysis the cytosol becomes acidic, thus limiting or blocking these mechanisms and exacerbating cellular dysfunction.
Essential Concept 1-3. Cell Death.
Severe or persistent injury can overwhelm the cell's capacity to restore homeostasis, in which case potentially reversible acute cell swelling can become irreversible and progress to cell death. The morphologic features of cell death change with the passage of time and depend on the manner of death (oncotic necrosis versus apoptosis) and the type of cell or tissue. Oncotic necrosis is a process of cell swelling and thereby distinct from cell death by apoptosis, which is a process of cellular shrinkage and fragmentation. If an acutely swollen cell fails to correct the electrolyte imbalance and loss of volume control, then potentially reversible cell injury can become the initial stage of oncotic necrosis. Once thought always to be unregulated, oncotic necrosis, like apoptosis, can be a programmed process (necroptosis). Programmed cell death, whether by necroptosis or apoptosis, has many extrinsic and intrinsic (acting mainly through mitochondria) triggers. Programmed cell death is a complex and varied process that includes stages of initiation, propagation, and execution. Cells that die by oncotic necrosis tend to do so in groups, whereas apoptosis commonly affects individual cells. Furthermore, oncotic necrosis results in rupture of cell membranes and release of cytoplasmic content into the extracellular matrix with ensuing inflammation. In contrast, the cell that dies by apoptosis shrinks and fragments, but the fragments remain membrane bound and therefore do not elicit an inflammatory response although they are marked for phagocytosis.
Disruption of the intracellular calcium ion balance (Fig. 1-14 ) is integral to the transition from potentially reversible acute cell swelling to irreversible injury and cell death. The intracellular concentration of calcium is generally one-fourth that of extracellular calcium. In a normally functioning cell, calcium is sequestered into three major compartments: the cytosol (low concentration), ER (midrange concentration), and mitochondria (high concentration). Each compartment has its own ATPase membrane pumps. Ischemia opens plasma membrane calcium channels, leading to increased intracellular calcium concentration in the cytosol, which activates protein kinase C, endonucleases, phospholipases, and various proteases, including calpains. Calpains abolish protein kinase C activity and cleave Na+/Ca2+ exchangers in mitochondrial and plasma membranes, leading to decreased calcium efflux and reuptake by the ER with ensuing calcium overload in the cytosol and, even worse, in the mitochondria. Although the timing of the point of no return remains elusive, if the cell fails to restore mitochondrial function, acute cell swelling becomes irreversible, leading to cell death.
Figure 1-14.
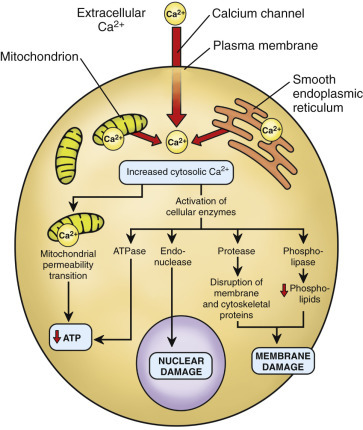
Sources and Consequences of Increased Cytosolic Calcium in Cell Injury.
ATP, Adenosine triphosphate; ATPase, adenosinetriphosphatase.
Paradoxically, restoration of blood flow and oxygen supply can exacerbate ischemic cell injury. This phenomenon is called ischemia-reperfusion injury, and it can continue for several days after reperfusion. It is attributed to “oxidative stress,” which involves the formation of ROS, calcium imbalance, opening of the mitochondrial permeability transition (MPT) pore, endothelial damage, thrombogenesis, and arrival of leukocytes in the damaged tissue. Reperfusion injury correlates with the duration of ischemia, but the susceptibility of organs (brain > heart > kidney > intestine > skeletal muscle) varies. The brain is exquisitely sensitive to ischemia because of its high metabolic activity, absolute requirement for glucose, high concentration of polyunsaturated fatty acids, and release of excitatory neurotransmitters. A less susceptible tissue (e.g., adipose tissue, fibrous tissue) can, to an extent, undergo atrophy or enter a quiescent state in response to decreased perfusion, using autophagy and apoptosis as means to remove effete organelles or dead cells, respectively.
Once considered an unregulated process, necrosis can, at least in some circumstances, be regulated by signaling pathways. In fact, regulated necrosis may be the predominant form of oncotic cell death. A regulated process of necrotic cell death begins with a trigger (e.g., binding of TNF or Fas ligand [FasL] to a death receptor [DR; i.e., transmembrane protein of the plasma membrane]), followed sequentially by initiation, propagation, and execution. A cell can respond to binding of TNF to its receptor in at least three different ways: (1) survival through activation of nuclear factor κB (NFκB), (2) apoptosis, or (3) necrosis. Apoptosis is directed by caspases. Interestingly, it was the discovery that inhibition of caspases, rather than protecting the cell from death, could redirect it from apoptosis to necrotic cell death. The myriad triggers of regulated necrosis include TNF, FasL, DNA damage, cluster of differentiation 3 (CD3) via the T lymphocyte receptor, lipopolysaccharide via Toll-like receptors, and interferon γ. The term necroptosis refers to the regulated necrotic cell death that begins with TNF receptor activation by TNF and is initiated by receptor-interacting protein-serine/threonine kinase (RIPK) 1. The ubiquitination status of RIPK1 determines whether it directs the cell toward survival, apoptosis, or necroptosis. Inhibition of caspase-8, in particular, is important in redirecting the cell from apoptosis toward necroptosis with assembly of the so-called necrosome, composed of RIPK1, RIPK3, and mixed lineage kinase domain-like (MLKL). Though much remains to be learned about the necroptosis pathway, MLKL has been proposed as the main mediator downstream of RIPK3.
Another pathway of regulated necrosis is initiated by opening of the MPT pore, which entails an increase in permeability of inner and outer mitochondrial membranes and leads to mitochondrial swelling, production of ROS, and oxidized nicotinamide adenine dinucleotide (NAD+) depletion. Mitochondrial production of ROS, mainly through reduced forms of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, is considered requisite to TNF-α–induced necrosis. ROS, along with Ca2+ dysregulation and depletion of NAD+ and ATP, propagates the signal in regulated necrosis. Finally, the execution phase, with its catastrophic ATP depletion, cell swelling, lipid peroxidation, and lysosomal membrane permeability with release of cathepsins, leads to irreversible cell injury and death.
Cell Membrane Injury Leading to Cell Death.
The failure to restore mitochondrial function and repair cell membrane damage is a critical component of irreversible cell injury. In particular, uncoupled oxidative phosphorylation and impaired mitochondrial calcium sequestration significantly increase the risk for cell death. Injured cell membranes have increased permeability, so when membrane ATPase ion pumps fail, extracellular calcium enters the cell. The calcium imbalance exacerbates the damage to mitochondria and to the cytoskeleton and activates endonucleases, proteases, and phospholipases. Phospholipase A catalytically hydrolyzes the phospholipids of the cell membranes, further exacerbating cell and mitochondrial membrane damage and the progression to irreversible cell injury.
Free Radical Injury.
Free radicals contribute to mitochondrial injury and to cell death by oncotic necrosis, especially when ischemia is followed by reperfusion (see earlier section that discusses ischemia-reperfusion injury). Free radicals damage cell lipids (especially the phospholipids of cell membranes), proteins, and nucleic acids (Fig. 1-15 ). A free radical is any molecule with an unpaired electron. Free radicals include ROS (e.g., the superoxide radical [O2 .]) and reactive nitrogen species (e.g., NO). Such molecules are highly reactive, short-lived products of oxidative metabolism and occur in membranes of mitochondria and other organelles. NADPH oxidase, an enzyme complex found in membranes of a variety of cell types, especially phagocytes, such as neutrophils and macrophages, functions in the production of ROS.
Figure 1-15.
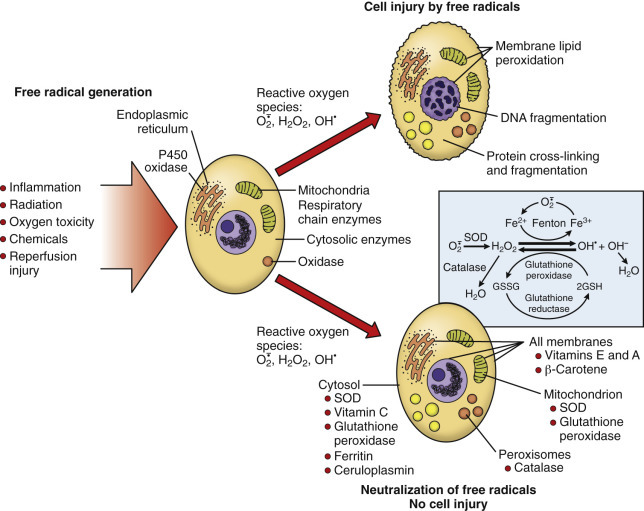
The Role of Reactive Oxygen Species in Cell Injury.
GSH, Reduced glutathione; GSSG, oxidized glutathione; SOD, superoxide dismutase.
Endogenous free radicals, such as reactive oxygen or nitrogen species, serve physiologic functions in cell signaling and in defense against microbes but also can harm cells, especially in the setting of ischemia/reperfusion injury. Free radicals, with their unpaired electron, are prone to extract a H+ from the polyunsaturated fatty acids in cell membranes. The fatty acid that loses a H+ becomes, itself, a free radical that can then be oxidized to an even more reactive radical that will extract a H+ from the neighboring fatty acid, propagating a chain reaction that leads to membrane disintegration. Antioxidants, such as superoxide dismutase (SOD), catalase, glutathione peroxidase, and vitamins A, C, and E, are protective because they scavenge free radicals and can break the chain reaction of lipid peroxidation.
Morphologic Appearance of Necrotic Cells and Tissues (Oncotic Necrosis).
The appearance of necrotic cells depends on the type of necrosis (see the next section), the tissue involved, the cause of cell death, and the time elapsed. In this chapter, necrosis (or necrotic) generally implies oncotic cell death.
Gross Appearance of Necrotic Tissue.
Soon after death of the cell, necrotic tissue may have the same macroscopic features (gross appearance) as those of acute cell swelling, namely, swelling and pallor. With time, necrosis becomes more obvious with a loss of structural detail and demarcation from adjacent viable tissue. Zonal necrosis, such as centrilobular hepatic necrosis (see Fig. 8-15) or renal proximal tubular necrosis (see Figs. 11-11 and 11-12), particularly if diffuse rather than segmental or focal, can be indistinguishable in its early stages from reversible degeneration. In contrast, unifocal or multifocal (randomly distributed) necrosis or segmental zonal necrosis is more easily recognized macroscopically precisely because it differs from adjacent viable tissue. Multifocal hepatic necrosis, for example, is recognizable in part because the necrotic foci differ from surrounding viable tissue and every hepatic lobule is not affected in the same manner. Likewise, segmental laminar cerebrocortical necrosis is recognized because only segments of the cerebral cortex are discolored or changed in texture or structure. An infarct, which is necrosis due to regional loss of blood supply, is recognized because it assumes the shape of the vascular field—rhomboidal in many tissues (e.g., lung or skin) or conical (wedge shaped in two dimensions) with its base at the edge of the spleen (see Fig. 13-64) or cortical surface of the kidney (see Figs. 2-37 and 2-38).
Histologic Changes in Necrosis (Oncotic Necrosis).
The light microscopic changes of necrosis (Fig. 1-16 ) were described in the nineteenth century by Rudolf Virchow. The hallmarks are pyknosis (nuclear condensation with shrinkage and intense basophilia), karyorrhexis (nuclear fragmentation), or karyolysis (nuclear dissolution or loss). Dead cells also tend to have intense cytoplasmic eosinophilia because of the denatured protein and loss of ribosomes, hence loss of basophilia. Later the dead cell may have cytoplasmic pallor and become swollen, rounded, and detached from the basement membrane or from neighboring cells.
Figure 1-16.
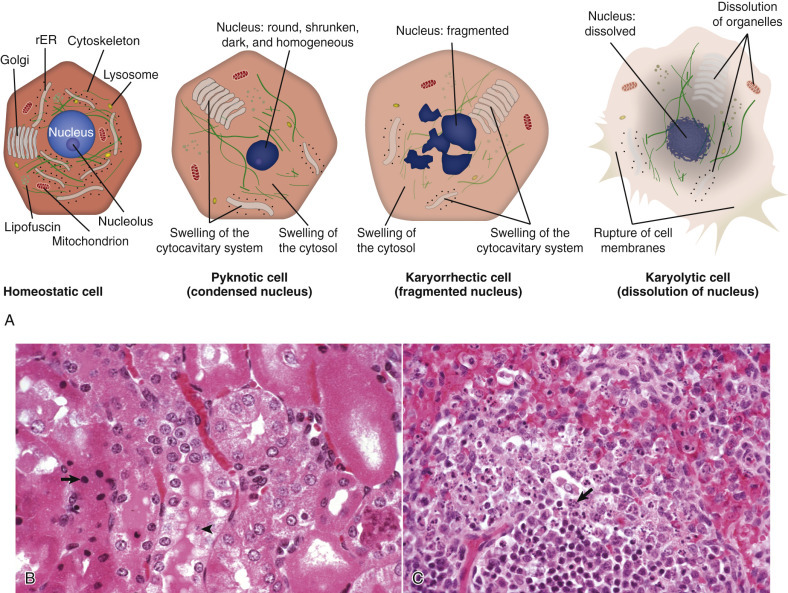
Cytoarchitecture of Cellular Necrosis.
A, Schematic representation of nuclear and cytoplasmic changes in the stages of necrosis. rER, rough endoplasmic reticulum. B, Pyknosis and karyolysis, renal cortex, chloroform toxicosis, mouse. Some tubular epithelial cells have undergone hydropic degeneration; others are necrotic with pyknosis (arrow) or karyolysis (arrowhead). H&E stain. C, Karyorrhexis, lymphocytes, spleen, dog. Necrotic lymphocytes have fragmented nuclei (arrow) because of parvovirus infection. H&E stain.
(A courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois; B and C courtesy Dr. L.H. Arp.)
Ultrastructure of Necrotic Cells (Oncotic Necrosis).
Initially the necrotic cell is swollen, rounded, and detached from adjacent cells and from the basal lamina, in the case of epithelium, or from the ECM, in the case of mesenchymal cells. Chromatin is clumped. The cytosol is electron lucent. Mitochondria are swollen and develop flocculent densities. The ER and the rest of the cytocavitary network swell and fragment into vesicles. Ultimately, cell swelling disrupts membranes, including the plasma membrane, at which point the cell literally explodes then collapses.
Types of Oncotic Necrosis.
It can be diagnostically useful, though somewhat arbitrary, to classify necrosis by its morphologic features in tissue sections. This classification depends on the tissue involved, the nature of the injurious agent, and the time elapsed after cell death. Necrosis has been classified traditionally as coagulative, caseous, liquefactive or lytic, and gangrenous. The student should remember that the morphologic appearance of necrotic cells and tissues changes with time. For example, the morphologic features of coagulative necrosis can progress to those of lytic necrosis with liquefaction, particularly in certain tissues or when leukocytes arrive.
Coagulative Necrosis.
The term coagulative necrosis refers to the denaturation of cytoplasmic proteins, which at the histologic level imparts an opaque and intense cytoplasmic eosinophilia to necrotic cells. Coagulative necrosis is a typical early response to hypoxia, ischemia, or toxic injury. It appears that the initial injury or the subsequent cellular acidosis denatures not only structural proteins, but also lysosomal enzymes in the affected cell. Normally, lysosomal enzymes would cause proteolytic disintegration of the entire cell, but as a result of this denaturation, proteolytic disintegration of the cell is delayed. However, the degradation of nucleic acids is not hindered. Thus a cell that has undergone coagulative necrosis has the expected nuclear features of cell death by oncosis (i.e., pyknosis, karyorrhexis, or karyolysis), but the cell outlines are still visible histologically (see Fig. 1-16). Coagulative necrosis is most easily recognized in the liver, kidney, myocardium, or skeletal muscle, in which the temporary preservation of cell outlines also preserves tissue architecture so that the outlines of hepatic plates, renal tubules, or muscle bundles are visible at the light microscopic level. Neurons also undergo coagulative necrosis before disappearing by lytic necrosis. Grossly, coagulative necrosis appears pale tan to pale gray, often sharply demarcated from the normal color of adjacent viable tissue, and solid (without apparent crumbling, sloughing, liquefaction, or other obvious loss of structure).
Infarction typically begins as coagulative necrosis, especially in tissues such as kidney (Fig. 1-17 ; E-Fig. 1-2), where scaffolding provided by tubular basement membranes and interstitial fibrous tissue maintains the tissue structure. Initially the tissue with loss of its blood supply is blanched, but within minutes blood enters the infarcted tissue because blood flow either was restored in the obstructed vessel or arrives from collateral circulation (therefore infarcts in organs with a dual blood supply, such as the lung, are typically hemorrhagic) or leaks from veins in unaffected tissue in and adjacent to the damaged tissue. In an end-artery organ, such as the kidney, macrophages remove the blood from acute hemorrhagic infarcts over the course of a few days, and the infarct becomes pale and sharply demarcated by a red rim, attributable to hyperemia, hemorrhage, and acute inflammation, from adjacent renal parenchyma.
Figure 1-17.
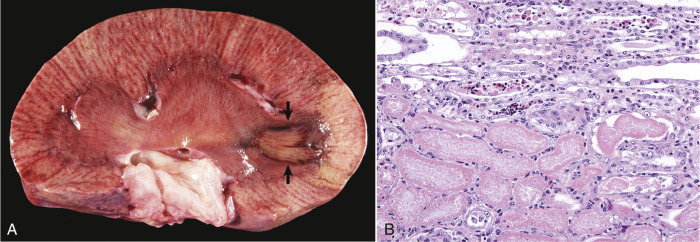
Coagulative Necrosis, Infarct, Kidney, Ox.
A, A pale tan wedge of coagulative necrosis extends from the medulla to the capsular surface of the kidney. The apical (medullary) portion of this renal infarct has a dark red border of reactive hyperemia and inflammation (arrows). B, Coagulative necrosis of renal tubular epithelial cells. Necrotic cells (lower half of figure) have homogeneous eosinophilic cytoplasm and pyknosis or karyolysis, but faint cell outlines and tubular architecture are retained. H&E stain.
(A courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University. B courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
E-Figure 1-2.
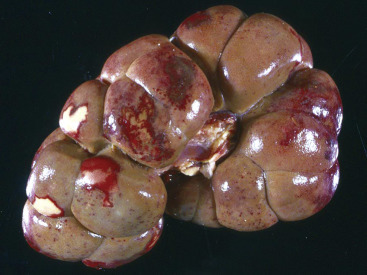
Coagulation Necrosis, Infarcts, Kidney, Cow.
Note the yellow-white areas of acute coagulation necrosis surrounded by a red rim of hyperemia and inflammation.
(Courtesy Dr. D.E. Tyler, College of Veterinary Medicine, The University of Georgia; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia.)
Caseous Necrosis.
Caseous, from the Latin word for cheese, refers to the curdled or cheeselike gross appearance of this form of necrosis. In comparison to coagulative necrosis, caseous necrosis is an older lesion with complete loss of cellular or tissue architecture (Fig. 1-18 ). Macroscopically, caseation may appear as crumbled, granular, or laminated yellow-white exudate in the center of a granuloma or a chronic abscess. Histologically, the lysis of leukocytes and parenchymal cells converts the necrotic tissue into a granular to amorphous—cell outlines are not visible—eosinophilic substance with basophilic nuclear debris. Calcification of the necrotic tissue can contribute to the basophilic granular appearance.
Figure 1-18.
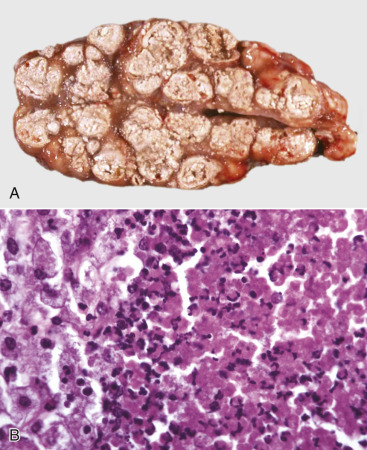
Tuberculosis (Caseous Necrosis), Lymph Node, Transverse Section, Ox.
A, The lymph node contains coalescing caseated granulomas. Caseous necrosis is characterized by off-white, crumbly exudate. B, Granulomatous inflammation in caseous necrosis. Cell walls are disrupted, and tissue architecture is lost. Degenerated or lysed leukocytes, including many neutrophils, are at the center (right) of a granuloma; note epithelioid macrophages at left. H&E stain.
(A courtesy Dr. M. Domingo, Autonomous University of Barcelona; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Caseous necrosis is prominent in the granulomas of bovine tuberculosis, caused by Mycobacterium bovis. M. bovis replicates within macrophages, protected by components of its cell wall from destruction by lysosomal enzymes until, with the development of cell-mediated (type IV) hypersensitivity, cytotoxic T lymphocytes destroy the infected macrophages, as well as parenchymal cells of the infected organ (see also Chapters 3, 4, and 5Chapter 3Chapter 4Chapter 5). Corynebacterium pseudotuberculosis, the cause of caseous lymphadenitis in sheep and goats, is another bacterium that can replicate in phagosomes of macrophages without being destroyed by lysosomal enzymes. The chronic stage of infection results in caseous abscesses in peripheral or internal lymph nodes (caseous lymphadenitis, see also Chapter 13 and Figs. 13-79 and 13-80) or other organs, such as the lungs.
Liquefactive Necrosis.
In liquefactive necrosis, cells are lysed, and the necrotic tissue is converted to a fluid phase. This manifestation is typically the final stage of necrosis in parenchyma of the brain (Fig. 1-19 ; see also Chapter 14) or spinal cord because of the lack of a fibrous interstitium to uphold tissue structure and because cells of the CNS tend to be rich in lipids and lytic enzymes. The term for the macroscopic (gross) appearance of necrosis in the brain and spinal cord is malacia. Neurons are generally the cells most susceptible to necrosis, especially from hypoxia or ischemia, and develop (early in the process of cell death) the morphologic features of coagulative necrosis. With time, however, the glial cells also undergo necrosis and liquefaction of the neuropil begins. Initially malacia may merely result in a translucency of affected tissue, but within a few days necrotic tissue undergoes yellowing, softening, or swelling. Liquefaction progresses with arrival of macrophages (gitter cells) to phagocytize the myelin debris and other components of the necrotic tissue. Eventually the parenchymal cells are completely lysed or phagocytized, and all that remains is the vasculature with intervening spaces that are partially filled with lipid- and debris-laden gitter cells. In organs or tissues outside the CNS, liquefactive necrosis is most commonly encountered as part of pyogenic (pus-forming) bacterial infection with suppurative (neutrophil-rich) inflammation (see also Chapter 3) and is observed at the centers of abscesses or other collections of neutrophils.
Figure 1-19.
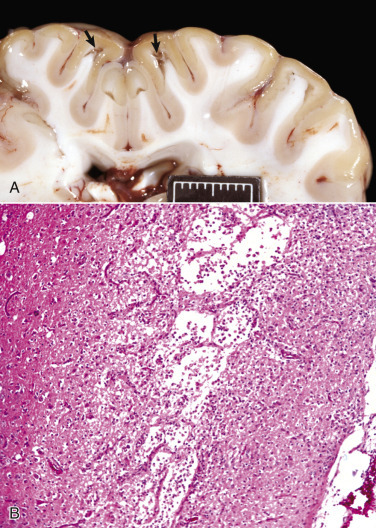
Liquefactive Necrosis.
A, Acute polioencephalomalacia, brain, goat. A thiamine deficiency has resulted in cerebrocortical malacia, which microscopically is liquefactive necrosis with focal tissue separation (arrows). Note yellow discoloration of affected cortex. Scale bar = 2 cm. B, Cortical necrosis, cerebrum, dog. The pale zone in deep laminae of the cerebral cortex is an area of liquefactive necrosis with loss of parenchyma. All that remains is the vasculature with gitter cells in intervening spaces. H&E stain.
(A courtesy Dr. R. Storts, College of Veterinary Medicine, Texas A&M University. B courtesy Dr. L.H. Arp.)
Gangrenous Necrosis.
Gangrene denotes a type of necrosis that tends to develop at the distal aspect of extremities, such as the limbs, tail, or pinnae, or in dependent portions of organs, such as the mammary glands or lung lobes. Gangrene can be designated as wet or dry; these forms are unrelated. If the dependent necrotic tissue is infected by certain bacteria, wet gangrene ensues. If those bacteria are gas forming (e.g., Clostridium spp.), then wet gangrene becomes gas gangrene. In the lung, wet gangrene is often a sequel to the lytic necrosis of aspiration pneumonia. The aspirated material could be foreign material (food or medicament) or gastric content (a mixture of ingesta and gastric secretions). Such materials can be caustic in their own right and are also likely to deliver bacteria from the environment or oropharynx into the lung. Staphylococcal infection of the ruminant mammary gland can result in gangrenous mastitis (Fig. 1-20, A ; E-Fig. 1-3), a form of wet gangrene. Grossly, tissues with wet gangrene are red-black and wet. Histologically, the lesion of wet gangrene resembles that of liquefactive necrosis but is usually accompanied by more numerous leukocytes, especially neutrophils.
Figure 1-20.
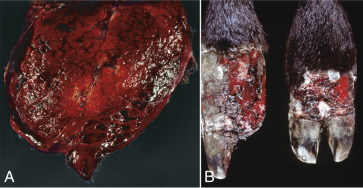
Gangrenous Necrosis.
A, Wet gangrene, mammary gland (longitudinal section through the teat), sheep. Staphylococcal infection caused the gangrenous mastitis in this ewe. Note wet and hemorrhagic necrosis of mammary tissue and overlying skin, especially at the distal (ventral) aspect of the udder. B, Dry gangrene, digits, ox. Vasoconstriction from ergot alkaloids produced by endophyte-infected fescue grass caused this ischemic necrosis of the distal aspects of the hind limbs. Note that one of the claws (left) has been lost due to the process.
(A courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University. B courtesy Dr. R.K. Myers, College of Veterinary Medicine, Iowa State University.)
E-Figure 1-3.
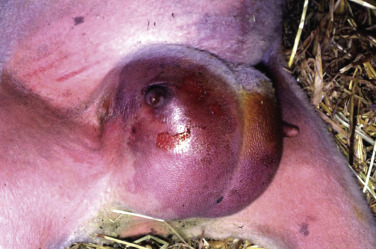
Gangrenous Necrosis.
Wet gangrene, udder, ewe. Skin is darkened and reddened over the necrotic mammary tissue. Note sharp line of demarcation of ventral necrotic tissue from adjacent viable tissue.
(Courtesy Dr. C. Wallace, College of Veterinary Medicine, University of Georgia; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia.)
Dry gangrene is the result of decreased vascular perfusion and/or loss of blood supply. It is a form of infarction resulting in coagulative necrosis that imparts a dry, leathery texture to the necrotic tissue, providing that it remains free of putrefactive bacteria. Arterial thrombosis (e.g., “saddle thrombus” formation at the iliac bifurcation of the aorta in cats) and frostbite are causes of dry gangrene of extremities. Dry gangrene is also the lesion of “fescue foot” in cattle (see Fig. 1-20, B ), caused by the vasoconstrictive effect of the ergot alkaloids produced by endophyte-infected fescue grass.
Necrosis of Epithelium.
Necrosis that develops in epithelial surfaces (e.g., epidermis or corneal epithelium) or epithelial linings (e.g., mucosal epithelium of the respiratory, digestive, or reproductive tracts) causes exfoliation or sloughing of dead cells, resulting in erosion of the epithelium, or, with full-thickness necrosis, in ulceration. Trauma, certain microbes (e.g., herpesviruses), and loss of blood supply are among the many causes of epithelial necrosis.
Necrosis of Adipose Tissue (Fat Necrosis).
Fat necrosis can be classified etiologically as nutritional, enzymatic, traumatic, and idiopathic (see also Chapter 7). Nutritional fat necrosis, also known as steatitis or yellow fat disease, is usually the result of feeding a diet high in unsaturated fatty acids and low in vitamin E or other antioxidants, setting the stage for ROS production and lipid peroxidation. Yellow fat disease is often seen in carnivores, such as cats or mink, on a fish-based diet. Affected adipose tissue is firm, nodular, and yellow-brown.
Enzymatic necrosis of fat is seen mainly in peripancreatic adipose tissue, where it is attributed to release of lipases from necrotic pancreatic acinar cells (Fig. 1-21 ; see Figs. 8-88 and 8-89). Grossly, necrotic adipose tissue becomes firm and nodular with off-white chalky deposits, the result of saponification (soap formation). Microscopically, fat necrosis elicits inflammation that consists mainly of lipid-laden macrophages and variable number of neutrophils. Lipids are removed by solvents during histologic processing, so the cytoplasm of normal adipocytes is not stained, whereas necrotic adipocytes tend to have pale eosinophilic to amphophilic cytoplasm with scattered intensely basophilic soap deposits.
Figure 1-21.
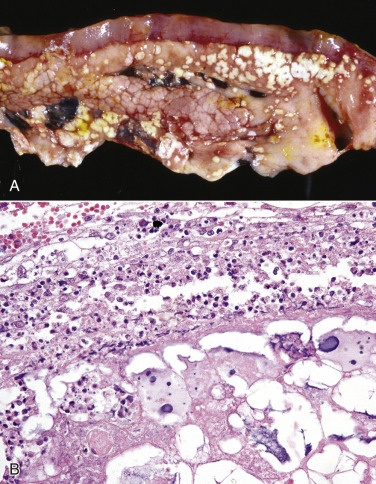
Fat Necrosis.
A, Enzymatic necrosis of fat (fat necrosis); duodenum, pancreas, and peripancreatic adipose tissue, dog. Recurrent bouts of pancreatitis with leakage of lipases and other enzymes causes saponification of necrotic adipose tissue, giving it a chalky, off-white appearance. B, Peripancreatic adipose tissue, dog. Note the necrotic adipose tissue (bottom) with saponification (basophilic areas) and the border of neutrophils and macrophages (top). H&E stain.
(A courtesy Dr. J. Wright, College of Veterinary Medicine, North Carolina State University; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Traumatic fat necrosis is typically the result of blunt trauma or chronic pressure on adipose tissue against bony prominences, such as the subcutaneous adipose tissue compressed against the sternum in recumbent cattle. Ischemia is thought to contribute to the cell injury. Inflammation and saponification are inconspicuous in this form of fat necrosis.
Necrosis of abdominal fat in cattle is an example of idiopathic fat necrosis. This lesion tends to develop in the abundant adipose tissue of the mesentery and retroperitoneal tissue of overconditioned cows. Some have attributed retroperitoneal fat necrosis to ischemia associated with consumption of endophyte-infected tall fescue grass. Idiopathic fat necrosis is also encountered in the ventral parietal peritoneum of horses and ponies (see Fig. 7-15).
Sequelae to Oncotic Necrosis.
Oncotic necrosis elicits an inflammatory reaction in most tissues. In the CNS the inflammatory reaction is slow to develop and consists mainly of an influx of macrophages that become gitter cells. In most other tissues a band of hyperemia (hemorrhage and acute inflammation) encircles the necrotic tissue and brings leukocytes to the site. The neutrophils and macrophages phagocytize and lyse the necrotic tissue, converting coagulative to liquefactive necrosis and hastening (in many cases) the removal of damaged tissue. In other cases, foreign material or bone fragments resist digestion and form a sequestrum. Smaller cavitations left by liquefactive necrosis may heal without scarring, depending on the regenerative capacity of the affected tissue. The liver is an organ with high regenerative capacity and, because of its dual blood supply, is not prone to infarction. In contrast, in renal infarcts the lost nephrons are seldom successfully repaired and are usually replaced by a fibrous scar.
Focal epithelial necrosis that results in ulceration can be repaired by hyperplasia of adjacent normal epithelial cells without scarring if the defect is small or shallow and if basal or other progenitor cells remain nearby to fill the gap (i.e., healing in coronavirus or parvovirus infections of the small intestine [see Figs. 4-39 and 7-180]). Adipose tissue, in contrast, is ill equipped to replace necrotic fat lobules because of the low regenerative capacity of adipocytes.
Morphologic Appearance of Postmortem Changes
Postmortem changes are the result of autolysis (postmortem decomposition) rather than of some pathologic process; thus they are not lesions. Although all cells in histologic sections are technically dead, the pathologist must distinguish between those that were viable and those that had already died before the tissue specimen was collected at biopsy or before the animal died in the case of postmortem examination. Antemortem cell death is recognized by the morphologic changes that follow the biochemical events of cell death and takes time (minutes to hours) to develop. Autolysis, in contrast, is the decomposition of cells that takes place after somatic death (i.e., death of the body) or after collection of a biopsy sample from a living animal. Autolysis results in morphologic changes in cells and tissues, and it is these changes that the pathologist must distinguish from those of necrosis. Ideally, autolysis is minimized by collecting tissues before (biopsy) or as close to the time of somatic death as possible and fixing them for histologic processing upon collection. This is especially important in tissues such as the intestine, in which autolysis develops within minutes because the mucosal epithelium is exposed to bacteria and digestive enzymes from the gut lumen. Autolyzed enterocytes have microscopic changes that can be mistaken for degenerative lesions, such as loss of the microvillous brush border, rounding, attenuation, and detachment from the basement membrane. The so-called cadaver bacilli invade other tissues, especially the liver with its portal blood supply, but can reach nearly every tissue in the body.
Postmortem decomposition proceeds at various rates depending on the tissue, the cause of death, body and environmental temperature, and microbial flora. Brain and spinal cord are quick to autolyze. The brain is also subject to another postmortem change, known as dark neurons, from handling in the early postmortem period. Dark neurons must be distinguished from neurons that died from ischemia or excitotoxicity (see Chapter 14). Skeletal muscle is not so quick to autolyze but retains the ability to contract after somatic death, especially when immersed in formalin, so muscle biopsy specimens must be clamped in a slightly stretched state before fixation to avoid contraction artifacts. Rigor mortis is a generalized contraction of skeletal muscle that commences 1 to 6 hours after death and can persist for 1 to 2 days. Muscle contraction and relaxation require ATP and glycogen, so once the stores are depleted, postmortem muscle contraction is reversible only by autolysis.
Rapid cooling of the carcass (by refrigeration, not freezing, which induces ice crystal formation) helps considerably to delay autolysis but is of limited value for adult herbivores, in which the rumen or equine cecum generates heat from fermentation long after death of the animal. Abundant adipose tissue or a heavy coat also impedes rapid cooling of the carcass. Autolysis progresses so rapidly in the rumen that its stratified squamous mucosal epithelium sloughs in broad sheets within a few hours of death. Autolysis is rapid throughout the gastrointestinal tract in all species, but especially that of herbivores. Gas production by enteric bacteria continues unabated in the postmortem period, distending the entire gastrointestinal tract, but especially the rumen and the equine cecum. Gaseous distension of the gastrointestinal tract can result in rupture, displacement of abdominal viscera, eversion of rectal mucosa through the anus, and even rupture of the diaphragm. In severely autolyzed carcasses, gas bubbles (postmortem emphysema) are found in most tissues.
Livor mortis, or hypostatic congestion, is the gravitational pooling of blood on the dependent (down) side of the carcass. This results in reddening of tissues that can be observed externally in pale-skinned, sparsely haired animals (E-Fig. 1-4) and internally in organs and tissues of the down side. Postmortem hypostatic congestion becomes permanent once the blood clots, so if an animal is moved subsequently, the pooled blood will remain on the original down side, a useful finding in forensic pathology.
E-Figure 1-4.
Livor Mortis, Pig.
Note red to purple discoloration of skin on the right side, the side on which the pig was lying when it died. This color change is termed livor mortis or hypostatic congestion. The pale areas are pressure points on the down side into which blood could not flow after death.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
A postmortem clot (E-Fig. 1-5) can be distinguished from an antemortem thrombus by its smooth shiny surface and lack of lamination or attachment to the endothelial surface of the vessel. However, depending on the erythrocyte sedimentation rate, a postmortem clot can organize within a vessel just as within a test tube, with erythrocytes at the bottom separated by a “buffy coat” of leukocytes from the serum at the top. The resemblance of clotted serum to avian adipose tissue has garnered the name of “chicken fat clot” for this postmortem clot that is often seen in horses because of their high erythrocyte sedimentation rate. Inflammation can accelerate the sedimentation rate. Anticoagulants or hereditary coagulopathies can delay or prevent postmortem clotting of blood.
E-Figure 1-5.
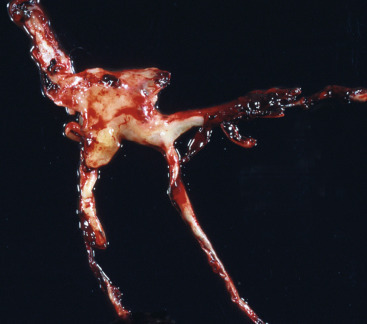
Postmortem Clot, Dog.
The postmortem clot is off-white to yellow (chicken fat clot) in some areas and shiny red (currant jelly clot) in others. Note how it conforms to the shape of the lumen of the vessels from which it was removed.
(Courtesy Dr. R.K. Myers, College of Veterinary Medicine, Iowa State University.)
Color changes in the autolyzed carcass must be distinguished from lesions. Hemoglobin imbibition is the reddish discoloration of tissue by hemoglobin from lysed erythrocytes. It is first noted in endocardium and intima of large vessels but eventually discolors surrounding tissues (E-Fig. 1-6). In dead fetuses that are retained in utero, hemoglobin imbibition imparts a pink-brown discoloration to all tissues. Bile imbibition is a greenish discoloration from leakage of bile through the wall of the gallbladder or bile ducts (E-Fig. 1-7). Pseudomelanosis is a blue-green to black discoloration of tissues, especially along the digestive tract, by iron sulfide deposits—there is no melanin in pseudomelanosis—that are formed by the reaction of hydrogen sulfide from putrefactive bacteria with the iron in hemoglobin from lysed erythrocytes. Postmortem proliferation of gas-forming bacteria results in distension of the gastrointestinal tract, formation of gas bubbles (emphysema) in tissues, and bloating of the carcass (E-Fig. 1-8). Localized postmortem bacterial proliferation in the liver can result in spots of pallor that resemble necrotic foci. Postmortem pressure on organs, such as the lung or liver, forces the blood out of underlying tissue, producing pale “imprints” (E-Fig. 1-9). Chilling or partial freezing of the carcass can make the lens opaque and white (E-Fig. 1-10). This can be confused with cataracts, but the lens reverts to normal transparency on warming.
E-Figure 1-6.
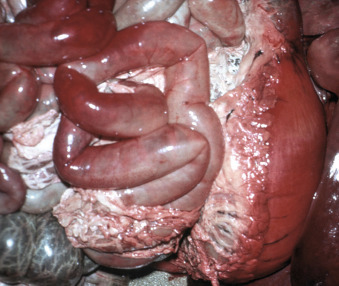
Imbibition of Hemoglobin, Viscera, Pig That Was Dead Several Hours Before Necropsy.
Note dark pink discoloration of serosal surfaces of the stomach and small intestine. This is termed hemoglobin imbibition and is due to staining by hemoglobin that has seeped out of autolyzed erythrocytes.
(Courtesy Dr. R.K. Myers, College of Veterinary Medicine, Iowa State University.)
E-Figure 1-7.
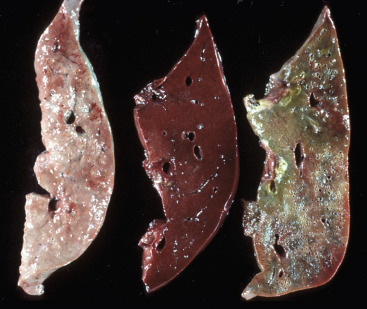
Postmortem Autolysis.
Cross-sections of livers from three pigs at different stages of postmortem autolysis. The slice on the right has green discoloration caused by leakage of bile into the surrounding parenchyma after death (bile imbibition). All three livers, especially the one on the left, were softer than normal, another characteristic of autolytic tissue.
(Courtesy Dr. R.K. Myers, College of Veterinary Medicine, Iowa State University.)
E-Figure 1-8.

Postmortem Bloat or Emphysema.
Cow killed by lightning several hours earlier. When animals, especially ruminants, die, the bacteria in the gastrointestinal tract continue to grow and produce gas. Rumen microbes may produce abundant gas, causing the carcass to swell tremendously.
(Courtesy Dr. W. Crowell, College of Veterinary Medicine, The University of Georgia; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia.)
E-Figure 1-9.
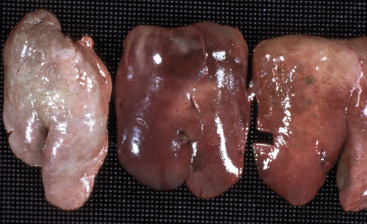
Postmortem Autolysis.
Pig livers at various intervals after death. Pale foci on the middle liver are due to blood being forced out of the parenchyma by intestinal swelling (intestinal imprints) and from pressure from the overlying ribs (rib imprints). Multiple small pale foci can sometimes be caused by colonies of postmortem bacteria and can be confused with antemortem necrosis.
(Courtesy Dr. R.K. Myers, College of Veterinary Medicine, Iowa State University.)
E-Figure 1-10.
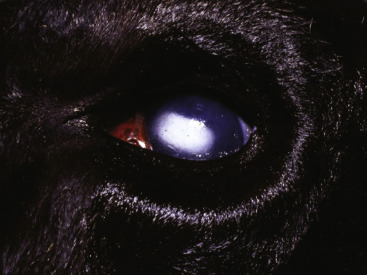
Postmortem Autolysis, Eye, Lens, Calf.
The cloudiness of the lens is due to cooling or freezing and is reversible as the carcass warms up. It should not be confused with cataracts.
(Courtesy Dr. P.N. Nation, University of Alberta; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia.)
Cell Death by Apoptosis
In contrast to oncotic necrosis, in which the dying cell swells until it literally bursts, apoptotic cell death is a process of condensation and shrinkage. Apoptosis is a form of programmed cell death that is important in embryologic development, homeostasis, and involution of organs or tissues deprived of hormonal stimulation or growth factors. It is also a regulated form of cell death that is directed by signaling pathways in response to certain types of injury.
Triggers of Apoptosis.
The triggers of apoptosis include binding of ligands such as TNF to cell surface DRs, various stresses or injury from toxins or ROS, nutrient deprivation or withdrawal of growth factors or hormones, DNA damage, or immune-mediated injury from cytotoxic T lymphocytes or NK cells. Apoptosis (Fig. 1-22 ) proceeds through an extrinsic pathway (initiated by the binding of a ligand to its DR) or an intrinsic pathway (initiated in mitochondria in response to various stresses or DNA damage) and almost always entails activation of caspases. Caspases are cysteine proteases that cleave peptides after aspartate residues. The initiator caspases that start the process of apoptosis include caspase-8 (activated by the death-inducing signaling complex (DISC) of the extrinsic pathway), caspase-9 (activated with the apoptosome in the intrinsic pathway), and caspase-2 (activated by p53 following DNA damage). The initiator caspases activate effector caspase-3, caspase-6, and caspase-7, which then execute apoptosis.
Figure 1-22.
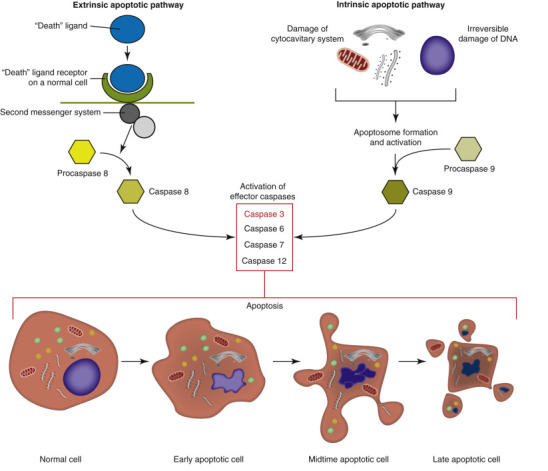
Apoptosis.
In the extrinsic pathway (left), apoptosis is triggered by binding of a ligand to a cell surface death receptor with subsequent formation of a cytoplasmic death-inducing signaling complex that activates an initiator caspase (e.g., caspase-8). The intrinsic pathway (right) of apoptosis is triggered by DNA damage or various cell stressors, especially those that result in permeabilization of the mitochondrial outer membrane, and leads to formation of the caspase-activating complex or apoptosome. The initiator caspase in the intrinsic pathway is usually caspase-9. In both the extrinsic and the intrinsic pathways, initiator caspases activate effector (executioner) caspases, resulting in cell death with the characteristic morphologic features of apoptosis (shown at bottom).
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
The Extrinsic (Death Receptor–Initiated) Pathway.
Extrinsic apoptosis (see Fig. 1-22) begins with ligand-induced trimerization of a cell surface DR. The DRs include Fas, tumor necrosis factor receptor (TNFR) 1, and TNF-related apoptosis-inducing ligand receptor (TRAILR). The next step is internalization and recruitment of the intermediate membrane proteins TNF receptor–associated death domain (TRADD), Fas-associated death domain (FADD), and caspase-8 to form the cytoplasmic DISC. Remember that RIPK1, depending on its ubiquitination status, can associate with the trimerized DR and direct the cell toward regulated necrosis (if caspases are inhibited) or toward survival via activation of NFκB, and has an N-terminal death domain (DD) that links it to the apoptotic pathway through adaptor proteins such as TRADD or FADD. TRADD interacts with FADD, which in turn activates procaspase-8. Sufficient active caspase-8 then activates effector (executioner) caspase-3 and caspase-7 to execute apoptosis. Caspase-8 can also truncate Bid, a proapoptotic Bcl-2 protein, which translocates to mitochondria to trigger intrinsic apoptosis (see the next section). Importantly, the protein FLIP blocks the extrinsic pathway by binding procaspase-8 without activating it. If caspase-8 activity is insufficient, DR-mediated apoptosis can be augmented by mitochondria, almost always through Bcl-2 proteins, such as the proapoptotic Bak (Bcl-2 antagonist/killer) and Bax (Bcl-2–associated X protein). Even cells that cannot initiate or propagate apoptotic signaling can still die, but do so via caspase-independent pathways of cell death, such as regulated necrosis.
The Intrinsic (Mitochondrial) Pathway.
The intrinsic or mitochondrial pathway of apoptosis (see Fig. 1-22) does not require ligation of a cell surface DR and can be triggered by a variety of cell stressors or by DNA damage that leads to activation of p53-upregulated modulator of apoptosis (PUMA). The key event of intrinsic apoptosis is mitochondrial outer membrane permeabilization (MOMP). MOMP can be triggered by activation, posttranslational modification, and upregulation of proapoptotic BH3-only proteins (e.g., PUMA protein). The BH3-only proteins usually induce MOMP via oligomerization of Bax and Bak to form channels in the outer mitochondrial membrane. This permeabilization of the outer mitochondrial membrane releases cytochrome c from the intermembrane space into the cytosol. Cytochrome c promotes the assembly of the caspase-activating complex or apoptosome, which consists of caspase-9 plus apoptotic protease activating factor 1 (Apaf-1). MOMP also releases the second mitochondrial activator of caspases (SMAC), as well as the catabolic hydrolases, apoptosis-inducing factor (AIF), and endonuclease G.
Recall from the section on regulated necrosis that opening of the MPT pore is a key event in cell death because it dissipates the proton gradient needed for oxidative phosphorylation. At low concentrations, opening of the MPT pore can induce protective autophagy to remove dysfunctional mitochondria. However, MOMP is a lethal permeabilization that initiates intrinsic apoptosis.
The Execution Phase of Apoptosis.
Initiator caspases (2, 8, 9, or 10) cleave the downstream effector (executioner) caspases (mainly 3, 6, and 7), which then execute apoptosis by cleaving cell proteins after aspartate residues. Granzyme B from cytotoxic T lymphocytes and NK cells can also trigger apoptosis by activating caspase-3 and caspase-7. Effector caspases cleave nuclear and cytoplasmic proteins, leading to disintegration of the nucleus and disruption of the cytoskeleton.
Morphologic Appearance of Apoptosis.
Morphologically, apoptotic cell death is a process of condensation and fragmentation of the nucleus (pyknosis and karyorrhexis) with blebbing of the plasma membrane to form membrane-bound apoptotic bodies that contain nuclear fragments, organelles, and condensed cytosol (Fig. 1-23 ; see Fig. 1-22). The plasma membrane that surrounds apoptotic bodies prevents the inflammation occurring with necrotic cell death but does express factors to attract phagocytes and stimulate heterophagy. Not surprisingly, apoptotic and necrotic cell death can coexist in the same tissue (E-Fig. 1-11).
Figure 1-23.
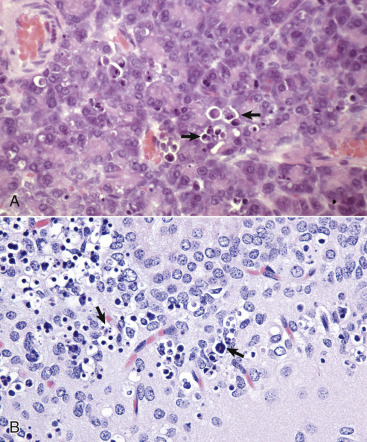
Apoptosis, Cytologic Features.
A, Pancreas, rat. Individual acinar cells are shrunken, condensed, and fragmented (arrows). Apoptotic bodies are in adjacent cells, but inflammation is absent. H&E stain. B, Hippocampus, brain, mouse. Individual neurons are shrunken, condensed, and fragmented (arrows). H&E stain.
(A courtesy Dr. M.A. Wallig, College of Veterinary Medicine, University of Illinois. B courtesy Drs. V.E. Valli and J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
E-Figure 1-11.
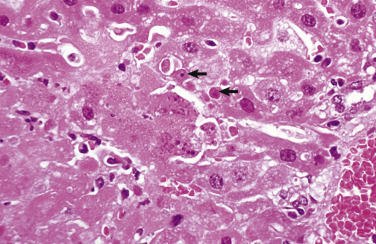
Necrosis and Apoptosis, Mouse Hepatitis Virus Infection, Liver, Mouse.
The virus causes hepatocellular death, typically by oncotic necrosis, but sometimes by apoptosis. Note coagulative necrosis (lower left) with lytic necrosis (center left) and individual cells with features of apoptosis (arrows). H&E stain.
(Courtesy Dr. R.K. Myers, College of Veterinary Medicine, Iowa State University.)
Chronic Cell Injury and Cell Adaptations
In the previous section we considered reversible injury with acute cell swelling and irreversible injury with cell death. In this section we examine chronic sublethal injury to which the cell may adapt by undergoing hypertrophy (increased cell size because of increase in the number and size of organelles), hyperplasia (increased number of cells due to proliferation of cells capable of mitosis), metaplasia (change in cell type), or dysplasia (development of cellular atypia). Alternatively, cells may undergo degenerative changes such as atrophy (diminished number and size of organelles with decreased cell size and tissue mass) or accumulation of normal or abnormal substances.
Cellular Survival during Sublethal Ischemia or Involution
Autophagy
Autophagy evolved as a cell survival mechanism during ischemia or involution in response to loss of growth factors or hormonal stimuli. In autophagy, cells consume their own damaged organelles, as a housekeeping function, and cytosolic proteins and carbohydrates, as a source of nutrients. Thus autophagy is distinct from heterophagy (Fig. 1-24 ), in which one cell phagocytizes another cell or parts thereof. Autophagy usually inhibits apoptosis; however, if uncontrolled, it can result in cell death. Autophagy can be categorized as macroautophagy, microautophagy (direct phagocytosis by the lysosome), and chaperone-assisted autophagy. In macroautophagy, portions of cytosol and organelles are enveloped in a double-membrane–bound autophagosome, which subsequently fuses with a lysosome to form a single-membrane–bound autophagolysosome.
Figure 1-24.
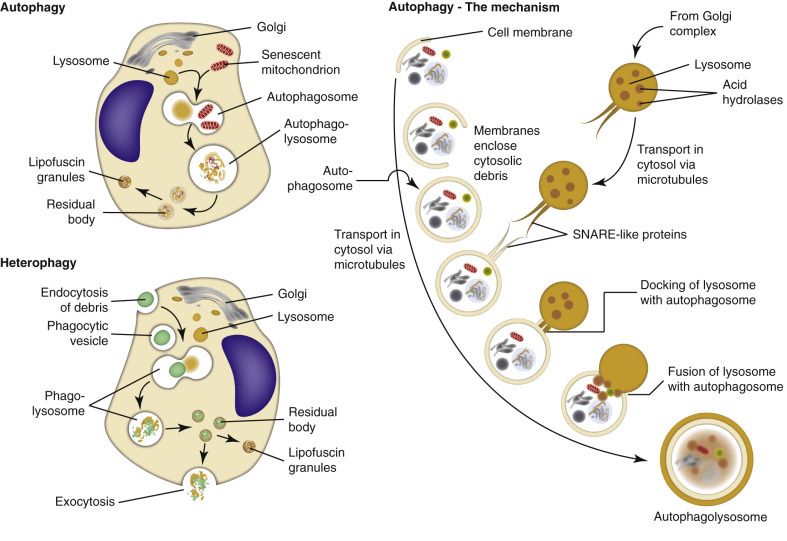
Autophagy and Heterophagy.
Schematic comparison of autophagy (top left) and heterophagy (lower left). The mechanism of autophagy is also illustrated. SNARE, Soluble NSF (N-ethylmaleimide–sensitive fusion protein) attachment protein receptor.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
The autophagy signaling pathway begins with formation of the ULK1 complex, composed of ULK1 (UNC-51–like kinase), FIP 200 (a kinase-interacting protein), and autophagy-related gene products (ATG) 13 and 101. The ULK1 complex drives the formation of the isolation membrane; mammalian target of rapamycin (mTOR), a protein-serine/threonine kinase, complex 1 inhibits the ULK1 complex. The Beclin 1-VPS34 (vacuolar protein sorting 34, a phosphatidylinositol-3 kinase) complex drives nucleation of the isolation membrane or phagophore, usually at the point of contact between mitochondria and ER, though other cell membranes may contribute. Transmembrane ATG9 and VMP1 (vacuolar membrane protein 1) recruit lipids to the isolation membrane. The double-layered isolation membrane wraps around a portion of cytosol with organelles. Two ubiquitin-like (UBL) protein conjugation systems—the ATG12-UBL system and the protein light chain (LC) 3-UBL system—cleave LC3 and catalyze the conjugation of ATG proteins. Finally, soluble NSF (N-ethylmaleimide–sensitive fusion protein) attachment protein receptor (SNARE)–like proteins are involved in docking and fusion of the lysosome to the autophagosome. The end result is a single membrane-bound autophagolysosome that contains a portion of the cytosol with dysfunctional organelles. See the later section on Intracellular Accumulations for the histologic appearance of autophagolysosomes.
In general, autophagy provides an escape from cell death by facilitating the removal of effete organelles and unnecessary cell proteins and by providing nutrients to the deprived cell. However, even when the autophagic cell dies (from apoptosis, oncotic necrosis, or uncontrolled autophagy), autophagy protects tissues from unnecessary inflammation by promoting the secretion of lysophosphatidylcholine, a chemotactic factor for phagocytes, and surface expression of phosphatidylserine, which marks the cell for heterophagy.
Adaptations That Change Cell Size, Number, or Appearance
Tissues adapt to chronic injury in positive or negative ways, depending on the nature of the injury and the type of cell (Essential Concept 1-4 ). Some changes, such as an increase in cell size (hypertrophy) or number (hyperplasia) can increase the function of the organ or tissue at least temporarily and are considered positive adaptations. In other cases, cells shrink (atrophy) and the organ or tissue has diminished function, but this seemingly negative adaptation can have the beneficial effect of avoiding cell death. A change in cell type (metaplasia) generally decreases normal cell function but can offer greater protection to underlying tissues. Dysplastic changes (dysplasia) in cell appearance, on the other hand, have little or no protective effect and can be a precursor to neoplasia. These changes are illustrated in Figure 1-25 .
Essential Concept 1-4. Adaptations to Chronic Cell Injury.
In the case of repetitive or continuous injury that is not inherently or immediately lethal, cells of many different types can survive, even without complete recovery, by adapting. Depending on the cell type—not all cells are capable of all possible responses—cellular adaptations to chronic injury include the following:
-
1.
Hypertrophy, an increase in cell size by virtue of an increase in number and size of organelles
-
2.
Hyperplasia, an increase in cell number that only those cells capable of mitosis can undergo
-
3.
Metaplasia, a change from one differentiated cell type to another of the same germ layer (e.g., from ciliated epithelium to stratified squamous epithelium in the respiratory tract)
-
4.
Dysplasia, abnormal differentiation with features of cellular atypia
-
5.
Atrophy, a decrease in cell size by virtue of a decrease in number and size of organelles
-
6.
Intracellular accumulations of endogenous or exogenous substances
Certain adaptations (e.g., myocardial hypertrophy) can increase the functional capacity of cells or tissues, at least temporarily, but more often cellular adaptations to chronic injury serve as means of protection (for example, keratinized stratified squamous epithelium offers more protection to underlying tissue than does pseudostratified ciliated epithelium) or survival (an alternative to cell death) and result in altered or diminished function of cells or tissues. Dysplasia is an adaptation without apparent advantages to the host. Indeed, dysplasia can be a precursor to malignant neoplasia (cancer).
Figure 1-25.
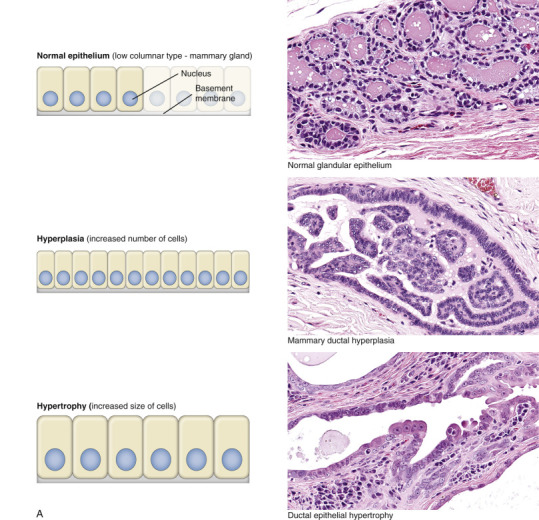
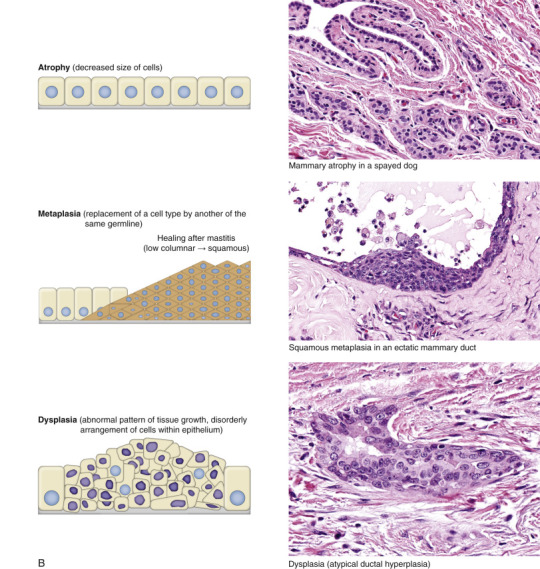
Adaptive Changes Illustrated in Canine Mammary Epithelium.
Schematic diagrams of epithelial adaptations paired with histologic examples from canine mammary glands. A, Normal epithelium, hyperplasia, and hypertrophy. B, Atrophy, metaplasia, and dysplasia. H&E stain.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Atrophy
Atrophy is the decrease in the mass of a tissue or organ due to decreased size and/or number of cells after it has reached its normal size (see Fig. 1-25, B). Atrophy must be distinguished from hypoplasia, the term applied to tissues or organs that are smaller than normal because they never developed completely. The shrinkage of atrophied tissue is caused by decreased size or loss of its principal cells. The causes of cellular or tissue atrophy include nutrient deprivation or loss of hormonal stimulation, decreased workload (disuse atrophy), denervation (especially in skeletal muscles), and compression (e.g., adjacent to neoplasms, other masses, or distended body cavities). Autophagy and apoptotic cell death can contribute to the shrinkage or loss of cells, respectively, in an atrophied organ. Histologically, the principal cells of the tissue are small with little to no mitotic activity. Ultrastructurally, atrophied cells have few mitochondria or other organelles.
Atrophy occurs in most organ systems of the animal body (see Pathology of Organ Systems chapters for details). Thyroid atrophy (Fig. 1-26 ) can be idiopathic or the result of autoimmune destruction of follicular cells (see Chapter 12). Because the portal vein provides most of the blood supply to the liver, a portosystemic shunt results in hepatic atrophy (E-Fig. 1-12; see also Chapter 8, Fig. 8-38). Atrophy can be particularly striking in the thymus, causing a rapid and drastic loss of tissue through apoptosis of lymphocytes. Thymic atrophy is so consistent and often severe in certain viral infections (e.g., canine distemper or canine and feline parvovirus infections) with a predilection for rapidly dividing cells that it serves as a diagnostically useful, but easily overlooked gross lesion (see also Chapter 13). The serous atrophy of fat in starving animals results in diminished volume and a translucent, semifluid to gelatinous appearance to adipose tissue throughout the body, but especially in the coronary groove of the heart (see Fig. 10-59) or in the marrow of long bones.
Figure 1-26.
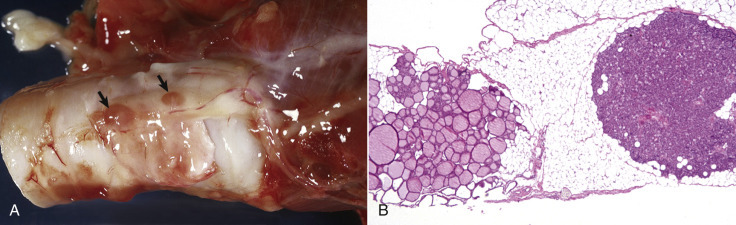
Atrophy, Thyroid Gland, Trachea, Dog.
A, The thyroid gland is thin, translucent, and barely discernable. Note grossly normal parathyroid glands (arrows).B, The atrophied thyroid follicles vary in size and colloid content but generally have a relative increase in luminal diameter and decrease in follicular epithelial height. Much of the supporting stroma has been replaced by adipose tissue. The parathyroid gland (right) is of normal size. H&E stain.
(A courtesy Dr. W. Crowell, College of Veterinary Medicine, The University of Georgia; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy College of Veterinary Medicine, University of Illinois.)
E-Figure 1-12.
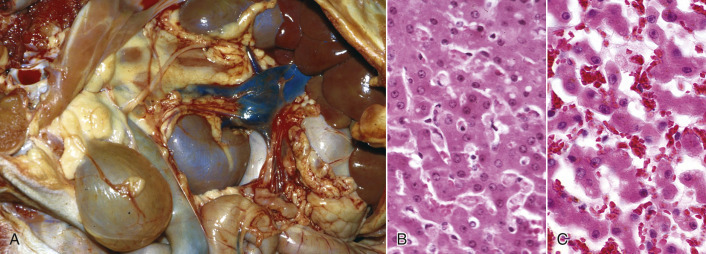
Atrophy, Liver, Dog.
A, Note the small size (up under the rib cage) but normal color of the liver in this dog and the anomalous portocaval shunt between the portal vein and the caudal vena cava. This shunt caused a reduction in blood flow to the liver and therefore atrophy of hepatocytes. B, Normal liver. H&E stain. C, Liver, atrophy. Hepatocytes are smaller, so hepatic cords are narrower than those in the normal liver. Consequently, the sinusoids are wider. H&E stain.
(A courtesy Dr. J. Sagartz, College of Veterinary Medicine, The Ohio State University; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee. C courtesy Dr. R.K. Myers, College of Veterinary Medicine, Iowa State University.)
Hypertrophy
Hypertrophy, from the Greek word for increased growth, refers to an increase in size and volume of a tissue or organ due to increase in cell size (see Fig. 1-25, A). Importantly, the increased tissue mass is due to increased size of the parenchymal cells rather than stromal cells or leukocytes. Hypertrophy often accompanies an increase in cell number (hyperplasia) due to cellular proliferation but as a stand-alone phenomenon is observed mainly in organs or tissues such as the heart (E-Fig. 1-13; see Chapter 10) or skeletal muscle (see Chapter 15), in which the principal cells are postmitotic and incapable of replication. When the term hypertrophy is applied at the cellular level, it denotes an increase in cell size because of an increase in size or number of organelles as distinguished from increased cell size from hydropic cell swelling (loss of volume control) or from accumulation of endogenous or exogenous substances. Cellular hypertrophy is the process by which postmitotic cells, such as cardiomyocytes or skeletal myocytes, can grow as the juvenile animal grows. It is also the physiologic response of striated muscle to increased workload such as occurs in training of race horses. Smooth muscle cells (e.g., in the tunica media of arteries) also undergo hypertrophy in response to increased workload. Although muscular hypertrophy increases functional capacity in the short term, accompanying changes, such as increased fibrous stroma or decreased vascular perfusion, in myocardium, for example, can lead to decompensation of the affected organ.
E-Figure 1-13.
Hypertrophy, Heart, Dog.
A, Narrowing of the pulmonary outflow tract caused by pulmonic valve stenosis has forced the right ventricle to contract with much more pressure. This increased workload has caused hypertrophy of the wall of the right ventricle, which is much thicker here than it would normally be. B, Note the increased size (hypertrophy) of myocytes in the overworked heart muscle.
(Courtesy Dr. L. Miller, Atlantic Veterinary College, University of Prince Edward Island; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia.)
Hyperplasia
Hyperplasia implies an increase in number of the principal cells of a tissue or organ (see Fig. 1-25, A). This response can occur only in a cell population that is capable of mitosis (see subsequent section on the Cell Cycle). Many epithelial cells (e.g., hepatocytes and epithelia of the epidermis and intestinal mucosae) are quick to undergo hyperplasia in response to hormonal stimulation, inflammation, or physical trauma. Hyperplasia of glandular epithelium (e.g., thyroid follicular epithelium) can be marked, resulting in striking gross enlargement of the thyroid gland (Fig. 1-27 ). Importantly, hyperplasia differs from neoplastic cellular proliferation in that it generally subsides if the stimulus is removed. Striated muscle and nervous system tissues have negligible capacity to proliferate and in general do not undergo hyperplasia. Other tissues, such as smooth muscle, bone, and cartilage, are intermediate in their ability to proliferate.
Figure 1-27.
Hyperplasia, Thyroid Gland, Goat.
A, Maternal iodine deficiency caused hyperplasia (and hypertrophy) of thyroid follicular epithelial cells in this neonatal goat, resulting in massive enlargement (goiter) of both lobes. B, Follicular epithelial cells from a normal thyroid gland. H&E stain. C, Thyroid follicular epithelial cells from a case of goiter. Note the increased number (and size) of the follicular epithelial cells. H&E stain.
(A courtesy Dr. O. Hedstrom, College of Veterinary Medicine, Oregon State University; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia. B and C courtesy Dr. B. Harmon, College of Veterinary Medicine, The University of Georgia; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia.)
Hyperplasia is considered physiologic when it is a response to cyclic hormonal stimulation as in the endometrial or mammary development of pregnancy and lactation, respectively. The hyperplasia of wound healing is not a normal event, but it is an appropriate and compensatory response of fibroblasts and endothelial cells to traumatic injury. Likewise, hyperplastic goiter is not a normal change in the thyroid gland (see Fig. 1-27; see also Chapter 12) but is an appropriate response to generate thyroid hormones in the face of iodine deficiency. Idiopathic (of unknown cause) nodular hyperplasia is encountered rather commonly in certain organs (e.g., liver, pancreas, or spleen), especially in older dogs, and often is of no clinical significance. In contrast, inappropriate elevation of trophic hormones or growth factors can lead to persistent hyperplasia that can be a precursor to neoplastic transformation (see Chapter 6).
Metaplasia
Metaplasia (see Fig. 1-25, B) is a change from one differentiated (mature) cell type to another differentiated cell type of the same germline. Typically, squamous metaplasia is a reparative response to chronic inflammation (e.g., in mammary ducts in chronic mastitis), hormonal imbalance (e.g., estrogen-induced squamous metaplasia in the prostate gland; see Fig. 19-26, C), vitamin A deficiency (E-Fig. 1-14), or trauma. Although stratified squamous epithelium creates a protective barrier between the irritant and underlying tissue, there are negative consequences. For example, squamous metaplasia of respiratory epithelium in the trachea or bronchi entails a loss of ciliated cells and goblet cells, which are important for mucociliary clearance and resistance to pneumonic diseases.
E-Figure 1-14.
Squamous Metaplasia, Esophagus, Parrot.
A, The esophageal mucosa has multiple white raised nodules from squamous metaplasia of mucosal glands. Metaplasia arose from the lack of dietary vitamin A (avitaminosis A). B, Note the squamous metaplasia of the esophageal glands. Vitamin A is necessary for maintenance of the normal epithelium. Avitaminosis A results in the replacement of normal mucosal epithelium and goblet cells in the glands by keratinized stratified squamous epithelium. H&E stain.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Dysplasia
Dysplasia (see Fig. 1-25, B) implies an abnormality in formation of a tissue. For example, renal dysplasia (see Chapter 11) is the abnormal formation of the kidney; hip dysplasia (see Chapter 16) is the abnormal formation of the coxofemoral joint. When applied to epithelium, dysplasia implies an increase in the number of poorly differentiated or immature cells and can be a precursor to neoplasia (see Chapter 6). Microscopically, dysplastic epithelial cells have atypical features, such as abnormal variation in size (anisocytosis) and shape (poikilocytosis), hyperchromatic nuclei, increased nuclear size (karyomegaly), and increased number of mitotic figures.
Intracellular Accumulations
Injured cells can accumulate endogenous by-products and exogenous substances because of metabolic abnormalities, genetic mutations, or exposure to an indigestible exogenous substance. Some of these accumulations are relatively harmless; others promote cellular degeneration and can lead to death of the cell.
Lipids
Lipidosis (steatosis) is the accumulation of lipids within parenchymal cells. Intracellular lipid accumulation can develop in many organs and tissues, but because the liver is so important in lipid metabolism, hepatic lipidosis is particularly common (see Chapter 8). The causes of hepatic lipidosis (Fig. 1-28 ) include increased mobilization of free fatty acids, abnormal hepatocellular metabolism (of fatty acids, triglycerides, and apoproteins), and impaired release of lipoproteins. Grossly, hepatic lipidosis results in a swollen, yellowed liver, with a greasy texture (Fig. 1-29, A ). Severe lipidosis can alter the specific gravity of hepatic parenchyma to the point that slices of liver float in formalin (or water). Histologically, lipid vacuoles (sharply defined and unstained because the lipid is leached by the solvents of histologic processing) distend the hepatocellular cytoplasm and displace the nucleus to the periphery of the cell (see Fig. 1-29, B).
Figure 1-28.
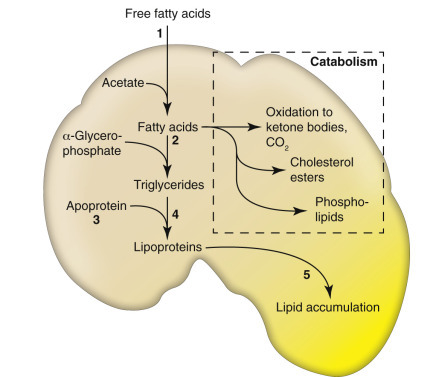
Hepatic Steatosis (Lipidosis).
Schematic of hepatic lipid metabolism (uptake, catabolism, and secretion) and possible mechanisms resulting in lipid accumulation. 1, Excessive delivery of free fatty acids (FFAs) from fat stores or diet. 2, Decreased oxidation or use of FFAs. 3, Impaired synthesis of apoprotein. 4, Impaired combination of protein and triglycerides to form lipoproteins. 5, Impaired release of lipoproteins from hepatocytes.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Figure 1-29.
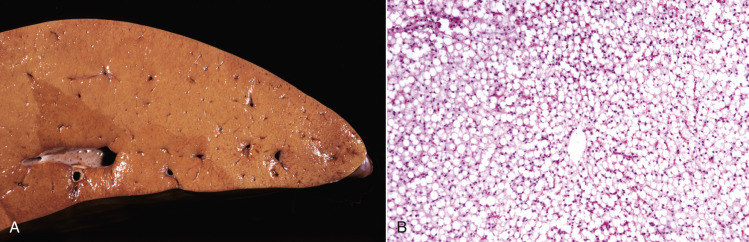
Steatosis (Fatty Liver, Fatty Change, Hepatic Lipidosis), Liver, Ox.
A, Note the uniformly pale yellow-tan color. The liver is enlarged with rounded edges, bulges on incision, and may feel greasy. B, In this severely affected liver, all hepatocytes contain unstained, sharply defined cytoplasmic lipid vacuoles that displace the nucleus to the periphery of the cell. H&E stain.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Glycogen
In homeostasis, glycogen is stored mainly in hepatocytes and in skeletal muscle cells, though the stores are often depleted in starving or sick animals. In contrast, glycogen accumulation can be excessive in certain metabolic abnormalities in skeletal muscle (see Chapter 15), in various organs or tissues in the rare glycogen storage diseases, and in the liver in diabetes mellitus or canine hyperadrenocorticism (see Chapter 12). The hepatic response to hyperadrenocorticism, called glucocorticoid hepatopathy, imparts a swollen, pale brown, and mottled appearance (Fig. 1-30, A ). Histologically, hepatocellular vacuoles of glycogen (see Fig. 1-30, B) are less sharply defined and more irregularly shaped than the vacuoles of hepatic lipidosis.
Figure 1-30.
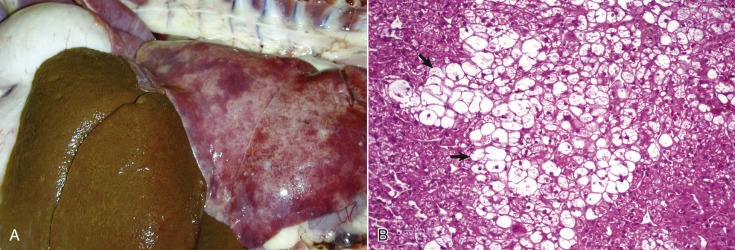
Glucocorticoid Hepatopathy, Liver, Dog.
A, Extensive hepatocellular accumulation of glycogen leads to an enlarged and pale brown liver in dogs with glucocorticoid excess from endogenous or exogenous sources. B, Note the swollen hepatocytes (arrows) with extensive cytoplasmic vacuolation. H&E stain.
(A courtesy Dr. K. Bailey, College of Veterinary Medicine, University of Illinois. B courtesy Dr. J.M. Cullen, College of Veterinary Medicine, North Carolina State University.)
The amount of glycogen that can be demonstrated in hepatocytes microscopically is a function of its original concentration, the delay between death and fixation (during which time the glycogen is metabolized), and the fixation procedure. Although alcoholic fixatives have been recommended to preserve glycogen, fixation in 10% neutral buffered formalin at 4° C retains most of the glycogen without the excessive shrinkage and distortion of tissue seen with alcoholic fixatives, and it avoids polarization of the glycogen to one side of the cell. The periodic acid–Schiff (PAS) histochemistry technique can be used to demonstrate glycogen (Fig. 1-31 ; E-Fig. 1-15 ). The PAS reaction breaks 1,2-glycol linkages to form aldehydes, which are then revealed by Schiff's reagent. The glycol linkages occur in substances other than glycogen, so the PAS technique is often used with and without diastase pretreatment. Diastase digests glycogen and removes it from the histologic section. Thus, if glycogen is the PAS-positive material, pretreatment with diastase will remove it and render the PAS test negative.
Figure 1-31.
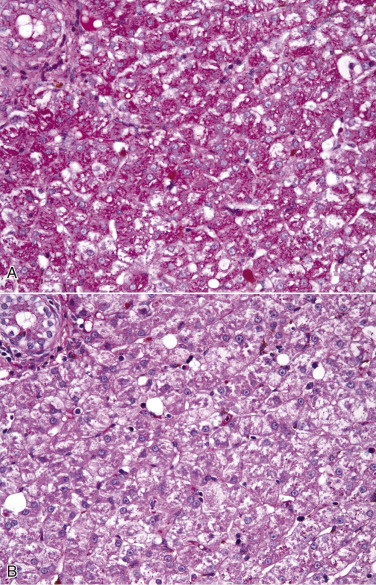
Glycogen Accumulation, Liver, Dog.
A, Glycogen, accumulated in the cytoplasm of hepatocytes, appears as magenta granules with the periodic acid–Schiff technique. B, Hepatocellular glycogen was removed from the histologic section by pretreatment with diastase before application of the periodic acid–Schiff technique.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University.)
E-Figure 1-15.
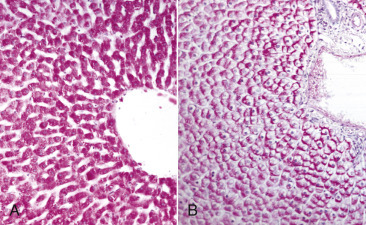
Glycogen, Liver, Dog.
A, Ten-percent buffered neutral formalin fixation at 4° C. Glycogen (purplish red) is uniformly dispersed throughout the cytoplasm of all hepatocytes. Periodic acid–Schiff technique. B, Absolute alcohol (ethanol) fixation at room temperature. The glycogen in each hepatocyte has been pushed to the side of the cell, so-called polarization of glycogen. Periodic acid–Schiff technique.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Proteins
Histologically, proteins are eosinophilic; thus, depending on their biochemical nature (i.e., levels of structural organization [primary through quaternary]), proteins are pink to orange to red in an H&E-stained section.2 In some diseases, proteins account for the “hyaline” appearance observed with H&E stain. The adjective hyaline is used to indicate a homogeneous, eosinophilic, and translucent appearance to a cellular or extracellular substance. Abnormal accumulations of intracellular hyaline proteins occur in various diseases. The protein resorption vesicles in the apical cytoplasm of proximal renal tubular epithelial cells in protein-losing nephropathy (see Chapter 11) appear as hyaline droplets (Fig. 1-32, A ). Hyaline accumulations may also be a normal finding in specific types of cells (e.g., the globular Russell bodies [immunoglobulin-containing protein in distended rER] of plasma cells).
Defects in Protein Folding.
After ribosomal synthesis, emerging proteins are moved into the ER lumen for folding and addition of disulfide bonds before translocation and packaging by the Golgi complex for secretion. Thus the ER is well developed in cells, such as hepatocytes, plasma cells, and pancreatic β cells that synthesize proteins for systemic export. Proteins can be folded into globular (e.g., myoglobin) conformation or exist in a relatively unfolded or disordered state (Fig. 1-33 ). Nevertheless, normal protein function requires correct three-dimensional conformation (i.e., correct folding in the ER and Golgi complex). Protein homeostasis is aided by molecular chaperones that foster the soluble and functional state of proteins, escort proteins to their site of action, assist in protein folding, target misfolded peptides for refolding or degradation, and generally protect against pathologic protein aggregation.
Figure 1-33.
Mechanisms of Protein Folding and the Unfolded Protein Response.
Proteins have different conformational states according to thermodynamic properties, rates of synthesis and degradation, interaction with chaperones, and posttranslational modifications. Amyloidosis is a protein folding disorder in which unfolded or partially unfolded peptides form fibrils that are rich in β-sheets and capable of self-replication.
Hepatocytes, plasma cells, pancreatic islet cells, and other “professional” secretory cells have a sophisticated system that responds to the presence of unfolded proteins. Protein folding disorders develop when an ineffective response to unfolded proteins occurs in these cells. Unfolded proteins can result in “loss-of-function disorders”3 and are usually managed and resolved by ubiquitination and degradation in a proteasome. In these “resolved” situations, there is no accumulation of protein. However, certain protein-folding disorders result in intracellular accumulation or extracellular deposition (see the later section on Extracellular Accumulations) of relatively insoluble proteins, some of which, such as amyloid, are toxic to cells or tissues (see Fig. 1-33).
Other Intracellular Inclusions
Autophagic Vacuoles.
Autophagy is a common response to sublethal cellular injury in which cell membranes are wrapped around portions of the cytoplasm to form an autophagosome. At the ultrastructural level, an autophagosome appears as a double- membrane–bound vesicle with a portion of cytosol and an organelle (e.g., mitochondrion) inside. At the light microscopic level, the autophagosome is an eosinophilic cytoplasmic inclusion. Subsequent fusion with a lysosome leads to at least partial digestion of the autophagolysosome. Residual material may be extruded from the cell or remain as lipofuscin (see later discussion of pigments).
Crystalline Protein Inclusions.
Rhomboidal crystalline protein inclusions, also known as crystalloids (see Fig. 1-32, B), are common in hepatocytes and renal tubular epithelial cells of older dogs. Their significance, other than as a marker of aging, is unknown.
Viral Inclusion Bodies.
Some types of viruses produce characteristic intranuclear or cytoplasmic inclusion bodies. Certain DNA viruses (e.g., herpesviruses, adenoviruses, and parvoviruses) exclusively produce intranuclear inclusions that are round to oval and vary from eosinophilic to basophilic or amphophilic. Other DNA viruses (e.g., poxviruses) produce large eosinophilic cytoplasmic inclusion bodies. The inclusion bodies of RNA viruses (e.g., rabies virus and canine distemper virus) are eosinophilic and cytoplasmic. The viral inclusions of rabies, called Negri bodies, are in the cytoplasm of neuronal soma. Canine distemper virus produces both cytoplasmic and intranuclear inclusions (see Fig. 1-32, C). The intranuclear location of inclusions in this RNA viral infection has been attributed to heat shock proteins.
Lead Inclusions.
In some cases of lead poisoning, intranuclear inclusions develop in renal tubular epithelial cells. The inclusions are a mixture of lead and protein and are more easily observed with acid-fast stains than in H&E-stained sections (see Fig. 1-32, D).
Extracellular Accumulations
Hyaline Substances
Proteins account for the “hyaline” appearance in H&E-stained sections. Proteins are eosinophilic (i.e., have affinity for the eosin dye of the H&E stain). The word hyaline is used for intracellular (see previous section) or extracellular proteinaceous substances that take up eosin dye homogeneously. A variety of extracellular proteinaceous accumulations have a hyaline appearance in histologic sections. Examples include protein casts (albumin, hemoglobin, or myoglobin) in the lumen of renal tubules; serum or plasma in blood vessels; plasma proteins in vessel walls; collagen fibers in some scars or collagen fibers encrusted with proteins from degranulated eosinophils; thickened basement membranes; the “hyaline membranes” of diffuse alveolar damage in acute respiratory distress syndrome (see Chapter 9); fibrin thrombi in the microvasculature in disseminated intravascular coagulation (see Chapter 2); and amyloid (described next).
Amyloid.
Increasingly diseases are recognized to be the result of misfolding of soluble and functional peptides or proteins, converting them into relatively insoluble and nonfunctional aggregates. Amyloidosis is one of the best-studied protein-misfolding disorders. Aggregated proteins can be rather amorphous ultrastructurally; however, in the case of amyloidosis, the misfolded and aggregated proteins have a characteristic highly organized fibrillar structure, even though their amino acid sequence varies. Thus amyloidosis is a biochemically diverse group of disorders that have a common pathogenesis (protein misfolding) and generic morphologic appearance. Not only is the biologic function of the misfolded protein generally lost, but the tissue in which amyloid is deposited may be damaged as well.
The mechanisms of amyloidosis (see Fig. 1-33) include (1) propagation of misfolded proteins that serve as a template for self-replication (e.g., prion diseases), (2) accumulation of misfolded precursor proteins due to failure to degrade them, (3) genetic mutations that promote misfolding of precursor proteins, (4) protein overproduction because of an abnormality or proliferation in the synthesizing cell (e.g., plasma cell dyscrasia or neoplasia), and (5) loss of chaperoning molecules or other essential components of the protein assembly process. Amyloid is typically formed from unfolded or partially unfolded proteins or peptide fragments and has a highly ordered, generic (independent of amino acid sequence) structure of fibrillar polypeptide chains that are rich in cross β-sheets (arranged perpendicular to the axis of the fibrils) and can self-replicate by virtue of this template formation. Amyloidosis was recognized as a disease by Virchow, who dubbed the offending material amyloid (starchlike) because the tissue deposits were stained with iodine. Iodine is still used on occasion as a gross technique to stain amyloid (see Fig. 11-40), even though amyloid deposits consist mainly of protein (typically associated with other molecules, such as carbohydrate moieties). If visible macroscopically, amyloid appears as yellow, waxy, coalescing nodular or amorphous deposits (Fig. 1-34 ). At the light microscopic level, amyloid is homogeneous to indistinctly fibrillar, and pale eosinophilic (Fig. 1-35, A ). With the Congo red stain, amyloid takes a more orange-red hue (i.e., congophilia) (see Fig. 1-35, B). Because of its molecular periodicity, amyloid is anisotropic. Anisotropic substances are birefringent (i.e., they can refract polarized light into two rays that vibrate in perpendicular waves). Thus, when histologic sections are viewed through the microscope with polarized light (achieved by inserting a polarizing filter between the light source and the histologic section), amyloid deposits or other anisotropic substances (e.g., crystals, collagen) can rotate the plane of light so that it passes through the analyzer (a second polarizing filter between the histologic section and the oculars), whereas negligible light is passed through isotropic substances (most of the rest of the section). Amyloid has characteristic apple-green birefringence in polarized light, especially with the Congo red stain (see Fig. 1-35, C). Ultrastructurally amyloid appears as extracellular bundles of nonbranching filaments that are 7 to 10 nm in diameter.
Figure 1-34.
Amyloidosis, Conjunctiva, Horse.
The palpebral conjunctiva (upper eyelid) in this horse with nasal and conjunctival amyloidosis is thickened by coalescing, waxy yellow nodules of amyloid in the subepithelial tissue.
(Courtesy Dr. E.D. Conway, College of Veterinary Medicine, Purdue University.)
Figure 1-35.
Equine Nasal Amyloidosis.
A, The amyloid appears as homogeneous to faintly fibrillar, pale eosinophilic deposits (arrows) in the nasal mucosal interstitium. H&E stain. B, Congophilic substances, such as amyloid, are red-orange with Congo red stain. C, Viewed with polarized light, amyloid is birefringent and “apple-green.” Congo red stain, same field and same magnification as in B.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University.)
Classification and Localization of Amyloidosis.
Amyloid can be classified by the biochemical identity of its precursor peptide or protein. AL amyloid consists of immunoglobulin light chains derived from plasma cells. In light chain (AL) amyloidosis, abnormal plasma cells secrete the light chain fragments into the circulation, and the amyloid can be deposited almost anywhere in the body. Amyloidosis is considered primary when dyscrasias or neoplastic proliferations of plasma cells (see also Chapter 5) are the source of the amyloid. AL amyloidosis can be systemic, but in some extramedullary (e.g., cutaneous) plasmacytomas, amyloid deposition is limited to the stroma of the neoplasm. The localized deposits in nasal amyloidosis of horses (see Fig. 1-35) consist of AL amyloid. The conjunctiva (see Fig. 1-34) and skin are also affected in some horses with nasal AL amyloidosis. AL amyloid retains its congophilia and apple-green birefringence after pretreatment with potassium permanganate.
In systemic amyloidosis associated with chronic inflammation, and therefore classified as secondary, serum amyloid A (AA) protein (produced mainly by hepatocytes) is cleaved into fragments that are deposited as amyloid fibrils in various tissues, particularly the kidney (especially renal glomeruli; E-Fig. 1-16; see Chapter 11 and Figs. 11-32, 11-33, and 11-34Fig. 11-32Fig. 11-33Fig. 11-34), liver (especially the space of Disse; see Chapter 8 and Fig. 8-44), and splenic white pulp (see Fig. 13-61). Hereditary or familial forms of AA amyloidosis are also recognized. In Shar-Pei dogs and Abyssinian cats, AA amyloid deposits are typically most abundant in the renal medullary interstitium, rather than in renal glomeruli. Amyloid A is sensitive to potassium permanganate (i.e., congophilia and apple-green birefringence are lost or diminished after potassium permanganate pretreatment).
E-Figure 1-16.
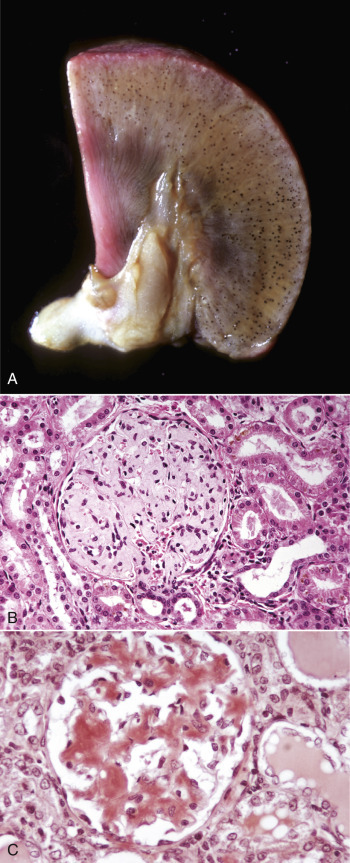
Amyloidosis, Kidney, Dog.
A, Note the blue-black foci, which are glomeruli containing amyloid stained by Lugol's iodine. B, The renal glomerulus contains extracellular deposits of pale homogeneous eosinophilic amyloid. H&E stain. C, The amyloid in the glomerulus stains red-orange. Congo red stain.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Amyloid deposits can be systemic (extracellular deposits in multiple organs or tissues, independent of the site of synthesis of the precursor protein) or localized (restricted to tissues in which the precursor protein or peptide is synthesized). Systemic amyloidosis is more likely to be life threatening, depending on the organs or tissues involved and on the volume of amyloid deposits. Thus diffuse and severe renal glomerular amyloidosis results in a protein-losing nephropathy (see Chapter 11).
In contrast to systemic amyloidosis, the severity of disease in localized amyloidosis may depend more on the biochemical nature of the amyloid fibrils. In fact, the precursor peptides or intermediate oligomers, rather than the mature amyloid fibrils, are thought to be the injurious agent at least in some forms of localized amyloidosis. The amyloid deposited in pancreatic islets of cats and human beings is derived from islet amyloid peptide and is secreted by the β cells. It can be associated with insulin-resistant (type 2) diabetes mellitus, but islet amyloidosis is also encountered in cats with normal glucose tolerance (see Chapter 12). Another example of localized amyloidosis is the accumulation of β-amyloid (Aβ) in the cerebral cortex of aged dogs with canine cognitive disorder and in human beings with Alzheimer's disease.
Other Extracellular Accumulations
Fibrinoid Change.
Fibrinoid change is the result of leakage of plasma proteins, such as immunoglobulin, complement, or fibrin, into the wall of a blood vessel. This lesion is observed in septic or immune-mediated vasculitis. Injury, such as that caused by viruses or endotoxin, to endothelial cells, basement membrane, or smooth muscle cells of the tunica media can activate the acute phase inflammatory response leading to circumferential deposition of plasma proteins in blood vessel walls. These proteins, especially fibrin, are intensely eosinophilic and can be accompanied by leukocytic infiltration (Fig. 1-36 ; see also Chapter 3).
Figure 1-36.
Fibrinoid Change, Artery.
Note the deeply eosinophilic circumferential deposits in the arterial tunica media. The fibrinoid change is accompanied by leukocytic infiltration and medial necrosis. H&E stain.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Collagen (Fibrosis).
Fibrosis is an excess in fibrous collagen, predominantly type I collagen fibers, in the interstitium of organs or tissues. Necrosis, especially necrosis that destroys epithelial basement membranes, but also necrosis of mesenchymal tissues, tends to induce proliferation of fibroblasts. In many injured tissues, especially beneath ulcers or in wound healing, fibroblastic proliferation is accompanied by endothelial proliferation with formation of granulation tissue (fibrosis plus neovascularization [see Chapters 3 and 17]). As granulation tissue matures, the neovascularization subsides, fibroblasts become quiescent, collagen fibers remain, and scar tissue is the end result. In the liver (see Chapter 8), stellate cells are the source of the collagen in fibrosis. Macrophages (i.e., Kupffer cells in the liver, histiocytes, or other tissue macrophages) direct fibrosis by release of cytokines and growth factors such as TNF-α and TGF-β.
Fatty Infiltration.
Fatty infiltration is an increase in the number and/or volume of adipocytes in the interstitium of an organ or tissue. Thus it is distinct from the intracellular accumulations known as lipidosis or steatosis. Normally adipocytes are present in small numbers in the myocardial interstitium, especially near the epicardium, and in skeletal muscle bundles. The adipocytes can increase in size and number in obesity and in certain cardiomyopathies (see Chapter 10) or skeletal myopathies (see Chapter 15 and Fig. 15-9). Adipocytes also accumulate in atrophied tissues, such as skeletal muscle (particularly when the result of denervation; see Chapter 15), thymus, and thyroid gland (see Fig. 1-26, B).
Gout
Gout is the deposition of sodium urate crystals or urates in tissue. It occurs in primates, birds, and reptiles but has not been reported in domestic mammals.
In the most common human form, urate crystals are deposited in the articular and periarticular tissues and elicit an acute inflammatory response characterized by the presence of neutrophils and macrophages and aggregates of urate crystals called tophi. Tophi may be visible grossly and are pathognomonic for gout. Later in the course of the disease, the inflammation becomes chronic, and a foreign body reaction to the tophi develops. Microscopically, urate crystals are acicular, and they, or (more accurately) the spaces left after they have been dissolved by histologic processing, are surrounded by neutrophils, macrophages, and multinucleated giant cells.
Birds and reptiles suffer two forms of gout, namely (1) the articular type, which is rare, and (2) a visceral type. The latter characteristically affects the visceral serosae, particularly the parietal pericardium, and the kidneys. The serosa is covered with a thin layer of gray granules, and the gross appearance is diagnostic. In the renal form, urate deposits are visible in renal tubules and ureters. Uric acid and urates are the end products of purine metabolism, and in birds and reptiles these products are eliminated as semisolid urates. Visceral gout is usually diagnosed at autopsy and is seen sporadically as the result of vitamin A deficiency, high-protein diets, and renal injury.
Pseudogout
Pseudogout is characterized by deposits of calcium pyrophosphate crystals. It is well recognized in human beings and has been reported but is rare in the dog. The pathogenesis of the canine disease is unknown, but in human beings, one form is inherited as an autosomal dominant trait. Grossly, there are chalky white deposits of the crystalline material in the joints, with a chronic reaction of macrophages and fibrosis. The disease can be differentiated from gout by chemical analysis of the crystalline deposits.
Cholesterol.
Cholesterol crystals are dissolved out of the tissue specimen during histologic processing, leaving characteristic acicular (needle-shaped) clefts in histologic sections. In three dimensions the crystals are thin rhomboidal plates with a notched corner. Cholesterol crystals often form in tissue at sites of hemorrhage or necrosis. They are present in atheromas (degenerated arterial intima plaques in atherosclerosis); however, with the exception of hypothyroid dogs, atherosclerosis is not common in domestic mammals. Cholesterol crystals typically elicit granulomatous inflammation (Fig. 1-37 ) and are common in the cholesterol granulomas (“cholesteatomas”) in the choroid plexus of old horses; these can become large enough to obstruct the flow of cerebrospinal fluid but more often are an incidental finding. Grossly, the cholesterol granuloma appears as friable pale yellow nodules in the choroid plexus of the lateral or fourth ventricles (see Chapter 14 and Fig. 14-87).
Figure 1-37.
Cholesterol Granuloma, Mammary Gland, Dog.
Note the acicular cholesterol clefts (cholesterol is removed from the histologic section in processing) with granulomatous inflammation. H&E stain.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University.)
Pathologic Calcification
Pathologic calcification refers to the deposition of calcium salts, typically as phosphates or carbonates, in soft tissues (i.e., tissues that would not be calcified in a healthy state). Soft tissue calcification as the result of elevated serum calcium concentration is termed metastatic calcification, whereas the calcification of dead tissue as part of the process of necrosis is called dystrophic calcification. If calcification is extensive, it appears grossly as chalky white deposits (Fig. 1-38 ) with a brittle or gritty texture. Calcium deposits that also contain hemosiderin or other blood pigments (see subsequent section on Pigments) may be discolored yellow-brown.
Figure 1-38.
Calcification, Vitamin E or Selenium Deficiency, Heart, Lamb.
The chalky white lesions are areas of myocardial necrosis that have been calcified.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Dystrophic Calcification
A review of the biochemical events in cell death explains dystrophic calcification. Recall that loss of the ability to regulate cellular Ca2+ balance is a critical turning point that converts reversible to irreversible injury. Ischemia opens membrane calcium channels, leading to increased intracellular calcium concentration, which is normally sequestered in the cytosol, ER, and mitochondria, each with its own Ca2+-ATPase membrane pumps. The increased intracellular calcium concentration activates calpains, which cleave Na+/Ca2+ exchangers in mitochondrial and other cell membranes, leading to decreased efflux of Ca2+ and decreased reuptake of Ca2+ by the ER. Thus calcium overload is an expected sequel to cell death. Dystrophic calcification is most prominent in mitochondria and is first evident histologically as a basophilic stippling of the dead cell. With increasing deposition of calcium salts, the entire cell and even extracellular tissue can be calcified, resulting in more intense and widespread basophilia. Calcification is the gross lesion for which the myocardial and skeletal muscle necrosis of vitamin E or selenium deficiency in ruminants was named white muscle disease (see Fig. 1-38). Calcification is also prominent in other forms of necrosis (e.g., in the caseous necrosis of tuberculoid granulomas), in parasitic granulomas, and in necrotic fat or lipomas (benign neoplasms of adipocytes).
Calcification of the skin (see Chapter 17) is categorized as (1) calcinosis cutis, a poorly understood form of epithelial and collagenous calcification seen mainly in canine hyperglucocorticoidism, and (2) calcinosis circumscripta. Calcinosis circumscripta is a localized deposit of calcium salts in the dermis or subcutis, and less often in other soft tissues or in the tongue. It is common over bony prominences of distal aspects of the limbs in young dogs of the large breeds but can occur in other species (e.g., horses). It is probably a form of dystrophic calcification and usually attributed to repetitive trauma.
Metastatic Calcification
Metastatic calcification targets the intima and tunica media of vessels, especially those in the lungs, pleura, endocardium, kidneys, and stomach. The primary defect is an imbalance in calcium and phosphate concentrations in the blood.
In chronic kidney disease, phosphate retention is the cause of the calcium-phosphate imbalance (see Chapter 11). In “uremic gastropathy” the damage to gastric arteries and arterioles results in ischemic injury and metastatic calcification in the gastric mucosa. The metastatic calcification of renal failure is also prominent in the lungs, pleura, and endocardium. In an H&E-stained section, metastatic calcification imparts a subtle basophilic stippling (Fig. 1-39, A ). The von Kossa histochemical technique blackens the calcium phosphate or calcium carbonate salts (see Fig. 1-39, B).
Figure 1-39.
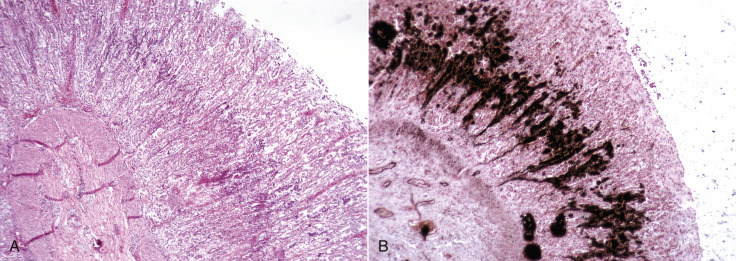
Uremic Calcification, Stomach, Dog.
A band of calcification is in the middle of the gastric mucosa. A, The calcium salts are basophilic (stained blue with hematoxylin). H&E stain. B, The calcium salts are black with the von Kossa technique for mineralization.
(A and B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Toxicosis with vitamin D or its analogues is also characterized by calcium-phosphate imbalance. Cestrum diurnum, a plant introduced from the West Indies to the Gulf Coast of the United States, is poisonous to herbivores because it contains glycosides of 1,25-dihydroxycholecalciferol (1,25-(OH)2D3) that cause elevated serum calcium concentration and often severe metastatic calcification of the lungs, kidney, and heart, especially the atrial endocardium and ascending aorta. Dogs and cats can be poisoned by consumption of rodenticides containing cholecalciferol.
Inappropriately elevated concentrations of parathyroid hormone (PTH) or secretion of PTH-related peptide cause hypercalcemia and metastatic calcification (see also Chapter 12). Primary hyperparathyroidism, usually the result of neoplasia of the parathyroid glands, is uncommon. Certain nonparathyroid neoplasms are associated with the so-called humoral hypercalcemia of malignancy (aka pseudohyperparathyroidism), either because the neoplastic cells secrete PTH-related peptide or because the neoplasm invades and lyses bone. Canine lymphoma and apocrine carcinoma of the anal sac glands are two tumors that can secrete PTH-related peptide.
Heterotopic Ossification
Heterotopic ossification is the formation of bony tissue at an extraskeletal site. It entails osteoid (bone matrix) deposition by osteoblasts with remodeling and mineralization to form bone. Although calcification is part of the process of ossification, whether skeletal or extraskeletal, and heterotopic ossification can develop in chronic lesions of soft tissue calcification, pathologic calcification of soft tissue does not necessarily entail ossification.
Heterotopic ossification appears grossly as hard spicules or nodules. Small bony spicules are commonly encountered as incidental findings in the pulmonary interstitium (Fig. 1-40 ) of old dogs. Nodular deposits of cartilage and bone may form the bulk of a canine mixed mammary tumor (see Chapter 18) in which the myoepithelial cells are thought to give rise to chondrocytes and osteoblasts.
Figure 1-40.
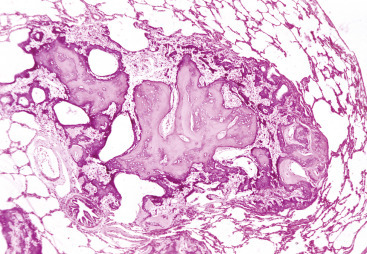
Ectopic Bone, Lung, Dog.
A nodule of mature bone in the connective tissue of the lung. H&E stain.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Pigments
Various exogenous and endogenous substances can alter the color of tissues. These color changes may be evident clinically or at least macroscopically at autopsy and can be diagnostically useful. Though some pigmented substances disappear from histologic sections, others remain and must be interpreted by the pathologist.
Exogenous Pigmented Substances
Carbon and Other Dusts.
Coal mine dust lung disease, also known as black lung, is the best-studied example of pneumoconiosis (lung disease due to inhalation of dusts; see Chapter 9). The major dust inhaled by coal mine workers is carbon, so this form of pneumoconiosis is called anthracosis. Carbon particles in the lung account for the black discoloration in anthracosis. Many cases, especially with the lower exposure in urban-dwelling people or animals that breathe polluted air, are not associated with clinical disease but impart a fine gray-black stippling to the lung (Fig. 1-41, A ), visible through the visceral pleura, plus a dark gray discoloration of tracheobronchial lymph nodes. Carbon particles deposited in alveolar spaces are phagocytized by macrophages and then transported to bronchus-associated lymphoid tissue and on to tracheobronchial lymph nodes. Histologically, the indigestible carbon particles and other inhaled dusts appear as fine black granular material and crystalline material in macrophages in extracellular tissues adjacent to intrapulmonary airways and vasculature (see Fig. 1-41, B). This finding is usually incidental in older animals, but coal dust and other mineral dusts, especially silica,4 can elicit an inflammatory response with release of TNF-α and interleukin 1 (IL-1) and interleukin 6 (IL-6). These cytokines can promote progressive fibrosis. Macrophages laden with carbon particles are also thought to have diminished capacity to phagocytize and destroy infectious agents.
Figure 1-41.
Anthracosis, Lung, Aged Dog.
A, The fine black subpleural stippling represents peribronchiolar deposits of carbon. B, Inhaled carbon (black) has been phagocytized by macrophages and transported to the peribronchial/peribronchiolar tissue. H&E stain.
(A and B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Carotenoid Pigments.
Carotenoid pigments, such as β-carotene, are abundant in leafy green plants and impart a yellow coloration to plasma, adipose tissue, and other lipid-laden cells. The deep yellow color of adipose tissue in herbivores on lush green pasture can be striking, especially in horses and dairy cattle of high milk-fat breeds, such as Jersey dairy cattle (Fig. 1-42 ). This discoloration is not a lesion but just a dietary indicator. Indeed, the carotenoids stored in fat are a source of antioxidants. Because carotenoids are fat soluble, they are removed from histologic sections by the solvents used in processing.
Figure 1-42.
Carotenosis, Kidney and the Perirenal Fat, Jersey Ox.
Accumulation of carotenoids in the adipocytes has colored the fat yellow to dark yellow.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Tetracycline.
The antibiotic tetracycline binds to calcium phosphate in teeth and bones. If administered to animals during the time of mineralization of the teeth, tetracycline results in permanent discoloration. Initially the staining is yellow, but after tooth eruption and exposure to light, oxidation changes the color to brown (Fig. 1-43 ). Yellowish discoloration (with bright yellow fluorescence under ultraviolet light) is also observed in bone.
Figure 1-43.
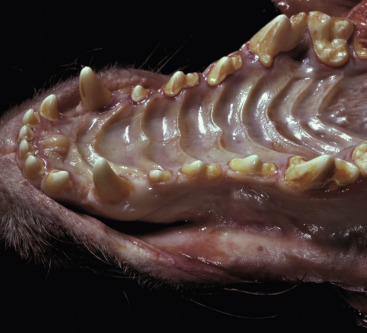
Tetracycline Staining, Teeth, Young Dog.
The yellow-brown discoloration of the permanent teeth is the result of tetracycline therapy during their development.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Nonhematogenous Endogenous Pigments
Melanin.
Melanin is the pigment responsible for the color of the hair, skin, and iris. It also colors the leptomeninges in black-faced sheep (Fig. 1-44 ) and cattle and may be present multifocally in oral mucosa in various species. Localized deposits of melanin (melanosis) are common in the aortic intima in ruminants with pigmented coats and in the lungs (Fig. 1-45 ) of red or black pigs. The localized deposits in congenital melanosis are merely a color change and not a lesion because they are not a response to injury and have no ill effect on the animal.
Figure 1-44.
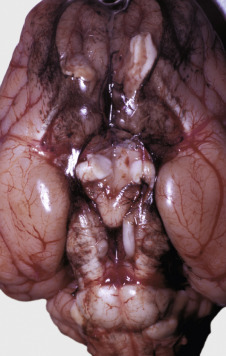
Congenital Melanosis, Leptomeninges, Suffolk Sheep.
The leptomeninges have scattered black areas of melanin. This pigmentation is normal in black ruminants.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Figure 1-45.
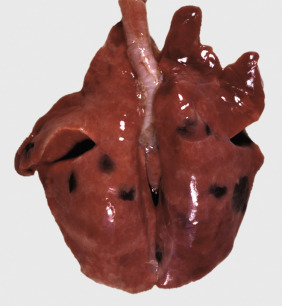
Congenital Melanosis, Lung, Pig.
Melanin deposits are subpleural and extend into pulmonary parenchyma. This pigmentation, seen mainly in red or black pigs, has no detrimental consequences.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
The melanocytes that synthesize and secrete melanin are derived from the neural crest and migrate to the site of pigment production during embryonic development of the structure. In the skin, melanocytes reside in the stratum basale of the epidermis and follicular epithelium. Melanin is formed in organelles called melanosomes, then transferred through dendritic cell processes to adjacent keratinocytes. In the keratinocyte, melanin granules are mainly in the apical cytoplasm, where they may shield the nucleus from ultraviolet light. Histologically, melanin granules are small (usually less than 1 µm in diameter), brown, and nonrefractile.
Melanin pigment can be diminished or excessive in disease. The first step in melanin synthesis is the conversion of tyrosine to dihydroxyphenylalanine (DOPA), catalyzed by the copper-containing enzyme, tyrosinase. Thus a lack of tyrosinase results in albinism (lack of melanin pigmentation), and sheep and cattle with copper deficiency have defective tyrosinase and fading of coat color. Partial albinism in Chédiak-Higashi syndrome (CHS) (recognized in people, mink, Persian cats, mice, and other species) is caused by a mutation of the LYST gene that codes for a lysosomal trafficking regulator protein. The mutation causes abnormal lysosomal structure and function in leukocytes and in melanocytes. The melanocytes of animals with CHS have enlarged melanosomes, but the melanin pigment is not transferred effectively to keratinocytes, so coat color is a pastel shade of what it should have been. Normally pigmented skin and hair can also become depigmented because of an immune-mediated attack on melanocytes (vitiligo) or basilar keratinocytes (see Chapter 17). The dead keratinocytes spill their melanin into adjacent dermis in a process called pigmentary incontinence, where it is phagocytized by macrophages (melanophages).
The term hyperpigmentation implies excessive melanin. This finding can be a common epidermal response to chronic injury and appears as darkened skin. Endocrine skin disease, especially hyperadrenocorticism, is often associated with hyperpigmentation. Histologically, melanin granules are numerous, not only in the basilar keratinocytes, but in all layers of the epidermis, even the stratum corneum. Neoplasms of melanocytes can be darkly pigmented or not pigmented at all (amelanotic) (see Chapters 6 and 17).
Lipofuscin and Ceroid.
Lipofuscin is a yellow-brown lipoprotein that accumulates as residual bodies in secondary lysosomes, especially in long-lived postmitotic cells, such as neurons and cardiac myocytes (Fig. 1-46 ), and especially in aged animals. It is known as a “wear and tear” pigment of aging—its accumulation in canine myocardium has a linear correlation with the age of the dog—and is generally thought to have little or no deleterious effect on the cell. Lipofuscin is autofluorescent with an excitation wavelength between 320 and 480 nm and emission wavelength between 460 and 630 nm. It is approximately two-thirds heterogeneous protein and one-third lipid (mainly triglycerides, free fatty acids, cholesterol, and phospholipids). Because of its lipid content, lipofuscin reacts with fat stains such as Sudan black B or Oil Red O; its carbohydrate moieties make it also PAS positive.
Figure 1-46.
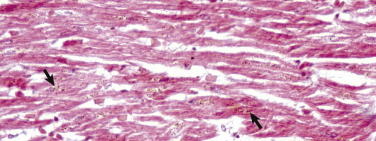
Lipofuscinosis, Heart, Dog.
Note the brown lipofuscin granules (arrows) in the cytoplasm of cardiac myocytes. H&E stain.
(Courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Ceroid is a lipofuscin-like (i.e., same morphologic appearance) pigment that accumulates in disease states, such as neuronal ceroid-lipofuscinosis (a group of hereditary lysosomal storage diseases), cachexia, vitamin E deficiency, or other oxidative stress. Ceroid can be grossly evident in the tunica muscularis of the small intestine of dogs with vitamin E deficiency (leiomyometaplasia [brown dog gut]; see Fig. 7-112) or dogs with ceroid-lipofuscinosis (Fig. 1-47 ).
Figure 1-47.
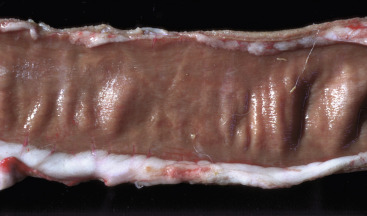
Ceroid, Intestine, Serosal Surface, Dog.
Note the brown discoloration of the muscular layer. The condition has been called intestinal lipofuscinosis but is not age-related.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Lipofuscin and ceroid have strikingly similar histologic and biochemical characteristics, yet are distinct. Both are autofluorescent lipoproteins with similar but not identical spectra. Ultrastructurally, lipofuscin has a granular appearance, whereas ceroid is more likely to form membranous stacks or whorls (“myelin figures”). Although both compounds are composed of proteins, lipids, dolichols, carbohydrates, and metals, their exact composition varies. Whereas the protein content of lipofuscin is heterogeneous, subunit c of mitochondrial ATP synthase is the predominant component of ceroid in neuronal ceroid-lipofuscinosis. Lectin histochemistry is useful to distinguish neuronal ceroid from lipofuscin by its sugar moieties.
Hematogenous Pigments
Hematogenous pigments are derived from erythrocytes. They include hemoglobin, hematins, hemosiderin, hematoidin, bilirubin, biliverdin, and porphyrins.
Hemoglobin.
The hemoglobin molecule consists of four globular protein subunits, each folded around and tightly associated with a central nonprotein, iron-containing heme group. Oxyhemoglobin, formed when oxygen binds to the heme group, gives oxygenated (arterial) blood its red color and imparts a pink tinge to well-perfused and well-oxygenated tissues. Deoxygenated hemoglobin explains the blue cast to venous blood and accounts for the blue to purple discoloration, known as cyanosis (Fig. 1-48 ), of hypoxic tissues. The word cyanosis comes from the Greek word for dark blue.
Figure 1-48.
Cyanosis, Paw, Cat.
The pads of the paw on the left are bluish due to deoxygenated hemoglobin, the result of obstruction of the iliac artery by a saddle thrombus at the aortic bifurcation. The pads of the normal paw (on the right) are pink.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Toxic or Other Metabolic Disorders of Hemoglobin
Cyanide.
Cyanide (CN−) is a toxic compound that, when ingested, blocks oxidative phosphorylation in mitochondria by binding cytochrome oxidase. As a result, cells cannot use the oxygen in hemoglobin, so venous blood in cases of cyanide poisoning tends to be as red as arterial blood. Cyanide poisoning in herbivores is usually the result of consumption of plants that contain cyanogenic glycosides.
Carbon Monoxide.
Hemoglobin has a much higher affinity for CO than for oxygen, so even a small amount of CO reduces oxygen transport capacity. When hemoglobin binds CO, it forms carboxyhemoglobin, which colors the blood bright cherry red and imparts a bright pink color to the tissues even in fatal cases of CO poisoning (E-Fig. 1-17).
E-Figure 1-17.
Carbon Monoxide (CO) Poisoning, Brain, Human.
The blood in the brain is cherry red from the carboxyhemoglobin formed by the inhalation of CO in exhaust gases.
(Courtesy Dr. J.C. Parker, School of Medicine, University of Louisville.)
Nitrite Poisoning.
Nitrite poisoning can be associated with consumption of nitrate-accumulating plants by livestock, usually ruminants, or from a water source contaminated with nitrate runoff from fertilized fields. Nitrate is converted in the rumen to nitrite, which can oxidize the iron in the heme group of the hemoglobin molecule to the Fe+3 (ferric) state, converting hemoglobin to methemoglobin, which has low affinity for oxygen. Methemoglobin turns the color of blood to a chocolate brown (Fig. 1-49 ).
Figure 1-49.
Methemoglobinemia, Experimental Nitrite Poisoning, Hind Quarters, Pig.
Left, The methemoglobin has discolored the blood and musculature chocolate brown. Right, Normal control.
(Courtesy Dr. L. Nelson, College of Veterinary Medicine, Michigan State University.)
Intravascular Hemolysis (Hemoglobinuria).
If erythrocytes are lysed within vessels (intravascular hemolysis), the released hemoglobin imparts a transparent pink tinge to the plasma or serum. In the kidneys, intravascular hemoglobin passes through glomerular capillaries into the urinary filtrate with the formation of hemoglobin “casts” in renal tubules and reddish discoloration of the urine. Hemoglobinuria turns the color of renal parenchyma a dark red to gunmetal blue (Fig. 1-50 ; see Fig. 11-39, A and B). A similar or browner discoloration of kidney and urine occurs with myoglobinuria; the myoglobin is derived from injured skeletal muscle fibers.
Figure 1-50.
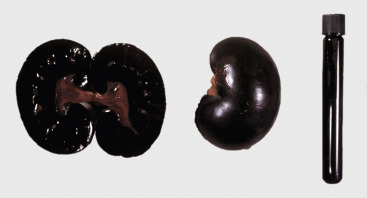
Hemolytic Crisis in Chronic Copper Poisoning, Kidneys and Urine, Sheep.
The dark bluish color of the kidney and the dark red of the urine are caused by hemoglobinuria (hemoglobin excreted via the kidney).
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Hematin.
Hematin is a brown-black, Fe+3-containing pigment formed by the oxidation of hemoglobin.
Acid Hematin (Formalin Pigment).
The “acid” hematin that forms in tissues fixed in unbuffered, and therefore acidic (pH < 6), formalin appears as dark brown to nearly black, granular or crystalline material mainly in vessels or other areas of the tissue section where erythrocytes (and hemoglobin) are numerous (Fig. 1-51 ). The presence of acid hematin is a postmortem change and therefore not a lesion, but rather an indicator that the formalin solution was not properly buffered. Correctly prepared phosphate-buffered 10% formalin should have a pH of 6.8. Acid hematin can be so abundant in congested tissues that it hinders histologic evaluation. In these cases, hematin can be removed by soaking the dewaxed tissue section before H&E staining in a saturated alcoholic solution of picric acid.
Figure 1-51.
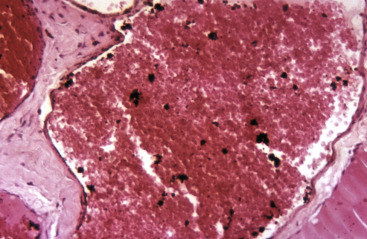
Formalin Pigment, Blood.
Note the black specks of acid hematin on and around erythrocytes, the result of fixation in unbuffered (acidic) formalin. H&E stain.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Parasitic Hematin.
Parasites that infect (e.g., Plasmodium spp.) or consume (e.g., Haemonchus contortus) erythrocytes liberate heme during the proteolysis of hemoglobin. Free heme is toxic, but the parasites have evolved to aggregate it into heme dimers called hemozoin or β-hematin. Hematin accounts for the blackening of the migration tracts of juvenile liver flukes (Fascioloides magna) in ruminants (Fig. 1-52 ; see also Figs. 8-60 and 8-61) and for the black speckling of the lungs in macaques infested with the lung mite, Pneumonyssus simicola.
Figure 1-52.
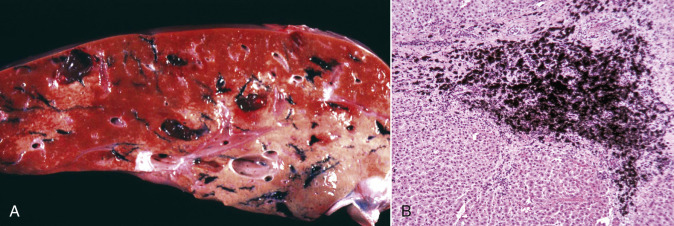
Hematin pigment from Fascioloides magna, Liver, Ox.
A, Blackened areas in the liver are the result of hematin pigment excreted by migrating trematode larvae. B, Hematin (black) pigment in a fluke migration tract. H&E stain.
(A courtesy Dr. J. Wright, College of Veterinary Medicine, North Carolina State University; and Noah's Arkive, College of Veterinary Medicine, The University of Georgia. B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Hemosiderin.
Free iron is toxic to cells because it catalyzes the formation of ROS via the Fenton reaction. However, ferritin, a globular iron storage protein present in all tissues and particularly in the liver, spleen, and bone marrow, binds free iron and stores it in a nontoxic form available for use by the cell. Ferritin is mainly an intracellular protein, but serum concentrations correlate with iron stores. Accumulations of ferritin bound with iron, mainly in macrophages, are converted to golden brown granules of hemosiderin (Fig. 1-53, A ). The Prussian blue reaction detects the iron in hemosiderin (see Fig. 1-53, B) in histologic tissue sections.
Figure 1-53.
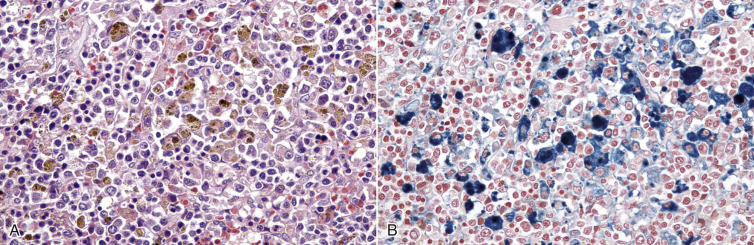
Hemosiderosis, Spleen, Dog.
A, Hemosiderin appears as golden brown granules in macrophages. H&E stain. B, Granules of hemosiderin are stained blue by the Prussian blue reaction, which is specific for iron. Prussian blue reaction.
(A and B courtesy Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Hemosiderin is an intracellular iron storage complex, especially common in macrophages and less so in hepatocytes and renal tubular epithelial cells. Iron stores are most conspicuous in the spleen and are excessive (hemosiderosis) when there is an increased rate of destruction of erythrocytes. Rarely, excess iron can be derived from the diet (e.g., hemochromatosis, a more severe iron storage disease) or other external sources. The presence of hemosiderin-laden macrophages can also be an indicator of chronic passive congestion (Fig. 1-54, A ). If abundant, hemosiderin imparts a brownish discoloration to tissues that should be pink (see Fig. 1-54, B). Hemosiderin is also one of the pigments that typifies a bruise (Fig. 1-55 ).
Figure 1-54.
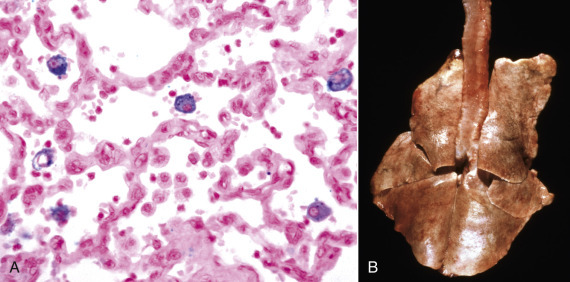
Chronic Passive Congestion, Lung, Dog.
A, Macrophages containing hemosiderin (blue) are in the alveolar spaces. Prussian blue reaction. B, Chronic passive congestion of the lungs results in brownish discoloration because of the numerous hemosiderin-laden alveolar macrophages. Inflammatory mediators produced by these macrophages induce interstitial fibrosis, which caused the failure of the lungs to collapse upon opening of the thoracic cavity. Note the striped appearance of the lungs from rib imprints.
(A courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee. B courtesy College of Veterinary Medicine, University of Illinois.)
Figure 1-55.
Subcutis, Old Bruise, Leg, Horse.
The display of colors—red, yellow, and brown—is due to hemoglobin, bilirubin, and hemosiderin, respectively, from the breakdown of the erythrocytes.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Hematoidin.
Hematoidin is a bright-yellow crystalline pigment that is derived from hemosiderin, presumably within macrophages, but is free of iron. It is similar or identical to bilirubin, biochemically, and is deposited in tissues at sites of hemorrhage.
Bilirubin.
Bilirubin is normally present in low amounts in the plasma as a breakdown product of erythrocytes (see Chapters 8 and 13). Effete erythrocytes are phagocytized and lysed by macrophages. The globular protein components of hemoglobin are broken down into amino acids. After removal of iron, the rest of the heme is converted by heme oxygenase to biliverdin, then by biliverdin reductase to bilirubin. The unconjugated bilirubin is released into the blood to be carried as an albumin-bilirubin complex to the liver for conjugation with glucuronic acid and secretion into the bile canaliculus, where it becomes a component of bile.
If the elevation in serum or plasma bilirubin level (hyperbilirubinemia) is sufficient, it will result in the yellow staining of tissues called icterus or jaundice. Icterus is often classified pathogenetically as prehepatic, hepatic, or posthepatic (see Chapter 8). Prehepatic icterus is caused by hemolysis or any process that increases the turnover of erythrocytes and delivers more unconjugated bilirubin to the liver than it can accommodate. Hepatic icterus is the result of hepatocellular injury that decreases the uptake, conjugation, or secretion of bilirubin. In posthepatic icterus, it is the outflow of bile from the liver into the intestine via the biliary system that is reduced by an obstruction.
Mutant Corriedale (Fig. 1-56 ) and Southdown sheep develop conjugated hyperbilirubinemia that is attributed to a defective ATP-dependent transport system for various organic anions, including bilirubin diglucuronide. Affected animals have a disease similar to the human Dubin-Johnson syndrome and can conjugate bilirubin but cannot secrete it into the bile efficiently.
Figure 1-56.
Defective Bilirubin Excretion, Mutant Corriedale Sheep, Animal Model for Dubin-Johnson Syndrome.
Note the faint yellow discoloration of the lung from bilirubin. The other tissues are discolored dark green from phylloerythrin, which also has a similar defect in excretion in the liver.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Grossly, the yellow discoloration of icterus is easiest to see in pale or colorless tissues, such as plasma, the sclera, intima of the great vessels, adipose tissue (unless it is already yellowed by carotenoids), and even in a pale liver (Figs. 1-57 and 1-58, A ). Icterus is not observed histologically but is often associated with cholestasis, the distension of canaliculi by yellow-brown “casts” of bile (see Fig. 1-58, B).
Figure 1-57.
Icterus, Hemolytic Anemia, Abdominal and Thoracic Viscera, Dog.
The yellow discoloration from the bilirubin is particularly evident in fat and mesentery.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Figure 1-58.
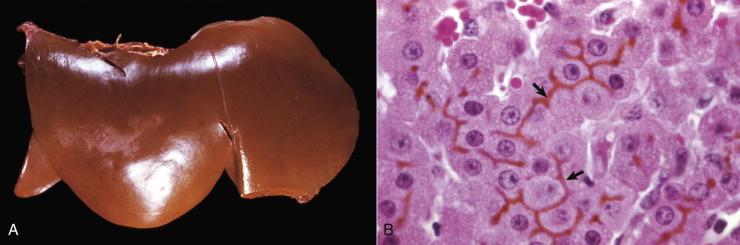
Icterus.
A, Icterus, liver, cat. The liver is swollen with rounded edges and orange-brown discoloration caused by retained bilirubin. B, Acute hemolytic anemia, babesiosis, liver, cow. Bile casts distend canaliculi (arrows). The cholestasis in this case was secondary to intravascular hemolysis with excessive delivery of unconjugated bilirubin to the liver. H&E stain.
(A courtesy College of Veterinary Medicine, University of Illinois. B courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Porphyria.
Porphyrias are heme synthesis disorders that result in deposition of porphyrin pigments in tissues. The porphyrin ring in the hemoglobin molecule is composed of four pyrrole moieties linked together around the central iron ion. Congenital erythropoietic porphyrias of calves, cats, and pigs are the result of genetic defects caused by a deficiency of uroporphyrinogen III synthase. The disease name pink tooth comes from the discoloration of dentin and bone (Fig. 1-59 ; see also Chapter 7). Teeth, bone, and urine of affected animals are red-brown and fluoresce red under ultraviolet light. The feline disease has been mapped to two missense gene mutations in uroporphyrinogen III synthase.
Figure 1-59.
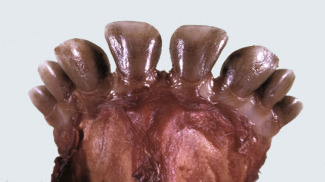
Pink Tooth, Congenital Porphyria, Mandibular Incisor Teeth, Ox.
The teeth are discolored brown from the accumulation of porphyrins in the dentin.
(Courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Cell Cycle
Study of the cell cycle is fundamental to understanding development, homeostasis, and cellular proliferation in response to physiologic or pathologic stimuli, genetic disease, and the effects of cellular aging that include both the uncontrolled cellular proliferation of neoplasia and the permanent cessation of cellular replication known as senescence. The cell cycle (E-Fig. 1-18) consists of interphase (G1, S, and G2) and mitosis (M). Interphase, depending on the cell type, usually lasts at least 12 to 24 hours; in contrast, mitosis can be completed in as little as an hour or two. Cells enter the cell cycle in Gap 1 (G1) in which they grow and produce protein, followed sequentially by the synthesis (S) phase in which DNA is replicated, a second (premitotic) gap (G2) for continued growth and protein production, and finally the M phase for mitosis and cytokinesis, with partitioning of cellular contents between two daughter cells.
E-Figure 1-18.
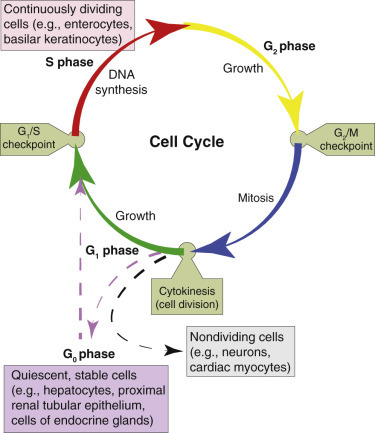
Cell Cycle Landmarks.
The cell cycle includes interphase (G1, S, and G2) and the mitotic M phase. Under appropriate conditions, actively dividing cells may exit the cell cycle to G0, a quiescent state. Nondividing cells in G0 may also reenter the cell cycle. The G1/S and G2/M checkpoints are sites at which cell cycle arrest in response to DNA damage may occur.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Because uncontrolled cellular replication perpetuates DNA damage and can lead to neoplasia, regulation of the cell cycle is essential. The cell cycle is controlled by a family of cyclin-dependent kinases (CDKs) that are activated by cyclins. Cells enter G1 in response to growth factors that also cause the sequential accumulation of cyclins whose roles are to modulate the progress of G1. Cyclin D activation of CDK4/6 results in phosphorylation of retinoblastoma (RB) protein, which in turn releases the transcription factor E2F and enables the cell to pass through the so-called restriction point in G1, after which the cell is independent of extracellular growth signals. This restriction point is near the G1 checkpoint, in which detection of damaged DNA results in growth arrest before S phase (i.e., before DNA replication). Other major checkpoints to interrupt the cell cycle occur in G2 and M phases, if DNA is incorrectly replicated in the S phase or if the mitotic spindle is not properly formed in the M phase.
Growth arrest during the cell cycle is directed by many factors operating at checkpoints, but p53 plays a key role. Growth arrest can be a pause for the cell to repair damaged DNA and then resume cell division. Alternatively, if DNA is irreparably damaged, the cell dies, usually by apoptosis, or enters senescence, which is a permanent growth arrest (see subsequent section on Cellular Aging). Importantly, mutations in p53 are a common event in cancer (see also Chapter 6) and partly explain the uncontrolled proliferation that is the essence of neoplasia.
In health, most mature tissues are a mixture of continuously dividing (labile) cells, quiescent cells, terminally differentiated (postmitotic) cells, and stem cells. Homeostasis is a balance among cellular replication (of stem cells, labile cells, and quiescent cells), cellular differentiation, and cellular death. Labile tissues, such as epidermis, mucosal epithelium, and hematopoietic tissue, have germinal cells that cycle continuously throughout the life of the animal. These labile tissues are therefore quick to respond to physiologic or pathologic stimuli with an increased rate of cell division (hyperplasia).
Quiescent or stable tissues consist mainly of cells (e.g., the parenchymal cells of many organs, mesenchymal cells, or resting lymphocytes) that do not divide continuously and are said to reside in G0 (i.e., outside the cell cycle). However, quiescent cells can reenter the cell cycle in response to hormonal stimuli or growth factors and are capable of striking proliferation in certain physiologic states (e.g., the pregnant uterus or the lactating mammary gland), as well as replacement of damaged tissue in disease (e.g., regeneration of hepatic tissue after lobectomy). The recruitment of quiescent cells into the cell cycle, a major mechanism to increase cellular replication, requires physiologic or pathologic signals to overcome barriers to proliferation.
Other adult tissues (e.g., the CNS, skeletal muscle, and myocardium) are composed mainly of terminally differentiated cells (e.g., neurons, skeletal myocytes, and cardiomyocytes) that no longer divide. Obviously, such cells must have a longer life span than the terminally differentiated cells of labile tissues, but if destroyed, they generally cannot be replaced by the same type of cell. That said, even relatively permanent tissues, such as those of the CNS, have stem cell niches.
The stem cells in adult tissues have an unlimited capacity to proliferate, although their rate of cell division is generally much lower than that of more differentiated cells. Importantly, stem cell division is asymmetric, producing one daughter cell that can differentiate into a variety of mature cell types and another daughter cell with stem cell properties.
The degree of cellular differentiation affects the size of a cell population and its proliferative potential. In labile tissues, such as bone marrow, epidermis, or mucosal epithelium, the mature cells are terminally differentiated, incapable of replication, and short-lived but are replaced by new cells arising from the germinal population, which cycles continuously. Most cells in stable tissues, such as hepatic or renal parenchyma, are in G0 but retain the ability to proliferate on demand. In contrast, relatively permanent tissues, such as the brain, spinal cord, myocardium, or skeletal muscle, are composed mainly of terminally differentiated cells that are incapable of replicating.
Cellular Aging
With advanced age the function of cells and tissues diminishes from the molecular to the organismal level. DNA, especially telomeric DNA (see the following section), and metabolic pathway components affect the life span of cells in tissue culture and in laboratory mice. The stem cell theory of aging postulates that critical shortening of telomeres results in a DNA damage response (DDR) that activates p53 and leads to growth arrest, senescence, or apoptosis of the affected cell. A theory that combines genetic and metabolic pathways postulates that even indirect DNA damage through epigenetic changes, oxidative injury, or other cellular stresses can initiate the DDR, and that persistent DDR causes mitochondrial injury that generates a feed-forward loop.5
Genetic Basis of Aging
Telomeres
Since the discovery that functional telomeres were the limiting factor for replication of fibroblasts in cell culture, telomeres have been at the forefront of research on cellular aging. Most somatic cells have a finite number of cell divisions that, at least in part, is determined by the length of telomeres. Telomeres are repetitive nucleotide (TTA-GGG) sequences that cap the ends of linear chromosomes, providing a template for complete replication of the chromosomal DNA and preventing the chromosomal ends from being misinterpreted as double-stranded DNA breaks. Telomeric DNA is protected from inappropriate repair by associated proteins that form the shelterin complex. Telomeres are truncated (shortened) with each cell division because the DNA polymerases require a leading primer and so cannot replicate all the way to the end of the DNA molecule. In “immortal” cells, such as germ cells or certain stem cells, leukocytes (e.g., activated T lymphocytes), or cancer cells, active telomerase replenishes telomeres (E-Fig. 1-19). Telomerase consists of an RNA subunit template component (TERC) and a catalytic component (TERT), which is a reverse transcriptase. Mutations of either component have been associated with aging syndromes and other disorders. Dysfunctional telomeres signal the DDR with activation of p53 and arrest of the cell cycle. Arrest of the cell cycle can be a temporary pause for DNA repair or can progress to senescence (an irreversible growth arrest) or to cell death through apoptosis. DNA repair pathways that are triggered by dysfunctional telomeres tend to result in abnormal repair (e.g., chromosomal fusions) that exacerbates the DNA damage and elicits a persistent DDR.
E-Figure 1-19.
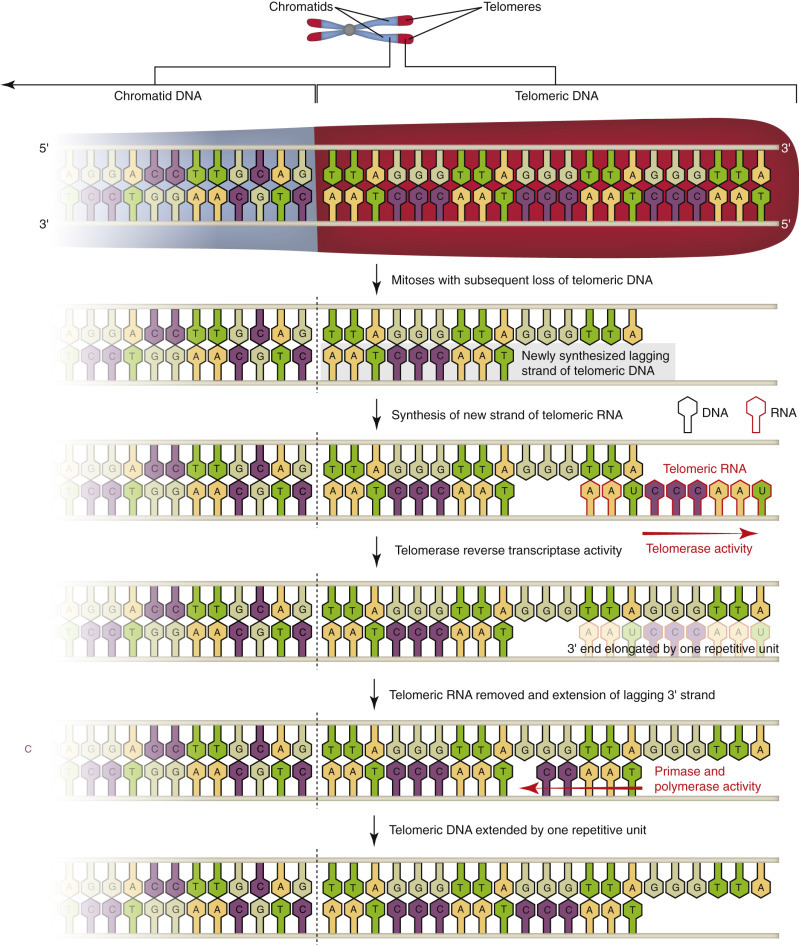
Maintenance of Telomeres in Germ Cells, Stem Cells, and Cancer Cells.
After repeated mitoses, somatic cells lose telomeric DNA (repetitive TTA-GGG sequences) and their ability to divide. In contrast, “immortal” cells with active telomerase can replenish telomeric DNA and divide indefinitely.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
A purely telomeric theory of aging does not explain the aging in tissues or organs composed mainly of quiescent postmitotic cells (e.g., neurons and muscle cells), in which telomeres would be less important. A broader theory combines DNA damage and metabolic abnormalities (E-Fig. 1-20) and proposes that endogenous and exogenous factors contribute to telomere dysfunction, impaired DDR, or increased ROS, each of which can independently activate p53, which in turn compromises mitochondrial function through repression of coactivators of peroxisome proliferator-activated receptor γ (PPARγ), a nuclear receptor that regulates many metabolic pathways. The interplay between the DDR and metabolism is complex; however, induction of a persistent DDR activates p53. Repression of PPARγ coactivators by p53 exacerbates oxidative injury and decreases energy production. Although p53 also represses the insulin/insulin-like growth factor-1 (IGF-1) and mTOR6 pathways, this repression can protect cells by activating forkhead box protein O (FOXO) transcription factors and PPARγ coactivators that promote oxidative phosphorylation, antioxidant production, and p53 inactivation.
E-Figure 1-20.
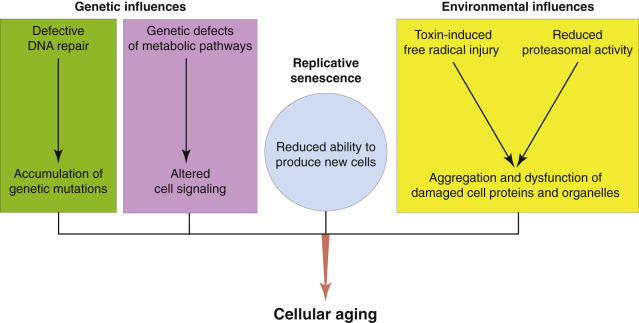
Genetic and Environmental Factors in Cellular Aging.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Cellular Senescence
The gradual decline in function in aging animals is associated with both degenerative and proliferative changes that are intricately linked to the stress response known as cellular senescence (E-Fig. 1-21). Genetically, senescence seems to be a situation of antagonistic pleiotropy, in which a group of genes are beneficial in early life, promoting survival during the reproductive years, yet the same genes contribute to debility and other diseases in aging animals.
E-Figure 1-21.
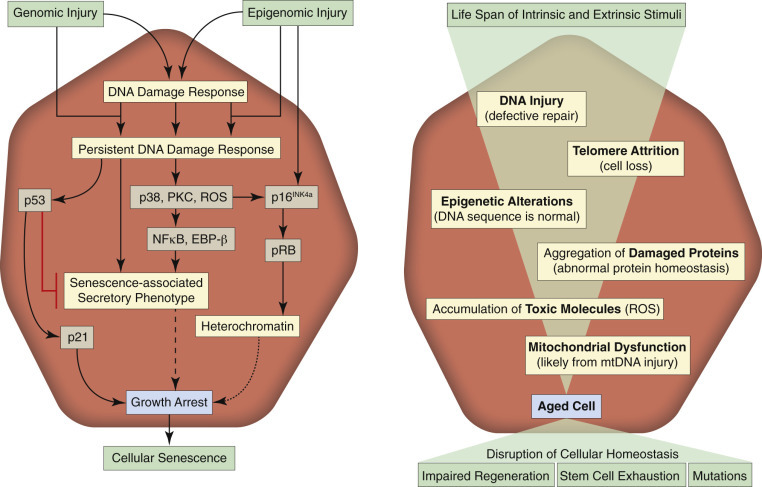
Cellular Senescence.
A persistent DNA damage response can induce a permanent growth arrest known as cellular senescence. The schematic on the left depicts the molecular sequence of events in the induction of senescence. The schematic on the right lists the corresponding mechanisms of injury. EBP-β, enhancer-binding protein-β; mtDNA, mitochondrial DNA; NFκB, nuclear factor κB; PKC, protein kinase C; pRB, retinoblastoma protein; ROS, reactive oxygen species.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
The stresses that typically cause senescence include DNA damage (especially shortening of telomeres), epigenomic damage, oncogenes and other mitogenic stimuli, and activation of certain tumor suppressor genes. Notably, oxidative stress can indirectly cause double-stranded DNA breaks, especially in the guanine-rich telomeric DNA. Cellular senescence is an essentially irreversible arrest of the cell cycle, regulated by two tumor suppressor pathways: p53-p21 and p16INK4a-RB. When the DDR becomes persistent, p53 causes growth arrest through the cell cycle inhibitor p21. A persistent DDR also, through p38 MAPK (a mitogen-activated protein kinase pathway component), protein kinase C, and ROS, activates p16INK4a, which in turn activates RB protein, halting the cell cycle.
The benefits of cellular senescence are that it can prevent the formation of neoplasms and promote wound healing with less scarring. However, cellular senescence can also promote the degenerative diseases of old age and, ironically, can contribute to tumor progression in aging animals (see Chapter 6).
Structural and Biochemical Changes with Cellular Aging
In long-lived postmitotic cells, such as neurons and striated muscle cells, lipofuscin tends to accumulate with advancing age. Senescent cells (i.e., cells that were mitotic but have ceased to divide because of accumulated DNA damage or other factors) have cytologically detectable heterochromatin foci, increased volume, and a flattened profile if adherent to a basement membrane or other scaffolding.
Biochemically, senescent cells are recognized in part by their lack of expression of proliferation markers. Senescent cells take on what is known as a senescence-associated secretory phenotype (SASP). They overexpress the acidic lysosomal enzyme β-galactosidase. Another commonly used marker of senescence is p16INK4a. The SASP is associated with secretion of numerous proinflammatory cytokines, as well as chemokines, growth factors, and proteases, including, as examples, growth-regulated oncogenes, vascular endothelial growth factor (VEGF), secreted frizzled related protein 1 that modulates Wnt, IL-6 and IL-8, and matrix metalloproteinases. Some SASP factors promote or inhibit proliferation depending on the setting. Other SASP factors can elicit inflammation or induce epithelial-to-mesenchymal transition, which can be part of the progression to invasive cancer. Importantly, not all senescent cells assume a SASP; it is mainly a response to DNA damage or epigenomic perturbations. NFκB has a positive effect on the SASP; p53, in contrast, restrains it.
Genetic Basis of Disease7
Genetic diseases are caused by alterations in the number, structure, and/or function of chromosomes and their genes and gene products (proteins). Genes determine the differentiation, development, maturation, and aging of the 200 to 210 cell types in an animal's body and the tissues and organ systems they form. Additionally, they establish (1) the structural and functional roles each of these cell types plays in forming barrier systems and defense mechanisms against noninfectious and infectious diseases and (2) how each of these cell types and their organelles respond in homeostasis and to cellular adaptation, injury, aging, and neoplasia. Alterations in the structure and/or function of genes and gene products can have serious outcomes on cells, tissues, and organ systems that are reflected in patterns of lesions unique to affected cells and thus clinical signs reflective of the disease. Specific diseases with genetic bases are discussed in more detail in the Pathology of Organ Systems chapters; this section provides an “E-content” overview of (1) the structure and function of chromosomes and genes, (2) some basic mechanisms of genetic disorders, and (3) the outcomes of specific genetic diseases and includes:
- Chromosome Structure and Function
- Nuclear Chromosomes
- Mitochondrial Chromosomes
Gene Structure and Function
- Mechanisms of Genetic Disorders
- Single-Gene Disorders
- Single-Gene Disorders of Somatic Cells
- Single-Gene Disorders of Germ Cells
- Autosomal Dominant Disorders
- Autosomal Recessive Disorders
- X-Linked Disorders
- Single-Gene Disorders of Mitochondria
- Chromosomal Disorders
- Errors in Cell Division
- Numeric Alterations
- Structural Alterations
- Complex Multigenic Disorders
The interaction of microbial genes with host genes in determining resistance to infectious diseases is discussed in Chapter 4. The role of genes in controlling immune responses and neoplastic transformation is discussed in Chapters 5 and 6, respectively. Examples of known or suspected genetic disorders in domestic animals are listed in E-Box 1-1 and are discussed in the chapters covering pathology of organ systems.
E-Box 1-1. Potential, Suspected, or Known Genetic Disorders*.
Chapter 7 Alimentary System and the Peritoneum, Omentum, Mesentery, and Peritoneal Cavity
-
•
Abnormal dentition (teeth abnormalities)
-
•
Agangliosis (dysautonomia)
-
•
Amyloidosis
-
•
Anasarca
-
•
Bloat
-
•
Cleft lip
-
•
Cleft palate
-
•
Coliform enteritis
-
•
Cricopharyngeal dysfunction
-
•
Dental malocclusion
-
•
Dysphagia
-
•
Elongated soft palate
-
•
Eosinophilic granuloma
-
•
Esophageal achalasia
-
•
Esophageal dilatation
-
•
Gastric dilatation and volvulus
-
•
Gastric volvulus
-
•
Gingival hyperplasia
-
•
Gluten-sensitive enteropathy
-
•
Granulomatous colitis
-
•
Hemorrhagic gastroenteritis
-
•
Histiocytic colitis
-
•
Inguinal hernia
-
•
Intestinal malabsorption (protein-losing enteropathy)
-
•
Intussusception
-
•
Ivermectin sensitivity
-
•
Lymphangiectasia
-
•
Malabsorption syndrome
-
•
Malocclusion
-
•
Megaesophagus
-
•
Melanoma
-
•
Oligodontia
-
•
Parotitis (parotiditis)
-
•
Prolapsed rectum
-
•
Protein-losing enteropathy
-
•
Pyloric stenosis
-
•
Ulcerative colitis
-
•
Umbilical hernia
-
•
Ventral ankyloglossia
-
•
Vitamin B12–responsive malabsorption
-
•
Wheat-sensitive enteropathy
Chapter 8 Hepatobiliary System and Exocrine Pancreas
-
•
Amyloidosis—familial in Shar-Pei dog, Oriental cat breeds (e.g., Abyssinian, Siamese)
-
•
Bilirubin metabolism defects in Corriedale and Southdown sheep
-
•
Congenital porphyria
-
•
Copper-associated hepatopathy—autosomal recessive in Bedlington terrier, familial in several other breeds
-
•
Exocrine pancreatic atrophy—German shepherd dog and rough-coated collie
-
•
Hepatocerebellar degeneration—autosomal recessive in Bernese mountain dog
-
•
Lysosomal storage disorders—can affect the liver and/or pancreas as part of multiorgan disease
-
•
Polycystic disease—Persian cats, Cairn terriers, West Highland white terriers
-
•
Portal vein hypoplasia—toy dog breeds predisposed
-
•
Portosystemic shunts—extrahepatic in small breeds (Cairn terriers predisposed) and intrahepatic in large breeds (Irish wolfhounds predisposed)
Chapter 9 Respiratory System, Mediastinum, and Pleurae
-
•
Aberrant cilia
-
•
Atresia of the nasolacrimal puncta
-
•
Brachycephalic airway syndrome
-
•
Ciliary dyskinesia (immotile cilia syndrome)
-
•
Collapsed trachea
-
•
Congenital melanosis
-
•
Dacryocystitis
-
•
Guttural pouch tympany
-
•
Histiocytic sarcoma
-
•
Hypoplasia of the larynx
-
•
Hypoplasia of the trachea
-
•
Lung torsion
-
•
Stenotic nares
-
•
Tracheal collapse
Chapter 10 Cardiovascular System and Lymphatic Vessels
-
•
Amyloidosis
-
•
Aortic body tumors
-
•
Aortic stenosis
-
•
Arrhythmogenic right ventricular cardiomyopathy
-
•
Arteriovenous fistula
-
•
Atrial septal defects
-
•
Bundle branch block
-
•
Bundle of His degeneration
-
•
Cardiac valvular disease
-
•
Cardiomyopathy—dilated
-
•
Cardiomyopathy—hypertrophic
-
•
Cardiomyopathy associated with muscular dystrophy
-
•
Carotid body tumors
-
•
Congenital heart defects
-
•
Conus septal defect
-
•
Cutaneous vasculopathy
-
•
Dilated cardiomyopathy
-
•
Endocardial fibroelastosis
-
•
Femoral artery occlusion
-
•
Hemangiosarcoma
-
•
Hepatic portosystemic shunt or arteriovenous fistula
-
•
Inherited ventricular tachycardia
-
•
Microvascular dysplasia
-
•
Mitral valve defects
-
•
Myxomatous valvular degeneration
-
•
Patent ductus arteriosus
-
•
Persistent right aortic arch
-
•
Portosystemic shunt
-
•
Pulmonic stenosis
-
•
Sinoatrial syncope
-
•
Subaortic stenosis
-
•
Subvalvular aortic stenosis
-
•
Taurine-deficient cardiomyopathy
-
•
Tetralogy of Fallot
-
•
Tricuspid dysplasia
-
•
Vasculitis
-
•
Ventricular septal defect
-
•
Ventricular tachycardia
Chapter 11 the Urinary System
-
•
Alport syndrome (hematuria, renal failure, and deafness)
-
•
Amyloidosis
-
•
Cystinuria
-
•
Cystitis and cystic calculi
-
•
Ectopic ureter
-
•
Fanconi syndrome
-
•
Glomerulopathy (due to collagen defects)
-
•
Hereditary nephropathy/nephritis
-
•
Kidney aplasia (renal agenesis)
-
•
Mononephrosis
-
•
Polycystic kidney disease
-
•
Protein-losing nephropathy
-
•
Renal cortical hypoplasia
-
•
Renal cystadenocarcinomas and nodular dermatofibrosis
-
•
Renal dysplasia
-
•
Renal hypoplasia
-
•
Renal tubular dysplasia (dysfunction)
-
•
Silica uroliths
-
•
Transitional cell carcinoma
-
•
Uric acid calculi
-
•
Uric acid excretion abnormalities
-
•
Urolithiasis
Chapter 12 Endocrine System
-
•
Addison's disease (hypoadrenocorticism)
-
•
Apocrine gland tumor
-
•
Chemodectoma
-
•
Cushing's disease (hyperadrenocorticism)
-
•
Diabetes mellitus
-
•
Dyshormonogenetic goiter
-
•
Hyperthyroidism
-
•
Hypoglycemia
-
•
Hypoparathyroidism
-
•
Hyposomatotropism
-
•
Hypothyroidism
-
•
Metabolic syndrome
-
•
Pituitary dwarfism
-
•
Pituitary tumor
-
•
Primary hyperparathyroidism
-
•
Thyroiditis
Chapter 13 Bone Marrow, Blood Cells, and the Lymphatic/Lymphoid System
-
•
Band 3 deficiency in Japanese black cattle
-
•
Bone marrow dyscrasia in poodle dogs
-
•
Calcium diacylglycerol guanine nucleotide exchange factor I (CalDAG-GEFI) thrombopathia
-
•
Cell-mediated immunodeficiency
-
•
Chédiak-Higashi syndrome
-
•
Cobalamin (vitamin B12) deficiency
-
•
Combined immunodeficiency
-
•
Complement deficiency
-
•
Cyclic hematopoiesis in gray collie dogs
-
•
Cytochrome b5 reductase deficiency
-
•
Dyserythropoiesis and alopecia in polled Hereford calves
-
•
Elliptocytosis
-
•
Erythropoietic porphyria
-
•
Factor I deficiency
-
•
Factor II deficiency
-
•
Factor VII deficiency
-
•
Factor VIII deficiency (hemophilia A)
-
•
Factor IX deficiency (hemophilia B)
-
•
Factor X deficiency
-
•
Factor XI deficiency
-
•
Factor XII deficiency
-
•
Flavin adenine dinucleotide deficiency
-
•
γ-Glutamyl carboxylase defect
-
•
Glanzmann's thrombasthenia
-
•
Glucose-6-phosphate dehydrogenase deficiency
-
•
Granulocyte dysfunction
-
•
Hemolytic anemia
-
•
Immunoglobulin A deficiency
-
•
Immunoglobulin G deficiency
-
•
Immunoglobulin M deficiency
-
•
Leukocyte adhesion deficiency
-
•
Lymphoma
-
•
Macrothrombocytopenia in Cavalier King Charles spaniel dogs
-
•
Pelger-Huët anomaly
-
•
Phosphofructokinase deficiency
-
•
Physiologic leukopenia
-
•
Physiologic thrombocytopenia
-
•
Pyruvate kinase deficiency
-
•
Scott syndrome
-
•
Severe combined immunodeficiency of Arabian foals, basset hounds (X-linked), Cardigan Welsh corgis, Jack Russell terriers
-
•
Splenic torsion
-
•
Stomatocytosis
-
•
Thrombocytopathy
-
•
von Willebrand disease
Chapter 14 Nervous System
-
•
Ataxia
-
•
Cerebellar and extrapyramidal abiotrophy
-
•
Cerebellar ataxia
-
•
Cerebellar cortical abiotrophy
-
•
Cerebellar degeneration
-
•
Cerebellar hypoplasia
-
•
Cerebrospinal demyelination
-
•
Cervical disk disease
-
•
Cervical vertebral malformation or instability
-
•
Compressive myelopathy
-
•
Degenerative myelopathy
-
•
Epilepsy
-
•
Facial nerve paralysis
-
•
Familial amaurotic idiocy
-
•
Fucosidosis
-
•
Globoid cell leukodystrophy
-
•
Hydrocephalus
-
•
Hypertrophic neuropathy
-
•
Hypomyelinogenesis
-
•
Juvenile amaurotic idiocy
-
•
Laryngeal paralysis
-
•
Lipidosis (GM-1 gangliosidosis)
-
•
Lissencephaly
-
•
Lysosomal storage diseases
-
•
Mucopolysaccharidosis
-
•
Necrotizing meningoencephalitis (pug dog encephalitis)
-
•
Necrotizing myelopathy
-
•
Neuronal ceroid lipofuscinosis
-
•
Oligodendroglioma
-
•
Optic nerve hypoplasia
-
•
Peripheral sensory neuropathy
-
•
Polyneuropathy
-
•
Polyradiculoneuritis
-
•
Progressive ataxia
-
•
Pug dog encephalitis (necrotizing meningoencephalitis)
-
•
Spinal cord demyelination (ataxia)
-
•
Stockard's paralysis
-
•
Syncope
-
•
Syringomyelia
-
•
Wobbler syndrome
Chapter 15 Skeletal Muscle
-
•
Arthrogryposis
-
•
Autosomal recessive myopathies, species and breed specific
-
•
Carnitine deficiency
-
•
Centronuclear myopathy
-
•
Congenital myasthenia gravis
-
•
Glycogen storage disease
-
•
Hereditary spinal muscular atrophy (see Chapter 15)
-
•
Hyperkalemic periodic paralysis
-
•
Malignant hyperthermia
-
•
Mitochondrial myopathy
-
•
Muscular dystrophy
-
•
Myasthenia gravis
-
•
Myostatic defect (double muscling)
-
•
Myotonia congenita
Chapter 16 Bones, Joints, Tendons, and Ligaments†
-
•
Anemia with chondrodysplasia
-
•
Anomaly of the third cervical vertebra
-
•
Avulsion fractures
-
•
Bithoracic ectromelia
-
•
Brachygnathism (undershot jaw)
-
•
Carpal subluxation
-
•Chondrodysplasia (many different types)
-
•Achondrogenesis
-
•Achondroplasia (dwarfism)
-
•Chondrodysplasia punctata
-
•Endochondrodystrophy
-
•Epiphyseal chondrodysplasia
-
•Hypochondroplasia
-
•Multiple epiphyseal dysplasia
-
•Oculoskeletal dysplasia (cataracts and skeletal deformity, primarily achondroplasia)
-
•Oculoskeletal dysplasia with or without hematologic abnormalities
-
•Osteochondrodysplasia
-
•Pseudoachondroplasia
-
•
-
•
Cranial cruciate ligament disease
-
•
Craniomandibular osteopathy
-
•
Cranioschisis
-
•
Deformed tail
-
•
Diffuse idiopathic skeletal hyperostosis
-
•
Dislocation of the shoulder
-
•
Dwarfism
-
•
Elbow dysplasia (including one of more of the following: osteochondrosis dissecans (OCD) of the medial humeral condyle, fragmentation of the medial coronoid process, and ununited anconeal process)
-
•
Elbow joint malformation
-
•
Elbow subluxation
-
•
Enlarged foramen magnum
-
•
Enostosis
-
•
Eosinophilic panosteitis
-
•
Epiphyseal dysplasia
-
•
Fracture
-
•
Fragmented coronoid process (see Elbow dysplasia)
-
•
Genu valgum (knock-knee)
-
•
Hemimelia (developmental anomaly characterized by the absence of all or part of the distal half of one or more limbs [e.g., fibular, radial, tibial, or ulnar])
-
•
Hemivertebra
-
•
Hip dysplasia
-
•
Hypoplasia of the dens
-
•
Idiopathic multifocal osteopathy
-
•
Intervertebral disk disease
-
•
Juvenile polyarthritis
-
•
Kinked tail
-
•
Legg-Calvé-Perthes disease
-
•
Metabolic bone disease
-
•
Metaphyseal osteopathy (previously known as hypertrophic osteodystrophy)
-
•Mucopolysaccharidosis; different types including the following:
-
•Type I (α-l-glucuronidase deficiency)
-
•Type VI (arylsulfatase B deficiency)
-
•Type VII (β-d-glucuronidase deficiency)
-
•
-
•
Multiple cartilaginous exostoses
-
•
Multiple epiphyseal dysplasia (see Chondrodysplasia)
-
•
Neurotropic osteopathy
-
•
Open fontanelle
-
•
Osteochondrosis (multiple anatomic sites, including elbow, shoulder, stifle, tarsus, sacrum, spine)
-
•
Osteodystrophy
-
•
Osteogenesis imperfecta
-
•
Osteopetrosis
-
•
Osteosarcoma (neoplasia)
-
•
Otocephalic syndrome
-
•
Panosteitis
-
•
Parosteitis
-
•
Patella luxation
-
•
Polydactyly (extra digits)
-
•
Polyostotic fibrous dysplasia
-
•
Prognathism (overshot jaw)
-
•
Screw tail
-
•
Short skull
-
•
Short spine syndrome
-
•
Shoulder abnormalities
-
•
Shoulder dysplasia
-
•
Spina bifida
-
•
Spinal dysraphism
-
•
Spinal process (vertebral) malformation
-
•
Spondylolisthesis (wobbler syndrome)
-
•
Spondylosis
-
•
Tail abnormalities (including brachyury, short tail; anury, absence of a tail)
-
•
Transitional vertebrae
-
•
Ununited anconeal process (see Elbow dysplasia)
-
•
Wobbler syndrome
Chapter 17 the Integument‡
-
•
Acral mutilation syndrome
-
•
Alopecias
-
•
Collagen dysplasias (cutaneous asthenia, dermatosparaxis, Ehlers-Danlos syndrome)
-
•
Cornification disorders
-
•
Curly coat
-
•
Dermatosis vegetans
-
•
Ectodermal dysplasias
-
•
Epidermolysis bullosa
-
•
Epitheliogenesis imperfecta (aplasia cutis)
-
•
Familial canine dermatomyositis
-
•
Follicular dysplasias
-
•
Follicular parakeratosis
-
•
Hypotrichoses
-
•
Ichthyoses
-
•
Inherited epidermal acantholyses
-
•
Nasal parakeratoses
-
•
Nodular dermatofibroses
-
•
Paw pad hyperkeratoses
-
•
Pigmentary disorders
-
•
Primary cornification disorders
-
•
Zinc-related disorders
Chapter 18 Female Reproductive System and Mammae
-
•
Cystic ovarian disease
-
•
Early embryonic mortality
-
•
Hermaphroditism
-
•
Prolonged gestation
-
•
Supernumerary papillae
-
•
Uterine segmental aplasia
-
•
XX SRY-negative males (sex reversal)
-
•
XY SRY-positive female (horses)
Chapter 19 Male Reproductive System
-
•
Ciliary dyskinesia
-
•
Cryptorchidism
-
•
Preputial eversion and prolapse
-
•
Segmental aplasia of the mesonephric duct
-
•
Spermatic granuloma of the epididymal head
-
•
Testicular hypoplasia
-
•
XX SRY-negative testicular disorders of sexual development, polled intersex syndrome
-
•
XY SRY-positive testicular disorders of sexual development, persistent müllerian duct syndrome.
Chapter 20 the Ear
-
•
Deafness
-
•
Necrotizing panotitis
-
•
Otitis externa
Chapter 21 the Eye
-
•
Cataracts
-
•
Cataracts with microphthalmia
-
•
Choroidal hypoplasia
-
•
Chronic superficial keratitis (pannus)
-
•
Collie eye anomaly
-
•
Coloboma
-
•
Congenital stationary night blindness
-
•
Corneal dystrophy
-
•
Corneal ulcer, indolent
-
•
Distichiasis
-
•
Ectropion
-
•
Entropion
-
•
Equine recurrent uveitis
-
•
Eversion of the vertical cartilage of the third eyelid
-
•
Glaucoma
-
•
Hemeralopia
-
•
Iris hypoplasia
-
•
Keratoconjunctivitis sicca (dry eye)
-
•
Lens luxation
-
•
Lenticonus/lentiglobus
-
•
Macroblepharon
-
•
Melanocytic neoplasia
-
•
Merle ocular dysgenesis
-
•
Microphthalmia
-
•
Ocular melanosis
-
•
Optic nerve hypoplasia
-
•
Persistent hyaloid vasculature/persistent tunica vasculosa lentis
-
•
Persistent pupillary membrane
-
•
Photoreceptor dysplasia
-
•
Pigmentary keratopathy
-
•
Pigmentary uveitis
-
•
Progressive retinal atrophy
-
•
Prolapse of the gland of the third eyelid
-
•
Retinal detachment
-
•
Retinal dysplasia
-
•
Scrolled cartilage of the third eyelid
-
•
Uveodermatologic syndrome
-
•
Vitreoretinal dysplasia
Chromosome Structure and Function
Nuclear Chromosomes
Every species of animal has a unique chromosomal (genetic) representation called a karyotype (i.e., the number and morphology of the chromosomes that make up its genome) (E-Box 1-2). With the exception of cells that develop into ova and spermatozoa (i.e., germline cells), all cells in the body are called somatic cells (from soma for body). The genome in the nucleus of somatic cells consists of chromosomes arranged in pairs. One member of each pair of chromosomes is inherited from the sire, the other from the dam. All pairs, except for one, are similar in males and females and are called autosomes. The remaining pair is the sex chromosomes: two X chromosomes in females, and an X chromosome and a Y chromosome in males.
E-Box 1-2. Karyotypes of Domestic Animal Species.
Horses—64 chromosomes
Cattle—60 chromosomes
Sheep—54 chromosomes
Goats—60 chromosomes
Pigs—38 chromosomes
Dogs—78 chromosomes
Cats—38 chromosomes
Chromosomes are not naked DNA double helices. The genome is packaged as chromatin, in which DNA is complexed with one or more of five types of chromosomal proteins called histones. Histones have large quantities of arginine and lysine, which are positively charged amino acids. This charge allows histones to bind tightly to negatively charged DNA, forming complexes called nucleosomes that are further condensed into chromosomes. This arrangement allows long strands of DNA to be condensed into molecules that easily fit into the nucleus (E-Fig. 1-22). However, for a gene to be active, the condensed DNA must revert to long strands of DNA for transcription into RNA.
Mitochondrial Chromosomes
Mitochondria are the cellular site of aerobic energy production. In highly active cells, such as type I skeletal muscle cells of equine athletes, up to 10,000 mitochondria may be present in the cytoplasm of a myocyte. Each mitochondrion contains a single chromosome formed by circular double-stranded DNA, called mitochondrial DNA (mtDNA). The genome of a mitochondrial chromosome encodes for 37 genes, including those for mRNAs, rRNAs, transfer RNAs (tRNAs), and 13 protein subunits for enzymes, such as cytochrome b and cytochrome oxidase, which are involved in oxidative phosphorylation. The mitochondrial genome also has distinct transcription and protein-synthesis (i.e., translation) systems. A specialized RNA polymerase, encoded in the nuclear genome, is used to transcribe the mitochondrial genome, and then the mitochondrial transcripts are processed to generate the various mitochondrial mRNAs, tRNAs, and rRNAs.
Gene Structure and Function
Normally the genes at each locus on each chromosome of a pair of homologous chromosomes are identical; however, in some instances a different form of a gene, called an allele, occurs at one or both loci, which can alter the gene product (i.e., protein). Alleles can also occur in groups of genes and affect the proteins and processes regulated by these genes and their alleles. Alleles result from mechanisms that create mutations in the DNA and will be discussed and illustrated in the remainder of this chapter. Alterations expressed by some alleles may not affect the function of the protein regulated by the gene pair; in other cases the expression of the allele (or a combination of alleles) may cause structural or cellular dysfunction and lesions in affected tissues and thus clinical signs and “genetic” disease.
On average, there are approximately 20,000 genes in the nucleus of an individual cell of the different domestic animal species. Genes, the heredity units of the genetic code, determine the structural and functional (i.e., enzymes) biologic traits (i.e., expression of genes) necessary to create and maintain cells, tissues, and organs and to pass the genetic code on to offspring. Additionally, genes, especially those of the major histocompatibility complex (see Chapter 5), play important roles in establishing the resistance (or susceptibility) of an animal to infectious diseases. Many biologic traits have a mendelian pattern of inheritance. Gene expression, however, depends on cell structure and function and the responses of cells to injury. Constitutively expressed genes, including those for tRNAs, rRNAs, cell membranes, and enzymes, are transcribed continually. In contrast, facultative or differentially expressed genes are transcribed only when prompted (e.g., in response to receptor binding by a hormone or growth factor) or to cellular perturbations as in neoplastic transformation (see Chapter 6). Tissue-specific genes are expressed only in a particular tissue such as the gene for galactocerebrosidase in myelinating cells of the nervous system (see section on Globoid Cell Leukodystrophy in Chapter 14). Tissue-specific genes may be either constitutively or differentially expressed.
In its simplest form a gene is a segment of a DNA molecule that contains the nucleotide code for the amino acid sequence of a protein (see E-Fig. 1-22). Genes are arranged in a linear order along each chromosome, each gene having a precise position (locus). The composition of genes is determined by the chromosomal DNA. In nuclear DNA each chromosome consists of a DNA double helix; in other words, each chromosome is a long, linear double-stranded DNA molecule, and the genome (and genes) consists therefore of DNA molecules totaling billions of nucleotides (pyrimidines: cytosine [C], thymine [T], uracil [U, found in RNA in place of T in DNA]; purines: adenine [A] and guanine [G]). Within a gene, nucleotides are arranged in triplets or codons. Each codon represents the code for a specific amino acid, and the linear sequence of many codons determines the amino acid sequence of the gene product (i.e., protein). Thus, with a codon of three nucleotides, there are 64 possible triplet combinations that constitute the genetic code and the basis for synthesis and assembly of proteins.
Certain genes may be active (i.e., differentially expressed) only during specific embryologic or fetal stages because specific proteins are required for development, differentiation, and growth. Other genes are expressed constitutively or facultatively postpartum and are involved in growth, homeostasis, or reproduction. Thus an important consideration in genetic diseases is the period during which a normal gene would be active. If a mutated gene is expressed during the active period for the normal gene, the outcome may be detrimental. Mutations in genes that are important for embryogenesis or organogenesis have earlier and generally more severe consequences than genetic mutations that are not expressed until sexual maturation or thereafter. Genetic mutations can result in congenital malformations (e.g., palatoschisis or cheiloschisis [see Chapter 7]) or metabolic dysfunction (e.g., multisystem neuronal degeneration [see Chapter 14]).
The process of synthesizing proteins from genes involves two steps, transcription in the nucleus and translation in the cytoplasm. In addition to codons, genes contain nucleotide sequences that determine when the gene is active. In active genes, RNA is synthesized from the DNA template through a process known as transcription (E-Fig. 1-23). The RNA carrying the coded information, called mRNA, is then transported from the nucleus to the cytoplasm where the RNA sequence is decoded, or translated, to determine the amino acid sequence of the protein gene product. The key to translation is the nucleotide sequence along the mRNA. Each set of three nucleotide bases (the triplet codon) in the mRNA encodes a particular amino acid that is inserted into the forming protein molecule during translation. In theory, an almost infinite variation in the arrangement of the bases along a polynucleotide chain is possible. The process of translation occurs on ribosomes, both free in the cytoplasm and on rER. Ribosomes consist of many different structural proteins in association with rRNA and have binding sites for all the interacting molecules of translation, including mRNA and tRNA. The function of tRNA is to transfer the appropriate amino acid from the cytosol to the codon in the base sequence of each mRNA bound to a ribosome.
E-Figure 1-23.
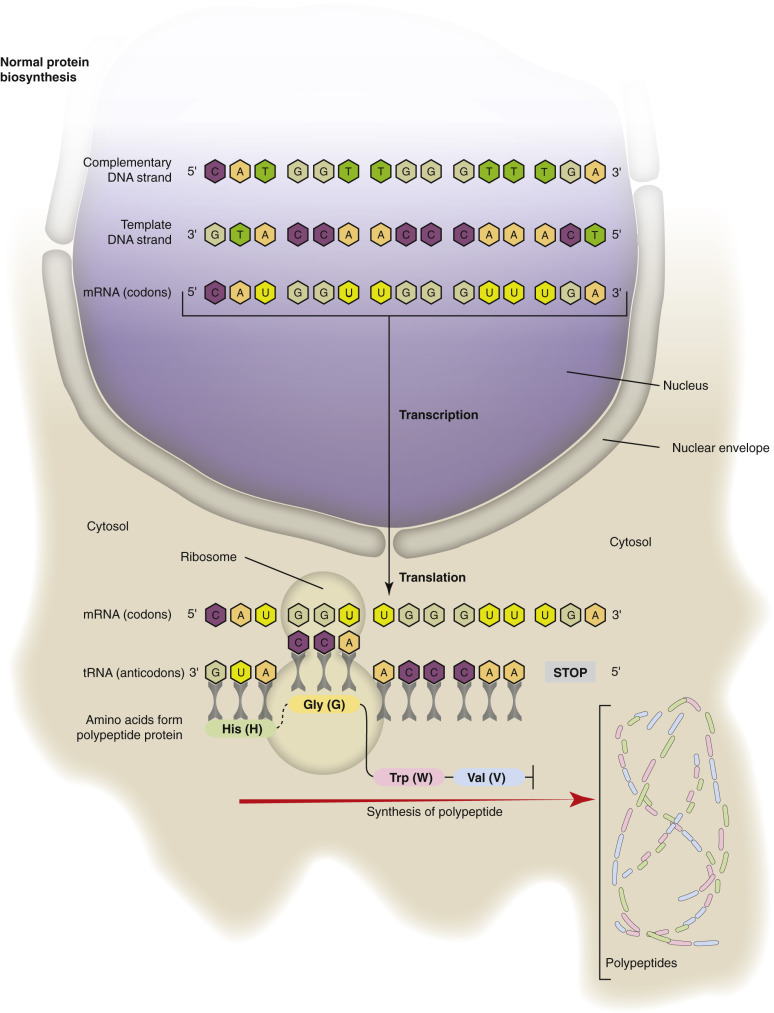
Protein Synthesis: Transcription and Translation.
Messenger RNA (mRNA), transcribed in the nucleus from one strand of DNA, is transported to the cytoplasm for translation into polypeptides. The amino acid product of the trinucleotide genetic code is attached to the complementary transfer RNA (tRNA).
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Protein gene products are responsible for the development (e.g., formation of structural proteins) and function (e.g., enzymes) of cells, tissues, and organs. Many genes belong to gene families, which share closely related DNA sequences and encode polypeptides with closely related amino acid sequences. However, if there were a simple one-to-one correspondence between genes and proteins, then the approximately 20,000 genes in an animal should give rise to approximately 20,000 different proteins. This number seems insufficient for cell structure and function. The answer to this conundrum is found in two features of genes. First, genes can generate multiple different proteins, not just one. Second, many proteins undergo posttranslational modifications, which can include chemical changes (e.g., hydroxylation, methylation) of amino acid side chains, the addition of carbohydrate moieties (e.g., glycosylation), or proteolytic cleavage of polypeptides (e.g., the conversion of proinsulin to insulin). The polypeptide chain that is the primary translation product is also folded and bonded into a specific three-dimensional structure determined by its amino acid sequence. Two or more polypeptide chains, products of the same or different genes, may combine to form a protein complex. Thus it has been estimated that 20,000 genes can encode as many as a million different proteins. The proteome includes all the proteins of a cell (cellular proteome), tissue, or animal (complete proteome). Individual proteins in a proteome do not function in isolation. They form elaborate networks, involving many different proteins and respond in a coordinated fashion to many different genetic, developmental, or environmental signals such as those that occur in homeostasis and in cellular adaptation, injury, aging, and neoplasia. Combinations of such protein networks result in an even greater diversity of cellular functions.
Mechanisms of Genetic Disorders
The effects of the estimated 20,000 genes in an animal's genome involve complex interrelationships among the factors controlling gene expression (by mechanisms of chromosome replication and segregation), gene structure, and finally, transcription, RNA splicing, mRNA stability, translation, protein processing, posttranslational modification, and protein degradation. For some genes, fluctuations in the concentration of a gene product, whether from an inherited structural variation in the gene or from nongenetic factors in the diet or environment, are of little clinical importance. However, for other genes, changes in the concentration of expression have dire clinical consequences, reflecting the importance of those gene products in specific biologic pathways. The nature of inherited variation in the structure and function of chromosomes and genes and the influence of this variation on the expression of specific biologic traits underlie the mechanisms of genetic disorders.
Genetic disorders can involve somatic cells (i.e., cells forming the structure of an animal) or germline cells (i.e., cells that give rise to gametes) and can be broadly classified into the following three categories:
-
1.
Single-gene disorders caused by mutations in DNA of a single gene such as point, frameshift, and trinucleotide-repeat mutations
-
2.
Chromosomal disorders caused by alterations in the number and/or structure of chromosomes (i.e., the karyotype)
-
3.
Complex multigenic disorders
Single-Gene Disorders
Somatic cells are mitotic cells, and disorders involving mitotic cells are not heritable but are important in the genesis of cancers and some congenital malformations. Most germline cells are meiotic cells, and disorders involving them can be inherited. Single-gene disorders can affect either somatic cells or germline cells and usually result from mutations in DNA from (1) environmental causes such as excessive exposure to ultraviolet light, radiation, or certain chemicals (i.e., mutagens) or (2) errors in cell division when somatic or germline cells copy their DNA in preparation for mitosis or meiosis, respectively. Most mutations are not perpetuated because cells have reparative mechanisms (i.e., base excision repair, nucleotide excision repair, and mismatch repair) and DNA repair proteins that correct mistakes caused by mutagens or other factors (E-Fig. 1-24). These repair mechanisms and proteins determine which nucleotide bases are paired incorrectly and then replace the incorrect base with the correct one(s). The effects of mutations can be considerable if not repaired by one of these mechanisms. Mutations of the genes for DNA repair proteins also can have serious outcomes, especially in neoplastic transformation of somatic cells.
E-Figure 1-24.
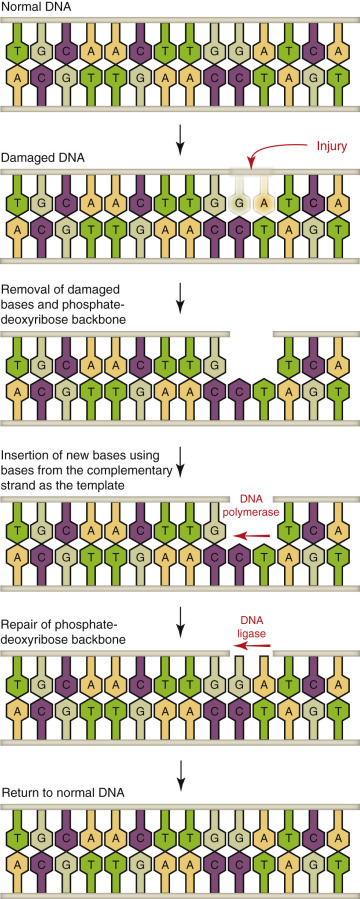
Repair of Damaged DNA.
Three pathways exist to repair single-stranded DNA damage: base excision repair, nucleotide excision repair, and DNA mismatch repair. Base excision repair can be used at any point in the cell cycle and is important in removing damaged bases that could cause mutations by mispairing or lead to breaks in DNA during replication. Nucleotide excision repair mends DNA damaged by chemicals, UV radiation, or other mutagens that cause formation of DNA adducts (DNA covalently bonded to a chemical). DNA mismatch repair mends erroneous insertion, deletion, or mismatched base pairs.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Single-gene disorders arise from a mutation in one gene and result in a permanent change of the cell's nuclear DNA (E-Figs. 1-25 and 1-26). Such mutations can affect protein synthesis by disrupting one or more steps in transcription or translation and lead to the following:
E-Figure 1-25.
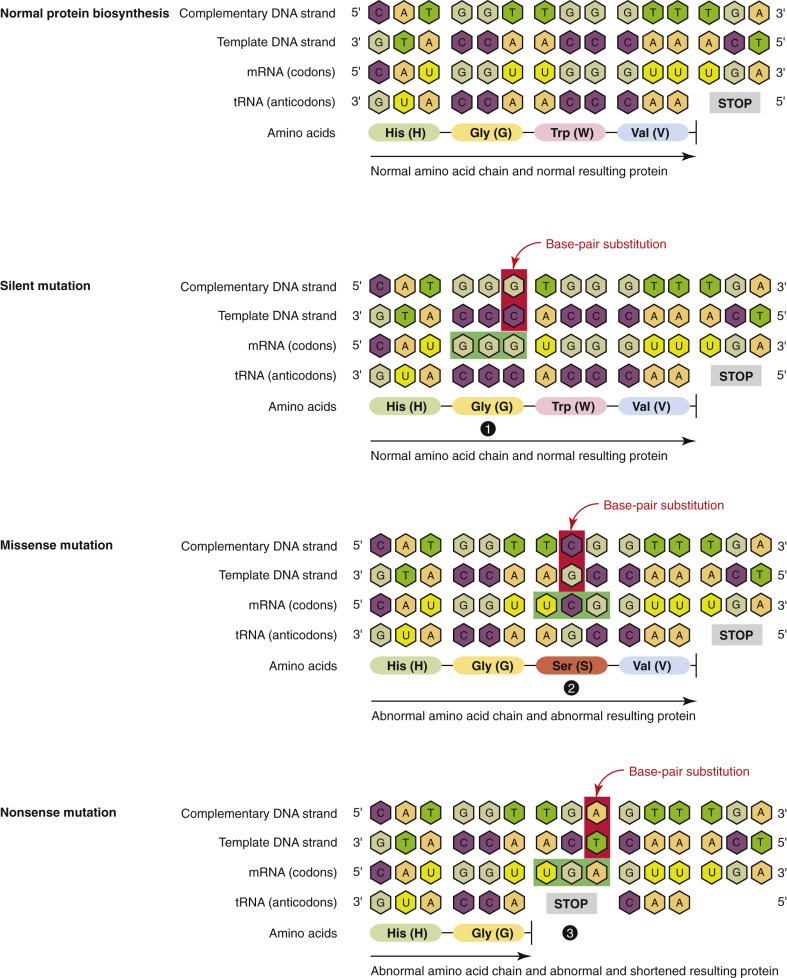
Types of Base-Pair Substitutions in Gene Mutations.
1, Silent mutations. Base-pair substitutions in silent mutations change the messenger RNA (mRNA) codons (GGU to GGG in the example); however, the mutated codons (GGG) code for the same amino acid as the original mRNA codons, so the synthesis of the polypeptide is unaffected. 2, Missense mutations. Base-pair substitutions in missense mutations change the mRNA codons (UGG to UCG in the example); the mutated codons (UCG) code for a different amino acid than the original mRNA codons, so the synthesis of the polypeptide is altered. 3, Nonsense mutations. Base-pair substitutions in nonsense mutations change the mRNA codons (UGG to UGA in the example); the mutated codons (UGA) are a “stop” code, prematurely halting polypeptide chain synthesis. tRNA, Transfer RNA.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
E-Figure 1-26.
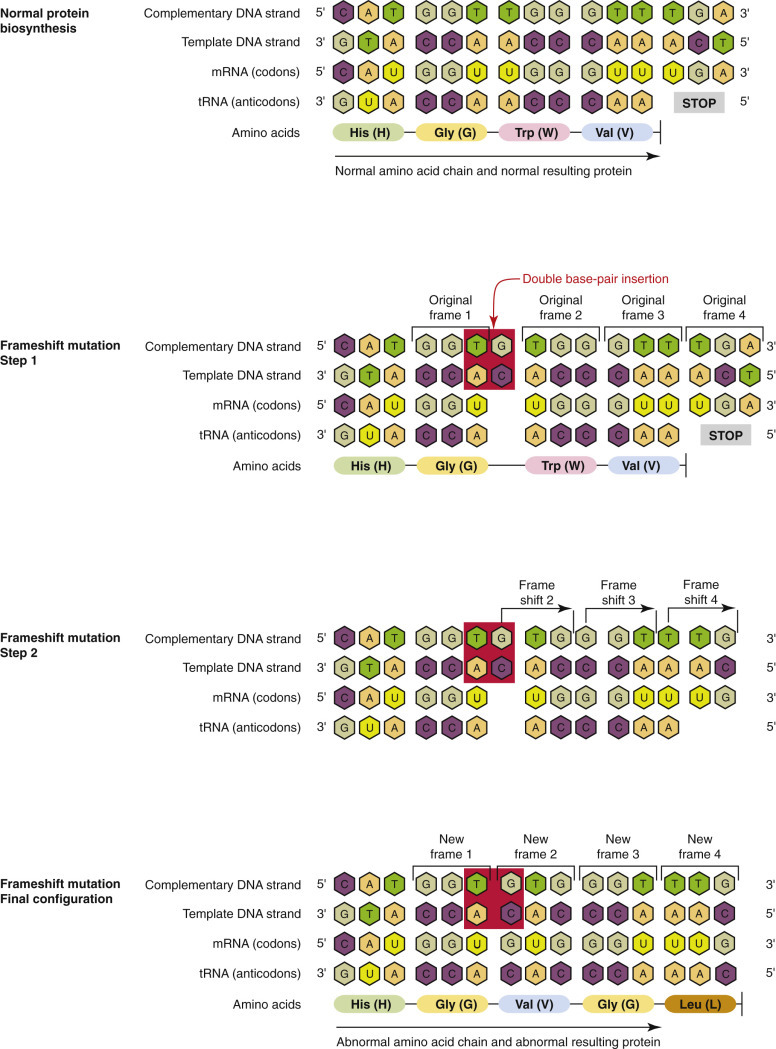
Mechanism of Frameshift Mutations.
Frameshift mutations are the result of insertions or deletions that alter the reading frame of the triplet codons, thereby altering translation and altering the structure and function of the protein product. mRNA, Messenger RNA; tRNA, transfer RNA.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
-
1.
The formation of an abnormal protein
-
2.
A reduction in the amount of protein synthesized
-
3.
The formation of abnormal proteins without impairing any step in protein synthesis
-
4.
Modification in the rate of synthesis, posttranslational mechanisms, or transporting of proteins out of the cell
Virtually any type of protein may be altered in single-gene disorders with the following adverse effects:
-
1.
Enzyme defects (i.e., lack of function)
-
2.
Defects in membrane receptors and transport systems
-
3.
Alterations in the structure, function, or quantity of nonenzyme proteins
-
4.
Unusual reactions to drugs
In enzyme defects, mutations may result in the synthesis of a defective enzyme with reduced activity or in a reduced amount of a normal enzyme. An enzyme defect may have deleterious effects by allowing intracellular accumulation of a substrate(s) or of products of an alternative pathway. Such metabolic by-products can be cytotoxic. An enzyme defect can also disrupt a metabolic pathway, resulting in insufficient end product for normal function.
Mutations resulting in the accumulation of complex substrates or the blockage of metabolic pathways are best illustrated by a group of diseases called storage diseases, in which defective processing of a metabolic substrate leads to the accumulation of the substrate in the cytoplasm or within lysosomes. Such diseases are discussed in the Pathology of Organ Systems chapters of this book (especially see Chapter 14). Storage diseases are commonly grouped as lysosomal storage diseases and glycogen storage diseases. Lysosomal storage diseases are characterized by a deficiency of lysosomal acid hydrolases with incomplete breakdown of their substrates, leading to accumulation of the partially degraded insoluble metabolite within lysosomes (E-Fig. 1-27). Lysosomal acid hydrolases break down a variety of complex macromolecules derived from the metabolic turnover of organelles or acquired from outside the cells by phagocytosis. Stuffed with incompletely digested macromolecules, the lysosomes become large and numerous enough to interfere with normal cell functions. Lysosomal storage diseases are exemplified by globoid cell leukodystrophy, in which a functional deficiency in lysosomal galactocerebroside β-galactosidase (galactosylceramidase) results in the accumulation of galactocerebroside in macrophages recruited into the CNS (see Chapter 14 and Fig. 14-64).
Glycogen storage diseases (glycogenoses) are caused by a deficiency of an enzyme involved in the synthesis or degradation of glycogen (E-Fig. 1-28; see Fig. 14-63). In glycogenosis type III (Cori's disease), deficient function of an amylo-1,6-glucosidase (debranching enzyme) results in the accumulation of a structurally abnormal glycogen within hepatocytes.
E-Figure 1-28.

Enzyme Deficiencies Involved in Glycogen Storage Diseases.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Other examples of single-gene disorders include X-linked muscular dystrophy, some types of collagen dysplasias, and drug transporter dysfunction. X-linked (recessive trait) muscular dystrophy (Duchenne's type) occurs in a variety of dog breeds and is characterized by defects in the dystrophin gene, which codes for a membrane-associated cytoskeletal protein in skeletal and cardiac muscle (see Chapter 15). In the absence of dystrophin, muscle fibers are more susceptible to repeated occurrences of necrosis and regeneration, potentially leading to cardiac fibrosis and progressive cardiomyopathy (see Chapter 10).
Collagen dysplasia (hyperelastosis cutis, dermatosparaxis, and cutaneous asthenia) occurs in most domestic animals and is caused by defects in genes for enzymes involved in collagen synthesis or processing (see Chapter 17). Skin tears easily, or is hyperextensible, but the severity and nature of lesions varies among species.
Mutation of the multidrug resistance 1 gene (MDR1), a homozygous dominant trait in collies and other canine breeds, causes a defect in a membrane P-glycoprotein that facilitates the transport of drugs, such as ivermectin and loperamide, out of the brain. These drugs accumulate in the brain and may result in neurologic injury and dysfunction.
The consequences of other mutations of genes for membrane receptors, transport systems, or nonenzyme proteins or mutations resulting in unusual drug reactions have not been adequately documented in animals.
Single-Gene Disorders of Somatic Cells.
Mutations that arise in somatic cells are not heritable but are important in the genesis of tumors (E-Fig. 1-29; see also Chapter 6) and some congenital malformations. Tumor-specific acquired single-gene mutations are expressed in some types of tumors. During the clinical management of cancer, such mutations can serve as a means to detect the growth of a tumor and monitor its response to therapy.
E-Figure 1-29.
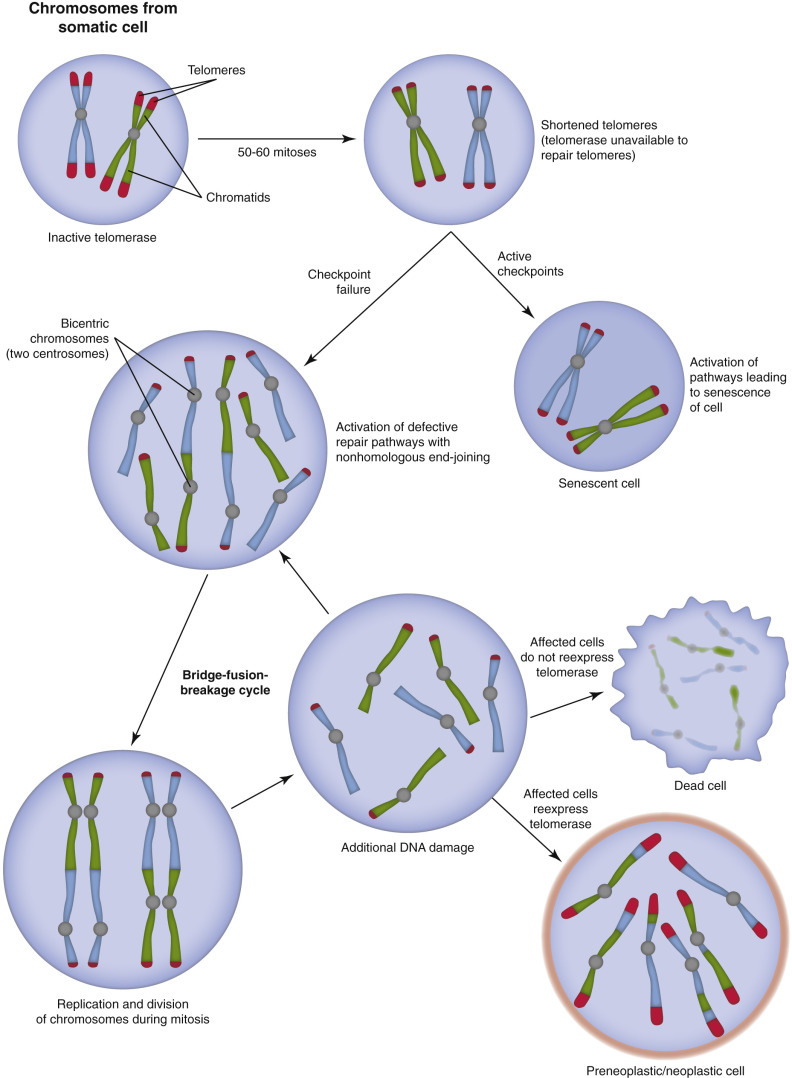
Sequence of Events in Tumorigenesis.
There are approximately 220 types of somatic cells in an animal's body. During their lifespan, somatic cells can divide up to 50 to 60 times. If they do not express telomerase, their telomeres are significantly shortened. A telomere is a region at the end of each chromatid that protects the end of the chromosome from deterioration and/or from fusion with adjacent “aged” chromosomes. In the presence of active checkpoints, cells become senescent. If these checkpoints fail, DNA-repair pathways are inappropriately activated and result in the formation of dicentric chromosomes. During mitosis, dicentric chromosomes are pulled apart, leading to random double-stranded breaks in affected chromatids. In response, DNA-repair pathways are again activated, resulting in the formation of additional dicentric chromosomes. Cells that go through repetitive sequences of this “bridge-fusion-breakage” cycle have substantial chromosomal instability and numerous mutations. Subsequently, if affected cells fail to express telomerase, they eventually die. If they express telomerase, the cells can escape the “bridge-fusion-breakage” cycle. However, their survival enhances tumorigenesis because of chromosomal instability and numerous mutations.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Single-Gene Disorders of Germ Cells.
Mutations that affect germ cells are transmitted to the progeny and can give rise to inherited diseases that usually follow the classic mendelian pattern of inheritance. Inherited mutations involving single genes typically follow one of three patterns: autosomal dominant, autosomal recessive, or X-linked.
Autosomal Dominant Disorders.
In autosomal dominant disorders, only one allele of a mutated gene is necessary for disease. This allele may come from the sire or from the dam; thus, if one parent carries even one mutated allele (heterozygous), each offspring has a 50% chance of inheriting the mutation. Examples of autosomal dominant disorders in animals include polycystic kidney disease (see Fig. 11-26, F), osteogenesis imperfecta (see Chapter 16), and chondrodysplasia (see Fig. 16-39). In autosomal dominant disorders, most mutations lead to reduced production of a protein or give rise to an inactive protein. The clinical effect of these loss-of-function mutations depends on the activity of the protein affected. If such mutations involve an enzyme, heterozygotes may be clinically normal because the normal allele can compensate for up to a 50% loss of enzymatic activity. In contrast, autosomal dominant disorders have serious effects on structural proteins, such as collagen or spectrin, even in heterozygotes with one normal allele. A 50% reduction in the amount of such proteins results in abnormal structure and assembly of collagen, and a spectrin deficiency causes osmotic fragility of erythrocytes and hereditary spherocytosis in golden retriever dogs.
Less common than loss-of-function mutations are gain-of-function mutations. In this type of mutation, the gene product acquires new biologic activities not usually associated with the normal-type protein.
Autosomal Recessive Disorders.
In autosomal recessive disorders, both alleles at a given gene locus must be mutated for an animal to be affected by the disorder. One mutated allele is provided by the sire and the other by the dam. Thus there is a 25% chance that each offspring from heterozygous parents will inherit both mutated alleles. Heterozygotes, with only one mutated allele, are clinically normal carriers of the trait. Homozygous animals usually have clinical disease, and the onset is usually early in life. Many of the mutated genes encode enzymes. Examples of autosomal recessive disorders in animals include lysosomal storage diseases (see E-Fig. 1-27 and Figs. 14-63 and 14-64), glycogen storage diseases (see Fig. 14-63) and mucopolysaccharidoses, and aminoacidopathies that affect organs such as the brain, spinal cord, skeletal muscle, liver, and kidney.
X-Linked Disorders.
All sex-linked disorders are X-linked, and almost all are recessive and caused by mutations in genes on the X chromosome. Male animals are more likely to be affected by X-linked recessive disorders because they have only one X chromosome. Females, unless they inherit the mutation from both parents, are typically clinically normal carriers of X-linked traits. Examples of X-linked recessive disorders in animals include Duchenne's muscular dystrophy (see Chapter 15) and agammaglobulinemia of the immune system (see Chapter 5).
Single-Gene Disorders of Mitochondria.
Some single-gene disorders have a nonmendelian pattern of inheritance and include disorders arising from mutations in mtDNA and those in which the transmission is influenced by trinucleotide-repeat mutations, genomic imprinting, or gonadal mosaicism. In such cases, mitochondrial mutations occur in mtDNA rather than in the nuclear genome. Diseases resulting from mitochondrial inheritance are rare, and many affect the nervous (e.g., mitochondrial encephalopathies; see Chapter 14) and muscular systems. There are only 37 mitochondrial genes, and a feature unique to mtDNA is maternal inheritance. Dams and only dams transmit mtDNA to their offspring, both male and female. Sires make no contribution of mtDNA to offspring. This peculiarity exists because ova contain numerous mitochondria within their cytoplasm, whereas spermatozoa contain few, if any. Thus the mtDNA complement of the fertilized ovum is derived entirely from the ovum.
Each of the 200 to 210 cell types of an animal's body requires a specific amount of ATP for normal function. Although, within individual cell types, some variation in ATP concentrations may be tolerated, there is typically a threshold concentration below which cells begin to degenerate and die. Thus cells with large ATP requirements tend to be the ones most seriously affected by mitochondrial diseases. Because mtDNA encodes enzymes involved in oxidative phosphorylation, mutations affecting these genes exert their deleterious effects primarily on the organs most dependent on oxidative phosphorylation such as the CNS, skeletal muscle (type II myofibers), cardiac muscle, liver, and kidneys. For example, the CNS produces approximately 20% of the body's total ATP and therefore is most often affected by mtDNA mutations. The mutation rate of mtDNA is approximately 10 times that of nuclear DNA. This difference is caused by a relative lack of DNA repair mechanisms in mtDNA and by damage from ROS released during oxidative phosphorylation.
Chromosomal Disorders.
Errors in Cell Division.
Most chromosomal disorders are caused by errors in cell division, which transfers the disorder within somatic and/or germline cells. Abnormalities of chromosome number and/or structure can arise in autosomes (chromosomes in somatic cells) or sex chromosomes (germline cells). There are two kinds of cell division, mitosis and meiosis (E-Fig. 1-30). Mitosis is somatic cell division by which the body grows and differentiates and tissues regenerate. Mitotic division results in two daughter cells, each with chromosomes and genes identical to those of the parent cell. In contrast, meiosis occurs only in cells of the germline and results in the formation of ova or spermatozoa, with, under normal conditions, each cell type having half of the normal karyotype (one of each kind of autosome and either an X or a Y chromosome).
E-Figure 1-30.
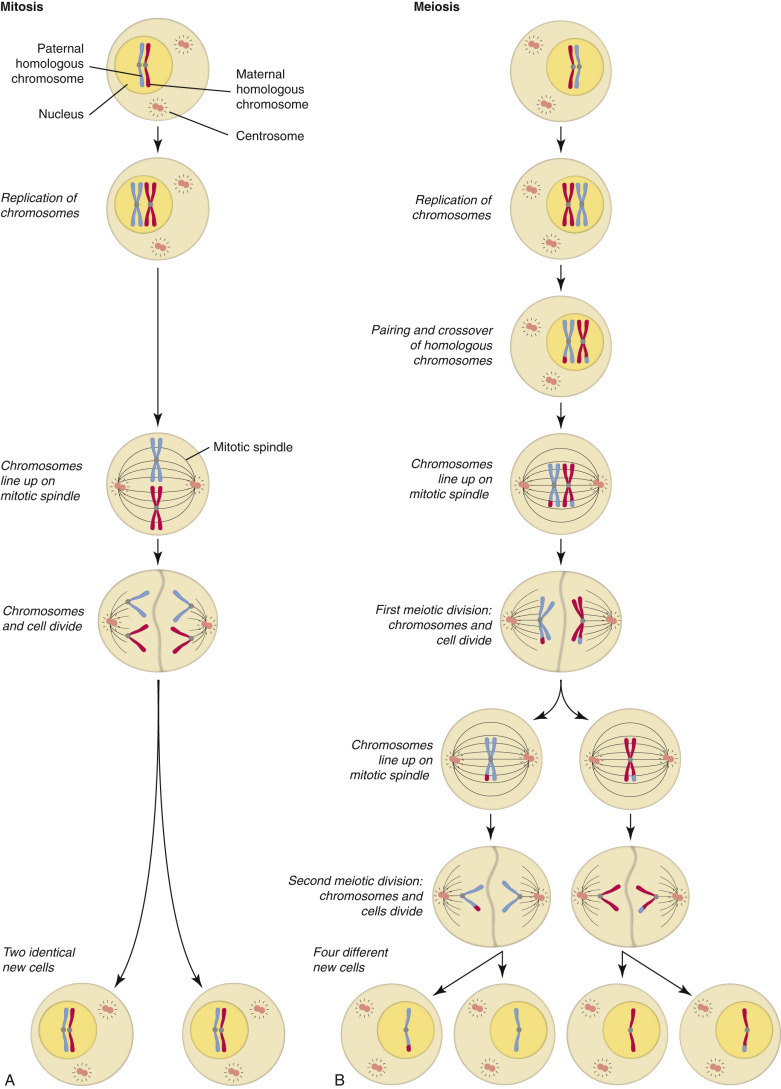
Mitosis and Meiosis.
A comparison of normal mitotic (A) and meiotic (B) cell division.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
After fertilization a single-cell zygote gives rise to all cells of the body (estimated at 1 × 1014 cells), which are derived from dozens or even hundreds of mitoses. The biologic significance of meiosis and mitosis lies in ensuring the constancy of chromosome number and thus the integrity of the genome from one cell to its progeny and from one generation to the next. The medical significance of these processes involves errors of cell division, which lead to the formation of an individual cell or of a cell lineage with an abnormal number of chromosomes and thus an inappropriate amount of genomic material. Such errors are called nondisjunctions and represent a failure of chromosome pairs to disjoin (separate) during cell division, and as a result both chromosomes go to one cell and none to the other. Meiotic nondisjunction, particularly in oogenesis, is a common mutational mechanism, responsible for chromosomally abnormal fetuses. In those fetuses that survive to term, chromosome abnormalities cause developmental defects, failure to thrive, and reduced mental function. Mitotic nondisjunctions can also be inherited. Nondisjunction soon after fertilization, either in the developing embryo or in extraembryonic tissues like the placenta, leads to chromosomal mosaicism that can be the basis for some genetic disorders. Additionally, abnormal chromosome segregation in rapidly dividing tissues can be a step in the development of tumors.
Numeric Alterations.
Cells with normal chromosome numbers have euploid karyotypes (i.e., normal number of chromosomes for the species). If an error occurs in meiosis or mitosis and a cell acquires a lesser or greater number of chromosomes, the abnormal karyotype is referred to as aneuploidy. One cause of aneuploidy is nondisjunction during meiosis (E-Fig. 1-31), resulting in either extra chromosomes (e.g., trisomy, tetrasomy) or one less chromosome (i.e., monosomy) (see E-Fig. 1-31). Fertilization of such ova by normal spermatozoa results in two types of zygotes, trisomic (or tetrasomic) or monosomic. Trisomic or tetrasomic offspring are extremely rare in domestic animals, but an autosomal trisomy has been reported in an Italian Friesian calf with malformed limbs, congenital opisthotonus, brachygnathia, blindness, and absence of external genitalia. Monosomic offspring are more common in domestic animals, in which an X chromosome monosomy (Turner-like syndrome) has been reported mainly in horses (see Chapter 18). The vulva, uterus, and ovaries in affected mares are smaller than normal; most fail to cycle or show estrous behavior.
E-Figure 1-31.
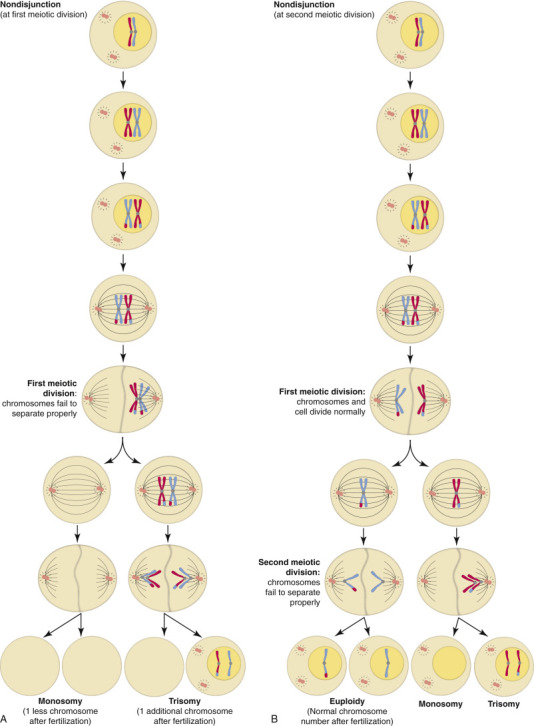
Nondisjunction.
Nondisjunction is the failure of homologous chromosomes (chromatids) to separate properly during meiotic cell division. A, Nondisjunction at the first meiotic division. B, Nondisjunction at the second meiotic division.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Occasionally, mitotic errors in early development give rise to two or more populations of cells with different chromosomal karyotypes in the same animal, a condition referred to as mosaicism. Mosaicism can result from mitotic errors during the division of the fertilized ovum or in somatic cells. Mosaicism affecting the sex chromosomes is relatively common. In the division of the fertilized ovum, an error may lead to one of the daughter cells receiving three sex chromosomes, whereas the other receives only one, yielding, for example, an n-1, X/n+1, XXX mosaic. All cells derived from each of these cells will have the same abnormal karyotype. An example of X (sex) chromosome mosaicism occurs in tortoiseshell and calico (tricolored) cats. In all female mammalian cells, the function of one X chromosome is inactivated through a random process called X chromosome inactivation. Approximately 50% of the cells of tricolored cats have inactivated paternal X chromosomes; the other 50% have inactivated maternal X chromosomes. Thus female tricolored cats have roughly equal populations of two genetically different cell types and are therefore a type of mosaic that is expressed in the patterns of hair coloration (orange, black, and white).
Autosomal mosaicism seems to be much less common than that involving the sex chromosomes. An error in an early mitotic division affecting the autosomes usually leads to a nonviable mosaic fetus.
Structural Alterations.
Changes in the structure of chromosomes are caused by deletion, inversion, duplication, or translocation of a portion of a sex or autosomal chromosome during cell division (E-Fig. 1-32). During embryogenesis, structural alterations of sex chromosomes are more common than those of autosomes and can result in some cells having XX and others having XY sex chromosomes. These cells coexist, so both male and female reproductive structures develop to varying degrees dependent on the expression of the sex chromosomes. As a result, these diseases are characterized by sexual ambiguity and include hermaphroditism and pseudohermaphroditism (see Chapters 18 and 19). Structural alterations also likely involve autosomes in animals, but their occurrence and significance have not been adequately characterized.
E-Figure 1-32.
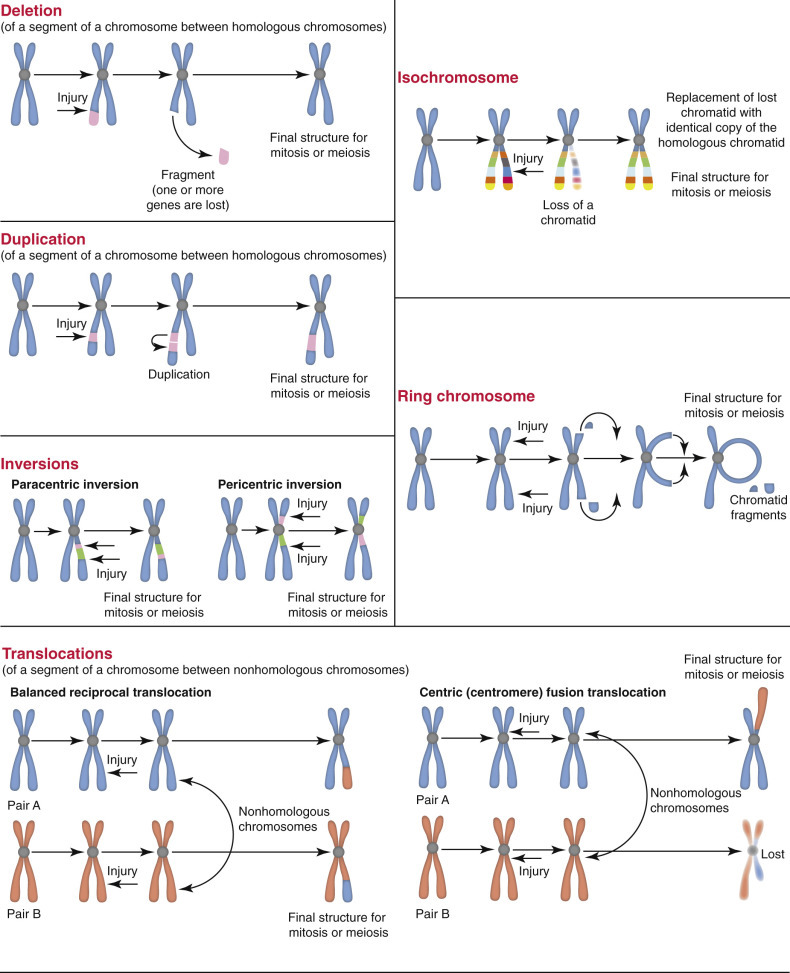
Forms of Chromosomal Rearrangements.
Chromosomal rearrangements are chromosome abnormalities characterized by structural changes in chromosomes such as missing, extra, or irregular segments of chromosomal DNA. They are caused by breakage of DNA double helices from errors in DNA replication and/or from damage caused by mutagens. These rearrangements include deletions, duplications, inversions, translocations, isochromosomes, and ring chromosomes.
(Courtesy Dr. M.A. Miller, College of Veterinary Medicine, Purdue University; and Dr. J.F. Zachary, College of Veterinary Medicine, University of Illinois.)
Complex Multigenic Disorders
Complex multigenic disorders are caused by interactions between variant forms of genes (e.g., polymorphisms) and environmental factors. Each variant gene confers a small increase in disease risk, but no single gene is necessary or able alone to produce disease. It is only when several polymorphisms are present in an animal that disease occurs, hence the terms multigenic or polygenic. Thus, unlike the single–mutant gene disorders (discussed previously) that commonly cause disease and give rise to mendelian-inherited disorders, each individual polymorphism has a small effect and rarely causes disease by itself. Because environmental interactions are important in the pathogenesis of these diseases and the complex traits do not follow a mendelian pattern of inheritance, the genes and polymorphisms that contribute to these diseases have been very difficult to determine. Assigning a disease to this mode of inheritance must be done with caution. Diagnosis of diseases that are considered complex multigenic disorders such as type 1 diabetes (a known human complex multigenic disorder) depends on many factors and the exclusion of mendelian (single-gene) and chromosomal modes of transmission. The occurrence of complex multigenic disorders in animals has not been demonstrated except in laboratory animal models of human disease, but their existence in domestic animal species is highly likely.
Types of Diagnoses
Diagnosis is a common goal in veterinary medicine and in pathology. Clinical diagnosis is based mainly on the signalment, history, and physical examination findings. Even with clinical pathology data or diagnostic imaging, the clinical diagnosis may be only tentative or presumptive.
Pathologists, especially anatomic pathologists, as the term implies, are particularly interested in morphologic diagnosis, which is based on the structural features of the observed lesions. A morphologic diagnosis should categorize the lesion—as degeneration or necrosis, a disturbance of growth, vascular disturbance, or inflammation—and indicate the affected organ(s) or tissue. The just-introduced word necrosis could be the part of a morphologic diagnosis that categorizes the lesion and then needs just one more word (e.g., hepatic) to indicate the location of the lesion. Thus hepatic necrosis is a complete morphologic diagnosis, though it may need an adjective or two for descriptive purposes. For example, multifocal (randomly distributed) hepatic necrosis implicates an infection, whereas a lobular pattern of hepatic necrosis is usually the result of ischemic, metabolic, or toxic injury (see also the section on Morphologic Appearance of Necrotic Cells and Tissues). A morphologic diagnosis can be based on gross (visible to the naked eye) or histologic (visible microscopically) lesions.
A differential diagnosis is a list of diseases or conditions that would explain the clinical findings or observed lesions. In some settings the morphologic diagnosis is the end point. More often it is the basis for formulation of the differential diagnosis. A differential diagnosis should not be an exhaustive list but rather should contain those diseases that are most likely to have caused the observed changes in an animal with the recorded signalment and history. Generally, a differential diagnosis is refined (shortened) as the diagnostician progresses from the clinical findings to postmortem or surgical biopsy pathology and supporting laboratory results.
The ultimate goal is the definitive diagnosis. The definitive diagnosis identifies the specific disease or condition. In some settings (e.g., in the naming of a neoplasm) a morphologic diagnosis is the definitive diagnosis. More often it is a starting point. For example, if a morphologic diagnosis of multifocal hepatic necrosis implicates an infectious disease, then the identification of the particular disease or the etiologic agent of that disease is the diagnostic end point.
Ancillary tests, such as immunohistochemistry, chemical analyses, and microbiology, are paramount in refining a differential diagnosis to reach the definitive diagnosis. Ancillary tests are selected based on the nature of the disease. For example, a poorly differentiated mast cell tumor might necessitate histochemistry, immunohistochemistry, or perhaps even molecular diagnostics, such as PCR amplification of nucleic acids, whereas the H&E histologic section may suffice for a well-differentiated mast cell tumor. Neoplastic disease is one of the few situations in which a morphologic diagnosis is also the definitive diagnosis. Lesions caused by trauma (e.g., fractures, hemorrhages) are additional examples in which morphologic diagnoses are definitive. Lesions of nutritional deficiencies or toxic diseases are seldom so specific and generally implicate chemical injury without incriminating a particular substance. Infectious diseases, unless the lesions are highly specific, likewise engender a list of probable causes; microbiologic (bacteriologic, mycologic, virologic, or parasitologic) assays are then required for identification of a particular cause (genus and species of the etiologic agent).
An etiologic diagnosis highlights the cause (rather than the morphologic features) of a disease. For example, in a foal with Tyzzer's disease, a morphologic diagnosis of multifocal necrotizing hepatitis, once the cause (Clostridium piliforme) is identified, could be replaced or supplemented with an etiologic diagnosis of clostridial hepatitis. Importantly, an etiologic diagnosis of clostridial hepatitis does not implicate a particular species of Clostridium and so is less useful than the definitive diagnosis of Tyzzer's disease (C. piliforme infection).
Summary
This chapter is focused on the response to injury at the cellular level, but the student must remember that an injured cell is affected not only by its direct injury but also by neighboring and distant cells, stroma, and vasculature, and that the injured cell in turn affects cells and tissues around it (and at distant sites). In subsequent chapters we will see how blood flow, the inflammatory response, the immune response, and other factors come into play and realize that the whole body, not just one or a few cells, responds to injury.
Suggested Readings
Suggested Readings are available at www.expertconsult.com.
Footnotes
For a glossary of abbreviations and terms used in this chapter see E-Glossary 1-1.
As a general rule in an H&E stain, hematoxylin dyes stain nucleic acids blue, and eosin dyes stain proteins red.
If a protein is not properly formed (folded), it is not able to complete its assigned function, and thus the outcome is called a “loss-of-function disorder.”
Silica crystals are colorless, so they are not an example of a pigmented substance.
A feed-forward loop is the positive or negative effect that a process or substance in a metabolic pathway may have on another substance or step in a process that occurs later in the pathway.
Mammalian target of rapamycin (mTOR) is a growth regulator that can be inhibited by caloric restriction (or by rapamycin, hence the name) with a protective effect on mitochondria.
To ensure accuracy in this section the contributors consulted Turnpenny PD, Ellard S: Emery's elements of medical genetics, ed 14, Edinburgh, 2011, Churchill Livingstone.
Data, in part, from Guide to congenital and heritable disorders in dogs, Davis, CA, May 2011, The Humane Society Veterinary Medical Association.
Data, in part, from Breur GJ, Lambrechts NE, Todhunter RJ: The genetics of canine orthopaedic traits. In Ostrander E, Ruvinsky A, editors: The genetics of the dog, ed 2, Cambridge, MA, 2012, CABI Publishing, pp 136-160.
See E-Box 17-2 for more detail. Therein, genetic diseases of the skin are organized by species, diagnoses, and mechanisms.
Suggested Readings
- Adkison L. ed 2. Saunders; St. Louis: 2011. Elsevier’s integrated review genetics. [Google Scholar]
- Bertoli C, Skotheim JM, de Bruin RAM. Control of cell cycle transcription during G1 and S phases. Nature Rev Mol Cell Biol. 2013;14:518–528. doi: 10.1038/nrm3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blancas-Mejía LM, Ramirez-Alvarado M. Systemic amyloidosis. Annu Rev Biochem. 2013;82:745–774. doi: 10.1146/annurev-biochem-072611-130030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubeck D. The making of a macromolecular machine: assembly of the membrane attack complex. Biochemistry. 2014;53:1908–1915. doi: 10.1021/bi500157z. [DOI] [PubMed] [Google Scholar]
- Buchakjian MR, Kornbluth S. The engine driving the ship: metabolic steering of cell proliferation and death. Nature Rev Mol Cell Biol. 2010;11:715–727. doi: 10.1038/nrm2972. [DOI] [PubMed] [Google Scholar]
- Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nature Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- Cassidy SKB, O'Riordan MXD. More than a pore: the cellular response to cholesterol-dependent cytolysins. Toxins. 2013;5:618–636. doi: 10.3390/toxins5040618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challa S, Chan FKM. Going up in flames: necrotic cell injury and inflammatory diseases. Cell Mol Life Sci. 2010;67:3241–3253. doi: 10.1007/s00018-010-0413-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Saikumar P, Weinberg JM. Calcium in cell injury and death. Annu Rev Pathol Mech Dis. 2006;1:405–434. doi: 10.1146/annurev.pathol.1.110304.100218. [DOI] [PubMed] [Google Scholar]
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Kroemer G. Mitochondria: master regulators of danger signaling. Nature Rev Mol Cell Biol. 2012;13:780–788. doi: 10.1038/nrm3479. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vanden Berghe T, Vanlangenakker N. Programmed necrosis: from molecules to health and disease. Int Rev Cell Mol Biol. 2011;289:1–35. doi: 10.1016/B978-0-12-386039-2.00001-8. [DOI] [PubMed] [Google Scholar]
- Geiger B, Bershadsky A, Pankov R. Transmembrane extracellular matrix—cytoskeleton crosstalk. Nature Rev Mol Cell Biol. 2001;2:793–805. doi: 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- Jorde LB, Carey JC, Bamshad MJ. ed 5. Mosby; St. Louis: 2015. Medical genetics. [Google Scholar]
- Kalogeris T, Baines CP, Krenz M. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229–317. doi: 10.1016/B978-0-12-394309-5.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Coulombe PA. Emerging role for the cytoskeleton as an organizer and regulator of translation. Nature Rev Mol Cell Biol. 2010;11:75–81. doi: 10.1038/nrm2818. [DOI] [PubMed] [Google Scholar]
- Knowler SP, McFadyen AK, Freeman C. Quantitative analysis of Chiari-like malformation and syringomyelia in the Griffon Bruxellois dog. PLoS One. 2014;9:e88120. doi: 10.1371/journal.pone.0088120. 1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles TPJ, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nature Rev Mol Cell Biol. 2014;15:384–396. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Vandenabeele P. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Abbas AK, Aster JC. ed 9. Elsevier Saunders; Philadelphia: 2015. Robbins & Cotran pathologic basis of disease. [Google Scholar]
- Levin S, Bucci TJ, Cohen SM. The nomenclature of cell death: recommendations of an ad hoc committee of the Society of Toxicologic Pathologists. Toxicol Pathol. 1999;27:484–490. doi: 10.1177/019262339902700419. [DOI] [PubMed] [Google Scholar]
- Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455–465. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockshin RA, Zakeri Z. Review: apoptosis, autophagy, and more. J Biochem Cell Biol. 2004;36:2405–2419. doi: 10.1016/j.biocel.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Longo VD, Lieber MR, Vijg J. Turning anti-ageing genes against cancer. Nature Rev Mol Cell Biol. 2008;9:903–910. doi: 10.1038/nrm2526. [DOI] [PubMed] [Google Scholar]
- Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nature Rev Mol Cell Biol. 2013;14:38–48. doi: 10.1038/nrm3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majno G, Joris I. ed 2. Oxford University Press; Oxford: 2004. Cells, tissues, and disease: principles of general pathology. [Google Scholar]
- Mariño G, Niso-Santano M, Baehrecke EH. Self-consumption: the interplay of autophagy and apoptosis. Nature Rev Mol Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxie MG, editor. Jubb, Kennedy & Palmer's pathology of domestic animals. ed 6. Elsevier; St. Louis: 2015. [Google Scholar]
- McCance KL, Huether SE, editors. Pathophysiology: The biologic basis for disease in adults and children. ed 7. Elsevier; St. Louis: 2015. [Google Scholar]
- McGavin MD. Factors affecting visibility of a target tissue in histologic sections. Vet Pathol. 2014;51:9–27. doi: 10.1177/0300985813506916. [DOI] [PubMed] [Google Scholar]
- Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583–596. doi: 10.1056/NEJMra023144. [DOI] [PubMed] [Google Scholar]
- Mohr U, Carlton WW, Dungworth DL, editors. Pathobiology of the aging dog. Iowa State University Press; Ames, IA: 2001. [Google Scholar]
- Nandakumar J, Cech TR. Finding the end: recruitment of telomerase to the telomere. Nature Rev Mol Cell Biol. 2013;14:69–82. doi: 10.1038/nrm3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Dugger DL, Wickliffe KE. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–1360. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- Nezelof C, Seemayer TA. The history of pathology: an overview. In: Damjanov I, Linder J, editors. Anderson's pathology. ed 10. Mosby; St. Louis: 1996. [Google Scholar]
- Nicholas FW. ed 3. Wiley-Blackwell; Hoboken, NJ: 2009. Introduction to veterinary genetics. [Google Scholar]
- Nussbaum RL, McInnes RR, Willard HF. ed 7. Saunders; Philadelphia: 2007. Thompson & Thompson's genetics in medicine. [Google Scholar]
- Oberdoerffer P, Sinclair DA. The role of nuclear architecture in genomic instability and ageing. Nature Rev Mol Cell Biol. 2007;8:692–702. doi: 10.1038/nrm2238. [DOI] [PubMed] [Google Scholar]
- Pearse AGE. Little, Brown, and Co; Boston: 1961. Histochemistry: theoretical and applied. [Google Scholar]
- Petsonk EL, Rose C, Cohen R. Coal mine dust lung disease. New lessons from old exposure. Am J Respir Crit Care Med. 2013;187:1178–1185. doi: 10.1164/rccm.201301-0042CI. [DOI] [PubMed] [Google Scholar]
- Porta EA. Pigments in aging: an overview. Ann N Y Acad Sci. 2002;959:57–65. doi: 10.1111/j.1749-6632.2002.tb02083.x. [DOI] [PubMed] [Google Scholar]
- Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5(11):897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, De Stefani D, Raffaello A. Mitochondria as sensors and regulators of calcium signaling. Nature Rev Mol Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- Rowland AA, Voeltz GK. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nature Rev Mol Cell Biol. 2012;13:605–625. doi: 10.1038/nrm3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin E, DePinho RA. Axis of ageing: telomeres and mitochondria. Nature Rev Mol Cell Biol. 2012;13:397–404. doi: 10.1038/nrm3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seehafer SS, Pearce DA. You say lipofuscin, we say ceroid: defining autofluorescent storage material. Neurobiol Aging. 2006;27:576–588. doi: 10.1016/j.neurobiolaging.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Sperka T, Wang J, Rudolph KL. DNA damage checkpoints in stem cells, ageing and cancer. Nature Rev Mol Cell Biol. 2012;13:579–590. doi: 10.1038/nrm3420. [DOI] [PubMed] [Google Scholar]
- Stoecklin G, Bukau B. Telling right from wrong in life—cellular quality control. Nature Rev Mol Cell Biol. 2013;14:613–615. doi: 10.1038/nrm3662. [DOI] [PubMed] [Google Scholar]
- Trump BF, Berensky IK. The reaction of cells to lethal injury: oncosis and necrosis—the role of calcium. In: Lockshin RA, Zakeri Z, Tilly J, editors. When cells die. Wiley-Liss; New York: 1998. [Google Scholar]
- Turnpenny P, Ellard S. ed 14. Churchill Livingstone; Philadelphia: 2012. Emery's elements of medical genetics. [Google Scholar]
- Vandenabeele P, Galluzzi L, Vanden Berghe T. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nature Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe T, Linkermann A, Jouan-Lanhouet S. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nature Rev Mol Cell Biol. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- Van Herreweghe F, Festjens N, Declercq W. Tumor necrosis factor-mediated cell death: to break or to burst, that is the question. Cell Mol Life Sci. 2010;67:1567–1579. doi: 10.1007/s00018-010-0283-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach D, Kang TB, Kovalenko A. Concepts of tissue injury and cell death in inflammation: a historical perspective. Nature Rev Immunol. 2014;14:51–59. doi: 10.1038/nri3561. [DOI] [PubMed] [Google Scholar]
- Weber LWD, Boll M, Stampfl A. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol. 2003;33:105–136. doi: 10.1080/713611034. [DOI] [PubMed] [Google Scholar]