

Key points.
-
•
Taxonomy is the classification, nomenclature and identification of microbes (algae, protozoa, slime moulds, fungi, bacteria, archaea and viruses). The naming of organisms by genus and species is governed by an international code.
-
•
Bacteria can be separated into two major divisions by their reaction to Gram's stain, and exhibit a range of shapes and sizes from spherical (cocci) through rod shaped (bacilli) to filaments and spiral shapes.
-
•
In clinical practice, bacteria are classified by macroscopic and microscopic morphology, their requirement for oxygen, and activity in phenotypic and biochemical tests.
-
•
Various diagnostic test systems are used to detect specific bacteria in clinical systems, including specific gene probes, reaction with antibodies in ELISA formats, immunofluorescence and, increasingly, PCR-based technology.
-
•
Different bacterial species often exhibit different population structures, highly diverse (panmictic) or relatively uniform (clonal) depending mainly on the frequency of gene recombination (from external sources).
-
•
Typing of bacterial isolates is necessary for epidemiological investigations in outbreaks and for surveillance, and a variety of phenotypic and genetic methods has evolved for the identification of strains.
Micro-organisms may be classified in the following large biological groups:
-
1
Algae
-
2
Protozoa
-
3
Slime moulds
-
4
Fungi
-
5
Bacteria
-
6
Archaea
-
7
Viruses.
The algae (excluding the blue–green algae), the protozoa, slime moulds and fungi include the larger eukaryotic (see Ch. 2) micro-organisms; their cells have the same general type of structure and organization as that found in plants and animals. The bacteria, including organisms of the mycoplasma, rickettsia and chlamydia groups, together with the related blue–green algae, comprise the smaller micro-organisms, with the form of cellular organization described as prokaryotic. The archaea are a distinct phylogenetic group of prokaryotes that bear only a remote ancestral relationship to other organisms (see Ch. 2). As the algae, slime moulds and archaea are not currently thought to contain species of medical or veterinary importance, they will not be considered further. Blue–green algae do not cause infection, but certain species produce potent peptide toxins that may affect persons or animals ingesting polluted water.
The viruses are the smallest of the infective agents; they have a relatively simple structure that is not comparable with that of a cell, and their mode of reproduction is fundamentally different from that of cellular organisms. Even simpler are viroids, protein-free fragments of single-stranded circular RNA that cause disease in plants. Another class of infectious particles are prions, the causative agents of fatal neurodegenerative disorders in animals and man. These are postulated to be naturally occurring host cell membrane glycoproteins that undergo conformative changes to an infectious isoform (see Ch. 60).
Taxonomy
Taxonomy consists of three components: classification, nomenclature and identification. Classification allows the orderly grouping of micro-organisms, whereas nomenclature concerns the naming of these organisms and requires agreement so that the same name is used unambiguously by everyone. Changes in nomenclature may give rise to confusion and are subject to internationally agreed rules. In clinical practice, microbiologists are generally concerned with identification – the correct naming of isolates according to agreed systems of classification. These components, together with taxonomy, make up the overarching discipline of systematics, which is concerned with evolution, genetics and speciation of organisms, and is commonly referred to as phylogenetics.
Protozoa, fungi and helminths are classified and named according to the standard rules of classification and nomenclature that have been developed following the pioneering work of the eighteenth century Swedish botanist Linnaeus (Carl von Linné). Large subdivisions (class, order, family, etc.) are finally classified into individual species designated by a Latin binomial, the first term of which is the genus, e.g. Plasmodium (genus) falciparum (species). Occasionally it is useful to recognize a biological variant with particular properties: thus, Trypanosoma (genus) brucei (species) gambiense (variant) differs from the variant T. brucei brucei in being pathogenic for man.
Bacteria are similarly classified, but bacterial diversity encompasses more variety than all the rest of cellular life put together and the natural capacity of bacteria for genetic and phenotypic variation and adaptation make rigid classifications difficult. To date, identification has predominantly been performed by the use of keys that allow the organization of bacterial traits based on growth or activity in systems that test their biochemical properties. Some tests are definitive of a genus or species, for example the universal production of catalase enzyme and cytochrome c, respectively, by Staphylococcus spp. and Pseudomonas aeruginosa. Other characters may be unique to individual species and serve to differentiate them from organisms with closely similar biochemical activity profiles. Some bacteria do not grow in the laboratory (leprosy bacillus, treponemes), and identification by genetic methods may be necessary. As the technologies for genetic analysis become more readily applicable in clinical labs, so they and other rapid analytical methods, such as those based on mass spectrometry, are coming to replace the traditional biochemical methods to achieve identification. The taxonomic ranks used in the classification of bacteria are (example in parentheses):
-
•
Kingdom (Prokaryotae)
-
•
Division (Gracilicutes)
-
•
Class (Betaproteobacteria)
-
•
Order (Burkholderiales)
-
•
Family (Burkholderiaceae)
-
•
Genus (Burkholderia)
-
•
Species (Burkholderia cepacia).
Some genera, such as Acinetobacter, have been subdivided into a number of genomic species by DNA homology analysis. Some are named and others are referred to only by a number. Many of the genomic species cannot be differentiated with accuracy by phenotypic tests. Another subgenus grouping in current usage recognizes species complexes, which are differentiated into genomovars by polyphasic taxonomic methods. A good example of this is the B. cepacia complex of organisms, which includes a very diverse group of organisms ranging from strict plant to human pathogens.
At present no standard classification of bacteria is universally accepted and applied, although Bergey's Manual of Determinative Bacteriology is widely used as an authoritative source. Bacterial nomenclature is governed by an international code prepared by the International Committee on Systematic Bacteriology and published as Approved Lists of Bacterial Names in the International Journal of Systematic and Evolutionary Microbiology; most new species are also first described in this journal, and a species is considered to be validly published only if it appears on a validation list in this journal.
The International Committee on Taxonomy of Viruses (ICTV) classifies viruses and publishes its reports in the journal Archives of Virology. Latin names are used wherever possible for the ranks family, subfamily and genus, but at present there are no formal categories higher than family and binomial nomenclature is not used for species. Viruses do not lend themselves easily to classification according to Linnaean principles, and vernacular names still have wide usage among medical virologists. Readers are referred to the standard work on virus taxonomy Classification and Nomenclature of Viruses and the ICTV database website.
Methods of classification
Adansonian or numerical classification
In most systems of bacterial classification, the major groups are distinguished by fundamental characters such as cell shape, Gram-stain reaction and spore formation; genera and species are usually distinguished by properties such as fermentation reactions, nutritional requirements and pathogenicity. The relative ‘importance’ of different characters in defining major and minor groupings is often purely arbitrary. The uncertainties of arbitrary choices are avoided in the Adansonian system of taxonomy. This system determines the degrees of relationship between strains by a statistical coefficient that takes account of the widest range of characters, all of which are considered of equal weight. It is clear, of course, that some characters, for instance cell shape or Gram-stain reaction, represent a much wider and permanent genetic commitment than other characters which, being dependent on only one or a few genes, may be unstable. For this reason, the Adansonian method is most useful for the classification of strains within a larger grouping that shares major characters.
By scoring a large number of phenotypic characters it is possible to estimate a similarity coefficient when shared positive characters are considered or a matching coefficient when both negative and positive shared characters (matches) are taken into account. This numerical taxonomy is best performed on a computer that can calculate degrees of similarity for a group of different organisms; these data are displayed as a similarity matrix or a dendrogram tree (Fig. 3.1 ) (see also Owen 2004 in Recommended reading list).
Fig. 3.1.
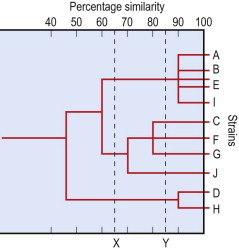
Hierarchical taxonomic tree (dendrogram) prepared from similarity matrix data. The dashed lines X and Y indicate levels of similarity at which separation into genera and species might be possible.
DNA composition
The hydrogen bonding between guanine and cytosine (G–C) base pairs in DNA is stronger than that between adenine and thymine (A–T). Thus, the melting or denaturation temperature of DNA (at which the two strands separate) is determined primarily by the G + C content. At the melting temperature, the separation of the strands brings about a marked change in the light absorption characteristics at a wavelength of 260 nm, and this is readily detected by spectrophotometry. There is a very wide range in the G + C component of bacterial DNA, varying from about 25–80% mol in different genera. However, for any one species, the G + C content is relatively fixed, or falls within a very narrow range, and this provides a basis for classification.
DNA homology
Another approach to classification is to arrange individual organisms into groups on the basis of the homology of their DNA base sequences. This exploits the fact that double strands re-form (anneal) from separated strands during controlled cooling of a heated preparation of DNA. This process can be readily demonstrated with suitably heated homologous DNA extracted from a single species, but it can also occur with DNA from two related species, so that hybrid pairs of DNA strands are produced. These hybrid pairings occur with high frequency between complementary regions of DNA, and the degree of hybridization can be assessed if labelled DNA preparations are used. Binding studies with messenger RNA (mRNA) can also give information to complement these observations, which provide genetic evidence of relatedness among bacteria. Organisms with different G + C ratios are unlikely to show significant DNA homology. However, organisms with the same, or close, G + C ratios do not necessarily show homology. A novel real-time polymerase chain reaction (PCR) (see pp. 31 and 79–80) has been described for estimation of G + C content.
Ribosomal RNA sequencing
The structure of ribosomal RNA (rRNA) appears to have been highly conserved during the course of evolution, and close similarities in nucleotide sequences reflect phylogenetic relationships. Advances in technology have made nucleotide sequencing relatively simple, and the rDNA sequences (and other genes) of most medically important bacterial species are available from a number of internet sites. Far fewer full-length gene sequences are known for 23S rRNA than for 16S rRNA and 16S–23S internal transcribed sequences. In practice the DNA of the test organism is extracted and amplified by PCR using universal primers. The DNA sequence of the product is determined and the sequence compared against databases to find the closest fit. It is generally accepted that sequence similarity between 0.5% and 1.0% (with the type species) is required for the identification of an unknown organism, and less than 97% similarity is a common cut-off point for the differentiation of species. However, different species of Mycobacterium may exhibit more than 97% similarity in rDNA sequence. Commercial systems are available for bacterial species identification (MicroSeq; Applied Biosystems, Foster City, California, USA).
Nucleotide sequence variation in ribosomal genes is sometimes insufficient to discriminate between closely related species. Other candidate genes have been explored but the recA gene, which encodes a protein essential for repair and recombination of DNA, appears to be one of the best suited for phylogenetic analysis in that it defines evolutionary trees consistent with those observed for rRNA genes. Additional housekeeping genes used for phylogenetic studies include rpoB (RNA polymerase), groEL (heat shock protein) and gyrB (DNA gyrase), among others.
Classification in clinical practice
The identification of micro-organisms in routine practice requires a pragmatic approach to taxonomy. The primary purpose of the names used is to communicate amongst the clinical and public health teams so that appropriate management of individuals and groups can be implemented. Unfortunately, as we learn more about the interrelatedness of microbes, largely through genome sequence data, certain reallocation and renaming becomes necessary in order to sustain a coherent relationship with our basic understanding. Table 3.1 outlines a simple, but practical, classification scheme in which organisms are grouped according to a few shared characteristics; as far as possible these have been reconciled with the relatively recently established phylogenetic nomenclature. Within these groups, organisms may be further identified, sometimes to species level, by a few supplementary tests. Protozoa, helminths and fungi can often be definitively identified on morphological criteria alone (see appropriate chapters).
Table 3.1.
Simplified classification of some cellular micro-organisms of medical importance
| Common group name | Normal genus names |
|---|---|
| Eukaryotes | |
| Protozoa | |
| Sporozoa | Plasmodium, Isospora, Toxoplasma, Cryptosporidium |
| Flagellates | Giardia, Trichomonas, Trypanosoma, Leishmania |
| Amoebae | Entamoeba, Naegleria, Acanthamoeba |
| Other | Babesia, Balantidium |
| Fungi | |
| Mould-like | Epidermophyton, Trichophyton, Microsporum, Aspergillus |
| Yeast-like | Candida |
| Dimorphic | Histoplasma, Blastomyces, Coccidioides |
| True yeast | Cryptococcus |
| Prokaryotes | |
| Bacteria | |
| Actinobacteria | (High G+C Gram positives) Actinomyces, Streptomyces, Corynebacterium, Nocardia, Mycobacterium, Micrococcus |
| Firmicutes | (Low G+C Gram positives) |
| Gram-positive bacilli | Listeria, Bacillus, Clostridium*, Lactobacillus*, Eubacterium* |
| Gram-positive cocci | Staphylococcus, Streptococcus, Enterococcus |
| Gram-negative cocci** | Veillonella*, Mycoplasma |
| Proteobacteria | (a very large group with 5 sub-divisions) |
| Gram-negative cocci | Neisseria, Moraxella |
| Gram-negative bacilli | Enterobacteria – Escherichia, Klebsiella, Proteus, Salmonella, Shigella, Yersinia |
| Pseudomonads – Pseudomonas, Burkholderia, Stenotrophomonas | |
|
Haemophilus, Bordetella, Brucella, Pasteurella Rickettsia, Coxiella | |
| Gram-negative curved and spiral bacilli | Vibrio, Spirillum, Campylobacter, Helicobacter |
| Bacteroidetetes | Bacteroides*, Prevotella* |
| Spirochaetes | Borrelia, Treponema, Brachyspira, Leptospira |
| Chlamydia | Chlamydia |
Anaerobic organism.
Exceptions in a predominantly Gram-positive group.
Protozoa
These are non-photosynthetic unicellular organisms with protoplasm clearly differentiated into nucleus and cytoplasm. They are relatively large, with transverse diameters mainly in the range of 2–100 µµm. Their surface membranes vary in complexity and rigidity from a thin, flexible membrane in amoebae, which allows major changes in cell shape and the protrusion of pseudopodia for the purposes of locomotion and ingestion, to a relatively stiff pellicle in ciliate protozoa, preserving a characteristic cell shape. Most free-living and some parasitic species capture, ingest and digest internally solid particles of food material; many protozoa, for instance, feed on bacteria. Protozoa, therefore, are generally regarded as the lowest forms of animal life, although certain flagellate protozoa are closely related in their morphology and mode of development to photosynthetic flagellate algae in the plant kingdom. Protozoa reproduce asexually by binary fission or multiple fission (schizogony), and some also by a sexual mechanism (sporogony). The most important groups of medical protozoa are the sporozoa (malaria parasites, etc.), amoebae and flagellates (see Ch. 62).
Fungi
These are non-photosynthetic organisms that possess relatively rigid cell walls. They may be saprophytic or parasitic, and take in soluble nutrients by diffusion through their cell surfaces.
Moulds grow as branching filaments (hyphae), usually 2–10 µm in width, which interlace to form a meshwork (mycelium). The hyphae are coenocytic (i.e. have a continuous multinucleate protoplasm), being either non-septate or septate with a central pore in each cross-wall. Moulds reproduce by the formation of various kinds of sexual and asexual spores that develop from the vegetative (feeding) mycelium, or from an aerial mycelium that effects their air-borne dissemination (see Ch. 61).
Yeasts are ovoid or spherical cells that reproduce asexually by budding and also, in many cases, sexually, with the formation of sexual spores. They do not form a mycelium, although the intermediate yeast-like fungi form a pseudo-mycelium consisting of chains of elongated cells. The dimorphic fungi produce a vegetative mycelium in artificial culture, but are yeast-like in infected lesions. The higher fungi of the class Basidiomycetes (mushrooms), which produce large fruiting structures for aerial dissemination of spores, are not infectious for human beings or animals, although some species are poisonous.
Bacteria
The main groups of bacteria are mostly distinguished by microscopic observation of their morphology and staining reactions. The Gram-staining procedure, which reflects fundamental differences in cell wall structure, separates most bacteria into two great divisions: Gram-positive bacteria and Gram-negative bacteria (see Ch. 2). Where bacteria can be Gram stained the cell shape and clustering are of practical value in identification. However, at this point the relationship of the older, largely phenotypic, classification system to the newer DNA sequence-based phylogenetic classification, which reflects the evolutionary relationships between groups, begins to break down and anomalies are apparent. Nonetheless, practical identification schemes used in clinical labs rely heavily on recognizing whether bacteria are Gram positive or negative, bacillary or coccal in shape, able to grow aerobically or anaerobically (see Ch. 4) and whether they form spores.
The major phylogenetic groupings of medically significant bacteria are:
-
•
Actinobacteria, these are also recognised as Gram-positve bacteria with a high G + C content, many of which are capable of filamentous growth with true branching and which may produce a type of mycelium. Many members of this group do not stain well with the Gram method and, importantly, they include the mycobacteria which can be recognized with acid fast stains (see Ch. 2).
-
•
Firmicutes, low G + C Gram-positive bacteria including bacilli, some of which are spore formers and cocci; the group includes most of the medically significant Gram-positives. Two groups that have presumably lost their Gram-positivity, Veillonella and Mycoplasma are included here.
-
•
Proteobacteria, a very large of group of Gram-negative bacteria (bacilli and cocci) with five subdivisions (alpha, beta, gamma, delta and epsilon).
-
•
Bacteroidetes, Gram-negative anaerobes.
-
•
Spirochaetes, possessing cells with a tight spiral shape and an internal flagellum.
-
•
Chlamydiae, which are strict intracellular parasites.
Actinobacteria
These include antibiotic producing bacteria and a small number of highly significant pathogens.
-
•
Actinomyces. Gram-positive, non-acid-fast, tend to fragment into short coccal and bacillary forms and not to form conidia; anaerobic (e.g. Actinomyces israelii).
-
•
Streptomyces. Vegetative mycelium does not fragment into short forms; conidia form in chains from aerial hyphae (e.g. Streptomyces griseus).
-
•
Mycobacterium. Acid-fast; Gram-positive, but does not readily stain by the Gram method; usually bacillary, rarely branching; make mycolic acids (e.g. Mycobacterium tuberculosis).
-
•
Nocardia. Similar to Actinomyces, but aerobic and mostly acid-fast; make mycolic acids (e.g. Nocardia asteroides).
-
•
Corynebacterium. Pleiomorphic (variably shaped) Gram-positive bacilli; make mycolic acids (e.g. Corynebacterium diphtheriae).
Firmicutes
-
•
Streptococcus. Gram-positive cocci, mainly adherent in chains due to successive cell divisions occurring in the same axis (e.g. Streptococcus pyogenes); sometimes predominantly diplococcal (e.g. Streptococcus pneumoniae).
-
•
Staphylococcus. Gram-positive cocci, mainly adherent in irregular clusters due to successive divisions occurring irregularly in different planes (e.g. Staph. aureus).
-
•
Mycoplasma and ureoplasma. Pleiomorphic cocci that do not make peptidoglycan.
-
•
Veillonella. Gram-negative; generally very small cocci arranged mainly in clusters and pairs; anaerobic (e.g. Veillonella parvula).
-
•
Gram-positive spore-forming bacilli. The key medically relevant genera here the genera Bacillus and Clostridium (anaerobic). They are Gram positive, but liable to become Gram negative in ageing cultures. The size, shape and position of the spore may assist recognition of the species, for example the bulging, spherical, terminal spore ('drumstick’ form) of Clostridium tetani.
-
•
Gram-positive non-sporing bacilli. These include several genera. Erysipelothrix and Lactobacillus are distinguished by a tendency to grow in chains and filaments, and Listeria by flagella that confer motility.
Proteobacteria
Gram-negative bacteria.
-
•
Alphaproteobacteria. Include the cell dependent Rickettsia group, the facultatively intracellular Brucella group and the Bartonella group.
-
•
Betaproteobacteria. Neisseria – cocci, mainly adherent in pairs and slightly elongated at right angles to axis of pairs (e.g. Neisseria meningitidis). Burkholderia – bacilli (Burkholderia pseudomallei).
-
•
Gammaproteobacteria. Bacilli including the enterobacteria (Escherichia coli), and the genera Pseudomonas, Legionella and the curved vibrios (e.g. Vibrio cholera).
-
•
Deltaproteobacteria. No medically significant bacteria.
-
•
Epsilonproteobacteria. Curved and loosely spiral bacilli including the genera Helicobacter and Campylobacter.
Bacteroidetes
Gram-negative non-sporing anaerobic bacilli.
-
•
Bacteroides, Prevotella and Porphyromonas are major genera.
Spirochaetales
These organisms differ from the other groups in being slender flexuous spiral filaments that, unlike the spirilla, are motile without possession of flagella. The staining reaction, when demonstrable, is Gram negative. The different varieties are recognized by their size, shape, waveform and refractility, observed in the natural state in unstained wet films by dark-ground microscopy. Genera of medical importance include Borrelia, Treponema and Leptospira.
Chlamydiae
Chlamydiae have a complex intracellular cycle (e.g. Chlamydia trachomatis).
Viruses
Viruses usually consist of little more than a strand of DNA or RNA enclosed in a simple protein shell known as a capsid. Sometimes the complete nucleocapsid may be enclosed in a lipoprotein envelope derived largely from the host cell. Viruses are capable of growing only within the living cells of an appropriate animal, plant or bacterial host; none can grow in an inanimate nutrient medium. The viruses that infect and parasitize bacteria are termed bacteriophages or phages. A simple classification of the viruses that are involved in human disease is shown in Table 3.2 .
Table 3.2.
Principal types of virus causing human disease
| Type of virus | Examples |
|---|---|
| RNA viruses | |
| Orthomyxoviruses | Influenza A, B and C viruses |
| Paramyxoviruses | Parainfluenza viruses, mumps virus, measles virus, respiratory syncytial virus, Hendra virus, Nipah virus, human metapneumovirus |
| Rhabdoviruses | Rabies virus |
| Arenaviruses | Lassa virus |
| Filoviruses | Marburg and Ebola viruses |
| Togaviruses | Many arboviruses, rubella virus |
| Flaviviruses | Yellow fever virus, Dengue virus, Japanese encephalitis virus, West Nile virus, hepatitis C virus |
| Bunyaviruses | Hantaan virus |
| Coronaviruses | Human coronavirus, 229E (group 1), OC43 (group 2), NL63 (group 1-like novel corona viruses), SARS (zoonosis) |
| Caliciviruses | Norwalk-like viruses, hepatitis E virus |
| Picornaviruses | Enteroviruses: poliovirus (3 types), echovirus (31 types), Coxsackie A virus (24 types), Coxsackie B virus (6 types), enterovirus types 68–71, hepatitis A virus (type 72), rhinovirus, many serotypes |
| Retroviruses | Human immunodeficiency virus types 1 and 2, human T-lymphotropic virus types I and II |
| Reoviruses | Rotaviruses |
| DNA viruses | |
| Poxviruses | Variola, vaccinia, molluscum contagiosum virus, Orf virus |
| Herpesviruses | Herpes simplex virus types 1 and 2, varicella-zoster virus, cytomegalovirus, Epstein–Barr virus, human herpesvirus types 6, 7 and 8 |
| Adenoviruses | Many serotypes |
| Papovaviruses | JC virus, BK virus, human papillomavirus |
| Hepadnaviruses | Hepatitis B virus |
| Parvoviruses | B 19 virus, new human parvovirus |
SARS, severe acute respiratory syndrome.
Identification of micro-organisms
Precise or definitive identification of bacteria is time consuming, contentious and best carried out in specialized reference centres. For most clinical purposes, clear, rapid guidance on the likely cause of an infection is required and, consequently, microbiologists usually rely on a few simple procedures, notably microscopy and culture, backed up, when necessary, by a few supplementary tests to achieve a presumptive identification. Microscopy is the most rapid test of all, but culture inevitably takes at least 24 hours, sometimes longer. More rapid tests are constantly being sought, and antigen detection methods and genetic detection methods are now well established (see below).
Most specimens for bacteriological examination, whether from human beings, animals or the environment, contain mixtures of bacteria, and it is essential to obtain pure cultures of individual isolates before embarking on identification. Non-cultural methods, such as antigen or nucleic acid based detection, do not have this disadvantage; however, they do have the potential limitation of being highly specific so that the investigator must know beforehand what it is necessary to look for.
Microscopy
Morphology and staining reactions of individual organisms generally serve as preliminary criteria to place an unknown species in its appropriate biological group. A Gram-stain smear suffices to show the Gram reaction, size, shape and grouping of the bacteria, and the arrangement of any endospores. An unstained wet film may be examined with dark-ground illumination in the microscope to observe the morphology of delicate spirochaetes; an unstained wet film, or ‘hanging-drop’, preparation is examined with ordinary bright-field illumination for observation of motility. Capsules surrounding bacterial cells are demonstrated by ‘negative staining’ with India ink; the capsules remain unstained against the background of ink particles. To identify mycobacteria, or other acid-fast organisms, a preparation is stained by the Ziehl–Neelsen method or one of its modifications (see p. 16). The microscopic characters of certain organisms in pathological specimens may be sufficient for presumptive identification, for example tubercle bacilli in sputum, or T. pallidum in exudate from a chancre. However, many bacteria share similar morphological features, and further tests must be applied to differentiate them.
Cultural characteristics
The appearance of colonial growth on the surface of a solid medium, such as nutrient agar, is often very characteristic. Attention is paid to the diameter of the colonies, their outline, their elevation, their translucency (clear, translucent or opaque) and colour. Changes brought about in the medium (e.g. haemolysis in a blood agar medium) may also be significant. The range of conditions that support growth is characteristic of particular organisms. The ability or inability of the organism to grow in the presence (aerobe) or absence (anaerobe) of oxygen, in a reduced oxygen atmosphere (micro-aerophile) or in the presence of carbon dioxide, or on media containing selective inhibitory factors (e.g. bile salt, specific antimicrobial agents, or low or high pH) may also be of diagnostic significance (see Table 4.1).
Biochemical reactions
Species that cannot be distinguished by morphology and cultural characters may exhibit metabolic differences that can be exploited. It is usual to test the ability of the organism to produce acidic and gaseous end-products when presented with individual carbohydrates (glucose, lactose, sucrose, mannitol, etc.) as the sole carbon source. Other tests determine whether the bacterium produces particular end-products (e.g. indole or hydrogen sulphide) when grown in suitable culture media, and whether it possesses certain enzyme activities, such as oxidase, catalase, urease, gelatinase or lecithinase. Traditionally, such tests have been performed selectively and individually according to the recommendations of standard guides, such as the invaluable Cowan and Steel's Manual for the Identification of Medical Bacteria. However, today most diagnostic laboratories use commercially prepared microgalleries of identification tests which, though expensive, combine simplicity and accuracy. Test kits are now available for a number of different groups of organisms, including enterobacteria, staphylococci, streptococci and anaerobes. Other kits facilitate the testing of carbon source utilization, the assimilation of specific substrates and the enzymes produced by an organism.
On occasion, more elaborate procedures may be used for the analysis of metabolic products or whole-cell fatty acids. Indeed, a fully automated, fatty acid-based identification system, which combines high-resolution gas chromatography and pattern recognition software, is widely used to identify a variety of aerobic and anaerobic bacterial species. New profiles are added to a computerized database, thus increasing the sensitivity of the system. Mass spectrometric methods show promise for rapid identification, particularly matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry. This offers the analysis of whole bacterial cultures for unique mass spectra from charged macromolecules by rapid, high-throughput testing with a rapidly growing database.
Indirect identification methods
Gene-targeted analyses
These are fragments of DNA that recognize complementary sequences within micro-organisms (see p. 79). Binding is detected by tagging the DNA with a radioactive label, or with a reagent that can be developed to give a colour reaction, such as biotin. By selecting DNA fragments specific for features characteristic of individual organisms, gene probes can be tailored to the rapid identification of individual species in clinical material. The disadvantages of this approach are that the organism itself may be dead and a viable isolate is not made available for subsequent tests of susceptibility to antimicrobial agents, toxin production or epidemiological investigation.
Culture and preliminary identification of bacteria in the laboratory is time consuming and relatively labour intensive. Bacteria may also be uncultivable, slow growing, or fastidious in nutrient requirements. Nucleic acid techniques for the detection and identification of bacteria have evolved against this background; today numerous commercially available systems have been developed and many are in use in diagnostic laboratories. The technologies fall into three basic groups:
-
1
Target amplification by PCR, transcription-mediated amplification, nucleic acid sequence-based amplification, etc.
-
2
Probe amplification using ligase chain reaction or Q-beta replicase.
-
3
Signal amplification, as in branched DNA assay.
It is possible to detect the presence of an increasing number of species either by PCR of universal or specific gene targets or by hybridization with specific probes. As with conventional methods, nucleic acid technology has its limitations, the most frequent being contamination of a sample by post-amplification products. Other factors include operator skill, primer design, stringency of assay, presence of inhibitory compounds in the specimen and the ubiquity of the organism sought. The latter is fundamental to the interpretation of results as many bacterial pathogens occur naturally as commensals in certain body sites. The new technologies do, however, offer a considerable advantage over phenotypic methods in terms of sensitivity, and many optimized PCR systems claim to be able to detect as few as two to ten bacteria per millilitre of specimen, which is far below the threshold of conventional culture.
Nucleic acid assays for antimicrobial resistance genes are also in use and development. A recently described innovation may have the potential to detect, identify the species, subtype the organism and identify resistance genes on microscope slide smears. The technique uses peptide nucleic acid (PNA, pseudopeptides) with DNA binding capacity. The PNA molecules have a polyamide backbone instead of the sugar phosphate of DNA and RNA, and nucleotide bases attached to this backbone are able to hybridize by specific base pairing with complementary DNA or RNA sequences. PCR amplification of target genes on conventionally stained microscope smears is also possible, and this accelerates the prospect of very rapid and sensitive test methods limited only by the specificity of the primer for the target.
The availability of numerous prokaryotic genomic sequences has enabled the development of high-density oligonucleotide arrays which consist of many thousands of different probes. These arrays are constructed by in-situ oligonucleotide synthesis on a glass support by photolithography or other methods. Hybridization of the prelabelled target nucleic acids to the bound probe is detected directly with fluorescein or radioactive ligands, or indirectly using enzyme conjugates. Areas of application for high-density arrays include DNA sequencing, strain genotyping, identifying gene functions, location of resistance genes, changes in mRNA expression and phylogenetic relatedness. Arrays with selected gene targets have recently been developed in an Eppendorf tube format. The chip is embedded in the bottom of the tube and carries optimized sets of oligonucleotide probes specific for certain organisms or for antimicrobial resistance genes or virulence factors. In this way chips can be customized for individual bacteria or groups of bacteria. All stages of the assay – sample preparation from agar-grown colonies, PCR amplification, hybridization, conjugation with reporter molecule and detection by automated image recording – are carried out in a single tube within 6–8 hours.
A widely used development is that of real-time PCR, which combines sample amplification with a means for detection of the specific product by fluorescence so that both steps take place conveniently in a single reaction tube. The system has significant advantages over conventional PCR in terms of rapidity, simplicity and number of manual procedures; contamination is effectively eliminated by the tube being closed after amplification. The DNA product may be detected with a fluorescent dye, or increased specificity obtained with hybridization with fluorescence-labelled sequence-specific oligonucleotide probes. Quantification of the target DNA is also possible with this system, allowing estimation of viral or bacterial numbers in a specimen (see p. 79–80).
Fluorescence in-situ hybridization (FISH) directed to the multiple copies of 16S ribosomal RNA has also been used to detect bacteria directly in clinical specimens. This technique utilizes probes specific for target organisms in the sample without the need for culture and allows quantification of the cell count and morphology. Sensitivity and specificity vary according to the probe used, but if the probe is optimized it can rival conventional culture methods.
Antibody reactions
Species and types of micro-organism can often be identified by specific serological reactions. These depend on the fact that the serum of an animal immunized against a micro-organism contains antibodies specific for the homologous species or type that react in a characteristic manner (e.g. agglutination or precipitation) with the particular micro-organism. Such simple in-vitro tests have been used for many years in microbiology, notably in the formal identification of presumptive isolates of pathogens (e.g. salmonellae) from clinical material. The specificity and range of antibody tests have been greatly improved by the availability of highly specific monoclonal antibodies. These are produced by the hybridoma technique in which individual antibody-producing spleen cells are fused with ‘immortal’ tumour cells in vitro. The progeny of these hybrid cells produce only the type of antibody appropriate to the spleen cell precursor (see p. 125).
Latex agglutination
By adsorbing specific antibody to inert latex particles, a visible agglutination reaction can be induced in the presence of homologous antigen. This principle can be applied in reverse to detect serum antibodies. Latex-based kits are widely used for serological grouping of organisms and detection of toxins produced by bacteria during growth.
Enzyme-linked immunosorbent assay
In enzyme-linked immunosorbent assay (ELISA), a specific antibody is attached to the surface of a plastic well and material containing the test antigen is added. After washing, the presence of the antigen is detected by addition of more of the specific antibody, this time labelled with an enzyme that can initiate a colour reaction when provided with the appropriate substrate. The intensity of the colour change is related to the amount of antigen bound. The ELISA method may also be used in the reverse manner for the quantitative detection of antibodies, by adsorbing purified antigen to the well before adding test serum; in this case the enzyme-linked system used to detect the antigen–antibody reaction is a labelled anti-human globulin. In immunoglobulin (Ig) M antibody capture ELISA (MAC-ELISA), widely used in virological diagnosis for the detection of IgM, anti-human µ-chain antibody (usually raised in goats) is bound to the well. The test serum is added, and any IgM binds to the capture reagent; after washing, purified antigen (e.g. rubella antigen) is added, and this can be detected with an appropriate labelled antibody.
Haemagglutination and haemadsorption
Certain viruses, notably the influenza viruses, have the property of attaching to specific receptors on the surface of appropriate red blood cells. In this manner the virus particles act as bridges linking the red cells in visible clumps. In tissue culture, such haemagglutinins may appear on the surface of cells infected with a virus. If red cells are added to the tissue culture, they adhere to the surface of infected cells, a phenomenon known as haemadsorption. Red blood cells can also be coated with specific antibody so that they agglutinate in the presence of the homologous virus particle in a manner similar to that described for latex agglutination above.
Fluorescence microscopy and immunofluorescence
When certain dyes are exposed to ultraviolet light, they absorb energy and emit visible light; that is, they fluoresce. Tissues or organisms stained with such a dye and examined with ultraviolet light in a specially adapted microscope are seen as fluorescent objects; for example, auramine can be used in this way to stain Mycobacterium tuberculosis. Antibody molecules can be labelled by conjugation with a fluorochrome dye such as fluorescein isothiocyanate (which fluoresces green) or rhodamine (orange–red). When fluorescent antibody is allowed to react with homologous antigen exposed at a cell surface, this direct immunofluorescence procedure affords a highly sensitive method for the identification of the particular antigen. For this procedure it is necessary to have a specific antibody conjugate for each antigen; however, unconjugated antibody can be used and the reaction then detected by the addition of an antiglobulin conjugate, which will react with any antibody from the species in which the antibody was raised.
Immuno-polymerase chain reaction
This technique arose out of the fusion of antibody technology with PCR methods with the aim of enhancing the capability of antigen detection systems. In immuno-PCR, a linker molecule with bispecific binding affinity for DNA and antibodies is used to attach a DNA molecule (marker) to an antigen–antibody complex. This produces a specific antigen–antibody–DNA conjugate. The attached DNA marker can be amplified by PCR with appropriate primers, and the presence of amplification products shows that the marker DNA is attached to antigen–antibody complexes, indicating the presence of antigen. The enhanced sensitivity of immuno-PCR achieved over ELISA is reported to be in excess of 105, theoretically allowing as few as 580 antigen molecules (9.6 × 10−22 moles) to be detected.
Typing of bacteria
Different bacterial species often exhibit different population structures. Some species are characterized by highly diverse populations at one extreme and closely similar members at the other. The frequency of recombination of chromosomal genes (see Ch. 6) is considered the major determinant of a population structure of a given species, and this frequency ranges from absent to low to very high. Highly recombining populations are termed panmictic, in contrast to clonal populations where recombination is infrequent (Table 3.3 ). Species such as Neisseria gonorrhoeae and Haemophilus influenzae are naturally transformable, that is, they are able to take up DNA (foreign and native) from their environment, and their populations are characterized by a high frequency of recombination, segregation of alleles and relatively low mutation. In clonal populations such as Salmonella enterica, recombination is rare and there is non-random association of alleles in a background of limited genetic exchange. Mutations occur as a result of natural and selective pressures, but these are not sufficient to disrupt the clonal lineage and daughter cells continue to resemble the ancestral parent. Bacterial clones are therefore not identical to their parents but display a number of characteristics in common with their ancestors. Many species are characterized by considerable genetic diversity but with clonal expansion of a subpopulation. Some of these clones may be transient, although others may persist and spread nationally and globally.
Table 3.3.
Panmictic versus clonal populations
| Reproduction | Recombination | Allele arrangement | Mutation | Selective pressures | |
|---|---|---|---|---|---|
| Panmictic | Sexual* | Frequent | Segregated | Normal | Natural selection |
| Clonal | Asexual | Rare | Non-random association | Normal | Environmental |
Refers to recombination between genetic elements from different organisms in bacteria.
By typing we identify a recognizable subdivision of a species that serves as a reference marker against which other isolates of the same species can be compared. A population of bacteria presumed to descend from a single bacterium, as found in a natural habitat, in primary cultures from the habitat, and in subcultures from the primary cultures, is called a strain. Each primary culture from a natural source is called an isolate. The distinction between strains and isolates may be important; for example, cultures of typhoid bacilli isolated from ten different patients should be regarded simply as ten different isolates unless epidemiological or other evidence indicates that the patients have been infected from a common source with the same strain. The ability to discriminate between similar strains may be of great epidemiological value in tracing sources or modes of spread of infection in a community or hospital ward, and various typing methods have been devised. Strains may be distinguishable only in minor characters and it is usually simpler to establish differences between isolates from a common source than unequivocally to prove their identity. Demonstration of an identical response by a single reproducible typing method is not proof that two strains are the same. However, the confidence with which similarity can be inferred is greatly increased if more than one typing method is used.
Typing may inform different levels of epidemiological investigation, ranging from micro-epidemiology (local investigation), macro-epidemiology (regional, national, international) to population structure analysis (evolution of strains and global patterns of spread). The data derived may assist in the control of infection by excluding sources, identifying carriers and establishing the prevalence of individual strains. Common reasons for microbial typing are to identify common or point sources, discriminate between mixed strain infections, distinguish re-infection from relapse, and occasionally to identify a type and disease association (e.g. Escherichia coli O157 and haemolytic uraemic syndrome, skin and throat types of group A Str. pyogenes, etc.).
Typing methods should be reproducible both in the laboratory and clinically. The former is easily established by repeated tests on a sample of experimental strains, but should also be established in vivo by examining multiple pairs of isolates from single sources to determine the stability of the strain characteristics probed by the typing method used. A typing method should also discriminate adequately and clearly between different populations and be comprehensive, that is, assign most populations to a type. The typing data should be in a format that is easily assimilated into databases and should be able to be incorporated into the national picture to inform other workers in the field. Very few, if any, single typing methods will meet these criteria and hence there is a need to utilize different methods, preferably directed at unlinked targets and always in the context of an epidemiological investigation.
Biotyping
Biochemical test reactions that are not universally positive or negative within a species may define biotypes of the species, and these may be efficient strain markers. In practice biotyping is often less discriminatory than other strain typing methods and may be unstable because of loss of the property. Differences among strains may also be detected by variations in sensitivity to fixed concentrations of chemicals such as heavy metals, a process known as resistotyping. The nutritional requirements of the isolate (amino acids) for growth may also be used to define the auxotype of an isolate.
Serotyping
Many surface structures of bacteria (lipopolysaccharide and outer membrane, flagella, capsule, etc.) are antigenic, and antibodies raised against them can be used to group isolates into defined serotypes. Some species are characterized by numerous antigenic types and serotyping for these species is highly discriminatory, whereas for others conservation of antigen epitopes renders serotyping of little value for epidemiological purposes. Members of the species Salmonella enterica are defined by their somatic and flagellar serotypes (see Ch. 24). Capsular antigens may be associated with pathogenicity of the organism, and many vaccines protect the individual against infection by stimulating antibodies to capsular antigen epitopes. Agglutination of bacterial suspensions with rabbit antibodies is the most commonly used method for typing, but other techniques such as precipitation in agar gels, ELISA and capsular swelling (Fig. 3.2 ) may be used.
Fig. 3.2.
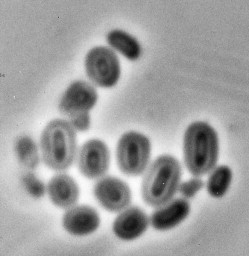
Capsular swelling reaction of Klebsiella pneumoniae. Antibody adsorbed to capsule alters the refractive index, allowing visualization of the capsule around the cell within.
Phage typing
Bacteria often show differential susceptibility to lysis by certain bacteriophages. The phage adsorbs to a specific receptor on the bacterial surface and injects its DNA into the host. Phage DNA may become stably integrated into the bacterial chromosome and this state is referred to as lysogeny; phages capable of this are called temperate phages. In lysogeny, a small proportion of host cells express the phage genes, and some cell lysis and liberation of phage progeny occurs. Alternatively, the phage DNA may enter a replicative cycle, leading to the death of the host and the production of new phage particles. These lytic or virulent wild phages lyse the bacterium at the end of the replicative cycle and release a large number of daughter phage particles that infect neighbouring cells.
This process leads to visible inhibition of the growth of the host cells (Fig. 3.3 ). The phage type of the culture is identified according to the pattern of susceptibility to a set of lytic and/or temperate phages. Lytic phages may be readily recovered from sewage, waste and river water, and temperate phages may be released from a lysogenic strain by induction with ultraviolet radiation or chemical mutagens.
Fig. 3.3.
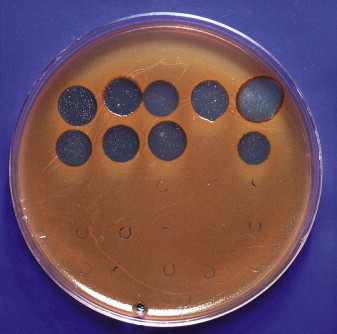
Phage-mediated lysis of red pigmented strain of Serratia marcescens.
The critical factors governing the interpretation of phage typing results are discrimination and reproducibility. If the system is both highly discriminatory and reproducible, any differences in lysis patterns of isolates will indicate that they represent different strains. Schemes that utilize adapted phages (a single phage propagated in different strains) that are specific for a particular receptor site such as the Vi polysaccharide of Salmonella enterica serotype Typhi are relatively reproducible, and minor differences are significant and reproducible. On the other hand, a phage set comprising random unrelated phages may adhere to a number of different receptor sites and have different biological properties, and so is unlikely to be highly reproducible but may be adequately discriminating. This lack of stability results in the definition of broad phage groups rather than defined types, and is the case for Staph. aureus where at least two strong lytic reactions have to be present in the patterns of isolates before they can be termed distinct strains.
Bacteriocin typing
Bacteriocins are naturally occurring antibacterial substances, elaborated by most bacterial species, that are active mainly against strains of the same genus as the producer strain. Bacteriocin typing may define the spectrum of bacteriocins produced by field strains, or the sensitivity of these strains to bacteriocins of a standard panel of strains. Patterns of production or susceptibility to bacteriocins allow the division of species into bacteriocin types.
Protein typing
Bacteria manufacture thousands of proteins that can be visualized by electrophoresis in acrylamide gels in the presence of a strong detergent. The proteins separate according to molecular size and, after staining with a dye, the pattern of bands from each isolate can be compared. This system has been used successfully to type many bacterial and fungal species, but lacks reproducibility. Investigation of microbial populations by gel electrophoresis of metabolic enzymes, which can then be detected by specific substrates, has also in the past been applied widely for clonal analysis within species.
Restriction endonuclease typing
Restriction endonucleases are a family of enzymes that each cut DNA at a specific sequence recognition site, which may be rare or frequent in the DNA of the species being examined. The frequency with which an enzyme cuts in a particular species is dependent on the oligonucleotide sequence, the frequency of the restriction site, and the percentage G + C content of the species. For example, the recognition site of enzyme SmaI is 5′-CCC↓GGG-3′ (↓ site of cleavage) and this cuts infrequently in the AT-rich genome of Staph. aureus, whereas enzyme XbaI (5′-T↓CTAGA-3′) is a rare cutter in most Gram-negative species with a high GC content. Both plasmid and chromosomal DNA can be analysed by this means. Frequent-cutting endonucleases generate numerous small fragments that can be resolved by conventional electrophoresis in agarose gel and detected by staining with a dye. The resolution of conventional agarose gel electrophoresis does not exceed 20 kb and optimal separation in standard length gels is achieved between 1 and 15 kb.
The large DNA fragments produced by infrequent-cutting enzymes need to be separated in special electrical fields with a pulsed current (pulsed-field gel electrophoresis; PFGE). In this technique, bacteria are encased in an agarose plug (to minimize shearing of DNA) and the cells are digested with proteinase K enzyme before the DNA is digested with the enzyme. By introducing a pulse or change in the direction of the electric field, fragments as large as 10 Mb can be separated. The time taken by fragments to reorient to the alternate electric field is proportional to their molecular size and where they migrate in the electric field. The most widely used apparatus is the contour-clamped homogeneous electric field (CHEF), which has 24 electrodes arranged in a hexagonal array. Run times are often of the order of 30–40 hours, but shorter, more rapid, protocols have been described. A number of factors influence the quality of results, including DNA quality and concentration, agarose concentration, voltage and pulse times, and buffer strength and temperature.
Interpretation of PFGE profiles can be problematic. For some species the criteria of Tenover (see recommended reading list) can be applied to establish the significance of differences in banding profiles of strains. As a rule of thumb, isolates from an incident under investigation that show no difference in profiles can be considered indistinguishable, those with one to three band differences as closely related, four to six bands as possibly related, and seven or more band differences as indicating distinct strains. However, this rule should be applied with a degree of caution as some species (e.g. Enterococcus faecium) can exhibit significant variation (six to ten band differences) apparently within members of the same clone. A number of computer-assisted analysis packages are available that calculate coefficients of similarity between strains and represent these as dendrograms (Fig. 3.4 ). Two commonly employed coefficients, the Jaccard and Dice, use the number of concordant bands in profiles and the total number of possible band positions to calculate the percentage similarity between the isolates. The Pearson coefficient gives the advantage that specific band positions do not have to be defined. A cut-off point of 85% similarity is often used but, as for the band difference rule, this should be set by experiment with related and unrelated strain sets.
Fig. 3.4.
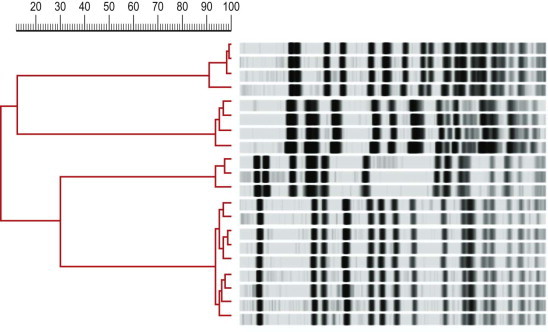
Pulsed-field gel electrophoresis profiles of Xbal digests of DNA of Ps. aeruginosa isolates. Dendrogram shows relative percentage similarity of profiles calculated with Pearson's coefficient.
Gene probe typing
DNA probes (see above) for strain typing consist of cloned specific, random or universal sequences that can detect restriction site heterogeneity in the target DNA. The detection of variation in rDNA gene loci is the basis of ribotyping, and this method has been universally applied to the typing of various species. Other commonly used probes are insertion sequences (lengths of DNA involved in transposition; see Ch. 6) that may define clonal structures of populations.
Polymerase chain reaction typing
PCR is a technique that allows specific sequences of DNA to be amplified. Multiple copies of regions of the genome defined by specific oligonucleotide primers are made by repeated cycles of amplification under controlled conditions. Such methods can be used to study DNA from any source. Several variations on the PCR theme have been described, and use of these techniques continues to expand and develop. PCR-mediated DNA fingerprinting makes use of the variable regions in DNA molecules. These may be variable numbers of tandem repeat regions or areas with restriction endonuclease recognition sequences. To perform PCR typing, it is necessary to know the sequences of the bordering regions so that specific oligonucleotide primers can be synthesized. Primers may be specific for a known sequence or be random. Random primers are extensively used in the techniques of random amplification of polymorphic DNA (RAPD) and arbitrarily primed PCR (AP-PCR). Both of these approaches have problems with reproducibility as a result of false priming, faint versus sharp bands and variation in electrophoretic migration of products. Repetitive sequence-based PCR (rep-PCR) indexes variation in multiple interspersed repetitive sequences in intergenic regions dispersed throughout the genome. An automated, standardized rep-PCR system has proven useful for strain typing of a number of species and is reported to give similar discrimination to PFGE (Bacterial Barcodes, Houston, Texas, USA). Amplified fragment length polymorphism is a DNA sequence-based technique that combines restriction endonuclease digestion with PCR. Incorporation of a fluorescent label and the use of a capillary DNA sequencer allows optimal standardization of reproducibility and resolution of single base-pair differences between genomes.
Multilocus sequence typing
This technique indexes allelic variation in several housekeeping genes by nucleotide sequencing rather than indirectly from the electrophoretic mobilities of their gene products, as was the case with its parent technique, multilocus enzyme electrophoresis. Housekeeping genes are not subject to selective forces as are variable genes and they diversify slowly. Multiple genes (usually seven) are employed to overcome the effects of recombination in a single locus, which might distort the interpretation of the relationship of the strains being compared. Multilocus sequence typing (MLST) can rightly be referred to as definitive genotyping as sequence data are unambiguous and databases of allelic profiles of isolates of individual species are accessible via the internet. The level of discrimination of MLST depends on the degree of diversity within the population to generate alleles at each locus, but some highly uniform species such as M. tuberculosis are not amenable to analysis by the technique. Recently, increased discrimination has been sought in virulence-associated genes necessary for survival and spread of the organism on the basis that these genes are exposed to frequent environmental changes and thus provide a higher degree of sequence variation. Intergenic regions of selected genes are amplified by PCR and a 500-bp internal fragment sequenced to identify allelic polymorphisms.
A variant of MLST termed multilocus restriction typing introduces restriction digestion of amplified housekeeping genes and removes the need for sequencing. The restriction fragment length polymorphisms (RFLPs) can be sorted into type patterns and reveal population structures similar to those with MLST.
Variable number tandem repeat analysis
Variable number tandem repeats (VNTRs) are short nucleotide sequences (20–100 bp) that vary in copy number in bacterial genomes. They are thought to arise through DNA strand slippage during replication and are of unknown function. Separate VNTR loci are identified from published sequences and are often located in intergenic regions and annotated open reading frames. Primers are designed to amplify five to eight loci and the products sequenced to generate a digital profile. VNTR typing is rapid and reproducible, and relatively simple to perform. Improved discrimination may be achieved by identification of more loci but there is debate about their stability over time.
Whole genome based typing
The advent of new high throughput DNA sequencing methods is ushering in a new era in which it is becoming feasible to compare and type bacteria based on entire genome sequence data. These massively parallel sequencing technologies produce relatively short nucleotide sequence reads but on such a scale that these can be assembled into a sequence matched against those obtained from previous isolates of that organism. This enables a genome-wide comparison to be made and a more or less definitive evolutionary relationship to be established to other contemporaneous and historical isolates. The costs of such analyses are rapidly becoming competitive with traditional typing methods. Such analyses have the potential to transform medical bacteriology by producing unambiguous epidemiological information and by identifying genetic elements such as those encoding antibiotic resistance and significant antigens under selection pressures.
Recommended reading
- Barrow GI, Feltham RKA, editors. Cowan and Steel's Manual for the Identification of Medical Bacteria. ed 3. Cambridge University Press; Cambridge: 1993. [Google Scholar]
- Garrity GM, editor. Bergey's Manual of Systematic Bacteriology. ed 2. Springer; New York: 2005. [Google Scholar]
- Kaufmann ME. Pulsed-field gel electrophoresis. In: Woodford N, Johnson AP, editors. Methods in Molecular Medicine, Vol. 15: Molecular Bacteriology: Protocols and Clinical Applications. Humana Press; Totowa, NJ: 1998. pp. 33–50. [Google Scholar]
- Murray PR, Baron EJ, Jorgensen JH, editors. Manual of Clinical Microbiology. ed 8. ASM Press; Washington, DC: 2003. [Google Scholar]
- Owen RJ. Bacterial taxonomics: finding the wood through the phylogenetic trees. Methods in Molecular Biology. 2004;266:353–384. doi: 10.1385/1-59259-763-7:353. [DOI] [PubMed] [Google Scholar]
- Schleifer KH. Classification of Bacteria and Archeaea: Past, present and future. Systematic and Applied Microbiology. 2009;32:533–542. doi: 10.1016/j.syapm.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Spratt BG, Feil EJ, Smith NH. Population genetics of bacterial pathogens. In: Sussman M, editor. Molecular Medical Microbiology. Academic Press; San Diego: 2002. pp. 445–484. [Google Scholar]
- Tenover FC, Arbeit RD, Goering RV. Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for strain typing. Journal of Clinical Microbiology. 1995;33:2233–2239. doi: 10.1128/jcm.33.9.2233-2239.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Regenmortel MHV, Fauquet CM, Bishop DHL, editors. Virus Taxonomy. Classification and Nomenclature of Viruses. Academic Press; San Diego: 2000. [Google Scholar]
- Woese CR. Interpreting the universal phylogenetic tree. Proceedings of the National Academy of Sciences of the USA. 2000;97:8392–8396. doi: 10.1073/pnas.97.15.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
Websites
- http://www.mlst.net/ Genotyping database at Oxford University.
- http://www.ncbi.nlm.nih.gov/Genbank/ National Center for Biotechnology information for rRNA sequence analysis.
- http://www.ridom-rdna.de/ Ribosomal differentiation of medical microorganisms for rRNA sequence analysis.
- http://www.ictvdb.org/ Universal virus database of the International Committee on Taxonomy of Viruses.