

Abstract
Translocator protein (TSPO), previously known as the peripheral benzodiazepine receptor (PBR), is an outer mitochondrial membrane protein. TSPO has been shown to cooperate with the steroidogenic acute regulatory protein (StAR) and function in the transport of cholesterol into mitochondria. TSPO has also been considered as a structural component of the mitochondrial permeability transition pore. However, recent advances have changed these views of TSPO functions and have prompted a re-evaluation of established concepts. This review summarizes the history of TSPO, key elements of the debate, and functional experiments that have changed our understanding. Moving forward, we examine how this fundamental change impacts understanding of TSPO and affects the future of TSPO as a therapeutic and diagnostic target.
Keywords: mitochondria, benzodiazepine, cholesterol, steroid hormone, permeability transition, therapy
TSPO: a protein with a long history
Function of the translocator protein (TSPO), previously known as the peripheral benzodiazepine receptor (PBR, see glossary), has been a topic of active research for the past 25 years. It was initially described in 1977 as a peripheral receptor for benzodiazepines, distinct from the central nervous system benzodiazepine receptor (γ-aminobutyric acid type A receptor/GABAA receptor), and its pharmacological characterization has been extensive ever since [1, 2]. Specificity of chemicals like PK11195 (an isoquinoline carboxamide derivative) and Ro5–4864 (4’-chlorodiazepam) that could bind to TSPO with high affinity, but not to the GABAA receptor, were exploited to study potential cellular actions mediated by TSPO such as in steroidogenesis and apoptosis [3]. Protein sequence for TSPO is fairly conserved from bacteria to humans (Figure 1), suggesting a possible evolutionarily conserved fundamental role for TSPO in cellular and organismal physiology [4].
Figure 1. Translocator protein amino acid sequence homology.
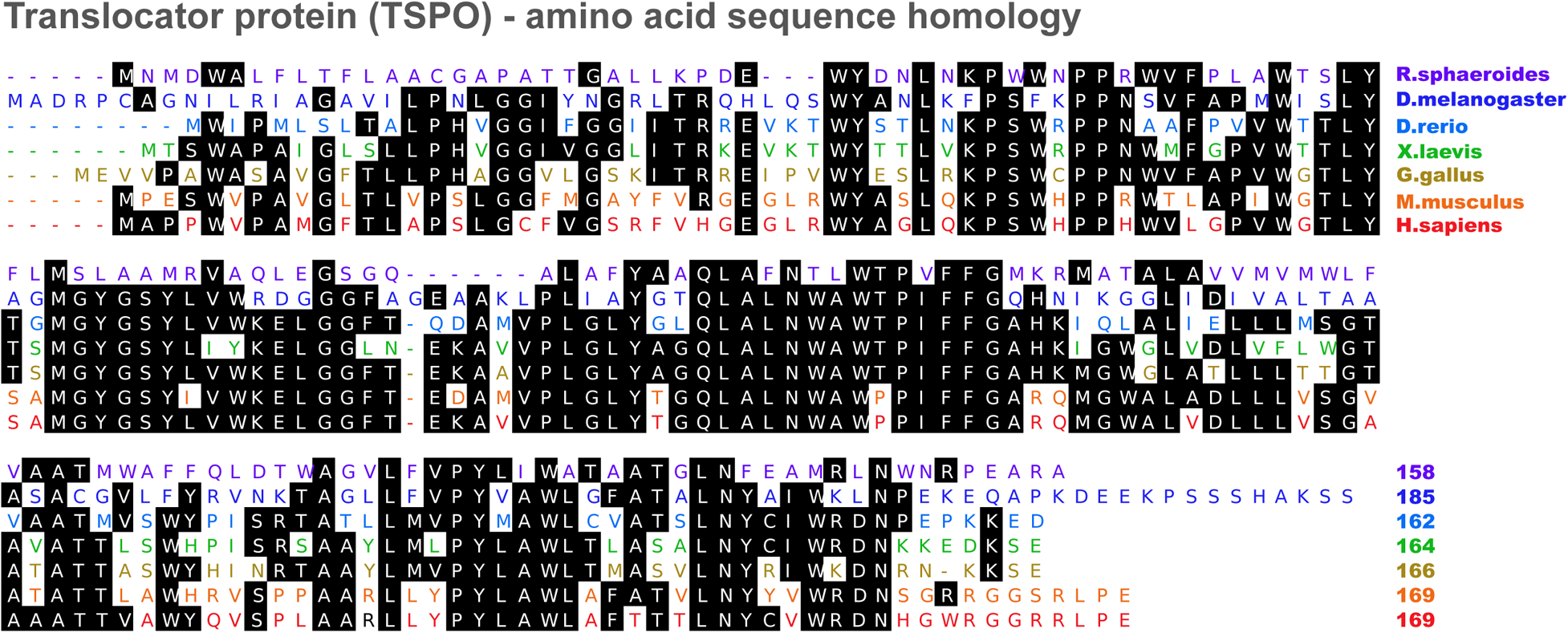
TSPO amino acid sequence comparisons showing fairly conserved consensus sequences (shaded) in different model organisms. Percent identity to Homo sapiens: Rhodobacter sphaeroides (33.5%), Drosophila melanogaster (42.6%), Danio rerio (54.3%), Xenopus laevis (57.3%), Gallus gallus (60.4%), and Mus musculus (81.1%).
Early studies highlighted predominant TSPO localization to the mitochondrial outer membrane. Although TSPO was widely detected in multiple organs, its expression appeared particularly high in steroid hormone producing cells of the adrenal glands, testes and ovaries. Two key studies linked TSPO function to steroid hormone biosynthesis: First, TSPO binding by PK11195 and Ro5–4864 could induce steroid hormone production in both adrenocortical and Leydig tumor cell lines [5, 6]. Second, TSPO knockdown or disruption by homologous recombination in rat Leydig tumor cells could decrease steroid hormone production [7, 8]. These studies were deemed highly significant because they demonstrated that TSPO effects were independent of the steroidogenic acute regulatory protein (StAR), a key player in mitochondrial cholesterol transport required for steroidogenesis (Box 1). As a result, TSPO was thought to function as the channel that receives cholesterol from StAR and mediates its transport to the mitochondrial inner membrane. This steroidogenic function for TSPO was the most studied among all its implicated properties. Given this putative link, up- or downregulation of TSPO expression in different tissues has been considered a direct indication of steroid hormone production in numerous studies [9–11]. Results that describe TSPO and steroidogenesis have been highlighted in recent literature reviews on this topic [12, 13].
Box 1. Mitochondrial cholesterol import and steroidogenesis.
The first enzymatic step in the biosynthesis of steroid hormones involves the cleavage of the side chain of cholesterol to form pregnenolone. The cytochrome P450 side chain cleavage (CYP11A1) that carries out this reaction resides on the matrix side of the inner mitochondrial membrane. To produce steroid hormones in response to trophic hormone stimulation, it is essential that cholesterol cross the outer mitochondrial membrane, the aqueous intermembrane space, and the inner mitochondrial membrane. This cholesterol import mechanism also forms a regulatory step that is rate limiting for steroid hormone production. It was discovered that rapid de novo synthesis of the steroidogenic acute regulatory protein (StAR) plays an indispensable role in mitochondrial cholesterol import required for adrenal and gonadal steroidogenesis (reviewed in [75]). Mutations in StAR were found to be the basis of lipoid congenital adrenal hyperplasia, and similar to this human condition, StAR gene-deletion in mice revealed an almost complete inability to synthesize steroid hormones. Nevertheless, the gap in understanding the precise mechanism by which StAR mediates mitochondrial cholesterol import, and the fact that StAR is not expressed in steroidogenic cells of the human placenta, led to speculations that another player that works at the level of the outer mitochondrial membrane in cooperation with StAR may be essential. TSPO filled the void in this model with overwhelming pharmacological indicators, but without direct evidence. Recent demonstration that TSPO may not be involved in steroidogenesis has reopened this informational gap in the cholesterol import model.
Co-purification of TSPO with key molecular components previously implicated to form the mitochondrial permeability transition pore (MPTP) resulted in its inclusion as a regulator of this process [14]. Mitochondrial permeability transition (MPT) refers to a sudden increase in permeability of the inner mitochondrial membrane, a mechanism that has been associated with cell death seen in different human pathologies (Box 2). Early biochemical studies seeking core MPTP components identified the voltage-dependent anion channel (VDAC) in the outer mitochondrial membrane, the adenine nucleotide transporter (ANT) in the inner mitochondrial membrane, and Cyclophilin D (CyPD) in the matrix [15]. Demonstration that TSPO could be co-purified with VDAC and ANT resulted in attempts to dissect the effects of TSPO binding chemicals as means to regulate MPT. The chemicals PK11195 and Ro5–4864 were found to affect MPTP opening [16, 17]. The endogenous protoporphyrin IX, a heme precursor that binds with high affinity to TSPO could also mediate MPTP opening [18]. As a result, the VDAC/ANT/TSPO model became widely accepted as the structure of the MPTP.
Box 2. Mitochondrial permeability transition (MPT).
The MPT refers to an increase in permeability of the inner mitochondrial membrane to any molecule <1.5 kDa due to opening of a non-specific pore [76–78]. Based on recent evidence, the MPT pore (MPTP) is formed by dimers of F0F1 ATP synthase. Opening of this MPTP results in equilibration of the mitochondrial matrix and the cytosol leading to uncoupling, osmotic swelling of the matrix, cristae unfolding and rupture of the outer mitochondrial membrane. The ensuing traumatic release of intermembrane space proteins such as cytochrome c and apoptosis inducing factor can trigger apoptosis in these cells. Matrix Ca2+ is essential for this process, and MPTP opening is triggered by elevated matrix Ca2+ concentration. Although transient opening of the MPTP may exist under physiological conditions, persistent opening of the MPTP has been associated with cell death seen in numerous pathological conditions.
Subsequent genetic analysis of the putative core components of the MPTP systematically excluded a role for all genes encoding VDAC [19] and ANT [20] in MPT. These findings set the field on a new course that led to the recent definition of the molecular nature of the MPTP, in that it is formed by dimers of F0F1 ATP synthase [21]. Although a role for TSPO was not discussed in the new model, it remains to be explained how TSPO binding chemicals could modulate cell death processes. The pharmacological evidence supporting a TSPO link to MPTP appears well documented [22–25]. Developments in the field of MPT have been extensively described in a recent literature review on this topic [26].
TSPO as a biomarker in human disease pathology
TSPO expression in the central nervous system is very low under normal physiological conditions and restricted to astrocytes and microglia. However, in response to brain injury and inflammation, TSPO levels dramatically increase in these glial cells [27]. First identified for its pharmacological binding profile, use of selective and high-affinity TSPO binding chemicals like PK11195 labeled with radioisotope tracers aided in visualization of affected brain regions in several neurodegenerative diseases [2, 28]. This prospect led to extensive efforts to develop novel synthetic chemicals that bind with high affinity and specificity to TSPO for diagnostic imaging in vivo [28]. Use of TSPO as a diagnostic marker has been reliable, and several TSPO binding chemicals as positron emission tomography (PET) tracers have progressed from preclinical studies to human clinical trials. Clinical trials using TSPO binding chemicals were focused on diagnosis of a variety of pathologies including: traumatic brain injury (NCT01547780), Alzheimer’s disease (NCT01209156; NCT01028209), Parkinson’s disease (NCT01527695; NCT01028209), multiple sclerosis (NCT01428505; NCT00432900; NCT01028209), encephalopathy (NCT00459693), autism (NCT0132255), neuroinflammation (NCT02062099; NCT02233868; NCT02009826; NCT01881646), neurodegeneration (NCT02086240), dementia (NCT00613119) and neurocysticercosis (NCT00526916) [Source: www.clinicaltrials.gov].
Identical to inflammation and injury seen in the brain, TSPO overexpression is also observed in cardiac pathologies [29, 30], and in different cancers [31]. TSPO binding chemicals for use as diagnostic PET tracers to locate cardiac conditions and track cancer development are also under development. Human clinical trials to detect cardiac sarcoidosis (NCT02017522), carotid atherosclerosis (NCT00547976), and squamous and basal cell carcinomas (NCT01265472) have either been completed or are currently underway.
Exploiting TSPO upregulation in pathologies for diagnostic imaging as described above has potential to grow and extend across multiple fields. Although imaging complications have been encountered as a result of in vivo metabolism of these TSPO binding PET tracers and aberrant signals contributing to non-specific noise in some cases, new synthetic TSPO binding chemicals are being developed to tackle these drawbacks [32]. Therefore, diagnostic imaging is probably the primary clinical value that TSPO research has to offer at this present time.
What is the therapeutic potential of TSPO binding chemicals?
Upregulation of TSPO expression at sites of inflammation and injury has often resulted in questions regarding its context in disease etiology. Effects observed as a result of TSPO binding have in fact led to several seemingly disparate biological interpretations of TSPO function [1, 3]. Nevertheless, treatment with TSPO binding drugs has proven efficacious in ameliorating disease pathology in numerous preclinical models. Examples in the central nervous system include: multiple sclerosis, traumatic injury, excitotoxicity, contusion, neuropathy, neuroinflammatory pain, and anxiety disorders (reviewed in [2]). In a recent study it was demonstrated that the TSPO binding drug Ro5–4864 could not only attenuate, but also reverse the pathology associated with Alzheimer’s disease in vivo [33]. A large part of these positive effects were attributed to the function for TSPO in steroid hormone production [34]. Human clinical trials were performed for a TSPO “agonist” XBD173 (AC-5216, Emapunil) for treatment of anxiety disorders by Novartis and Dainippon Sumitomo Pharma (NCT00108836). According to the proof of concept: XBD173 can bind to TSPO and induce production of neurosteroids, which in turn can potentiate GABAA receptor-based neurotransmission and bring about anxiolytic effects. Promising outcomes were reported using a high dose of XBD173 in an induced model of panic disorder, but at lower doses XBD173 effects showed no efficacy and were not different from placebo [35]. High TSPO binding variability across human subjects was observed on subsequent testing of XBD173 [36]. Currently, XBD173 is not in the drug development pipeline of either of these companies.
TSPO binding drugs have also been demonstrated to cause death of cancer cells, and as a result they are also considered as potential anti-cancer therapeutics (reviewed in [31]). This cell death mediated by TSPO binding drugs in cancer cells has been linked to their putative role in activating MPT. Linked to a similar mechanism, use of TSPO binding drugs after ischemia-reperfusion injury in the heart offered protection to cardiomyocytes and reduced infarct size [37, 38].
Even without a direct etiological link, this quest for TSPO binding therapeutics has been quite extensive in recent years and applied to a variety of diseases. These investigations, prompted by the pressing need for drug discovery, were based on the established mitochondrial function of TSPO and its upregulation in pathological lesions. However, as the landscape in TSPO physiology is rapidly changing, these TSPO attributes need to be carefully re-evaluated.
Is there a role for TSPO in steroidogenesis?
Recent studies examining TSPO function using genetic approaches have changed our views of its involvement in steroidogenesis. Using a conditional knockout mouse with TSPO deletion in Leydig cells, it was demonstrated that lack of TSPO did not compromise testosterone production in vivo [39]. This was a surprising result [40], and it raised the question of whether findings in Leydig cells could be extrapolated to other steroidogenic cell types [41]. Subsequently, another study also demonstrated that global TSPO deletion (TSPO−/−) in mice did not affect viability, fertility and the ability to generate steroid hormones [42]. TSPO−/− mice did not show any apparent abnormalities and produced both gonadal and adrenal steroid hormones at levels similar to control mice. These results were contradictory to results from a previous report showing that TSPO knockout in mice was early embryonic lethal [43]. In order to understand these conflicting findings, additional experiments were performed including knockdown of TSPO expression using siRNA in different steroidogenic cell lines (Mouse Leydig: MA-10 and MLTC; Mouse adrenal: Y1; Rat Leydig: R2C) to test for steroid hormone production. In this experimental setting, steroidogenesis was not affected [42], contradictory to a previous result suggesting that TSPO knockdown inhibited steroidogenic capacity in MA-10 cells [8]. In fact, it was observed that in one of the cell lines utilized, the human adrenal H295R cell line, there was no expression of TSPO mRNA or protein, yet this cell line can produce levels of steroid hormones equivalent to wild type cells, clearly supporting the notion that TSPO is not involved in steroidogenesis [42]. Another discordant finding was with regards to the impact of TSPO on cell viability. It was previously shown that cells with TSPO knockdown of >70% cannot survive [44, 45], however, in later studies, in vitro TSPO knockdown of >80% in different steroidogenic cell lines showed no effect on viability [42]. In strong corroboration of evidence against TSPO in steroidogenesis, a subsequent recent study described an independently generated TSPO−/− mouse to have no defects in steroid hormone biosynthesis [46]. This study also showed that isolated steroidogenic mitochondria devoid of TSPO did not have any deficits in cholesterol transfer suggesting that TSPO was not involved in this mitochondrial process [46].
In addition, another recent study demonstrated that complete disruption of TSPO in the MA-10 cell line (MA-10:TspoΔ/Δ) using CRISPR/Cas9-mediated mutagenesis did not have any effect on viability of the cells or steroidogenesis [47]. This result was again not consistent with another early report that mono-allelic disruption of TSPO in the R2C Leydig cell line obliterated its steroidogenic potential and caused phenotypic abnormality [7]. However, changes in phenotype or issues with viability were not observed in three independent clones of the MA-10:TspoΔ/Δ cells examined in the recent study [47], suggesting that TSPO deletion does not affect essential cellular functions. Moreover, when MA-10:TspoΔ/Δ cell clones were used to examine the steroidogenic ability of the TSPO binding chemical PK11195, both control and TSPO deficient cells showed identical increases in steroid hormone production [47]. This finding indicated that the effect of PK11195 on steroidogenesis in MA-10 cells is not mediated through TSPO, suggesting that previous studies reporting the pharmacological link between TSPO and steroidogenesis in this cell line and at the same range of concentrations [5, 6], were in all probability, off-target effects.
It has been recently proposed that alteration of physical membrane properties due to incorporation of TSPO binding chemicals into the membrane bilayer could contribute to some TSPO-independent effects [48]. Other studies have also associated effects of TSPO binding chemicals to membrane cholesterol accumulation/modulation [49, 50] and steroidogenesis [51]; but in the light of recent shift in understanding, whether these observations are indeed mediated through TSPO is a question that needs confirmation.
Collectively, recent results uncouple TSPO from steroidogenesis and cell viability thus opening the question of what is the true function of the protein.
TSPO and the mitochondrial permeability transition
A recent study using liver and heart specific conditional TSPO knockout mice concluded that TSPO is not involved in the regulation of MPT, or structure of the MPTP [52]. Results from this study showed that MPT could occur in the complete absence of TSPO. The link between TSPO and MPT was established by tests using TSPO binding chemicals that demonstrated an effect on MPT. However, these results published in numerous manuscripts over a period of 20 years indicate that effects mediated by the TSPO binding chemicals including PK11195 and Ro5–4864 were highly variable, and depend on experimental setting as well as the concentration used (nanomolar to micromole), and in some cases could even be biphasic [16, 17, 45, 53]. In addition, different classes of TSPO binding chemicals bind to different regions in the TSPO structure with variable specificity. Nevertheless, these effects were considered to be specific, as it was demonstrated that anti-TSPO antibodies could block MPT [23] induced by the endogenous TSPO binding heme precursor, protoporphyrin IX [18, 54]. But, recent experiments using TSPO−/− cells indicated that the effects of both the endogenous protoporphyrin IX and the synthetic PK11195 and Ro5–4864 TSPO ligands were not mediated through TSPO [52], as shown previously [54], and that the mechanism by which outer mitochondrial membrane regulates MPT did not involve TSPO [52]. As a result, these observations have challenged previous findings implicating TSPO in MPT opening for a variety of pathological processes.
The mechanism underlying how MPT could still be activated by TSPO binding chemicals remains an interesting question. The new knowledge that the F0F1 ATP synthase forms the MPTP [21], and that TSPO binding chemicals like PK11195 can interact with, and inhibit F0F1 ATP synthase [55–57], suggests that the effects observed with these chemicals on MPT may be independent of TSPO [26].
Seeking a function for TSPO
The experimental evidence linking TSPO function to steroidogenesis and MPT over the past 25 years is substantial, but it should be noted that most studies were performed using TSPO binding chemicals. The finding that TSPO might not be involved in steroidogenesis was surprising. Similarly, excluding TSPO involvement in MPT was also unexpected. In both cases, genetic deletion of TSPO showed no effect on the purported functions [39, 42, 46, 52], and that the effects of TSPO binding chemicals on both steroidogenesis and MPT are not mediated through TSPO [47, 52]. These results refute a function for TSPO in both steroidogenesis and MPT and the core pharmacological basis for both models (Fig 2). Although the outcome for TSPO in these contexts can be considered negative, these developments are a substantial leap for research progress in these fields.
Figure 2. Reexamining the functional network of TSPO.
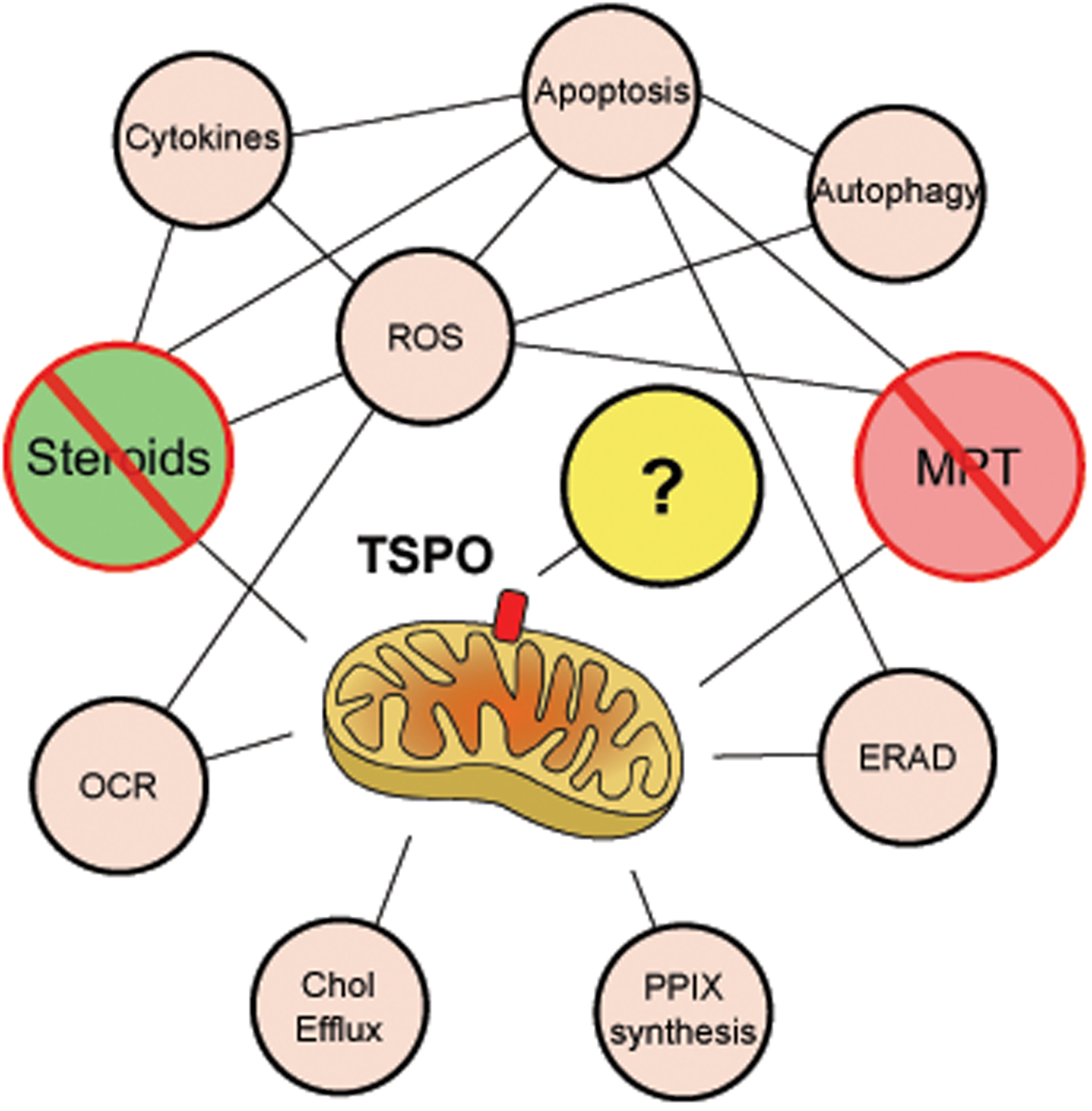
Steroidogenesis and mitochondrial permeability transition, the two major putative TSPO functions associated with its structural properties, and linked to multifarious downstream effects have been refuted. Responses to TSPO knockdown or knockout in cells have recorded changes in reactive oxygen species (ROS) production, cytokine production, apoptosis, autophagy, endoplasmic reticulum associated protein degradation (ERAD), protoporphyrin IX (PPIX) synthesis, cholesterol efflux and mitochondrial oxygen consumption rate (OCR). However, precise function of TSPO in these observations remain to be elucidated.
At the present time, we propose that mammalian TSPO is left without a known function. Although studies have elaborated TSPO’s structural, biochemical and pharmacological properties, we believe that its precise physiological and pathological functions remain unknown. Future research will need to focus on examining mitochondrial functions in a comprehensive way including respiration, bioenergetics, metabolism, and stress responses, and also cautiously take into account distinct yet unexplained pharmacological observations [58]. Recent observations correlated with TSPO knockdown in different cells like activation of endoplasmic reticulum-associated protein degradation [59], inhibition of autophagy [60], decrease of cholesterol efflux [50], and increase pro-inflammatory cytokine production [61], also need careful consideration to identify the core structure-function relationship to TSPO (Fig 2). In addition, information from TSPO in lower organisms that include functions like oxygen sensing in photosynthetic bacteria [62], stress regulation in plants [63], apoptosis mediation and longevity in drosophila [64], and erythropoiesis regulation in zebrafish [65], should be evaluated for an evolutionary link when studying mammalian systems. However, it should also be taken into account that TSPO homology and organelle localization in these organisms may vary. Understanding the exact physiological mechanism of mammalian TSPO action is imperative to delineating its organismal function(s), which will lead to better comprehension of its pharmacology (Box 3), and will unlock its potential as a diagnostic/therapeutic target.
Box 3. Are TSPO binding chemicals true ligands?
The word ligand used loosely in TSPO literature refers to all substances that bind to TSPO at different distinct sites; the findings about responses to ligand binding have also been inconsistent and diverse in the TSPO literature ranging from steroid hormone production to cell proliferation and apoptosis. By definition, ligand in biochemistry and pharmacology refers to a small molecular substance that binds to a target biomolecule (or a complex) and elicits/modulates a specific biological response (agonistic or antagonistic) that is directly associated with the target’s function. With genetic models showing that physiological TSPO function does not correspond to pharmacological effects recorded in experiments using TSPO binding chemicals, it is reasonable to propose that these chemicals are not acting as ligands. Moving forward, it will be prudent to interpret binding results carefully and tie their pharmacologic effects to physiologic ones.
Concluding remarks and future perspectives
In recent years, poor and frequently questionable validation of targets under investigation has led to drop in success rates of Phase II clinical drug trials from 28% to 18% [66]. A majority of these cases have resulted from the irreproducibility of published results that describe key findings [67]. This emphasizes the need to thoroughly investigate the pathways being targeted for drug discovery. With regards to TSPO, key findings supporting the TSPO-steroidogenesis model by knockdown in cell lines [8], gene deletion in cell lines [7], and gene deletion in mice [43] have not been reproducible in the same homologous systems [39, 46, 68], raising the question of whether mechanisms of functional redundancy are at play. We believe that if redundant or alternative mechanisms exist to accomplish TSPO function as recently suggested [69], they should have become evident in earlier studies. Thus, one must be open-minded to the possibility that TSPO function is yet to be identified. Previous studies using TSPO binding chemicals have indeed reported non-TSPO mediated effects [70–72]. We can only speculate that these effects represent non-specific integration of the chemicals perturbing cellular membrane bilayers [48] and/or unknown off-target effects in the case of steroidogenesis, or binding of TSPO ligands to F0F1 ATP synthase [55–57] in the case of MPT. It is also possible that these observations may be due to unknown effects due to complex crosstalk across multiple distinct pathways [73, 74]. So without understanding physiological function, it might never be possible to determine the exact pharmacological effects of specific TSPO binding chemicals (Box 4). We believe and hope that the emergence of viable TSPO−/− animals and TSPO−/− cell lines will inject new enthusiasm into the field and will offer an opportunity to re-evaluate the physiology of TSPO. These TSPO null models represent a new experimental approach to delineate the specific TSPO-based effects mediated by TSPO binding chemicals that will allow for a more focused understanding of its therapeutic potential.
Box 4. What does TSPO translocate?
In 2006, appeal from a scientific consortium (supported by Novartis Pharmaceuticals, Basel, Switzerland) [79], resulted in renaming of the peripheral benzodiazepine receptor (PBR) to the translocator protein (TSPO). The basis for this change in nomenclature was prompted by a desire to better reflect the structure and molecular “function” of this protein [79]. At that time, experimental homology modeling of the structure of mammalian TSPO was interpreted as a channel-like structure with a hydrophobic interior core lined by the cholesterol recognition amino acid consensus (CRAC) motif for putative translocation of lipophilic substrates. The key substrate in question was cholesterol, but translocation of other substances like porphyrins, and translocation of a variety of molecules during mitochondrial permeability transition (MPT) was also considered a possibility. Although cholesterol transport was considered the preeminent function for TSPO, direct physiological proof for such a function does not exist. A more recent high-resolution NMR structure of TSPO showed that the side chains of the CRAC motif (Figure I), considered central for cholesterol translocation, is located on the outside pointing towards the membrane environment [80] suggesting that previous homology models are inaccurate. In this study, structural evidence for TSPO homo-oligomerization, considered to be an important feature for cholesterol transport, was not observed. This supported other observations in the literature that challenged this concept [39, 68]. Moreover, recent physiological data show that TSPO is neither involved in mitochondrial cholesterol import required for steroidogenesis, nor in the regulation of mitochondrial permeability transition [39, 42, 52]. Besides steroidogenesis and MPT, it remains to be confirmed if mammalian TSPO plays a role in porphyrin transport and heme synthesis. Therefore, despite the name change, potential ‘translocator’ functions for TSPO still remain as unproven hypotheses.
BOX 4, Figure I. TSPO structure showing distinct cholesterol and PK11195 binding sites.
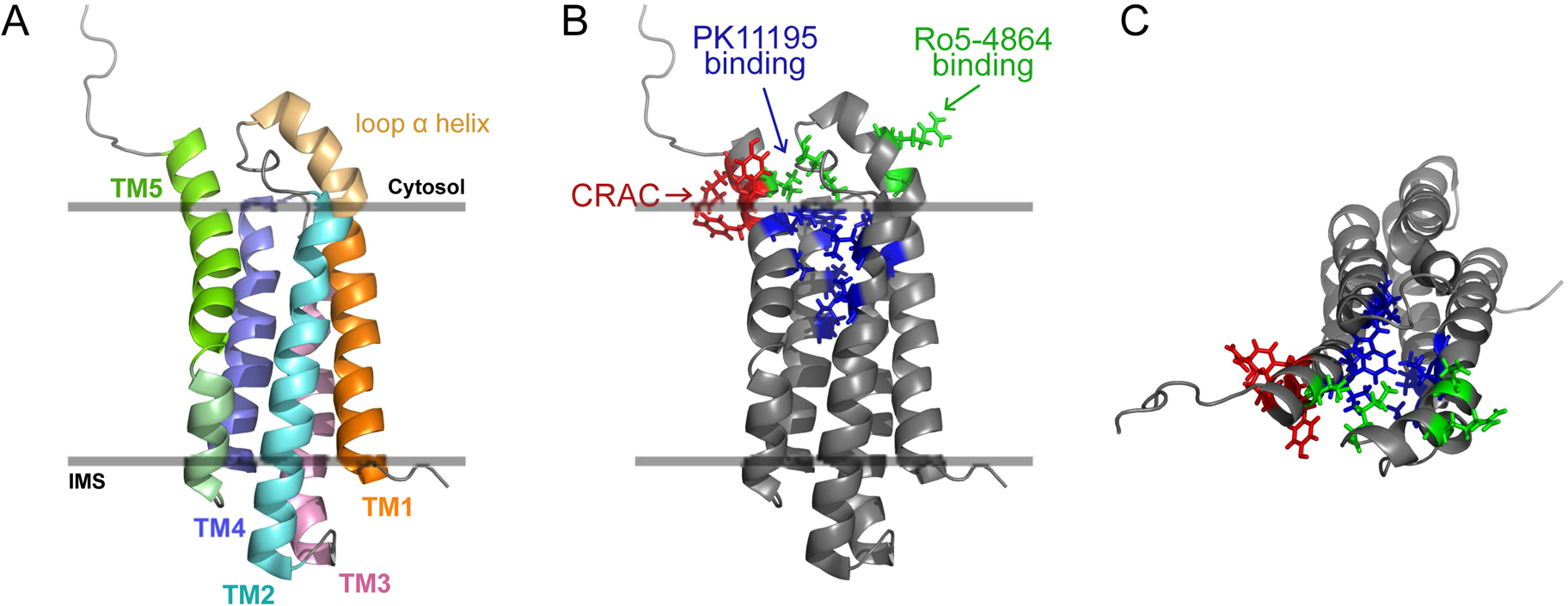
(A) Structure of TSPO in the outer mitochondrial membrane (side view) showing the five α-helix transmembrane structure (TM1–5; IMS - inter-membrane space).
(B) TSPO structure showing: (1) location of the cholesterol recognition amino acid consensus (CRAC) motif at the C terminus (residues 147–159); side chains Tyr152, Tyr153 and Arg156 (in red), essential for cholesterol binding are located on the outside of the TSPO structure and point toward the membrane environment. (2) Side chains of PK11195 binding pocket formed by residues Ala23, Val26, Leu49, Ala50, Ile52, Trp107, Ala110, Leu114, Ala147 and Leu150 (in blue), that do not involve side chains of CRAC motif amino acids. (3) Binding site for Ro5–4864 is distinct from that of PK11195 and was identified to include residues Glu29, Arg32, Lys39 and Val154 (in green) [81].
(C) Top view: Cholesterol binding side chains of the CRAC motif pointing outside of the TSPO structure and do not interfere with internal PK11195 binding site. Images were constructed from the high-resolution NMR structure PDB ID: 2MGY [80].
As we look into the future, several important questions for TSPO remain (Box 5). Should investment in drug discovery based on TSPO continue? As a major part of TSPO research involves diagnostic in vivo imaging for changes in TSPO expression as indicators of inflammation and injury, it seems reasonable that even without a functional definition for TSPO, clinical trials assessing the specificity and diagnostic capabilities of TSPO binding chemicals should proceed. However, for meaningful therapeutic targeting of TSPO, mechanistic understanding of the protein function followed by designing of true (specific) ligands that act as agonists or antagonists to TSPO’s yet to be uncovered function is warranted. Deciphering complex off-target effects of TSPO binding chemicals from existing literature/datasets to glean meaningful information may not be a pragmatic approach to address this problem. But as a practical twist, alternative targets that may mediate some of these beneficial effects could be identified to open new areas of exploration in drug discovery. Conclusively, understanding the precise physiological function for TSPO will be necessary to fully realize its potential as a therapeutic target.
Box 5. Outstanding questions.
What is the precise function of TSPO?
In steroidogenesis, what is the mechanism that cooperates with StAR for mitochondrial cholesterol import?
In MPTP, does TSPO pharmacology still have a case for regulation of MPT?
What is the pathologic basis for TSPO upregulation in different inflammatory pathologies?
Without a defined physiological function for TSPO can we consider TSPO a valid therapeutic target? Are the beneficial effects of TSPO binding chemicals specific? Can candidate TSPO binding drugs enter human clinical trials?
Highlights.
Recent data question the role of TSPO in steroidogenesis and mitochondrial permeability transition and suggest that TSPO is not involved.
Exploiting TSPO upregulation in pathologies for diagnostic imaging has potential to grow and extend across multiple fields.
The physiological function of TSPO remains undefined and its future as a therapeutic target needs to be carefully reevaluated.
Acknowledgements
We apologize to those whose work could not be cited directly due to space constraints. This manuscript was possible through funding from the Cornell Center for Vertebrate Genomics to VS, National Institutes of Health grant HD-17481 and Robert A. Welch Foundation to DMS.
Glossary
- Benzodiazepines
Refers to a class of drugs that are clinically used as muscle relaxants, anticonvulsants and sedative-hypnotics. Pharmacology of benzodiazepines is primarily mediated by its binding to the ‘central’ benzodiazepine receptor (γ-aminobutyric acid type A receptor/GABAA receptor) located in the central nervous system and potentiating inhibitory neurotransmission.
- Mitochondrial cholesterol transport
Mitochondria are cellular organelles that consist of a double membrane creating two distinct internal compartments. For steroid hormone production, cholesterol must be transported through the outer mitochondrial membrane across the aqueous inter-membrane space to the matrix side of the inner mitochondrial membrane before it can be converted to pregnenolone. This is an essential and rate-limiting step in steroid hormone biosynthesis.
- Mitochondrial Permeability Transition
Refers to an increase in permeability of the inner mitochondrial membrane to any molecule <1.5 kDa due to opening of a non-specific pore. Mitochondrial permeability transition (MPT) has been associated with cell death seen in numerous pathological conditions.
- Mitochondrial Permeability Transition Pore
Refers to the structural components that form the pore in the inner mitochondrial membrane during MPT. Recent evidence suggests that this is formed by dimers of the F0F1 ATP synthase.
- Peripheral benzodiazepine receptor
Benzodiazepines were observed to also occupy ‘peripheral’ sites distinct from the ‘central’ GABAA receptor on the outer mitochondrial membrane. The peripheral benzodiazepine receptor (PBR) was the protein characterized based on this distinct pharmacology, which was subsequently renamed as the translocator protein (TSPO).
- Steroidogenesis
Refers to the biosynthesis of steroid hormones. In this process, steroid hormone producing cells convert cholesterol into pregnenolone in an enzymatic step catalyzed by CYP11A1/P450scc that occurs in the mitochondrial matrix. Pregnenolone is then converted into different classes of steroid hormones by subsequent enzymatic steps that happen within the endoplasmic reticulum and the mitochondria.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Gavish M, et al. (1999) Enigma of the peripheral benzodiazepine receptor. Pharmacol Rev 51, 629–650. [PubMed] [Google Scholar]
- 2.Rupprecht R, et al. (2010) Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov 9, 971–988. [DOI] [PubMed] [Google Scholar]
- 3.Scarf AM, et al. (2012) Is there any correlation between binding and functional effects at the translocator protein (TSPO) (18 kDa)? Curr Mol Med 12, 387–397. [DOI] [PubMed] [Google Scholar]
- 4.Fan J, et al. (2012) Structural and functional evolution of the translocator protein (18 kDa). Curr Mol Med 12, 369–386. [DOI] [PubMed] [Google Scholar]
- 5.Mukhin AG, et al. (1989) Mitochondrial benzodiazepine receptors regulate steroid biosynthesis. Proc Natl Acad Sci U S A 86, 9813–9816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Papadopoulos V, et al. (1990) The peripheral-type benzodiazepine receptor is functionally linked to Leydig cell steroidogenesis. J Biol Chem 265, 3772–3779. [PubMed] [Google Scholar]
- 7.Papadopoulos V, et al. (1997) Targeted disruption of the peripheral-type benzodiazepine receptor gene inhibits steroidogenesis in the R2C Leydig tumor cell line. J Biol Chem 272, 32129–32135. [DOI] [PubMed] [Google Scholar]
- 8.Hauet T, et al. (2005) Peripheral-type benzodiazepine receptor-mediated action of steroidogenic acute regulatory protein on cholesterol entry into leydig cell mitochondria. Mol Endocrinol 19, 540–554. [DOI] [PubMed] [Google Scholar]
- 9.Daugherty DJ, et al. (2013) A TSPO ligand is protective in a mouse model of multiple sclerosis. EMBO Mol Med 5, 891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lassance L, et al. (2014) Obesity-induced downregulation of the mitochondrial translocator protein (TSPO) impairs placental steroid production. J Clin Endocrinol Metab, jc20142792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen C, et al. (2014) Estradiol modulates translocator protein (TSPO) and steroid acute regulatory protein (StAR) via protein kinase A (PKA) signaling in hypothalamic astrocytes. Endocrinology 155, 2976–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papadopoulos V and Miller WL (2012) Role of mitochondria in steroidogenesis. Best Pract Res Clin Endocrinol Metab 26, 771–790. [DOI] [PubMed] [Google Scholar]
- 13.Miller WL (2013) Steroid hormone synthesis in mitochondria. Mol Cell Endocrinol 379, 62–73. [DOI] [PubMed] [Google Scholar]
- 14.McEnery MW, et al. (1992) Isolation of the mitochondrial benzodiazepine receptor: association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc Natl Acad Sci U S A 89, 3170–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zamzami N and Kroemer G (2001) The mitochondrion in apoptosis: how Pandora’s box opens. Nat Rev Mol Cell Biol 2, 67–71. [DOI] [PubMed] [Google Scholar]
- 16.Kinnally KW, et al. (1993) Mitochondrial benzodiazepine receptor linked to inner membrane ion channels by nanomolar actions of ligands. Proc Natl Acad Sci U S A 90, 1374–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chelli B, et al. (2001) Peripheral-type benzodiazepine receptor ligands: mitochondrial permeability transition induction in rat cardiac tissue. Biochem Pharmacol 61, 695–705. [DOI] [PubMed] [Google Scholar]
- 18.Pastorino JG, et al. (1994) Protoporphyrin IX, an endogenous ligand of the peripheral benzodiazepine receptor, potentiates induction of the mitochondrial permeability transition and the killing of cultured hepatocytes by rotenone. J Biol Chem 269, 31041–31046. [PubMed] [Google Scholar]
- 19.Baines CP, et al. (2007) Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 9, 550–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kokoszka JE, et al. (2004) The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giorgio V, et al. (2013) Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A 110, 5887–5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costa B, et al. (2014) TSPO ligand residence time influences human glioblastoma multiforme cell death/life balance. Apoptosis. [DOI] [PubMed] [Google Scholar]
- 23.Azarashvili T, et al. (2014) Carbenoxolone induces permeability transition pore opening in rat mitochondria via the translocator protein TSPO and connexin43. Arch Biochem Biophys 558, 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gemelli C, et al. (2014) Cytotoxic effect of hemin in colonic epithelial cell line: involvement of 18 kDa translocator protein (TSPO). Life Sci 107, 14–20. [DOI] [PubMed] [Google Scholar]
- 25.Daniele S, et al. (2014) Apoptosis therapy in cancer: the first single-molecule co-activating p53 and the translocator protein in glioblastoma. Sci Rep 4, 4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bernardi P (2013) The mitochondrial permeability transition pore: a mystery solved? Front Physiol 4, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen MK and Guilarte TR (2008) Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacol Ther 118, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chauveau F, et al. (2008) Nuclear imaging of neuroinflammation: a comprehensive review of [11C]PK11195 challengers. Eur J Nucl Med Mol Imaging 35, 2304–2319. [DOI] [PubMed] [Google Scholar]
- 29.Qi X, et al. (2012) Translocator protein (18 kDa): a promising therapeutic target and diagnostic tool for cardiovascular diseases. Oxid Med Cell Longev 2012, 162934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fairweather D, et al. (2014) Sex differences in translocator protein 18 kDa (TSPO) in the heart: implications for imaging myocardial inflammation. J Cardiovasc Transl Res 7, 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Austin CJ, et al. (2013) The translocator protein (TSPO): a novel target for cancer chemotherapy. Int J Biochem Cell Biol 45, 1212–1216. [DOI] [PubMed] [Google Scholar]
- 32.Liu GJ, et al. (2014) The 18 kDa translocator protein, microglia and neuroinflammation. Brain Pathol 24, 631–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barron AM, et al. (2013) Ligand for translocator protein reverses pathology in a mouse model of Alzheimer’s disease. J Neurosci 33, 8891–8897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rone MB, et al. (2009) Cholesterol transport in steroid biosynthesis: role of protein-protein interactions and implications in disease states. Biochim Biophys Acta 1791, 646–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rupprecht R, et al. (2009) Translocator protein (18 kD) as target for anxiolytics without benzodiazepine-like side effects. Science 325, 490–493. [DOI] [PubMed] [Google Scholar]
- 36.Owen DR, et al. (2011) Variation in binding affinity of the novel anxiolytic XBD173 for the 18 kDa translocator protein in human brain. Synapse 65, 257–259. [DOI] [PubMed] [Google Scholar]
- 37.Xiao J, et al. (2010) 4’-Chlorodiazepam, a translocator protein (18 kDa) antagonist, improves cardiac functional recovery during postischemia reperfusion in rats. Exp Biol Med (Maywood) 235, 478–486. [DOI] [PubMed] [Google Scholar]
- 38.Schaller S, et al. (2010) TRO40303, a new cardioprotective compound, inhibits mitochondrial permeability transition. J Pharmacol Exp Ther 333, 696–706. [DOI] [PubMed] [Google Scholar]
- 39.Morohaku K, et al. (2014) Translocator protein/peripheral benzodiazepine receptor is not required for steroid hormone biosynthesis. Endocrinology 155, 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stocco DM (2014) The Role of PBR/TSPO in Steroid Biosynthesis Challenged. Endocrinology 155, 6–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Papadopoulos V (2014) On the Role of the Translocator Protein (18-kDa) TSPO in Steroid Hormone Biosynthesis. Endocrinology 155, 15–20. [DOI] [PubMed] [Google Scholar]
- 42.Tu LN, et al. (2014) Peripheral Benzodiazepine Receptor/Translocator Protein Global Knockout Mice are Viable with no Effects on Steroid Hormone Biosynthesis. J Biol Chem 289, 27444–27454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Papadopoulos V, et al. (1997) Peripheral benzodiazepine receptor in cholesterol transport and steroidogenesis. Steroids 62, 21–28. [DOI] [PubMed] [Google Scholar]
- 44.Amri H, et al. (1999) The peripheral-type benzodiazepine receptor and adrenal steroidogenesis. Current Opinion in Endocrinology and Diabetes 6, 179–184. [Google Scholar]
- 45.Veenman L, et al. (2007) Channel-like functions of the 18-kDa translocator protein (TSPO): regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr Pharm Des 13, 2385–2405. [DOI] [PubMed] [Google Scholar]
- 46.Banati RB, et al. (2014) Positron emission tomography and functional characterization of a complete PBR/TSPO knockout. Nat Commun 5, 5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tu LN, et al. (2014) PK11195 effect on steroidogenesis is not mediated through the translocator protein (TSPO). Endocrinology, en20141707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hatty CR, et al. (2014) Investigating the interactions of the 18kDa translocator protein and its ligand PK11195 in planar lipid bilayers. Biochim Biophys Acta 1838, 1019–1030. [DOI] [PubMed] [Google Scholar]
- 49.Paradis S, et al. (2013) Cardioprotection by the TSPO ligand 4’-chlorodiazepam is associated with inhibition of mitochondrial accumulation of cholesterol at reperfusion. Cardiovasc Res 98, 420–427. [DOI] [PubMed] [Google Scholar]
- 50.Taylor JM, et al. (2014) Targeting mitochondrial 18 kDa translocator protein (TSPO) regulates macrophage cholesterol efflux and lipid phenotype. Clin Sci (Lond) 127, 603–613. [DOI] [PubMed] [Google Scholar]
- 51.Chung JY, et al. (2013) Drug ligand-induced activation of translocator protein (TSPO) stimulates steroid production by aged brown Norway rat Leydig cells. Endocrinology 154, 2156–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sileikyte J, et al. (2014) Regulation of the Mitochondrial Permeability Transition Pore by the Outer Membrane does not Involve the Peripheral Benzodiazepine Receptor (TSPO). J Biol Chem 289, 13769–13781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kugler W, et al. (2008) Ligands of the mitochondrial 18 kDa translocator protein attenuate apoptosis of human glioblastoma cells exposed to erucylphosphohomocholine. Cell Oncol 30, 435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sileikyte J, et al. (2011) Regulation of the inner membrane mitochondrial permeability transition by the outer membrane translocator protein (peripheral benzodiazepine receptor). J Biol Chem 286, 1046–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson KM, et al. (2006) Mechanistic basis for therapeutic targeting of the mitochondrial F1F0-ATPase. ACS Chem Biol 1, 304–308. [DOI] [PubMed] [Google Scholar]
- 56.Cleary J, et al. (2007) Inhibition of the mitochondrial F1F0-ATPase by ligands of the peripheral benzodiazepine receptor. Bioorg Med Chem Lett 17, 1667–1670. [DOI] [PubMed] [Google Scholar]
- 57.Seneviratne MS, et al. (2012) PK11195 inhibits mitophagy targeting the F1Fo-ATPsynthase in Bcl-2 knock-down cells. Curr Mol Med 12, 476–482. [DOI] [PubMed] [Google Scholar]
- 58.Gut P, et al. (2013) Whole-organism screening for gluconeogenesis identifies activators of fasting metabolism. Nat Chem Biol 9, 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou T, et al. (2014) The mitochondrial translocator protein, TSPO, inhibits HIV-1 envelope glycoprotein biosynthesis via the endoplasmic reticulum-associated protein degradation pathway. J Virol 88, 3474–3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gatliff J, et al. (2014) TSPO interacts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Autophagy, 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bae KR, et al. (2014) Translocator protein 18 kDa negatively regulates inflammation in microglia. J Neuroimmune Pharmacol 9, 424–437. [DOI] [PubMed] [Google Scholar]
- 62.Yeliseev AA, et al. (1997) A mammalian mitochondrial drug receptor functions as a bacterial “oxygen” sensor. Proc Natl Acad Sci U S A 94, 5101–5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vanhee C, et al. (2011) The Arabidopsis multistress regulator TSPO is a heme binding membrane protein and a potential scavenger of porphyrins via an autophagy-dependent degradation mechanism. Plant Cell 23, 785–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lin R, et al. (2014) Genetic analysis of dTSPO, an outer mitochondrial membrane protein, reveals its functions in apoptosis, longevity, and Aβ42-induced neurodegeneration. Aging Cell 13, 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rampon C, et al. (2009) Translocator protein (18 kDa) is involved in primitive erythropoiesis in zebrafish. Faseb J 23, 4181–4192. [DOI] [PubMed] [Google Scholar]
- 66.Arrowsmith J (2011) Trial watch: Phase II failures: 2008–2010. Nat Rev Drug Discov 10, 328–329. [DOI] [PubMed] [Google Scholar]
- 67.Prinz F, et al. (2011) Believe it or not: how much can we rely on published data on potential drug targets? Nat Rev Drug Discov 10, 712. [DOI] [PubMed] [Google Scholar]
- 68.Morohaku K, et al. (2013) Developmental expression of translocator protein/peripheral benzodiazepine receptor in reproductive tissues. PLoS One 8, e74509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Midzak A and Papadopoulos V (2014) Binding Domain-Driven Intracellular Trafficking of Sterols for Synthesis of Steroid Hormones, Bile Acids and Oxysterols. Traffic. [DOI] [PubMed] [Google Scholar]
- 70.Gonzalez-Polo RA, et al. (2005) PK11195 potently sensitizes to apoptosis induction independently from the peripheral benzodiazepin receptor. Oncogene 24, 7503–7513. [DOI] [PubMed] [Google Scholar]
- 71.Hans G, et al. (2005) Peripheral benzodiazepine receptor (PBR) ligand cytotoxicity unrelated to PBR expression. Biochem Pharmacol 69, 819–830. [DOI] [PubMed] [Google Scholar]
- 72.Walter RB, et al. (2005) PK11195, a peripheral benzodiazepine receptor (pBR) ligand, broadly blocks drug efflux to chemosensitize leukemia and myeloma cells by a pBR-independent, direct transporter-modulating mechanism. Blood 106, 3584–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li L, et al. (2008) The peripheral benzodiazepine receptor ligand 1-(2-chlorophenyl-methylpropyl)-3-isoquinoline-carboxamide is a novel antagonist of human constitutive androstane receptor. Mol Pharmacol 74, 443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lockhart A, et al. (2003) The peripheral benzodiazepine receptor ligand PK11195 binds with high affinity to the acute phase reactant alpha1-acid glycoprotein: implications for the use of the ligand as a CNS inflammatory marker. Nucl Med Biol 30, 199–206. [DOI] [PubMed] [Google Scholar]
- 75.Stocco DM (2001) Tracking the role of a star in the sky of the new millennium. Mol Endocrinol 15, 1245–1254. [DOI] [PubMed] [Google Scholar]
- 76.Hunter DR and Haworth RA (1979) The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch Biochem Biophys 195, 453–459. [DOI] [PubMed] [Google Scholar]
- 77.Hunter DR and Haworth RA (1979) The Ca2+-induced membrane transition in mitochondria. III. Transitional Ca2+ release. Arch Biochem Biophys 195, 468–477. [DOI] [PubMed] [Google Scholar]
- 78.Haworth RA and Hunter DR (1979) The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys 195, 460–467. [DOI] [PubMed] [Google Scholar]
- 79.Papadopoulos V, et al. (2006) Translocator protein (18kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci 27, 402–409. [DOI] [PubMed] [Google Scholar]
- 80.Jaremko L, et al. (2014) Structure of the mitochondrial translocator protein in complex with a diagnostic ligand. Science 343, 1363–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Farges R, et al. (1994) Site-directed mutagenesis of the peripheral benzodiazepine receptor: identification of amino acids implicated in the binding site of Ro5–4864. Mol Pharmacol 46, 1160–1167. [PubMed] [Google Scholar]