

Abstract
Background
Reproductive performance of livestock is an economically important aspect of global food production. The Chinese Meishan pig is a prolific breed, with an average of three to five more piglets per litter than European breeds; however, the genetic basis for this difference is not well understood.
Results
In this study, we investigated copy number variations (CNVs) of 32 Meishan pigs and 29 Duroc pigs by next-generation sequencing. A genome-wide analysis of 61 pigs revealed 12,668 copy number variable regions (CNVRs) that were further divided into three categories based on copy number (CN) of the whole population, i.e., gain (n = 7,638), and loss (n = 5,030) CNVRs. We then compared Meishan and Duroc pigs and identified 17.17 Mb of 6,387 CNVRs that only existing in Meishan pigs CNVRs that overlapped the reproduction-related gene encoding the aryl hydrocarbon receptor (AHR) gene. We found that normal AHR CN was more frequent than CN loss in four different pig breeds. An association analysis showed that AHR CN had a positive effect on litter size (P < 0.05) and that a higher CN was associated with higher total number born (P < 0.05), number born alive (P < 0.05), number of weaned piglets, and birth weight.
Conclusions
The present study provides comprehensive CNVRs for Meishan and Duroc pigs through large-scale population resequencing. Our results provide a supplement for the high-resolution map of copy number variation in the porcine genome and valuable information for the investigation of genomic structural variation underlying traits of interest in pig. In addition, the association results provide evidence for AHR as a candidate gene associated with reproductive traits that can be used as a genetic marker in pig breeding programs.
Keywords: AHR, Copy number variation, Meishan, Next-generation sequencing, Pig, Reproduction
Background
Sow reproductive performance is an important factor for the profitability of pig production [1]. Litter traits mainly include total number of piglets born (TNB), number of piglets born alive (NBA), number of weaned piglets (NWP), birth weight (BW), gestation length (GL), and number of stillborn piglets. Genomic variation such as single nucleotide polymorphisms can affect reproductive traits by controlling gene expression levels [2]. Copy number variation (CNV) is defined as a variable copy number of DNA segments ranging from 50 bp to several megabases (Mb) compared with a reference genome [3]. CNVs are useful molecular markers that influence gene expression and phenotype through various mechanisms such as gene dosage modification, gene structure disturbance, and loss of regulatory elements or polymorphisms [4].
The functional relevance of CNVs to genetic diseases [5], immunity [6], and reproduction has been investigated in several studies [7, 8], which have revealed new markers for complex traits in humans and important economics traits in domestic animals. In humans, several studies of CNV have shown that it is associated with susceptibility to Mendelian diseases and complex genetic diseases such as cancer [9] and various congenital defects [10]. In cattle, CNV of the PRAMEY gene was found to be associated with testis size and bull fertility in Holstein [7], and a deletion-type CNV encompassing ANXA10 was shown to be critical for embryonic development in Japanese Black cattle [8]. In pigs, previous studies used array-based comparative genomic hybridization (aCGH), SNP-array or next-generation sequencing methods to detect CNVs. For instance, Chen et al. identified 1,315 putative CNVs belonging to 565 CNVRs in 1,693 pigs from 18 diverse populations using Porcine SNP60 BeadChip and PennCNV algorithm and revealed 7 copy number variable genes as candidate genes related to carcass length, backfat thickness, abdominal fat weight, length of scapular [11]. Revilla et al. [12] identified 1,279 CNVs and 540 CNVRs using whole genome data and they provided candidate genes for fatty acid composition and growth traits. Jiang et al. [13] indicated the total CNVRs amounted to 4.0% based on the porcine genome (Sus scrofa build10.2) and most CNVRs fell into the interval between 10 kb and 20 kb. Paudel et al. identified 1,408 regions, comprising 17.83 Mb of the porcine genome (Sus scrofa build10.2). Many of the identified CNVRs are relatively small, the size of CNVRs ranges from 6 to 98 kb, 78% of the CNVRs that were identified is between 6 and 15 kb. CNVRs covered 0.7% of the porcine genome [14]. Although the size ranges and coverage of CNVR detected in previous swine studies were different, the functional genes were consistent, such as olfactory receptor, which is known to play a prominent role in food foraging and mate recognition in Sus. Keel et al. [15] using three methods to identify CNVRs covered 0.94% of the porcine genome and the number of CNVRs per animal ranged from 0 to 348, with a mean of 157.8. However, the CNVRs were most overlapped with reproductive traits [15]. A limitation in most of the aforementioned CNV studies in swine is using the Sscrofa 10.2 genome builds.
Candidate CNVs and genes associated with complex traits have also been reported [11, 16, 17]. A CNV of the MSRB3 gene encoding methionine sulfoxide reductase has been shown to increase porcine ear size [18]. CNV of the MTHFSD gene affects litter size in the Chinese indigenous Xiang pig [19]. Meanwhile, MCHR1, PPARα, SLC5A1, and SLC5A4 CNVs have been implicated in fat-related functions [16]. Meishan is a Chinese swine breed that it is well known for its high prolificacy; however, the genetic basis for this high fecundity is largely unknown. In addition, the using of next-generation sequencing (NGS) in this study allows to identify a wide range of CNV, especially many small CNVs that are missed when using SNP chips.
To address these issues, in this study we performed a genome-wide CNV analysis in Meishan pigs by next-generation sequencing (NGS). We also performed a comparative analysis with Duroc pigs to identify the putative CNV regions (CNVRs) only existed in Meishan as well as an association study between genes in CNVRs and pig reproductive traits.
Methods
Animal ethics
Animal care and next-generation sequencing were approved by the Institutional Animal Care and Use Committee of China Agricultural University (Beijing, People’s Republic of China; permit No. DK1023).
Sample collection
A total of 61 pigs were analyzed by NGS including 32 Meishan pigs from Kunshan City and 29 Duroc pigs from Yancheng City of Jiangsu Province. For qPCR analysis, we obtained 853 blood samples from Beijing Liuma Technology Co. (Beijing, China) and Meishan Pig Conservation Breeding Co. (Kunshan, China). The samples were from four pig breeds: Duroc (n = 171), Landrace (n = 176), Yorkshire (n = 478), and Meishan (n = 28). Reproductive data such as TNB and NBA were available for all sampled individuals.
Genomic DNA was isolated from pig blood samples using the Genomic DNA Extraction Kit Qiagen DNeasy Tissue kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. DNA quality was verified with a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and by 0.8% agarose gel electrophoresis.
Re-sequencing and CNV detection
Next-generation sequencing library preparation involves generating a collection of DNA fragments for sequencing. In the library preparation, 50 ng genomic DNA was fragmented in 16 μL of TE. After fragmentation, samples were end-repaired using the New England Biolabs (NEB) sample preparation kit and protocol (NEB Next DNA Sample prep, E6000S), with incubation time of 30 min. Following end-repair, a single dA was added to the end of each Blunted. After A-tailing, DNA ligation was performed. Once ligation had been assessed, the adapter ligated library was PCR amplified. Finally, the libraries generated for 61 pigs were sequenced on an Illumina HiSeq2000 platform at Novogene (Beijing, China). All paired-end reads reached the length of 125 bp, with an average insert size of 460–490 bp and the standard deviation of 11–14 bp estimated for all samples.
In the preprocessing of CNV detection, all the data were removed adapters and low quality reads (the quality score lower than 20) using NGSQC Toolkit [20]. The filtered reads were further aligned to pig reference genome (Sus sacrofa 11.1) by Burrows-Wheeler Aligner (BWA) with the default parameter. The average of read mapping ratio was 96% for 61 pig samples.
The CNVs were detected using CNVnator (v0.3.3) software [21] and CNVcaller (RRID:SRC 015752) [22]. The CNVnator captured the read-depth signal in genomic regions with different CNs and genotyped both deletions and duplications with the correction for GC bias. For each pig, the aligned files were processed to identify genome-wide CNVs (except those on the X and Y chromosome) with standard parameters and 200-bp bins [23]. The CNVcaller applies robust signal detection and noise deduction methods on basis of RD algorithm to increase the computational efficiency in complex genomes. We ran the CNVcaller by population levels for Meishan and Duroc breeds with the default arguments [22].
After the CNV detection, all CNVs of each individual detected by CNVnator and CNVcaller were merged by one-to-one correspondence when the overlap is of at least 1 bp by bedtools [24]. Then we merged the CNVs of Meishan and Duroc into CNVRs by breed. For each breed, the CNVRs were defined as the CNVs identified in three or more individuals when the overlap is of at least 1 bp.
For CNVs that overlapped among different individuals, CNVRuler software [25] (http://www.ircgp.com/CNVRuler/?ckattempt=1) was used to define two types of common CNVRs, loss and gain, along with fragmented CNVs. CNVRs were used to construct a CNV map for Meishan and Duroc pigs, and fragmented CNVs were used for subsequent CNV comparisons in all pigs.
To detect CNVs between Meishan and Duroc populations, we used the relative frequency difference (RFD value) [26] to assess CNV diversity within each breed based on fragmented CNV frequency. Fragmented CNV means that a single large CNV is fragmented into multiple smaller calls [27], which is mainly used in CNV comparison among different populations. The RFD of the Meishan population relative to the Duroc population was calculated as follows: RFDMeishan-Duroc = (FMeishan − FDuroc)/FMeishan-Duroc, where FMeishan, FDuroc, and FMeishan-Duroc represent the fragmented CNV frequency in the Meishan population (with CNV discarded for both FMeishan and FDuroc < 0.05). We calculated both deletion and duplication of RFD values for mixed CNVs in all pigs of both breeds.
GO enrichment analysis and functional classification
KOBAS 3.0 is a web server for gene/protein functional annotation (Annotate module) and functional gene set enrichment (Enrichment module). Thus, to provide insight into the functional enrichment of the CNVRs, we performed gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses for the genes in CNVRs using KOBAS 3.0 (http://kobas.cbi.pku.edu.cn/kobas3/?t=1) and PANTHER 15.0 classification system (http://www.pantherdb.org).
CNV type assay
We evaluated CNV of the AHR gene using qPCR and 2−ΔΔCT method. Primers used for qPCR were designed using Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast). We selected one segment of the GCG gene as the reference locus since this gene is highly conserved across species and is present as a single copy in animals [28]. Primer sequences for AHR and GCG are shown in Table 1. To ensure comparability between AHR and GCG, we first determined the amplification efficiency of each assay using serial dilutions of 100 ng DNA prepared in triplicate. The threshold amplification efficiency of primers used in this study was 1.99–2.01.
Table 1.
Primer sequences for transcripts used in real-time quantitative PCR
| Gene | Primer sequence (5′→3′) | Annealing temperature, °C | Product length, bp |
|---|---|---|---|
| AHR | Forward: ACTACCACCCATCTTCACCCG | 60 | 183 |
| Reverse: CAACACACATCAATGCTTCCC | |||
| GCG | Forward: GAATCAACACCATCGGTCAAAT | 60 | 147 |
| Reverse: CTCCACCCATAGAATGCCCAGT |
CNVs in 853 samples were detected by qPCR on a LightCycler 480 Real-Time PCR System (Roche, Basel, Switzerland) using DNA according to the manufacturer’s protocol. PCR amplifications were performed in a total volume of 20 μL consisting of the following reagents: 1 μL DNA (around 50 ng), 1 μL (20 pmol/μL) of both forward primer and reverse primer, 10 μL of Master Mix (2×) and water (Roche Applied Science). PCRs were run as follows: 5 min at 95 °C followed by 40 cycles at 95 °C for 10 s and 60 °C for 10 s. All PCRs were performed in 96-well clear reaction plates (Roche Applied Science). Relative expression levels were estimated with the cycle threshold (2−ΔΔCt) method [29], which compares the ΔCt value (Ct of the target − Ct of the control region) of samples with CNV to that of the calibrator sample. The CN of the AHR gene was confirmed based on the assumption that there were two copies of the DNA segment in calibrator animals. The CNV type of the AHR gene was defined as loss (fewer than two gene copies) and normal (two gene copies as in the positive control) according to previous studies [30, 31]. The qPCR assays for each individual were performed in triplicate.
Association analysis
To determine the effect of CNV on pig reproductive traits, we performed an association analysis using SAS v.9.2 software (SAS Institute, Cary, NC, USA) according to the model:
yijklmn = μ + Hi + Yj + Sk + Pl + CNVm + eijklmn.
where yijklmn is the phenotypic value of each trait in pigs; μ is overall population mean; Hi, Yj, and Sk are fixed effects of farm (two farms), year (3 years), and season (four seasons), respectively; Pl is parity; CNVm is genotype effect; and eijklmn is the random residual with e~N (0, Iσe2) (where I is a diagonal matrix and σe2 is the residual error variance). A P value < 0.05 was considered statistically significant for each test. Five reproductive traits including TNB, NBA, NWP, BW, and GL were examined in the association study of Landrace (n = 176) and Yorkshire (n = 478) pigs. We used the false discovery rate (FDR) test for significance threshold. And 5% FDR as guideline to control overall false positive during multiple testing.
Results
Sequencing and CNV detection
To detect genome-wide CNVs in Meishan and Duroc pigs, we performed whole-genome resequencing of 32 unrelated Meishan and 29 Duroc pigs. The mapped read depth ranged from 6.08 to 10.96, with an average depth per sample of 8.20, which was calculated using SAMtools [32] software (Table S1).
We identified 8,282 CNVRs in the Meishan pigs, including 3,724 deletions and 4,558 duplications (Tables S2–5). And we identified 6,700 CNVRs in the Duroc pigs, consisting of 2,029 deletions, 4,670 duplications and 1 mixed (Tables S2, S3 and S6). Among them, the median number of duplications was 1,999 and the median number of deletions was 1,999. These CNVs are located in all 18 autosomal chromosomes with a mean size of 3,721.53 bp ranging from 199 bp to 279,799 bp of Meishan pig (Table 2). On average, we identified 258 and 231 variants each animal of Meishan and Duroc pigs, respectively. The CNVRs covered 1.10% and 0.99% of the porcine genome (Sscrofa 11.1) in Meishan and Duroc pigs, respectively (Table 2). All the CNVR maps for Meishan and Duroc pigs were showed in Fig. 1. In addition, we also calculated the relationship between sequence coverage and the number of CNVs identified in each individual (Fig. 2).
Table 2.
Descriptive statistics of copy number variant identified for two breeds
| Breeds | No. CNVR | No. losses | No. gains | CNV min, bp | CNV max, bp | CNV mean, bp | CNV median, bp | Coverage, kp | Coverage, % |
|---|---|---|---|---|---|---|---|---|---|
| Meishan | 8,282 | 3,724 | 4,558 | 199 | 279,799 | 3,721.53 | 1,999 | 30,821.72 | 1.10% |
| Duroc | 6,700 | 2,029 | 4,670 | 199 | 598,399 | 4,164.97 | 1,999 | 27,905.32 | 0.99% |
Fig. 1.

The overall CNVR maps for Meishan and Duroc pigs in the 18 autosomal chromosomes. Two types of CNVR were identified including gain (red), and loss (light blue)
Fig. 2.

Link between the number of variants and the sequence coverage for each animal
Frequency of variants across animals
We calculated the allele frequencies of the CNVRs in the Duroc and Meishan pigs separately (Fig. 3). Results showed the detecting frequency for duplication was higher than that for deletion. For Meishan pigs, the percentage of carriers for each variant varied from 12.5% (4 animals out of 32) to 100% (32 animals out of 32) and 34.22% of the detected CNVRs were observed in 4 (frequency 12.5%) to 6.4 (allele frequency 20%) animals. Such pattern also observed in Duroc pigs (34.69% of the identified CNVRs were existed in 4 to 5.8 animals).
Fig. 3.

The allele frequencies of variants in the Duroc and Meishan pigs (n= 61)
Population structure analysis of Meishan and Duroc pigs
We analyzed the population structure of the sequenced Meishan and Duroc pigs by using Principal Components Analysis (PCA). Results showed that there was a clear distinction between Meishan and Duroc pigs based on two principal components (the variance ratio of the two major components is 23.9% and 15.4%) (Fig. 4).
Fig. 4.
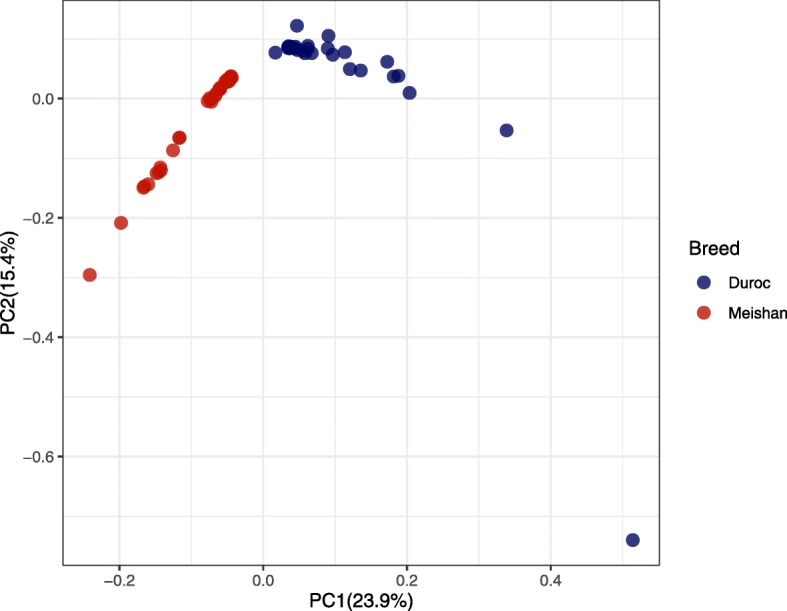
PCA plot based on the first two principal components in the Meishan and Duroc pigs. The two breeds were clustered to two groups
Comparison with CNVRs identified in previous reports
We compared our results of CNVRs to those identified in several previous swine studies. Results showed varying levels of overlapping CNVRs between our studies. Here, we used a stringent definition to identify overlapping CNVRs, i.e., where two CNVRs were considered overlapped when they shared at least 75% bases.
The highest percentage of overlap in CNV events identified between this work and previous studies was 4.62% (Table 3). The average overlap was 2.13%, which was lower than that in previous study (average of 4.33% overlap) [15].
Table 3.
Comparison of CNVRs identified in this study to results from other studies (based on the Sscrofa 11.1 genome assembly)
| Platform | Study | Breeds | Samples | No. of CNVRs (NO. before mapping) | No. of overlapped CNVRs from this study | Percent of overlapped CNVRs from this study |
|---|---|---|---|---|---|---|
| Next-generation sequencing | [12] | 2 | 7 | 416 (540) | 107 | 0.84% |
| Next-generation sequencing | [34] | 2 | 16 | 2,265 (3,118) | 262 | 2.07% |
| Next-generation sequencing | [13] | 10 | 13 | 1,903 (3,131) | 585 | 4.62% |
| Porcine SNP60 | [11] | 18 | 1693 | 243 (565) | 153 | 1.21% |
| Next-generation sequencing | [14] | 7 | 14 | 754 (1,408) | 254 | 2.00% |
| 1 M aCGH | [35] | 9 | 12 | 436 (758) | 180 | 1.42% |
| Next-generation sequencing | [36] | 13 | 49 | 1,906 (3,131) | 172 | 1.36% |
| Next-generation sequencing | [15] | 3 | 240 | 3,538 | 449 | 3.54% |
| This Study | 12,668 |
Note: CNVRs were converted to Sscrofa 11.1 coordinates using the liftOver tool. Successfully mapped CNVRs are shown in the CNVRs column with the original number of published CNVRs (Sscrofa 10.2) shown in parentheses
Gene annotation and functional analysis of the CNVRs
A total of 3,554 genes from the Ensembl annotation of the Sscrofa 11.1 genome were identified to be overlapping with our detected 12,668 CNVRs (Table S7), including 1,914 known genes and 1640 unknown genes (NA). Using PANTHER’s statistical overrepresentation test to inspect GO terms mapping to CNV-overlapped genes, we identified that CNVRs enriched for genes related to sensory perception, detection of stimulus, development, metabolic and nervous system process for biological process, which is consistent with previous studies [13]. Molecular function terms were related to G protein-coupled receptor activity, catalytic activity, sensory organ development and cation binding were significantly overrepresented in the genes overlapped by CNVR (Table S8), which were also observed by Paudel et al. [14].
KEGG analysis using KOBAS 3.0 for the total 3,514 Sus scrofa genes showed CNVRs are significantly enriched in Pathways related to disease and immunity (including IFN, ILR and TNF genes), reproduction, and development (Table S9, such as Pathways in cancer (corrected P value = 5.02E-07), Rap1 signaling pathway (corrected P value = 2.11E-05), Wnt signaling pathway (corrected P value = 0.024) as well as MAPK signaling pathway (corrected P value = 3.1706E-04). We found 11 genes were in the olfactory transduction pathway, however, the P value was more than 0.5.
Considering that the limited annotation for pig, we also performed KEGG analysis based on Homo sapiens (on the bottom of Table S9). We found the olfactory transduction pathway was over-represented, including 27 genes (on the bottom of Table S9).
Comparative analysis of CNVRs in Meishan and Duroc
To determine whether the CNVs that we identified in Meishan differed from those in Duroc, we identified 6,387 CNVRs comprising 17.17 Mb in Meishan (Table S10). These CNVRs were further divided based on CN into gain (n = 3,348) and loss (n = 3,039), comprising 8.21 Mb and 8.95 Mb, respectively. The Ensembl gene annotation set (http://www.ensembl.org/) facilitated identification of a total of 6,387 CNVRs overlapping 2,610 genes.
To evaluate the contribution of CNVRs to the high prolificacy of Meishan pigs, we compared Meishan CNVRs of Meishan pigs with those of Duroc pigs and identified the regions that differed significantly between the two breeds. We extracted 17.17 Mb regions (Table S10) that was not found in the Duroc group. This region included 6,387 CNVRs overlapping 2,610 genes, of which AHR, ESR2, STAT3 and FSHR are closely associated with reproductive traits. For example, the AHR gene has been linked to a larger litter size in European pigs [33]. These results demonstrate that Meishan pigs have given CNVRs comparing to Duroc pigs that are potentially associated with prolificacy.
We used the statistical parameter RFD [26] to detect selective sweeps in Meishan pigs based on fragmented CNV frequencies (split with CNVRuler software) between Meishan and Duroc pigs. The highest 10% absolute RFD value for each breed was used as the threshold (Meishan, 1.66; Duroc, 1.89) to identify fragmented CNVs. In total, we found 1,099 fragmented CNVs (11.7 kb on average) overlapping 443 genes. Six of the genes were closely related to reproduction, including the AHR gene encoding aryl hydrocarbon receptor.
AHR CNV is associated with pig reproductive traits
Previous studies have employed qPCR to evaluate CNVs and their effects in Chinese bulls [37]; for instance, CNV of the TSPY gene was detected in 14 different cattle breeds [38]. The AHR gene is known to play a critical role in regulating reproductive lifespan and fertility and establishing an optimal environment for fertilization [39], as well as is important in ovarian function. AHR mRNA and protein expression varies according to the reproductive tissue and estrous cycle phase, suggesting their involvement in the regulation of reproductive function in female pigs [40]. Indeed, the loss of AHR gene expression in mutant mice and AHR overexpression can lead to adverse phenotypes in the female reproductive organs and impaired reproductive function [41–43]. On the basis of these observations, we speculate that AHR CNVs may affect reproductive performance in pigs.
To investigate the functional significance of different CNV types of the AHR gene in terms of pig reproductive traits, we evaluated AHR CN in four pig breeds (Meishan, Duroc, Landrace, and Yorkshire) by quantitative (q)PCR. The porcine AHR gene is located at chr9: 86,511,369 to 86,555,943 (Sscrofa 11.1) and overlapped with the CNV region: chr9: 86,518,401–86,520,000 (Table S10). We designed primer to amplify AHR and GCG gene. The primer pair for AHR started from 86,553,480 to 86,553,662 and the detection sequence size is 183 bp. The detection sequence is located in the eleventh exon of AHR gene. We assessed the efficiency of amplification and calculated the correlation coefficient for the target gene AHR and the reference glucagon gene (GCG); the results showed a high degree of precision in the determination of relative CN. In our study, CNVs of high quality were detected in four pig breeds through qPCR, demonstrating that this approach could be useful for other CNV studies in pigs.
The 2−ΔΔCT value in all breeds ranged from 0.5–2.5 (Table S11). The pigs were divided into two classes: those with 2−ΔΔCT values ranging from 0.5–1.5 as one copy (loss type) and those with values of 1.5–2.5 as two copies (normal type). Among the 853 samples analyzed, both normal and loss types were observed in Duroc, Landrace, and Yorkshire whereas normal CN was observed in Meishan. The frequency of the two types also differed across breeds: the rank order of proportion of individuals with normal CN was Duroc (46.1%) < Yorkshire (66.9%) < Landrace (67.0%) < Meishan (100%).
CNVs may affect the phenotype by altering the transcription of genes within or adjacent to a CNVR, which ultimately affects protein levels. We evaluated the association between CNV type and pig reproductive traits (i.e., TNB, NBA, NWP, BW, and GL) in Landrace and Yorkshire pigs, using a general linear model (Table 4). We found that TNB and NBA were significantly associated with the AHR gene CNV type in the Yorkshire breed (P < 0.05). Moreover, TNB and NBA were higher in individuals with the normal type compared to the loss type (P < 0.01); this trend was also observed in Landrace pigs, with a higher TNB in normal- than in loss-type individuals (P < 0.05). These findings suggest that the AHR gene CNV positively affects TNB and NBA in pigs.
Table 4.
Association analysis of AHR CNV types with reproductive traits
| Breeds | CNV type | TNB | NBA | BW | NWP | GL |
|---|---|---|---|---|---|---|
| Yorkshire | Loss | 11.55 ± 0.28b | 10.84 ± 0.19b | 16.80 ± 0.27a | 9.81 ± 0.17a | 115.15 ± 0.10a |
| Normal | 12.00 ± 0.24a | 11.46 ± 0.13a | 16.90 ± 0.19a | 9.88 ± 0.12a | 115.02 ± 0.08a | |
| P-value | 0.002 | 0.0002 | 0.017 | 0.085 | 0.332 | |
| Landrace | Loss | 11.22 ± 0.30b | 10.26 ± 0.29a | 16.10 ± 0.47a | 9.00 ± 0.29a | 115.79 ± 0.32a |
| Normal | 11.75 ± 0.25a | 10.72 ± 0.24a | 16.54 ± 0.40a | 9.21 ± 0.24a | 115.56 ± 0.27a | |
| P-value | 0.039 | 0.061 | 0.270 | 0.408 | 0.408 |
Note: Values with different superscripts (a,b) within the same line differ significantly at P < 0.05
TNB total number born, NBA number born alive, NWP number of weaned piglets, BW birth weight, GL gestation length
Discussion
In this study, we detected 6,700 and 8,282 CNVRs in Duroc and Meishan pigs, respectively, accounting for approximately 0.99% and 1.10% of the reference pig genome (Sus scrofa 11.1). To the best of our knowledge, the present study is the first analysis of CNVRs for Meishan pigs through large-scale population resequencing. Notably, compared with Duroc pigs, Meishan pigs show an excess of CNVRs. The Meishan pig breed is well known for its high prolificacy. We identified CNV of the AHR gene as being potentially related to the prolificacy of Meishan pigs. In addition, qPCR analysis of the AHR gene in a large population of multiple pig breeds suggested that CNV of the AHR gene is associated with TNB and NBA. Furthermore, TNB was higher in Yorkshire and Landrace pigs with normal CN as compared to those with CN loss. These findings suggest that molecular marker-based breeding can improve pig production.
Characteristic and functional analysis of the CNVRs
The average frequency of duplications was higher than that of deletions in both Duroc and Meishan pigs in the present study. Revilla et al. also found duplications showed a higher average frequency than did deletions (106 vs. 77) in a global analysis of CNVs in swine [12]. Similar pattern was also observed in another CNV study in porcine using a 60 k SNP BeadChip, the predicted status for the CNVRs in this study was 38.7% for gain, and 16.3% for loss [44]. This proportion may be related to natural selection, as it is assumed that the genome is more tolerant to duplications than to deletions [45]. We found the sequence coverage was not correlated with the number of identified CNVs in each animal, which is distinct from this results in cattle [46].
The concordance in this study between previous CNV studies is limited. Potential reasons for the differences between our results and these studies may be due to the difference in population size and genetic background between our study and others, different call algorithms for CNV detecting. In addition, our results were based on the Sscrofa11.1 genome assembly, while most of the previous works were based on Sscrofa 10.2. Warr et al. indicated there was dramatically difference between Sscrofa11.1 and Sscrofa 10.2 version and many problems in the10.2 version have been solved in the 11.1 version [47].
Genes located in CNV regions have a wide spectrum of molecular functions and provide a resource for investigating the biological relationship of CNVs with the genetic basis of phenotypic variations. The GO enrichment and KEGG analysis revealed that genes in CNVRs participated in G protein-coupled receptor activity, sensory organ development, and olfactory transduction, which were related to the olfactory receptors (OR). OR gene family is the most well characterized CNV-related genes in humans [48] and one of the largest gene families in porcine [49]. Besides the OR gene family, we also found some genes involved in immunity and cytochrome P450, such as CYP4A24, CYP2C42 and CYP3A29. These results were observed in previous swine CNV studies [12, 14], and together with ORs, CNV in CYP450 genes suggests a relevant role of these genes in the organism’s adaptation to rapid changes in the environment [14]. Among KEGG pathways, we found the Ras signaling pathway and MAPK signaling pathway were included. The mitogen-activated protein kinase (MAPK) pathway is known to have an important role in numerous male reproductive processes, including spermatogenesis [50], sperm maturation and activation, capacitation and acrosome reaction, before fertilization of the oocyte [51]. P38 MAPK, one of the major family of protein kinases, might be involved in FSH-induced meiotic resumption of oocytes [52]. These findings provided insight for the function of pig CNVs.
The copy number of AHR gene is associated with pig’s litter traits
The difference in AHR CN among Meishan and three other pig breeds (Landrace, Yorkshire, and Duroc) may be attributable to their diverse genetic backgrounds. Similar findings were reported in studies of CNVs in bovine populations [53]. We found that the AHR CN was normal in all tested Meishan individuals and that CN loss was non-existent, unlike in the other three pig breeds. Meishan has one of the highest rates of fecundity and the highest TNB among pig breeds in China. Among the European commercial breeds Duroc, Landrace, and Yorkshire, two AHR gene copies were detected in 46.1, 67.0, and 66.9%, respectively, of the tested population. Previous studies indicated the Landrace and Large White were much more closely related than Duroc at the genome-wide level [54, 55], thus the higher proportion in Landrace and Yorkshire compared to Duroc could be attributed to the differences in the genome level among the three breeds. Landrace and Yorkshire were commonly used as maternal pigs, which are selected in the artificial breeding process based on large litter size. In contrast, Duroc is used as a paternal pig and is therefore selected for different traits, which may also be associated with the different proportions of AHR gene copies among these breeds. These observations reveal the potential role of AHR in influencing the phenotype of different pig breeds and we speculated that altering the AHR CN may increase litter size.
Numerous studies have reported that CNVs can affect the production and reproductive traits of livestock animals [37, 56]. For instance, CNVRs encompassing multiple genes associated with cattle production such as milk fat and protein yield have been reported [57], and the MTHFSD gene was found to be associated with litter size in Xiang pigs [19]. In the present study, an association was established between AHR CNV and reproductive traits such as TNB and NBA in Landrace and Yorkshire breeds, although no significant association was observed between CN and BW, or GL. AHR is a ligand-activated nuclear transcription factor that can transduce extracellular signals through DNA binding-dependent and -independent mechanisms [58]. In mammals, AHR plays an important role in primary follicle formation and regulation of follicle number [41], and can affect the follicle growth rate by regulating estradiol in mice [59]. In pigs, the AHR gene is known to be associated with reproductive traits [40] and litter size [33]; this was confirmed by the observation of the present study that CNV of the AHR gene was associated with TNB and NBA. We speculate that CNV affects the expression of the AHR gene in a dose-dependent manner, which in turn affects follicular proliferation and promotes ovulation, thereby increasing TNB. Taken together, our findings demonstrate that AHR CN is a useful marker for improving pig productivity; however, additional studies are required to confirm the molecular basis for the relationship between AHR gene CNV and reproductive traits.
Conclusions
NGS-based analysis has been widely applied to identify CNVs and has led to significant progress in porcine CNV detection. We identified 12,668 CNVRs with an average size of 3.78 kb comprising 47.93 Mb of the porcine genome, which cover a small (1.71%) fraction of the pig genome. Moreover, we found the small size CNVs (<10 kb) were abundant, which accounts for almost one-half of all the CNVs. The inferred CNV regions include 3,554 genes providing an important resource for future analyses on phenotypic variation in pigs. In addition, we identified AHR gene, which showed associations with several of the reproductive traits. Association analysis study indicated that AHR CN had a positive effect on litter size and that a higher CN was associated with higher total number born, number born alive, and birth weight. We believe that our study makes a significant contribution to the literature as it provides information for the investigation of genomics structural variation underlying traits of interest in the Meishan pig, which is one of the most prolific pig breeds. Although the genetic basis for their fecundity is not well understood, molecular marker-based breeding can improve pig productivity. These findings contribute to facilitate the further identification of trait-related CNVRs.
Supplementary information
Additional file 1 Table S1. Overall details of all pigs and their classification. Table S2. Overview of deletions for two pig breeds. Table S3. Overview of duplications for two pig breeds. Table S4. Detection of CNVRs in Meishan population. Table S5. Overview of CNVRs for Meishan population. Table S6. Detection of CNVRs in Duroc population. Table S7. Annotation of all the CNVRs for Meishan and Duroc pigs. Table S8. GO Enrichment Analysis for CNVR gene set by PANTHER. Table S9. KEGG and GO Enrichment Analysis for CNVR gene set by KOBAS. Table S10. Overview of given CNVRs only found in Meishan pigs. Table S11. CNV types of individuals on AHR CNV in four pig breeds.
Acknowledgements
The authors would like to acknowledge the funding bodies of this research.
Abbreviations
- AHR
Aryl hydrocarbon receptor
- BW
Birth weight
- CN
Copy number
- CNV
Copy number variation
- CNVRs
CNV regions
- GL
Gestation length
- NBA
Number of piglets born alive
- NGS
Next-generation sequencing
- NWP
Number of weaned piglets
- TNB
Piglets born
Authors’ contributions
J-F.L. conceived and designed the experiments. X.Z. performed CNV, qPCR and pathway analysis, graphic design and wrote the manuscript. P.Z. performed CNV analysis. K.Y. and H.F. performed qPCR assay. C.N. performed association analysis. X.Z., K.Y. and P.Z. collected samples and prepared for sequencing. J-F.L., X.Z., P.Z., K.Y., and L.Z. wrote and revised the paper. All authors read and approved the final manuscript.
Funding
This work was financially supported by National Natural Science Foundations of China (31661143013), National Natural Science Foundation of China (No.31790414), National Key R&D Program of China (2018YFD0501200), and Jinxinnong Animal Science Development Foundation.
Availability of data and materials
A total 61 pig samples with 1,403.35Gbases were uploaded to NCBI with BioProject ID: PRJNA378496.
Ethics approval and consent to participate
Animal care and sequencing were approved by the Institutional Animal Care and Use Committee of China Agricultural University (Beijing, People’s Republic of China; permit no. DK1023).
Consent for publication
All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Xianrui Zheng, Pengju Zhao and Kaijie Yang contributed equally to this work.
Contributor Information
Xianrui Zheng, Email: Zxrdk2015@cau.edu.cn.
Pengju Zhao, Email: zhaopengju@cau.edu.cn.
Kaijie Yang, Email: kjyang@cau.edu.cn.
Chao Ning, Email: ningchao@cau.edu.cn.
Haifei Wang, Email: hyfiwang@yzu.edu.cn.
Lei Zhou, Email: leiz@cau.edu.cn.
Jianfeng Liu, Email: liujf@cau.edu.cn.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s40104-020-00442-5.
References
- 1.Onteru SK, Ross JW, Rothschild MF. The role of gene discovery, QTL analyses and gene expression in reproductive traits in the pig. Soc Reprod Fertil Suppl. 2009;66:87–102. [PubMed] [Google Scholar]
- 2.Rempel LA, Nonneman DJ, Wise TH, Erkens T, Peelman LJ, Rohrer GA. Association analyses of candidate single nucleotide polymorphisms on reproductive traits in swine. J Anim Sci. 2010;88(1):1–15. doi: 10.2527/jas.2009-1985. [DOI] [PubMed] [Google Scholar]
- 3.Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Gene. 2011;12(5):363–76. [DOI] [PMC free article] [PubMed]
- 4.Bickhart DM, Liu GE. The challenges and importance of structural variation detection in livestock. Front Genet. 2014;18(5):37. doi: 10.3389/fgene.2014.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weischenfeldt J, Symmons O, Spitz F, Korbel JO. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat Rev Genet. 2013;14(2):125–138. doi: 10.1038/nrg3373. [DOI] [PubMed] [Google Scholar]
- 6.Liu GE, Brown T, Hebert DA, Cardone MF, Hou YL, Choudhary RK, et al. Initial analysis of copy number variations in cattle selected for resistance or susceptibility to intestinal nematodes. Mamm Genome. 2011;22(1–2):111–121. doi: 10.1007/s00335-010-9308-0. [DOI] [PubMed] [Google Scholar]
- 7.Yue XP, Dechow C, Chang TC, DeJarnette JM, Marshall CE, Lei CZ, et al. Copy number variations of the extensively amplified Y-linked genes, HSFY and ZNF280BY, in cattle and their association with male reproductive traits in Holstein bulls. BMC Genomics. 2014;15:113. doi: 10.1186/1471-2164-15-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sasaki S, Ibi T, Akiyama T, Fukushima M, Sugimoto Y. Loss of maternal ANNEXIN A10 via a 34-kb deleted-type copy number variation is associated with embryonic mortality in Japanese black cattle. BMC Genomics. 2016;17(1):968. doi: 10.1186/s12864-016-3312-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shlien A, Malkin D. Copy number variations and cancer. Genome Med. 2009;1(6):62. doi: 10.1186/gm62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sailani MR, Makrythanasis P, Valsesia A, Santoni FA, Deutsch S, Popadin K, et al. The complex SNP and CNV genetic architecture of the increased risk of congenital heart defects in Down syndrome. Genome Res. 2013;23(9):1410–1421. doi: 10.1101/gr.147991.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen C, Qiao R, Wei R, Guo Y, Ai H, Ma J, et al. A comprehensive survey of copy number variation in 18 diverse pig populations and identification of candidate copy number variable genes associated with complex traits. BMC Genomics. 2012;13:733. doi: 10.1186/1471-2164-13-733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Revilla M, Puig-Oliveras A, Castello A, Crespo-Piazuelo D, Paludo E, Fernandez AI, et al. A global analysis of CNVs in swine using whole genome sequence data and association analysis with fatty acid composition and growth traits. PLoS One. 2017;12(5):e0177014. doi: 10.1371/journal.pone.0177014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang J, Wang J, Wang H, Zhang Y, Kang H, Feng X, et al. Global copy number analyses by next generation sequencing provide insight into pig genome variation. BMC Genomics. 2014;15:593. doi: 10.1186/1471-2164-15-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paudel Y, Madsen O, Megens HJ, Frantz LA, Bosse M, Crooijmans RP, et al. Copy number variation in the speciation of pigs: a possible prominent role for olfactory receptors. BMC Genomics. 2015;16:330. doi: 10.1186/s12864-015-1449-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keel BN, Nonneman DJ, Lindholm-Perry AK, Oliver WT, Rohrer GA. A survey of copy number variation in the porcine genome detected from whole-genome sequence. Front Genet. 2019;10:737. doi: 10.3389/fgene.2019.00737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fowler KE, Pong-Wong R, Bauer J, Clemente EJ, Reitter CP, Affara NA, et al. Genome wide analysis reveals single nucleotide polymorphisms associated with fatness and putative novel copy number variants in three pig breeds. BMC Genomics. 2013;14:784. doi: 10.1186/1471-2164-14-784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hay EHA, Choi I, Xu L, Zhou Y, Rowland RRR, Lunney JK, et al. CNV analysis of host responses to porcine reproductive and respiratory syndrome virus infection. J Genomics. 2017;5:58–63. doi: 10.7150/jgen.20358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen CY, Liu CL, Xiong XW, Fang SM, Yang H, Zhang ZY, et al. Copy number variation in the MSRB3 gene enlarges porcine ear size through a mechanism involving miR-584-5p. Genet Sel Evol. 2018;50(1):72. doi: 10.1186/s12711-018-0442-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ran XQ, Pan H, Huang SH, Liu C, Niu X, Li S, et al. Copy number variations of MTHFSD gene across pig breeds and its association with litter size traits in Chinese indigenous Xiang pig. J Anim Physiol An N. 2018;102(5):1320–1327. doi: 10.1111/jpn.12922. [DOI] [PubMed] [Google Scholar]
- 20.Patel RK, Jain M, Liu Z. NGS QC Toolkit: A Toolkit for Quality Control of Next Generation Sequencing Data. PLoS One. 2012;7(2):e30619. [DOI] [PMC free article] [PubMed]
- 21.Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21(6):974–984. doi: 10.1101/gr.114876.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, Zheng Z, Cai Y, Chen T, Li C, Fu W, et al. CNVcaller: highly efficient and widely applicable software for detecting copy number variations in large populations. Gigascience. 2017;6(12):1–12. doi: 10.1093/gigascience/gix115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pirooznia M, Goes FS, Zandi PP. Whole-genome CNV analysis: advances in computational approaches. Front Genet. 2015;6:138. doi: 10.3389/fgene.2015.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JH, Hu HJ, Yim SH, Bae JS, Kim SY, Chung YJ. CNVRuler: a copy number variation-based case-control association analysis tool. Bioinformatics. 2012;28(13):1790–1792. doi: 10.1093/bioinformatics/bts239. [DOI] [PubMed] [Google Scholar]
- 26.Zhou Z, Jiang Y, Wang Z, Gou Z, Lyu J, Li W, et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat Biotechnol. 2015;33(4):408–414. doi: 10.1038/nbt.3096. [DOI] [PubMed] [Google Scholar]
- 27.Legault MA, Girard S, Lemieux Perreault LP, Rouleau GA, Dube MP. Comparison of sequencing based CNV discovery methods using monozygotic twin quartets. PLoS One. 2015;10(3):e0122287. doi: 10.1371/journal.pone.0122287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ballester M, Castello A, Ibanez E, Sanchez A, Folch JM. Real-time quantitative PCR-based system for determining transgene copy number in transgenic animals. BioTechniques. 2004;37(4):610–613. doi: 10.2144/04374ST06. [DOI] [PubMed] [Google Scholar]
- 29.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 30.Ferreira ID. Do Rosario VE, Cravo PVL. Real-time quantitative PCR with SYBR green I detection for estimating copy numbers of nine drug resistance candidate genes in plasmodium falciparum. Malar J. 2006;5:1. doi: 10.1186/1475-2875-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Passon N, Pozzo F, Molinis C, Bregant E, Gellera C, Damante G, et al. A simple multiplex real-time PCR methodology for the SMN1 gene copy number quantification. Genet Test Mol Biomarkers. 2009;13(1):37–42. doi: 10.1089/gtmb.2008.0084. [DOI] [PubMed] [Google Scholar]
- 32.Etherington GJ, Ramirez-Gonzalez RH, MacLean D. Bio-samtools 2: a package for analysis and visualization of sequence and alignment data with SAMtools in ruby. Bioinformatics. 2015;31(15):2565–2567. doi: 10.1093/bioinformatics/btv178. [DOI] [PubMed] [Google Scholar]
- 33.Bosse M, Megens HJ, Frantz LA, Madsen O, Larson G. Paudel Y, et al. Genomic analysis reveals selection for Asian genes in European pigs following human-mediated introgression. Nat Commun. 2014;5:4392. doi: 10.1038/ncomms5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paudel Y, Madsen O, Megens HJ, Frantz LA, Bosse M. Bastiaansen JW, et al. Evolutionary dynamics of copy number variation in pig genomes in the context of adaptation and domestication. BMC Genomics. 2013;14:449. doi: 10.1186/1471-2164-14-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Jiang J, Wang H, Kang H, Zhang Q, Liu JF. Improved Detection and Characterization of Copy Number Variations Among Diverse Pig Breeds by Array CGH. G3 (Bethesda). 2015;5(6):1253–61. [DOI] [PMC free article] [PubMed]
- 36.Wang H, Wang C, Yang K, Liu J, Zhang Y, Wang Y, et al. Genome wide distributions and functional characterization of copy number variations between Chinese and Western pigs. PLoS One. 2015;10(7):e0131522. [DOI] [PMC free article] [PubMed]
- 37.Zhang LZ, Jia SG, Yang MJ, Xu Y, Li CJ, Sun JJ, et al. Detection of copy number variations and their effects in Chinese bulls. BMC Genomics. 2014;15(1):480. [DOI] [PMC free article] [PubMed]
- 38.Hamilton CK, Favetta LA, Di Meo GP, Floriot S, Perucatti A, Peippo J, et al. Copy number variation of testis-specific protein, Y-encoded (TSPY) in 14 different breeds of cattle (Bos taurus) Sex Dev. 2009;3(4):205–213. doi: 10.1159/000228721. [DOI] [PubMed] [Google Scholar]
- 39.Hernandez-Ochoa I, Karman BN, Flaws JA. The role of the aryl hydrocarbon receptor in the female reproductive system. Biochem Pharmacol. 2009;77(4):547–559. doi: 10.1016/j.bcp.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jablonska O, Piasecka J, Ostrowska M, Sobocinska N, Wasowska B, Ciereszko RE. The expression of the aryl hydrocarbon receptor in reproductive and neuroendocrine tissues during the estrous cycle in the pig. Anim Reprod Sci. 2011;126(3–4):221–228. doi: 10.1016/j.anireprosci.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 41.Benedict JC, Lin TM, Loeffler IK, Peterson RE, Flaws JA. Physiological role of the aryl hydrocarbon receptor in mouse ovary development. Toxicol Sci. 2000;56(2):382–388. doi: 10.1093/toxsci/56.2.382. [DOI] [PubMed] [Google Scholar]
- 42.Gray LE, Jr, Ostby JS. In utero 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) alters reproductive morphology and function in female rat offspring. Toxicol Appl Pharmacol. 1995;133(2):285–294. doi: 10.1006/taap.1995.1153. [DOI] [PubMed] [Google Scholar]
- 43.Baba T, Mimura J, Nakamura N, Harada N, Yamamoto M, Morohashi K, et al. Intrinsic function of the aryl hydrocarbon (dioxin) receptor as a key factor in female reproduction. Mol Cell Biol. 2005;25(22):10040–10051. doi: 10.1128/MCB.25.22.10040-10051.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramayo-Caldas Y, Castello A, Pena RN, Alves E, Mercade A, Souza CA, et al. Copy number variation in the porcine genome inferred from a 60 k SNP BeadChip. BMC Genomics. 2010;11:593. doi: 10.1186/1471-2164-11-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Conrad DF, Hurles ME. The population genetics of structural variation. Nat Genet. 2007;39(7 Suppl):S30–S36. doi: 10.1038/ng2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Letaief R, Rebours E, Grohs C, Meersseman C, Fritz S, Trouilh L, et al. Identification of copy number variation in French dairy and beef breeds using next-generation sequencing. Genet Sel Evol. 2017;49(1):77. [DOI] [PMC free article] [PubMed]
- 47.Warr A, Robert C, Hume D, Archibald AL, Deeb N, Watson M. Identification of Low-Confidence Regions in the Pig Reference Genome (Sscrofa10.2). Front Genet. 2015;6:338. [DOI] [PMC free article] [PubMed]
- 48.Young JM, Endicott RM, Parghi SS, Walker M, Kidd JM, Trask BJ. Extensive copy-number variation of the human olfactory receptor gene family. Am J Hum Genet. 2008;83(2):228–242. doi: 10.1016/j.ajhg.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nguyen DT, Lee K, Choi H, Choi MK, Le MT, Song N, et al. The complete swine olfactory subgenome: expansion of the olfactory gene repertoire in the pig genome. BMC Genomics. 2012;13:584. doi: 10.1186/1471-2164-13-584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crepieux P, Marion S, Martinat N, Fafeur V, Vern YL, Kerboeuf D, et al. The ERK-dependent signalling is stage-specifically modulated by FSH, during primary Sertoli cell maturation. Oncogene. 2001;20(34):4696–4709. doi: 10.1038/sj.onc.1204632. [DOI] [PubMed] [Google Scholar]
- 51.Almog T, Naor Z. Mitogen activated protein kinases (MAPKs) as regulators of spermatogenesis and spermatozoa functions. Mol Cell Endocrinol. 2008;282(1–2):39–44. doi: 10.1016/j.mce.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 52.Villa-Diaz LG, Miyano T. Activation of p38 MAPK during porcine oocyte maturation. Biol Reprod. 2004;71(2):691–696. doi: 10.1095/biolreprod.103.026310. [DOI] [PubMed] [Google Scholar]
- 53.Xu Y, Zhang LZ, Shi T, Zhou Y, Cai HF, Lan XY et al. Copy number variations of MICAL-L2 shaping gene expression contribute to different phenotypes of cattle. Mamm Genome. 2013;24(11–12):508–16. [DOI] [PubMed]
- 54.Traspov A, Deng W, Kostyunina O, Ji J, Shatokhin K, Lugovoy S, et al. Population structure and genome characterization of local pig breeds in Russia, Belorussia. Kazakhstan and Ukraine Genet Sel Evol. 2016;48:16. doi: 10.1186/s12711-016-0196-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mujibi FD, Okoth E, Cheruiyot EK, Onzere C, Bishop RP, Fevre EM, et al. Genetic diversity, breed composition and admixture of Kenyan domestic pigs. PLoS One. 2018;13(1):e0190080. doi: 10.1371/journal.pone.0190080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang MJ, Lv JQ, Zhang LZ, Li MX, Zhou Y, Lan XY, et al. Association study and expression analysis of CYP4A11 gene copy number variation in Chinese cattle. Sci Rep. 2017;7:46599. doi: 10.1038/srep46599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ben Sassi N, Gonzalez-Recio O, de Paz-Del Rio R, Rodriguez-Ramilo ST, Fernandez AI. Associated effects of copy number variants on economically important traits in Spanish Holstein dairy cattle. J Dairy Sci. 2016;99(8):6371–6380. doi: 10.3168/jds.2015-10487. [DOI] [PubMed] [Google Scholar]
- 58.Rowlands JC, Gustafsson JA. Aryl hydrocarbon receptor-mediated signal transduction. Crit Rev Toxicol. 1997;27(2):109–134. doi: 10.3109/10408449709021615. [DOI] [PubMed] [Google Scholar]
- 59.Barnett KR, Tomic D, Gupta RK, Miller KP, Meachum S, Paulose T, et al. The aryl hydrocarbon receptor affects mouse ovarian follicle growth via mechanisms involving estradiol regulation and responsiveness. Biol Reprod. 2007;76(6):1062–1070. doi: 10.1095/biolreprod.106.057687. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1 Table S1. Overall details of all pigs and their classification. Table S2. Overview of deletions for two pig breeds. Table S3. Overview of duplications for two pig breeds. Table S4. Detection of CNVRs in Meishan population. Table S5. Overview of CNVRs for Meishan population. Table S6. Detection of CNVRs in Duroc population. Table S7. Annotation of all the CNVRs for Meishan and Duroc pigs. Table S8. GO Enrichment Analysis for CNVR gene set by PANTHER. Table S9. KEGG and GO Enrichment Analysis for CNVR gene set by KOBAS. Table S10. Overview of given CNVRs only found in Meishan pigs. Table S11. CNV types of individuals on AHR CNV in four pig breeds.
Data Availability Statement
A total 61 pig samples with 1,403.35Gbases were uploaded to NCBI with BioProject ID: PRJNA378496.