

Abstract
Rapid advances have been made in developing analytical technologies for characterization of highly heterogeneous active ingredients of complex drugs, such as pentosan polysulfate (PPS), active ingredient of the drug Elmiron®, approved by the Food and Drug Administration and marketed in the United States to treat interstitial cystitis. PPS sulfated polysaccharides comprise of a repeat unit of β(1—4)-D-xylopyranoses randomly substituted by 4-O-methyl-glucopyranosyluronic acid. To define the critical quality attributes (CQAs) of such a complex drug, it is critical to develop an approach that integrates data from orthogonal analytical methodologies. Here, we developed an approach integrating diverse analytical tools including gel permeation chromatography, LC/ESI-MS and NMR to measure CQAs of PPS. The proposed mathematical framework integrates the data from these diverse analytical methods as function of PPS chain length and building blocks. Our approach would facilitate in establishing a scientific foundation for comparative characterization of drug products with complex active ingredients.
Keywords: pentosan polysulfate, sulfated polysaccharide, structural elucidation, NMR, LC-MS, molecular weight distribution
1. Introduction
Pentosan polysulfate sodium (PPS) is a complex mixture of heterogeneous sulfated polysaccharides and therefore several studies have compared this mixture to heparin and heparin sulfate glycosaminoglycans (HSGAGs) in terms of similarity in anticoagulant, anti-inflammatory and anti-allergic properties (Fisher et al., 1982; Mulholland, Hanno, Parsons, Sant, & Staskin, 1990; Parsons et al., 1993; Sanden et al., 2017). However, unlike HSGAGs which are naturally sulfated, PPS is generated through a semisynthetic process of sulfation of 4-methyl glucuronoxylans derived from German beechwood. The PPS backbone comprises of repeating β-D-xylopyranose (Xyl) units linked 1-4 to each other and this linear polyxylose backbone is interspersed with a 4-O-methyl-glucuronic acid (MGA) that is α1-2 linked to the xylose sugar. The 2-O and 3-O hydroxyl positions of the Xyl residues in xylans derived from the beechwood can also be acetylated (Scheller & Ulvskov, 2010). The MGA can be considered as a monosaccharide branch from linear xylan backbone and its content can vary from source to source (Wang, Lee, & Armstrong, 1999; Jacobs, Larsson, & Dahlman, 2001). PPS is produced by an exhaustive and non-selective O-sulfation of hardwood glucuronoxylan leading to the prevalent formation of PPS chains with 2,3-di-O-sulfated Xyl/MGA sugars (Fig.S1). In some of the Xyl sugars including those with α1-2-linked MGA, the O-acetylation (specifically at the 3-O position) from the parent backbone xylans is likely to be retained (De Ferra, Naggi, Zenoni, & Pinto, 2014). The high degree of sulfation of PPS makes it a highly anionic polymer.
The anionic nature of PPS has led to tapping its potential for use in hydration of joints and enhancing mechanical strength to treat diseases such as osteoarthritis in animals (Read, Cullis-Hill, & Jones, 1996; Budsberg, Bergh, Reynolds, & Streppa, 2007; Kramer et al., 2014). Given the high degree of sulfation, PPS is also being investigated for its potential application to restore the extracellular matrix in cartilage in viral infection-induced arthritis (Herreroa et al., 2015).
PPS is the active pharmaceutical ingredient of Elmiron® which is an oral capsule drug approved by the FDA to treat interstitial cystitis / Bladder Pain Syndrome (IC/BPS), a chronic condition that causes painful urinary symptoms. The mechanism of action of PPS in the treatment of IC is not well understood. It has been proposed that the charge distribution properties from sulfation of PPS plays a role in replacing or repairing the GAGs on bladder epithelia and minimizes permeation of urinary bacteria or toxins (Schamhardt, de Boer, & Kurth, 1994). PPS-based Elmiron® drug for IC treatment was initially approved in the USA by FDA in 1996 (FDA approved drugs: Pentosan polysulfate sodium, NDA reference number 020193. (1996), available from https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm), and recently the US FDA has published a product specific guidance for developing generic version of PPS (FDA, 2012, https://www.govinfo.gov/content/pkg/FR-2012-09-20/pdf/2012-23177.pdf). This guidance was revised from the original draft guidance with emphasis on demonstrating sameness at various levels of heterogeneity, including at least three criteria: (1) physiochemical properties, (2) monosaccharide building blocks and MGA branching, and (3) properties of the xylan starting material.
The analytical methods for detailed characterization of PPS in the context of its heterogeneity are not as well developed as those employed for characterizing heparins and low molecular weight heparins (LMWHs). The only published method to distinguish PPS from different manufacturers is based on capillary zone electrophoresis which does not capture the fine structural nuances in the heterogeneity of PPS (Degenhardt, Benend, & Wätzig, 1998; Degenhardt, Ghosh, & Wätzig, 2001). The use of one- and two-dimensional NMR spectroscopy to monitor the process for generating sulfated xylans (including preservation of O-acetyl groups) and analyzing the xylan backbone of PPS after its chemical desulfation have been described in patent applications (De Ferra et al., 2012). Also, several analytical methods such as colorimetric analyses, size exclusion chromatography (SEC), NMR and MALDI-TOF analyses have been developed for detailed structural characterization of xylans from various plant sources (Teleman, Nordstrom, Tenkanen, Jacobs, & Dahlman, 2003; Urbanowicz et al., 2012; Zhong, Teng, Lee, & Ye, 2014) and many of these methods can be applied to characterization of PPS. Recently, a MS/MS approach was applied to PPS analysis (Lin et al., 2019).
In this paper we describe in detail different analytical methods and the orthogonal measurements on key structural attributes of PPS across different layers of heterogeneity including physiochemical properties, individual monosaccharide building blocks and higher order structures for the first time. Specifically, we show deep cross-correlations between key structural attributes between orthogonal measurements such as LC-MS, NMR analyses of the entire mixture and its preparative size fractions These cross correlations critically capture structural fingerprints of the process steps to generate PPS. In summary, the results presented here offer detailed scientific insights and considerations for characterization of PPS.
2. Materials and methods
2.1. Starting materials and reagents
Three commercial Elmiron® batches (PPS 1 lot 14KG449 and PPS 2 lot 14JG315, PPS 3 lot 14NG729) were purchased in the United States and were used in this study. Ammonium acetate, sodium azide, sodium nitrate, sodium dihydrogen phosphate monohydrate, sodium hydrogen phosphate dihydrate, trimethylsilyl-3-propionic acid (TSP), dibutylamine (DBA, ≥99.5%, LC-MS grade), glacial acetic acid (99.9% LC-MS grade), methanol (LC-MS grade), ammonium chloride, sodium nitrite (>95%), sodium tetraborate, hydrochloric acid (≥37%), trifluoroacetic acid (≥99.5%), sulfuric acid (95-97%) were purchased from Sigma Aldrich (Milan, Italy); calcium acetate (>97%) from BDH; sodium acetate and NaOH from Merck (Kenilworth, NJ, USA); Amberlite IR 120 H+,0.1 M NaOH and dimethylsulfoxide from Fluka Analytical (Milan, Italy); Amberlite IR45 from Carlo Erba (Milan, Italy) pyridine from Riedel-de Haën (Germany); phenol (>99.5%). Ethanol (96%) was purchased from Girelli Alcool (Milan, Italy); deuterated ethylenediaminetetraacetic acid (EDTA-d16) from Cambridge Isotope Laboratories (Tewksbury, MA, USA) and deuterium oxide (99.9%) from Euriso-top (Saint-Aubin, France). Deionized water (conductivity less than 0.1 μS) was prepared with an osmosis inverse system (Culligan, Milan, Italy). PolyCAL ™ Pullulan std-57k (Malvern Instruments LtD, Malvern, United Kingdom). When not specified, the reagents are ≥98%.
2.2. Sample preparation to extract PPS from Elmiron drug capsule
Prior to structural characterization, each Elmiron® sample underwent a water extraction procedure in order to separate PPS material from insoluble cellulose component of the drug (the presence of semicrystalline cellulose was independently confirmed by solid state NMR, not shown). The content of one capsule (about 250 mg) was suspended in 10 ml of water and stirred at room temperature for 16 h. After filtration on a mixed cellulose esters membrane with 0.22 μm cut-off (Millipore GSWP04700), water soluble portion of each sample was freeze dried for further analysis. The amount of the extracted PPS was about 100 mg.
2.3. Determining degree of sulfation using conductometric titration
The sulfation degree of all samples, expressed as sulfate-to-xylose molar ratio, was determined in duplicate by conductimetric titration adapting the method proposed by Casu and Gennaro (Casu & Gennaro, 1975). Experimental details are reported in the footnote to Table S1.
2.4. Determination of total sugar and glucuronic acid content by colorimetric assays
A phenol assay was adapted (Dubois, Gilles, Hamilton, Rebers, &. Smith, 1951) for determining the total sugar content in each PPS sample, using xylose monosaccharide solution for calibration.
Glucuronic acid content was determined following a previously developed method (Bitter & Muir, 1962). Experimental details are reported in the footnote to Table S1.
2.5. Molecular weight distribution by size exclusion chromatography with triple detector array (HP-SEC-TDA)
Measurements for molecular weight distribution were performed on a Viscotek 305 HPLC system (Houston, Texas) equipped with a triple detector array exploiting simultaneous action of refraction index detector (RI), viscometer and Right Angle Laser Light-scattering (RALLS) detector, using a method adapted from Bisio et al. (2015).
Prior to the analysis of three PPS batches RI increment dn/dc was determined in order to convert the RI response to concentration (Bertini, Bisio, Torri, Bensi, & Terbojevich, 2005). For this type of polysaccharide dn/dc was found to be 0.093 ml/g.
2.6. Preparative size exclusion chromatography
In order to isolate size specific fractions of PPS about 120 mg of water soluble PPS material dissolved in 2.5 ml of water were loaded onto a chromatographic Knauer Bioline system equipped with two Superdex S30 columns (48cm x 3 cm, and 83cm x 2 cm, GE Healthcare Life Sciences) and 0.25 M ammonium chloride as eluent. Separation was performed at room temperature and a flow-rate of 5 ml/min. UV detection at 260 nm was used to monitor the fractionation profile. For each fractionated PPS sample thirteen fractions with decreasing molecular weight from A1 to N were recovered (Fig.S2). Each fraction was desalted on a Biofox 40/100 SEC Agarose column (3.0 cm x 90 cm, Knauer) using 10% aqueous ethanol with a flow-rate of 15 ml/min and UV monitoring at 210 nm.
2.7. Sodium hydroxide treatment of PPS sample
About 40 mg of PPS material were dissolved in 2 ml of 0.01M NaOH. After 16 h under stirring at room temperature, the solution was neutralized with 0.1M HCl.
2.8. 1D and 2D NMR analyses
Spectra were obtained with a Bruker AVANCE IIIHD 500 MHz spectrometer (Bruker, Karlsruhe, Germany) equipped with a 5 mm TCI cryo-genic probe, at 303K. Spectra were processed with BrukerTopspin software version 3.2. About 20 mg PPS samples were dissolved in 0.6 ml of deuteriumoxide. 1H NMR spectra were aquired with presaturation of residual HDO, using 16 scans, 12s relaxation delay, number of time domain points of 32k. COSY and TOCSY were acquired with mixing time of 90 ms. HSQC and HMBC data were acquired in the phase-sensitive mode by using time proportional phase incrementation (TPPI) or States-TPPI experiment. Spectra were collected as 256/512 TD experiments each with 2k complex data points and 8 scans over a spectral width of 5 kHz.
2.9. Quantitative HSQC NMR
Between 45 and 55 mg of PPS samples were dissolved in 0.6 ml 0.15 mmol phosphate buffer solution (pH 7.1) containing 0.3 mmol EDTA d-16 in deuterium oxide with 0.002% TSP.
Quantitative HSQC experiment set up was based on the peak volume, which is dependent on the correlation peak according methods previously described on heparin (Guerrini, Naggi, Guglieri, Santarsiero, & Torri, 2005; Mauri et al., 2017). A 8s relaxation delay and 0.1s acquisition time were found sufficient to provide quantitative results. 1JC-H tune value was set to 170Hz. CVs were calculated from pooled standard deviation by two sets of determinations, each obtained from different operator or from replicates.
2.10. Reverse phase HPLC with ion pairing agent coupled to ESI (Q-TOF and FT-ICR) MS analyses of intact, low and high molecular weight components of PPS
The intact PPS samples and high molecular weight fractions, obtained by preparative SEC, were analysed on a HPLC system (Agilent 1100) coupled with an ESI-ICR-FT mass-spectrometer Solarix 7T (Bruker Daltonics). The chromatographic separation was performed using a Kinetex C18 analytical column (100 × 2.1 mm I.D., 2.6 μm particle size, Phenomenex). Eluent A (10 mM DBA, 10 mM acetic acid in water) and eluent B (10 mM DBA and 10 mM acetic acid in methanol) were delivered at 0.1 ml/min. The separation gradient was optimized for the separation of oligosaccharides from degree of polymerization (dp) 1 to 15 for intact PPS (up to at least dp 26 for high molecular weight PPS fraction). The final gradient was following: the solvent composition was held at 35% B for the first 3 minutes, then increased to 55% B over 2 min, and to 75 % B over another 35 min, where it was held at 75% for 20 min; afterwards, the content of the eluent B was increased to 98 % in 30 min, where it was held for 10 min in order to elute the longest components; finally, it was returned to 35% B over 2 min, and held for the last 23 min for equilibrating the chromatographic column before the injection of the next sample. The injection volume was 5 μl.
MS detection was performed using the following parameters: ESI in negative ionization mode, drying gas temperature +180°C and flow-rate 3.7 l/min, nebulizer pressure 0.9 bar; and capillary voltage +3.2 kV. The mass spectra of the oligosaccharides were acquired in a full scan mode (m/z scan range 200 – 3000) with time domain 1M and transient length 0.98 sec; average spectra parameter was 2. An external calibration method using sodium formate clusters as calibrants was used to achieve a good accuracy: the major species were detected with the accuracy < 5 ppm.
The monoisotopic m/z values of oligosaccharides with polymerization degree <6 and average mass m/z values of higher oligomers were used for obtaining extraction ion chromatograms (EICs) for various structural families.
Low molecular weight fraction, obtained by preparative SEC and enriched in oligomers with dp 3, was analyzed on a HPLC system Ultimate 3000 (Dionex) coupled with ESI-Q-TOF mass detector MicrOTOFq (Bruker Daltonics), using the same chromatographic column and eluents as the intact samples. Separation gradient was optimized for separating low molecular weight components (dp2-dp4) as following: the eluent composition was held at 37% B for the first 5 min, then increased to 46% B over 15 min, where it was held for other 15 min; afterwards, the content of the eluent B was increased to 90% in 55 min, where it was held for 10 min; finally, it was returned to 37% B over 2 min, and held for the last 23 min for equilibrating the chromatographic column before the injection of the next sample.
MS spectra were acquired in negative ion mode with m/z scan range 200 – 2000, drying gas temperature +180°C and flow-rate 7.0 l/min, nebulizer pressure 0.9 bar, and capillary voltage +3.2 kV. An external calibration with sodium formate clusters was used to achieve a good accuracy (< 5 ppm).
2.11. Estimating PPS attributes within credible intervals using a mathematical model.
Within the constrained optimization framework, the chain length model can be stated as follows: given the constraints imposed by the fractionation and a suitable loss function, find the possible distribution(s) satisfying the constraints. In general, there still may be a range of chain length distributions that explain the data well, and this range (rather than a single distribution) is considered to characterize the mixture along this dimension. The loss function chosen for the PPS mixture finds the smoothest function satisfying the constraints:
A is a (number of Measurements X f modeled chain lengths) matrix relating the quantity of interest a to the measurements, and b is the observed measurements. The experimental constraints for this model are obtained from the molecular weight distribution from SEC-TDA analyses. There are multiple possible objective functions (to use in place of 4) for the constrained minimization problem for determining the optimal value of a, including an objective function that gives bounds to the possible values of ai. The modifications can be treated at the level of xylose residue which can be modified at the reducing (RE) and nonreducing end (NRE), with sulfation/acetylation or MGA branching. Each of these modifications can be modeled as a stochastic feature function M1’(L;λ1), M2’,3’(L;λ2’,3’), M4’(L;λ4) where L is the chain length and λ are the parameters of the model associated with the distribution. The experimental data on relative abundances corresponding to all the observed modifications are available only for smaller oligosaccharide chain lengths (2-12 mers) and therefore only these datasets will be used to estimate the parameters of the feature functions. We use a Bayesian framework to obtain distributions over the parameters λ_, making each M_ a Bayesian hierarchical model relating the experimental observations to the parameters. The mean squared error (MSE) estimator for the value of the parameters is then determined using optimization, for example in the case of 1’ modifications:
Here Obs1’(L) is the experimental data for length L, and the expectation is taken over the prior distribution of the parameters π(λ1).
To demonstrate the value of orthogonal datasets using a mathematical model, we estimated representative structural attributes using data from orthogonal measurements. The specific structural attributes considered in this model are MGA-Xyl[3S], MGA-Xyl[3A], or Xyl[2S,3{S,A}], and the model uses a Bayesian framework to estimated parameters using the observed LC-MS and NMR datasets.
The model establishes a relationship between the relative bulk mixture quantities of the building blocks MGA-Xyl[3A] ( fGA), MGA-Xyl[3S] (fGS), and Xyl[2S,3{S,A}] (fX) as observed by NMR, and the fraction of chains at various number of sugars (L), cGA, cGS, cX respectively as observed by LC-MS, using the following equations:
| (1) |
| (2) |
| (3) |
In (1)-(3), N[L] is the total number of chains of length L. Equation (2) uses the assumption that there is at most 1 MGA per chain in the given chain lengths that we are simulating (up to 12 sugar units). A hierarchical Bayesian framework was used to enable the characterization of variation at both the lot-lot and measurement levels Fig. S3.
Given that all observed variables are compositional in nature, an isometric log ratio transformation is used to transform the data from compositional space to coordinate space so that distributions on RD can be used. Briefly, the coordinates for each lot are assumed to be drawn from a normal distribution with mean μ and covariance Σ, each of which are drawn from distributions with hyperparameters as shown in Fig. S3. LC-MS (yLCMS ) and NMR (yNMR) observations are made on each lot, and these observations are a function of the lot coordinates . The LC-MS coordinates are calculated from the lot coordinates (with using equation (3). Given the small sample size, point estimates based on the data are used for the covariance matrices Σnmr and Σlcms.
The model was simulated using Markov Chain Monte Carlo (MCMC), and posterior distributions for the parameters and posterior predictive samples of the NMR and LC-MS data were recorded. MCMC was performed using the Metropolis algorithm in pymc3. 10000 samples were generated, and the only the final 4000 used for analysis to ensure sampling of the highest probability region. Code for the data ingestion, simulation, and data visualization was all written in python, and on python3.6.5.
3. Results
As PPS is a complex mixture of sulfated polysaccharides, the attributes that provide a detailed picture on its heterogeneity can be described at different levels of structural complexity. In the following we present the analyses and the attributes measured at three levels for PPS extracted from three different lots of the Elmiron drug and answering the first two criteria mentioned above: (1) physiochemical properties, (2) monosaccharide building blocks and MGA branching. As for other polysulfated polysaccharide enoxaparin, at the first, and highest, level are the gross physicochemical attributes such as molecular weight distribution, total sugar content and degree of sulfation (Lee et al., 2013). At the second or middle are the individual monosaccharide building blocks, their modifications and relative abundance. Finally, the third level is how the attributes are distributed in different polysaccharide chains that constitute the mixture.
3.1. Gross physiochemical attributes of PPS
3.1.1. Total sugar and glucuronic acid content and overall sulfation degree
Using colorimetric assays for determining total sugar and glucuronic acid content and conductometric titration to measure overall degree of sulfation, it was demonstrated that there is a tight range in the variation of these attributes in PPS across different drug lots (Table S1). The uronic acid content represents the total MGA content, which is about 5 (%, w/w), the total sugar content is about 40% and finally the sulfation degree, expressed as molar ratio of SO3Na substituents obtained from conductometric titration, and xylose content, obtained from the difference of the total sugar amount (phenol assay) and the uronic acid content (carbazole assay) is around 2.2.
3.1.2. Molecular weight distribution of PPS
Size Exclusion Chromatography (or HP-SEC-TDA) coupled with analysis on multi-detector systems to capture light scattering, refractive index and viscosity a has been used extensively to obtain molecular weight distribution of oligosaccharides such as Low molecular weight heparins, and polysaccharide such as unfractionated heparins (Bertini et al., 2017; Bisio et al., 2015). An important consideration in accurately defining parameters that capture molecular weight distribution by light scattering is the dn/dc value. Since there is no published data on molecular weight distribution analysis for PPS, this value was independently determined in this study and presented for the first time (see section 2.5). The Mw (weight average molecular weight), Mn (number average molecular weight) and polydispersity (expressed as Mw/Mn ratio) values obtained for three PPS samples are reported in Table 1. Additionally, the w/w percentages of different size species with the selected Mw limits (i.e. <3kDa, 3-8kDa >8 kDa) were also compared for all the analyzed PPS samples (Table 1), following the strategy analogues to the one that European Pharmacopeia applies for heparin/LMWH characterization (Bertini et al., 2017; European Pharmacopeia, 2014). Such an approach could provide more information about molecular weight distribution for such a complex polymer mixture and, especially, allow more detailed comparison between different drug preparations. Results reported in Table 1 reveal small variations in molecular weight distribution between three analyzed samples, considering the method repeatability, evaluated as is 1.6% and 3.2% for Mw and Mn respectively, evaluated by validation of HP-SEC-TDA (data not shown).
Table 1.
Molecular weight distribution of three PPS obtained by HP-SEC-TDA
| Sample | Mw, kDa | Mn kDa | Pd (Mw/Mn) | % for the selected Mw limits (Da) |
||
|---|---|---|---|---|---|---|
| <3000 | 3000-8000 | >8000 | ||||
| PPS 1 | 6.6 | 4.1 | 1.6 | 29.3 | 47.9 | 22.9 |
| PPS 2 | 6.6 | 4.0 | 1.7 | 26.7 | 50.8 | 22.6 |
| PPS 3 | 6.3 | 3.9 | 1.6 | 28.1 | 50.3 | 21.6 |
Mw – weight average molecular weight, Mn – number average molecular weight, Pd – polydispersity
The obtained Mw ad Mn values were rounded to the nearest 100 Da.
Results refer to the mean values of duplicate injections
3.2. Individual monosaccharide building blocks, their modifications and relative abundance
3.2.1. Peak assignments from 1-D 1H and 13C and 2-D NMR spectroscopic analysis of PPS
The 1H and 1H-13C HSQC PPS spectra are shown in Fig. 1, respectively while 1H and 13C chemical shifts of the assigned monosaccharide residues of PPS are reported in Table 2. As expected, major signals correspond to the prevalent repeating unit: 2,3-di-O-sulfate β–(1, 4) xylose, including the signals at 5.39 and 5.10 ppm assigned to its unsubstituted hydroxyl group in α and β anomers (De Ferra et al., 2014). Several minor signals associated with particular structural features of PPS were also detected in the 1H NMR. Among those, signals were assigned to a modification of the RE xylose involving an unsaturated linkage between C2 and C3 (ΔXylred) and its corresponding open ring aldehyde - ΔXylred(CHO) by HSQC, H2BC and HMBC (Fig.S4a). After a treatment of PPS with sodium hydroxide, an increase of signals attributed to H1 of ΔXylred and ΔXylred(CHO) was observed (Fig.S4b). At the same time, the H1 Xylα signal dropped, confirming that the observed modification is related to the RE unit. These data are in agreement with the LC-MS data described in section 3.3.
Fig.1.
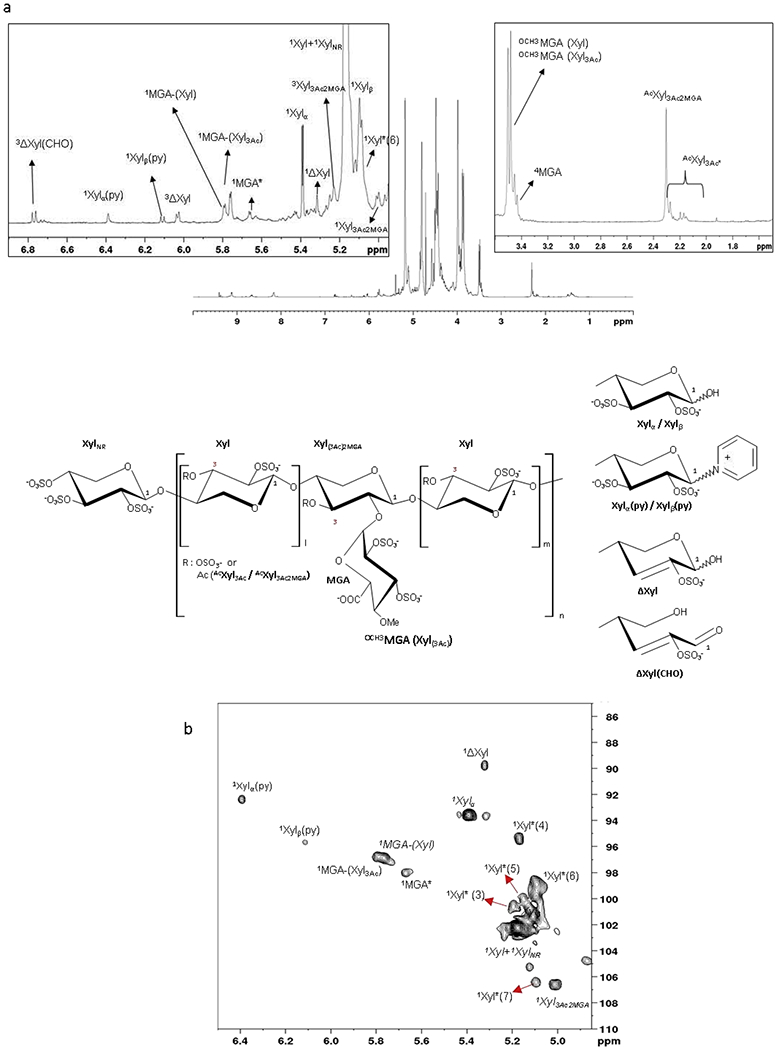
1H NMR and HSQC NMR spectra of PPS
a. 1H NMR spectrum with anomeric region and O-methyl -acetyl region expansions
b. Anomeric region of HSQC NMR spectrum
Apex number (e.g. 1Xyl) shows the number of the carbon observed in signal. Xyl – sulfated xylose (all position except specified), MGA – branching 4-O-methyl-2,3-di-O-sulfated glucuronic acid, Ac – acetyl group, Me – methyl group, Py – pyridine moiety, ΔXyl – C2-C3 double bond, * different from MGA-Xyl due to sequence effects. Xyl*(n) xylose residues related to sequence effects not fully elucidated.
Table 2.
1H and 13C chemical shifts of the assigned monosaccharide residues of PPS
| Monosaccharide residue | Chemical shift (ppm) H/C |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 5’ | 6 | Ac/Me | |||
| Xyl | 5.17 102.5 |
4.45 75.5 |
4.83 75.1 |
4.00 77.2 |
4.42 62.0 |
3.84 - |
||||
| XylNR | 5.17 102.5 |
4.49 75.4 |
4.86 73.6 |
4.58 74 74.0 |
4.54 61.0 |
3.84 - |
||||
| Xylα | 5.39 93.3 |
4.35 77.8 |
4.80 78.3 |
3.9 77.5 |
3.95 64.0 |
3.94 - |
||||
| Xylβ | 5.10 101.3 |
4.42 75.9 |
4.75 75.5 |
3.97 76.7 |
4.29 61.8 |
3.86 - |
||||
| Xyl3Ac-2MGA | 5.00 106.4 |
3.99 76.3 |
5.27 75.5 |
4.14 76.4 |
4.27 65.3 |
3.7 - |
2.31(CH3) 23.7(CH3) |
- 176.1(co) |
Ac | |
| Xylα(Py) | 6.40 92.7 |
n.a. | n.a. | n.a. | n.a. | n.a. | ||||
| Xylβ(Py) | 6.11 95.3 |
n.a. | n.a. | n.a. | n.a. | n.a. | ||||
| ΔXylred | 5.34 89.7 |
- 150.2 |
6.06 111.6 |
4.34 72.1 |
4.13 63.9 |
4.04 | ||||
| ΔXylred(CHO) | 9.38 193.2 |
- 151.9 |
6.78 141.3 |
5.24 74.2 |
4.37 | 4.27 | ||||
| MGA-(Xyl) | 5.80 96.9 |
4.36 78.2 |
4.97 80.0 |
3.47 84.0 |
4.38 74.8 |
- | - 178.9 |
3.50 63.2 |
Me | |
| MGA-(Xyl3Ac) | 5.77 97.0 |
4.34 77.8 |
4.44 80.5 |
3.47 84.0 |
3.94 75.5 |
- | - 178.4 |
3.48 63.6 |
Me | |
| MGA*-(Xyl) | 5.68 98.1 |
4.36 n.a. |
4.83 80.1 |
3.45 n.a. |
4.32 74.9 |
- | - n.a. |
|||
Xyl – sulfated xylose (all position except specified), MGA – branching 4-O-methyl-2,3-di-O-sulfated glucuronic acid, Ac – acetyl group, Me – methyl group, Py – pyridine moiety, ΔXylred – C2-C3 double bond, ΔXylred(CHO) – open ring aldehyde with C2-C3 double bond,
different from MGA-Xyl due to sequence effects.
The other RE modification assigned by combining NMR and LC-MS data includes the attachment of pyridine (Py) to the RE xylose, likely to occur during sulfation reactions of polysaccharides (Daus et al., 2011). As described below in detail, LC-MS analysis indicated the generation of a covalent carbon-nitrogen bond (see section 3.3). The HMBC NMR allowed to recognize the correlation between the ortho protons of pyridine and anomeric protons of α and β anomers of Xyl linked to pyridine (5.39 and 5.10 ppm), confirming this hypothesis (HSQC, H2BC and HMBC, Fig.S5). The molar ratio of Xylα(Py) and Xylβ(Py) anomers calculated by HSQC NMR was 60/40. This result is in agreement with what was previously observed during sulfation reaction of beech xylan (Daus et al., 2011).
The signals related to H1 of MGA residues at 5.80 and 5.77 ppm when bound to 3-O-sulfated xylose (MGA-(Xyl)) or 3-O-acetylated xylose (MGA-(XylAc)), respectively (Fig.1, Table 2), were confirmed by NOESY NMR (Fig.S6). Further TOCSY NMR permitted the assignment of the signal at 5.68 ppm to a MGA residue (Fig.S7), distinct from MGA-(Xyl) and MGA-(XylAc) and believed to be caused by heterogeneity in the environment of MGA, i.e. MGA attached to a NRE or RE xylose (which might have methyl or pyridine or sulfate group attached to the RE), but was not fully elucidated.
The 1H NMR spectra pointed out different signals related to acetyl group (Fig.1a). The major signal at 2.31ppm corresponds to the acetyl in position 3 of Xyl bearing a MGA. Other minor signals between 2.25 and 2.10ppm were observed by HMBC NMR (Fig. S8) as acetyl substituents in position 3 of Xyl residues not completely explained.
Several minor cross peaks pointed with asterisk in Fig. 1 were attributed by HSQC-TOCSY experiment (Fig.S9) to xylose residues, whose presence could be related to some sequence effects not fully elucidated. Relying on the LC-MS data described below, some of these Xyl* signals can be related to methylated RE xyloses. Minor signals at about 1.3ppm in the 1H NMR spectra (Fig.1) could be correlated at the methyl group of rhamnose residues that were described as components of the RE structure of xylans in conifers and several dicots (Scheller & Ulvskov, 2010). The HMBC NMR highlighted correlations of the methyl group with signals at 70.9 and 80.3ppm that could be related to rhamnose C5 and C4, respectively (Fig. S8).
3.2.2. Determination of relative abundance of monosaccharide building blocks of PPS by quantitative 1D and 2D NMR
The monosaccharide composition was determined by 1H NMR (Fig.1a, Table 3). The obtained data provided quantitative information about some structural features of PPS that can be directly related to xylan source (e.g. MGA content) and its preparation process (minor modifications, mainly present at the RE of PPS chains, e.g. formation of double bond, Py-adduct, possible methylation, as well as acetylation). The RE signal of β-xylose at 5.1 ppm is partially overlaid with a xylose residue at 5.08ppm not fully elucidated yet (Xyl*6), so that its value was calculated from the α/β ratio (35/65) obtained by 2D NMR (see 3.2.1). ΔXylred(CHO) was calculated from the integral of H3 signal at 6.78ppm, while Xyl3Ac-2MGA residue was calculated from acetyl signal at 2.31 ppm (Fig.1a). Finally, degree of acetylation was determined by calculating the ratio of the sum of acetyl group between 2.35 and 2.1ppm and the sum of xylose related signals.
Table 3.
Monosaccharide compositional analysis of three PPS samples by 1H NMR and HSQC NMR
| Quantitative 1H NMR analysis | ||||
|---|---|---|---|---|
| Monosaccharide residue | PPS 1 | PPS 2 | PPS 3 | |
| Xyl+XylNR | 72.6 | 72.7 | 73.2 | |
| XylRE | 10.5 | 10.6 | 11.0 | |
| Xyl3Ac-2MGA | 2.2 | 2.2 | 2.1 | |
| ΔXyl | 1.1 | 1.2 | 1.1 | |
| ΔXylred(CHO) | 1.1 | 1.1 | 1.0 | |
| Xylα(Py) + Xylβ(Py) | 0.9 | 0.9 | 1.0 | |
| Xyl*6 (5.08 ppm) | 6.7 | 6.2 | 5.8 | |
| MGA-(Xyl3Ac) | 2.1 | 2.1 | 2.1 | |
| MGA-(Xyl) | 1.7 | 1.7 | 1.6 | |
| MGA*-(Xyl) | 1.2 | 1.3 | 1.1 | |
| Degree of acetylation | 3.6 | 3.5 | 3.6 | |
| AcXyl3Ac-2MGA/Ac.tot | 66 | 66 | 63 | |
| Quantitative HSQC NMR analysis | ||||
| Monosaccharide residue | CV% | PPS 1 | PPS 2 | PPS 3 |
| Xyl+XylNR | 0.5 | 73.62 | 74.10 | 73.11 |
| Xylα | 3.8 | 3.98 | 4.00 | 3.53 |
| Xylβ | 1.2 | 6.35 | 6.36 | 6.20 |
| Xyl3Ac-2MGA | 1.0 | 1.75 | 1.78 | 1.58 |
| ΔXylred | 11.4 | 0.97 | 0.95 | 1.08 |
| Xylα(Py) | 15.9 | 0.52 | 0.40 | 0.56 |
| Xylβ(Py) | 3.6 | 0.23 < LOQ | 0.28 < LOQ | 0.35 < LOQ |
| MGA-(Xyl3Ac) + MGA-(Xyl) | 1.9 | 3.75 | 3.68 | 3.85 |
| MGA*-(Xyl) | 6.2 | 0.93 | 0.99 | 0.99 |
| Xyl*2 (6.0/116.2) | 11.2 | 0.20 < LOQ | 0.13 < LOQ | 0.25 < LOQ |
| Xyl*3 (5.21/100.4) | 8.8 | 0.80 | 0.65 | 0.96 |
| Xyl*4 (5.19/95.2) | 4.7 | 1.15 | 0.99 | 1.04 |
| Xyl*5 (5.16/99.9) | 8.8 | 0.86 | 0.71 | 1.25 |
| Xyl*6 (5.10/98.8) | 1.8 | 4.88 | 4.95 | 5.24 |
| Xyl*7 (5.10/106.3) | 3.7 | 0.98 | 0.97 | 1.09 |
| MGA tot (from C1) | 1.6 | 4.69 | 4.67 | 4.84 |
| MGA tot (from C4) | 2.0 | 4.69 | 4.95 | 4.80 |
| 3ΔXyl (CHO) | 12.6 | 0.84 | 0.46 | 1.00 |
- related to substituents not fully elucidated
Degree of acetylation - ratio of acetyl groups ((2.35–2.10 ppm)/3) *100/ sum of xylose signals
Ac Xyl3Ac-2MGA/Ac.tot - ratio of AcXyl3Ac-2MGA(2.31ppm) *100/Ac.tot (2.35-2.13 ppm)Calculation – monosaccharide residue (C1)*100/C1-SUM, Xyl* – C1 signals related to substituents not fully elucidated (1H-13C ppm), MGA tot (from C1)– sum of MGA residues, MGA tot (from C4)– 4MGA(3.46–83.8 ppm)*100/ C1-SUM, 3ΔXyl(CHO) – 3ΔXyl(CHO) (6.78-141.3ppm)*100/ C1-SUM. Apex number (e.g. 3Xyl) shows the number of the carbon observed in signal.
Since the enhanced signal dispersion in HSQC NMR was shown to enable of resolving additional minor signals (Fig.1b), an analytical procedure for the quantitative monosaccharide composition of PPS was developed also by HSQC NMR. The strategy for the evaluation of the monosaccharide composition by calculation from 2D-NMR signal volumes was described in previous papers (Guerrini et al., 2005; Guerrini, Guglieri, Naggi, Sasisekharan, & Torri, 2007). Sample concentration, pH, 1Jtune, relaxation delay (D1), temperature, spectral width and pulse sequence were optimized in order to set up the quantitative HSQC NMR method (see section 2.9). Molar percentages of each residue (Table 3) were calculated as the abundance of corresponding cross peak volumes normalized to the sum of all the cross peak volumes.
The results of quantitative analysis of three Elmiron batches obtained by 1D and 2D NMR (Tables 3) are comparable. The xylose content is about 84%, with approximately 10% of RE units. The total MGA content is about 5%, in agreement with colorimetric assay results. Degree of 3-O-acetylation of MGA-containing PPS chains was revealed by 1D NMR (Table 3). The amount of the minor structures formed during sulfation reaction (C2-C3 double bond and pyridine adduct formation) is less than 3%.
3.3. Capturing microheterogeneity at oligomeric level through LC-MS
3.3.1. LC-MS analyses of Intact PPS
The LC-MS method was optimized to achieve sufficient separation and detection of different PPS components and provide a characteristic LC-MS fingerprint of intact PPS sample. Ion-pair reversed phase HPLC (IPRP HPLC) was chosen as a separation method since it enables a good separation of sulfated components and can be easily coupled with ESI-MS (Langeslay et al., 2013). Given its high structural heterogeneity, intact PPS was expected to show a complex and multi-component LC-MS profile. In fact, several overlapped species belonging to various structural families were detected (Fig.2a–f, Fig.S10).
Fig.2.
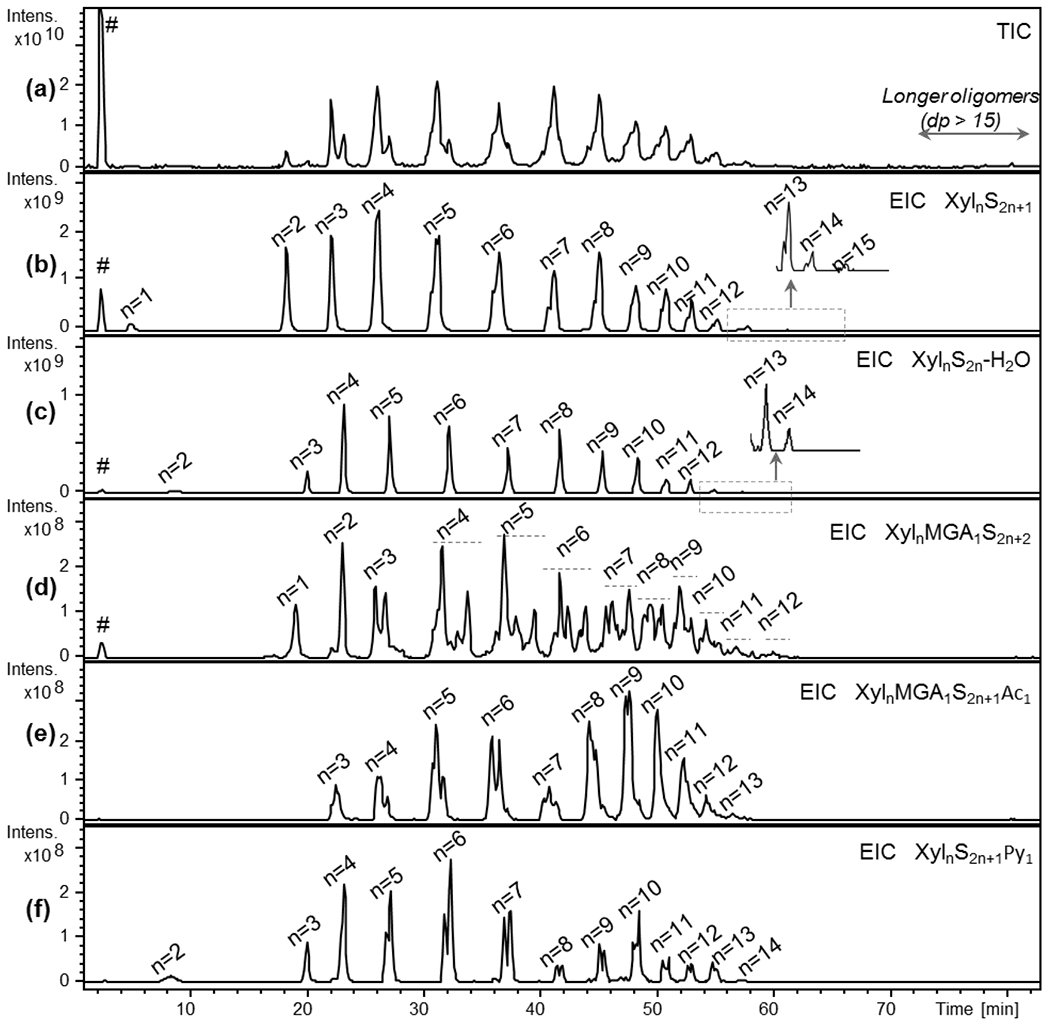
TIC (a) and EIC profiles (b-f) of prevalent structural families found in PPS by IPRP-HPLC/ESI-ICRFT-MS (a) – TIC, (b) – EIC of XylnS2n+1 structures (n: 1-15), (c) – EIC of XylnS2n-H2O structures (n: 2-14), (d) – EIC of XylnMGA1S2n+2 structures (n: 1-12), (e) – EIC of XylnMGA1S2n+1Ac1 structures (n: 3-13), (f) – EIC of XylnS2n+1Py1 structures (n: 2-14)
m/z values used for obtaining EIC profiles are those of the most abundant ion form reported in Table 6 Xyl – xylose repeating unit, MGA – branching 4-O-methyl-glucuronic acid, S – sulfate group, Ac – acetyl group, Py – pyridine moiety, # - salt clusters
Table 4 reports the m/z values of the prevalent ion forms observed for each detected component with dp from 1 to at least 14. Longer oligomers were also observed, their structural assignment was performed by the analysis of higher molecular weight fractions (see section 3.3.2).
Table 4.
Mass-to-charge (m/z) values of the principal PPS components, belonging to various structurally different groups detected by LC/ESI-FT-MS
| n | XylnS2n+1 | XylnS2n-H2O | XylnMGA1S2n+2 | XylnMGA1S2n+1Ac1 | XylnS2n+1Py1 * | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| m/z | Ion form | m/z | Ion form | m/z | Ion form | m/z | Ion form | m/z | Ion form | |
| 1 | 518.068 | [M-H+1A]1- | nd | - | 917.224 | [M-H+2A]1- | nd | - | nd | - |
| 2 | 1197.479 | [M-H+4A]1- | 970.360 | [M-H+3A]1- | 668.662 | [M-2H+3A]2- | nd | - | 1129.359 | [M-H+3A]1- |
| 3 | 744.214 | [M-2H+4A]2- | 630.654 | [M-2H+3A]2- | 943.792 | [M-2H+5A]2- | 860.243 | [M-2H+4A]2- | 710.154 | [M-2H+3A]2- |
| 4 | 1083.919 | [M-2H+7A]2- | 970.360 | [M-2H+6A]2- | 1283.497 | [M-2H+8A]2- | 1199.948 | [M-2H+7A]2- | 1049.859 | [M-2H+6A]2- |
| 5 | 1359.049 | [M-2H+9A]2- | 1245.489 | [M-2H+8A]2- | 1558.627 | [M-2H+10A]2- | 1476.081 | [M-2H+9A]2- | 1324.989 | [M-2H+8A]2- |
| 6 | 1635.182 | [M-2H+11A]2- | 1521.622 | [M-2H+10A]2- | 1834.758 | [M-2H+12A]2- | 1751.211 | [M-2H+11A]2- | 1601.122 | [M-2H+10A]2- |
| 7 | 1910.310 | [M-2H+13A]2- | 1796.752 | [M-2H+12A]2- | 1363.539 | [M-3H+13A]3- | 2026.840 | [M-2H+13A]2- | 1876.250 | [M-2H+12A]2- |
| 8 | 1413.907 | [M-3H+14A]3- | 1338.201 | [M-3H+13A]3- | 1546.959 | [M-3H+15A]3- | 1491.260 | [M-3H+14A]3- | 2151.880 | [M-2H+14A]2- |
| 9 | 1597.327 | [M-3H+16A]3- | 1521.620 | [M-3H+15A]3- | 1730.712 | [M-3H+17A]3- | 1675.013 | [M-3H+16A]3- | 1574.620 | [M-3H+15A]3- |
| 10 | 1781.080 | [M-3H+18A]3- | 1705.374 | [M-3H+17A]3- | 1914.132 | [M-3H+19A]3- | 1858.433 | [M-3H+18A]3- | 1758.373 | [M-3H+17A]3- |
| 11 | 1964.500 | [M-3H+20A]3- | 1888.794 | [M-3H+19A]3- | 2097.886 | [M-3H+21A]3- | 2041.853 | [M-3H+20A]3- | 1941.793 | [M-3H+19A]3- |
| 12 | 2148.253 | [M-3H+22A]3- | 2072.547 | [M-3H+21A]3- | 2281.306 | [M-3H+23A]3- | 2225.606 | [M-3H+22A]3- | 2125.547 | [M-3H+21A]3- |
| 13 | 2331.673 | [M-3H+24A]3- | 2255.967 | [M-3H+23A]3- | nd | - | 2409.026 | [M-3H+24A]3- | 2308.967 | [M-3H+23A]3- |
| 14 | 2515.427 | [M-3H+26A]3- | 2439.720 | [M-3H+25A]3- | nd | - | nd | - | 2492.720 | [M-3H+25A]3- |
| 15 | 2698.847 | [M-3H+28A]3- | nd | - | nd | - | nd | - | nd | - |
This abbreviation includes the loss of one water molecule (see text)
Xyl – xylose repeating unit, MGA – branching 4-O-methyl-glucuronic acid, S – sulfate group, Ac – acetyl group, Py – pyridine moiety, A – amine used as ion pairing agent (DBA)
The monoisotopic m/z values of oligosaccharides with degree of polymerization <6 and average mass m/z values of higher oligomers, used for EICs shown in Fig.5, are reported in this table
The complexity of the LC-MS chromatogram required the use of EIC data processing for better representation of the obtained results (Fig.2b–f). The regular structures, containing only sulfated Xyl residues (XylnS2n+1, where S symbol represents SO3 groups), appeared prevalent among the other structural PPS families. Their sulfation pattern suggests that these components possess one unsubstituted hydroxyl-group within their backbone, presumably at the RE based on NMR data (see section 3.2). The second most prevalent structural family corresponds to desulfated and dehydrated oligomers XylnS2n-H2O (where S symbol represents SO3 groups), that is in agreement with C2-C3 unsaturated linkage annotated in the 1D and 2D NMR spectra (see section 3.2).
Among the less abundant species, MGA-containing families with different sulfation/acetylation pattern (XylnMGA1S2n+2 and XylnMGA1S2n+1Ac1, where S and Ac symbols represent SO3 and acetyl groups, respectively) were found (Fig.2d, e). The terminal Xyl residues are supposed to be unsubstituted at C-1 as in the case of regular XylnS2n+1 oligomers, while branched MGA-residues are likely to be 2,3-di-O-sulfated. Interestingly, only mono-acetylated MGA-containing oligomers were observed, that is in agreement with the aforementioned partial preservation of 3-O-acetylation. Additionally, the oligosaccharide distribution is shifted to higher masses in the case of acetylated species when compared with XylnMGA1S2n+2 family (see discussion, section 4). Worthy of note, higher complexity of the EIC profiles of the MGA-containing structural groups, likely to be associated with the presence of different positional isomers, can be noticed (Fig.2d, e).
Minor pyridine containing oligomers XylnS2n+1Py1 were also detected by LC-MS (Fig.2). Notably, all the observed species of this family are characterized by an absorbance at 254 nm and a loss of one water molecule and were assigned to the structures with a covalent bond between the anomeric carbon of the RE xylose and nitrogen atom of the pyridine moiety. This hypothesis was conceivably in accordance with the presence of two XylnS2n+1Py1 isomers (most likely, α- and β-anomers) well-distinguishable by LC-MS (Fig.2f). The presence of aromatic pyridine fragment is in agreement with lower retention of XylnS2n+1Py1 in ion pair separation mode with respect to the regular XylnS2n+1. The observation of oligomers comprising of pyridine attached to RE xylose is also consistent with the annotation and quantification of NMR signals associated with this moiety (see section 3.2).
To a smaller extent than pyridine containing components, also regular structures with a +14Da mass shift were observed among the low abundance species in PPS, indicating a conceivable presence of a methyl group presumably at the RE. These components were analyzed and discussed in detail in section 3.3.2.
3.3.2. Analysis of low and high molecular weight fractions of PPS
To further augment information obtained on intact PPS mixture, the LC-MS analyses were independently performed on high and low molecular weight fractions (A1 and H in Fig. S2). The use of high resolution ESI-ICRFT-MS allowed to detected ion forms with (5−) negative charge (Table S2) and, consequently, to find oligomers with dp up to 26 (Fig.3a).
Fig.3.
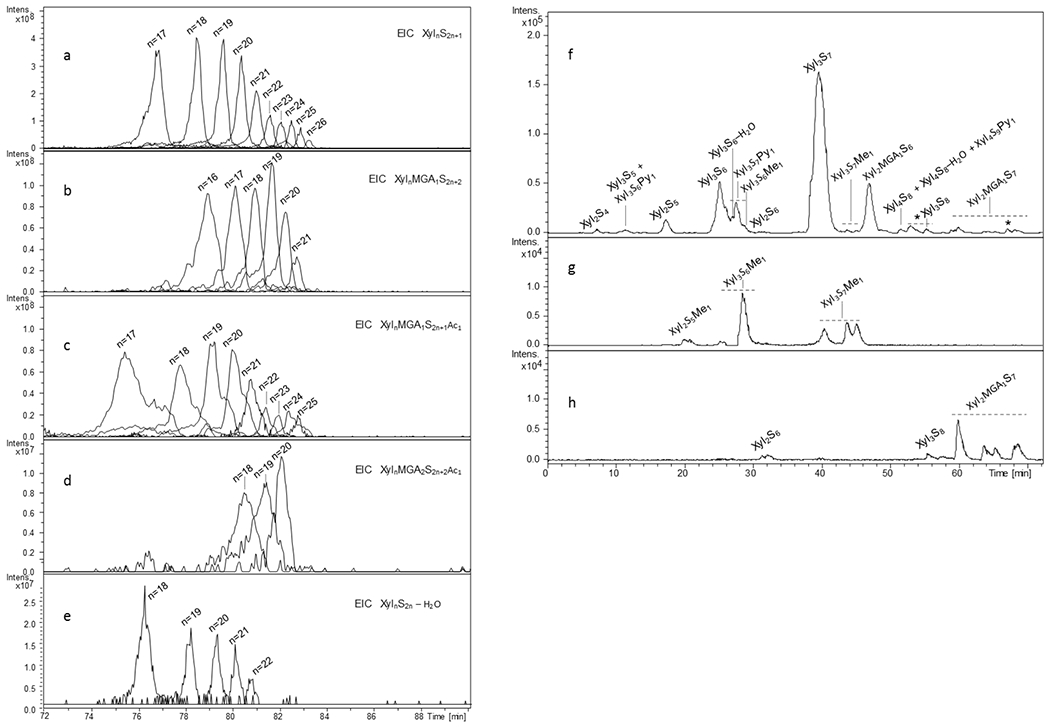
LC-MS profiles of different structural families of PPS detected in high molecular weight (a-e) and low molecular weight (f-h) fractions of PPS by LC/ESI-FT-MS
(a) – EIC of XylnS2n+1 structures (n: 17-16), (b) –EIC of XylnMGA1S2n+2 structures (n: 16-21), (c) – EIC of XylnMGA1S2n+1Ac1 structures (n: 17-25), (d) – EIC of minor XylnMGA2S2n+2Ac1 structures (n: 18-20), (e) - XylnS2n-H2O structures (n: 18-22) , (f) basepeak chromatogram of a small size (dp 3) fraction of PPS, (g) and (h) - EIC profiles of some minor methylated and completely sulfated components, respectively
m/z values used for obtaining EIC profiles are those of the most abundant ion form reported in Tables S2 and S3 Xyl – xylose repeating unit, MGA – branching 4-O-methyl-glucuronic acid, S – sulfate group, Py – pyridine moiety, Me – methyl group
The prevalent components belong to XylnS2n+1 structural group as in the case of intact PPS samples. However, relative abundance of MGA-containing species XylnMGA1S2n+2/XylnMGA1S2n+1Ac1 appeared higher than for oligomers observed within intact PPS samples, suggesting higher branching and acetylation degree for longer PPS chains (Fig.2,3). Notably, only several minor mono-acetylated oligomers containing two MGA residues, Xyl18MGA2S38Ac1 , Xyl19MGA2S40Ac1 and Xyl20MGA2S42Ac1 were also detected (Fig.3d). Interestingly, their non-acetylated and di-acetylated structural analogues were not detected. This is in agreement with the fact that acetyl groups are mainly present within MGA-branched xyloses as observed by NMR (see section 3.2).
The LC-MS analysis of low molecular weight fraction, enriched in structures with dp 3 (Fig. 3f–h), was carried out using an ESI-Q-TOF-MS instead of ESI-ICRFT-MS since its resolution and accuracy are enough for detection of small PPS components ionized prevalently as singly or doubly charged ions (Table S3). The optimized separation conditions allowed detection of regular XylnS2n+1, branched XylnMGA1S2n+2 and modified at their RE (XylnS2n–H2O, XylnS2n+1Py1, XylnS2n+1Me1) structures. Moreover, several minor and trace compounds with different sulfation/acetylation degree, e.g. oversulfated XylnS2n+2/XylnMGA1S2n+3 and less sulfated XylnS2n/XylnS2n-1, as well as traces of mono-acetylated Xyl3S6Ac1-H2O and Xyl3S6Ac1 (Table S3) were also found in the analyzed fraction (see Discussion, section 4). Their low abundance is in accord with the overall low acetylation of PPS (Table 3) and its prevalence within MGA-branched chains. Interestingly, the oversulfated Xyl2MGA1S7 is represented by four peaks in the range of 59-69 min (Fig.3h, Table S3) that is believed to be related to the presence of both positional (MGA at the NRE/RE xylose) and anomeric (α/β isomers of 1-O-sulfated RE xylose) isomers. The Xyl3S7Me1 appeared as three isomers (Fig.3f–h, S10) that can be explained by the presence of both possible α- and β-anomers of 1-O-methylated trisaccharide and rhamnose containing species (Scheller & Ulvskov, 2010), compatible with NMR data.
The developed LC-MS method was applied to all the three commercial PPS lots, both unfractionated and low molecular weight SEC fractions. The structural families reported in Table 4 were detected in all the three commercial samples and the EIC profiles of each structural group did not shown noticeable differences (data not shown), that is in accord with small variations detected by other aforementioned methods. As expected, the SEC fractionation prior to LC-MS analysis allowed to simplify the sample complexity, to better separate the fraction components and to observe some variations in the LC-MS profiles between different batches. It is worth noting that these variations are mainly related to small differences in the intensities of low abundance species (Fig.S10).
3.4. Cross-integration of data into a mathematical model
The detailed description of the attributes measured provide a comprehensive picture of the PPS heterogeneity. Towards integration of the datasets from the aforementioned analyses, a mathematical model was developed to represent the mixture of oligosaccharides that make up the drug substance in PPS. Table 5 illustrates how the proposed mathematical model incorporating a priori knowledge of the PPS structure space, and constrained by orthogonal data can provide insight into the behavior of different chain signatures (rows) across the chain length distribution (columns). The full extent of model parameterization and estimation used to populate Table 5 may be found in section 2.11.
Table 5.
Distribution of key attributes of structural heterogeneity along the chain length of PPS
| S4-X | 0.08 | 0.93 | 0.93 | 1.83 | 2.73 | 3.54 | 3.69 | 3.45 | 3.02 | 2.47 | 1.91 | 1.33 | 0.92 | 0.63 | 0.44 | 0.31 | 0.22 | 0.16 | 0.10 |
| S4-X-1DH | 0.01 | 0.10 | 0.10 | 0.19 | 0.29 | 0.37 | 0.39 | 0.36 | 0.32 | 0.26 | 0.20 | 0.14 | 0.10 | 0.07 | 0.05 | 0.03 | 0.02 | 0.02 | 0.01 |
| S4-X-1M | 0.01 | 0.15 | 0.15 | 0.29 | 0.44 | 0.57 | 0.59 | 0.56 | 0.49 | 0.4 | 0.31 | 0.21 | 0.15 | 0.10 | 0.07 | 0.05 | 0.03 | 0.03 | 0.02 |
| S4-X-1P | 0.01 | 0.14 | 0.14 | 0.28 | 0.42 | 0.55 | 0.57 | 0.53 | 0.46 | 0.38 | 0.29 | 0.20 | 0.14 | 0.10 | 0.07 | 0.05 | 0.03 | 0.02 | 0.02 |
| S4-X-1S | 0.01 | 0.07 | 0.07 | 0.14 | 0.20 | 0.26 | 0.28 | 0.26 | 0.23 | 0.18 | 0.14 | 0.10 | 0.07 | 0.05 | 0.03 | 0.02 | 0.02 | 0.01 | 0.01 |
| S4-X-G | 0.00 | 0.08 | 0.08 | 0.24 | 0.49 | 0.84 | 1.11 | 1.29 | 1.38 | 1.38 | 1.29 | 1.09 | 0.92 | 0.77 | 0.66 | 0.58 | 0.53 | 0.51 | 0.49 |
| S4-X-G-1DH | 0.00 | 0.01 | 0.01 | 0.04 | 0.09 | 0.15 | 0.20 | 0.23 | 0.24 | 0.24 | 0.23 | 0.19 | 0.16 | 0.14 | 0.12 | 0.10 | 0.09 | 0.09 | 0.09 |
| S4-X-G-1S | 0.00 | 0.03 | 0.03 | 0.06 | 0.08 | 0.11 | 0.11 | 0.10 | 0.09 | 0.07 | 0.06 | 0.04 | 0.03 | 0.02 | 0.01 | 0.01 | 0.01 | 0.00 | 0.00 |
| S4-X-GA | 0.01 | 0.10 | 0.10 | 0.28 | 0.56 | 0.93 | 1.20 | 1.38 | 1.47 | 1.45 | 1.34 | 1.12 | 0.94 | 0.79 | 0.67 | 0.59 | 0.53 | 0.51 | 0.50 |
| X | 0.01 | 0.09 | 0.09 | 0.17 | 0.26 | 0.33 | 0.35 | 0.32 | 0.28 | 0.23 | 0.18 | 0.12 | 0.09 | 0.06 | 0.04 | 0.03 | 0.02 | 0.01 | 0.01 |
| X-1DH | 0.02 | 0.18 | 0.18 | 0.35 | 0.52 | 0.68 | 0.70 | 0.66 | 0.58 | 0.47 | 0.36 | 0.25 | 0.18 | 0.12 | 0.08 | 0.06 | 0.04 | 0.03 | 0.02 |
| X-GA | 0.00 | 0.01 | 0.01 | 0.03 | 0.05 | 0.09 | 0.11 | 0.13 | 0.14 | 0.14 | 0.13 | 0.11 | 0.09 | 0.08 | 0.06 | 0.06 | 0.05 | 0.05 | 0.05 |
| X-GA - 1DH | 0.00 | 0.01 | 0.01 | 0.03 | 0.05 | 0.09 | 0.11 | 0.13 | 0.14 | 0.14 | 0.13 | 0.11 | 0.09 | 0.08 | 0.06 | 0.06 | 0.05 | 0.05 | 0.05 |
| X-GS | 0.00 | 0.01 | 0.01 | 0.03 | 0.05 | 0.08 | 0.11 | 0.12 | 0.13 | 0.13 | 0.12 | 0.10 | 0.08 | 0.07 | 0.06 | 0.05 | 0.05 | 0.05 | 0.04 |
| X-GS - 1DH | 0.00 | 0.01 | 0.01 | 0.04 | 0.08 | 0.12 | 0.16 | 0.19 | 0.20 | 0.19 | 0.18 | 0.15 | 0.13 | 0.11 | 0.09 | 0.08 | 0.07 | 0.07 | 0.07 |
X: Xyl[2S,3S]; S4-X: Xyl[2S,3S,4S]; 1DH: dehydration at RE xylose; 1M: methylation of RE xylose; 1P: Pyridine linkage at RE xylose; X-1S: RE Xyl[1S,2S, 3S]; G: MGA[2S,3S]α1-2Xyl[3S]; GA- MGA[2S,3S]α1-2Xyl[3Ac]. The columns show the relative abundance of the different units across chain length which includes xylose and MGA units.
The estimation of the parameters that capture the distribution of sulfation and reducing end modifications found in PPS in different chain lengths (Table 5) is consistent with the experimentally determined values. For example, the prevalence of additional sulfate group in the 4-O position of the nonreducing end xylose (S4-X in Table 5) across all the chain lengths is consistent with experimentally measured degree of sulfation greater than 2 (Table S1) given that 2-O and 3-O position of both 4MGA and internal xylose (apart from small percentage that is 3-O acetylated) is predominantly sulfated. Another specific example of model parameter estimation relating to MGA branching, and corresponding xylose acetylation, is shown below.
LC-MS and NMR data provide distinct, but correlated information regarding the MGA distribution across the chain length distribution. The function relating the fraction of chains containing MGA (as observed by LC-MS) to the total amount of MGA in the mixture was hypothesized to be linear in chain length (for chain lengths <10 as had sufficient LC-MS data), and, the longer the chains, the more likely a chain is to contain MGA. Using this relationship, a Bayesian Hierarchical model was built to estimate the relative abundance of building block components and their batch-to-batch variation (see section 2.11). The goal was to understand how the posterior distribution of the parameters changed when using NMR alone, LC-MS alone, or a combination of the LC-MS and NMR datasets.
The combined model shows several advantages of using orthogonal measurements to characterize mixture properties. Firstly, as shown in Fig. 4, the combined model fits the observed LC-MS data, while only estimating the three mixture level parameters (rather than new parameters for each chain length). Secondly, the combined model is able to find the parameters that best fit both datasets (Fig. 4). Finally, the combined parameter estimate is using more samples at different levels and so, intuitively, we would expect a less variable estimate of the model parameters. Using the 90% highest density interval (HDI) (i.e. width of the distribution) as a metric for variability, the combined model has smaller than or equal width when compared with either model individually (Table S4).
Fig.4.
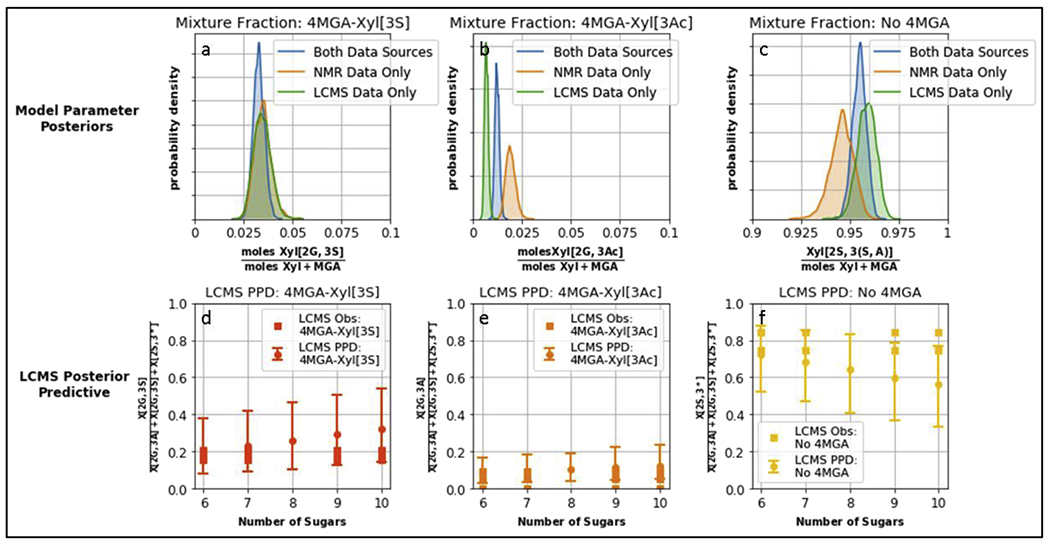
Top row: posterior distribution of building block fractions Xyl[2G,3S], Xyl[2G, 3Ac], Xyl[2S, 3(S,A)], as estimated when using either one or both of the data sources. Width of the distribution indicates the variability in estimating the parameter. Bottom row: the model provides a posterior predictive distribution for the LC-MS measurements, plotted with 90% credible intervals. An advantage of using a Bayesian framework is that we can simulate the distribution of measurement values in addition to the unobserved parameter values. Note that the credible intervals in panels d,e,f incorporate both the parameter variability shown in figures a,b,c respectively as well as a component of measurement variability.
4. Discussion
The US FDA has published a product specific guidance (FDA, 2012, https://www.govinfo.gov/content/pkg/FR-2012-09-20/pdf/2012-23177.pdf) for developing generic PPS. It recommends, given the complexity associated with PPS, to use orthogonal analytical methods specific and sensitive to key structural attribute(s). The orthogonality of the analytical methods employed in our study and the cross-correlation of attributes derived by these orthogonal measurements is evident at multiple instances. Firstly, the cross-validation of sulfation/acetylation pattern (XylnMGA1S2n+2 and XylnMGA1S2n+1Ac1 families), branching (MGA-containing oligomers) and minor process-related modifications (XylnS2n+1Py1, XylnS2n+1Me1, XylnS2n-H2O families) are borne out by integration of LC-MS and NMR analyses. Secondly, specific attributes (such as degree of sulfation, MGA content) are correlated across the different levels of analysis. Additionally, several process related characteristic fingerprints of PPS (extraction of xylan from wood, sulfation, depolymerization, etc.) are revealed from the present detailed structural characterization.
The degree of sulfation and molecular weight distribution at the overall physiochemical level taken together with distribution of sulfate groups in the monosaccharide building blocks level provides critical insights into understanding relationship between sulfation and depolymerization procedures. Both NMR and LC-MS, together with results of colorimetric assays, suggest that majority of the xylose and MGA sugars are O-sulfated. On the other hand, the MGA content (determined by both colorimetric method and NMR) and MGA distribution including the variety of positional isomers shown by LC-MS analysis, represent a characteristic signature of the xylan source material.
Additionally to MGA distribution, 3-O-acetylation was shown to be another important process related signature. LC-MS analysis revealed acetylated oligomers only for MGA-containing structural group, except for several compounds detectable in trace only when SEC fractionation was performed prior to the LC-MS analysis (Xyl3S6Ac1 and Xyl3S6Ac1-H2O, Table S3). Quantitative NMR confirms the prevalence of 3-O-acetylation in the MGA-containing chains. Altogether, these data suggest that 3-O-acetyl group at the 2-O-MGA-bearing xylose can be partially and randomly preserved during the extraction process presumably by MGA-substituent. Their presence and distribution within the PPS chains can be traced back to the xylans that are extracted from hardwood. Several parameters such as temperature, pH, acid or base and solvent impact the acetyl content of xylan which in turn impacts acetyl content of final PPS (De Ferra et al., 2016; Junior et al., 2010). There is not much known publicly about the properties of xylans that are used to make PPS or on standardized extraction methods that would help control for these properties. The insights on acetyl content taken together with molecular weight distribution and other properties would facilitate identifying optimal key properties of xylan.
The crossed NMR and LC-MS data revealed also two minor structural features observed through all the analyzed commercial PPS samples, which are mainly related to the manufacturing process: (i) the C2-C3 double bond formation at the level of RE xyloses, hypothesizing possible desulfation that happens during depolymerization; (ii) pyridine adducts with a carbon-nitrogen covalent bond formation between the RE xylose and pyridine to their structure.
Additionally, the set up and validation of 1D and 2D quantitative NMR allowed to provided even more fine structural details (Fig.5).
Fig.5.
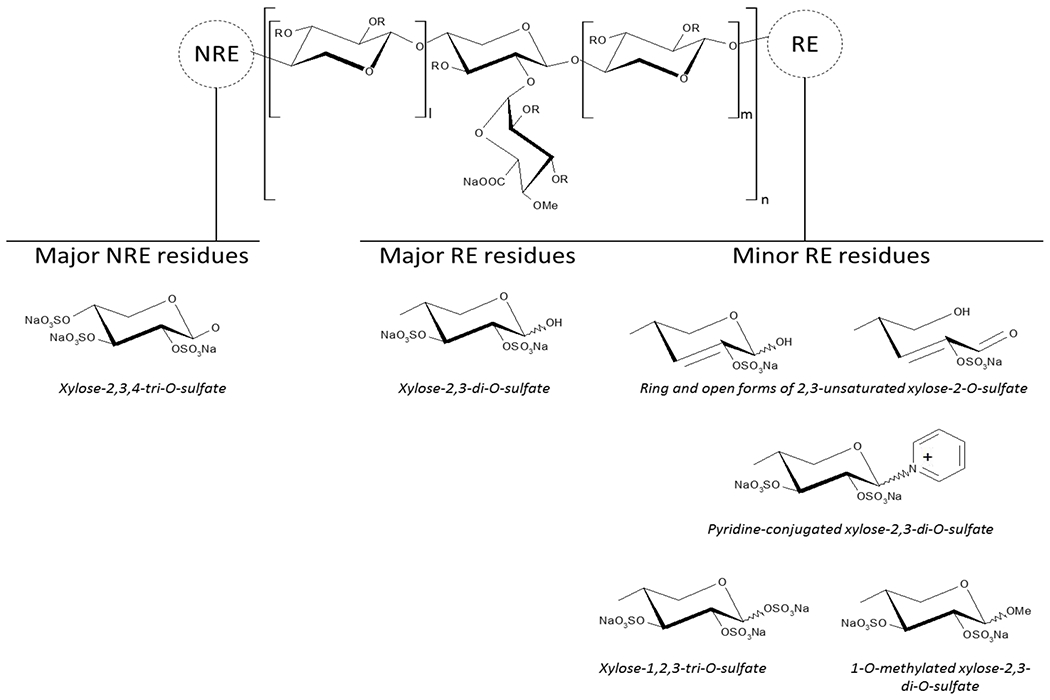
PPS prevalent structural formula and minor RE modifications identified; I and m >/= 0, n >/= 1.
The RE xylose residues were found in a 10% range, while minor RE modifications, e.g. the formation of C2-C3 double bond and pyridine adduct, are present in relatively low amount (totally approx. 3%, Table 3). It is worth noting that the narrow range of RE residues content obtained by NMR is in agreement with a small variation in molecular weight distribution through the lots (Table 1). Notably, 1D and 2D NMR methods provided comparable and complementary data: the 1D analysis underlined the complexity in the evaluation of some signals i.e. Xylβ and Xyl3Ac-2MGA that can be resolved by 2D NMR, however, it allowed to determine the important 3-O-acetylation signature.
The information contained in the top-level physiochemical property of molecular weight distribution is substantially amplified upon fractionation of the mixture and characterization of the fractions. LC-MS analyses of the high and low molecular weight fractions provide a diversity of details on the structural attributes that can be captured at these two ends of the distribution. Analysis of a trisaccharides PPS fraction allowed to detect trace totally sulfated structures Xyl2S6, Xyl3S8 and Xyl2MGA1S7 as well as those with low sulfation degree, e.g. Xyl2S4 , Xyl3S5 and Xyl3S6 , otherwise difficult to detect in intact PPS samples (Fig. 3f–h). In contrast, the LC-MS analysis of the highest molecular weight portion of PPS (Fig.3a–e) mainly provides information about most abundant components due to limitations in ionization and detection of longer chain oligomers. However, it allowed to obtain a characteristic LC-MS profiles of components with dp > 16, particularly, the aforementioned distribution of MGA branched structures and their acetylation pattern. Worthy of note, the choice of the MS detector may play a crucial role in feasibility of the analysis of longer chain oligosaccharides. For example, Table S2 shows that the prevalent ion form of oligomers with dp ~ 21 is characterized by 5 negative charge, allowing their detection with a good accuracy only when a high resolution mass spectrometer, such as FT-MS, is implicated.
Overall, the obtained results offer a set of potential critical quality attributes of PPS and the method developed herein can be considered as an analytical tool for their evaluation. Additionally, the proposed mathematical model could provide additional characteristics of interest about such complex products by estimating confidence intervals not only for individual measurements but also combining data across measurements. Building a model that captures detailed sequence information of all oligosaccharide chain lengths and their relative abundances is an extremely daunting task given that there are no precise enzymatic tools that act on PPS.
5. Conclusions
We have developed an unbiased comprehensive approach to define several critical structural attributes of PPS at various levels ranging from gross physiochemical characteristics to detailed analysis of constituent oligosaccharides at different size fractions. These structural attributes reflect key fingerprints that are imprinted on the PPS from the beechwood xylan starting material and from the process chemistry. The sulfation of PPS is such that almost all of the internal xylose and 4MGA are sulfated at the 2-O and 3-O positions. This indicates that the sulfation reaction in the process is nearly complete. The exception to this situation is the predominant acetylation of the 3-O position of xylose to which 4-MGA is linked at the 2-O position. This shows that acetylation of xylose building block is a possible fingerprint of the beechwood xylan and the abundance of the 3-O acetylation of xylose is controlled by the extraction process of xylan from beechwood and the resistance of the 3-O position of xylose to which 4MGA is linked at the 2-O position to be deacetylated and sulfated as a part of the process chemistry. The detailed characterization of size fractionated oligosaccharides demonstrates that longer chains of xylan are more likely to have 4MGA attached to Xylose compared to the shorter chains. This shows that the interspersing of 4MGA linkage is not completely random and there is a biosynthetic machinery in the beechwood plant source for the xylan from which PPS is made that controls the minimum chain length required for transferring 4-MGA to the 2-O position of xylose. Finally, our in-depth characterization of PPS also identified of minor species (which can be categorized as process related impurities) in the reducing end including 2,3 unsaturated xylose, sulfation, methylation and pyridinium complex at the anomeric position of the reducing end.
The mathematical model is a new and elegant approach to capture the diverse analytical datasets in an integrated fashion to define the PPS mixture. The mathematical model also demonstrates that using multiple orthogonal datasets such as NMR and LC-MS improve the confidence interval in estimating critical structural signatures such as 4MGA or acetylation content over the estimates from using each analytical method independently to generate the model.
To our knowledge this is the first study that reports an approach for integrated characterization of PPS using diverse analytical methods and a mathematical model. We believe that such an approach would facilitate in developing a foundational scientific framework for defining key criteria to demonstrate comparability between different PPS drug products similar to what was done earlier for enoxaparin (Lee et al., 2013). The results reported herein can be amplified by further studies and analysis of larger number of the samples in order to develop a reference analytical approach for PPS characterization.
Supplementary Material
Highlights.
The combination of several analytical methods to characterize PPS is reported
An in-depth characterization of PPS is reported for the first time.
The integrative approach reveals major and minor quality attributes of PPS.
This approach represents a tool for comparative studies of Pentosan samples.
Acknowledgements
This work was supported by grant 1 U01 FD005291-01 (National Institutes of Health and United States Food and Drug Administration). We like to acknowledge Dr. Stancanelli (Ronzoni Institute) for NMR experiment work, Dr. Boccardi (Ronzoni Institute) for statistical contribution to validations and Dr. Risi (Ronzoni Institute) SEC results.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bertini S, Bisio A, Torri G, Bensi D, & Terbojevich M (2005). Molecular weight determination of heparin and dermatan sulfate by size exclusion chromatography with a triple detector array. Biomacromolecules, 6, 168–173. [DOI] [PubMed] [Google Scholar]
- Bertini S, Risi G, Guerrini M, Carrick K, Szajek AY, & Mulloy B (2017). Molecular weights of bovine and porcine heparin samples: comparison of chromatographic methods and results of a collaborative survey. Molecules, 22, 1214 Doi.org/10.3390/molecules22071214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisio A, Mantegazza A, Vecchietti D, Bensi D, Coppa A, Torri G, & Bertini S (2015). Determination of the molecular weight of low-molecular-weight heparins by using high-pressure size exclusion chromatography on line with a triple detector array and conventional methods. Molecules, 20, 5085–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitter T, & Muir HM (1962). Quantitative determination of uronic acids with m-hydroxydiphenyl. Analytical Biochemistry, 4, 330–334. [DOI] [PubMed] [Google Scholar]
- Budsberg SC, Bergh MS, Reynolds LR, & Streppa HK (2007). Evaluation of pentosan polysulfate sodium in the postoperative recovery from cranial cruciate injury in dogs: a randomized, placebo-controlled clinical trial. Veterinary Surgery, 36, 234–244. [DOI] [PubMed] [Google Scholar]
- Casu B, & Gennaro UA (1975). Conductimetric method for the determination of sulfate and carboxyl groups in heparin and other mucopolysaccharides. Carbohydrate Research, 39, 168–176. [DOI] [PubMed] [Google Scholar]
- Daus S, Petzold-Welcke K, Kotteritzsch M, Baumgaertel A, Schubert US, & Heinze T (2011). Homogeneous sulfation of xylan from different sources. Macromolecular Materials and Engineering, 296, 551–561. [Google Scholar]
- Degenhardt M, Benend H, & Wätzig H (1998). Quality control of pentosan polysulfate by capillary zone electrophoresis using indirect detection. Journal of Chromatography A, 817, 297–306. [DOI] [PubMed] [Google Scholar]
- Degenhardt M, Ghosh P, & Wätzig H (2001). Studies on the structural variations of pentosan polysulfate sodium (NaPPS) from different sources by capillary electrophoresis. Archiv der Pharmazie, 334, 27–29. [DOI] [PubMed] [Google Scholar]
- Dubois M, Gilles K, Hamilton JK, Rebers PA, & Smith FA (1951). Colorimetric method for the determination of sugars. Nature, 168, 167. [DOI] [PubMed] [Google Scholar]
- European Pharmacopeia (8th ed.). (2014). Strasburg: Council of Europe; (Volume 2). [Google Scholar]
- De Ferra L, Naggi A, Zenoni M, & Pinto B (2014). Patent WO 2014114723 Al: Method for the qualification of preparations of pentosan polysulfate, raw materials and production processes thereof.
- De Ferra L, Ammirati E, Andreassi S, Annibaldi M, Mandelli L, Pinto B, & Stracqualursi F (2016) Patent WO 2016184887 Al: Process for the preparation of polysaccharides.
- Fischer AM, Merton RE, Marsh NA, Williams S, Gaffney PJ, Barrowcliffe TW, & Thomas DP (1982). A comparison of pentosan polysulphate and heparin. II: Effects of subcutaneous injection. Thrombosis and Haemostasis, 30, 109–113. [PubMed] [Google Scholar]
- Guerrini M, Naggi A, Guglieri S, Santarsiero R, & Torri G (2005). Complex glycosaminoglycans: profiling substitution patterns by two-dimensional nuclear magnetic resonance spectroscopy. Analytical Biochemistry, 337, 35–47. [DOI] [PubMed] [Google Scholar]
- Guerrini M, Guglieri S, Naggi A, Sasisekharan R, & Torri G (2007). Low molecular weight heparins: structural differentiation by bidimensional nuclearmagnetic resonance spectroscopy. Seminars in Thrombosis and Hemostasis, 33, 478–486. [DOI] [PubMed] [Google Scholar]
- Herreroa LJ, Fooa S-S, Shenga Κ-C, Chena W, Forwoodb MR, Bucalac R, & Mahalingama S (2015). Pentosan polysulfate: a novel glycosaminoglycan-like molecule for effective treatment of alphavirus-induced cartilage destruction and inflammatory disease. Journal of Virology, 89, 8063–8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs A, Larsson PT, & Dahlman O (2001). Distribution of uronic acids in xylans from various species of soft- and hardwood as determined by MALDI mass spectrometry. Biomacromolecules, 2, 979–990. [DOI] [PubMed] [Google Scholar]
- Junior DL, Ayoub A, Venditti RA, Jameel H, Colodette JL, & Chang H (2013). Ethanol precipitation of hetero-polysaccharide material from hardwood by alkaline extraction prior to the kraft cooking process. BioResources, 8, 5319–5332. [Google Scholar]
- Kramer CM, Tsang AS, Koenig T, Jeffcott LB, Dart CM, & Dart AJ (2014). Survey of the therapeutic approach and efficacy of pentosan polysulfate for the prevention and treatment of equine osteoarthritis in veterinary practice in Australia. Australian Veterinary Journal, 92, 482–487. [DOI] [PubMed] [Google Scholar]
- Langeslay DJ, Beecher CN, Naggi A, Guerrini M, Torri G, & Larive CK (2013). Characterizing the microstructure of heparin and heparan sulfate using N-sulfoglucosamine 1H and 15N NMR chemical shift analysis. Analytical Chemistry, 85, 1247–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Raw A, Yu L, Lionberger R, Ya N, Verthelyi D, Rosenberg A, Kozlowski S, Webber K, & Woodcock J (2013). Scientific considerations in the review and approval of generic enoxaparin in the United States. Nature Biotechnology, 31, 220–226. [DOI] [PubMed] [Google Scholar]
- Lin L, Yu Y, Zhang F, Xia K, Zhang X, & Linhardt RJ (2019). Bottom-up and top-down profiling of pentosan polysulfate. Analyst, doi: 10.1039/c9an01006h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauri L, Boccardi G, Torri G, Karfunkle M, Macchi E, Muzi L, Keire D, & Guerrini M (2017). Qualification of HSQC methods for quantitative composition of heparin and low molecular weight heparins. Journal of Pharmaceutical and Biomedical Analysis, 136, 92–105. [DOI] [PubMed] [Google Scholar]
- Mulholland SG, Hanno P, Parsons CL, Sant GR, & Staskin DR (1990). Pentosan polysulfate sodium for therapy of interstitial cystitis: a double-blind placebo-controlled clinical study. Urology, 35, 552–558. [DOI] [PubMed] [Google Scholar]
- Parsons CL, Benson G, Childs SJ, Hanno P, Sant GR, & Webster G (1993). A quantitatively controlled method to study prospectively interstitial cystitis and demonstrate the efficacy of pentosanpolysulfate. Journal of Urology, 150, 845–848. [DOI] [PubMed] [Google Scholar]
- Read RA, Cullis-Hill D, & Jones MP (1996). Systemic use of pentosan polysulphate in the treatment of osteoarthritis. Journal of Small Animal Practice, 37, 108–114. [DOI] [PubMed] [Google Scholar]
- Sanden C, Mori M, Jogdand P, Jönsson J, Krishnan R, Wang X, & Erjefält JS (2017). Broad Th2 neutralization and anti-inflammatory action of pentosan polysulfate sodium in experimental allergic rhinitis. Immunity, Inflammation and Disease, 5, 300–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schamhardt DH, de Boer EC, & Kurth KH (1994). Interaction between bacteria and the luminal bladder surface: modulation by pentosane polysulfate, an experimental and theoretical approach with clinical implication. World Journal of Urology, 12, 27–37. [DOI] [PubMed] [Google Scholar]
- Scheller H, & Ulvskov P (2010). Hemicelluloses. Annual Review of Plant Biology, 61, 263–289. [DOI] [PubMed] [Google Scholar]
- Teleman A, Nordström M, Tenkanen M, Jacobs A, & Dahlman O (2003). Isolation and characterization of O-acetylated glucomannans from aspen and birch wood. Carbohydrate Research, 338, 525–534. [DOI] [PubMed] [Google Scholar]
- Urbanowicz BR, Peña MJ, Ratnaparkhe S, Avci U, Backe J, Steet HF, Foston M, Li H, O’Neill MA, Ragauskas AJ, Darvill AG, Wyman C, Gilbert HJ, & York WS (2012). 4-O-methylation of glucuronic acid in Arabidopsis glucuronoxylan is catalyzed by a domain of unknown function family 579 protein. Proceedings of the National Academy of Sciences of the United States of America, 109, 14253–14258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lee J-T, & Armstrong DW (1999). Separation of enantiomers by capillary electrophoresis using pentosane sulfate. Electrophoresis, 20, 162–170. [DOI] [PubMed] [Google Scholar]
- Zhong R, Teng Q, Lee C, & Ye Z-H (2014). Identification of a disaccharide side chain 2-O-α-D-galactopyranosyl-α-D-glucuronic acid in Arabidopsis xylan. Plant Signaling and Behavior, 9, e27933. doi: 10.4161/psb.27933 [DOI] [PMC free article] [PubMed] [Google Scholar]
Web references
- FDA approved drugs: Pentosan polysulfate sodium, NDA reference number 020193. (1996), available from https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm
- FDA. Draft guidance for industry on bioequivalence recommendations for pentosan polysulfate sodium capsule availability. (2012). Federal Register Notices, 77,183 https://www.govinfo.gov/content/pkg/FR-2012-09-20/pdf/2012-23177.pdf [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.