

Abstract
3D multicellular aggregates, and more advanced organotypic systems, have become central tools in recent years to study a wide variety of complex biological processes. Most notably, these model systems have become mainstream within oncology (multicellular tumor spheroids) and regenerative medicine (embryoid bodies) research. However, the biological behavior of these in vitro tissue surrogates is extremely sensitive to their aggregate size and geometry. Indeed, both of these geometrical parameters are key in producing pathophysiological gradients responsible for cellular and structural heterogeneity, replicating in vivo observations. Moreover, the fabrication techniques most widely used for producing these models lack the ability to accurately control cellular spatial location, an essential component for regulating homotypic and heterotypic cell signaling. Herein, we report on a 3D bioprinting technique, laser direct-write (LDW), that enables precise control of both spatial patterning and size of cell-encapsulating microbeads. The generated cell-laden beads are further processed into core-shelled structures, allowing for the growth and formation of self-contained, self-aggregating cells (e.g., breast cancer cells, embryonic stem cells). Within these structures we demonstrate our ability to produce multicellular tumor spheroids (MCTSs) and embryoid bodies (EBs) with well-controlled overall size and shape, that can be designed on demand. Furthermore, we investigated the impact of aggregate size on the uptake of a commonly employed ligand for receptor-mediated drug delivery, Transferrin, indicating that larger tumor spheroids exhibit greater spatial heterogeneity in ligand uptake. Taken together, these findings establish LDW as a versatile biomanufacturing platform for bioprinting and patterning core-shelled structures to generate size-controlled 3D multicellular aggregates.
Keywords: Tissue Engineering, 3D Bioprinting, Multicellular Tumor Spheroid, Embryoid Body, Microcapsule
Graphical Abstract
Multicellular 3D aggregates are powerful in vitro models used to study a wide variety of complex biological processes, particularly within oncology and regenerative medicine. These tissue surrogates are fabricated using environments that encourage cellular self-assembly. However, specific applications require control of aggregate size and position to recapitulate key in vivo parameters (e.g., pathophysiological gradients and homotypic/heterotypic cell signaling). Herein, we demonstrate the ability to create and spatially pattern size-controlled embryoid bodies and tumor spheroids, using laser-based 3D bioprinting. Furthermore, we investigated the effect of tumor spheroid size on internalization of transferrin, a common ligand for targeted therapy, finding greater spatial heterogeneity in large aggregates. Overall, this technique offers incredible promise and flexibility for fabricating idealized 3D in vitro models.
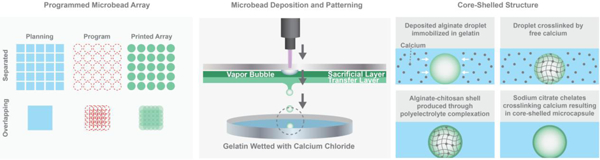
1. Introduction
The development of biomimetic 3D in vitro models has been a major research focus over the last decade. Such models are expected to play a key role in numerous biomedical fields, including in vitro diagnostics, high throughput drug-screening, personalized cell-based therapies, and reveal key attributes of various pathogenesis. However, to create models with well-controlled features that mimic in vivo conditions, as closely as possible, it is imperative to develop biomanufacturing methodologies that enable fabrication of spatially-defined 3D cultures or engineered constructs, using scalable, high-throughput platforms. Currently, much of the research in 3D cell cultures is led by the re-emergence of multicellular aggregates, and more complex organoids [1]. These models utilize self-aggregating cells in conjunction with methods designed to encourage their cell-to-cell interaction, (e.g., seeding cells within non-adherent conical wells (liquid overlay technique), hanging droplets, or microcapsules [2–5]), to form 3D cellular aggregates with self-derived matrices [6]. These models have become an important tool for self-assembling cell lines, such as cancer, liver, and stem cells, leading to the generation of matured 3D aggregates termed tumor spheroids, hepatocyte spheroids, and embryoid bodies, respectively [7–11]. Though, only recently, more attention has been devoted to characterizing the interplay between the morphology of these 3D aggregates and biological characteristics.
Undeniably, the 3D geometry and structural characteristics of these models are critical to their overall function, and physiological relevance. For both multicellular tumor spheroids (MCTSs) and embryoid bodies (EBs), the aggregate size plays a key role in the phenotype of the individual cells, and the overall model behavior. This is, at least in part, explained by the development of nutrient (e.g., oxygen and glucose) and cellular-signaling gradients within the aggregates. Within EBs, this phenomena is reflected in the preferential differentiation to specific cell lineages based on the size of the progenitor EB [12–15]. For MCTSs, the size has profound influence on the biological function due to its changes in the availability of oxygen, nutrients, and biochemical messengers throughout the spheroid. Small MCTSs (100–300 m diameter) exhibit no gradient-related challenges in cell survival, and demonstrate different 3D cell-to-cell and cell-to-matrix interactions, expression profiles, and proliferation rates as compared to 2D cultures [16,17]. This is a large step towards greater physiological relevance, while maintaining a size-scale compatible with conventional microscopy techniques [18]. At large sizes (500–1000 m diameter) spheroids often develop necrotic cores due to lack of sufficient nutrient/waste transport. This model may no longer be representative of in vivo conditions, where embedded vasculature prevents this extreme cell death from occurring. The size of these models also excludes them from analysis via microscopy. Thus, medium sized MCTSs (300–500 m diameter) provide a unique balance between relevant physiological features and the potential to be non-invasively assessed by contemporary imaging modalities. Indeed, medium sized MCTSs develop biochemical gradients with discrete radial cell zones (i.e., periphery cells are an active proliferating layer, and most inward are quiescent and/or hypoxic) [19,20]. The high level of hypoxia at their core enables in vitro mimicking of the pre-metastatic signaling found in vivo. This finding has led to the hypothesis that aggregate heterogeneity is a factor of increasing spheroid size, and that medium-sized tumor spheroids provide a more accurate surrogate for avascular solid tumors; which are of significant interest for screening due to the shortcomings in traditional therapeutics [21–23]. Unfortunately, with this increase in aggregate size over small spheroids, these models have still proven difficult, if not impossible, to assess by conventional brightfield or wide-field fluorescent microscopy.
Beyond size governing microenvironmental gradients, the shape of cellular aggregates is also an important factor in model development. Size-related heterogeneity is, in part, due to spatiotemporal signaling effects, which are influenced by aggregate shape. Often, it is assumed that cellular aggregates take on a spherical morphology, which is approximated by maximum intensity projection (MIP) images in the x-y plane taken using brightfield microscopy. Mounting evidence has shown that various aggregate models can have vastly different sizes and shapes, which can only be appreciated when analyzed in 3D [24]. Thus, aggregate shape is a crucial factor that can influence how the models behave, with important implications in drug screening applications, and the development of the desired pathophysiological gradients [25].
Beyond the effects of size and geometry of the spheroid, some advanced in vitro models seek to produce spatially-defined aggregate cultures or engineered constructs to study the influence of cell signaling; both homotypic signaling between neighboring aggregates, and heterotypic cross-talk between cellular aggregates and ancillary cells. Cell-cell signaling contains a significant element of spatial-temporal sensitivity, which requires fabrication tools with precise spatial-patterning capabilities. Additive manufacturing technologies, such as 3D bioprinting, show great promise to achieve the desired size, geometric, and spatial control of cellular aggregates, with the added benefit of being built on CAD/CAM technology that lends itself to scaling for high-throughput applications.
3D bioprinting is a powerful tool in tissue engineering, regenerative medicine, biosensing, and in vitro diagnostics [26–28]. While creating functional organs and tissue replacements is a compelling future goal, much of bioprinting’s current impact can be realized in in vitro applications, where its fabrication versatility, control, and throughput can be leveraged to produce sophisticated cell-based constructs to model pathologies, simulate injury and healing, personalize diagnostics, and create “organ-on-a-chip” models [29–33]. Of the various bioprinting techniques, laser-based bioprinting has emerged as a powerful, noncontact method to enable high-resolution spatial patterning of a variety of biomaterials (e.g., hydrogel solutions, microbeads), cells, proteins, and nucleic acids [34–37].
One such technique, laser-direct write (LDW) bioprinting, is a nozzle-free, forward transfer technique that uses a pulsed laser to deposit material from a ‘print ribbon’ to a ‘receiving substrate’. LDW falls in a class of bioprinting with other laser-based techniques, such as laser-induced forward-transfer (LIFT), AFA-LIFT, and biological laser-printing (BioLP), which have the advantage of high-spatial resolution patterning, the ability to dispense viscous and/or small volumes, and the precision to prescribe fine geometric features. The flexibility and performance of these techniques have been demonstrated in the printing of a variety of different architectures, using a multitude of natural and synthetic bioinks [38]. Of these bioinks, alginate serves as an exceptional material for bioprinting and cellular encapsulation, due to its biocompatibility and inert nature. Previously, we have shown our ability to use LDW for single-step fabrication and patterning of alginate microbeads, and process these beads into core-shelled microcapsules [39,40]. Herein, we utilize LDW bioprinting to pattern size-controlled alginate microbeads that can be processed into core-shelled structures. Within these structures, aggregating and self-assembling cells, namely cancer cells and stem cells, are shown to survive with high viability, and self-assemble to form 3D aggregates that grow to match the geometry of their printed construct. Tight size-regulation is demonstrated, and utilized to show that aggregate size influences the model’s biologic function, with larger tumor spheroids exhibiting greater spatial heterogeneity in protein ligand uptake. Overall, LDW’s ability to produce size-controlled 3D aggregates (e.g., MCTS, EB) in spatially-defined cultures or engineered constructs, makes it a powerful biofabrication method for applications in tissue engineering, in vitro diagnostics, and regenerative medicine.
2. Methods
2.1. Materials
For use as a transfer material, alginate was prepared as a solution of 2% (w/v) alginate (Sigma Aldrich, St. Louis, MO) and 0.9% (w/v) NaCl within cell culture grade dH2O. Solutions were subsequently filtered through 0.8 and 0.2 m filters, to ensure sterility. For use as a sacrificial layer, gelatin powder was added to warm filtered alginate to produce a solution with 2% alginate-10% gelatin (w/v). Chitosan (Spectrum, New Brunswick, NJ), for use in the processing of alginate to core-shelled structures, was dissolved in glacial acetic acid, and neutralized to a pH of 6.2 and 0.5% (w/v).
2.2. Cell maintenance
GFP labeled MDA-MB-231 human breast cancer cells (ATCC, Manassas, VA) were grown in standard cell culture conditions (37°C, 5% CO2, 95% RH) in growth medium consisting of Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 100 U mL-1 penicillin/streptomycin, and 2mM L-glutamine.
CCE Mouse Embryonic Stem Cells (mESCs) (StemCell Technologies, Vancouver, BC, Canada) were grown in standard ES maintenance media with 15% fetal bovine serum, 1mM sodium pyruvate, 100 U mL-1 penicillin/streptomycin, 2 mM L-Glutamine, 0.1 mM MEM non-essential amino acids, 10 ng mL-1 leukemia inhibitory factor (LIF), and 100 M monothioglycerol in DMEM high glucose. Cells were passaged every 2–3 days using 0.1% trypsin/EDTA onto 0.1% gelatin coated tissue cultured flasks [41].
2.3. Laser direct write (LDW) bioprinting
Single-step fabrication and patterning of alginate microbeads via LDW bioprinting is described in detail in our previous work, and demonstrated in Figure 1 [39]. Briefly, the LDW system (Teosys LLC, Crofton MD) consists of an ArF excimer laser source, with independently CAD/CAM-controlled printing ribbon and receiving substrate stages. To prepare a print ‘ribbon’, a UV-transparent quartz disk (Edmond Optics, Barrington, NJ) is spincoated with a thin, ‘sacrificial layer’ of alginate-gelatin, and a subsequent layer of ‘transfer material’ containing the biopayload is added via pipette. For these experiments, a fixed-density of cells suspended in alginate served as the transfer material, and a Petri dish, or a glass bottom MaTek plate (when needed for confocal microscopy), spincoated with a thin film of 10% gelatin-2% CaCl2, was utilized as the receiving substrate.
Figure 1:

Schematic of LDW (a), representative diagrams of desired geometry (blue), programmed print arrays (red), and depiction of how the printed array would appear (b), 3D view of patterned beads (c), their processing into core-shelled structures (d), and aggregate formation (e).
To fabricate and pattern a microbead (i.e., “print a microbead”), a pulse from the ArF excimer laser (193nm) passes through a variable iris and beam-delivery optics, hitting the UV-transparent print ribbon where it volatizes the sacrificial layer, forming a vapor pocket that ejects a spot of transfer material (e.g., cell-loaded alginate), from the print ribbon to the underlying receiving substrate. As the material leaves the print ribbon, it forms a droplet, taking on a spherical shape through cohesion and surface tension, then lands in the gelatin film on the receiving substrate, where it rapidly crosslinks in situ due to the free calcium on the substrate [39]. This crosslinking event occurs very rapidly, such that the resultng microbead retains the spherical shape of the droplet [39]. Immediately after the microbead is printed, the receiving substrate is automatically moved to the next programmed position within the printed array, the ribbon stage moves to utilize a new loacation on the print ribbon, and the laser is again pulsed to creat a new microbead. This process continues until the entire pattern is complete, at which point the printed microbead construct is incubated in 2% CaCl2 solution for 15 minutes to provide additional crosslinking, then switched to cell culture medium.
2.4. Control and characterization of microbeads (size and cell density)
In our LDW system, the beam diameter can be adjusted by an intra-cavity variable aperture before the laser objective, to change the laser spot size, which we utilize to control the size of the printed microbead. We also adjust the cell seeding densities on our print ribbon in an effort to change the cell density in the printed microbeads. To determine whether we could utilize these to control the size and cell density of the resulting microbeads, respectively, we created 5x5 microbead arrays at two discrete beam diameters (small ~100 μm, and larger ~175 μm), for five different initial ribbon cell seeding densities (1.0, 2.5, 5, 7.5, and 10 M/ml). Beam size was measured by pulsing the laser on an ink-covered glass slide, and laser energy was recorded using an Opher energy meter, and printed microbead arrays were analyzed for size and cell number, as described below.
After crosslinking of the alginate, images were acquired using a Zeiss Z1 microscope with Axiovision software (Carl Zeiss, Thornwood, NY). Each alginate microbead was approximated as spherical, and the diameter of beads was analyzed within Axiovision software or ImageJ (US National Institutes of Health Bethesda, MD, USA). Cells were counted within collected images using ImageJ (US National Institutes of Health Bethesda, MD, USA) with the cell count plugin.
2.5. Processing alginate-based structures to core-shelled constructs
Within this work, the printed constructs were typically 5x5 arrays of individual alginate microbeads. To explore larger aggregates, the nodal spacing between patterned alginate spots was collapsed, such that microbeads were printed in overlapping fashion to create a single, rectangular alginate structure, or mat. As illustrated in Figure 1, to create a larger rectangular mat, a 5x5 array was printed with bead diameters twice the size of the between-bead distance.
Fabricated alginate microbeads and microstructures were given 1 hour in cell culture medium prior to processing into core-shelled constructs [40], then washed thrice with 0.9% NaCl to remove any excess media from substrates. Next, a 0.5% chitosan solution was washed over the alginate structures for 7 minutes; during which the chitosan complexes to alginate structure periphery, in a process known as polyelectrolyte complexation. After the chitosan treatment, 0.9% NaCl is washed thrice again used to remove excess solution. The chitosan-shelled alginate structures were then washed with 0.1% sodium citrate solution (a calcium sequesterate), and incubated for 2 minutes to liqeufy their cores; producing a polymer-shelled microenvironment with a liquid core; termed a core-shelled strucutre. Excess sodium citrate was then removed, and the samples were washed with NaCl before cell culture medium was added to the construct.
2.6. Cell viability analysis
An analysis of cellular viability was performed after each of the following steps: loading of alginate cell suspension to the print ribbon, deposition of a microbead, and after processing to a core-shelled microcapsule (for MDA-MB-231 and CCE cells), using Live-Dead Stainning Kit (Enzo Life Sciences, Farmingdale, NY), following the maufacturers staining procedure. Multichannel fluorescent imaging was performed, in triplicate, on microbeads and microcapsule arrays (n=36), using a Zeiss Z1 microscope and Axiovision software (Carl Zeiss, Thornwood, NY). The number of red (dead) and green (live) cells were independently quantified, and the average viability was calculated as an overall percentage at each time point.
2.7. Optical coherence tomography
OCT is a well-established technique that enables 3D imaging of biologial structural features, at micrometer resolution. OCT has been recently proposed as a powefull tool to assess biomanufactured constructs non-invasively [42,43]. Herein, we employed this imaging modality to structurally assess laser-fabricated core-shelled structures. OCT imaging was performed using a commercial spectral domain OCT system (TEL220C1; Thorlabs Inc.), having a maximum sensitivity of 101 dB and a maximum A-scan line rate of 76 kHz. For all imaging, the 5.5 kHz line rate (highest sensitivity) was utilized, with 1024 A-scans per B-scan. The OCT probe was positioned orthogonally above the sample in the experimental design. Cellular aggregates contained within core-shelled structures were visualized and quantified using Imaris image analysis software (v9.2, Bitplane USA, Concord, MA) [44].
2.8. Cell staining and confocal microscopy
Core-shelled structures, containning GFP stably transfected MDA-MB-231 cells, were grown on 1.5 (25 mm) glass coverslips within 6-well plates, for a 14-day period to completely fill the structure. For Transferrin (Tf) uptake, the samples were washed with Dulbecco modified-Phosphate buffer saline (DPBS), incubated for one hour at 37°C in serum-free imaging medium (DHB) to deplete unlabelled endogenous Tf (DHB: phenol red-free DMEM, 5 mg/mL bovine serum albumin (Sigma), 4 mM L-glutamine, 20 mM HEPES (Sigma) pH 7.4). Then, samples were loaded for 1 h at 37°C with 40 μg/mL iron-loaded Tf-Alexa Fluor 555 (Life Technologies) in DHB medium. Samples were then washed twice with DPBS and fixed with 4% Paraformaldehyde for 15 min, followed by two additional PBS washes. Cells were counterstained for 15 minutes with 0.1 μg/mL DAPI (4’,6-diamidino-2-phenylindole) solution for nuclear visualization, and washed again with DPBS. Prepared samples were stored in DPBS until imaging. Imaging was performed using a Zeiss LSM510 META-NLO multiphoton confocal laser microscope. Images were acquired with a 25x oil immersion lens. To visualize the 3D cellular aggregate within a microcapsule, up to 120 Z-stacks, 1 μm z-interval, were collected for 3D rendering using Imaris software.
2.9. Imaris: structural and functional quantification
Collected OCT data were imported into Imaris; there, segmentation of the sample region of interest was performed via intensity-based thresholding. The ‘Add Surface’ function was used to trace the aggregate slice-by-slice. The resulting volume was stitched together using the ‘Mask all’ tool to enable 3D visualization and quantification of the aggregates. Metrics pertinent to aggregate physiology, such as aggregate sphericity and bulk volume, were reported using the ‘Statistics’ tool.
Collected fluorescence data were similarly imported into Imaris, for both large and small MCTSs. Samples were analyzed for their uptake of Tf using the 3D rendering “surface” Imaris module and a quantitative method for determining regional fluorescence intensity levels of Tf and GFP. Using the “line intensity” Imaris module, 8 lines were selected to cross the whole 3D aggregate volume, from top center to different points at bottom periphery (lines 1–8). Data from each of the channels (GFP & Tf) were removed when GFP fluorescence intensity <100 arbitrary units, prior to calculation of Tf/GFP ratio.
2.10. Statistical analysis
All statistical analyses were performed within MATLAB (MathWorks Inc., Natick, MA). Exploratory analyses utilized bar graphs and scatter plots to visualize the means, variance, and distribution of the data. Data were further assessed for normality using Anderson-Darling tests. All data sets examined were normally distributed, with groups of comparable sizes, and equal variances, justifying the use of parametric statistical analyses. Data were analyzed by one-way ANOVA (analysis of variance, using the built-in “anova1” function) to identify any significant differences observed between variables, with post-hoc analysis (Tukey’s Honestly Significant Difference) to determine which variables had significant effects on these values. In all cases, p < 0.05 for statistical significance.
Frequency distribution analysis of the Tf/GFP ratios was performed using GraphPad Prism (GraphPad Software, Inc). Results were plotted as Tf/GFP ratio of fluorescence intensity vs. distance from top point to bottom periphery points (lines 1–8), to identify regional differences in Tf internalization within a single aggregate, and to qualitatively compare uptake between small and large MCTSs.
3. Results
3.1. Ribbon density and printed bead characteristics
Laser energy characterized for the small and large beam sizes was measured during printing (frequency of 1Hz) for all patterns, revealing mean energies of 3.05 0.66 and 5.01 1.21 μJ for the small and large beams, respectively (Fig. 2a), with Gaussian distribution profiles (Fig. 2b). This resulted in alginate beads with diameters of 200 μm for small beam sizes (202 30 μm), and 400 μm for large beam sizes (410 60.0 μm) (Fig. 2c,d), illustrating the ability to control microbead size via beam diameter.
Figure 2:

The average recorded energy and distribution of 100 μm (small) and 175 μm(large) beam size (n = 100 pulses) (a,b), and representative fabricated microbeads at the small (c) and large (d) beam diameters. Scale bars are 200 μm.
However, for a given beam diameter, the microbead sizes appeared to vary slightly with changes in ribbon seeding density, although no clear trend in microbead diameter with ribon seeding density was observed (Fig. 3a,b). The number of cells per bead was positively correlated with ribbon seeding density (Fig. 3), with roughly 3–21 cells in small beads (Fig. 3a), and 15–157 cells in large beads (Fig. 3b). When this count was normalized to bead volume, we found printed microbead cell densities ranging from 0.69 (0.48) to 5.69 (1.84) M/mL in small beads, and 0.20 (0.07) to 3.24 (0.64) M/mL in large beads (Fig. 3e). Thus, small bioprinted microbeads displayed cell densities closely resembling that of the print ribbon, whereas cell densities in the larger printed beads were roughly half that of the ribbon seeding density (Fig. 3e).
Figure 3:

The influence of ribbon density on the resultant bead diameter (a-b), the number of cells per microbead (c-d), and the overall cell density per bead volume (e) (n = 48 microbeads).
3.2. Viability of cells printed with LDW
Short-term cell viability was assessed within printed alginate microbeads and subsequently processed microcapsules, containing either GFP-expressing MDA-MB-231 breast cancer cells, or CCE stem cells. Cell viability was reported on the print ribbon (control), within alginate microbeads 1 h after printing, and within LDW-printed microcapsules 1 h after processing (Fig. 4). Cancer cells were 97.6 2.1% viable on the print ribbon, which reduced to 83.9 8.1% after printing as a microbead, and 75.5 7.2% within a processed microcapsule. For stem cells, this viability was 93.0 2.0%, 76.3 7.3%, and 70.2 6.6%, for ribbon, microbead, and microcapsule, respectively. Both cell types exhibited a significant drop in viability from print ribbon to microbead, and from microbead to microcapsule, yet maintained high viability throughout processing. In addition, viability appears to be lower in stem cells than tumor cells at every stage (i.e., ribbon, microbead, and microcapsule).
Figure 4:

Viability assessment throughout fabrication process. Stem cells’ and tumor cells’ viability was assessed upon loading on the ribbon (control), immediately after printing and fabrication into a microbead, and upon processing into a microcapsule. Stem cells appear to be more sensitive to this processing, but overall both cell types maintain high viability.
3.3. Customizable core-shelled structures
Core-shelled microcapsule arrays (5 x 5 arrays with 600-μm centroid-to-centroid spacing) were produced containning mESCs, at both small and large microbead sizes. The time course of stem cell growth and aggregation within these capsules can be visualized in Figure 5. Small aggregates begin to form within the capsule after 1 day in culture, then merge into a larger single aggregate by day 7, which continues to grow until it fills the entire capsule volume by day 14. This was repeated in similar 5 x 5 arrays of cancer cells, printed with reduced array spacing (200-μm centroid-to-centroid) to demonstrate the spatial control of microcapsules created by LDW bioprinting (Fig. 5). Similar growth and aggregation was observed for the cancer cells over the 14-day time course; aggregating, merging, and forming a single large aggregate filling the 3D volume of each microcapsule.
Figure 5:

Representative images of various print configurations of microbeads (5x5 array and rectangular mat) at day 0, at 1, 7 and 14 days after their processing into a core-shelled structures. Over the 14-day period, both stem cells (upper rows) and tumor cells (lower rows) formed single aggregates which grew to fill the volume of each core-shelled structure. Scale bars are 500 μm (rows 1and 4) and 200 μm (row 2 and 3).
In the larger-volume custom geometries, created by printing the beads in overlapping fashion to form a rectangular micromat (Fig. 1b,c), the structure and geometry was maintained throughout the core-shelling process. Over a 14-day time course, the encapsulated MDA-MB-231 cancer cells proliferated and self-assembled to form a large, single aggregate that filled the entirety of the shelled-volume in which they were encapsulated (Fig. 5).
3.4. Structural evaluation of cell constructs
OCT provided a label-free, non-invasive method to image the aggregates grown within the core-shelled environments. When processed within Imaris, OCT images granted visualization and quantification of the 3D geometries (Fig. 6). Core-shelled structures of different sizes (i.e., small microcapsule, large microcapsule, and rectangular mat) produced aggregates of significantly different sizes, such that larger core-shelled microenvironments yielded larger 3D aggregates, for both stem cells and tumor cells. Embryoid bodies grown in small microcapsules (volume = 3.68 0.51), were signifincantly smaller than those grown in larger microcapsules (volume = 56.5 8.9 x106 μm3) (Fig. 7c). The same was true for cancer cells, where aggregate volumes increased significantly when grown in larger core-shelled strucutres; small microcapsules, large microcapsules, and a rectangular mat, produced tumor aggregate volumes of 3.38 0.74 x106 μm3, 56.4 10.2 x106 μm3, and 173 62 x106 μm3, respectively (Fig. 7c). Thus, the size of the aggregate, for both multicellular tumor spheroids (MCTS) and embryoid bodies (EBs), could be controlled by the size of the core-shelled structure. An additional important note, when grown in similarly-sized microcapsules, cancer cells and stem cells produced 3D aggregates of similar volume.
Figure 6:

Volumetric scan of a 14-day EB grown in large (400 μm) capsule, collected via OCT and processed using Imaris analysis software.
Figure 7:

OCT images processed in Imaris of a small microcapsule (ai), a large microcapsule (aii), and a rectangular mat (aiii). The overall volume of the structures (b) and sphericity (c) of the aggregates grown within small and large microcapsule. Scale bars are 200 μm.
In addition to volume, Imaris also enabled measurements of aggregate sphericity. Stem cell aggregates formed in small and large microcapsules were highly spherical, with sphericities of 0.986 0.006 and 0.917 0.017, respectively. Tumor aggregates also displayed a high sphericity when grown in small microcapsules (sphericity = 0.976 0.059), which was significantly lower in large microcapsules (0.822 0.049), and further reduced when grown in the core-shelled micromat (Fig. 7b). Aggregates contained within the micromats displayed a significantly lower sphericity than all other samples. While all of the microcapsule structures had overall high sphericity (> 0.8), smaller microcapsules produced aggregates with significantly greater sphericity than those grown in the larger microcapsules. This is likely due to differences in the amount of flatening the alginate microbead experinces during crosslinking, with increased flattening occuring at larger bead sizes, which would be reflected in the resultant processed microcapsule. Interestingly, stem cells and tumor cells produced aggregates with similarly high spherecity when grown in small microcapsules, however, when grown in larger microcapsules, stem cell aggregates (i.e., EBs) had notably higher sphericity than analogous cancer cell aggregates (i.e., MCTS).
3.5. Molecular imaging of core-shelled structures
Confocal microscopy was performed on aggregates grown within small and large microcapsules (Fig. 8) to visualize the cellularity of the aggregate, as well as its overall 3D morphology. We observed high cellularity on the aggregate edges, demonstrated by the relative intensity of GFP and DAPI signal. However, significant attenuation of GFP and DAPI was observed towards the center of the aggregates (both small and large) within the cross-sectional images (Fig. 8a,b). Two main issues may be at the basis of this fluorescence intensity decrease towards the center of the aggregates. Firstly, confocal microscopy has been shown to have difficulty in imaging the core of tumor spheroid due to reduced light penetration [45]. Secondly, DAPI visualization is negatively affected by dye delivery issues across the cellular aggregates. Utilizing a mean intensity projection (MIP), we were able to visualize the aggregate clearly within the x-y and x-z plane (Fig. 8c,d). Although the MIP provided evidence of general aggregate sphericity, the aggregate size (z-direction) appeared to exceed the depth capabilities of the confocal microscope to produce an overall 3D image, as seen within the saggital view.
Figure 8:

Confocal microscopy images of representative MDA-MB-231 3D aggregates within small (a,c) and large (b,d) microcapsules at day 14. Aggregates were GFP-transfected (green) and nuclear stained with DAPI (blue). Cross-sections (a,b), and mean intensity projections (MIP) (c,d) demonstrate the shape of the aggregates, as well as the limitations of confocal imaging for capturing the full 3D geometry of the larger aggregates.
In addition to 3D morphology within the tumor spheroid aggregates, we sought to validate the functional status of the cellular aggregates within the microcapsules by evaluating their ability to take in iron-loaded Transferrin (Tf) conjugated with Alexa Fluor 555. In both small and large MCTSs, Tf-containing structures can be detected across x, y, and z-directions, as shown by their typical endosomal punctate staining (Figure 1 supplementary). To identify differences in Tf uptake heterogeneity, between the large and small aggregates, we analyzed the confocal z-stacks using 3D rendering Imaris software. In both small and large aggregates, Tf signal appears to be stronger on the periphery of the spheroids, which reflects the decreased diffusion rate towards the core of the aggregates, as well the limitations of confocal imaging in reaching the core of the aggregates (Fig. 9). However, in the small aggregate, Tf signal is more evenly distributed than in the large aggregate, which shows areas of reduced Tf uptake at its core (Fig. 9A). Eight lines were established across the radius of the aggregates, and subsequently analyzed using the intensity module of Imaris to extract their respective Tf and GFP fluorescence intensity levels, as described in Methods (Figure 9A). In Figure 9B, the Tf/GFP ratio was used to determine the heterogeneity of Tf uptake in cells expressing GFP in the small and large aggregates. Since areas of very low Tf and GFP fluorescence intensity may reflect technical limitations of confocal microscopy in reaching across the 3D z-axis of the cell aggregates, we have removed data that displays very reduced Tf and GFP levels (GFP <100 A.U.), prior to the Tf/GFP ratio calculation. To evaluate heterogeneity of Tf uptake across small vs. large aggregates, lower Tf/GFP ratios (< 0.5) indicate reduced level of Tf uptake in cells expressing detectable levels of GFP, whereas, higher Tf/GFP ratios (> 0.5) indicate higher Tf levels in cells expressing GFP. The smaller aggregate displays higher Tf/GFP ratios across its z-axis, whereas the larger aggregate shows a reduced frequency of elevated Tf/GFP ratios and an increased frequency of low Tf/GFP ratios, suggesting that aggregate size may play an important role in determining Tf uptake levels.
Figure 9:

3D rendering of Tf-containing endocytic structures using Imaris “surface” module in cells expressing GFP. Panel A: Tf-containing 3D rendered structures display mean intensity values (a, c, I, k) and z-position (b, d, j, l) in aggregates with GPF-cytosolic staining (e, g, m, o) to indicate the presence of cells expressing GFP, and Tf fluorescence intensity to indicate Tf uptake (f, h, n, p). The 8 lines (gray lines) across Tf-containing endocytic structures are shown for the smaller and larger aggregates. Scale bar: 50 m. Panel B: In panels a and c, heat maps of frequency % of Tf/GFP ratios are shown for large and small capsules, respectively. In panels b and d, Tf/GFP intensity ratio vs. distance ( m) charts are shown using the Imaris “line intensity” module.
4. Discussion
There is a compelling need for more advanced, physiologically-representative 3D in vitro models to overcome limitations presented by simplified culture platforms and complicated in vivo testing and translation. Cellular aggregates provide a promising framework to bridge simple 2D models, that fail to recapitulate the 3D microenvironment and key biological phenomena, to xenograft models, which are costly (i.e., time and resources), as well as difficult to reproduce. Additive manufacturing technologies, such as LDW bioprinting, offer the potential for enhancing the cellular aggregate model to a more scalable, sophisticated tissue-engineered model. We have previously shown that LDW can be used to fabricate and spatially pattern cell-loaded microbeads [39], which can be utilized to create customizable, cell-loaded, core-shelled structures [40]. Core-shelled microcapsules provide an effective method for generating 3D cellular aggregates [46,47]. When compared to other cell encapsulation and core-shelled techniques (i.e., electrostatic bead generators and microfluidic channels), LDW bioprinting has the added benefit of being built on scalable, CAD/CAM patterning technology, making it possible to produce size-controlled spatially-defined constructs, ultimately in high-throughput fashion. These two, seemingly disparate techniques (LDW and cellular aggregates), together have the potential for advancing our current control and capabilities for in vitro models. Herein, we applied LDW to fabricate and pattern core-shelleds structures, containing either cancer or stem cells, demonstrating our ability to control aggregate size, spatial placement, and overall geometry.
A key aspect of embryoid bodies’ and tumor spheroids’ performance is their size. For embryoid bodies, their differentiation potential has been shown to be strongly influenced by the resultant size of the aggregate [14,48,49]. We have previously shown that embryoid body size can be manipulated with 2D gelatin-based LDW, via changes in printed cellular density [50]. Aggregate size also has important implications for tumor spheroids; small spheroids remain healthy, missing a hypoxic core, which is a hallmark of avascular solid tumors [51–53], whereas large spheroids develop overly necrotic cores that precludes their use for modeling in vivo conditions. On the other hand, medium-sized MCTSs capture the characteristic hypoxic core, allowing investigations into cancer cell signaling. Thus, aggregate size control is critical, and highly desired in any biofabrication or biomanufacturing method. We were able to control final aggregate size using LDW bioprinting, by adjusting the beam diameter of the laser to create size-controlled microbeads (e.g., small and large scale), which were further processed into microcapsules that would constrain the final aggregate geometry. While the total laser energy was significantly greater for the large beam diameter (on average 40% higher than the small diameter), there was overlap in the tails of the energy distributions. Interestingly, the beads produced at the large beam diameter were twice the size of the small beam diameter, with no observable overlap. This implies that beam diameter in LDW bioprinting plays a more significant role in determining the size of deposited alginate droplets, than the total laser energy.
With LDW, we controlled the initial number of cells deposited within each bead (and its associated microcapsule), as well as the initial cellular density of LDW-printed structures (e.g., micromat), through manipulating the seeding density of cells suspended on the print ribbon. This allowed for great flexibilty, enabling controlled fabrication of cellular aggregates over a broad range of initial growth conditions. Within the printed microbeads, high cellular viability was observed for both cell types, however, there was a significant drop in viability at each step of the process (i.e., suspending cells to the ribbon, printing microbeads, and processing to core-shelled structures). This reduction in viability was greater in stem cells than the cancer cells. Despite drops in viability during the process, the overall cell viability within the final bioprinted structure was still high (>80%), and similar to previous printing work with embryonic stem cell [54,55].
Within core-shelled structures, tumor spheroids and embryoid bodies were observed to form and grow over a 14-day time course. The core-shelled structures were able to constrain the growing aggregates within, allowing for control over the final size, and shape, of the 3D cellular aggregate. Aggregates within core-shelled microcapsules exhibited high sphericity, which reduced slightly with increasing aggregate size, possibly due to flattening occurring with larger droplet sizes (either at initial microbead fabrication, during processing into microcapsules, or over the time of aggregate formation). Because our volume calculations assume a spherical geometry, these differences in microbead sphericity (between small and large beads) may have had a minor effect on bead density values, which would lead to slight overestimation of bead volume, and underestimation of cell density in the larger microbeads. Small and large tumor spheroids were examined with confocal microscopy, revealing the high cellularity of aggregates grown within microcapsules. Heterogeniety within the aggregates was probed through the use of fluorescently-labeled Tf, whose receptor is often overexpressed in cancer cells [56–58]. Tf has been widely used in anti-cancer drug/siRNA delivery systems [59–66], and in molecular imaging of tumors [67,68]. The aggregates of both sizes appeared to have greater internalization of Tf closer to the edges compared to the core. Most likely, this Tf distribution pattern is a result of protein diffusion rate, since Tf receptor is upregulated by hypoxia, and its density would be expected to be higher in the center cells [69]. When comparing these regional differences of Tf internalization between aggregates, each size appeared to have a unique intensity-position plot profile, with the larger aggregate having greater distribution variability. Whereas the Tf signal is detected at similar levels from the edge to the center of the smaller aggregate, the larger aggregate shows clear visible areas of reduced Tf uptake at its core. These results suggest that larger aggregates present an improved model for studies of drug distribution and delivery.
Beyond functional quantification, confocal microscopy was similarly used to gauge the overall 3D structure of spheroids. However, there appeared to be significant amounts of GFP signal attenuation toward the center of the aggregate when viewed in cross-section (Fig. 8), likely due to their high cellular density. Furthermore, it appeared that there were limitations in DAPI diffusion into the aggregate core, because DAPI could only be visualized at depths within ~100 microns of the aggregate surface. The MIP 3D renderings of these large aggregates hinted that they were spherically-shaped, however the size and signal attenuation in the 3D aggregates prevented confocal microscopy from accurately estimating their size and sphericity. Typically, such measurements are made from 2D projections in brightfield microscopy, under the assumption that the aggregates are spherical, which is often not the case. We utilized OCT imaging and Imaris image analysis to accurately quantify aggregate size and geometry, showing LDW’s strong volumetric control of aggregates, in numerous individual microcapsules, for both stem cell and cancer cell lines. Additionally, aggregates formed within these structures displayed high sphericity, indicating a truly 3D geometry. Sphericity appeared to decrease within larger microcapsules, especially for tumor cell aggregates, which may imply that stem cells posess stronger cues for self-assembly to generate a spherical aggregate.
In addition to size-control, our LDW method provides spatial control of the aggregates within cultures, bioprinted arrays, or 3D tissue-engineered constructs. Leveraging this combination of size control and bead/capsule localization, we were able to print cell-loaded beads, in overlapping fashion, to create large custom geometries, such as micromats. These cell-loaded micromats could then be processed into core-shelled structures in which the cells could proliferate and aggregate to fill the entire continuous geometry. With this method, production of an aggregate beyond the size of a single microcapsule was possible (macrocapsule). This could not have been done without the size control and placement precision that is uniquely afforded by the direct-writing technique. By using cell-loaded microbeads as building blocks, large 3D aggregates with custom geometries can be fabricated to a desired shape by planar (2D) patterning, while maintaining a highly favorable surface area to volume ratio. Extensions of this work have great potential for the addition of patterning in 3D, (layer-by-layer) to produce larger more sophisticated, core-shelled geometries and resulting aggregates. Using such models, we can explore whether cellular aggregates of the same volume, but different 3D geometries, exhibit metabolic differences, due to differences in nutrient/waste diffusion rates. Moreover, future work can utilize various core-shelled geometries to probe whether tumor spheroids behave in characteristic fashion for their volume, when the aggregate is no longer spherically shaped. This could elucidate important volume- and surface-based properties of tumor spheroids, and the influence of size and shape on their cellular behaviors and response to drugs.
5. Conclusion
Herein, we demonstrate the ability to produce and spatially pattern, size-controlled 3D cellular aggregates, by creating core-shelled microenvironments of aggregating cell types, using LDW bioprinting. Within these structures, both cancer cells and stem cells remain highly viable, and self-assemble to form 3D aggregates (tumor spheroids and embyroid bodies, respectively), that have sizes corresponding to the dimensions of the fabricated capsule. Finally, we can leverage LDW’s unique size- and spatial-control to print cell-loaded microbeads, in overlapping fashion, to build larger, continous alginate structures, of custom geometries, which can be processed into core-shelled structures to create large, size-controlled, and shape-controlled, 3D aggregates. This ability to create size-controlled 3D aggregates, in spatially-defined cultures or engineered constructs, opens exciting possibilities for next generation in vitro models in oncology, regenerative medicine, and diagnostics.
ACKNOWLEDGMENT
This work was supported by the National Institute of Health (NIH) BRG CA207725 (DTC, MB, XIN); RPI’s Knowledge and Innovation Program (KIP) seed program (DTC, XIN). We would like to thank Christopher Sloat for his help with the Imaris software, Dana Lee Church and Jason Acuesta for their assistance with the Teosys laser system, Brad Tricomi for his work on microbead cellular density, and Summer Jenning for her help with the fabrication schematic. We would also like to thank the AMC imaging core for the use of Zeiss LSM confocal microscopes and Dr. L. Wang for her help with the image analysis in Figure 9.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none
Part of the Cell and Tissue Biofabrication Special Issue, edited by Professors Guohao Dai and Kaiming Ye.
Contributor Information
David M. Kingsley, Department of Biomedical Engineering, Rensselaer Polytechnic Institute, 110 Eighth St., Troy, NY 12180, USA
Cassandra L. Roberge, Department of Biomedical Engineering, Rensselaer Polytechnic Institute, 110 Eighth St., Troy, NY 12180, USA
Alena Rudkouskaya, Department of Molecular and Cellular Physiology, Albany Medical College, Albany, NY, 12208, USA.
Denzel E. Faulkner, Department of Biomedical Engineering, Rensselaer Polytechnic Institute, 110 Eighth St., Troy, NY 12180, USA
Margarida Barroso, Department of Molecular and Cellular Physiology, Albany Medical College, Albany, NY, 12208, USA.
Xavier Intes, Department of Biomedical Engineering, Rensselaer Polytechnic Institute, 110 Eighth St., Troy, NY 12180, USA.
David T. Corr, Department of Biomedical Engineering, Rensselaer Polytechnic Institute, 110 Eighth St., Troy, NY 12180, USA.
REFERENCES
- [1].Sachs N, Clevers H, Organoid cultures for the analysis of cancer phenotypes., Curr. Opin. Genet. Dev 24 (2014) 68–73. doi: 10.1016/j.gde.2013.11.012. [DOI] [PubMed] [Google Scholar]
- [2].Fennema E, Rivron N, Rouwkema J, Van Blitterswijk C, De Boer J, Spheroid culture as a tool for creating 3D complex tissues, Trends Biotechnol 31 (2013) 108–115. doi: 10.1016/j.tibtech.2012.12.003. [DOI] [PubMed] [Google Scholar]
- [3].Tung Y-C, Hsiao AY, Allen SG, Torisawa Y, Ho M, Takayama S, High-throughput 3D spheroid culture and drug testing using a 384 hanging drop array., Analyst 136 (2011) 473–8. doi: 10.1039/c0an00609b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Perez RA, Kim H, Core – shell designed scaffolds for drug delivery and tissue engineering, Acta Biomater 21 (2015) 2–19. doi: 10.1016/j.actbio.2015.03.013. [DOI] [PubMed] [Google Scholar]
- [5].Liu Z, Takeuchi M, Nakajima M, Hasegawa Y, Huang Q, Shape-controlled high cell-density microcapsules by electrodeposition, Acta Biomater 37 (2016) 93–100. doi: 10.1016/j.actbio.2016.03.045. [DOI] [PubMed] [Google Scholar]
- [6].Friedrich J, Seidel C, Ebner R, Kunz-Schughart LA, Spheroid-based drug screen: considerations and practical approach, Nat. Protoc 4 (2009) 309–324. doi: 10.1038/nprot.2008.226. [DOI] [PubMed] [Google Scholar]
- [7].Zaytseva-Zotova DS, Udartseva OO, Andreeva ER, Bartkowiak A, Bezdetnaya LN, Guillemin F, Goergen J-L, Markvicheva E. a, Polyelectrolyte microcapsules with entrapped multicellular tumor spheroids as a novel tool to study the effects of photodynamic therapy., J. Biomed. Mater. Res. B. Appl. Biomater 97 (2011) 255–62. doi: 10.1002/jbm.b.31808. [DOI] [PubMed] [Google Scholar]
- [8].Zhang X, Wang W, Yu W, Xie Y, Zhang X, Zhang Y, Ma X, Development of an in Vitro Multicellular Tumor Spheroid Model Using Microencapsulation and Its Application in Anticancer Drug Screening and Testing, Biotechnol. Prog 21 (2005) 1289–1296. [DOI] [PubMed] [Google Scholar]
- [9].Wang X, Wang W, Ma J, Guo X, Yu X, Ma X, Proliferation and differentiation of mouse embryonic stem cells in APA microcapsule: A model for studying the interaction between stem cells and their niche., Biotechnol. Prog 22 (2006) 791–800. doi: 10.1021/bp050386n. [DOI] [PubMed] [Google Scholar]
- [10].de Vos P, Faas MM, Strand B, Calafiore R, Alginate-based microcapsules for immunoisolation of pancreatic islets., Biomaterials 27 (2006) 5603–17. doi: 10.1016/j.biomaterials.2006.07.010. [DOI] [PubMed] [Google Scholar]
- [11].Zhang W, Zhao S, Rao W, Snyder J, Choi JK, Wang J, Khan IA, Saleh NB, Mohler PJ, Yu J, Hund TJ, Tang C, He X, A Novel Core-Shell Microcapsule for Encapsulation and 3D Culture of Embryonic Stem Cells, J Mater Chem B Mater Biol Med 2013 (2013) 1002–1009. doi: 10.1039/C2TB00058J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cha JM, Bae H, Sadr N, Manoucheri S, Edalat F, Kim K, Kim SB, Kwon IK, Hwang YS, Khademhosseini A, Embryoid body size-mediated differential endodermal and mesodermal differentiation using polyethylene glycol (PEG) microwell array, Macromol. Res 23 (2015) 245–255. doi: 10.1007/s13233-015-3034-0. [DOI] [Google Scholar]
- [13].Schukur L, Zorlutuna P, Cha JM, Bae H, Khademhosseini A, Directed Differentiation of Size-Controlled Embryoid Bodies Towards Endothelial and Cardiac Lineages in RGD-Modified Poly(Ethylene Glycol) Hydrogels, Adv. Healthc. Mater 2 (2013) 195–205. doi: 10.1002/adhm.201200194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bauwens CL, Peerani R, Niebruegge S, Woodhouse KA, Kumacheva E, Husain M, Zandstra PW, Control of human embryonic stem cell colony and aggregate size heterogeneity influences differentiation trajectories., Stem Cells 26 (2008) 2300–10. doi: 10.1634/stemcells.2008-0183. [DOI] [PubMed] [Google Scholar]
- [15].Peerani R, Bauwens C, Kumacheva E, Zandstra PW, Patterning Mouse and Human Embryonic Stem Cells Using Micro-contact Printing, in: Stem Cells Regen. Med, 2009: pp. 21–33. doi: 10.1007/978-1-59745-060-7. [DOI] [PubMed] [Google Scholar]
- [16].Kosaka T, Tsuboi S, Fukaya K, Pu H, Ohno T, Tsuji T, Miyazaki M, Namba M, Spheroid Cultures of Human Hepatoblastoma Cells ( HuH-6 Line ) and Their Application for Cytotoxicity Assay of Alcohols Spheroid Cultures of Human Hepatoblastoma Cells ( HuH-6 Line ) and Their Application for Cytotoxicity Assay of Alcohols, Acta Med. Okayama 50 (1999) 1–6. [DOI] [PubMed] [Google Scholar]
- [17].Kelm JM, Timmins NE, Brown CJ, Fussenegger M, Nielsen LK, Method for generation of homogeneous multicellular tumor spheroids applicable to a wide variety of cell types, Biotechnol. Bioeng 83 (2003) 173–180. doi: 10.1002/bit.10655. [DOI] [PubMed] [Google Scholar]
- [18].Robertson FM, Ogasawara MA, Ye Z, Chu K, Pickei R, Debeb BG, Woodward WA, Hittelman WN, Cristofanilli M, Barsky SH, Imaging and Analysis of 3D Tumor Spheroids Enriched for a Cancer Stem Cell Phenotype, 15 (2010) 820–829. doi: 10.1177/1087057110376541. [DOI] [PubMed] [Google Scholar]
- [19].Jiang Y, Pjesivac-grbovic J, Cantrell C, Freyer JP, A Multiscale Model for Avascular Tumor Growth, Biophys. J 89 (2005) 3884–3894. doi: 10.1529/biophysj.105.060640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lorenzo C, Frongia C, Jorand R, Fehrenbach J, Weiss P, Maandhui A, Gay G, Ducommun B, Lobjois V, Live cell division dynamics monitoring in 3D large spheroid tumor models using light sheet microscopy, Cell Div 6 (2011) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wenning LA, Murphy RM, Coupled Cellular Trafficking and Diffusional Limitations in Delivery of Immunotoxins to Multicell Tumor Spheroids, Biotechnol. Bioeng 62 (1999) 562–575. [PubMed] [Google Scholar]
- [22].De Sousa F, Melo E, Vermeulen L, Fessler E, Medema JP, Cancer heterogeneity – a multifaceted view, in: EMBO Rep, 2018: pp. 686–695. [DOI] [PMC free article] [PubMed]
- [23].Friedrich J, Ebner R, Kunz-Schughart L. a, Experimental anti-tumor therapy in 3-D: spheroids--old hat or new challenge?, Int. J. Radiat. Biol 83 (2007) 849–871. doi: 10.1080/09553000701727531. [DOI] [PubMed] [Google Scholar]
- [24].Froehlich K, Haeger J, Heger J, Pastuschek J, Photini SM, Yan Y, Lupp A, Pfarrer C, Mrowka R, Schleußner E, Markert UR, Schmidt A, Generation of Multicellular Breast Cancer Tumor Spheroids : Comparison of Different Protocols, J. Mammary Gland Biol. Neoplasia (2016) 89–98. doi: 10.1007/s10911-016-9359-2. [DOI] [PubMed]
- [25].Raghavan S, Mehta P, Horst EN, Ward MR, Katelyn Rowley R, Mehta G, Comparative analysis of tumor spheroid generation techniques for differential in vitro drug toxicity, Oncotarget 7 (2016) 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Murphy SV, Atala A, 3D bioprinting of tissues and organs, Nat. Biotechnol 32 (2014) 773–785. doi: 10.1038/nbt.2958. [DOI] [PubMed] [Google Scholar]
- [27].Dias A, Kingsley DM, Corr D, Recent Advances in Bioprinting and Applications for Biosensing, Biosensors 4 (2014) 111–136. doi: 10.3390/bios4020111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lee VK, Dai G, Printing of Three-Dimensional Tissue Analogs for Regenerative Medicine, Ann. Biomed. Eng 45 (2017) 115–131. doi: 10.1007/s10439-016-1613-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Arslan-yildiz A, El Assal R, Chen P, Guven S, Inci F, Demirci U, Towards artificial tissue models : past, present, and future of 3D bioprinting, Biofabrication 8 (2016) 14103. [DOI] [PubMed] [Google Scholar]
- [30].Dias AD, Kingsley DM, Corr DT, Engineering 2D and 3D microenvironments using laser direct-write, in: Zhang LG, Fisher JP, Leong KW (Eds.), 3D Bioprinting Nanotechnol. Tissue Eng. Regen. Med, New York: Elsevier, 2015: pp. 105–127. [Google Scholar]
- [31].Nguyen DG, Funk J, Robbins JB, Crogan-Grundy C, Presnell SC, Singer T, Roth AB, Bioprinted 3D primary liver tissues allow assessment of organ-level response to clinical drug induced toxicity in vitro, PLoS One 11 (2016) 1–17. doi: 10.1371/journal.pone.0158674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kural MH, Dai G, Niklason LE, Gui L, An Ex Vivo Vessel Injury Model to Study Remodeling, Cell Transplant (2018) 1–15. doi: 10.1177/0963689718792201. [DOI] [PMC free article] [PubMed]
- [33].Ye K, Kaplan DL, Bao G, Bettinger C, Forgacs G, Dong C, Khademhosseini A, Ke Y, Leong K, Sambanis A, Sun W, Yin P, Advanced Cell and Tissue Biomanufacturing, ACS Biomater. Sci. Eng 4 (2018) 2292–2307. doi: 10.1021/acsbiomaterials.8b00650. [DOI] [PubMed] [Google Scholar]
- [34].Vinson BT, Phamduy TB, Shipman J, Riggs B, Strong AL, Sklare SC, Murfee WL, Burow ME, Bunnell BA, Huang Y, Chrisey DB, Laser direct-write based fabrication of a spatially-defined, biomimetic construct as a potential model for breast cancer cell invasion into adipose tissue, Biofabrication 9 (2017) 25013. doi: 10.1088/1758-5090/aa6bad. [DOI] [PubMed] [Google Scholar]
- [35].Koch L, Deiwick A, Franke A, Schwanke K, Haverich A, Laser bioprinting of human induced pluripotent stem cells—the effect of printing and biomaterials on cell survival, pluripotency, and differentiation, Biofabrication 10 (2018) 35005. [DOI] [PubMed] [Google Scholar]
- [36].Schiele NR, Koppes RA, Corr DT, Ellison KS, Thompson DM, Ligon LA, Lippert TKM, Chrisey DB, Laser direct writing of combinatorial libraries of idealized cellular constructs: Biomedical applications, Appl. Surf. Sci 255 (2009) 5444–5447. doi: 10.1016/j.apsusc.2008.10.054. [DOI] [Google Scholar]
- [37].Guillemot F, Souquet A, Catros S, Guillotin B, Lopez J, Faucon M, Pippenger B, Bareille R, Rémy M, Bellance S, Chabassier P, Fricain JC, Amédée J, High-throughput laser printing of cells and biomaterials for tissue engineering., Acta Biomater 6 (2010) 2494–500. doi: 10.1016/j.actbio.2009.09.029. [DOI] [PubMed] [Google Scholar]
- [38].Zhang Z, Jin Y, Yin J, Xu C, Xiong R, Christensen K, Ringeisen BR, Chrisey DB, Huang Y, Zhang Z, Jin Y, Yin J, Xu C, Xiong R, Christensen K, Evaluation of bioink printability for bioprinting applications, Appl. Phys. Rev 41304 (2018). doi: 10.1063/1.5053979. [DOI] [Google Scholar]
- [39].Kingsley DM, Dias AD, Chrisey DB, Corr DT, Single-step laser-based fabrication and patterning of cell-encapsulated alginate microbeads, Biofabrication 5 (2013) 45006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kingsley DM, Dias AD, Corr DT, Microcapsules and 3D customizable shelled microenvironments from laser direct-written microbeads, Biotechnol. Bioeng 113 (2016) 2264–2274. doi: 10.1002/bit.25987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kang X, Xie Y, Powell HM, James Lee L, Belury MA, Lannutti JJ, Kniss DA, Adipogenesis of murine embryonic stem cells in a three-dimensional culture system using electrospun polymer scaffolds, Biomaterials 28 (2007) 450–458. doi: 10.1016/j.biomaterials.2006.08.052. [DOI] [PubMed] [Google Scholar]
- [42].Jung Y, Klein OJ, Wang H, Evans CL, Longitudinal, label-free, quantitative tracking of cell death and viability in a 3D tumor model with OCT, Sci. Rep 6 (2016) 1–11. doi: 10.1038/srep27017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Huang Y, Wang S, Guo Q, Kessel S, Rubinoff I, Chan LLY, Li P, Liu Y, Qiu J, Zhou C, Optical coherence tomography detects necrotic regions and volumetrically quantifies multicellular tumor spheroids, Cancer Res 77 (2017) 6011–6020. doi: 10.1158/0008-5472.CAN-17-0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Faulkner DE, Roberge C, Kingsley DM, Sloat CJ, Corr DT, Intes X, Improving optical coherence tomography contrast for assessing biologic constructs via TiO2, in: Opt. InfoBase Conf. Pap, 2018: pp. 5–6. doi: 10.1364/TRANSLATIONAL.2018.JTh3A.47. [DOI]
- [45].Leary E, Rhee C, Wilks BT, Jeffrey R Morgan, Quantitative Live-Cell Confocal Imaging of 3D Spheroids in a High-Throughput Format, SLAS Technol. Transl. Life Sci. Innov (2018) 2472630318756058. [DOI] [PMC free article] [PubMed]
- [46].Agarwal P, Zhao S, Bielecki P, Rao W, Choi JK, Zhao Y, Yu J, Zhang W, He X, One-step microfluidic generation of pre-hatching embryo-like core–shell microcapsules for miniaturized 3D culture of pluripotent stem cells, Lab Chip 13 (2013) 4525–4533. doi: 10.1039/c3lc50678a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Alessandri K, Sarangi BR, Gurchenkov VV, Sinha B, Kießling TR, Fetler L, Rico F, Scheuring S, Lamaze C, Simon A, Geraldo S, Vignjevic D, Doméjean H, Rolland L, Funfak A, Bibette J, Bremond N, Nassoy P, Cellular capsules as a tool for multicellular spheroid production and for investigating the mechanics of tumor progression in vitro., Proc. Natl. Acad. Sci. U. S. A 110 (2013) 14843–8. doi: 10.1073/pnas.1309482110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yanai A, Laver CRJ, Joe AW, Viringipurampeer IA, Wang X, Gregory-Evans CY, Gregory-Evans K, Differentiation of Human Embryonic Stem Cells Using Size-Controlled Embryoid Bodies and Negative Cell Selection in the Production of Photoreceptor Precursor Cells, Tissue Eng. Part C Methods 19 (2013) 755–764. doi: 10.1089/ten.tec.2012.0524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sasaki D, Shimizu T, Masuda S, Kobayashi J, Itoga K, Tsuda Y, Yamashita JK, Yamato M, Okano T, Mass preparation of size-controlled mouse embryonic stem cell aggregates and induction of cardiac differentiation by cell patterning method, Biomaterials 30 (2009) 4384–4389. doi: 10.1016/j.biomaterials.2009.05.003. [DOI] [PubMed] [Google Scholar]
- [50].Dias AD, Unser AM, Xie Y, Chrisey DB, Corr DT, Generating size-controlled embryoid bodies using laser direct-write., Biofabrication 6 (2014) 25007. doi: 10.1088/1758-5082/6/2/025007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Cheng G, Tse J, Jain RK, Munn LL, Micro-Environmental Mechanical Stress Controls Tumor Spheroid Size and Morphology by Suppressing Proliferation and Inducing Apoptosis in Cancer Cells, PLoS One 4 (2009) e4632. doi: 10.1371/journal.pone.0004632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Korhonen RK, Laasanen MS, Töyräs J, Rieppo J, Hirvonen J, Helminen HJ, Jurvelin JS, Comparison of the equilibrium response of articular cartilage in unconfined compression, confined compression and indentation, J. Biomech 35 (2002) 903–909. doi: 10.1016/S0021-9290(02)00052-0. [DOI] [PubMed] [Google Scholar]
- [53].Gutenberg-universitat J, On the relationship between size of necrosis and diameter of tumor spheroid, Int. J. Radiat. Oncol. Biol. Phys 34 (1996) 395–401. [DOI] [PubMed] [Google Scholar]
- [54].Raof NA, Schiele NR, Xie Y, Chrisey DB, Corr DT, The maintenance of pluripotency following laser direct-write of mouse embryonic stem cells., Biomaterials 32 (2011) 1802–8. doi: 10.1016/j.biomaterials.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ouyang L, Yao R, Mao S, Chen X, Na J, Sun W, Three-dimensional bioprinting of embryonic stem cells directs highly uniform embryoid body formation, Biofabrication 7 (2015) 44101. doi: 10.1088/1758-5090/7/4/044101. [DOI] [PubMed] [Google Scholar]
- [56].Ball G, Green ÆAR, Aleskandarany ÆM, Paish EC, Macmillan ÆRD, Nicholson ÆRI, Ellis IO, Gee ÆJMW, Transferrin receptor ( CD71 ) is a marker of poor prognosis in breast cancer and can predict response to tamoxifen, Breast Cancer Res. Treat 119 (2010) 283–293. doi: 10.1007/s10549-009-0345-x. [DOI] [PubMed] [Google Scholar]
- [57].Talati R, Vanderpoel A, Eladdadi A, Anderson K, Abe K, Barroso M, Automated selection of regions of interest for intensity-based FRET analysis of transferrin endocytic trafficking in normal vs. cancer cells, Methods 66 (2014) 139–152. doi: 10.1016/j.ymeth.2013.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML, The transferrin receptor part I : Biology and targeting with cytotoxic antibodies for the treatment of cancer, Clin. Immunol 121 (2006) 144–158. doi: 10.1016/j.clim.2006.06.010. [DOI] [PubMed] [Google Scholar]
- [59].Bartlett DW, Davis ME, Impact of Tumor-Specific Targeting and Dosing Schedule on Tumor Growth Inhibition After Intravenous Administration of siRNA-Containing Nanoparticles, Biotechnol. Bioeng 99 (2008) 975–985. doi: 10.1002/bit. [DOI] [PubMed] [Google Scholar]
- [60].Yoon DJ, Liu CT, Quinlan DS, Nafisi PM, Kamei DT, Intracellular Trafficking Considerations in the Development of Natural Ligand-Drug Molecular Conjugates for Cancer., Ann. Biomed. Eng 39 (2011) 1235–51. doi: 10.1007/s10439-011-0280-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Choi CHJ, Alabi CA, Webster P, Davis ME, Mechanism of active targeting in solid tumors with transferrin-containing gold nanoparticles, Proc. Natl. Acad. Sci. U. S. A 107 (2010) 1235–1240. doi: 10.1073/pnas.0914140107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Davis ME, The First Targeted Delivery of siRNA in Humans via a Nanoparticle : From Concept to Clinic, Mol. Pharm 6 (2009) 659–68. doi: 10.1021/mp900015y. [DOI] [PubMed] [Google Scholar]
- [63].Pun SH, Tack F, Bellocq NC, Cheng J, Grubbs BH, Jensen GS, Davis ME, Brewster M, Janicot M, Janssens B, Floren W, Bakker A, Targeted delivery of RNA-cleaving DNA enzyme (DNAzyme) to tumor tissue by transferrin-modified, cyclodextrin-based particles, Cancer Biol. Ther 3 (2004) 641–650. doi: 10.4161/cbt.3.7.918. [DOI] [PubMed] [Google Scholar]
- [64].Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME, Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging, Proc. Natl. Acad. Sci. U. S. A 104 (2007) 15549–15554. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Davis ME, Zuckerman JE, Choi CHJ, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A, Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles, Nature 464 (2010) 1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Daniels TR, Bernabeu E, Rodríguez JA, Patel S, Kozman M, Chiappetta DA, Holler E, Ljubimova JY, Helguera G, Penichet ML, The transferrin receptor and the targeted delivery of therapeutic agents against cancer., Biochim. Biophys. Acta 1820 (2012) 291–317. doi: 10.1016/j.bbagen.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Shan L, Hao Y, Wang S, Korotcov A, Zhang R, Wang T, Califano J, Gu X, Sridhar R, Bhujwalla ZM, Wang PC, Visualizing Head and Neck Tumors In Vivo Using Near- Infrared Fluorescent Transferrin Conjugate, Mol. Imaging 7 (2008) 42–49. doi: 10.2310/7290.2008.0006. [DOI] [PubMed] [Google Scholar]
- [68].Using T, Non-Invasive In Vivo Imaging of Near Infrared-labeled Transferrin in Breast Cancer Cells and Tumors Using Fluorescence Lifetime FRET, PLoS Biol 8 (2013) e80269. doi: 10.1371/journal.pone.0080269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lok CN, Ponka P, Identification of a Hypoxia Response Element in the Transferrin Receptor Gene *, J. Biol. Chem 274 (1999) 24147–24152. [DOI] [PubMed] [Google Scholar]