

Abstract
The actin cortex that lines the plasma membrane of most eukaryotic cells resists external mechanical forces and plays critical roles in a variety of cellular processes including morphogenesis, cytokinesis, and cell migration. Despite its ubiquity and significance, we understand relatively little about the composition, dynamics, and structure of the actin cortex. Adenomatous polyposis coli (APC) proteins regulate the actin and microtubule cytoskeletons through a variety of mechanisms, and in some contexts, APC proteins are cortically enriched. Here we show that APC2 regulates cortical actin dynamics in the follicular epithelium and the nurse cells of the Drosophila ovary and in addition affects the distribution of cortical actin at the apical side of the follicular epithelium. To understand how APC2 influences these properties of the actin cortex, we investigated the mechanisms controlling the cortical localization of APC2 in S2 cultured cells. We previously showed that the N-terminal half of APC2 containing the Armadillo repeats and the C-terminal 30 amino acids (C30) are together necessary and sufficient for APC2’s cortical localization. Our work presented here supports a model that cortical localization of APC2 is governed in part by self-association through the N-terminal APC Self-Association Domain (ASAD) and a highly conserved coiled-coil within the C30 domain.
Keywords: Armadillo repeats, coiled-coil, follicular epithelium, FRAP, nurse cell, self-association
1 |. INTRODUCTION
The cell cortex is a 50–100 nm thick mesh of filamentous actin, myosin, and actin binding and regulatory proteins that are cross-linked like a “dynamic shell” tethered to the plasma membrane (Clark, Dierkes, & Paluch, 2013; Salbreux, Charras, & Paluch, 2012). This actin-rich cortex plays critical roles in the cell cycle, in resistance to external mechanical forces, and in cell shape changes that occur during migration, cytokinesis, and morphogenesis (Heng & Koh, 2010; Woodham et al., 2017). Despite its apparent ubiquity and essential functions, many fundamental properties of the cortex including its molecular composition, three-dimensional structural organization, regulation of assembly, and mechanical properties are not well understood (Fritzsche, Erlenkamper, Moeendarbary, Charras, & Kruse, 2016; Salbreux et al., 2012). Changes in cellular mechanics that are tightly controlled by the structure of the actin cortex are being increasingly recognized as a factor in diseases including cancer, where there is a correlation between malignancy and cortical tension (Cross, Jin, Rao, & Gimzewski, 2007; Salbreux et al., 2012).
The cortical actin network is composed of both linear and branched actin filaments, and these two populations can differ significantly in abundance, length, and stability (Charras & Paluch, 2008; Cramer, 2010; Fritzsche et al., 2016; Fritzsche, Lewalle, Duke, Kruse, & Charras, 2013; Pollard & Borisy, 2003). In HeLa and M2 (melanoma) cells, formin-assembled linear actin filaments are roughly 10 times longer than Arp2/3-nucleated branched filaments, resulting in a 20× smaller turnover rate and thus significantly greater stability compared to branched filaments (Fritzsche et al., 2016). While these linear filaments make up only ~10% of the total number of filaments at the cortex, with their long length, they contribute to approximately 20–25% of the cortical actin network and are key determinants of cortical elasticity (Fritzsche et al., 2016). Their length and stability have suggested that formin-dependent linear filaments contribute to “cortical integrity” (Fritzsche et al., 2016), while the more abundant and dynamic branched actin filaments contribute to processes like protrusion and endocytosis (Fritzsche et al., 2016; Liu et al., 2008; Yamaguchi & Condeelis, 2007).
The formins are unusual among actin assembly factors in that they can both nucleate and elongate linear actin filaments in vitro (Chesarone, DuPage, & Goode, 2010). Drosophila Diaphanous (Dia), the first formin identified, was originally shown to be essential for the formation of cytokinetic furrows (Castrillon & Wasserman, 1994). We now know that Dia and other formins play key roles in the ass and lamellipodia, and many others (Bohnert, Willet, Kovar, & Gould, 2013; Homem & Peifer, 2008, 2009; Kage et al., 2017; Roy, Huang, Liu, & Kornberg, 2014; Young, Heimsath, & Higgs, 2015). Although Dia can independently nucleate and elongate actin filaments in vitro, mounting evidence suggests that in vivo Dia activity is regulated by binding partners at many steps during filament assembly. For example, binding to Rho-GTP relieves autoinhibition stimulating Dia activity (Goode & Eck, 2007), Bud6 collaborates with formins to promote the formation of a nucleation seed (Graziano et al., 2011), profilin binding is required for formin-mediated filament elongation (Paul & Pollard, 2008), and the microtubule plus-TIP protein CLIP-170 controls Dia’s elongation rate (Henty-Ridilla, Rankova, Eskin, Kenny, & Goode, 2016). New regulators and novel mechanisms continue to be discovered suggesting that there are many gaps in our understanding of formin activity and regulatory mechanisms.
The multifunctional Adenomatous polyposis coli (APC) proteins affect the actin and microtubule cytoskeletons through diverse and distinct mechanisms and negatively regulate the Wnt signaling pathway (Logan & Nusse, 2004; McCartney & Näthke, 2008). More than 80% of colorectal cancers are initiated by mutations in APC, and its role as a tumor suppressor is clearly linked to disruption of Wnt regulation (Cadigan & Peifer, 2009; Fodde, Smits, & Clevers, 2001). The connection between APC’s cytoskeletal functions and cancer initiation and progression are not well understood. Both vertebrate (vAPC) and its Drosophila homologue APC1 contain a C-terminal basic domain (Figure 1a) that directly binds both Dia and actin monomers, and together this “super nucleator” overcomes the inhibitory effects of Capping protein and Profilin to drive actin filament nucleation and Dia-dependent filament elongation in vitro (Breitsprecher et al., 2012; Jaiswal et al., 2013; Okada et al., 2010). Furthermore, a point mutation in vAPC that specifically abolishes its actin nucleation activity without altering its interactions with microtubules is associated with defective actin assembly at focal adhesions (Juanes et al., 2017). APC proteins localize to cortical actin in a cell type specific way (Harris & Nelson, 2010; Langford, Askham, Lee, Adams, & Morrison, 2006b; Langford, Lee, Askham, & Morrison, 2006a; McCartney et al., 1999; Rosin-Arbesfeld, Ihrke, & Bienz, 2001; Zhou, Kunttas-Tatli, Zimmerman, Zhouzheng, & McCartney, 2011), suggesting that the cortical association of APC proteins may be dependent on the composition and organization of the network.
FIGURE 1.
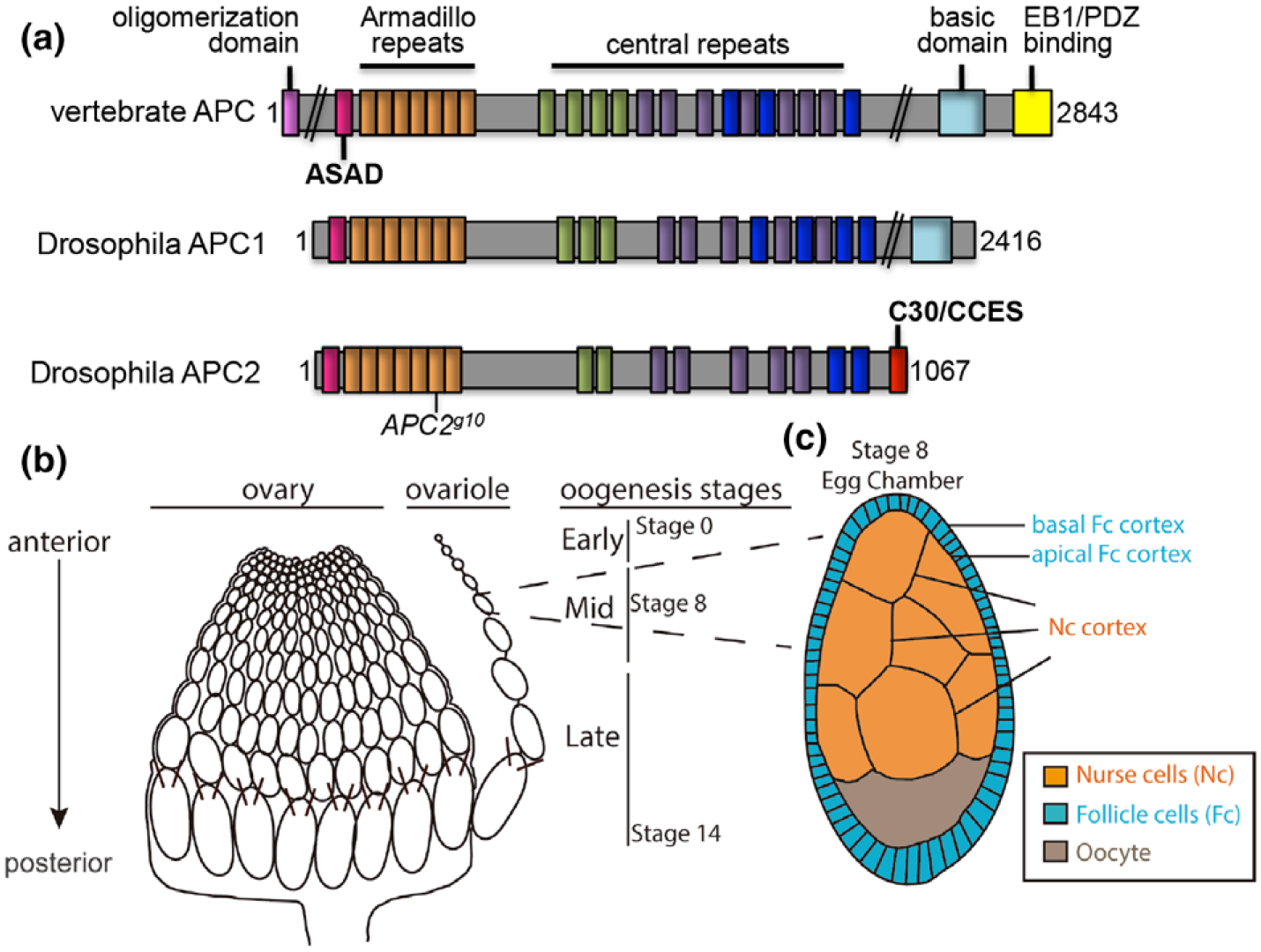
Schematics of the APC proteins and the Drosophila ovary. (a) APC family proteins contain many functional domains that are well conserved between vertebrates and Drosophila, and some that are unique: Oligomerization domain (magenta); ASAD, APC self-association domain (pink); Armadillo repeats (orange); the central repeats include the 15 amino acid repeats (green), the 20 amino acid repeats (purple), and the SAMP repeats (dark blue); C30, C-terminal 30 amino acids and CCES, C-terminal cortical enrichment sequence (red); basic domain (light blue); EB1/PDZ binding domain (yellow). APC2g10 is a nonsense mutation at the position indicated. (b) the Drosophila ovary is composed of multiple ovarioles, each of which is made up of individual egg chambers in a developmental gradient from the youngest at the anterior to fertilizable eggs at the posterior. (c) Schematics of a stage 8 egg chamber illustrate the three different cell types within each egg chamber and their physical arrangement: the germline derived NCs and oocyte, and the somatic follicular epithelium. The apical side of the FCs is adjacent to the NC cortex
Both vertebrates and Drosophila contain a second APC comprised of many of the same conserved domains (McCartney et al., 1999; van Es et al., 1999). Drosophila APC2 contains the core domains found in all known APC proteins, including the N-terminal APC self-association domain (ASAD), the Armadillo (Arm) repeats, the 15 and 20 amino acid repeats, and the SAMP repeats; however, it lacks the C-terminal basic domain known to promote actin assembly (Figure 1a). Instead, we have shown that APC2 contains a 30 amino acid C-terminal domain (C30) required for its cortical localization and function (Zhou et al., 2011). APC2 and Dia bind directly through the C-terminal halves of both proteins, and together they are required for the elongation of actin-based pseudocleavage furrows in the early fly embryo (Webb, Zhou, & McCartney, 2009). While we and others have shown that Drosophila APC2 is strongly enriched at the actin cortex in a wide variety of cell types throughout development (McCartney et al., 1999; Townsley & Bienz, 2000; Yu & Bienz, 1999), the cortical localization of APC2 is actin dependent in S2 cells and in intact tissues (Townsley & Bienz, 2000; Zhou et al., 2011), APC2’s cortical role is largely unknown. Some reports suggest that cortical APC2 contributes to Cadherin-based adhesion and tethering the mitotic spindle to the adherens junction in some cell types (Hamada & Bienz, 2002; McCartney et al., 2001; Townsley & Bienz, 2000; Yamashita, Jones, & Fuller, 2003). In other cell types, APC2 does not appear to significantly contribute to Cadherin-based adhesion (McCartney et al., 2006). The scope of APC’s cortical functions and the molecular mechanisms underlying them is not known.
Here we show that cortical APC2 is required to maintain wild type cortical actin dynamics in two distinct cell types in the Drosophila ovary, the nurse cells and the follicle cells, and to promote wild type cortical actin distribution in the follicle cells. To uncover the mechanisms governing APC2’s cortical localization, we dissected the domain requirements in Drosophila S2 cells and identified 15 amino acids within the required C30 domain that together with the N-terminal half of the protein are necessary and sufficient for APC2’s cortical localization. We have termed this sequence as the C-terminal Cortical Enrichment Sequence or CCES (Figure 1a). Computational prediction and targeted mutagenesis together strongly suggest that the CCES is a heptad repeat that forms a coiled-coil. Our previous work demonstrated that the N-terminal half of APC proteins contains another short heptad repeat (ASAD, APC Self-Association Domain, Figure 1a; Kunttas-Tatli, Roberts, & McCartney, 2014) that is necessary for APC2’s self-association. Here we show that co-expression of chimeric proteins containing the C30 fused to the inducible dimerization domains FKBP or FRB localize to the actin cortex in S2 cells similar to wild-type APC2. This suggests that a role of the N-terminal half of APC2 is to promote APC2 self-association through the ASAD, and that this self-association contributes to the cortical localization activity of the CCES.
2 |. RESULTS AND DISCUSSION
2.1 |. APC2 exhibits cell type specific requirements for cortical localization
Each ovary contains 15–25 ovarioles, strands of developing egg chambers with the youngest and smallest at the anterior end of the strand and mature, and fertilizable eggs at the posterior end (Figure 1b). Each egg chamber is composed of three distinct cell types: the oocyte and the nurse cells (NCs), both germline derived, and the somatic epithelial follicle cells (FCs) that completely surround the germ cells during early and mid-oogenesis (Figure 1c). Consistent with what we and others have observed in many other cell types and in the ovary, endogenous APC2 (McCartney et al., 1999; Roberts, Pronobis, Poulton, Kane, & Peifer, 2012) and APC2-GFP driven by the native promoter (McCartney et al., 2006; Figure 2a,b yellow arrows) are strongly enriched at the NC cortex. We previously showed that the N-terminal APC Self-Association Domain (ASAD) and the C-terminal 30 amino acids (C30) are both necessary for APC2’s cortical enrichment in S2 cultured cells and in the embryonic epithelium (Kunttas-Tatli et al., 2014; Zhou et al., 2011); in the absence of either the ASAD or the C30, cortical enrichment in those cells and tissues was significantly reduced. To examine localization requirements in the ovary, we expressed GFP-tagged APC2FL (full length APC2) or the cortical localization mutants APC2ΔC30 and APC2ΔASAD in an APC2g10 background. We previously showed that APC2g10 is a null allele resulting from a nonsense mutation at amino acid 383 (McCartney et al., 2006).
FIGURE 2.
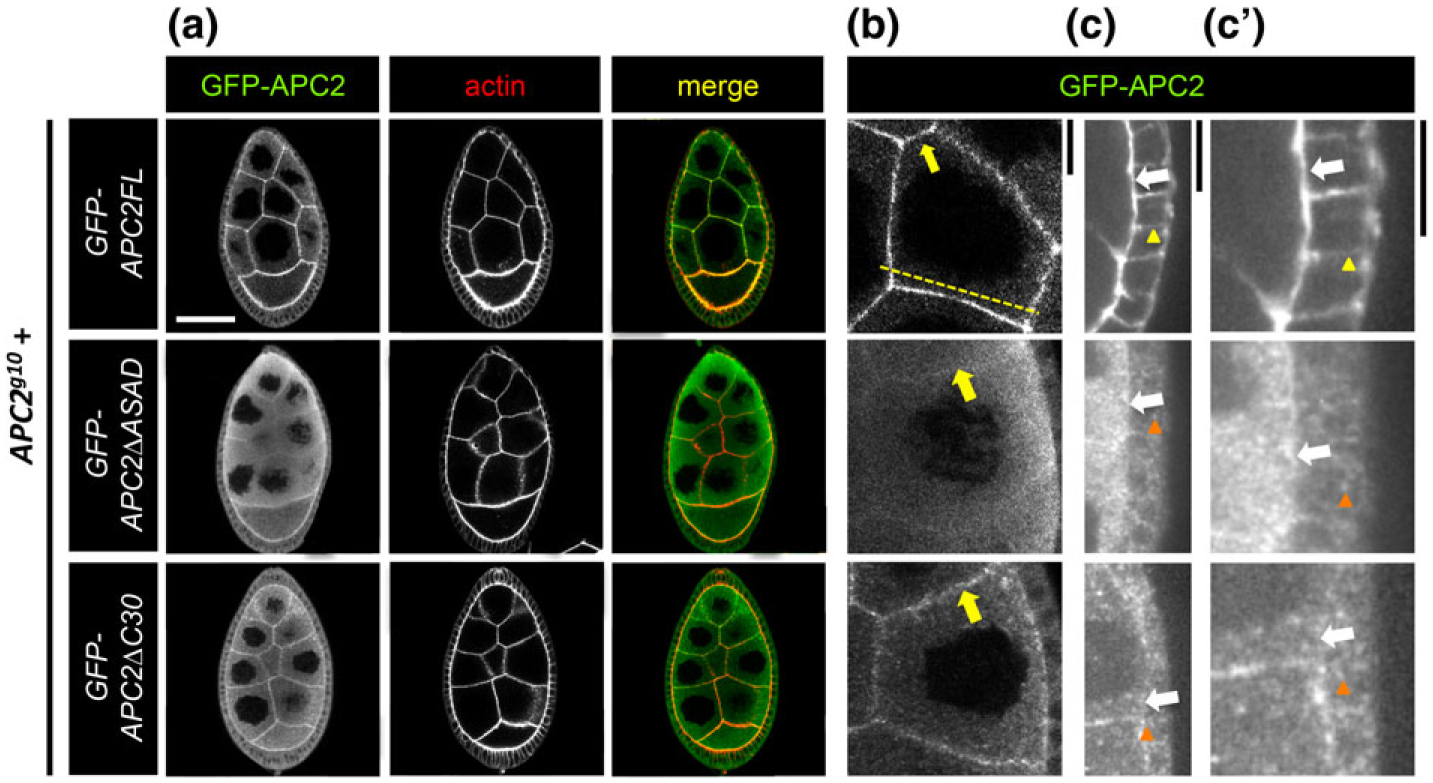
APC2 localization to the nurse cell and follicle cell cortices depends on ASAD and C30. (a-c’) Localization of GFP-tagged APC2FL, APC2ΔASAD, and APC2ΔC30 in the NCs and FCs of the Drosophila egg chamber detected in fixed tissue with anti-GFP. At higher magnification, we saw that APC2FL is cortically enriched in both NCs (b, yellow arrow), and FCs (c,c’, white arrows). APC2FL appears enriched at the apical cortex in FCs (c,c’ white arrow), compared to the basolateral cortex (c,c’ yellow arrowhead), and appears uniform at the NC cortex (b, yellow arrow). Both mutant proteins accumulate significantly in the cytoplasm of NCs and FCs as puncta (a–c’, orange arrowheads). Neither mutant retains significant cortical localization in the FCs (c,c’ white arrows). While the extent of cortical enrichment of the mutant proteins at the NC cortex was variable, APC2ΔASAD tended to have less cortical enrichment than APC2ΔC30 (b, yellow arrows). The dashed line in (b) illustrates the position of a representative line scan shown in Supporting Information Figure S1. Scale bars: (a) 40 μm; (b,c’) 10 μm
In NCs, the clear accumulation of cytoplasmic protein in the mutants compared to APC2FL (Figure 2a–c) is consistent with a defect in cortical enrichment. This is similar to what we observed in S2 cells and in the embryo (Kunttas-Tatli et al., 2014; Zhou et al., 2011). Both APC2ΔASAD and APC2ΔC30 retained some cortical enrichment (Figure 2a,b arrows), while APC2ΔC30 appeared to be more cortically enriched than APC2ΔASAD (Figure 2b, arrows). Representative line scans of fluorescence intensity across the NCs illustrate the differences in cortical enrichment between APC2FL and the two mutants (Supporting Information Figure S1); the difference in intensity between the cortex and the cytoplasm is most pronounced in APC2FL and APC2ΔC30 (Supporting Information Figure S1a,b), while APC2ΔASAD exhibits less distinction between cortex and cytoplasm in the NCs (Supporting Information Figure S1c).
The FCs are a polarized epithelium whose apical ends are adjacent to the NCs (Figure 1c, 2c,c’, white arrows), where wild type APC2 appears to accumulate more strongly than at the basolateral cortex (Figure 2c,c’, yellow arrowhead in GFP-APC2FL). In contrast to the NCs, loss of either ASAD or C30 in the FCs appeared to displace APC2 very strongly from the apical and basolateral cortex (Figure 2c, c’, white arrows at apical cortex). Instead, the mutant APC2 is primarily localized in cytoplasmic puncta in the FCs compared to APC2FL (Figure 2c’, orange arrowheads). The results of our localization studies suggest that there is context dependence for APC2’s domain requirements for cortical association. This suggests that the specific composition, organization, and/or dynamics of the cortical actin network may contribute to APC2’s cortical association.
2.2 |. APC2’s cortical localization promotes a dynamic cortical actin network
To begin to understand how the properties of the cortical actin network might affect the localization of APC2, and how the localization of APC2 influences the cortical actin network, we assessed turnover dynamics at the NC and FC cortices at stage 8 of oogenesis using Fluorescence Recovery After Photobleaching (FRAP) of the actin-binding domain of Moesin fused to GFP (MoeGFP; (Edwards, Demsky, Montague, Weymouth, & Kiehart, 1997). MoeGFP turnover has been shown to be a good proxy for actin turnover (Cao, Albertson, Riggs, Field, & Sullivan, 2008). Because we cannot detect GFP signal in live tissue expressing GFP-APC2 driven by the native promoter (wild type or mutants; data not shown), in all cases, we were measuring the turnover of MoeGFP rather than that of GFP-APC2. Because the cortex is likely composed of both linear and branched actin, and MoeGFP does not have any reported preference, the FRAP analysis is assessing turnover of a mixture of filament types. At the apical FC cortex, complete loss of APC2 or disruption of its cortical localization with the APC2ΔC30 mutant significantly reduced the mobile fraction (Figure 3a,c). Expression of APC2FL in the APC2 null rescued this defect, increasing the mobile fraction to wild type levels (Figure 3a,c). The rate of fluorescence recovery as assessed by the t1/2 (time to half of the full recovery) was unchanged in the APC2 mutants (Figure 3c). Thus, there are fewer mobile MoeGFP molecules in the apical FC cortex in APC2 mutants, indicative of greater stability in the network, but the MoeGFP molecules that are mobile turnover at the same rate as in wild type.
FIGURE 3.
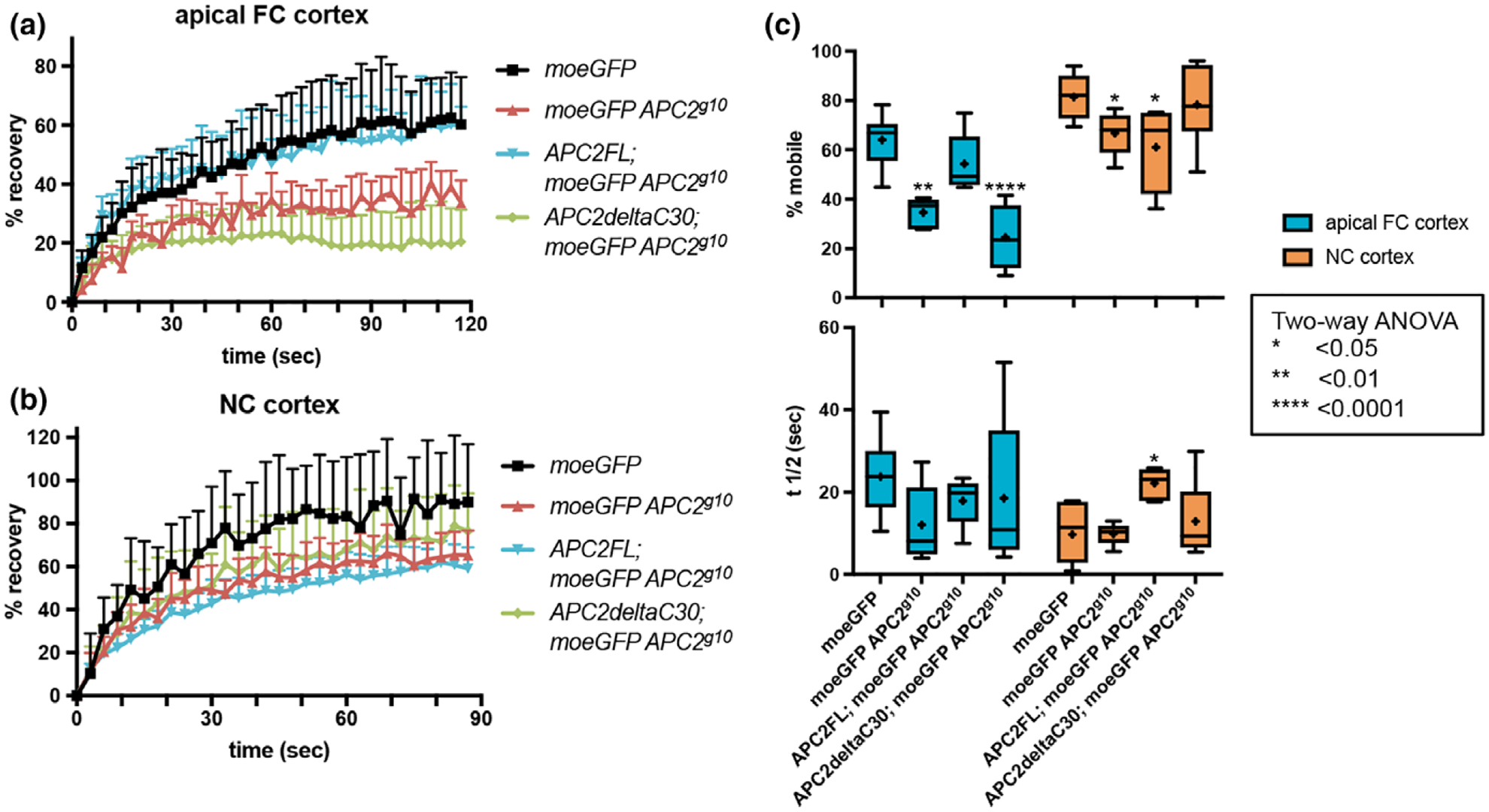
Disruption of APC2’s cortical localization alters cortical dynamics. (a, b) Normalized raw fluorescence recovery data for the indicated genotypes at the apical FC cortex (a) and the NC cortex (b). Each line represents the average of the raw data for the genotype indicated with the error bars illustrating the standard deviation. (c) the percent mobile fraction (top) and t1/2 (bottom) at the apical FC cortex (blue) and NC cortex (orange) in the genotypes indicated. Each box extends from the 25th to 75th percentiles, and the whiskers indicate the minimum and maximum values. The line within each box is the median, and the + indicates the mean. At the apical FC cortex, complete loss of APC2 (APC2g10) or deletion of the C30 domain (APC2ΔC30) significantly reduced the mobile fraction. Expression of APC2-FL rescued this defect. The t1/2 did not differ significantly between the genotypes. At the NC cortex we saw a similar decrease in mobile fraction in the APC2 null, and this was not rescued by APC2ΔC30. Expression of APC2FL resulted in a significantly reduced mobile fraction like the null at the NC cortex, and a significantly increased t1/2 unlike any other genotype
In wild type tissue, the dynamics of the NC cortex are distinct from that of the apical FC cortex with a higher mobile fraction and a lower t1/2, both indicative of increased dynamics (Figure 3c, p < .05 Two-way ANOVA; for clarity this statistic is not indicated in the figure). At the NC cortex, we found that complete loss of APC2 also resulted in a significant decrease in the mobile fraction, but here the APC2ΔC30 mutant NCs had a mobile fraction indistinguishable from wild type (Figure 3b,c). The simplest interpretation of our FRAP results is that APC2 is needed for wild-type cortical dynamics in both NCs and FCs. In NCs, APC2ΔC30 rescues the reduced mobile fraction because the mutant protein retains some cortical localization (Figure 2b,c’), but in FCs where the localization defect is more severe (Figure 2b,c’), APC2ΔC30 cannot regulate cortical dynamics (Figure 3a–c). This simple interpretation is complicated by what we observed when we expressed APC2FL in the APC2 null. While APC2FL completely rescued the APC2 null reduction in mobile fraction in the FCs (Figure 3a,c), in the NCs, we observed a significantly reduced mobile fraction like the APC2 null and a significantly increased t1/2 unlike any other genotype (Figure 3c). One interpretation of the unexpected effects of APC2FL is that this protein has impaired function in some contexts. We previously showed that while APC2FL appeared to completely rescue APC2 activity in the Wnt pathway (McCartney et al., 2006), it had reduced actin-associated functions in the embryo (McCartney et al., 2006; Webb et al., 2009). Though we cannot explain the anomalous behavior of APC2FL in NCs, the FRAP data are most consistent with a role for APC2 in promoting a dynamic cortical actin network in both FCs and NCs.
In actin furrow extension in the embryo, APC2 appears to promote Dia’s activity (Webb et al., 2009). If the same mechanism explains APC2’s role at the cortex, loss of APC2 would lead to less Dia-dependent linear actin in the cortical network. If the linear network is more stable than the branched network as in HeLa cells (Fritzsche et al., 2016), loss of APC2 and consequent loss of linear actin would produce a more dynamic cortex compared to wild type. Contrary to that prediction, the cortex is less dynamic in the absence of APC2 (Figure 3c). One possible explanation for this conundrum is the complex regulatory interaction between Dia and the Ena/VASP protein Enabled (Bilancia et al., 2014; Nowotarski, McKeon, Moser, & Peifer, 2014); loss of Dia-mediated filament assembly in APC2 mutants could increase the activity of the elongation factor Enabled, in turn altering the dynamic properties of the actin network. Future work will be needed to dissect these regulatory interactions at the cortex. Alternatively, APC2 may function at the cortex in part to restrict Dia’s actin assembly activity, resulting in a lower linear to branched ratio in the network and thus a more dynamic cortex. To test this hypothesis, more work is needed to uncover the consequences of the molecular interactions between APC2 and Dia (Webb et al., 2009).
2.3 |. Cortical actin distribution and cortical structure in follicle cells and nurse cells
At stage 8, the NC cortex has an overall gauze-like appearance without significant structural definition (Figure 4a). Because of the reduced mobile fraction at the cortex in the APC2 null (Figure 3), we predicted that we might also observe an organizational change visible by confocal microscopy. We did not see any consistent differences in the actin intensity or gauze-like nature of the cortex when we manipulated APC2 (Figure 4b–e). In projections of wild-type egg chambers, the NC cortex tends to be regular and planar (Figure 4a, arrowhead), and the same was generally true in the APC2 mutants (Figure 4b–e). In all of the genotypes, we observed occasional crinkles or buckles in the cortex that we collectively refer to as “waves” (for example Figure 4d, arrowhead). We speculate that these changes could result from the contractions of the muscular sheath surrounding each ovariole (Andersen & Horne-Badovinac, 2016; Middleton et al., 2006). We found that the frequency of these cortical waves was significantly different between wild type and all of the APC2 genotypes (Figure 4f). Although there was no clear pattern implicating APC2 in either promoting or suppressing these waves, we cannot rule it out completely. Thus, while APC2 is regulating cortical dynamics in the NCs (Figure 3), that change in dynamics in the APC2 null was not correlated with a detectable change in overall cortical architecture. Lastly, we examined ring canals, actin-based channels embedded within the NC cortex (Figure 4a, arrow). Ring canals are formed through incomplete cytokinesis early in oogenesis and are required for the transport of molecules and organelles from the NCs into the developing oocyte. While the phalloidin staining in the ring canals was on average slightly brighter in the APC2 null compared to wild type (Figure 4a,b, arrows and G), the APC2 null was not significantly different from either APC2FL; APC2g10, or APC2ΔC30; APC2g10, and neither of these genotypes was significantly different from wild type (Figure 4g). Although we cannot rule out a role for APC2 in ring canal structure or function, we did not see a compelling difference in actin fluorescence intensity in this cortically associated structure.
FIGURE 4.
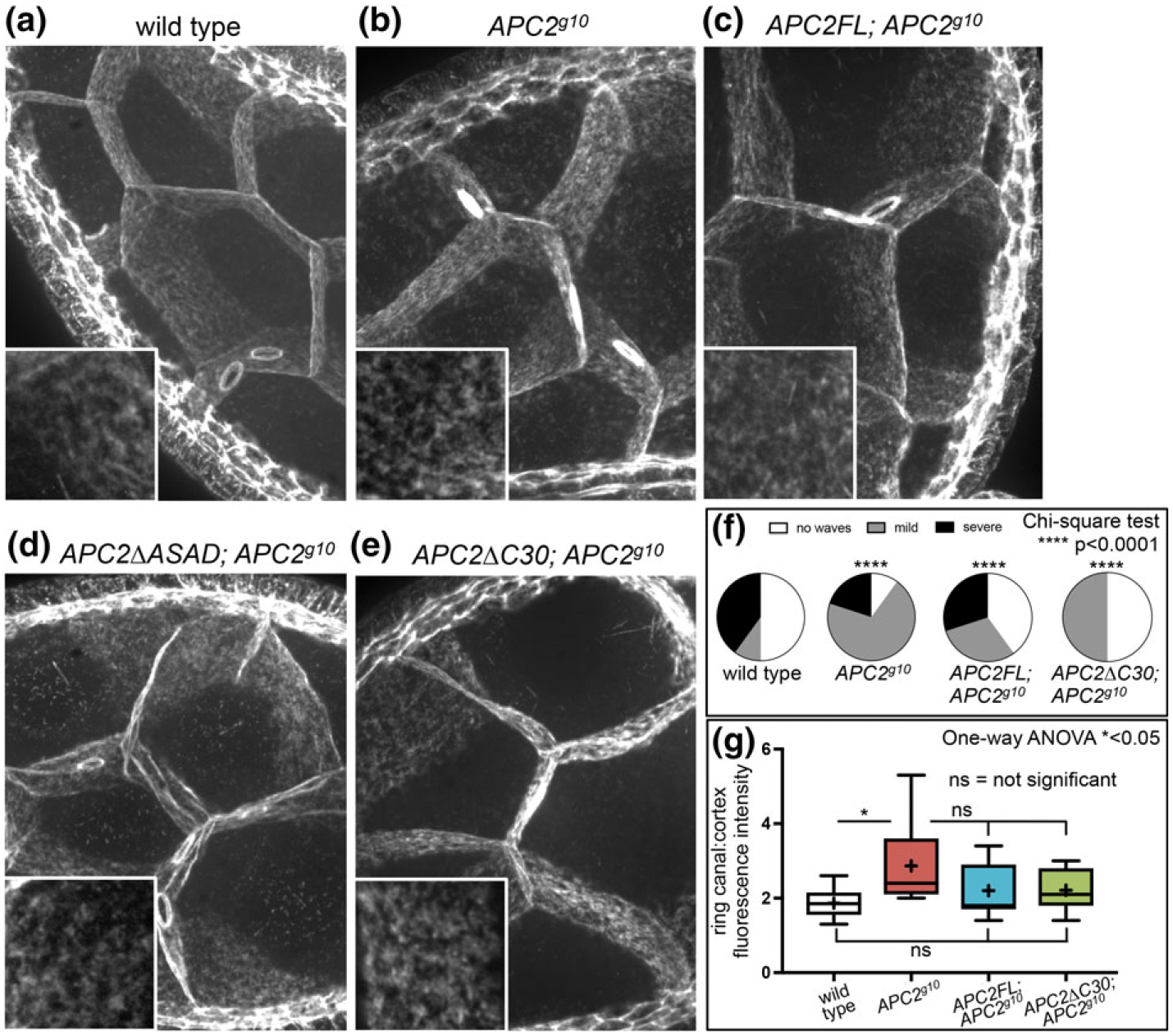
Disruption of APC2 in the nurse cells does not significantly alter cortical actin accumulation or overall cortical structure. (a–e) projections of 30 0.2 μm z-slices of phalloidin stained stage 8 egg chambers reveal the basic organization of the NC cortex in the genotypes indicated. Insets show higher magnification views of the cortical actin network in NCs. While the cortical network typically appears uniform and planar (a, arrowhead), we did observe irregularities like the waves in (d, arrowhead) in wild type and mutant NCs. Scale bar, 5 μm; inset, 1 μm (f) the percent of egg chambers with cortical waves. The frequency and severity of cortical waves was highly variable with every genotype significantly different from the wild type. N = 10 for each genotype. (g) the relative fluorescence intensity of the ring canals in the APC2 null was significantly greater than in the wild type (a, b arrows). The relative intensity in APC2FL; APC2g10 and APC2ΔC30; APC2g10 was not significantly different from either the wild type or from APC2g10. Each box extends from the 25th to 75th percentiles, and the whiskers indicate the minimum and maximum values. The line within each box is the median, and the + indicates the mean. For wild type, the median and the mean are the same. N = 10 for each genotype
In contrast to the homogenous pattern of actin accumulation at the NC cortex, in the wild-type follicular epithelium, cortical actin is enriched in apical puncta that largely correspond to the tricellular junctions (Figure 5a, blue circles, and b, white arrow) with significantly less accumulation along the cortex between those junctions (Figure 5a, yellow line, b, yellow arrow). To quantify this pattern, we measured pixel intensities along the cortex from junction to junction and normalized each pixel to the maximum pixel value along that line. This illustrates the peaks of intensity at the tricellular junctions and the dip in intensity between them (Figure 5e). In APC2 null FCs, actin remains enriched at the tricellular junctions (Figure 5c, white arrow), but there is also significant accumulation between the junctions (Figure 5c, yellow arrow and Figure 5e, red line). Expression of APC2FL or either of the cortical localization mutants restored the wild type pattern of apical actin enrichment (Figure 5e). Because disruption of APC2’s cortical localization with APC2ΔC30 resulted in a more stable apical FC cortex like the null mutant (Figure 3), and a cortical actin distribution like the wild type (Figure 5e), we conclude that the change in cortical dynamics we see in the mutants does not cause the defect in cortical actin distribution. This suggests that APC2 may have more than one function at the apical cortex: one to promote turnover and cortical dynamics and a second to promote the local accumulation of cortical actin, or to block accumulation outside of the tricellular junctions.
FIGURE 5.
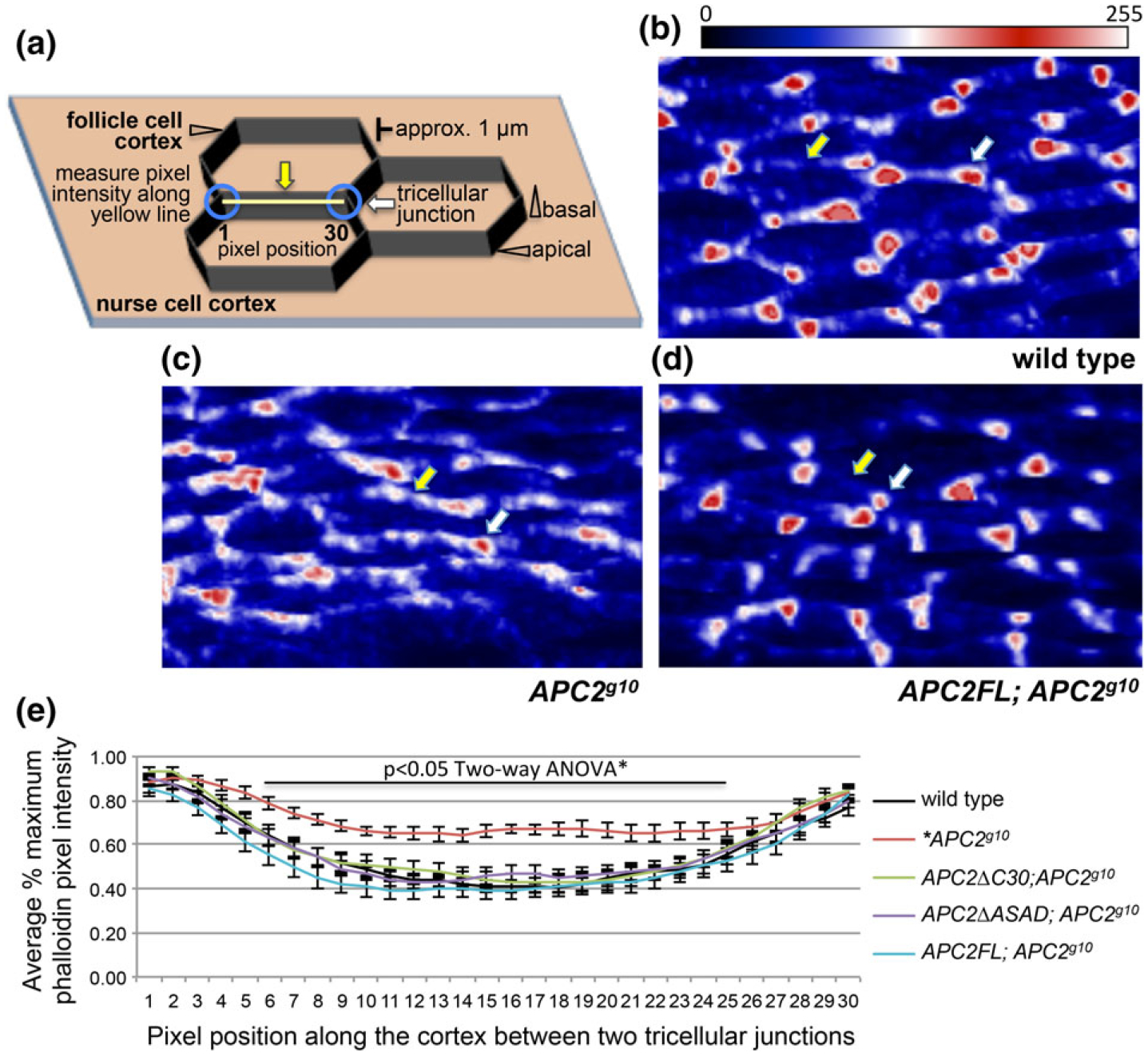
Complete loss of APC2 disrupts the heterogeneous accumulation of actin at the apical cortex in the follicle cells. (a) Schematic of the cellular arrangement, landmarks, and the quantification strategy employed for (b–e). The white arrow indicates a tricellular junction, and the yellow arrow indicates the apical cortex between two tricellular junctions (blue circles). For the analysis in e, pixel intensity was measured for 30 pixels along the cortex (yellow line) between two tricellular junctions. (b–d) rotated projections of six 0.2 μm optical sections of the apical ends of phalloidin stained FCs of the indicated genotypes. As in (a), the white arrow indicates a tricellular junction, and the yellow arrow indicates the apical cortex between two tricellular junctions. The images have been pseudo-colored using the look-up table shown (Fiji) to highlight the relative intensity differences between the tricellular junctions and the intervening cortex within an image (see methods for details). In wild type FCs (b), and in APC2 null FCs expressing APC2FL (d), actin at the apical cortex is enriched at the tricellular junctions (b,d white arrow) compared to the intervening cortex (b,d yellow arrow). In the APC2 null mutant (c), the actin distribution appears more homogenous with more actin along the intervening cortex (c, yellow arrow). (e) Quantification of pixel intensities as described in (a) for the genotypes as shown. This illustrates the distinction between apical cortical actin distribution in wild type FCs (black) and APC2g10 FCs (red). The APC2 null defect in apical cortical actin distribution was rescued by APC2FL (blue), APC2ΔC30 (green), and APC2ΔASAD (purple). N = 16–24 lines/genotype scale bar = 5 μm
2.4 |. A conserved 15 amino acids in the C30 domain is predicted to form a coiled-coil and is necessary for APC2’s cortical enrichment
To understand the mechanisms by which APC2 affects cortical actin structure and function we asked how APC2 localizes to the actin cortex. We previously demonstrated that the N-terminal domain of APC2 (N-term), containing the ASAD (APC Self-Association Domain) and the Armadillo (Arm) repeats, and the C-terminal 30 amino acids (C30) are each necessary, and together sufficient, for the cortical localization of APC2 (Kunttas-Tatli et al., 2014; McCartney et al., 2006; Zhou et al., 2011). We show some of those findings again here (Figure 6c,d) as a starting point for further dissecting these requirements. To identify the mechanism by which C30 promotes cortical localization, we compared the sequence of these 30 amino acids across Drosophila species and identified a highly conserved 17 amino acid sequence (Figure 6a). Deletion of the central 15 conserved amino acids (Figure 6a, yellow highlighted sequence; Figure 6b, APC2-N-C30-divergent) abolished cortical localization in S2 cells (Figure 6c,d), while replacing the C30 with only the central 15 amino acids of the conserved sequence (APC2-N-C30-conserved) was sufficient for wild-type cortical localization (Figure 6b–d). We confirmed the importance of this sequence using an independent deletional approach (Supporting Information Figure S2). Thus, in combination with APC2-N, this conserved sequence (the C-terminal Cortical Enrichment Sequence, or CCES) is necessary and sufficient for cortical localization.
FIGURE 6.
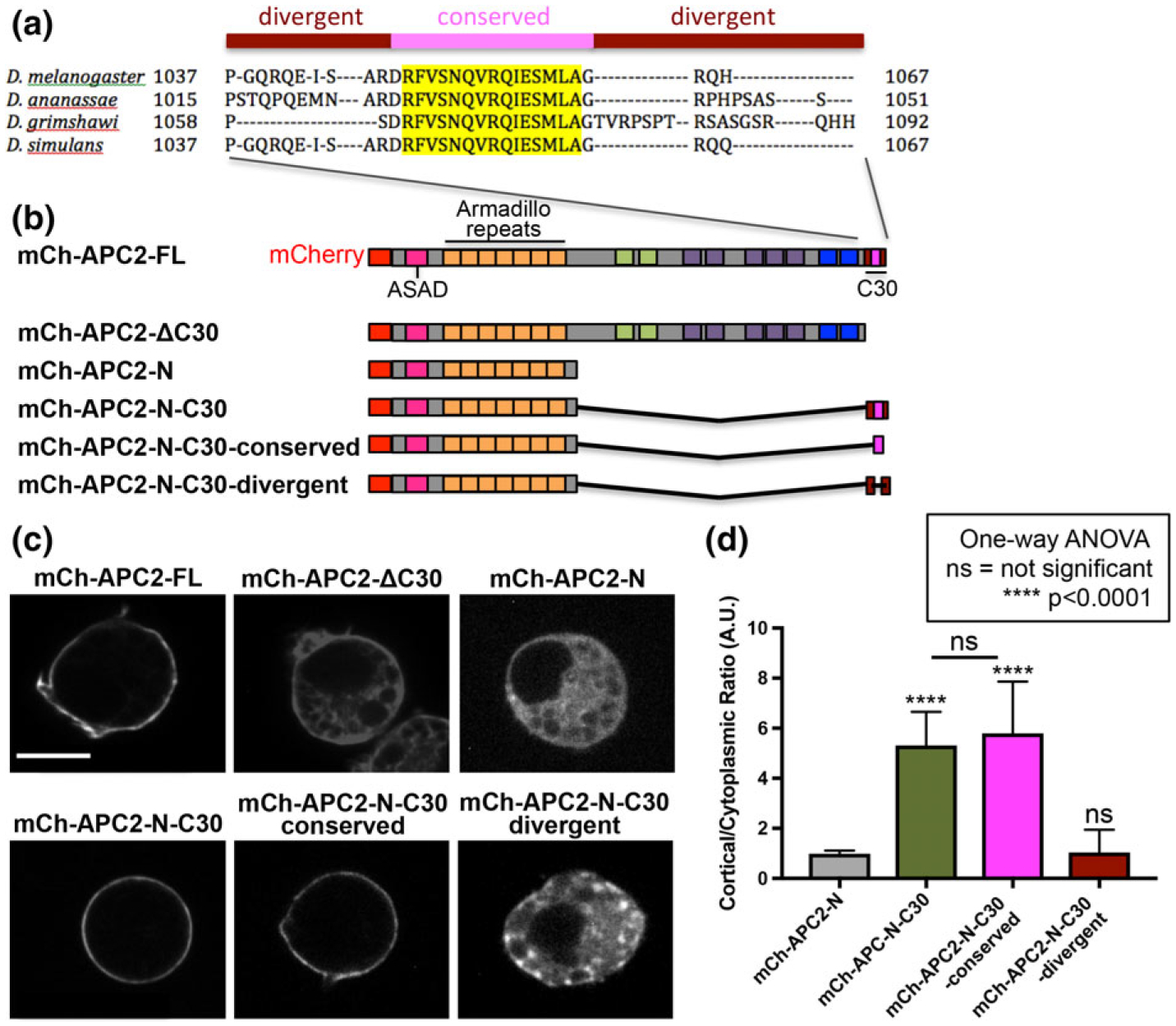
A conserved 15 amino acid sequence within C30 is necessary for cortical localization. (a) Multiple sequence alignment of the C-terminal 30 amino acids (C30) from four Drosophila species revealed a highly conserved sequence (yellow). (b) Schematics of mCherry-tagged (mCh) APC2 proteins expressed in S2 cells. (c) S2 cells transfected with the indicated mCh-APC2 constructs imaged live to visualize the mCh tag. APC2FL is localized to the cell cortex while mutants that deleted either the entire C30 (APC2ΔC30) or the conserved region (APC2-NC30-divergent) localized throughout the cytoplasm. Scale bar = 5 μm. (d) We used the cortical to cytoplasmic pixel intensity ratio of APC2 to quantify cortical enrichment (as in Zhou et al., 2011). Ratios close to 1 indicate no difference between APC2 accumulation at the cortex and in the cytoplasm. Ratios higher than 1 indicate cortical enrichment compared to the cytoplasm. The conserved region alone of APC2 C30 retains the complete cortical enrichment activity of C30, while the divergent region alone has no cortical enrichment activity. Error bars are standard deviation. N = 15 for each condition. All conditions were compared to mCh-APC2-N using one-way ANOVA with results as shown
To ask if the CCES forms a secondary structure relevant to APC2’s cortical localization mechanism, we used COILs (Alva, Nam, Söding, & Lupas, 2016; Lupas, Van Dyke, & Stock, 1991). This predicted that the CCES is a HEPTAD repeat that may form a coiled-coil (Figure 7a). Coiled-coils are generated by the association of two or more alpha-helices to create a stable zipper-like conformation that can play critical roles in protein–protein interactions and protein self-association (Lupas & Bassler, 2017). In fact, the ASAD that facilitates APC self-association also appears to form a coiled-coil (Kunttas-Tatli et al., 2014). To test the hypothesis that the CCES may form a coiled-coil necessary for cortical enrichment, we disrupted the putative coiled-coil by changing four key hydrophobic amino acids within the HEPTAD repeat (the a and d positions) to proline disrupting the formation of α-helices and coiled-coils (Figure 7a, blue stars). This resulted in the complete loss of APC2’s cortical enrichment in S2 cells (Figure 7b,c) and was indistinguishable from a complete deletion of the conserved residues (Figure 6c,d). Consistent with this, a single point mutation changing hydrophobic valine to hydrophilic threonine at the d position (Figure 7a, yellow star) likewise abolished cortical enrichment (Figure 7b,c). Taken together, the computational prediction and mutational analysis are consistent with the model that the CCES forms a coiled-coil that is necessary for the cortical enrichment of APC2.
FIGURE 7.
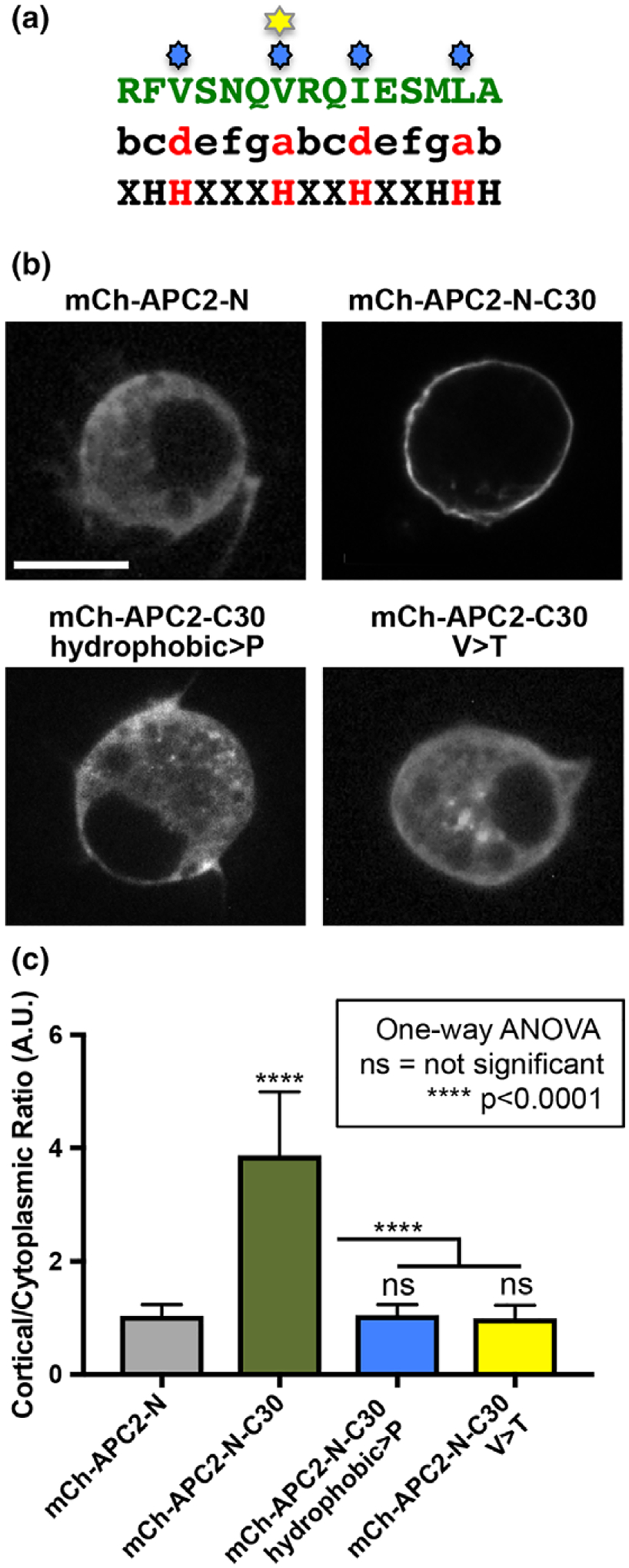
Mutational analysis supports the model that formation of a coiled-coil in C30 is necessary for the cortical localization mechanism of APC2 at the cortex. (a) the CCES (green) aligned to the amino acid pattern of a HEPTAD repeat. The hydrophobic amino acids at positions a and d are in red. Blue stars indicate the residues changed to proline, and the yellow star indicates the hydrophobic valine that was changed to a hydrophilic threonine. (b) S2 cells expressing mCh-tagged APC2 proteins as indicated imaged live. Altering all (hydrophobic>P), or just one (V > T), of the key hydrophobic amino acids abolished APC2’s cortical enrichment. (c) Quantification of APC2’s cortical enrichment demonstrates that mutations disrupting the putative coiled-coil eliminate the cortical enrichment of APC2. Error bars are standard deviation. N = 15 for each condition. All conditions were compared to mCh-APC2-N using one-way ANOVA with results as shown
2.5 |. Artificial dimerization using FRB and FBKP drives the C30 domain of APC2 to the cortex
Because the CCES alone is not sufficient for cortical enrichment and the required N-terminal half of the protein contains the ASAD (Kunttas-Tatli et al., 2014), we speculated that a role of the N-terminal half of APC2 is to drive efficient self-association via the ASAD to promote the assembly of the short CCES coiled-coil. To test if a role of the N-terminal half of APC2 is to drive the self-association necessary for cortical localization, we asked whether dimerization of C30 containing the CCES through the rapamycin inducible dimerization of FRB and FKBP (Figure 8a,b) (Muthuswamy, Gilman, & Brugge, 1999; Spencer, Wandless, Schreiber, & Crabtree, 1993) could promote cortical enrichment. Expression of either mCh-FKBP-C30 or EGFP-FRB-C30 alone, with or without rapamycin (Figure 8c,d,f), resulted in no significant cortical enrichment, similar to expression of the C-terminal half of APC2 or the C30 alone (Zhou et al., 2011). Coexpression of the chimeras without rapamycin also resulted in no cortical enrichment (Figure 8e,f). In contrast, coexpression of EGFP-FRB-C30 and mCh-FKBP-C30 with rapamycin resulted in significantly enhanced cortical enrichment of the chimeric proteins (Figure 8e,f). The reciprocal experiments using mCh-FRB-C30 and EGFP-FKBP-C30 yielded similar results (not shown). Our data suggest a model that self-association of APC2 through the N-terminal ASAD is necessary for the cortical localization activity of the CCES. Consistent with this model, deletion of ASAD alone significantly disrupts the cortical enrichment of APC2 in S2 cells and embryonic epithelia (Kunttas-Tatli et al., 2014).
FIGURE 8.
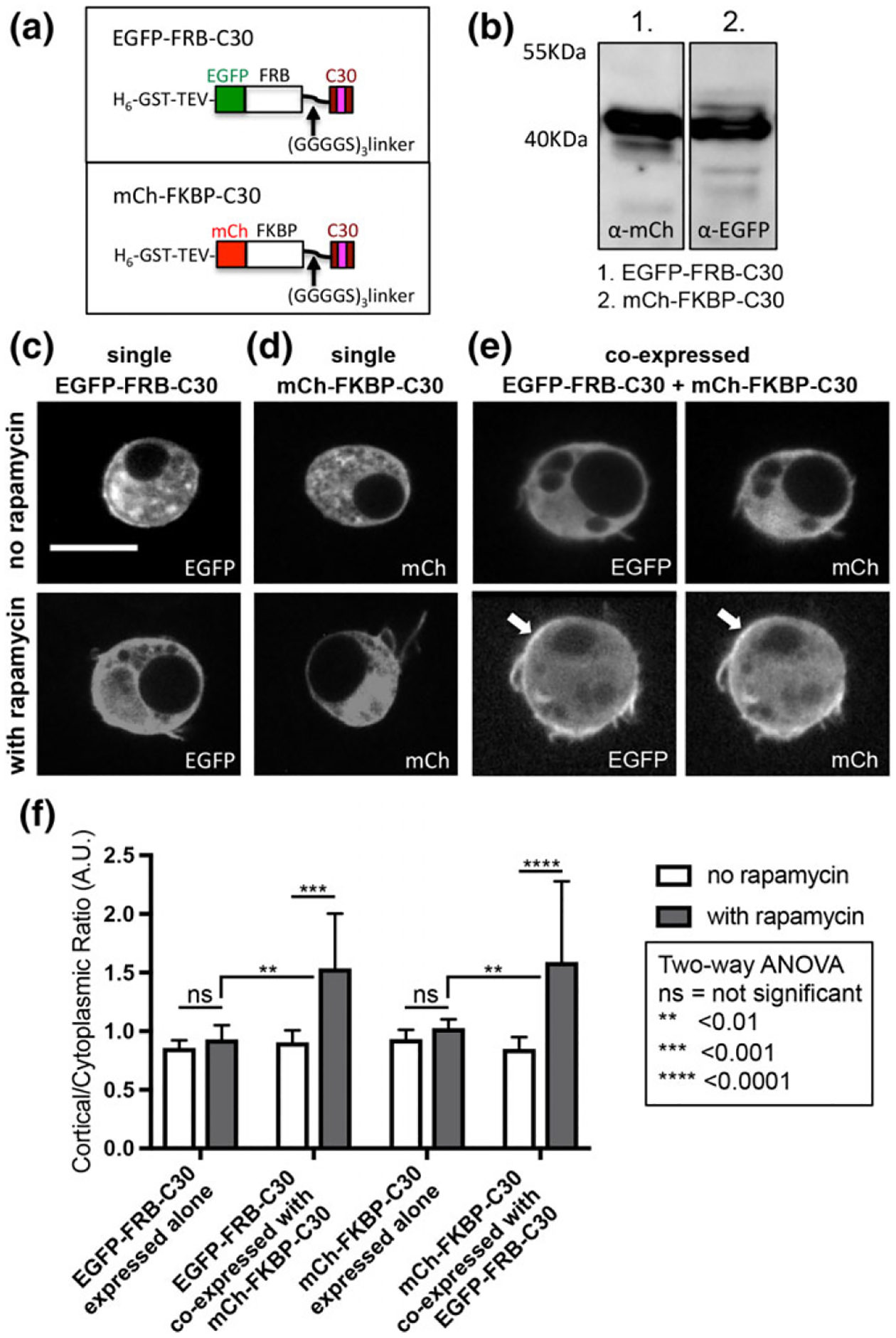
Dimerization of C30 via FKBP and FRB promotes cortical enrichment. (a) Schematics of FRB and FKBP chimeras with C30 tagged with EGFP and mCh, respectively. (b) Immunoblots of EGFPFRB-C30 and mCh-FKBP-C30 from S2 cells demonstrate that these two chimeras are expressed at comparable levels. (c-e) S2 cells expressing one or both chimeras were imaged live to visualize EGFP and/or mCh. When the chimeras are individually expressed (c, d) either with or without rapamycin, they did not show any apparent cortical enrichment. When EGFP-FRB-C30 and mCh-FKBP-C30 are coexpressed without rapamycin neither showed any cortical enrichment (e, top panels), but when EGFP-FRB-C30 and mCh-FKBPC30 were coexpressed with rapamycin both showed cortical enrichment (e, bottom panels, white arrows). Scale bar = 5 μm. (f) Plot of cortical to cytoplasmic ratios to quantify the localization of each FRB and FKBP chimera, expressed alone or co-expressed, with or without rapamycin. N = 8 for each condition. Error bars depict standard deviation. Statistical differences calculated using two-way ANOVA as indicated
While coexpression of the chimeras in the presence of rapamycin did drive cortical enrichment (Figure 8e,f), it did not reconstitute wild type levels of enrichment: APC2-N-C30 has a cortical to cytoplasmic ratio of approximately 5 (Figure 6d), while the co-expressed chimeras have a ratio of approximately 1.5 (Figure 8f). To test the possibility that mCh and EGFP sterically hinder FRB-FKBP binding, we repeated the experiments substituting mCh with a 6xHis-tag (Supporting Information Figure S3a–d). When we co-expressed EGFP-FKBP-C30 and 6xHis-FRB-C30 in the presence of rapamycin, we still observed a significant pool of cytoplasmic EGFP-FKBP-C30 (Supporting Information Figure S3d). Thus, exchanging the bulky mCh tag for the smaller 6xHis tag did not result in a dramatic increase in the cortical enrichment. The reduced cortical enrichment of the chimeras compared to a more wild-type APC2 could reflect the need for the formation of higher order oligomers of C30 rather than dimers. Additionally or alternatively, it may reflect the need for something in the N-terminal domain in addition to the ASAD. Our earlier work demonstrated that APC2 proteins with missense mutations in the Arm repeats exhibit reduced cortical enrichment (McCartney et al., 1999, 2006), consistent with the idea that the Arm repeats play a role in cortical localization. Arm repeats are a known protein–protein interaction domain (Tewari, Bailes, Bunting, & Coates, 2010), and the Arm repeats of vertebrate APC bind to several cytoskeletal regulators, including Striatin, ASEF, KAP3, and IQGAP (Breitman, Zilberberg, Caspi, & Rosin-Arbesfeld, 2008; Jimbo et al., 2002; Kawasaki et al., 2000; Kawasaki, Sato, & Akiyama, 2003; Watanabe et al., 2004). Interestingly, we showed that deletion of the ASAD and consequent loss of APC2 self-association enhanced KAP3 (Kinesin-associated protein 3) binding by APC2 (Kunttas-Tatli et al., 2014), suggesting that the self-association of APC2 can modulate the binding of Arm repeat partners. The results presented here, together with the results of our earlier work, support a model where the ASAD and the CCES cooperate to drive APC2 localization to the actin-rich cortex and suggest that an Arm repeat binding protein (s) could tether, enhance, and/or stabilize the interaction of APC2 with cortical actin. It is interesting to note that loss of ASAD appears to disrupt APC2’s localization to the NC cortex more significantly than loss of C30 and the CCES (Figure 2 and Supporting Information Figure S1). If the ASAD is promoting the activity of the CCES and potentially modulating the binding interactions of the Arm repeats, it is not surprising that loss of the ASAD might have a stronger effect on localization.
3 |. CONCLUSION
Here we showed that APC2 plays roles in cortical dynamics in NCs and in epithelial FCs and in actin distribution in FCs. APC2’s localization to the cortex requires actin itself (Zhou et al., 2011), and the Arm repeats (McCartney et al., 1999, 2006), and here we show that it depends on the activity of the ASAD and the CCES. Whether self-associated APC2 binds to cortical actin directly and/or through cortical actin binding partners and whether this interaction is influenced by the mechanical properties of the cortex is not known. Once associated with the cortex, we predict that APC2 is regulating cortical properties at least in part through its association with Dia. While our initial work suggested that APC2 promotes Dia activity (Webb et al., 2009), the change in cortical actin dynamics in the APC2 mutants (Figure 3) suggests that the regulatory relationship between APC2 and Dia may be more complex. Formins are known to assemble the linear filament component of the cortex in several cell types (Bovellan et al., 2014; Fritzsche et al., 2016). However, we know little about how formins might be regulated in this context to achieve the optimal ratio of linear to branched filaments that appear to play a significant role in modulating the mechanical properties of the cortex (Fritzsche et al., 2016). Future study of the APC-Dia collaboration at the cortex should provide important insight into the mechanisms governing the properties of the cortical actin network.
4 |. MATERIALS AND METHODS
4.1 |. DNA constructs
Deletion fragments and point mutations were synthesized by PCR site-directed mutagenesis using mCh-APC2-N-C30 in the metallothionein inducible pRmHa3 plasmid (Zhou et al., 2011). The following constructs were generated: mCh-APC2-N-C30-divergent (aa 1–490, aa 1,038–48, aa 1,064–67), mCh-APC2-N-C30-conserved (aa 1–490, aa 1,049–63), mCh-APC2-N-C30-hydrophobic>P (aa 1–490, aa 1,038–1,067 [V1051P, V1055P, I1058P, L1062P]), and mCh-APC2-N-C30-V > P (aa 1–490, aa 1,038–1,067 [V1055P]). To generate the FRB and FKBP dimerization constructs, we used pMoMo, a plasmid derived from the backbone of pKM263 and modified to include two cloning modules (N-terminal and C-terminal) separated by a 15 aa linker (GGGGS3) (a gift from Dr. Jon Jarvik). EGFP and mCherry (mCh) were PCR amplified with Sfi (NEB) overhangs, shuttled through pGEM-T for sequencing, and cloned into the N-terminal module of pMoMo. The plasmids containing FRB and FKBP (a gift from Dr. Jon Jarvik) were digested with Sfi and ligated in frame with EGFP or mCh to an AlwN1 (NEB) digested pMoMo-EGFP or pMoMo-mCh. The C30 was then PCR amplified with PflMI restriction site overhangs and ligated in frame into the C-terminal module of the EGFP and mCh versions of pMoMo-FRB or pMoM-FKBP. We then used pMoMo-EGFP-FRB-C30/pMoMo-mCh-FRB-C30 and pMoMo-EGFP-FKBP-C30/pMoMo-mCh-FKBP-C30 as PCR templates with 5’-Kpn1 and 3’-BamH1 ends to shuttle the entire fusion protein cassettes into pRmHa3.
4.2 |. S2 cell transfection, imaging, and analysis
S2 cells were transfected with our constructs using Effectene (Qiagen). All constructs were driven by the metallothionein promoter, and expression was induced 24 hours posttransfection with CuSO4 at a final concentration of 40 μM. For the FRB and FKBP experiments, expression was induced at 10 μM CuSO4, and the cells were incubated with rapamycin (100 μM) or without rapamycin for 6 hours before imaging. Images were acquired with a spinning-disc confocal microscope with a Yokagawa scan head (Solamere Technology Group) and a QICAM-IR camera (Qimaging) on a Zeiss Axiovert 200 M using QED InVivo software. All S2 cells were imaged live 14–16 hours postinduction. We quantified the APC2 cortical to cytoplasmic ratio as in Zhou et al., 2011. Briefly, we fixed the S2 cells with 4% formaldehyde in 1X phosphate buffered saline (PBS) for 10 min followed by blocking in PNT (PBS, 1% NGS, and 0.1% Triton-X100) and incubation with Alexa546-phalloidin at 1:1000 (Invitrogen). Using ImageJ, we drew a line bisecting each cell and obtained pixel intensities of all the channels along that line. We then used actin as marker for the cortex to calculate the cortical to cytoplasmic ratio of different APC2 proteins. Representative images of actin in S2 cells expressing different variants of APC2 do not reveal any apparent differences in the localization of cortical actin (Supporting Information Figure S3).
4.3 |. Fly stocks
The null allele, w1118; APC2g10/TM6Tb (McCartney et al., 2006), was used for our complete loss of function experiments. For localization and rescue experiments, P[endoP-EGFP-APC2-FL] (McCartney et al., 2006), P[endoP-EGFP-APC2-ΔC30] (Zhou et al., 2011), and P[endoP-EGFP-APC2-ΔASAD] (Kunttas-Tatli et al., 2014) were crossed into w1118; APC2g10/TM6 Tb. For the FRAP analysis, we used moeGFP (Edwards et al., 1997), and generated moeGFP APC2g10, P[endoP-EGFP-APCFL]; moeGFP APC2g10, and P[endo-PEGFP-APC2-ΔC30]; moeGFP APC2g10 using standard recombination and crossing methods. We could not recover a stock containing both P[endoP-EGFP-APC2-ΔASAD] and moeGFP APC2g10 preventing FRAP analysis of this mutant.
4.4 |. Drosophila egg chamber fixation and immunostaining
To optimize egg chamber yield per fly, female flies 3–4 days old were collected and yeasted overnight. Ovaries were dissected in Grace’s Insect Medium with 10% FBS and 0.1% Penicillin/Streptomycin and combed through with fine forceps to separate the ovarioles and remove the muscular sheath. For cortical actin assessment in fixed tissue, the ovaries were fixed with 10% formaldehyde with 1X PBS supplemented with 0.01% Tween-20 and Alexa488-phalloidin at 1:200 (Invitrogen) for 30 minutes. Following four washes with PBS and 0.01% Tween-20, the tissue was incubated with PBS and Alexa488-phalloidin (1:200) for 1 hour while rocking at room temperature. DAPI (1:10,000) was added for the last 5 minutes of the incubation, and the tissue was washed two times before mounting in AquaPoly/Mount (Polysciences). For immunolocalization, the ovaries were fixed and stained as in McCartney et al. (2006). Briefly, ovaries were fixed in fresh 4% paraformaldehyde in PBS for 18 minutes at room temperature, followed by four washes with PBS and a 1 hour block in PNT (PBS, 1% Normal Goat Serum [NGS], and 0.03% Triton-X 100). The tissue was incubated with rocking overnight at 4°C with anti-GFP (Abcam) that had been pre-absorbed at 1:5000 in PNT against wild-type ovaries overnight at 4°C to reduce background. Ovaries were incubated in Alexa-488 secondary antibody at 1:1000 (Invitrogen), Alexa546-phalloidin (1:500), and DAPI (1:10,000) before washing and mounting in Aqua-Poly/Mount.
4.5 |. Image acquisition and analysis of actin in stage 8 egg chambers
Images were acquired on a spinning-disc confocal microscope with a Yokagawa scan head (Solamere Technology Group) and a QICAM-IR camera (Qimaging) on a Zeiss Axiovert 200 M using QED InVivo software. Z-stacks were acquired at 63× with a 0.2-μm step size. Laser power, exposure time, and gain were kept constant for each data set, and the levels of all images were adjusted identically. To control for potential differences in cortical organization and dynamics due to developmental stage, the length and width of each stage 8 egg chamber was measured using Fiji (ImageJ). Only those approximately 520–620 pixels in length by 280–380 pixels in width were used for analysis. These measurements appeared to correspond to approximately mid-stage 8. To evaluate the NC cortex (Figure 4), 30 0.2-μm en-face z-sections were projected using Fiji (ImageJ). This stack represented the majority of the lateral cortex from the outer surface in contact with the FCs to the inner surface in contact with other NCs. We refer to any distortions of the NC cortex as “waves” (e.g., Figure 4d, arrow) and qualitatively assessed whether NCs had no waves, or mild or severe waves, by visual inspection of the projections described above (Figure 4f). If any sections of the cortex deviated from a smooth appearance, that egg chamber was categorized as “mild”, and egg chambers with greater than 2 sections of the cortex with deviations was characterized as “severe”. To determine the relative fluorescence intensity of the ring canals (Figure 4g), for each projection, we calculated the average fluorescence intensity of the ring canals from four 8 × 8 pixel regions of interest (ROIs), and the average fluorescence intensity of the cortex from four 8 × 8 ROIs. Using these two averages, we calculated the ring canal to cortex fluorescence intensity ratio for each projection. To calculate average percentage maximum pixel intensity of apical actin in the FCs (Figure 5e), six 0.2 μm en-face z-sections were projected, and a line was drawn from the center of one tricellular junction along the cortex to the center of the next tricellular junction. For each cortical line measuring 30 pixels (16–24 lines for each genotype), the maximum pixel intensity value was set to 100%, and the intensity of each pixel along that line was adjusted relative to that maximum.
4.6 |. Fluorescence recovery after photobleaching (FRAP)
Female flies 3–4 days old were collected and yeasted overnight. Ovaries were dissected in Grace’s Insect Medium with 10% FBS and 0.1% Penicillin/Streptomycin and combed through with fine forceps to separate the ovarioles and to remove the muscular sheath. Single ovarioles were removed from the ovary and when possible young egg chambers were completely isolated from the ovarioles and incubated in fresh Grace’s Insect medium in a 35-mm glass-bottom dish (MatTek Corporation). FRAP experiments were carried out using a 63 × 1.4 NA oil immersion objective on a Zeiss LSM880 confocal microscope with ZEN imaging software. Following 10 prebleach scans, MoeGFP in the cortical region of interest was photobleached using the 488-nm laser at 100% power for 50 iterations. After photobleaching, the fluorescence recovery was monitored every 1 s for 50 frames. FRAP was completed within 45 minutes of the initial dissection. We found that after this point, the variability increased significantly. FRAP data was analyzed using the FRAP module in the ZEN Black software. In brief, the fluorescence intensity of each region was first adjusted by subtracting the background intensity and then normalized to the intensity at the initial time point. The mobile fraction was then calculated as Imax – I0/(1 – I0), where Imax is the maximal fluorescence recovery and I0 is the initial fluorescence intensity immediately after photobleaching. FRAP recovery curve analysis and values for t1/2 were calculated using the ZEN FRAP calculator module. To display recovery curves of the raw data (Figure 3a,b), the raw fluorescence data for each photobleached spot were normalized relative to the average fluorescence intensity for the spot at the start of imaging, and relative to the average fluorescence intensity for the spot at the time of bleaching. The normalized fluorescence intensities for each genotype were averaged and plotted, where the point of bleaching was set to zero on the y-axis (% recovery) and on the x-axis (time).
4.7 |. Statistical analysis
All graphing and statistical analyses were conducted with GraphPad Prism 7.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank T. Wong, and M. Bernsten for their initial work on the project, J. Minden, J. Woolford, M. Blundon and other members of the McCartney lab for useful discussions, A. Leslie for manuscript review, G. Rule for guidance on the mutational strategy, and H. Yang for assistance with FRAP. This work was supported in part by National Institutes of Health grant R01 GM073891 to B. McCartney. P. Rudich was supported by an HHMI Undergraduate Education Award, grant number 52006917. None of the authors have any conflicts of interest.
REFERENCES
- Alva V, Nam S-Z, Söding J, & Lupas AN (2016). The MPI bioinformatics toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Research, 44, W410–W415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen D, & Horne-Badovinac S (2016). Influence of ovarian muscle contraction and oocyte growth on egg chamber elongation in drosophila. Development, 143, 1375–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilancia CG, Winkelman JD, Tsygankov D, Nowotarski SH, Sees JA, Comber K, … Peifer M (2014). Enabled negatively regulates diaphanous-driven actin dynamics in vitro and in vivo. Developmental Cell, 28, 394–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnert KA, Willet AH, Kovar DR, & Gould KL (2013). Formin-based control of the actin cytoskeleton during cytokinesis. Biochemical Society Transactions, 41, 1750–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovellan M, Romeo Y, Biro M, Boden A, Chugh P, Yonis A, … Charras G (2014). Cellular control of cortical actin nucleation. Current Biology, 24, 1628–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitman M, Zilberberg A, Caspi M, & Rosin-Arbesfeld R (2008). The armadillo repeat domain of the APC tumor suppressor protein interacts with Striatin family members. Biochimica et Biophysica Acta, 1783, 1792–1802. [DOI] [PubMed] [Google Scholar]
- Breitsprecher D, Jaiswal R, Bombardier JP, Gould CJ, Gelles J, and Goode BL (2012). Rocket launcher mechanism of collaborative actin assembly defined by single-molecule imaging. Science (80-.). 336, 1164–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadigan KM, & Peifer M (2009). Wnt signaling from development to disease: Insights from model systems. Cold Spring Harbor Perspectives in Biology, 1, a002881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Albertson R, Riggs B, Field CM, & Sullivan W (2008). Nuf, a Rab11 effector, maintains cytokinetic furrow integrity by promoting local actin polymerization. The Journal of Cell Biology, 182, 301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrillon DH, & Wasserman SA (1994). Diaphanous is required for cytokinesis in drosophila and shares domains of similarity with the products of the limb deformity gene. Development, 120, 3367–3377. [DOI] [PubMed] [Google Scholar]
- Charras G, & Paluch E (2008). Blebs lead the way: How to migrate without lamellipodia. Nature Reviews. Molecular Cell Biology, 9, 730–736. [DOI] [PubMed] [Google Scholar]
- Chesarone MA, DuPage AG, & Goode BL (2010). Unleashing formins to remodel the actin and microtubule cytoskeletons. Nature Reviews. Molecular Cell Biology, 11, 62–74. [DOI] [PubMed] [Google Scholar]
- Clark AG, Dierkes K, & Paluch EK (2013). Monitoring actin cortex thickness in live cells. Biophysical Journal, 105, 570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer LP (2010). Forming the cell rear first: Breaking cell symmetry to trigger directed cell migration. Nature Cell Biology, 12, 628–632. [DOI] [PubMed] [Google Scholar]
- Cross SE, Jin YS, Rao J, & Gimzewski JK (2007). Nanomechanical analysis of cells from cancer patients. Nature Nanotechnology, 2, 780–783. [DOI] [PubMed] [Google Scholar]
- Edwards KA, Demsky M, Montague RA, Weymouth N, & Kiehart DP (1997). GFP-moesin illuminates actin cytoskeleton dynamics in living tissue and demonstrates cell shape changes during morphogenesis in drosophila. Developmental Biology, 191, 103–117. [DOI] [PubMed] [Google Scholar]
- van Es JH, Kirkpatrick C, van de Wetering M, Molenaar M, Miles A, Kuipers J, … Clevers H (1999). Identification of APC2, a homologue of the adenomatous polyposis coli tumour suppressor. Current Biology, 9, 105–108. [DOI] [PubMed] [Google Scholar]
- Fodde R, Smits R, & Clevers H (2001). APC, signal transduction and genetic instability in colorectal cancer. Nature Reviews. Cancer, 1, 55–67. [DOI] [PubMed] [Google Scholar]
- Fritzsche M, Lewalle A, Duke T, Kruse K, & Charras G (2013). Analysis of turnover dynamics of the submembranous actin cortex. Molecular Biology of the Cell, 24, 757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsche M, Erlenkamper C, Moeendarbary E, Charras GT, & Kruse K (2016). Actin kinetics shapes cortical network structure and mechanics. Science Advances, 112, 453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode BL, & Eck MJ (2007). Mechanism and function of formins in the control of actin assembly. Annual Review of Biochemistry, 76, 593–627. [DOI] [PubMed] [Google Scholar]
- Graziano BR, DuPage AG, Michelot A, Breitsprecher D, Moseley JB, Sagot I, … Goode BL (2011). Mechanism and cellular function of Bud6 as an actin nucleation-promoting factor. Molecular Biology of the Cell, 22, 4016–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada F, & Bienz M (2002). A drosophila APC tumour suppressor homologue functions in cellular adhesion. Nature Cell Biology, 4, 208–213. [DOI] [PubMed] [Google Scholar]
- Harris ES, & Nelson WJ (2010). Adenomatous polyposis coli regulates endothelial cell migration independent of roles in beta-catenin signaling and cell-cell adhesion. Molecular Biology of the Cell, 21, 2611–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng Y-W, & Koh C-G (2010). Actin cytoskeleton dynamics and the cell division cycle. The International Journal of Biochemistry & Cell Biology, 42, 1622–1633. [DOI] [PubMed] [Google Scholar]
- Henty-Ridilla JL, Rankova A, Eskin JA, Kenny K, and Goode BL (2016). Accelerated actin filament polymerization from microtubule plus ends. Science (80-.). 352, 1004–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homem CC, & Peifer M (2008). Diaphanous regulates myosin and adherens junctions to control cell contractility and protrusive behavior during morphogenesis. Development, 135, 1005–1018. [DOI] [PubMed] [Google Scholar]
- Homem CC, & Peifer M (2009). Exploring the roles of diaphanous and enabled activity in shaping the balance between filopodia and lamellipodia. Molecular Biology of the Cell, 20, 5138–5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal R, Stepanik V, Rankova A, Molinar O, Goode BL, & McCartney BM (2013). Drosophila homologues of adenomatous polyposis coli (APC) and the formin diaphanous collaborate by a conserved mechanism to stimulate actin filament assembly. The Journal of Biological Chemistry, 288, 13897–13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimbo T, Kawasaki Y, Koyama R, Sato R, Takada S, Haraguchi K, & Akiyama T (2002). Identification of a link between the tumour suppressor APC and the kinesin superfamily. Nature Cell Biology, 4, 323–327. [DOI] [PubMed] [Google Scholar]
- Juanes M, Bouguenina H, Eskin JA, Jaiswal R, Badache A, & Goode BL (2017). Adenomatous polyposis coli nucleates actin assembly to drive cell migration and microtubule-induced focal adhesion turnover. The Journal of Cell Biology, 216, 3327–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kage F, Winterhoff M, Dimchev V, Mueller J, Thalheim T, Freise A, … Rottner K (2017). FMNL formins boost lamellipodial force generation. Nature Communications, 8, 14832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Senda T, Ishidate T, Koyama R, Morishita T, Iwayama Y, … Akiyama T (2000). Asef, a link between the tumor suppressor APC and G-protein signaling. Science, 289, 1194–1197. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Sato R, & Akiyama T (2003). Mutated APC and Asef are involved in the migration of colorectal tumour cells. Nature Cell Biology, 5, 211–215. [DOI] [PubMed] [Google Scholar]
- Kunttas-Tatli E, Roberts DM, & McCartney BM (2014). Self-association of the APC tumor suppressor is required for the assembly, stability, and activity of the Wnt signaling destruction complex. Molecular Biology of the Cell, 25, 3424–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langford KJ, Lee T, Askham JM, & Morrison EE (2006a). Adenomatous polyposis coli localization is both cell type and cell context dependent. Cell Motility and the Cytoskeleton, 63, 483–492. [DOI] [PubMed] [Google Scholar]
- Langford KJ, Askham JM, Lee T, Adams M, & Morrison EE (2006b). Examination of actin and microtubule dependent APC localisations in living mammalian cells. BMC Cell Biology, 7, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu AP, Richmond DL, Maibaum L, Pronk S, Geissler PL, & Fletcher DA (2008). Membrane-induced bundling of actin|[nbsp]|filaments. Nature Physics, 4, 789–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan CY, & Nusse R (2004). The WNT signaling pathway in development and disease. Annual Review of Cell and Developmental Biology, 20, 781–810. [DOI] [PubMed] [Google Scholar]
- Lupas AN, & Bassler J (2017). Coiled coils – A model system for the 21st century. Trends in Biochemical Sciences, 42, 130–140. [DOI] [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, & Stock J (1991). Predicting coiled coils from protein sequences. Science, 252, 1162–1164. [DOI] [PubMed] [Google Scholar]
- McCartney BM, & Näthke IS (2008). Cell regulation by the Apc protein. Apc as master regulator of epithelia. Current Opinion in Cell Biology, 20, 186–193. [DOI] [PubMed] [Google Scholar]
- McCartney BM, Dierick HA, Kirkpatrick C, Moline MM, Baas A, Peifer M, & Bejsovec A (1999). Drosophila APC2 is a cytoskeletally-associated protein that regulates wingless signaling in the embryonic epidermis. The Journal of Cell Biology, 146, 1303–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney BM, McEwen DG, Grevengoed E, Maddox P, Bejsovec a., & Peifer M (2001). Drosophila APC2 and armadillo participate in tethering mitotic spindles to cortical actin. Nature Cell Biology, 3, 933–938. [DOI] [PubMed] [Google Scholar]
- McCartney BM, Price MH, Webb RL, Hayden M. a., Holot LM, Zhou M, … Peifer M (2006). Testing hypotheses for the functions of APC family proteins using null and truncation alleles in drosophila. Development, 133, 2407–2418. [DOI] [PubMed] [Google Scholar]
- Middleton CA, Nongthomba U, Parry K, Sweeney ST, Sparrow JC, & Elliott CJ (2006). Neuromuscular organization and aminergic modulation of contractions in the drosophila ovary. BMC Biology, 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthuswamy SK, Gilman M, & Brugge JS (1999). Controlled dimerization of ErbB receptors provides evidence for differential signaling by homo- and heterodimers. Molecular and Cellular Biology, 19, 6845–6857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotarski SH, McKeon N, Moser RJ, & Peifer M (2014). The actin regulators enabled and diaphanous direct distinct protrusive behaviors in different tissues during drosophila development. Molecular Biology of the Cell, 25, 3147–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada K, Bartolini F, Deaconescu AM, Moseley JB, Dogic Z, Grigorieff N, … Goode BL (2010). Adenomatous polyposis coli protein nucleates actin assembly and synergizes with the formin mDia1. The Journal of Cell Biology, 189, 1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul AS, & Pollard TD (2008). The role of the FH1 domain and profilin in formin-mediated actin-filament elongation and nucleation. Current Biology, 18, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, & Borisy GG (2003). Cellular motility driven by assembly and disassembly of actin filaments. Cell, 112, 453–465. [DOI] [PubMed] [Google Scholar]
- Roberts DM, Pronobis MI, Poulton JS, Kane EG, & Peifer M (2012). Regulation of Wnt signaling by the tumor suppressor adenomatous polyposis coli does not require the ability to enter the nucleus or a particular cytoplasmic localization. Molecular Biology of the Cell, 23, 2041–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosin-Arbesfeld R, Ihrke G, & Bienz M (2001). Actin-dependent membrane association of the APC tumour suppressor in polarized mammalian epithelial cells. The EMBO Journal, 20, 5929–5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Huang H, Liu S, and Kornberg TB (2014). Cytoneme-mediated contact-dependent transport of the drosophila decapentaplegic signaling protein. Science (80-.). 343, 1244624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salbreux G, Charras G, & Paluch E (2012). Actin cortex mechanics and cellular morphogenesis. Trends in Cell Biology, 22, 536–545. [DOI] [PubMed] [Google Scholar]
- Spencer DM, Wandless TJ, Schreiber SL, & Crabtree GR (1993). Controlling signal transduction with synthetic ligands. Science, 262, 1019–1024. [DOI] [PubMed] [Google Scholar]
- Tewari R, Bailes E, Bunting KA, & Coates JC (2010). Armadillo-repeat protein functions: Questions for little creatures. Trends in Cell Biology, 20, 470–481. [DOI] [PubMed] [Google Scholar]
- Townsley FM, & Bienz M (2000). Actin-dependent membrane association of a drosophila epithelial APC protein and its effect on junctional armadillo. Current Biology, 10, 1339–1348. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Wang S, Noritake J, Sato K, Fukata M, Takefuji M, … Kaibuchi K (2004). Interaction with IQGAP1 links APC to Rac1, Cdc42, and actin filaments during cell polarization and migration. Developmental Cell, 7, 871–883. [DOI] [PubMed] [Google Scholar]
- Webb RL, Zhou M-N, & McCartney BM (2009). A novel role for an APC2-diaphanous complex in regulating actin organization in drosophila. Development, 136, 1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodham EF, Paul NR, Tyrrell B, Spence HJ, Swaminathan K, Scribner MR, … Machesky LM (2017). Coordination by Cdc42 of actin, contractility, and adhesion for Melanoblast movement in mouse skin. Current Biology, 27, 624–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, & Condeelis J (2007). Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochimica et Biophysica Acta, 1773, 642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita YM, Jones DL, and Fuller MT (2003). Orientation of asymmetric stem cell division by the APC tumor suppressor and centrosome. Science (80-.). 301, 1547–1550. [DOI] [PubMed] [Google Scholar]
- Young L, Heimsath E, & Higgs H (2015). Cell type dependent mechanisms for formin-mediated assembly of filopodia. Molecular Biology of the Cell, 26, 4646–4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, & Bienz M (1999). Ubiquitous expression of a drosophila adenomatous polyposis coli homolog and its localization in cortical actin caps. Mechanisms of Development, 84, 69–73. [DOI] [PubMed] [Google Scholar]
- Zhou M-N, Kunttas-Tatli E, Zimmerman S, Zhouzheng F, & McCartney BM (2011). Cortical localization of APC2 plays a role in actin organization but not in Wnt signaling in drosophila. Journal of Cell Science, 124, 1589–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.