

Abstract
Emerging resistance to current antimalarial medicines underscores the importance of identifying new drug targets and novel compounds. Malaria parasites are purine auxotrophic and import purines via the Plasmodium falciparum Equilibrative Nucleoside Transporter Type 1 (PfENT1). We previously showed that PfENT1 inhibitors block parasite proliferation in culture. Our goal was to identify additional, possibly more optimal chemical starting points for a drug discovery campaign. We performed a high throughput screen (HTS) of GlaxoSmithKline’s 1.8 million compound library with a yeast-based assay to identify PfENT1 inhibitors. We used a parallel progression strategy for hit validation and expansion, with an emphasis on chemical properties in addition to potency. In one arm, the most active hits were tested for human cell toxicity; 201 had minimal toxicity. The second arm, hit expansion, used a scaffold-based substructure search with the HTS hits as templates to identify over 2000 compounds; 123 compounds had activity. Of these 324 compounds, 175 compounds inhibited 3D7 strain P. falciparum parasite proliferation with IC50 values between 0.8 to ~180 μM. One hundred forty-two compounds inhibited PfENT1 knockout (pfent1Δ) parasite growth indicating they also hit secondary targets. Thirty-two hits inhibited growth of 3D7 but not pfent1Δ parasites. Thus, PfENT1 inhibition was sufficient to block parasite proliferation. Therefore, PfENT1 may be a viable target for antimalarial drug development. Six compounds with novel chemical scaffolds were extensively characterized in yeast-, parasite-, and human erythrocyte-based assays. The inhibitors showed similar potencies against drug sensitive and resistant P. falciparum strains. They represent attractive starting points for development of novel antimalarial drugs.
Keywords: malaria, purine, transporter, drug discovery, high throughput screen
Graphical Abstract
Malaria is a major public health problem that disproportionately affects developing countries. The World Health Organization estimated that in 2016, 3.5 billion people were at risk for contracting malaria.1 There were over 200 million clinical cases of malaria that resulted in about a half-million deaths. Most deaths occurred in sub-Saharan Africa among children under the age of five years old and pregnant women. Infection with Plasmodium species unicellular, eukaryotic protozoan parasites cause malaria. Although five Plasmodium species infect humans, infection by P. falciparum and P. vivax cause about 90% of cases. Most deaths result from P. falciparum infection.1
Over the past decade, a multifaceted approach to control malaria, which included the use of Artemisinin-based Combination Therapies (ACT) as first-line drugs, led to a major reduction in the number of cases and deaths. However, this success has been threatened by the development of P. falciparum parasites with delayed clearance in response to ACT therapy and their spread throughout Southeast Asia to East India.2–4 More recently, delayed clearance in response to artemisinin has been reported in parasites from sub-Saharan Africa that lack mutations in the K13-propeller domain protein, which had been associated with delayed clearance by artemisinin-based drugs in southeast Asia.5, 6 The emergence and spread of parasites with delayed clearance to artemisinin treatment makes it imperative to develop new antimalarial drugs against novel therapeutic targets.7–9 In the current work we utilized a target-based approach to identify small molecule inhibitors that target the malaria parasite purine import pathway.
Malaria parasites are purine auxotrophic organisms – they lack the enzymes for de novo purine biosynthesis. Purines are essential for parasite proliferation both for nucleic acid synthesis and for other metabolic processes. Plasmodium species import purines, mostly hypoxanthine, from the host erythrocyte via Equilibrative Nucleoside Transporters (ENT).10–13 The imported purine nucleosides and nucleobases are transformed by purine salvage pathway enzymes into purine nucleotides.14–16 The P. falciparum and P. vivax genomes contain four ENT homologues.17–19 Studies in P. falciparum indicate that PfENT1 is the primary purine import transporter. Knockout of the PfENT1 gene (pfent1Δ) is conditionally lethal - pfent1Δ parasites are not viable in culture media that contains purines at concentrations similar to those found in human blood (< 10 μM).20–23 It should be noted that standard P. falciparum culture media contains 50 μg/ml hypoxanthine (367 μM), a concentration that is ~100-fold higher than the concentration found in human blood.20 However, pfent1Δ parasite growth can be rescued with media containing purine concentrations > 50 μM, significantly greater than that found in human blood.20 This suggests that the parasites have a secondary, lower affinity, purine import pathway. The molecular identity of the secondary transporters is unknown, but may include the other ENT isoforms, the AMP import pathway, or other unknown transporters.19, 24 Furthermore, a recent study used transposon insertion mutagenesis to identify essential genes in blood stage parasites: By the authors’ criteria, PfENT1 is an essential gene.25 Collectively these results suggest that inhibition of PfENT1 may be a good target for antimalarial drugs.
Development of assays for a high throughput screen (HTS) to identify inhibitors of equilibrative membrane transport proteins has been difficult because most functional assays involve radioactive substrate uptake. Previously, we developed a yeast-growth based HTS assay to screen for PfENT1 inhibitors that is illustrated schematically in Figure 1A.23 The assay depends on the fact that 1) PfENT1 also transports pyrimidines, including the cytotoxic uridine analog 5-fluorouridine (5-FUrd), 2) in wild type Saccharomyces cerevisiae yeast, 5-FUrd is transported into the cell via the endogenous FUI1 uridine transporter, metabolized in the cell where it exerts toxic effects on nucleic acid synthesis, and 3) FUI1 knockout yeast (fui1Δ) are resistant to killing by 5-FUrd (Figure 1A).26, 27 In PfENT1-expressing fui1Δ yeast, 5-FUrd can enter via PfENT1, and cell killing is observed. With 5-FUrd in the growth media, PfENT1-expressing fui1Δ yeast can only grow if an inhibitor of PfENT1 is present in the media to block 5-FUrd uptake (Figure 1A). Because hits are identified by cell growth, compounds that are cytotoxic to yeast, and might be generally cytotoxic, are eliminated. This provides a robust positive selection for PfENT1 inhibitors that are not cytotoxic to yeast. Hereafter this assay is referred to as the “Yeast HTS Growth Assay”.
Figure 1.
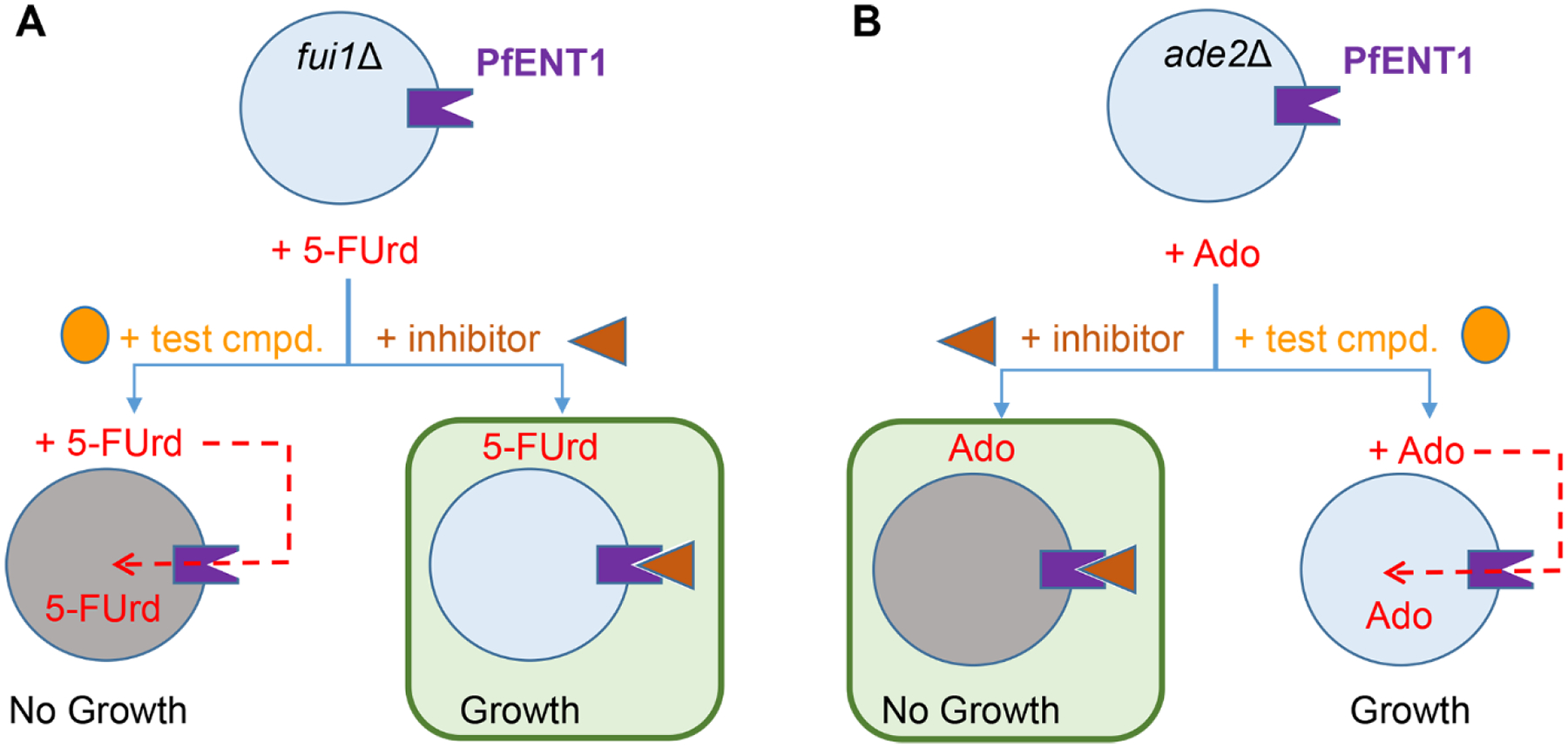
Schematic illustrating the two yeast-based assays used to identify and validate inhibitors of PfENT1. The green boxes indicate the outcomes that identified a hit. (A) In the primary HTS assay, referred to as the Yeast HTS Growth Assay, PfENT1-expressing yeast were grown in the presence of 5-FUrd and the test compound. If the test compound is not an inhibitor of PfENT1, 5-FUrd enters the yeast through PfENT1 and kills the yeast. If the test compound is a PfENT1 inhibitor, it blocks 5-FUrd entry and the yeast grow. Thus, in the Yeast HTS Growth Assay, hits were identified as compounds that permitted yeast growth. Compound potency is reported as EC50 values. (B) In the secondary assay, referred to as the Yeast Kill Assay, PfENT1 was expressed in purine auxotrophic yeast. With adenosine (Ado) as the sole purine source, these yeast can grow because Ado can enter via PfENT1. In this assay, a PfENT1 inhibitor blocks Ado entry and the yeast cannot grow. In the Yeast Kill Assay hits were identified as compounds that inhibited yeast proliferation. Compound potency is reported as IC50 values. In both assays, inhibitor concentration can be varied to determine the concentration dependence of the effects.
To validate hits from the Yeast HTS Growth Assay, we developed a secondary, orthogonal assay based on adenosine-dependent growth (illustrated in Figure 1B).23, hereafter referred to as the “Yeast Kill Assay”. We generated purine auxotrophic yeast by knockout of, ADE2, which codes for phosphoribosylaminoimidazole carboxylase, an essential enzyme in the purine biosynthetic pathway.28 These purine auxotrophic yeast can grow on adenine as the sole purine source because they have an endogenous adenine transporter. However, they lack an endogenous adenosine transporter and therefore, the purine auxotrophic yeast cannot grow in media when adenosine is the sole purine source. In contrast, PfENT1-expressing purine auxotrophic yeast can grow with adenosine as the sole purine source because adenosine can enter yeast cells via PfENT1.23 In contrast to the Yeast HTS Growth Assay, in this secondary assay PfENT1 inhibitors prevent the growth of PfENT1-expressing purine auxotrophic yeast in media with adenosine as the sole purine source. The only common element identifying hits between the Yeast HTS Growth Assay and the orthogonal adenosine dependent growth Yeast Kill Assay is inhibition of PfENT1. In the Yeast HTS Growth Assay, hits are identified by yeast proliferation because PfENT1 inhibitors block the uptake of the cytotoxic drug 5-FUrd (Figure 1A). In contrast, in the secondary Yeast Kill Assay, PfENT1 inhibitors block yeast cell growth by preventing the uptake of adenosine, an essential metabolic substrate for the purine auxotrophic yeast (Figure 1B). It is important to note that both yeast-based assays are designed to identify compounds that inhibit PfENT1 through complementary, but independent pathways. They do not screen out compounds that might also interact with secondary targets that might also inhibit parasite proliferation.
As proof of principle, we previously used the Yeast HTS Growth Assay (Figure 1A) to screen an approximately 64,500 compound Chembridge library.23 We identified 171 hits.23 Nine of the hits were characterized in a cascading series of secondary assays. The nine compounds displayed concentration-dependent effects in both the Yeast HTS Growth Assay (Figure 1A) and the Yeast Kill Assay (Figure 1B). The nine hits also blocked [3H]adenosine uptake into PfENT1-expressing yeast and into red blood cell (RBC) free P. falciparum parasites with IC50 values in the 5–50 nM range. The nine compounds blocked the proliferation of P. falciparum parasites during in vitro parasite culture with IC50 values in the 5–50 μM range. While the hits from this preliminary screen provided proof that PfENT1 was a potential target for antimalarial drug development, the nine hits did not have potent effects against P. falciparum cultures. Furthermore, the nine compounds had physicochemical and structural properties that made them less attractive as starting points for medicinal chemistry efforts.
The goal of the current work was to use the Yeast Growth HTS Assay to screen a large chemical library to identify compounds with improved characteristics as starting points for medicinal chemistry efforts to develop PfENT1 inhibitors as novel antimalarial drugs. As part of the 2013 GlaxoSmithKline (GSK) Discovery Fast Track Competition (http://openinnovation.gsk.com/news-2013-11-06/), these assays were modified to 1536-well format and used to screen GSK’s 1.8 million compound library. To increase the probability of finding suitable chemical starting points for PfENT1 inhibitors, we devised a parallel hit strategy to limit and refine the number of HTS hits to test in downstream assays in parasites and erythrocytes. Figure 2 illustrates the critical path for validation of the hits in a series of downstream assays. Multiple chemotypes were identified as PfENT1 inhibitors that inhibit P. falciparum parasite proliferation in culture. Six compounds representing distinct chemical scaffolds with generally favorable physicochemical properties were extensively characterized. These six compounds are potential therapeutic leads for medicinal chemistry efforts to develop novel antimalarial drugs.
Figure 2. Parallel hit progression strategy.
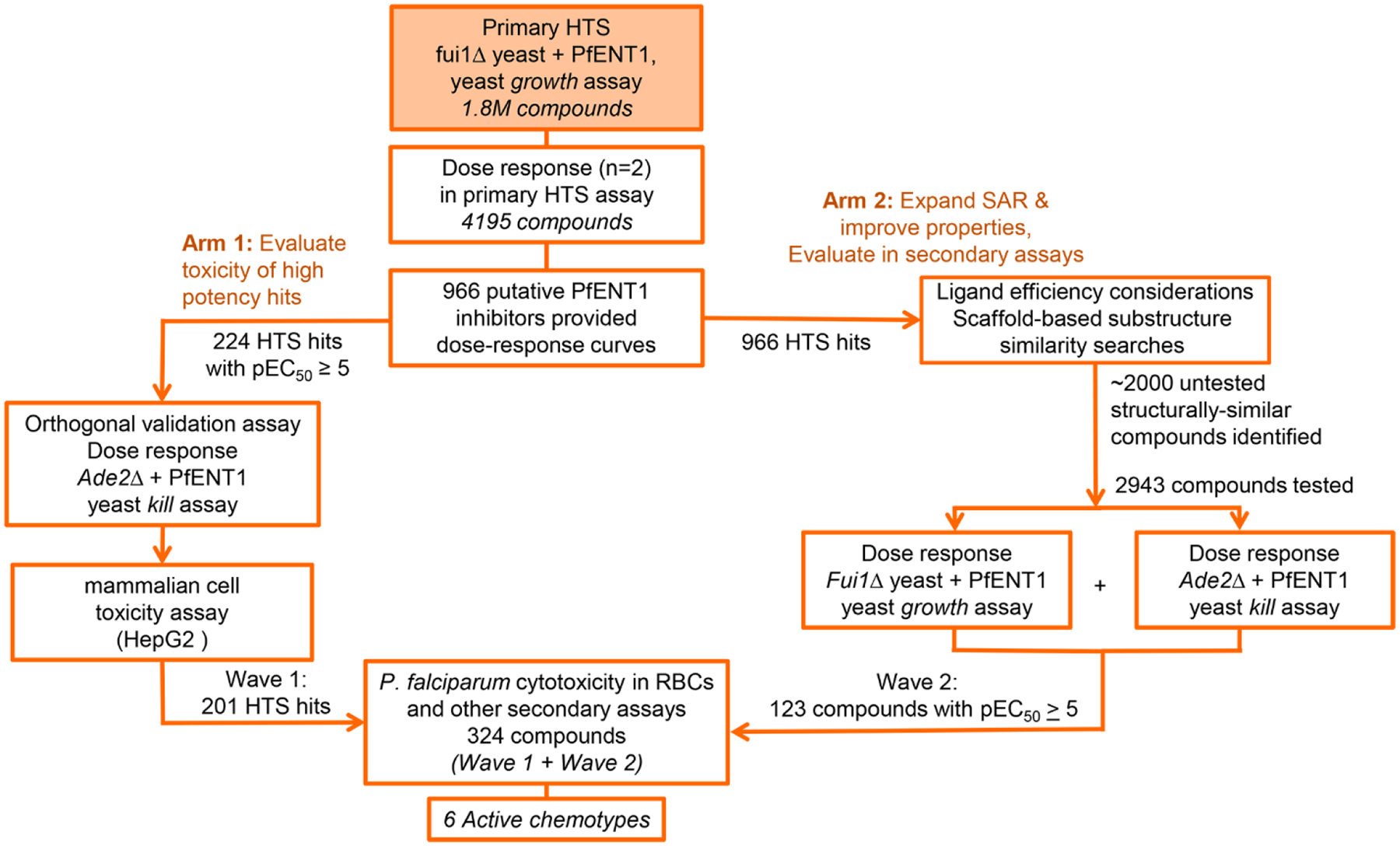
Critical path used to identify, validate, and characterize 6 chemotypes that inhibit PfENT1.
RESULTS AND DISCUSSION
HTS optimization and validation.
The first step was to transition the Yeast HTS Growth Assay from 384-well to 1536-well plates. An obstacle in the transition to 1536-well plates was that the yeast produced CO2. This resulted in the formation of gas bubbles which were often trapped in the corners of standard square-well plates (Figure 3A). Because yeast proliferation was assessed spectrophotometrically by optical density at 620 nm (OD620), the gas bubbles had a significant effect on the reliability of yeast proliferation measurements. The use of round-well, flat-bottom Corning #3893 type 1536-well plates solved this problem.
Figure 3.
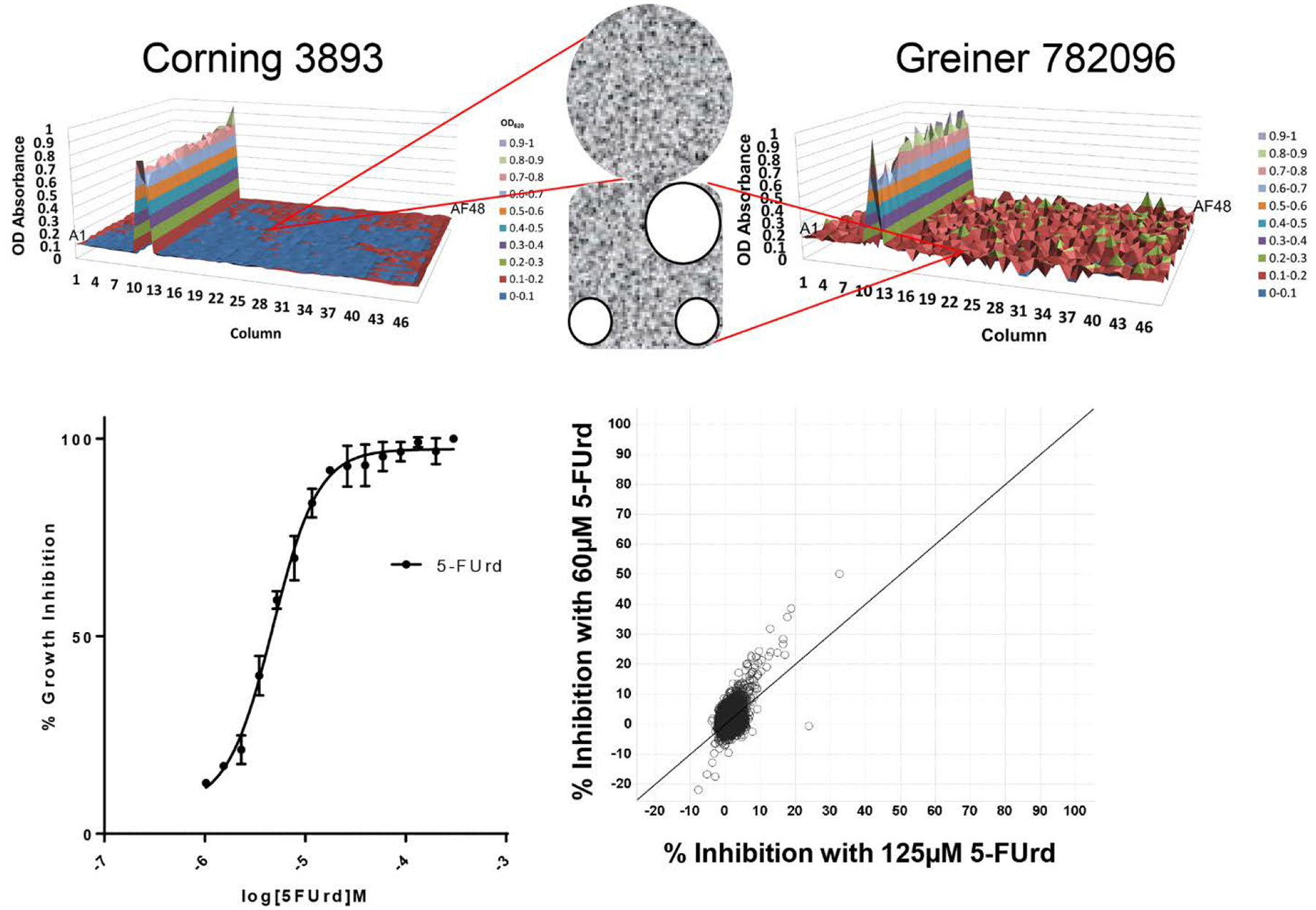
HTS assay optimization and validation. (A) Illustrates the effect of CO2 gas bubble formation in square well plates on background uniformity. X- and Y-axes indicate well coordinates and Z-axis indicates optical density (OD). Different colors indicate different level thresholds of positivity. Insert shows micrograph of gas bubbles (three round white circles) trapped in the corners of the square well from a Greiner 782096 plate and the lack of bubbles in the round well from the Corning 3893 plate. (B) Effect of 5-FUrd concentration on inhibition of PfENT1-expressing fui1Δ yeast growth. The IC50 for inhibition of yeast growth was 4.8 μM. (C) Averaged triplicate percent inhibition values for a 10 K validation compound set tested at 60 μM (y-axis) versus 125 μM (x-axis) 5-FUrd. Note that there was only 1 hit >30% inhibition at 125 μM 5-FUrd, while 60 μM 5-FUrd yielded 5 hits.
We evaluated several parameters to optimize the sensitivity vs the robustness of the HTS assay. These included the initial yeast culture seeding density, the duration of growth prior to OD620 measurement, effect of DMSO concentration, and the concentration of 5-FUrd. We tested three initial yeast culture seeding densities, OD620 (measured in a cuvette with 1 cm path length) 0.05, 0.075, and 0.1, which corresponded to 4000 to 8000 cells per well. The signal to background ratio decreased with higher seeding density, but the Z’ score was similar at all three densities (data not shown). Given the starting yeast cell densities, a 17 hour growth duration allowed maximal growth that did not saturate the spectrophotometric detector. Corrections were developed to account for non-uniform evaporative loss from edge wells during the 17-hour growth period. The assay was insensitive to DMSO concentrations below 2% (data not shown). We determined the concentration dependence of yeast killing by 5-FUrd (Figure 3B). The 5-FUrd IC50 was 4.8 μM (Figure 3B). The robustness of the assay was evaluated using 1408 compounds at 10 μM concentration and two 5-FUrd concentrations, 60 and 125 μM, both effectively IC100 concentrations chosen to minimize background yeast growth. The Z’ score was similar at both 5-FUrd concentrations, 0.68 and 0.70, respectively. The signal to background ratio with 125 μM 5-FUrd was 7, higher than with 60 μM 5-FUrd where it was 4 (data not shown). Validation testing was performed with 9854 compounds in triplicate at 10 μM concentration at both 60 and 125 μM 5-FUrd (Figure 3C). Validation statistics were acceptable. Based on these results, 60 μM 5-FUrd provided maximum sensitivity for the assay with minimal background yeast growth (data not shown). Maximal yeast growth was determined in the absence of 5-FUrd.
High Throughput Screen.
The HTS was run with 1,792,272 compounds at a concentration of 10 μM (Supplementary Table S1). The average Z’ score for the 1350 plates was 0.70. Robust active compounds would typically be defined as those with yeast growth greater than three standard deviations (SD) above the mean sample population per test occasion. Due to the low variability and high quality of the assay, the 3 SD cutoff corresponded to 9.1% of the maximal growth signal and included 70,648 compounds (Figure 4). By raising the cutoff to 28% of maximal growth (10 SD), a manageable set of 4104 primary hits was obtained. Medicinal chemists also reviewed the subset of compounds with maximal growth signals between 9–28% and selected an additional 191 primary screening hits based on considerations of low molecular weight and structural diversity. This increased the diversity of the chemical scaffolds to be tested in the downstream assays and gave a total of 4195 primary hits. Most of these primary hits (4195 compounds) were available in sufficient quantity to enable dose response measurements.
Figure 4.
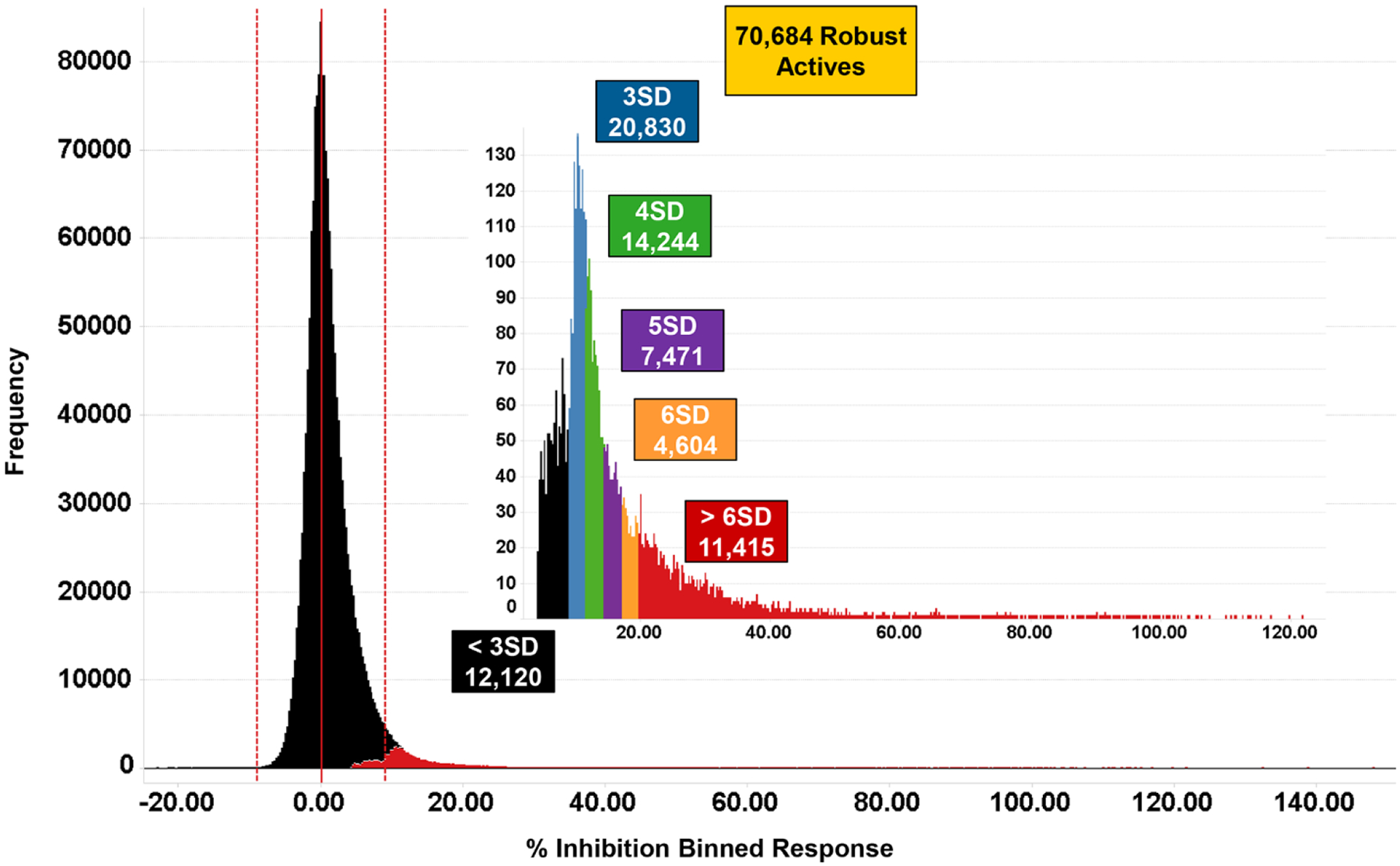
Performance of PfENT1 primary HTS. Histograms of the distribution of responses for the PfENT1 HTS (1.8 million compounds, 10 μM test concentration) showing the number of compounds observed at a range of statistical cut-offs. The colored boxes indicate the number of test compounds that exceed cut-offs defined as the indicated multiples of the robust standard deviation of the data population.
Each of the 4195 primary HTS hits was assayed in duplicate using the Yeast HTS Growth Assay to determine the concentration dependence of its effects. Only 966 of the 4195 primary hits yielded data sufficient to calculate dose-response curves in the concentration range tested. The pEC50 potency distribution for the 966 compounds is shown in Table 1. An Inhibition Frequency Index (IFI) value29 was calculated for each of the 966 screening hits that produced a dose response curve (Figure 5). IFI measures the frequency that a compound is found to be a robust active in multiple HTS campaigns (i.e., the number of times a compound is found to be a robust active hit divided by the number of times it has been screened in HTS campaigns at GlaxoSmithKline). The results from this heuristic index suggested that the vast majority of putative PfENT1 HTS hits were not promiscuous or nuisance compounds which repeatedly show up in HTS screening hit lists.
Table 1. Potency distribution for the 4195 HTS primary actives that were progressed to dose-response experiments (Yeast HTS Growth Assay).
Potency ranges are given as negative log EC50 (i.e., pEC50) from four parameter logistic fits of 11-point dose-response curves.
| pEC50 range | # of Cmpds | % of Cmpds |
|---|---|---|
| <4 | 3299 | 77% |
| 4 < × < 4.99 | 741 | 18% |
| 5 < × < 5.49 | 156 | 3.7% |
| 5.5 < × < 5.99 | 53 | 1.3% |
| 6 < × < 6.49 | 13 | 0.3% |
| 6.5 < × < 6.59 | 2 | 0.05% |
Figure 5.
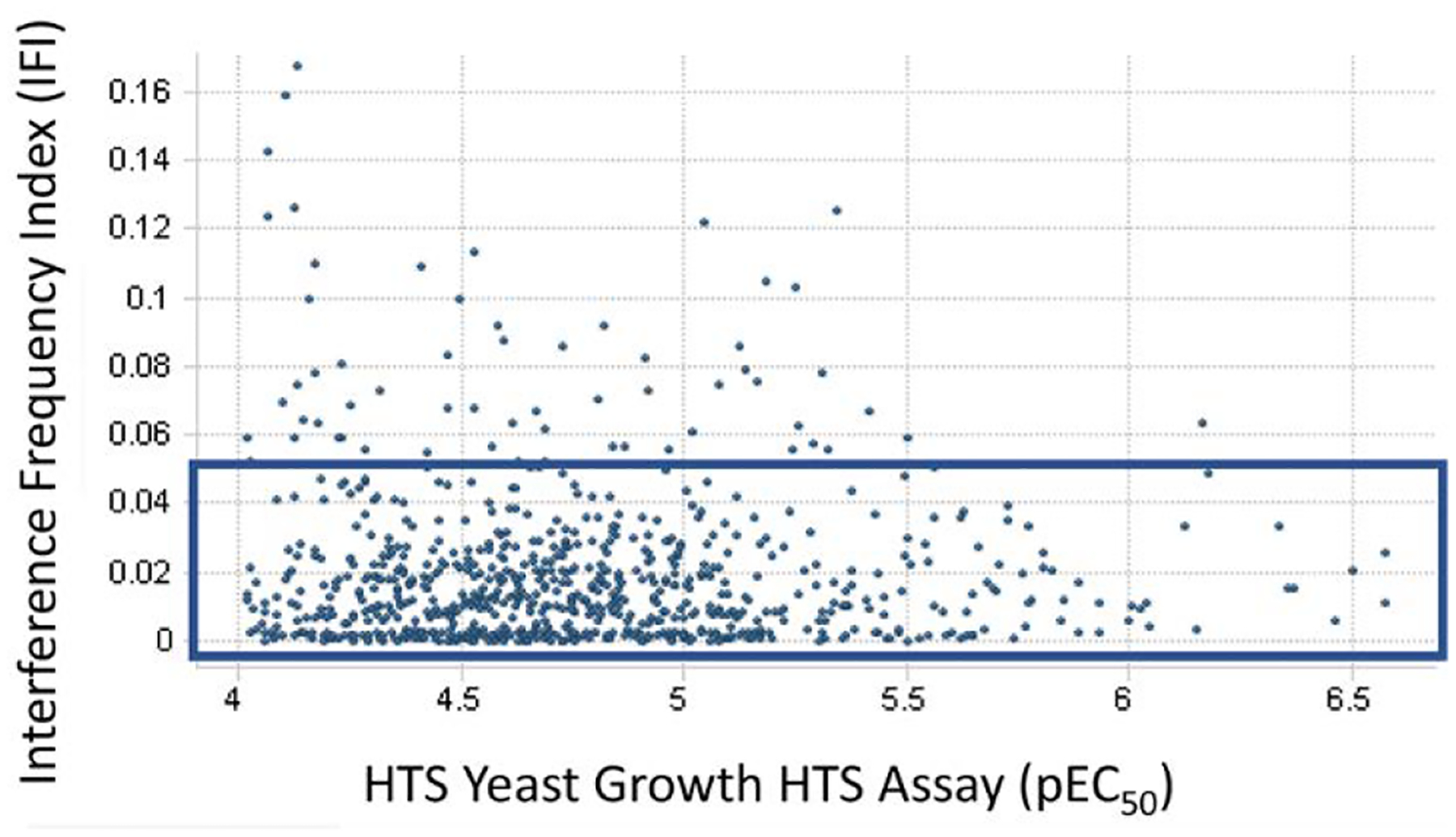
Interference Frequency Index (IFI) as a function of the compound pEC50 value for the 966 compounds that had sufficient potency to allow calculation of the EC50 values. Blue box indicates hits with IFI < 0.05. IFI measures the frequency that a compound is found to be a robust active in multiple HTS campaigns (i.e., the number of times a compound is found to be a robust active hit divided by the number of times it has been screened in HTS campaigns at GlaxoSmithKline).
Parallel Hit Progression Strategy.
A two-arm strategy was employed to quickly evaluate the PfENT1 HTS hits in secondary assays. In the first arm, the highest activity hits (224 compounds with pEC50 ≥ 5 in the Yeast HTS Growth Assay) were tested in a human hepatoma cell line (HepG2) acute cytotoxicity assay to identify HTS hits that were cytotoxic to mammalian cells. Hits that were highly cytotoxic to HepG2 cells were eliminated because they would not progress in the drug development process and might likely be false positives in the downstream P. falciparum growth inhibition assay. In the second arm, hit expansion, additional compounds were identified computationally based on a scaffold-based substructure search with the 966 HTS hits as templates. The goals were to expand the structure-activity relationship (SAR) information on the primary hits and to potentially identify novel compounds that might inhibit PfENT1. The efficacy of these additional, computationally-identified compounds was then assessed in the yeast-based assays and the most potent compounds were then tested in P. falciparum growth inhibition assays.
Arm 1 - Human hepatoma HepG2 cell toxicity – Identification of Wave 1 compounds.
Early in the critical path of Arm 1 (Figure 2, left pathway), the highest activity hits (224 hits with pEC50 ≥ 5 in the Yeast HTS Growth Assay) were tested in a HepG2 cytotoxicity assay to identify HTS hits that were potentially cytotoxic to human cells. While none of the primary HTS hits were cytotoxic to yeast at concentrations up to 100 μM (because hits enabled yeast growth in the HTS assay), more than a third of the hits (38%) showed some cytotoxicity to HepG2 cells with growth inhibition pIC50 values that ranged 4.0 to 6.8 (Supplementary Table S2). The most cytotoxic hits in the HepG2 assay (8 compounds with pIC50 > 5,) and hits containing known structural liabilities were removed to provide the set of 201 hits (Wave 1) for further validation in the Akabas lab. Of the 201 compounds in Wave 1, 132 showed no effects on HepG2 cell viability up to the maximum concentration tested, 100 μM. Three compounds were not tested for effects on HepG2 cells. Thus, for two-thirds of the compounds HepG2 cell cytotoxicity was not a major liability for further studies (Supplementary Figure S1).
Arm 2 - Hit expansion – Identification of Wave 2 compounds.
The purpose of Arm 2 (Figure 2, right path) was to further expand structure-activity-relationships (SAR) and to identify analogs with improved properties. About 10 structural hit families (> 3 analogs per hit family) were noted among the 966 screening hits. Unfortunately, many of the higher activity hits, like GSK-1 (Figure 6), had multiple undesirable properties for screening hits (i.e. low ligand efficiency ≤ 0.25, high molecular weight >450, >3 aromatic rings, or property forecast index (PFI) values >8).30, 31 Compounds with PFI values > 7 (chromatographic log D7.4 + aromatic ring count) are much more likely to be problematic in terms of physicochemical properties and other drug development issues (i.e. membrane permeability, metabolism/clearance, protein binding, hERG ion channel binding). In fact, a study of 240 oral drugs, all with oral bioavailability > 30%, revealed that 89% of them have PFI values < 7.31 About 50% of the higher activity PfENT1 HTS hits (pEC50 values ≥ 5 in the Yeast HTS Growth Assay) were predicted to have PFI values equal to or greater than 8.
Figure 6.
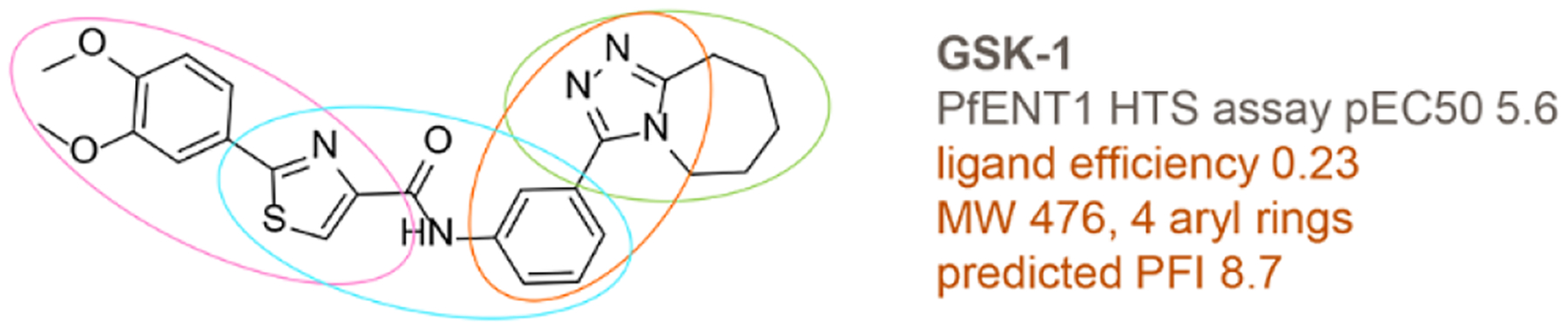
Chemical structure of representative HTS hit GSK-1. Potential liabilities are highlighted in orange text. Colored ovals show representative simpler scaffolds that were used as seeds for scaffold-based substructure searching. The simpler scaffolds were derived from the virtual deconstruction process.
To expand SAR of the hit families and to find new analogs with improved properties, several strategies were used to identify additional unscreened, structurally-related analogs from GSK compound inventories and external compound collections. To find compounds with improved properties, ligand efficiency calculations32–35 and plots were used to identify the more ligand efficient hits to seed scaffold-based substructure searches (Figure 7).36, 37 Ligand efficient hits of both low activity (pEC50 4–5) and higher activity (pEC50 >5) were used as seeds. In a second strategy, chemical structures of higher molecular weight hits, like GSK-1 (Figure 6), were computationally deconstructed via the NCATS R-group tool (https://tripod.nih.gov/?p=46) to simpler scaffolds that seeded automated substructure searches within the HTS dataset to aid identification of new structure-activity relationships. These strategies provided about 2000 additional compounds for screening from GSK and external compound collections.
Figure 7.
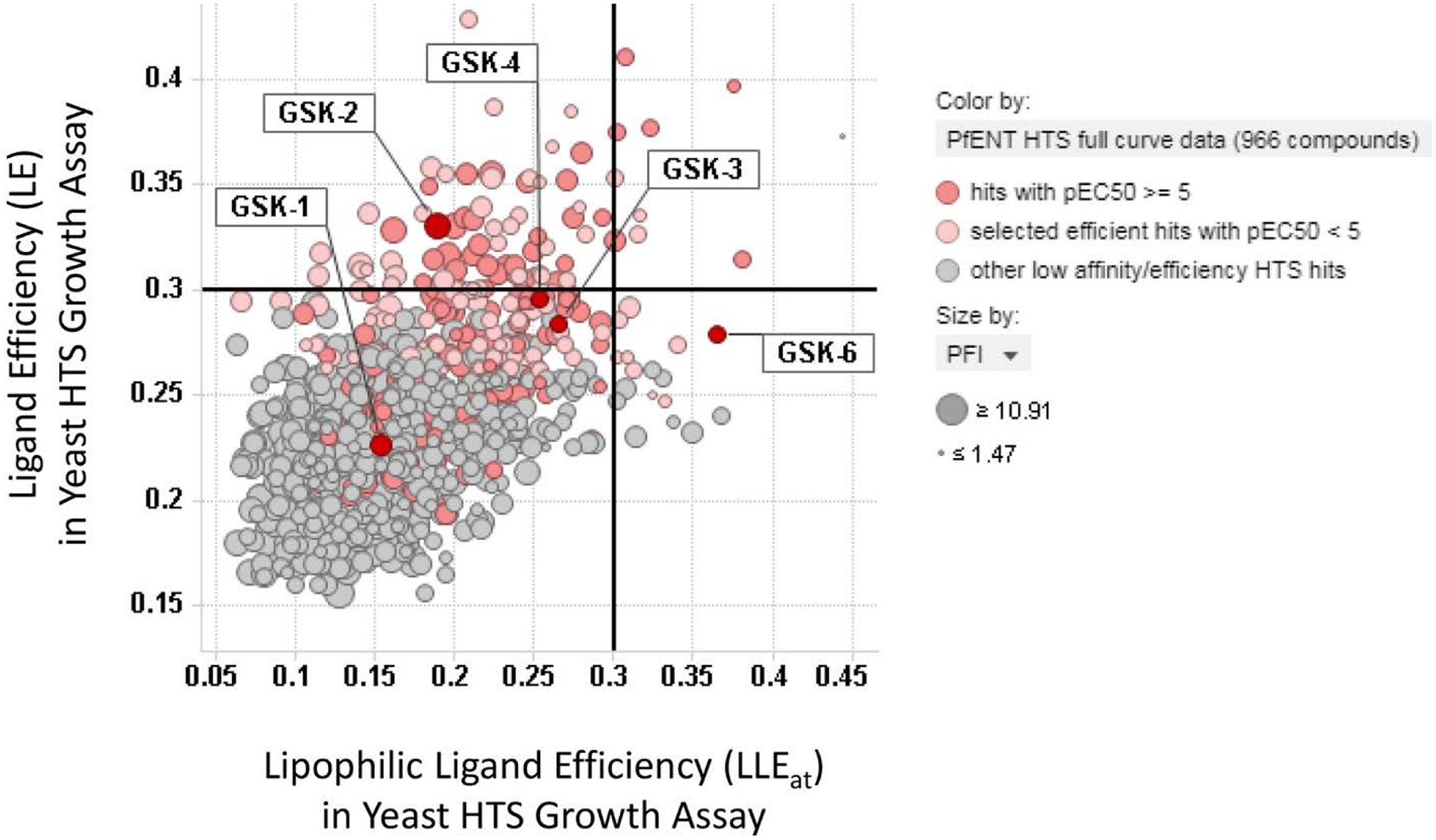
Scatterplot of ligand efficiency (LE) versus lipophilic ligand efficiency (LLEat) for the 966 putative hits identified in the Yeast HTS Growth Assay. In general, the more ligand efficient hits (LE values >0.25, light red highlight) and higher activity hits (pEC50 5–7, dark red highlight) were used to seed scaffold-based substructure searches. The data points are sized by Property Forecast Index (PFI). Labels highlight extensively characterized compounds GSK-1, GSK-2, GSK-3, GSK-4, GSK-6.
The 966 HTS hits and the ~2000 additional structurally-related compounds were then tested in both the Yeast HTS Growth Assay and the Yeast Kill Assay. The common element between these assays is inhibition of PfENT1. Figure 8 shows the correlation of pEC50 in the Yeast HTS Growth Assay and the pIC50 in the Yeast Kill Assay. Of the 966 HTS hits that gave dose response curves in the Yeast HTS Growth Assay, 98% of the compounds also provided dose response curves in the orthogonal Yeast Kill Assay. On average, the responses (pIC50) in the Yeast Kill Assay were about 0.5 log units more potent than seen in the Yeast HTS Growth Assay. This difference is believed to be due to the relative sensitivity of the experimental conditions in the two assays. The fact that most of the 966 HTS hits were active in both yeast based assays provides support for the hypothesis that their mechanism of action is via inhibition of PfENT1.
Figure 8.
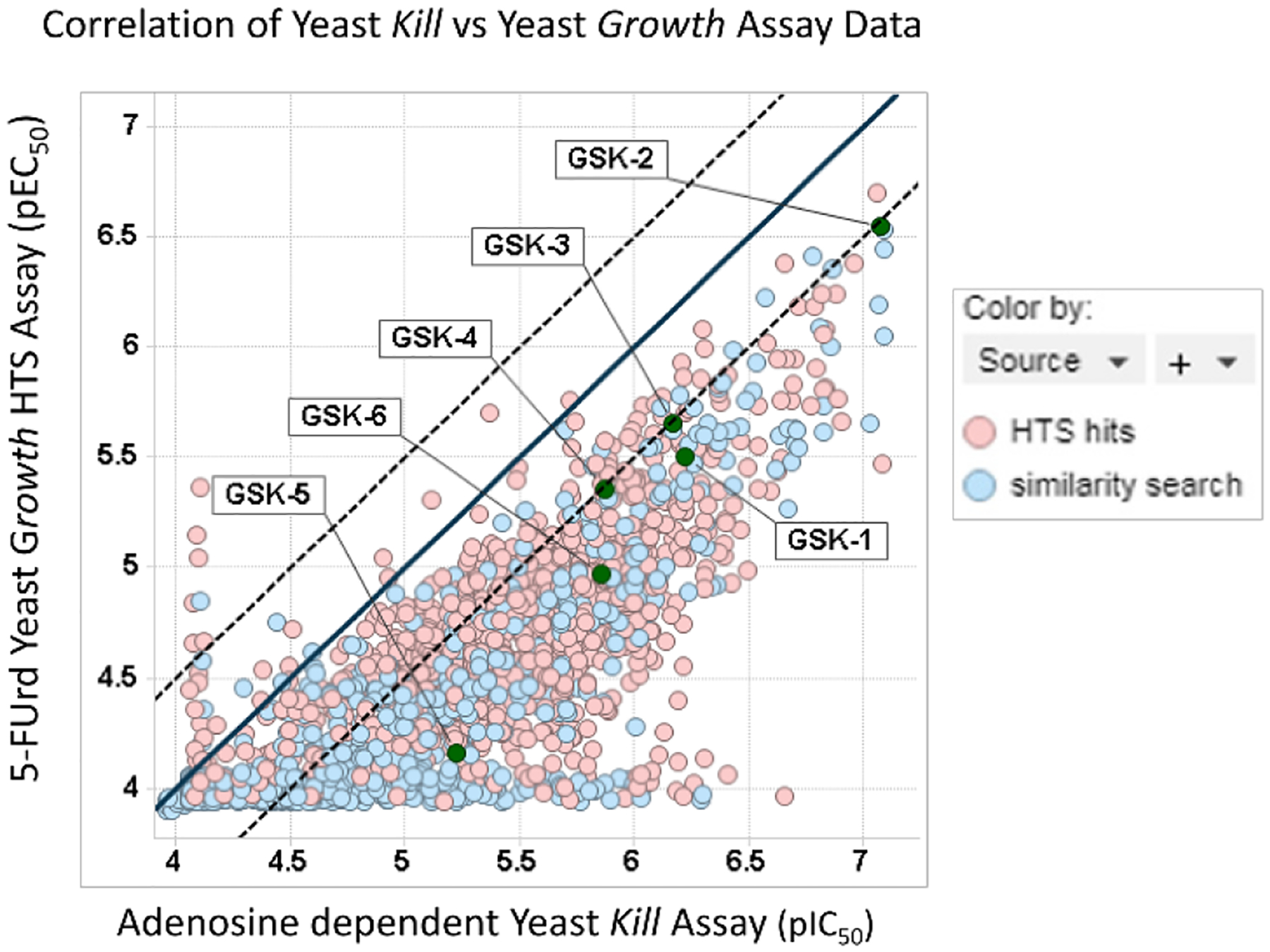
Scatterplot shows good activity correlation between the Yeast HTS Growth Assay (pEC50) and the orthogonal Yeast Kill Assay (pIC50). The unity line (x=y) is shown as a black line and parallel dotted lines represent ± 0.5 log units. The source of the hit is identified by color highlight (HTS hit, red; scaffold-based substructure search, blue). Labels highlight extensively characterized compounds GSK-1 through GSK-6.
The correlation plot also highlights the effectiveness of the scaffold-based substructure search strategies that identified numerous additional actives throughout the observed potency range (Figure 8, HTS hits, red symbols; scaffold-based substructure search, blue symbols). These additional analogs helped, in some cases, to identify active congeners with improved physicochemical properties. Labels also highlight the locations of the extensively characterized compounds described in more detail below (GSK-1 through GSK-6). Using available data (i.e. potency, ligand efficiency, physical properties, SAR trends and structural liabilities), medicinal chemists then selected and forwarded a set of 123 additional compounds to the Akabas lab for further characterization in downstream assays as “Wave 2” set of compounds. To expedite testing the Wave 2 compounds in the P. falciparum parasite proliferation assay, none of the 123 hit expansion Wave 2 compounds were tested for effects on HepG2 cell viability.
Further characterization of Wave 1 and Wave 2 compound sets.
The parallel hit progression strategy yielded a total of 324 compounds. The “Wave 1” compounds were comprised of the top 201 HTS hits that showed little or no toxicity to mammalian HepG2 cells. The “Wave 2” hit expansion compounds contained compounds identified by scaffold-based substructure searches (i.e. GSK-5).
Activity of Wave 1 compounds in yeast-based assays.
For each compound, the potency for inhibition of [3H]adenosine uptake into PfENT1-expressing yeast in a 15 minute assay was determined. The calculated compound IC50 values ranged from 2.8 nM to no effect at 25 μM, the highest compound concentration tested (Supplementary Table S2). Fourteen of the 201 compounds (7%) either did not cause any inhibition or did not cause sufficient inhibition of [3H]adenosine uptake to permit calculation of an IC50 value.
We also validated the potency of the 201 Wave 1 compounds in the Yeast Kill Assay in the Akabas lab. The IC50 values ranged from 1.2 nM to no effect at 25 μM, the highest concentration tested (Supplementary Table S2). Four of the 201 compounds had no effect at a 25 μM compound concentration. One of these compounds had no effect in both the [3H]adenosine uptake assay and in the Yeast Kill Assay. Perhaps this compound may have affected the binding and/or transport of 5-FUrd, but not adenosine.
The potency of the compounds in the yeast growth assays is complicated by the presence in the growth media of competing substrates for the PfENT1 transporter, 5-FUrd in the case of the Yeast HTS Growth Assay and adenosine in the case of the Yeast Kill Assay. We utilized a more direct assay of inhibitor function, inhibition of [3H]adenosine uptake into PfENT1-expressing yeast to further characterize the potency of the hits and their mechanism of action. The IC50 values for each of the 201 compounds in the [3H]adenosine uptake assay and in the Yeast Kill Assay are plotted in Figure 9. The scatter plot shows a lack of correlation between potency of individual compounds in the two assays. The explanation for the lack of correlation is uncertain because both assays require inhibition of adenosine transport via PfENT1 expressed in the yeast. However, there are many differences between the two assays that could provide some explanation. Both the adenosine concentration and the duration of the assays are quite different. For PfENT1, the EC50 for adenosine uptake is 650 μM.13 The adenosine concentration differs by 20,000 fold in the two assays, at 50 nM in the [3H]adenosine uptake assay and 1 mM in the Yeast Kill Assay. For some of the compounds, adenosine may compete for a binding site, especially in the Yeast Kill Assay. In the [3H]adenosine uptake assay the adenosine concentration is comparable to or lower than the compound IC50 values, whereas in the Yeast Kill Assay the adenosine concentration is orders of magnitude greater than the compound IC50 values. We did not investigate the dependence of the compound IC50 values on the adenosine concentration in either assay. Additionally, the duration of the assays differs 76-fold, 15 minutes vs 19 hours. Another difference is that in the [3H]adenosine uptake assay the buffer pH is 7.4, while the pH of yeast growth media is ~4.5. We do not think that the pH difference had a major effect on the transporter, because the transporter is not significantly pH dependent over the range from 5 to 8.13 However, some of the compounds might have differences in protonation state in this pH range that might contribute to the differences in potency in the two assays. Finally, one assay measured the direct uptake of radio-labeled adenosine, while the other assay measured the impact of inhibition of adenosine uptake on cell growth. Given these different endpoints, and the involvement of many processes in cell growth, one would not necessarily expect the two assays to correlate perfectly.
Figure 9.
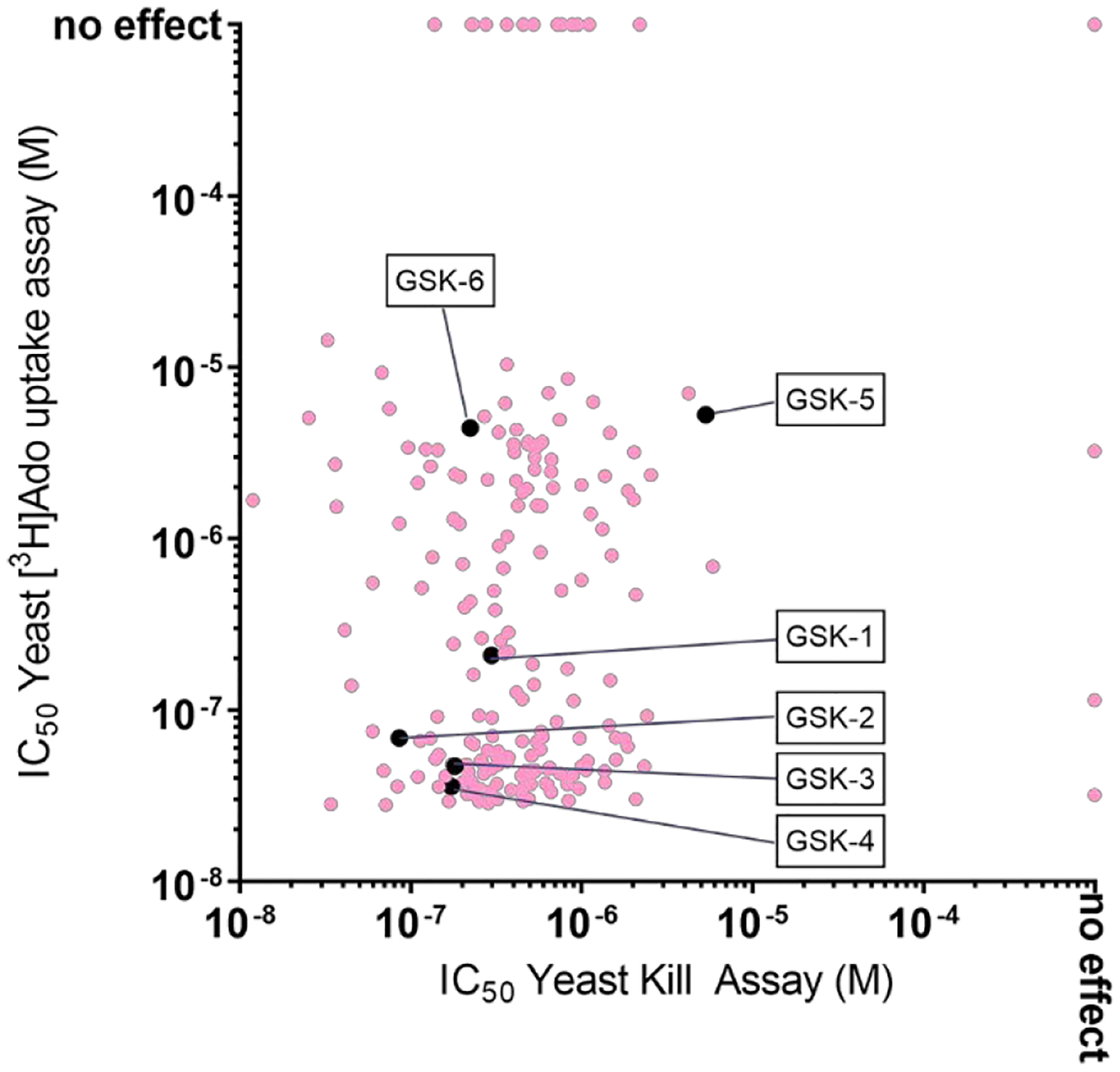
Scatterplot of the potency of Wave 1 compounds in the [3H]adenosine uptake inhibition and Yeast Kill Assays. For each of the 201 HTS hits, the IC50 value in the 15 minute inhibition of [3H]adenosine uptake assay (y-axis) is plotted as a function of the IC50 value in the 19 hr Yeast Kill Assay (x-axis). Compounds for which there was no effect at the highest concentration tested, 100 μM, are plotted at ”no effect”. The pale red circles indicate the blinded hits. The black symbols indicate the six extensively characterized compounds. GSK-5 was identified during hit expansion.
Inhibition of P. falciparum parasite proliferation by Wave 1 compounds.
We tested the ability of the 201 compounds to inhibit P. falciparum parasite proliferation using a 48 hour in vitro culture cytotoxicity assay. Compounds were tested over concentrations ranging from 34 nM to 75 μM. Two parasite strains were used, the wild type 3D7 reference strain, and a PfENT1-knockout strain, pfent1Δ, in the Dd2 strain background.23 Knockout of the gene encoding PfENT1 is conditionally lethal for P. falciparum parasites in culture. The pfent1Δ parasites are not viable at purine concentrations found in human blood (below 10 μM hypoxanthine).20 However, the pfent1Δ parasites can proliferate in media that contains greater than 50 μM hypoxanthine.21–23
The IC50 values for the 201 compounds against the two parasite strains are shown in Figure 10A and in Supplementary Table S2. The hits could be subdivided into four categories based on whether or not they had an effect or no effect on WT and pfent1Δ parasites (Figure 10B). For the 3D7 parasites, 175 compounds inhibited proliferation with IC50 values that ranged from 0.84 μM to 186 μM (Figure 10A and in Supplementary Table S2). Twenty-seven compounds either had no effect (NE) on parasite growth even at the highest concentration tested, 75 μM or an effect insufficient to calculate an IC50 value (low activity) (Supplementary Table S2). The fact that some compounds were active in the yeast-based assays but were not active against malaria parasites may relate to the compound’s ability to cross the RBC cell membrane. In the yeast-based assays the compounds may have direct access to PfENT1 in the yeast cell plasma membrane. In contrast, in the parasite growth assay, the compounds must cross the RBC plasma membrane and the parasitophorous vacuole membrane to reach PfENT1 in the parasite plasma membrane. Similarly, the compounds are subject to effects of RBC cytosol, such as binding to other proteins or chemical modification.
Figure 10.
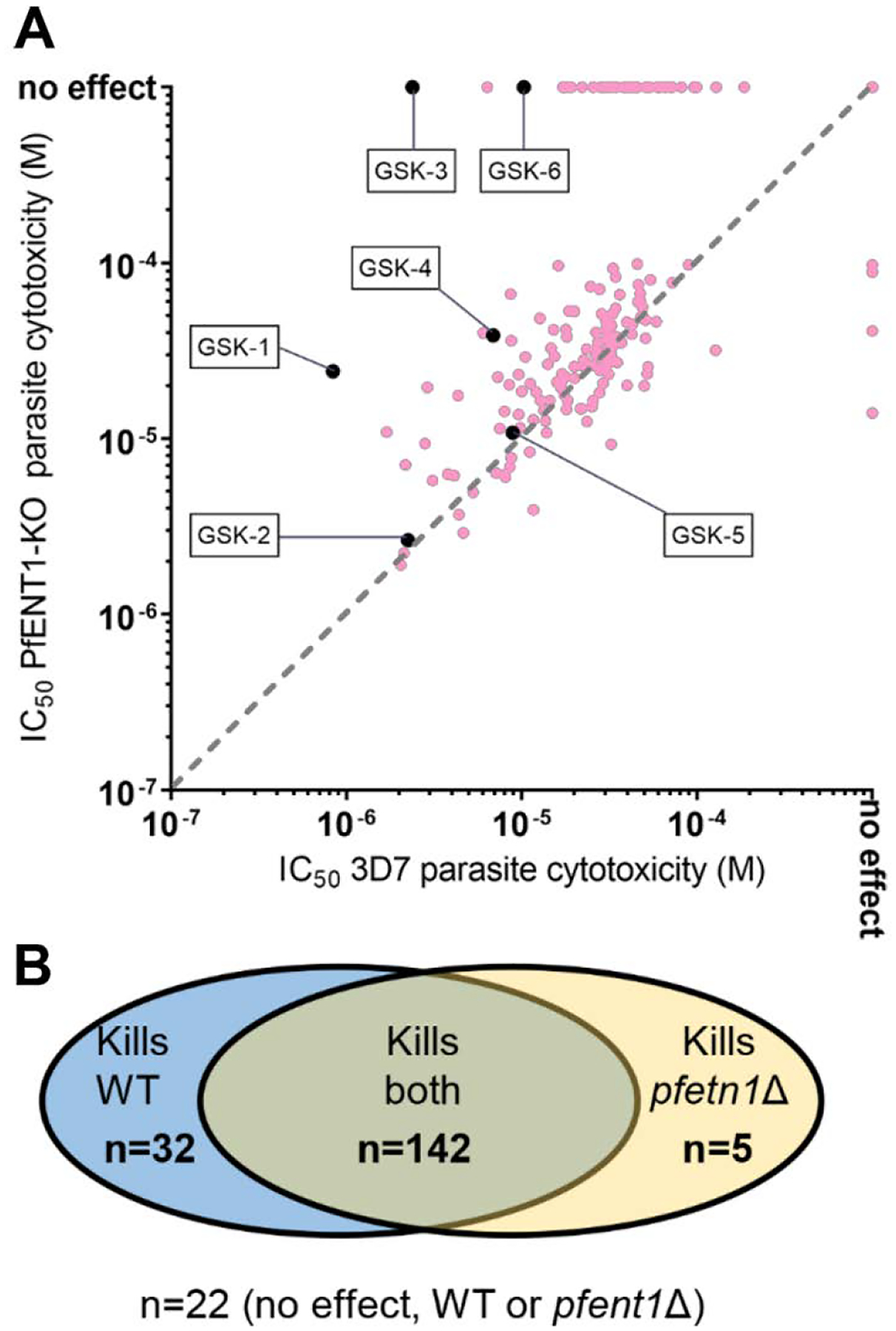
Comparison of the potency of the 201 compounds to inhibit proliferation of WT 3D7 parasites as compared to pfent1Δ parasites. (A) For each of the 201 HTS hits, the IC50 value for inhibition of proliferation of the PfENT1 knockout parasites (y-axis) is plotted as a function of the IC50 value for inhibition of proliferation of wild-type 3D7 strain P. falciparum parasites (x-axis). Compounds for which there was no effect at the highest concentration tested, 75 μM, are plotted at ”no effect”. The pale red circles indicate blinded hits. The black symbols indicate the six extensively characterized compounds. GSK-5 was identified during hit expansion. Note that each circle may represent multiple hits with identical IC50 values in the two assays. For example, the point at IC50 = 10−3 M in both assays, i.e., no effect, represents 22 hits. One compound was not tested in pfent1Δ parasites and is not included in the figure. (B) Comparison of the effect of the 201 HTS hits on wild type (WT) vs pfent1Δ parasites. For the purposes of this figure, if a compound had a measurable IC50 value, it was assumed to inhibit parasite proliferation. If a compound had no effect up to the maximum tested concentration (75 μM), it was considered to have no effect. n = number of compounds in each category. One compound was not tested in pfent1Δ parasites and was not included in the figure.
One hundred forty-seven compounds inhibited proliferation of PfENT1-knockout parasites with IC50 values that ranged from 1.91 μM to 359 μM (Supplementary Table S2). Fifty-four compounds had no effect (or insufficient activity to calculate an IC50 value) on pfent1Δ parasite proliferation. Of these 54 compounds, 32 compounds inhibited proliferation of WT 3D7 parasites with IC50 values that ranged from 2.4 μM to 186 μM (Supplementary Table S2). Thus, a subset of the compounds inhibited proliferation of 3D7 wild-type parasites, but did not inhibit proliferation of pfent1Δ parasites. This implies that they inhibited 3D7 parasite proliferation by inhibition of purine uptake via PfENT1. This supports the hypothesis that PfENT1 may be an effective therapeutic target for the development of novel antimalarial drugs.
Twenty-seven compounds had no effect on the growth of 3D7 parasites. Of these, 22 had no effect on pfent1Δ parasites and five inhibited proliferation of pfent1Δ parasites (Figure 10 and Supplementary Table S2). As noted above, the failure to act in the parasite-based assays may be due to limited membrane permeability. The five compounds that did not inhibit proliferation of 3D7 parasites but did inhibit proliferation of pfent1Δ parasites, presumably inhibited proliferation of pfent1Δ parasites through interactions with a secondary target(s) that becomes essential in the PfENT1-knockout parasites. The secondary target(s) is/are unknown.
It is notable that most of the compounds inhibited the growth of both 3D7 and pfent1Δ parasites. Figure 10 plots the IC50 values for each compound for 3D7 and pfent1Δ parasites. The compounds are not intrinsically cytotoxic, at least for yeast and human HepG2 cells. The fact that some compounds inhibited proliferation of pfent1Δ parasites that lack PfENT1 suggests that they also interact with other parasite targets. At this time, the identity of those secondary targets are unknown, but might include other parasite purine transporters such as PfENT4 or the AMP transport pathway.19, 24
Of the 132 compounds with no HepG2 cell cytotoxicity, 107 were active against 3D7 parasites and 25 had no effect on 3D7 parasite proliferation. Sixty-six of the 201 compounds were cytotoxic for HepG2 cells with IC50 values between 7.9 and 126 μM (Supplementary Table S2). Of these 66 compounds with measurable HepG2 cell toxicity, 65 were active against 3D7 parasites and one had no effect on 3D7 parasite growth. Thirteen of these compounds inhibited HepG2 cell growth more potently than they inhibited 3D7 parasite proliferation.
Validation of Wave 2 set.
Scaffold-based substructure search strategies and the orthogonal Yeast Kill Assay results provided an additional set of putative PfENT1 inhibitors (Wave 2). To expedite the characterization of these compounds, none were tested for effects on HepG2 cell growth. This set of 123 compounds was also sent to the Akabas lab for further characterization. We tested the potency of these additional compounds to inhibit P. falciparum 3D7 parasite proliferation. After 48 hours of growth in the presence of the compounds, IC50 values could be calculated for 82 of the compounds. The IC50 values ranged from 4.3 μM to 262 μM (Supplementary Table S3). The other 41 compounds did not sufficiently inhibit parasite proliferation at 75 μM to allow us to calculate an IC50 value (Supplementary Table S3).
We also compared the IC50 values for the Wave 1 and 2 compound sets in the 3D7 parasite cytotoxicity assay and in the Yeast Kill Assay (Supplementary Figure S2). There was little correlation between the IC50 values in the two assays. There are many differences between the assays including media pH, need for compounds to cross the red blood cell membrane to reach PfENT1 in the parasite assay, potential differences in glycosylation, phosphorylation, and interactions with other proteins. Despite all of these differences between the yeast and parasite assays, most of the compounds are active in both assays.
Extensive characterization of six selected compounds.
Six compounds were selected for more in depth profiling. The selected compounds had more optimal physicochemical properties based on potency, ligand efficiency considerations, physical properties, SAR trends, and availability. Each compound represented one of six prominent hit families. The chemical structures of the representative compounds are shown in Figure 11. Structure confirmation data for each compound (1H & 13C nuclear magnetic resonance spectra) are provided in the Supplementary Information.
Figure 11.
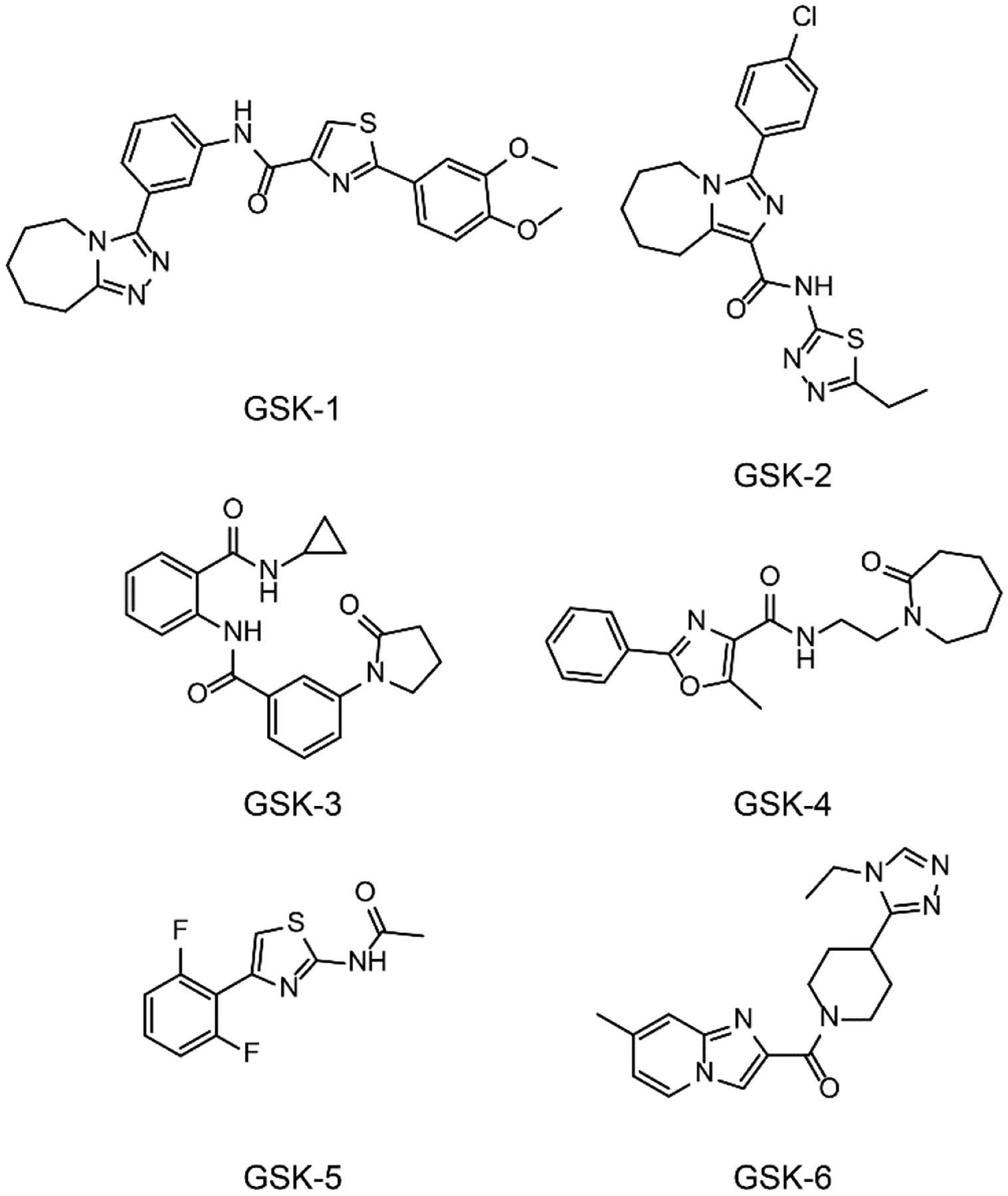
Chemical structures of the six HTS hits that were extensively characterized in multiple assays in this work. GSK-1, 2-(3,4-Dimethoxyphenyl)-N-[3-(6,7,8,9-tetrahydro-5H-[1,2,4]triazolo[4,3-a]azepin-3-yl)phenyl]-1,3-thiazole-4-carboxamide. GSK-2, 3-(4-Chlorophenyl)-N-(5-ethyl-1,3,4-thiadiazol-2-yl)-6,7,8,9-tetrahydro-5H-imidazo[1,5-a]azepine-1-carboxamide. GSK-3, N-Cyclopropyl-2-{[3-(2-oxo-1-pyrrolidinyl)benzoyl]amino}benzamide. GSK-4, 5-Methyl-N-[2-(2-oxo-1-azepanyl)ethyl]-2-phenyl-1,3-oxazole-4-carboxamide. GSK-5, N-[4-(2,6-Difluorophenyl)-1,3-thiazol-2-yl]acetamide. GSK-6, [4-(4-Ethyl-4H-1,2,4-triazol-3-yl)-1-piperidinyl](7-methylimidazo[1,2-a]pyridin-2-yl)methanone.
The EC50 and IC50 values in the yeast based assays are shown in Table 2. The EC50 and IC50 values in the two yeast-based assays were consistently different. The EC50 values in the primary screen Yeast HTS Growth Assay, were higher than the IC50 values in the secondary, Yeast Kill Assay. This is due to the difference in the stringency of the assay conditions. The 60 μM 5-FUrd concentration used in the HTS assay was effectively an IC100 concentration for inhibition of yeast proliferation, which was the basis of the assay robustness. However, it limited the sensitivity of the assay. We noted this difference in sensitivity of the two yeast-based growth assays in our original paper.23
Table 2.
EC50 and IC50 values for the six selected compounds in various yeast, parasite, red blood cell, and physicochemical assays.a
| Compounds | ||||||
|---|---|---|---|---|---|---|
| GSK-1 | GSK-2 | GSK-3 | GSK-4 | GSK-5 | GSK-6 | |
| Yeast HTS Growth Assay-GSK (μM) | 3.16 | 0.25 | 2.51 | 3.98 | 63.1 | 10 |
| Yeast Kill Assay-GSK (μM) | 0.63 | <0.08 | 0.63 | 1.26 | 6.31 | 1.26 |
| Yeast Kill Assay-Akabas lab (μM) | 0.54 ± 0.20 | 0.11 ± 0.04 | 0.46 ± 0.30 | 0.43 ± 0.30 | 4.0 ± 1.3 | 1.2 ± 1.1 |
| PfENT1 [3H]Ado uptake (μM) | 0.12 ± 0.08 | 0.03 ± 0.03 | 0.03 ± 0.01 | 0.02 ± 0.01 | 2.59 ± 2.36 | 3.98 ± 1.35 |
| PvENT1 [3H]Ado uptake (μM)b | 0.14 ± 0.07 | 0.04 ± 0.03 | 0.04 ± 0.01 | 0.02 ± 0.04 | 2.71 ± 0.25 | NEd |
| PbENT1 [3H]Ado uptake (μM)c | 0.13 ± 0.05 | 0.05 ± 0.03 | 0.07 ± 0.04 | 0.01 ± 0.01 | 3.09 ± 2.70 | NEd |
| 3D7 (μM) | 0.78 ± 0.2 | 2.25 ± 0.4 | 2.38 ± 0.5 | 6.20 ± 1.9 | 8.72 ± 1.6 | 10.3 ± 4.7 |
| PfENT1-KO (μM) | 17 ± 4.8 | 2.66 | Low activity/NEe | 29.4 ± 0.5 | 10.8 | NEe |
| Dd2 (μM) | 0.11 ± 0.009 | 1.89 ± 0.2 | 0.59 ± 0.05 | 1.90 ± 0.4 | 10.6 ± 2.1 | 3.3 ± 1.7 |
| HB3 (μM) | 0.34 ± 0.1 | 3.55 ± 0.7 | 2.21 ± 0.6 | 6.16 ± 2.5 | 9.09 ± 4.5 | 19.7 ± 4.1 |
| 7G8 (μM) | 0.48 ± 0.2 | 4.52 ± 0.1 | 6.08 ± 1.3 | 8.44 ± 2.4 | 16.1 ± 5.5 | 19.0 ± 3.5 |
| Cam3.II Artemisinin resistant (μM) | 0.16 ± 0.09 | 1.37 ± 0.2 | 0.79 ± 0.2 | 3.02 ± 1.6 | 8.37 ± 1.4 | 6.07 ± 1.0 |
| hENT1 [3H]adenosine uptake into RBC (μM) | 0.99 ± 0.81 | 24.5 ± 24.8 | 72.4 ± 19.3 | 51.0 ± 27.7 | Low activity/NEd | NEd |
| hFNT [3H]hypoxanthine uptake into RBC (μM) | 0.38 ± 0.1 | NEd | 9.93 ± 4.1 | NEd | 3.54 ± 1.5 | NEd |
| HepG2 cell cytotoxicity (μM)f | >100 | >100 | >100 | >100 | >100 | >100 |
| Molecular weight | 475 | 402 | 363 | 341 | 254 | 338 |
| Solubility (μg/mL) | 10.5 | 2.5 | 112 | 156 | 16 | >133 |
| ChromLogDpH7.4g | 4.8 | 6.8 | 4 | 4.5 | 3.7 | 1.2 |
| PFIh | 8.8 | 9.6 | 6 | 6.5 | 5.7 | 4.2 |
| HSA Column bindingi | 94% | 97% | 84% | 92% | 90% | 48% |
Values are mean ± SD. N ≥ 3 for all experiments except for the “5-FUrd growth assay-GSK” and the “Ado dependent growth-GSK columns”, where N=2, and for the PfENT1-KO experiments with compounds GSK-2 and GSK-5, where N=1.
PvENT1, yeast expressing the P. vivax ENT1 homologue
PbENT1, yeast expressing the mouse malaria model P. berghei ENT1 homologue
NE, no effect at the maximum concentration tested, 100 μM. Low activity/NE, indicates that at the highest concentration tested there seemed to be an effect, but it was not sufficient to calculate an IC50 value.
NE, no effect at the maximum concentration tested, 74.2 μM. Low activity/NE, indicates that at the highest concentration tested there seemed to be an effect, but it was not sufficient to calculate an IC50 value.
For the HepG2 cell cytotoxicity data, and >100 indicates no cytotoxicity up to 100 μM.
HSA, human serum albumin.61
We tested the ability of the compounds to inhibit the uptake of [3H]adenosine into PfENT1-expressing yeast. This assay provides a more direct test of the function of the identified compounds relative to the 19 hour growth assays. It also allows a more straightforward comparison of the potency of the compounds against homologous transporters from P. vivax and P. berghei expressed in yeast. These experiments involved 15 minute uptake experiments as opposed to 19 hour growth experiments. The six compounds inhibited [3H]adenosine uptake with IC50 values between 0.02 and 3.98 μM (Table 2). As discussed above the EC50 and IC50 values in the yeast-based growth assays were mostly higher than the IC50 values in the [3H]adenosine uptake assays.
We also tested the efficacy of the compounds on the homologous ENT1 transporters PvENT1 from P. vivax, the most common cause of human malaria, and, PbENT1 from P. berghei, a widely used mouse malaria model. PvENT1 and PbENT1 are, respectively, 75% and 60% amino acid sequence identical to PfENT1.38, 39 PvENT1 and PbENT1 were expressed in the same purine auxotrophic yeast background as PfENT1. For five of the compounds, the IC50 values for inhibition of [3H]adenosine uptake were within a factor of 2 of the values obtained with PfENT1-expressing yeast. This suggests that these compounds should work on P. vivax and could be tested in the P. berghei mouse malaria model. In contrast, GSK-6 did not inhibit either PvENT1 or PbENT1 up to the highest concentration tested, 99 μM (Table 2). This suggests that non-conserved amino acids in PvENT1 and PbENT1 may inhibit GSK-6 binding. Only 54% of the amino acids are identical among these three ENT1 homologues.
Potency against other P. falciparum strains.
We used the PfENT1 amino acid sequence from the 3D7 parasite strain (PF3D7_1347200, http://plasmodb.org/plasmo/app/record/gene/PF3D7_1347200) in our yeast constructs.23, 40 The IC50 values for the six compounds with 3D7 strain parasites ranged from 0.78 μM to 10.3 μM (Table 2). Of note, none of the six compounds was identified among the 13,533 compounds in the Tres Cantos Antimalarial TCAMS dataset.41 However, it should be noted that in the TCAMS phenotypic screen, the parasite growth media contained 150 μM hypoxanthine which functions as a competitor and diminishes the efficacy of these PfENT1 inhibitors.
Genome sequencing of other parasite strains has identified non-synonymous polymorphisms that change the PfENT1 amino acid sequence. In HB3, PfENT1 has the mutations F36S and V129I, and the 7G8 strain has a Q284E mutation. The efficacy of the compounds was determined against several P. falciparum parasite strains including the drug sensitive strains 3D7 from Africa and HB3 from Central America, and various drug resistant strains, including Dd2 from Southeast Asia, 7G8 from South America, and an artemisinin-resistant strain (ARTR) from Southeast Asia, Cam3.II.42 For all of these P. falciparum strains, the IC50 values were within a factor of three of those obtained with 3D7 parasites (Table 2). In general, the Dd2 and ARTR strains were more sensitive to inhibition by the six compounds. The HB3 and 7G8 strains were slightly less sensitive to several of the compounds. Thus, resistance to other antimalarial medicines does not appear to affect the parasite cytotoxic efficacy of PfENT1 inhibitors. Furthermore, it does not appear that these non-synonymous polymorphisms have a major impact on the compound potency values.
Efficacy against PfENT1 knockout (PfENT1-KO) parasites.
We used a pfent1Δ knockout parasite line constructed in the Dd2 background to test whether inhibition of PfENT1 was necessary and sufficient to kill P. falciparum parasites in culture.23 We measured the ability of the six compounds to inhibit proliferation of pfent1Δ knockout parasites (Figure 12). GSK-6 had an IC50 value of 1.89 μM against Dd2 parasites. In contrast, GSK-6 had no effect on the proliferation of PfENT1-KO parasites up to100 μM, the maximum concentration tested (Figure 12, Table 2). Furthermore, for GSK-1, GSK-3 and GSK-4, the IC50 values were 10- to 100-fold higher for the PfENT1-KO parasites compared to the IC50 values for Dd2 wild-type parasites (Figure 12, Table 2). This provides definitive evidence that inhibition of PfENT1 is sufficient to kill P. falciparum parasites in culture.
Figure 12.
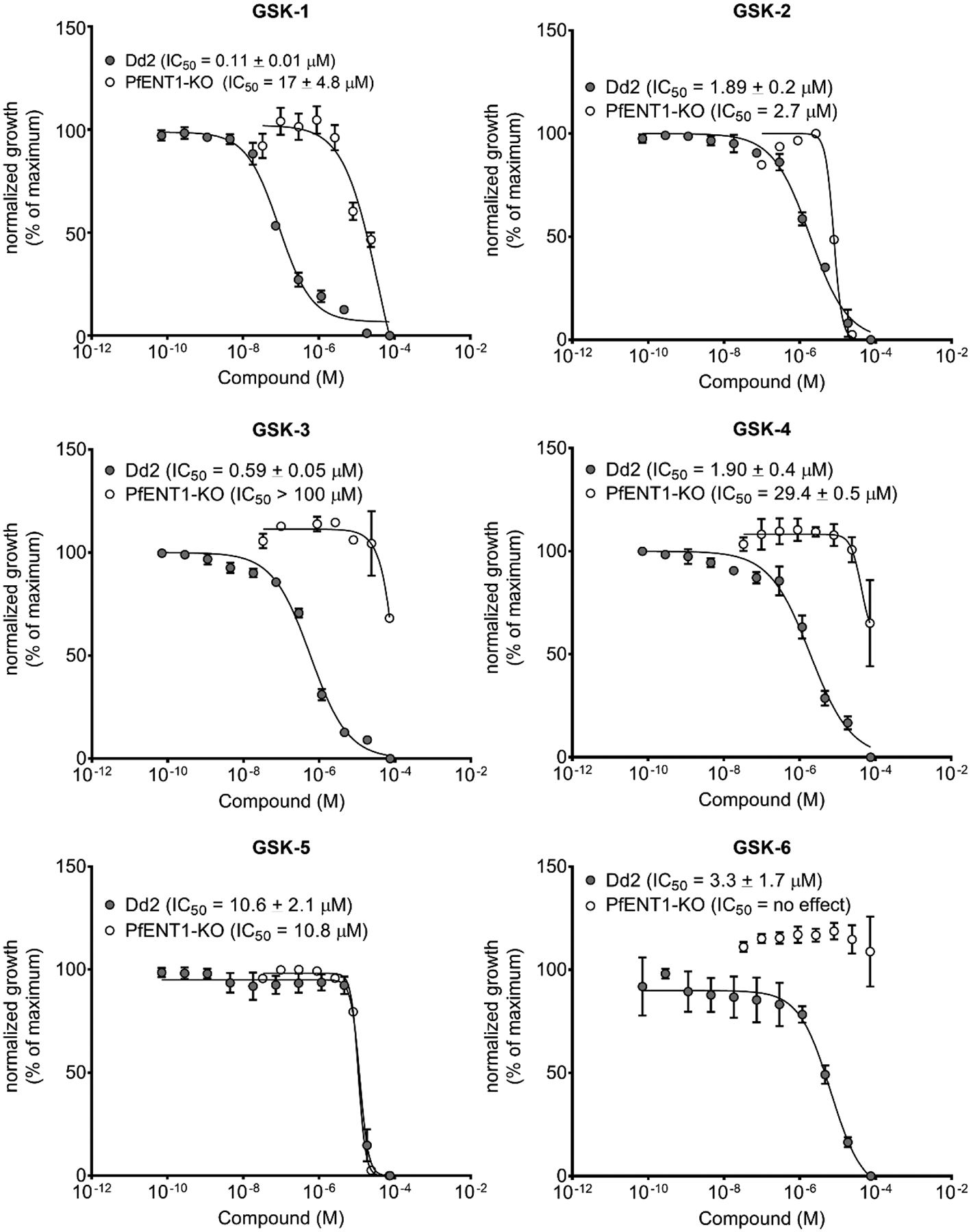
Normalized parasite growth inhibition effects for the six HTS hits, GSK-1 to GSK-6, for the inhibition of proliferation of Dd2 strain (filled circles) and PfENT1-knockout (open circles) parasites.
Interestingly, GSK-2 and GSK-5 had similar IC50 values against both Dd2 wild-type and PfENT1-KO parasites (Table 2). Both GSK-2 and GSK-5 inhibited PfENT1 in the yeast-based assays, so each must also hit one or more secondary targets in P. falciparum parasites with similar potency as their interaction with PfENT1. We had previously observed that PfENT1 inhibitors identified in our preliminary screen also hit one or more secondary targets in the malaria parasite with similar potency.23 The identity of these secondary targets is unknown at present.
Efficacy against human red blood cell purine transporters.
Purines must enter red blood cells (RBCs) before they can be transported into the malaria parasites. Human RBCs have two purine transport pathways. Nucleoside transport is mediated by the human Equilibrative Nucleoside Transporter Type 1 (hENT1), which is homologous to PfENT1, but with only 17% amino acid sequence identity. Nucleobase transport is mostly mediated by an unknown gene product named the human facilitated nucleobase transporter (hFNT).16, 43–48 hENT1 mainly transports nucleosides and hFNT only transports nucleobases. The nucleobase hypoxanthine is present in human blood at a concentration below 10 μM.20 Adenosine is intermittently released as a local vasodilator and is removed from plasma by uptake into RBCs and conversion via purine salvage pathway enzymes to either hypoxanthine or AMP.49 We examined whether the six PfENT1 inhibitors affected the human RBC purine transporters. To assess the effects of the six compounds on these transporters, we measured the concentration dependent inhibition of 1) [3H]adenosine uptake into RBC to determine interactions with hENT1, and 2) [3H]hypoxanthine uptake into RBC to determine interactions with hFNT using assays described previously38.
Both GSK-1 and GSK-3 inhibited uptake of [3H]adenosine and [3H]hypoxanthine into RBCs in a concentration range similar to the range at which they killed parasites in culture (Table 2). In contrast, GSK-6 did not inhibit RBC uptake of either [3H]adenosine or [3H]hypoxanthine (Table 2). Furthermore, GSK-2 and GSK-4 did not inhibit RBC [3H]hypoxanthine uptake and GSK-5 did not inhibit [3H]adenosine uptake (Table 2). Given that hypoxanthine is the only purine in our parasite culture media, this implies that the parasiticidal effects of the PfENT1 inhibitors were due to inhibition of PfENT1, and not due to inhibition of hypoxanthine entry into the RBC. Further support for this conclusion is provided by the fact that the PfENT1 inhibitors had different IC50 values with different parasite strains (Table 2). The strain dependent IC50 values would not be expected if the compound target was the human RBC purine transporters, hFNT or hENT1. These results provide further support for the therapeutic hypothesis that PfENT1 is a viable target for the development of novel antimalarial drugs.
Of note, inhibition of hENT1 may not cause significant side effects. Dipyridamole, which targets hENT1, is an FDA approved drug that is used as an antiplatelet agent and vasodilator. In addition to other activities, it inhibits hENT1 with nanomolar affinity.50–55 Dipyridamole is extensively used clinically without significant adverse effects.56, 57
Human cell cytotoxicity.
We evaluated the toxicity of the six compounds on human hepatoma HepG2 cells. None of the six compounds displayed significant HepG2 cell cytotoxicity up to a concentration of 100 μM, the highest concentration tested (Table 2).
Physiochemical properties.
Several physiochemical properties are shown in Table 2. Compounds GSK-3, GSK-4 and GSK-6 have favorable aqueous solubility, ChromLogDpH7.4, PFI values, and human serum albumin binding parameters. The lower solubility of the other three, and the higher PFI values of GSK-1 and GSK-2 are challenges that medicinal chemistry hit optimization efforts will need to overcome.30
CONCLUSIONS
We conducted a high throughput screen of the GSK compound library using a yeast-based assay to identify inhibitors of PfENT1. We identified 966 hits that gave rise to dose-response curves in the Yeast Growth Assay. Our parallel hit progression strategy allowed us to: 1) focus on hits that had high activity and low toxicity; and 2) expand the chemical space to structural analogs that were not identified in the original screen. By using this strategy we limited the amount of testing on parasites and erythrocytes. The combination of our screen, hit progression strategy, and downstream assays lead to the identification of effective and diverse chemical scaffolds that are new starting points for optimization and development of novel antimalarial drugs. Key points for this work include:
A yeast-based HTS based on positive selection for yeast growth successfully identified inhibitors of PfENT1. The screen eliminated compounds that were intrinsically cytotoxic to yeast.
A subset of the compounds inhibited proliferation of WT parasites but not PfENT1-knockout parasites. These data imply that inhibition of PfENT1 is sufficient to inhibited proliferation of malaria parasites in culture supporting the hypothesis that PfENT1 is a viable therapeutic target for antimalarial drug development.
Three of the highlighted compounds inhibited proliferation of malaria parasites but did not inhibit hFNT. This result implies that the compounds act by inhibiting PfENT1 not by inhibiting the human RBC purine transporters. However, we cannot rule out that inhibition of these transporters may be beneficial for the efficacy of some of the compounds. Regarding potential side effects, some inhibition of hENT1, or hFNT, is probably not problematic for use in humans because dipyridamole is an FDA approved anti-platelet agent and vasodilator that inhibits hENT1 with a nanomolar IC50 value. Furthermore, all six characterized compounds have no toxic effects on human HepG2 cells.
The PfENT1 inhibitors displayed similar IC50 values against a variety of P. falciparum strains, including strains that are sensitive and strains that are resistant to chloroquine and artemisinin. This suggests that parasite strains resistant to other antimalarial drugs should not be resistant to PfENT1 inhibitors.
EXPERIMENTAL SECTION
Yeast strains.
The yeast strains used were as described previously.23 The background yeast strain was “fui1Δ” in the BY4741 background (genotype MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, fui1Δ::kanMX4) from the knock-out collection.58 A yeast codon optimized gene encoding PfENT1 was inserted into the FUI1 genomic locus by double homologous recombination to generate the fui1Δ::PfENT1 strain for the use in the primary screen Yeast HTS Growth Assay.
Purine auxotrophic yeast used in the orthogonal Yeast Kill Assay and for the yeast [3H]adenosine uptake experiments. They were constructed in the fui1Δ::kanMX4 background by disrupting the ADE2 gene using the hphNT1 marker from the pFA6a-hphNT1 plasmid (ade2Δ::hphNT1), which confers resistance to hygromycin B as described previously.23 PfENT1 expression in the purine auxotrophic yeast was obtained by transforming them with the episomal plasmid PfENT1-pCM189m. Yeast growth media was as described previously.23 The yeast strains expressing PvENT1 and PbENT1 were described previously.38, 39
5-FUrd Yeast media.
YPD media contained 1% (w/v) yeast extract, 2% (w/v) peptone, and 2% (w/v) dextrose.
Yeast were assayed in synthetic defined media (SDM) containing 0.67% (w/v) yeast nitrogen base (US Biologicals cat# Y2025), 0. 2% (w/v) drop-out mix (US Biologicals cat# D9515; without nitrogen base), 2% (w/v) dextrose.
High Throughput Screen.
The GSK Small Molecule Chemical Library was used in the HTS. The 1,792,272 compounds in the library were selected to provide maximum diversity and minimal false positives using the principles outlined in Chakravorty et al. (2018)29 (Supplementary Table S1). The HTS was performed in round-well, flat-bottom, optically clear Corning #3893 type 1536 well plates (Supplementary Table S1). Frozen yeast cells were thawed and grown overnight in YPD media at 30 °C, shaking at 200 RPM. OD620 was measured and the yeast were diluted in SDM to an OD620 = 0.05. CHAPS was added to give a final concentration of 0.5 mM. A fraction of the culture was removed to the maximal growth (no 5-FUrd) control wells. 5-FUrd was added to the remaining yeast culture to a final concentration of 60 μM. Each well was preloaded with 0.08 μL of 1 mM test compound in DMSO. The final volume of each test well was 8 μL (10 μM test compound, 1% DMSO v/v, 60 μM 5-FUrd, 0.5 mM CHAPS, in SDM), and ~8,000 cells. Plates were spun at 500 rpm for 1–2 min and allowed to equilibrate at room temperature (RT) for 30 min. Plates were placed in a humidified incubator at 30 °C for 16 h. Plates were removed from the incubator and allowed to equilibrate at RT for at least 30 min and then OD620 was measured using an EnVision plate reader (Model #2102). Negative control wells contained no test compound and represented the 5-FUrd inhibited growth baseline. Positive control wells contained no 5-FUrd or test compound and represented maximal growth.
Compounds were considered “robust actives” if the OD620 values obtained for a well were > 3 SD above the mean OD620 values of the mean sample population. Ultimately, hits were considered compounds with OD620 values >10 SD above the mean sample population, 28% of maximal growth. This assay is referred to as the Yeast HTS Growth Assay in the text.
Yeast HTS Growth Assay concentration dependence.
Concentration dependence of compounds was determined using the HTS assay conditions described above. The compounds were serially diluted 1:3 with 100 μM being the highest concentration tested. Assays were performed in duplicate. This assay is referred to as the Yeast HTS Growth Assay in the text. Because hits in this assay are identified by yeast growth, compound potency is referred to as EC50 or pEC50 values in the text.
Ade2Δ Yeast media.
Yeast were assayed in synthetic defined media (SDM) containing 0.67% (w/v) yeast nitrogen base (US Biologicals cat# Y2025), 0. 2% (w/v) drop-out mix (US Biologicals cat# D9517–04A; without histidine, leucine, adenine), 2% (w/v) dextrose (Sigma cat# G7021), 0.004% L-Histidine (Sigma cat# H8000), 0.004% L-Leucine (Sigma cat# L8000), 1 mM adenosine (Sigma cat# A9251).
Secondary Assay – Yeast Kill Assay.
Assay was performed in round-well, flat-bottom, optically clear Corning #3893 type 1536 well plates. Frozen yeast were thawed and grown overnight in SDM media, supplemented with 1 mM adenosine (Sigma cat# A9251) at 30 °C, shaking at 200 RPM. Following the overnight incubation, the culture was diluted by a factor of 2 using SDM media, supplemented with 1 mM adenosine and incubated at 30 °C for 2–3 additional hours. Following incubation, 10 mL of culture was removed to prepare low (no growth) control, centrifuged at 2000 rpm for 2 min, media aspirated, and replaced with SDM without adenosine. This wash procedure was repeated 2 additional times. OD620 was measured for low control and source cultures, and then diluted to an OD620 = 0.15 in SDM without adenosine and SDM with 1 mM adenosine respectively. CHAPS was added to both cultures to give a final concentration of 0.5 mM. Each assay well was preloaded with 0.1 μL of 1 mM test compound in DMSO. The final volume of each test well was 10 μL (10 μM test compound, 1% DMSO). Plates were centrifuged at 500 rpm for 1–2 min and allowed to equilibrate at room temperature (RT) for 30 min. Plates were placed in a humidified incubator at 30 °C for 24 h. Plates were removed from the incubator and allowed to equilibrate at RT for at least 30 min and then OD620 was measured using an EnVision plate reader (Model #2102). Low control wells contained SDM with no adenosine, 1% DMSO and represented no yeast growth. Positive control wells contained SDM with 1% adenosine, 1% DMSO and represented maximal growth.
Compounds were considered “hits” if the OD620 values obtained for a well were > 3 SD above the mean OD620 values of the negative control wells. This assay is referred to as the Yeast Kill Assay in the text. Because hits in this assay are identified by inhibition of yeast growth, compound potency is referred to as IC50 or pIC50 values in the text.
[3H]Adenosine uptake into PfENT1-expressing yeast.
The assay to measure the concentration dependence of compound inhibition of [3H]adenosine uptake was conducted as described previously.23 96-well plates were preloaded with 100 μL of 100 nM [3H]adenosine ([2,8-3H]adenosine, 35 Ci/mmol, Moravek Biochemicals, Brea, CA) and 0.5 μL compound serially diluted in DMSO such that the final compound concentrations ranged from 11 nM to 25 μM. 100 μL yeast (2 × 108 cells mL−1) were added to each well to give a final 50 nM [3H]adenosine concentration and incubated at RT for 15 min. After 15 min, yeast were collected on glass fiber filtermats (Filtermat, GF/C; Perkin Elmer, Waltham, MA) using a TomTec 96 well cell harvester (#96-3-469, Hamden, CT) and washed extensively. Filtermats were dried for > 1 h and sealed in plastic bags containing 5 ml Betaplate Scint LSC (Perkin Elmer). Filtermats were counted using 1450 Microbeta Trilux (Perkin Elmer).
HepG2 Cytotoxicity Assay.
Assay plates (Greiner cat# 781091) were pre-dispensed with 0.25 μL of 10 mM compound in DMSO. Actively growing HepG2 cells (ATCC cat# HB-8065) were removed from T-175 cm2 culture flasks (Corning cat# 431080) using 5 mL cell dissociation media (Gibco# 14190) and diluted in 500 mL assay media Eagles MEM (Gibco# 31095–029, 10% FBS Gibco# 10100), 1% non-essential amino acids (Gibco# 11140–035), 1% penicillin + Streptomycin (Gibco# 10082–147) to achieve a cell density of 1.2 × 105 cells/mL. 0.025 mL per well of cells were dispensed to multiwell assay plates, followed by incubation at 37 °C, 5% CO2 for 48 hours. Next CellTiter-Glo® (Promega cat# G7571), was prepared according to manufacturer’s instructions, and 0.025 mL of was added to each assay well. Following a 10-minute incubation at room temperature, the cell viability was measured on a plate reader equipped with luminescence detection. High and low assay controls were established using DMSO (100% survival) and 100 μM Digitoxin (Sigma cat# D5878; 0% survival) respectively and all data were normalized to these controls and expressed as percent response.
Characterization of physicochemical properties.
Kinetic solubility was determined by GSK in-house assay: 5 μL of 10 mM DMSO stock solution diluted to 100 μL with pH 7.4 phosphate buffered saline, equilibrated for 1 hr at RT, filtered through Millipore MultiscreenHTS-PCF filter plates (MSSL BPC). The eluent was quantified by suitably calibrated flow injection CLND (Chemiluminescent nitrogen detector). The ChromoLogD7.4 and HSA measurements were determined using methodology that has been previously described.59–61
P. falciparum growth and cytotoxicity assays.
3D7 strain parasites were grown in erythrocytes (RBCs) acquired from healthy human volunteers with consent (Albert Einstein College of Medicine IRB protocol #2013–2227). The parasites were maintained in continuous culture at 4% hematocrit in complete malaria culture medium (MCM): one liter contained: 10.4 g RPMI 1640 (Gibco# 31800–014), 11 mM glucose, 27 mM NaHCO3, 25 mM HEPES, 0.5% Albumax-II (w/v) (Gibco# 11021–037), 20 mg gentamicin, 10 μM hypoxanthine, pH 7.4.23 pfent1Δ parasites were grown in the same medium except supplemented with 75 μM hypoxanthine.23 Cultures were gassed with 90% N2/5% CO2/5% O2 and grown under continuous agitation at 37 °C. Compounds were diluted 1:3 and the concentrations ranged from 34 nM to 75 μM. The DMSO concentration was <1%. P. falciparum growth was quantified using SYBR Green I (Thermo-Fisher Scientific #S7585) nucleic acid stain.62
Calculation of EC50 or IC50 values in all assays.
EC50 or IC50 values were calculated using GraphPad Prism log(inhibitor) vs. response - Variable slope (four parameters) function, Y=Ymax/(1+10^((LogXC50-[X])*nH)) where Y = % of maximal signal, Ymax = 100% or maximal signal, XC50 is either EC50 or IC50 depending on the assay, [X] = concentration of compound X, nH = Hill slope.
Human RBC purine uptake assays.
Assays for inhibition of uptake of [3H]adenosine into human RBCs to test for effects on hENT1 were performed as described previously38. For the [3H]hypoxanthine uptake assay, 100 μL of Silicone oil (AR200; Fluka; density 1.049) was pre-loaded into 0.2 mL strip PCR tubes (Fischer Scientific). 96-well plates (clear flat-bottom 96-well polystyrene, nonsterile plate; Fischer Scientific) were preloaded with 2 μL compound (serially diluted 4-fold in DMSO from a 10 mM stock) and 100 μL of 400 nM [3H]hypoxanthine (Hypoxanthine monohydrochloride [3H(G)]; Perkin Elmer) in Ringers solution. Uninfected RBCs were washed five times with Ringers solution (123 mM NaCl, 4.93 mM KCl, 0.85 mM CaCl2, 1.23 mM MgSO4, 5 mM glucose, 5 mM glutamine, 10 mM HEPES, 10 mM MES, pH 7.4) and resuspended at 4% hematocrit in Ringers solution. 100 μL of RBCs (4% hematocrit) were added to each well containing compound and radiolabel, resuspended, and incubated at room temperature for 15 minutes. At the end of the time course contents of each well were resuspended 3 times, removed 175 μL and added it to the pre-loaded silicone oil PCR tubes. The PCR tubes were centrifuged at 16,300 x g for 30 seconds and supernatant including excess silicone was removed. The RBC pellet was resuspended 3 times with 100 μL of 5% SDS (w/v) for RBC lysis. Each PCR tube containing lysed RBCs was inserted into scintillation vials and mixed with 3 mL scintillation fluid (UltimaGold; Perkin Elmer). Samples were counted for 1 min each using a Wallac TriCarb liquid scintillation counter (Perkin Elmer).
Source of six extensively characterized compounds.
All six compounds are commercially available and were purchased from the indicated supplier. GSK-1, 2-(3,4-Dimethoxyphenyl)-N-[3-(6,7,8,9-tetrahydro-5H-[1,2,4]triazolo[4,3-a]azepin-3-yl)phenyl]-1,3-thiazole-4-carboxamide; (Enamine # Z115045922). GSK-2, 3-(4-Chlorophenyl)-N-(5-ethyl-1,3,4-thiadiazol-2-yl)-6,7,8,9-tetrahydro-5H-imidazo[1,5-a]azepine-1-carboxamide; (ChemDiv # E711–0141). GSK-3, N-Cyclopropyl-2-{[3-(2-oxo-1-pyrrolidinyl)benzoyl]amino}benzamide (Enamine # Z95754787). GSK-4, 5-Methyl-N-[2-(2-oxo-1-azepanyl)ethyl]-2-phenyl-1,3-oxazole-4-carboxamide (Enamine # Z407267740). GSK-5, N-[4-(2,6-Difluorophenyl)-1,3-thiazol-2-yl]acetamide (Enamine # Z384175608). GSK-6, [4-(4-Ethyl-4H-1,2,4-triazol-3-yl)-1-piperidinyl](7-methylimidazo[1,2-a]pyridin-2-yl)methanone (Enamine # Z1082942462).
NMR structural confirmation of six extensively characterized compounds.
The structures of all six compounds (GSK-1 to GSK-6) were confirmed by 1H and 13C NMR using both 1D and 2D methods (Supplementary Information).
Supplementary Material
Acknowledgements.
We thank Dr. David Fidock (Columbia Univ.) for the Artemisinin-resistant Cam3.II parasite strain and for the HB3 and 7G8 strains. Vanessa Barroso-Poveda (GSK) performed the HepG2 cytotoxicity assays. This work was supported in part by a grant from the National Institutes of Health NIAID RO1-AI116665 (MHA) and by an unrestricted grant from GSK to MHA to support work performed at Einstein. All work performed by GSK employees was supported by funds from GlaxoSmithKline. YS was supported in part by training grant T32-AI070117 and by NIH NRSA individual fellowship F31-AI136488. RD and IJF were supported in part by NIGMS Medical Scientist Training Grant T32-GM007288.
Abbreviations.
- 5-FUrd
5-flurouridine
- ACT
Artemisinin-based Combination Therapy
- Ado
adenosine
- ARTR
artemisinin-resistant P. falciparum strain
- EC50
effective concentration causing 50% of maximal effect
- ENT1
Equilibrative Nucleoside Transporter Type 1
- HTS
high throughput screen
- hENT1
human ENT1
- hFNT
human facilitated nucleobase transporter
- IC50
concentration causing 50% inhibition of maximum
- IFI
Inhibition Frequency Index
- OD620
optical density at 620 nm
- PbENT1
Plasmodium berghei ENT1
- PfENT1
Plasmodium falciparum ENT1
- PvENT1
Plasmodium vivax ENT1
- PfENT1-KO
PfENT1 knockout
- RBC
red blood cell
- SAR
structure-activity-relationships
- SD
standard deviation
- SDM
synthetic defined media
- WT
wild type
Footnotes
Supporting Information. Supporting Information includes tables with the EC50 and IC50 values in the yeast- and parasite-based assays for the 201 Wave 1 compounds and the 123 Wave 2 compounds identified during hit expansion, and NMR data to validate the structures of the six compounds.
References.
- (1).World Health Organization. (2016) WHO Global Malaria Programme: World Malaria Report 2016, p 186, World Health Organization, Switzerland. [Google Scholar]
- (2).Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han KT, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, Rahman MR, Hasan MM, Islam A, Miotto O, Amato R, MacInnis B, Stalker J, Kwiatkowski DP, Bozdech Z, Jeeyapant A, Cheah PY, Sakulthaew T, Chalk J, Intharabut B, Silamut K, Lee SJ, Vihokhern B, Kunasol C, Imwong M, Tarning J, Taylor WJ, Yeung S, Woodrow CJ, Flegg JA, Das D, Smith J, Venkatesan M, Plowe CV, Stepniewska K, Guerin PJ, Dondorp AM, Day NP, and White NJ (2014) Spread of artemisinin resistance in Plasmodium falciparum malaria, N Engl J Med 371, 411–423. 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Fairhurst RM (2015) Understanding artemisinin-resistant malaria: what a difference a year makes, Curr Opin Infect Dis 28, 417–425. 10.1097/qco.0000000000000199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Das S, Saha B, Hati AK, and Roy S (2018) Evidence of Artemisinin-Resistant Plasmodium falciparum Malaria in Eastern India, N Engl J Med 379, 1962–1964. 10.1056/NEJMc1713777 [doi]. [DOI] [PubMed] [Google Scholar]
- (5).Sutherland CJ, Lansdell P, Sanders M, Muwanguzi J, van Schalkwyk DA, Kaur H, Nolder D, Tucker J, Bennett HM, Otto TD, Berriman M, Patel TA, Lynn R, Gkrania-Klotsas E, and Chiodini PL (2017) pfk13-Independent Treatment Failure in Four Imported Cases of Plasmodium falciparum Malaria Treated with Artemether-Lumefantrine in the United Kingdom, Antimicrob Agents Chemother 61, e02382–02316. AAC.02382–16 [pii] 10.1128/AAC.02382-16 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lu F, Culleton R, Zhang M, Ramaprasad A, von Seidlein L, Zhou H, Zhu G, Tang J, Liu Y, Wang W, Cao Y, Xu S, Gu Y, Li J, Zhang C, Gao Q, Menard D, Pain A, Yang H, Zhang Q, and Cao J (2017) Emergence of Indigenous Artemisinin-Resistant Plasmodium falciparum in Africa, N Engl J Med 376, 991–993. 10.1056/NEJMc1612765 [doi]. [DOI] [PubMed] [Google Scholar]
- (7).Burrows JN, van Huijsduijnen RH, Mohrle JJ, Oeuvray C, and Wells TN (2013) Designing the next generation of medicines for malaria control and eradication, Malar J 12, 187 10.1186/1475-2875-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Katsuno K, Burrows JN, Duncan K, Hooft van Huijsduijnen R, Kaneko T, Kita K, Mowbray CE, Schmatz D, Warner P, and Slingsby BT (2015) Hit and lead criteria in drug discovery for infectious diseases of the developing world, Nat Rev Drug Discov 14, 751–758. 10.1038/nrd4683. [DOI] [PubMed] [Google Scholar]
- (9).Wells TN, Hooft van Huijsduijnen R, and Van Voorhis WC (2015) Malaria medicines: a glass half full?, Nat Rev Drug Discov 14, 424–442. 10.1038/nrd4573. [DOI] [PubMed] [Google Scholar]
- (10).Carter NS, Ben Mamoun C, Liu W, Silva EO, Landfear SM, Goldberg DE, and Ullman B (2000) Isolation and functional characterization of the PfNT1 nucleoside transporter gene from Plasmodium falciparum, J Biol Chem 275, 10683–10691. 10.1074/jbc.275.14.10683 [DOI] [PubMed] [Google Scholar]
- (11).Parker MD, Hyde RJ, Yao SY, McRobert L, Cass CE, Young JD, McConkey GA, and Baldwin SA (2000) Identification of a nucleoside/nucleobase transporter from Plasmodium falciparum, a novel target for anti-malarial chemotherapy, Biochem J 349, 67–75. 10.1042/0264-6021:3490067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Downie MJ, Saliba KJ, Howitt SM, Broer S, and Kirk K (2006) Transport of nucleosides across the Plasmodium falciparum parasite plasma membrane has characteristics of PfENT1, Mol Microbiol 60, 738–748. 10.1111/j.1365-2958.2006.05125.x [doi]. [DOI] [PubMed] [Google Scholar]
- (13).Riegelhaupt PM, Cassera MB, Frohlich RF, Hazleton KZ, Hefter JJ, Schramm VL, and Akabas MH (2010) Transport of purines and purine salvage pathway inhibitors by the Plasmodium falciparum equilibrative nucleoside transporter PfENT1, Mol Biochem Parasitol 169, 40–49. 10.1016/j.molbiopara.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Downie MJ, Kirk K, and Mamoun CB (2008) Purine salvage pathways in the intraerythrocytic malaria parasite Plasmodium falciparum, Eukaryot Cell 7, 1231–1237. 10.1128/ec.00159-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ducati RG, Namanja-Magliano HA, and Schramm VL (2013) Transition-state inhibitors of purine salvage and other prospective enzyme targets in malaria, Future Med Chem 5, 1341–1360. 10.4155/fmc.13.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Frame IJ, Deniskin R, Arora A, and Akabas MH (2015) Purine import into malaria parasites as a target for antimalarial drug development, Ann N Y Acad Sci 1342, 19–28. 10.1111/nyas.12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Martin RE, Henry RI, Abbey JL, Clements JD, and Kirk K (2005) The ‘permeome’ of the malaria parasite: an overview of the membrane transport proteins of Plasmodium falciparum, Genome Biol 6, R26 10.1186/gb-2005-6-3-r26 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Downie MJ, El Bissati K, Bobenchik AM, Nic Lochlainn L, Amerik A, Zufferey R, Kirk K, and Ben Mamoun C (2010) PfNT2, a permease of the equilibrative nucleoside transporter family in the endoplasmic reticulum of Plasmodium falciparum, J Biol Chem 285, 20827–20833. 10.1074/jbc.M110.118489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Frame IJ, Merino EF, Schramm VL, Cassera MB, and Akabas MH (2012) Malaria parasite type 4 equilibrative nucleoside transporters (ENT4) are purine transporters with distinct substrate specificity, Biochem J 446, 179–190. 10.1042/bj20112220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Traut TW (1994) Physiological concentrations of purines and pyrimidines, Mol Cell Biochem 140, 1–22. [DOI] [PubMed] [Google Scholar]
- (21).El Bissati K, Zufferey R, Witola WH, Carter NS, Ullman B, and Ben Mamoun C (2006) The plasma membrane permease PfNT1 is essential for purine salvage in the human malaria parasite Plasmodium falciparum, Proc Natl Acad Sci U S A 103, 9286–9291. 10.1073/pnas.0602590103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).El Bissati K, Downie MJ, Kim SK, Horowitz M, Carter N, Ullman B, and Ben Mamoun C (2008) Genetic evidence for the essential role of PfNT1 in the transport and utilization of xanthine, guanine, guanosine and adenine by Plasmodium falciparum, Mol Biochem Parasitol 161, 130–139. 10.1016/j.molbiopara.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Frame IJ, Deniskin R, Rinderspacher A, Katz F, Deng SX, Moir RD, Adjalley SH, Coburn-Flynn O, Fidock DA, Willis IM, Landry DW, and Akabas MH (2015) Yeast-based high-throughput screen identifies Plasmodium falciparum equilibrative nucleoside transporter 1 inhibitors that kill malaria parasites, ACS Chem Biol 10, 775–783. 10.1021/cb500981y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Cassera MB, Hazleton KZ, Riegelhaupt PM, Merino EF, Luo M, Akabas MH, and Schramm VL (2008) Erythrocytic adenosine monophosphate as an alternative purine source in Plasmodium falciparum, J Biol Chem 283, 32889–32899. 10.1074/jbc.M804497200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zhang M, Wang C, Otto TD, Oberstaller J, Liao X, Adapa SR, Udenze K, Bronner IF, Casandra D, Mayho M, Brown J, Li S, Swanson J, Rayner JC, Jiang RHY, and Adams JH (2018) Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis, Science 360, eaap7847 10.1126/science.aap7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Vickers MF, Yao SY, Baldwin SA, Young JD, and Cass CE (2000) Nucleoside transporter proteins of Saccharomyces cerevisiae. Demonstration of a transporter (FUI1) with high uridine selectivity in plasma membranes and a transporter (FUN26) with broad nucleoside selectivity in intracellular membranes, J Biol Chem 275, 25931–25938. 10.1074/jbc.M000239200 [doi]. [DOI] [PubMed] [Google Scholar]
- (27).Zhang J, Smith KM, Tackaberry T, Sun X, Carpenter P, Slugoski MD, Robins MJ, Nielsen LP, Nowak I, Baldwin SA, Young JD, and Cass CE (2006) Characterization of the transport mechanism and permeant binding profile of the uridine permease Fui1p of Saccharomyces cerevisiae, J Biol Chem 281, 28210–28221. 10.1074/jbc.M605129200. [DOI] [PubMed] [Google Scholar]
- (28).Kokina A, Kibilds J, and Liepins J (2014) Adenine auxotrophy--be aware: some effects of adenine auxotrophy in Saccharomyces cerevisiae strain W303–1A, FEMS Yeast Res 14, 697–707. 10.1111/1567-1364.12154. [DOI] [PubMed] [Google Scholar]
- (29).Chakravorty SJ, Chan J, Greenwood MN, Popa-Burke I, Remlinger KS, Pickett SD, Green DVS, Fillmore MC, Dean TW, Luengo JI, and Macarron R (2018) Nuisance Compounds, PAINS Filters, and Dark Chemical Matter in the GSK HTS Collection, SLAS Discov, 2472555218768497. 10.1177/2472555218768497. [DOI] [PubMed] [Google Scholar]
- (30).Young RJ, Green DV, Luscombe CN, and Hill AP (2011) Getting physical in drug discovery II: the impact of chromatographic hydrophobicity measurements and aromaticity, Drug Discov Today 16, 822–830. 10.1016/j.drudis.2011.06.001 [doi]. [DOI] [PubMed] [Google Scholar]
- (31).Young RJ (2015) Physical Properties in Drug Design, In Tactics in Contemporary Drug Design (Meanwell NA, Ed.), pp 1–68, Springer Berlin Heidelberg. [Google Scholar]
- (32).Mortenson PN, and Murray CW (2011) Assessing the lipophilicity of fragments and early hits, J Comput Aided Mol Des 25, 663–667. 10.1007/s10822-011-9435-z. [DOI] [PubMed] [Google Scholar]
- (33).Meanwell NA (2016) Improving Drug Design: An Update on Recent Applications of Efficiency Metrics, Strategies for Replacing Problematic Elements, and Compounds in Nontraditional Drug Space, Chem Res Toxicol 29, 564–616. 10.1021/acs.chemrestox.6b00043. [DOI] [PubMed] [Google Scholar]
- (34).Scott JS, and Waring MJ (2018) Practical application of ligand efficiency metrics in lead optimisation, Bioorg Med Chem 26, 3006–3015. 10.1016/j.bmc.2018.04.004. [DOI] [PubMed] [Google Scholar]
- (35).Young RJ, and Leeson PD (2018) Mapping the Efficiency and Physicochemical Trajectories of Successful Optimizations, J Med Chem 61, 6421–6467. 10.1021/acs.jmedchem.8b00180. [DOI] [PubMed] [Google Scholar]
- (36).Stumpfe D, and Bajorath J (2011) Similarity searching, WIREs Comput Mol Sci. 1, 260–282. 10.1002/wcms.23. [DOI] [Google Scholar]
- (37).Willett P, Barnard JM, and Downs GM (1998) Chemical Similarity Searching, J Chem Inf Comput Sci. 38, 983–996. [Google Scholar]
- (38).Arora A, Deniskin R, Sosa Y, Nishtala SN, Henrich PP, Kumar TR, Fidock DA, and Akabas MH (2016) Substrate and Inhibitor Specificity of the Plasmodium berghei Equilibrative Nucleoside Transporter Type 1, Mol Pharmacol 89, 678–685. 10.1124/mol.115.101386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Deniskin R, Frame IJ, Sosa Y, and Akabas MH (2016) Targeting the Plasmodium vivax equilibrative nucleoside transporter 1 (PvENT1) for antimalarial drug development, Int J Parasitol Drugs Drug Resist 6, 1–11. 10.1016/j.ijpddr.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DM, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, and Barrell B (2002) Genome sequence of the human malaria parasite Plasmodium falciparum, Nature 419, 498–511. 10.1038/nature01097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Gamo FJ, Sanz LM, Vidal J, de Cozar C, Alvarez E, Lavandera JL, Vanderwall DE, Green DV, Kumar V, Hasan S, Brown JR, Peishoff CE, Cardon LR, and Garcia-Bustos JF (2010) Thousands of chemical starting points for antimalarial lead identification, Nature 465, 305–310. 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- (42).Straimer J, Gnadig NF, Witkowski B, Amaratunga C, Duru V, Ramadani AP, Dacheux M, Khim N, Zhang L, Lam S, Gregory PD, Urnov FD, Mercereau-Puijalon O, Benoit-Vical F, Fairhurst RM, Menard D, and Fidock DA (2015) K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates, Science 347, 428–431. 10.1126/science.1260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Cass CE, and Paterson AR (1973) Mediated transport of nucleosides by human erythrocytes. Specificity toward purine nucleosides as permeants, Biochim Biophys Acta 291, 734–746. 10.1016/0005-2736(73)90477-x [DOI] [PubMed] [Google Scholar]
- (44).Domin BA, Mahony WB, and Zimmerman TP (1988) Purine nucleobase transport in human erythrocytes. Reinvestigation with a novel “inhibitor-stop” assay, J Biol Chem 263, 9276–9284. [PubMed] [Google Scholar]
- (45).Giacomello A, and Salerno C (1979) Hypoxanthine uptake by human erythrocytes, FEBS Lett 107, 203–204. 10.1016/0014-5793(79)80495-0 [DOI] [PubMed] [Google Scholar]
- (46).Lassen UV (1967) Hypoxanthine transport in human erythrocytes, Biochim Biophys Acta 135, 146–154. 10.1016/0005-2736(67)90017-x [DOI] [PubMed] [Google Scholar]
- (47).Plagemann PG (1986) Transport and metabolism of adenosine in human erythrocytes: effect of transport inhibitors and regulation by phosphate, J Cell Physiol 128, 491–500. 10.1002/jcp.1041280319. [DOI] [PubMed] [Google Scholar]
- (48).Plagemann PG, Woffendin C, Puziss MB, and Wohlhueter RM (1987) Purine and pyrimidine transport and permeation in human erythrocytes, Biochim Biophys Acta 905, 17–29. 10.1016/0005-2736(87)90004-6 [DOI] [PubMed] [Google Scholar]
- (49).de Fazio A, Kosic JD, Moir RD, and Bagnara AS (1980) Evidence against the compartmentation of adenosine kinase and adenosine deaminase activities in human erythrocytes, FEBS Lett 113, 215–217. 10.1016/0014-5793(80)80594-1 [DOI] [PubMed] [Google Scholar]
- (50).Dawicki DD, Agarwal KC, and Parks RE Jr. (1985) Role of adenosine uptake and metabolism by blood cells in the antiplatelet actions of dipyridamole, dilazep and nitrobenzylthioinosine, Biochem Pharmacol 34, 3965–3972. 10.1016/0006-2952(85)90373-9 [DOI] [PubMed] [Google Scholar]
- (51).Gresele P, Arnout J, Deckmyn H, and Vermylen J (1986) Mechanism of the antiplatelet action of dipyridamole in whole blood: modulation of adenosine concentration and activity, Thromb Haemost 55, 12–18. [PubMed] [Google Scholar]
- (52).Jarvis SM (1986) Nitrobenzylthioinosine-sensitive nucleoside transport system: mechanism of inhibition by dipyridamole, Mol Pharmacol 30, 659–665. [PubMed] [Google Scholar]
- (53).Woffendin C, and Plagemann PG (1987) Interaction of [3H]dipyridamole with the nucleoside transporters of human erythrocytes and cultured animal cells, J Membr Biol 98, 89–100. [DOI] [PubMed] [Google Scholar]
- (54).Ferrandon P, Barcelo B, Perche JC, and Schoffs AR (1994) Effects of dipyridamole, soluflazine and related molecules on adenosine uptake and metabolism by isolated human red blood cells, Fundam Clin Pharmacol 8, 446–452. [DOI] [PubMed] [Google Scholar]
- (55).Visser F, Vickers MF, Ng AM, Baldwin SA, Young JD, and Cass CE (2002) Mutation of residue 33 of human equilibrative nucleoside transporters 1 and 2 alters sensitivity to inhibition of transport by dilazep and dipyridamole, J Biol Chem 277, 395–401. 10.1074/jbc.M105324200 [doi]. [DOI] [PubMed] [Google Scholar]
- (56).Balakumar P, Nyo YH, Renushia R, Raaginey D, Oh AN, Varatharajan R, and Dhanaraj SA (2014) Classical and pleiotropic actions of dipyridamole: Not enough light to illuminate the dark tunnel?, Pharmacol Res 87, 144–150. 10.1016/j.phrs.2014.05.008 [doi]. [DOI] [PubMed] [Google Scholar]
- (57).Ciacciarelli M, Zerbinati C, Violi F, and Iuliano L (2015) Dipyridamole: a drug with unrecognized antioxidant activity, Curr Top Med Chem 15, 822–829. CTMC-EPUB-65322 [pii]. [DOI] [PubMed] [Google Scholar]
- (58).Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M’Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M, and Davis RW (1999) Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis, Science 285, 901–906. 10.1126/science.285.5429.901 [DOI] [PubMed] [Google Scholar]
- (59).Valko K, Bevan C, and Reynolds D (1997) Chromatographic Hydrophobicity Index by Fast-Gradient RP-HPLC: A High-Throughput Alternative to log P/log D, Anal Chem 69, 2022–2029. 10.1021/ac961242d. [DOI] [PubMed] [Google Scholar]
- (60).Valko K, My Du C, Bevan C, Reynolds DP, and Abraham MH (2001) Rapid method for the estimation of octanol/water partition coefficient (log P(oct)) from gradient RP-HPLC retention and a hydrogen bond acidity term (zetaalpha(2)(H)), Curr Med Chem 8, 1137–1146. [DOI] [PubMed] [Google Scholar]
- (61).Valko K, Nunhuck S, Bevan C, Abraham MH, and Reynolds DP (2003) Fast gradient HPLC method to determine compounds binding to human serum albumin. Relationships with octanol/water and immobilized artificial membrane lipophilicity, J Pharm Sci 92, 2236–2248. 10.1002/jps.10494. [DOI] [PubMed] [Google Scholar]
- (62).Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, and Riscoe M (2004) Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening, Antimicrob Agents Chemother 48, 1803–1806. 10.1128/aac.48.5.1803-1806.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Hill AP, and Young RJ (2010) Getting physical in drug discovery: a contemporary perspective on solubility and hydrophobicity, Drug Discov Today 15, 648–655. 10.1016/j.drudis.2010.05.016 [doi]. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.