

Abstract
Histone deacetylases (HDACs) are part of the epigenetic machinery that regulates transcriptional processes. The current paradigm is that HDACs silence gene expression via regulation of histone protein lysine deacetylation, or by forming corepressor complexes with transcription factors. However, HDACs are more than just nuclear proteins, and they can interact and deacetylate a growing number of non-histone proteins to regulate cellular function. Cancer-field studies have demonstrated that deranged HDAC activity results in uncontrolled proliferation, inflammation, and fibrosis; all pathologies that may also occur in kidney disease. Over the past decade, studies have emerged suggesting that HDAC inhibitors may prevent and potentially treat various models of acute kidney injury. This review focuses on the physiology of kidney HDACs and highlights the recent advances using HDAC inhibitors to potentially treat kidney disease patients.
Keywords: histone deacetylase, HDAC, lysine acetylation, acute kidney injury, nephron
In 1942 C.H. Waddington coined the term “epigenetics” as a means to characterize the connection between the genotype (inherited material) and the phenotype (adult characteristics) of an organism.1 Epigenetics is now defined as the nongenetic regulation of gene expression with a potential for inheritance.2 There are three known global epigenetic mechanisms that can regulate gene expression: 1) DNA methylation,3 2) histone modification,4,5 3) non-coding RNA.6 These epigenetic mechanisms involve a number of proteins that modify transcriptional processing. The focus of this review is specifically on the histone modifiers, histone deacetylases (HDACs).
In 1964, Allfrey et al.4 published that histone proteins could be modified and speculated that this was a way to regulate RNA transcription. They provided evidence that histone proteins could be dynamically acetylated/deacetylated on their lysine ε-amino group.7 In the 1980’s the first acetyltransferases were purified8–10 and later cloned in 1995.11 This family of enzymes was termed the “histone acetyltransferases” (HAT) because of their functions in regulating histone structure. Shortly after the cloning of HAT1, the first mammalian deacetylase was cloned (name histone deacetylase-1, HDAC1).12 Currently, there are at least 21 acetyltransferases and 18 deacetylases found in mammals (Figure 1).13
Figure 1:
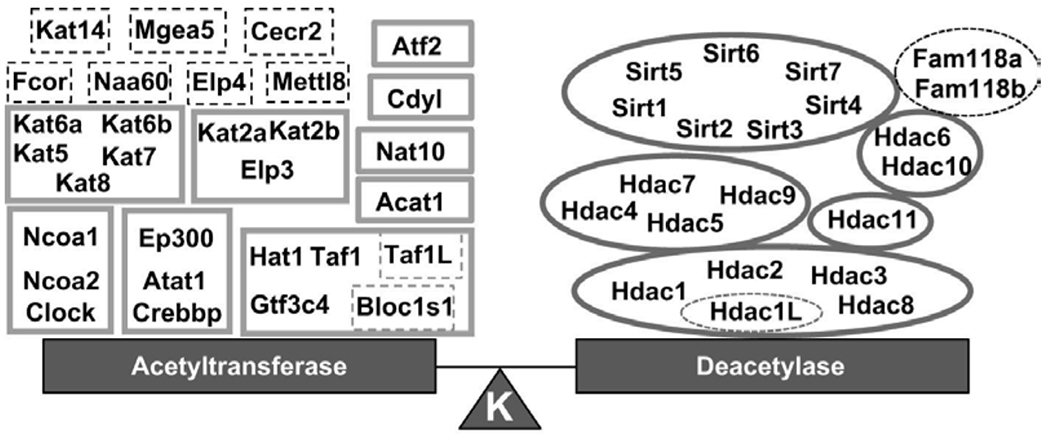
The lysine acetyltransferases and deacetylases found in mammals, grouped by their phylogenetic relationships. Dash lines represent predicted enzymes however whether they directly modify lysines is elusive. From Hyndman et al.13
The HDACs are organized into classes based upon phylogeny, structure and localization. Class I, II, and IV HDACs are the zinc dependent enzymes and class III are the NAD+ dependent sirtuins (SIRTs) (Figure 1).14 Current HDAC inhibitors (HDACis) do not directly affect SIRT activity 15 and thus SIRTs will not be further discussed in this review. Class I HDACs are predominantly nuclear (Figure 2); however, studies have demonstrated their ability to translocate into the cytosol.16 Class II and IV HDACs translocate between the nucleus and cytosol (Figure 2). HDACs function in the post-translational modification of lysine residues of over 3500 histone and non-histone proteins.17–19 Although HDACs are classically defined as epigenetic regulators, recent studies have highlighted that dynamic lysine acetylation controls a variety of processes including metabolism, redox homeostasis, actin cytoskeleton, and ion transport.17,19,20 The importance of non-histone lysine acetylation was recently reviewed.20
Figure 2:

Histone deacetylases (HDAC) are classified based upon phylogenetic relationship. Class I HDACs are predominantly expressed in the nucleus, while class II and IV HDACs can translocate between the nucleus and cytosol.
The HDACs have garnered a lot of attention because deranged HDAC activity is associated with cancer.21 There are four Food and Drug Administration (FDA) approved HDACis used to treat T-cell lymphomas and myelomas and hundreds of cancer based clinical trials using HDACis (Table 1). Because HDACi use can lead to reduced inflammation, angiogenesis, proliferation, and fibrosis; it is hypothesized that perhaps HDACis could be repurposed to prevent or treat cardiovascular disease 22,23 and kidney diseases.24–26 Although deranged HDAC activity is associated with variety of disease, the focus of this review is on acute kidney injury.
Table 1:
Food and Drug Administration (FDA) approved histone deacetylase inhibitors.
| Inhibitor | Class | FDA treatment approval | target | FDA approval Date | Terminal half life T1/2 (h) | Ref. |
|---|---|---|---|---|---|---|
| Vorinostat/SAHA | hydroxamic acid | T cell lymphoma | pan | 2006 | 1.05-1.44 | 89.90 |
| Belinostat | hydroxamic acid | T cell lymphoma | pan | 2015 | 0.45-2.9 | 91,92 |
| Panobinostat | hydroxamic acid | multiple myeloma | pan | 2015 | 13-29.3 | 50 |
| Romidepsin | cyclic tetrapeptide | cutaneous T cell lymphoma | I | 2009 | 1.1-8.1 | 93,94 |
| Valproic acid | short chain fatty acids | seizures | I, IIA | 1978* | 10-20 | 95 |
| Phenylbutyric acid | short chain fatty acids | hyperammonaemia | I,II | 1996* | <1 | 96 |
Footnote: Ref. - reference
HDACs and the regulation of transcription
Generally, HDACs are considered transcriptional repressors. The classical paradigm is that HDACs remove acetyl groups from lysine residues of core histone proteins27 thereby restoring the positive charge of the histone lysine. The negatively charged DNA can then bind tightly to the positively charged histone protein. HDACs also form complexes with DNA binding proteins (HDACs do not bind directly to DNA28) such as nuclear receptor corepressor 1 (NCOR1)29 and mammalian SIN3.30 Recruitment of these complexes in the nucleus leads to chromatin remodeling and transcriptional repression.31 However, perhaps paradoxical, HDAC activity can also result in transcriptional activation. For example, in the developing kidney, genetic deletion of HDAC1 and HDAC2 from the ureteric bud resulted in 226 transcripts significantly upregulated and 270 transcripts downregulated.32 Greer et al.33 reported that HDACs positively regulate transcriptional elongation; a key process involved in RNA polymerase transcription of target genes. They demonstrated that HDAC inhibitors cause a loss of enhancer RNA synthesis and elongation repression, which resulted in reduced gene transcription.33 Furthermore, HDACs can also activate transcription factors in the cytosol through lysine deacetylation and thereby indirectly leading to activation of transcription. An example of this is HDAC9 which interacts and deacetylates the transcription factor Forkhead box O 1 (FoxO1) in the cytosol.34 Once deacetylated, FoxO1 translocates to the nucleus where it increases the transcription of target genes such as the glucocorticoid receptor.34 Thus, HDACs function in variety of pathways to either negatively and positively regulate gene transcription.
HDAC expression and physiological function in the kidney
Kidneys are rich in acetylated lysine proteins17,19 and express many of the acetyltransferases and deacetylases in the nephron (Table 2).13 In RNA sequencing data studies of isolated tubule segments,35 all 11 HDACs have been shown to be expressed in the tubules. At the RNA level, HDACs are enriched in the thick ascending limb through to the inner medullary collecting duct.13 Interestingly, rats have a duplicate HDAC1 gene named HDAC1-like (NM_053446), which is also expressed at the RNA level in the loop and collecting duct.13 It shares 476/484 identity at the amino acid level with rat HDAC1, but whether this is a functional protein remains to be determined. Similar to the rat, the mouse single-tubule RNA-seq dataset36,37 and the mouse kidney single cell transcriptomics dataset38 provide evidence of all 11 HDACs in the kidney (Table 2). In human subjects, Class I and IV HDAC expression was also found in all tubule segments by serial analysis of gene expression (SAGE).39,40 Thus, at least over the short evolutionary scale from rodents to man, the kidney HDAC transcripts are highly abundant and conserved in their expression in the kidney.
Table 2:
Histone deacetylase (HDAC) RNA expression from the rat transcriptome35 or mouse single-cell transcriptome38 in the healthy kidney.
| Glom | Podo | PT | SDL | LDLOM | LDLIM | tAL | mTAL | cTAL | DCT | CNT | CCD | OMCD | IMCD | Endo | Fib | Macro | Neut | B lymph | T lymph | NK | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hdac1 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Hdac2 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| Hdac3 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||
| Hdac4 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |||||||||||||
| Hdac5 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||
| Hdac6 | ✓ | ||||||||||||||||||||
| Hdac7 | ✓ | ✓ | ✓ | ||||||||||||||||||
| Hdac8 | ✓ | ✓ | |||||||||||||||||||
| Hdac9 | ✓ | ✓ | ✓ | ||||||||||||||||||
| Hdac10 | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||||||||||||
| Hdac11 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||||||
| Rat Hdac 11 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
Glom, glomerulus; podo, podocyte; PT, proximal tubule; SDL, short descending limb of the loop of Henle; LDLOM, long descending limb of the loop of Henle in the outer medulla; LDLIM, long descending limb of the loop of Henle in the inner medulla; tAL, thin ascending limb of the loop of Henle; mTAL, medullary thick ascending limb of the loop of Henle; cTAL, cortical thick ascending limb of the loop of Henle; DCT, distal convoluted tubule; CNT, connecting tubule; CCD, cortical collecting duct; OMCD, outer medullary collecting duct; IMCD, inner medullary collecting duct; Endo, endothelium; Fib, fibroblast; Macro, macrophage; Neutro, neutrophil; B lymph, B lymphocytes; T lymph, T lymphocytes; NK, natural killer cells.
HDAC1-10 protein abundance was detected in rat or mouse, freshly isolated inner medullary collecting ducts using protein mass spectrometry (see http://helixweb.nih.gov/ESBL/Database/ The Kidney Systems Biology project of The Epithelial Systems Biology Laboratory, National Heart Lung and Blood Institute). Similarly, the Human Protein Atlas (http://www.proteinatlas.org),41 provides evidence of the 11 HDACs in the human kidney and localization of all but HDAC7. We recently reported the immunolocalization of the 11 HDACs in the adult mouse kidney.42 Compared to the single-cell transcriptomics of the mouse kidney,38 there is a strong agreement between our immunolocalization and the RNA sequencing of different kidney cell types. For example, HDAC1 localizes to the nucleus of nearly all cells in the kidney.42 HDAC1 RNA is found in all cells including endothelium, podocytes, epithelium, and interstitial cells (fibroblasts, neutrophils, and lymphocytes).38 HDAC9 immunolocalizes to kidney interstitial cells,42 and HDAC9 RNA is found predominantly in fibroblasts, macrophages, and B lymphocytes (Table 2).38 HDACs are expressed in all cell types of the kidney and this would suggest the function in diverse physiological processes.
Surprisingly, information is limited about the substrates that HDACs may regulate, and therefore the physiological pathways under their control in the kidney remain undiscovered. One hypothesis is that HDACs regulation water and ion transport processes in the kidney. For example, HDAC7 is expressed in the cortical collecting duct where it is proposed to be a negative regulator of the epithelial sodium channel (ENaC).43 With an increase in HDAC7, there were significant reductions in ENaC abundance and current that were associated with increased lysine ubiquitination.43 Butler et al.43 speculated that lysine acetylation of ENaC would stabilize the channel and lead to sodium reabsorption. The putative lysines that may be acetylated and whether they are substrates for HDAC7 remains unknown. A recent report demonstrated that pan-HDAC inhibition with trichostatin A (TSA) prevented aldosterone-mediated ENaC activity,44 and the authors suggested that lysine acetylation of the aldosterone receptor (mineralocorticoid receptor) disrupts the sodium retaining effect of aldosterone. This finding is in agreement with studies measuring blood pressure, which have proposed that HDACi may prevent the development of salt-sensitive hypertension.45 Thus, these studies suggest that specific HDAC isoforms may regulate cellular functions in distinct manners and that pan-HDACi reveal the complexity of these signaling cascades. There remains a large gap in our understanding of the physiological role of kidney HDACs, especially at the individual isoform level. We 19 and others17 have sequenced kidney acetylomes that identified substrates for HDACs to deacetylate, but the cell type, HDAC isoform, and ultimately physiological effect of regulating lysine acetylation all are understudied.
HDAC Inhibitors
Although HDAC isoform and cellular specific functions may not be clearly defined in the kidney under physiological conditions, the use of HDACis has demonstrated that HDACs may become dysfunctional or deranged in disease states. Most of what we know about HDACi comes from cancer-focused studies, but over the past decade, the therapeutic potential of inhibiting kidney HDACs has emerged as a result of studying different models of acute kidney injury (AKI). Current commercially available HDACis are generally pan-inhibitors that target Class I, II, and IV; or are Class I-selective inhibitors (Table 1). There are four FDA approved HDACis to treat cancers (Table 1). There are also two other compounds with deacetylase inhibitor activity, valproic acid (VPA) and phenylbutyric acid (PBA), which are FDA approved to treat seizures and hyperammonaemia, respectively. These compounds are also being tested in cancer trials.15 HDACis are given orally or by i.v. infusions, and each has a very different terminal half-life (Table 1). The pharmacokinetics may also differ between preclinical models and humans. For example, entinostat (also called MS-275) has a half-life of >30 h in humans46,47 but <1 h in rodents.46 It is unknown how entionostat is metabolized and removed from the body.48 Entinostat also binds plasma proteins in a species-specific manner,46 which may explain the significant differences in the pharmacokinetic profiles. This species-specific variability is important to consider when designing and analyzing preclinical studies.
Another large gap that requires attention is the in vivo specificity and pharmacokinetics of HDACi, especially in kidney disease patients. The main clearance pathways of HDACis are excretion of metabolites in the urine and/or feces.49–51 Even with different levels of kidney function, the terminal half-life of panobinostat was measured between 27.5-29.5 h.50 Only small traces of parent drug were found in the urine, yet up to 50% of the metabolites of panobinostat were found in the urine of all patients including those with impaired creatinine clearance.50,51 Adverse events were no worse among the subjects with reduced kidney function.50 Thus, panobinostat was concluded to be well tolerated and safe for people with reduced kidney function although subjects with end-stage renal disease have not been studied. The adverse events associated with HDACis include vomiting, diarrhea, hyponatremia, hypokalemia, and changes in blood pressure (both hypertension and hypotension),52–54 which need to be carefully monitored especially in people with reduced kidney function. These types of studies are necessary to determine if HDACis can be repurposed to treat kidney disease.
HDAC dysfunction in models of Acute Kidney Injury
There are many preclinical models of AKI.55 In the cisplatin-induced nephrotoxicity model of AKI in male mice, whole kidney mRNA for class I HDACs was increased.56 In the unilateral ureteral obstruction model, kidney abundance of HDAC1-6 and -10 were significantly increased 2 weeks after obstruction.57 In the bilateral model of ischemia-reperfusion injury (IRI), cortical HDAC abundance and localization was dynamic.42 For example, HDAC1 was expressed in the nucleus and within 1 h following IRI nuclear abundance was decreased but then progressively increased over the next 3 days. HDAC4 was found in a low abundance in the healthy kidney, but following IRI, it was clearly present in the nucleus 1 h post ischemia but highly expressed in the proximal tubules24-72 h later.42 When healthy to injured mouse kidneys were compared, many of the HDACs were significantly affected in their localization.42 The consequences of these changes remains to be determined. Although the specific HDAC isoform and its cellular source (epithelial, endothelia, interstitial cell etc.) that is deranged, in most AKI models is unknown, studies using HDACis have revealed some interesting mechanisms of AKI.
Preventative HDACi approaches in:
Sepsis:
Uncontrolled systemic inflammation or “sepsis”, is a clinically relevant problem that unfortunately can lead to AKI and increased mortality. In mice, sepsis can be modeled by the cecal ligation puncture (CLP) procedure.58 Valproic acid (VPA) given at the time of CLP, prevented the rise in plasma creatinine, reduced inflammatory cytokines, and limited kidney damage at 24 h post CLP.59 Sepsis can also be modeled by intraperitoneal injections of lippopolysacchride (LPS) leading to AKI. Following LPS injection, there was significant mortality within days, but this was significantly improved by a 5 day pretreatment with MS-275. Preventative MS-275 treatment attenuated the rise in serum creatinine, ameliorated kidney damage, and suppressed kidney reactive oxygen species.60 Thus, preventative HDACi use can limit sepsis-induced AKI; however, long-term outcomes and treatment regimens require further investigation.
Unilateral Ureteral Obstruction(UUO):
Ureteral obstruction can occur from kidney stones or scarring and lead to an inability to properly drain urine from the kidney to the bladder. If left untreated it causes profound kidney damage. This is modeled by ligating one of the ureters and leaving the other intact. Within days this model leads to severe kidney damage, inflammation, and fibrosis. When trichostatin A (a pan-HDACi) was given by i.p. daily for 6 days beginning at the time of obstruction, it significantly prevented kidney interstitial fibrosis and prevented epithelial apoptosis.61 Similarly, pretreatment with VPA (i.p. daily) for 5 days prior to 14 days of UUO, attenuated interstitial fibrosis, kidney inflammation, and kidney transforming growth factor (TGF)-β concentration.62
Cisplatin and other nephrotoxic drugs.
Unfortunately, some drugs can accumulate in the kidney, especially in the proximal tubules, resulting in nephrotoxicity.63 Cisplatin, a chemotherapy drug, is frequently used to model nephrotoxic-induced AKI. In cisplatin-induced AKI, pretreatment with TSA (daily i.p.) significantly attenuated the renal damage and rise in plasma creatinine.56 Liu et al.64 demonstrated in this same model that TSA was renoprotective via enhancing proximal tubule autophagy. Specifically, HDAC6 has been implicated in being deranged in cisplatin-induced AKI, and inhibition of HDAC6 with tubastatin A also increased autophagy and reduced kidney injury.65 This was also true for rhabdomyolysis-induced AKI, which is caused by nephrotoxic levels of myoglobin released from necrotic muscle. Inhibition of HDAC6 with tubastatin A resulted in an attenuation in the rise of serum creatinine and kidney damage 48 h after induction.66 This was associated with a decrease in kidney inflammation and oxidative stress and preserving theexpression of superoxide dismutase, a free radicle scavenger.66
Ischemia-reperfusion-injury (IRI).
If there is low perfusion of the kidneys, IRI can occur. IRI is prevalent in cardiac, vascular, and cardiopulmonary surgery because blood flow is reduced during the surgical procedures.67–69 To model this, either one or both of the renal pedicles are clamped resulting in ischemia followed by removal and reperfusion. This leads to kidney damage that is dependent upon the time of ischemia. In the warm (body temperature maintained at 36°C), unilateral model of IRI in female mice, an injection of TSA 16 h prior to and again at time of unilateral IRI surgery (ischemia = 28 min), a quicker recovery of plasma blood-urea-nitrogen (BUN) was found. This ultimately lead a to better 30-day survival than vehicle treated controls.70 MS-275 was also administered 16 h prior to and at time of unilateral IRI, and although the 24-96 h post injury recovery of plasma BUN was not improved, kidney interstitial fibrosis was still significantly attenuated.70 Interestingly, using HDAC6 null mice, Levine et al.70 demonstrated that the change in BUN was similar to control mice and interpreted this as the renoprotective effects of TSA and MS275 on inhibiting Class I HDACs and not Class II. This is distinct from nephrotoxic induced-AKI mentioned above, where deranged HDAC6 activity was inhibiting autophagy and leading to worsened outcomes.
Levine et al.70 also demonstrated that in cold IRI kidney transplant studies in female mice, TSA given as a single i.p. to both the donor and the recipient resulted in an improved 30-day survival compared to vehicle-treated mice. This was associated with attenuated interstitial fibrosis.70 From this study, we can conclude that two preventative i.p. doses of TSA can improve renal outcomes in both the warm IRI and cold IRI transplant models in females. This is likely due to inhibition of deranged Class I HDACs, although deeper investigation is needed.
We recently took a different approach. To better mimic the human pharmacokinetics of TSA and MS725 (Table 1), we implanted i.p. osmotic minipumps to deliver vehicle, TSA (1 mg/kg/day) or MS275 (20mg/kg/day) for 3 days prior to warm, bilateral IRI in male mice.42 Surprisingly, we found that HDACis led to an exacerbated increase in plasma creatinine in the IRI mice. This was associated with greater kidney tubular damage from histological evaluation, and greater kidney cortical proximal tubule injury markers compared to controls or shams.42 Although kidneys were evaluated at 72 h after ischemia, we detected a small but significant reduction in interstitial fibrosis in the HDACi treated mice. This is consistent with the aforementioned AKI studies in which HDAC inhibition prevents interstitial fibrosis. Thus, although interstitial fibrosis may be prevented (a benefit) by continual HDACi exposure, it may also lead to exacerbated tubular injury (an adverse effect). We speculate that part of the increased injury was from proximal tubule HDAC4 inhibition (with TSA) leading to an attenuated cell cycle and reduced proliferation.42 These findings suggest that changes in HDAC activity following AKI may be both beneficial by promoting cellular repair but also detrimental by potentiating injury and increasing scarring. We hypothesize that the cellular source and HDAC isoform activated leads to divergent functions in the kidney.
Interventional approaches:
The antibiotic gentamicin can cause AKI in larval zebrafish.71 Using this model, Cianciolo and colleagues71 demonstrated that their novel compound, methyl-4-(phenylthio)butanoate (m4PTB), significantly improved survival two days after gentamicin injection. Moreover, m4PTB treated zebrafish had increased proximal tubule proliferation that was further enhanced in the AKI larvae.71 Next, they treated two different IRI mouse models. First was a uninephrectomy followed by contralateral IRI (26 min) and treatment with vehicle of m4PTB 24 h post-surgery. Over the next 2 weeks, the m4PTB mice had significantly improved kidney function as measured by serum creatinine but still presented with significant tubular damage.71 The second model was a 30 min ischemia, followed by contralateral nephrectomy 8 days later. Mice were treated 24 h after IRI and presented with an attenuated increase in serum creatinine compared to vehicle-treated mice 24 h after nephrectomy. This model also presented with reduced kidney injury and less outer medullary interstitial fibrosis when treated with m4PTB.71 Similarly, m4PTB treatment 4 days after initiation of aristolochic acid nephropathy (AAN) in male mice improved kidney function, reduced proximal tubule injury, and prevented interstitial fibrosis.72 Recently, new analogs of 4-(phenylthio) butanoic acid (PTBA) have also been tested in zebrafish and mouse models.73 Skrypnyk et al.73 demonstrated that the prodrug, UPHD186, was metabolized in the liver to form active PTBA and can accelerate recovery when administered 3 days after injury. Thus, this novel HDAC inhibitor is able to treat AKI; however, the HDACs that it specifically targets have not yet been reported.
Adrianmycin infusion in mice causes focal segmental glomerulosclerosis leading to proteinuria and renal failure.74 Ten days after adrianmycin treatment of female mice, when proteinuria was at its peak, VPA treatment (given in the drinking water) lead to a return of proteinuria to baseline levels following 3 days of treatment (controls had significantnt proteinuria). Proteinuria remained suppressed over the two week treatment period in the VPA treated mice.74,75 This was associated with an attenuated rise in serum creatinine, less interstitial fibrosis, and less kidney inflammation. Ultimately, these studies demonstrate that in different models of established AKI, HDAC inhibitors may attenuate the progression of kidney disease.
In agreement with these preclinical studies, a recent clinical study suggested that VPA may also slow the decline of estimated glomerular filtration rate (eGFR) decline in human subjects. Using data from the Veterans Aging Cohort study (a male cohort), Inoue et al.,75 reported that the mean rate in eGFR decline was -0.94 ml/1.72 m2/yr (n= 122,870) in this population. Of these, n= 2269 were exposed to VPA as prescribed for psychiatric disorders. Exposure was defined as “presence of a VPA prescription for more than 50% of the days between 2 consecutive creatinine measurements.” VPA exposure resulted in a 35% reduction in the decline of eGFR (−0.61 ml/1.72 m2/yr). Moreover, in some individuals there were data before and after VPA exposure, and again, the decline in eGFR was slowed (−0.93 ml/1.72 m2/yr and -0.32 ml/1.72 m2/yr, respectively).75 Finally, this beneficial effect of VPA exposure was most pronounced in individuals with severe proteinuria.75
All of these different preventative and interventional studies in AKI models highlight how drug specificity, dosing, timing of treatment, and pharmacokinetics can all be significant contributors to whether HDACi are renoprotective or perhaps lead to severe adverse events. Considering the excitement and positive analysis from the Veterans Aging Cohort study,75 hopefully future retrospective analyses and encouragement leading to prospective kidney disease-focused clinical trials with HDACi.
Mechanisms of the renoprotective effects of HDACi in AKI
The HDACi-dependent mechanisms of renoprotection or potentiating injury are understudied. In all of the preclinical studies presented thus far, common pathways such as anti-fibrosis and anti-inflammation have emerged. Especially evident in the preventative strategy preclinical studies, pan- or Class I- selective HDACis attenuated the production of TGF-β, the master regulator of myofibroblast activation,76 reduced myofibroblast markers (e.g. vimentin alpha smooth muscle actin), and reduced extracellular matrix (e.g. collagen, fibronectin). Although it is unclear whether this is via direct or indirect inhibition of HDAC-mediated signaling cascade leading to less interstitial fibrosis, studies have demonstrated that HDACis prevent the phosphorylation of SMAD3 (Mothers against decapentaplegic homolog 3).61,62,77 in the presence of TGF-β, SMAD3 is phosphorylated, translocates to the nucleus, and increases transcription of target genes encoded for extracellular matrix proteins.78 How HDACis prevent the phosphorylation of SMAD3 is not clear. Whether it is through activation of a phosphatase, by directly affecting SMAD3 lysine acetylation, or via another pathway remains to be determined. Direct lysine acetylation of SMADs is an intriguing hypothesis because an ortholog of SMAD3, SMAD7, is lysine acetylated resulting in a stabilized protein that inhibits TGF-β signaling.79 SMAD7 acetylation of K161 was detected in the inner medullary collecting duct acetylome.19 Whether Smad3 is also lysine acetylated and the functional consequence of this posttranslational modification require further investigation.
HDACis also reduce inflammation and proinflammatory cytokines.62,72,77,80,81 M4PTB prevented CD68-positive macrophage infiltration at 7 and 21 days post injury, and significantly reduced chemokines in the AAN model.72 Likewise, the PTBA prodrug, UPHD25, significantly reduced AKI-dependent M1 but not M2 macrophages suggesting that PTBA likely isn’t involved in macrophage polarization.82 TSA significantly reduced kidney macrophage infiltration in the UUO model and attenuated the chemokine, colony-stimulating factor-1.81 Marumo et al.81 went on to demonstrate that HDAC1 and HDAC2 activity is necessary for Tumor Necrosis Factor (TNF)-stimulated CSF-1 production; however the mechanism was not elucidated. In studies with dendritic cells, SAHA induced indoleamine 2,3-dioxygenase (IDO), an enzyme that suppresses dendritic cell function, through increasing acetylation of histone H4 at the IDO promoter.83 Whether similar mechanisms occur in kidney immune cells with HDACi is unknown.
Kidney and splenic T-cells are also significantly affected by HDACi and UUO. Daily TSA by i.p. to UUO mice resulted in increased infiltrating T regulatory cells (Tregs; CD4+FOXP3+) and decreased inflammatory T helper 17 cells (Th17; CD4+IL-17+).84 The HDAC-mediated effects on T cell development were recently reviewed85 and revealed a complex system of inhibition and activation of various T cell populations through lysine acetylation, metabolism, and cofactor availability. Detailed studies are needed to understand AKI-induced, HDAC-mediated kidney immune responses.
Stimulation of regenerative pathways in the kidney are critical to promote healing. Extended cell cycle arrest in G2/M leads to injury and kidney fibrosis.86 HDACis may enhance the regenerative capacity of the injured kidneys via regulation of the cell cycle. Kidneys from IRI mice treated with m4PTB have significantly higher cell cycle-related gene expression, and more actively cycling cells with fewer cells arrested in the G2/M phase.71 Likewise, m4PTB treatment to AAN injured mice resulted in better regenerative capacity with less cells arrested in G2/M.72 However, this is controversial as class I HDACs may be required for proximal tubule proliferation through activation of the epithelial growth factor receptor (EGFR)/signal transducers and activators of transcription 3 (STAT3).87,88 We also reported that HDAC4 activation may be necessary for proximal tubule proliferation following IRI.42 Thus, the mechanisms of HDAC-mediated cell cycle regulation and regeneration are complex and require deeper investigation.
Perspective
A common criticism of studies utilizing HDACis is that because HDACs regulate transcriptional processes and posttranslational lysine acetylation, intervening could potentially lead to a high number of dysregulated genes and pathways. This may be one reason as to why there are many significant adverse events associated with the current pan- and class I-selective HDACis. This in itself is an important reason for defining the role of HDACs in the body, especially in the kidney. With over 500 clinical trials using HDACi to treat different cancers, increasing numbers of patients will be exposed to HDACi so it is necessary to understand HDAC function in the kidney in order to not cause further harm critically ill patients. HDACi have shown a great deal of promise in studies for preventing and even treating kidney injury and preserving kidney function. Still, there remains large gaps in our understanding of the physiological function of HDACs in the kidney (and other organs). Future studies determining HDAC isoform activity, localization, and downstream targets will be key to fully revealing the therapeutic potential of HDACis in the prevention and treatment of AKI. The resulting information will be useful for developing isoform selective, low toxicity compounds; particularly compounds that do not accumulate in the kidney and lead to nephrotoxicity. Finally, considering biological variables such as sex and time of day of dosing, especially given the varied terminal half-life of HDACis, may be key to developing successful therapeutic regiments to prevent and treat AKI.
Acknowledgments
Financial Support: The National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under award numbers K01DK105038 and R03DK120503, the University of Alabama at Birmingham Pittman Scholarship, and the UAB-UCSD O’Brien Center P30 DK 079337.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: None
References
- 1.Waddington CH. The epigenotype. 1942. Int J Epidemiol. 2012;41:10–3. [DOI] [PubMed] [Google Scholar]
- 2.Surani MA. Reprogramming of genome function through epigenetic inheritance. Nature. 2001;414:122–8. [DOI] [PubMed] [Google Scholar]
- 3.Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187:226–32. [PubMed] [Google Scholar]
- 4.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of rna synthesis. Proc Natl Acad Sci U S A. 1964;51:786–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allfrey VG, Mirsky AE. Structural modifications of histones and their possible role in the regulation of rna synthesis. Science. 1964;144:559. [DOI] [PubMed] [Google Scholar]
- 6.Lee JT. Epigenetic regulation by long noncoding rnas. Science. 2012;338:1435–9. [DOI] [PubMed] [Google Scholar]
- 7.Gershey EL, Vidali G, Allfrey VG. Chemical studies of histone acetylation. The occurrence of epsilon-n-acetyllysine in the f2a1 histone. J Biol Chem. 1968;243:5018–22. [PubMed] [Google Scholar]
- 8.Wiktorowicz JE, Campos KL, Bonner J. Substrate and product inhibition initial rate kinetics of histone acetyltransferase. Biochemistry. 1981;20:1464–7. [DOI] [PubMed] [Google Scholar]
- 9.Wiktorowicz JE, Bonner J. Studies on histone acetyltransferase. Partial purification and basic properties. J Biol Chem. 1982;257:12893–900. [PubMed] [Google Scholar]
- 10.Belikoff E, Wong LJ, Alberts BM. Extensive purification of histone acetylase a, the major histone n-acetyl transferase activity detected in mammalian cell nuclei. J Biol Chem. 1980;255:11448–53. [PubMed] [Google Scholar]
- 11.Kleff S, Andrulis ED, Anderson CW, Sternglanz R. Identification of a gene encoding a yeast histone h4 acetyltransferase. J Biol Chem. 1995;270:24674–7. [DOI] [PubMed] [Google Scholar]
- 12.Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator rpd3p. Science. 1996;272:408–11. [DOI] [PubMed] [Google Scholar]
- 13.Hyndman KA, Knepper MA. Dynamic regulation of lysine acetylation: The balance between acetyltransferase and deacetylase activities. Am J Physiol Renal Physiol. 2017;313:F842–F6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seto E, Yoshida M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb Perspect Biol. 2014;6:a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Z, Zeng C, Villar VA, Chen SY, Konkalmatt P, Wang X, et al. Human grk4gamma142v variant promotes angiotensin ii type i receptor-mediated hypertension via renal histone deacetylase type 1 inhibition. Hypertension. 2016;67:325–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lundby A, Lage K, Weinert BT, Bekker-Jensen DB, Secher A, Skovgaard T, et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012;2:419–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–40. [DOI] [PubMed] [Google Scholar]
- 19.Hyndman KA, Yang CR, Jung HJ, Umejiego EN, Chou CL, Knepper MA. Proteomic determination of the lysine acetylome and phosphoproteome in the rat native inner medullary collecting duct. Physiol Genomics. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol. 2019;20:156–74. [DOI] [PubMed] [Google Scholar]
- 21.Suraweera A, O’Byrne KJ, Richard DJ. Combination therapy with histone deacetylase inhibitors (hdaci) for the treatment of cancer: Achieving the full therapeutic potential of hdaci. Front Oncol. 2018;8:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li P, Ge J, Li H. Lysine acetyltransferases and lysine deacetylases as targets for cardiovascular disease. Nat Rev Cardiol. 2019. [DOI] [PubMed] [Google Scholar]
- 23.Felisbino MB, McKinsey TA. Epigenetics in cardiac fibrosis: Emphasis on inflammation and fibroblast activation. JACC Basic Transl Sci. 2018;3:704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen S, El-Dahr SS. Histone deacetylases in kidney development: Implications for disease and therapy. Pediatr Nephrol. 2013;28:689–98. [DOI] [PubMed] [Google Scholar]
- 25.Hadden MJ, Advani A. Histone deacetylase inhibitors and diabetic kidney disease. Int J Mol Sci. 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhuang S Epigenetic targeting for acute kidney injury. Nephrology (Carlton). 2018;23 Suppl 4:21–5. [DOI] [PubMed] [Google Scholar]
- 27.Drazic A, Myklebust LM, Ree R, Arnesen T. The world of protein acetylation. Biochim Biophys Acta. 2016;1864:1372–401. [DOI] [PubMed] [Google Scholar]
- 28.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heinzel T, Lavinsky RM, Mullen TM, Soderstrom M, Laherty CD, Torchia J, et al. A complex containing n-cor, msin3 and histone deacetylase mediates transcriptional repression. Nature. 1997;387:43–8. [DOI] [PubMed] [Google Scholar]
- 30.Laherty CD, Yang WM, Sun JM, Davie JR, Seto E, Eisenman RN. Histone deacetylases associated with the msin3 corepressor mediate mad transcriptional repression. Cell. 1997;89:349–56. [DOI] [PubMed] [Google Scholar]
- 31.Dhordain P, Lin RJ, Quief S, Lantoine D, Kerckaert JP, Evans RM, et al. The laz3(bcl-6) oncoprotein recruits a smrt/msin3a/histone deacetylase containing complex to mediate transcriptional repression. Nucleic Acids Res. 1998;26:4645–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen S, Yao X, Li Y, Saifudeen Z, Bachvarov D, El-Dahr SS. Histone deacetylase 1 and 2 regulate wnt and p53 pathways in the ureteric bud epithelium. Development. 2015;142:1180–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greer CB, Tanaka Y, Kim YJ, Xie P, Zhang MQ, Park IH, et al. Histone deacetylases positively regulate transcription through the elongation machinery. Cell Rep. 2015;13:1444–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen J, Zhang Z, Wang N, Guo M, Chi X, Pan Y, et al. Role of hdac9-foxo1 axis in the transcriptional program associated with hepatic gluconeogenesis. Sci Rep. 2017;7:6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JW, Chou CL, Knepper MA. Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J Am Soc Nephrol. 2015;26:2669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clark JZ, Chen L, Chou CL, Jung HJ, Lee JW, Knepper MA. Representation and relative abundance of cell-type selective markers in whole-kidney rna-seq data. Kidney Int. 2019;95:787–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen L, Lee JW, Chou CL, Nair AV, Battistone MA, Paunescu TG, et al. Transcriptomes of major renal collecting duct cell types in mouse identified by single-cell rna-seq. Proc Natl Acad Sci U S A. 2017;114:E9989–E98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science. 2018;360:758–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheval L, Pierrat F, Rajerison R, Piquemal D, Doucet A. Of mice and men: Divergence of gene expression patterns in kidney. PLoS One. 2012;7:e46876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chabardes-Garonne D, Mejean A, Aude JC, Cheval L, Di Stefano A, Gaillard MC, et al. A panoramic view of gene expression in the human kidney. Proc Natl Acad Sci U S A. 2003;100:13710–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- 42.Hyndman KA, Kasztan M, Mendoza LD, Monteiro-Pai S. Dynamic changes in histone deacetylases following kidney ischemia-reperfusion injury are critical for promoting proximal tubule proliferation. Am J Physiol Renal Physiol. 2019;316:F875–F88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Butler PL, Staruschenko A, Snyder PM. Acetylation stimulates the epithelial sodium channel by reducing its ubiquitination and degradation. J Biol Chem. 2015;290:12497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mansley MK, Roe AJ, Francis SL, Gill JH, Bailey MA, Wilson SM. Trichostatin a blocks aldosterone-induced na(+) transport and control of serum- and glucocorticoid-inducible kindas 1 in cortical collecting duct cells. Br J Pharmacol. 2019;in Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee HA, Lee DY, Cho HM, Kim SY, Iwasaki Y, Kim IK. Histone deacetylase inhibition attenuates transcriptional activity of mineralocorticoid receptor through its acetylation and prevents development of hypertension. Circ Res. 2013;112:1004–12. [DOI] [PubMed] [Google Scholar]
- 46.Acharya MR, Sparreboom A, Sausville EA, Conley BA, Doroshow JH, Venitz J, et al. Interspecies differences in plasma protein binding of ms-275, a novel histone deacetylase inhibitor. Cancer Chemother Pharmacol. 2006;57:275–81. [DOI] [PubMed] [Google Scholar]
- 47.Ryan QC, Headlee D, Acharya M, Sparreboom A, Trepel JB, Ye J, et al. Phase i and pharmacokinetic study of ms-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J Clin Oncol. 2005;23:3912–22. [DOI] [PubMed] [Google Scholar]
- 48.Knipstein J, Gore L. Entinostat for treatment of solid tumors and hematologic malignancies. Expert Opin Investig Drugs. 2011;20:1455–67. [DOI] [PubMed] [Google Scholar]
- 49.Kim M, Thompson LA, Wenger SD, O’Bryant CL. Romidepsin: A histone deacetylase inhibitor for refractory cutaneous t-cell lymphoma. Ann Pharmacother. 2012;46:1340–8. [DOI] [PubMed] [Google Scholar]
- 50.Sharma S, Witteveen PO, Lolkema MP, Hess D, Gelderblom H, Hussain SA, et al. A phase i, open-label, multicenter study to evaluate the pharmacokinetics and safety of oral panobinostat in patients with advanced solid tumors and varying degrees of renal function. Cancer Chemother Pharmacol. 2015;75:87–95. [DOI] [PubMed] [Google Scholar]
- 51.Clive S, Woo MM, Nydam T, Kelly L, Squier M, Kagan M. Characterizing the disposition, metabolism, and excretion of an orally active pan-deacetylase inhibitor, panobinostat, via trace radiolabeled 14c material in advanced cancer patients. Cancer Chemother Pharmacol. 2012;70:513–22. [DOI] [PubMed] [Google Scholar]
- 52.Moore DC, Arnall JR, Harvey RD. Incidence and management of adverse events associated with panobinostat in the treatment of relapsed/refractory multiple myeloma. J Oncol Pharm Pract. 2019;25:613–22. [DOI] [PubMed] [Google Scholar]
- 53.Cavenagh JD, Popat R. Optimal management of histone deacetylase inhibitor-related adverse events in patients with multiple myeloma: A focus on panobinostat. Clin Lymphoma Myeloma Leuk. 2018;18:501–7. [DOI] [PubMed] [Google Scholar]
- 54.Subramanian S, Bates SE, Wright JJ, Espinoza-Delgado I, Piekarz RL. Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals (Basel). 2010;3:2751–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fu Y, Tang C, Cai J, Chen G, Zhang D, Dong Z. Rodent models of aki-ckd transition. Am J Physiol Renal Physiol. 2018;315:F1098–F106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ranganathan P, Hamad R, Mohamed R, Jayakumar C, Muthusamy T, Ramesh G. Histone deacetylase-mediated silencing of amwap expression contributes to cisplatin nephrotoxicity. Kidney Int. 2016;89:317–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Choi SY, Piao ZH, Jin L, Kim JH, Kim GR, Ryu Y, et al. Piceatannol attenuates renal fibrosis induced by unilateral ureteral obstruction via downregulation of histone deacetylase 4/5 or p38-mapk signaling. PLoS One. 2016;11 :e0167340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, et al. Acute renal failure in critically ill patients: A multinational, multicenter study. JAMA. 2005;294:813–8. [DOI] [PubMed] [Google Scholar]
- 59.Zheng Q, Liu W, Liu Z, Zhao H, Han X, Zhao M. Valproic acid protects septic mice from renal injury by reducing the inflammatory response. J Surg Res. 2014;192:163–9. [DOI] [PubMed] [Google Scholar]
- 60.Zhang H, Zhang W, Jiao F, Li X, Zhang H, Wang L, et al. The nephroprotective effect of ms-275 on lipopolysaccharide (lps)-induced acute kidney injury by inhibiting reactive oxygen species (ros)-oxidative stress and endoplasmic reticulum stress. Med Sci Monit. 2018;24:2620–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pang M, Kothapally J, Mao H, Tolbert E, Ponnusamy M, Chin YE, et al. Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol. 2009;297:F996–F1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nguyen-Thanh T, Kim D, Lee S, Kim W, Park SK, Kang KP. Inhibition of histone deacetylase 1 ameliorates renal tubulointerstitial fibrosis via modulation of inflammation and extracellular matrix gene transcription in mice. Int J Mol Med. 2018;41:95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perazella MA, Shirali AC. Nephrotoxicity of cancer immunotherapies: Past, present and future. J Am Soc Nephrol. 2018;29:2039–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu J, Livingston MJ, Dong G, Tang C, Su Y, Wu G, et al. Histone deacetylase inhibitors protect against cisplatin-induced acute kidney injury by activating autophagy in proximal tubular cells. Cell Death Dis. 2018;9:322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tang J, Shi Y, Liu N, Xu L, Zang X, Li P, et al. Blockade of histone deacetylase 6 protects against cisplatin-induced acute kidney injury. Clin Sci (Lond). 2018;132:339–59. [DOI] [PubMed] [Google Scholar]
- 66.Shi Y, Xu L, Tang J, Fang L, Ma S, Ma X, et al. Inhibition of hdac6 protects against rhabdomyolysis-induced acute kidney injury. Am J Physiol Renal Physiol. 2017;312:F502–F15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lagny MG, Jouret F, Koch JN, Blaffart F, Donneau AF, Albert A, et al. Incidence and outcomes of acute kidney injury after cardiac surgery using either criteria of the rifle classification. BMC Nephrol. 2015;16:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.O’Neal JB, Shaw AD, Billings FTt. Acute kidney injury following cardiac surgery: Current understanding and future directions. Crit Care. 2016;20:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Evans RG, Lankadeva YR, Cochrane AD, Marino B, Iguchi N, Zhu MZL, et al. Renal haemodynamics and oxygenation during and after cardiac surgery and cardiopulmonary bypass. Acta Physiol (Oxf). 2017. [DOI] [PubMed] [Google Scholar]
- 70.Levine MH, Wang Z, Bhatti TR, Wang Y, Aufhauser DD, McNeal S, et al. Class-specific histone/protein deacetylase inhibition protects against renal ischemia reperfusion injury and fibrosis formation. Am J Transplant. 2015;15:965–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cianciolo Cosentino C, Skrypnyk NI, Brilli LL, Chiba T, Novitskaya T, Woods C, et al. Histone deacetylase inhibitor enhances recovery after aki. J Am Soc Nephrol. 2013;24:943–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Novitskaya T, McDermott L, Zhang KX, Chiba T, Paueksakon P, Hukriede NA, et al. A ptba small molecule enhances recovery and reduces postinjury fibrosis after aristolochic acid-induced kidney injury. Am J Physiol Renal Physiol. 2014;306:F496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Skrypnyk NI, Sanker S, Skvarca LB, Novitskaya T, Woods C, Chiba T, et al. Delayed treatment with ptba analogs reduces postinjury renal fibrosis after kidney injury. Am J Physiol Renal Physiol. 2016;310:F705–F16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Van Beneden K, Geers C, Pauwels M, Mannaerts I, Verbeelen D, van Grunsven LA, et al. Valproic acid attenuates proteinuria and kidney injury. J Am Soc Nephrol. 2011. ;22:1863–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Inoue K, Gan G, Ciarleglio M, Zhang Y, Tian X, Pedigo CE, et al. Podocyte histone deacetylase activity regulates murine and human glomerular diseases. J Clin Invest. 2019;129:1295–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Humphreys BD. Mechanisms of renal fibrosis. Annu Rev Physiol. 2018;80:309–26. [DOI] [PubMed] [Google Scholar]
- 77.Liu N, He S, Ma L, Ponnusamy M, Tang J, Tolbert E, et al. Blocking the class i histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating tgf-beta and egfr signaling. PLoS One. 2013;8:e54001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leivonen SK, Hakkinen L, Liu D, Kahari VM. Smad3 and extracellular signal-regulated kinase 1/2 coordinately mediate transforming growth factor-beta-induced expression of connective tissue growth factor in human fibroblasts. J Invest Dermatol. 2005;124:1162–9. [DOI] [PubMed] [Google Scholar]
- 79.Simonsson M, Heldin CH, Ericsson J, Gronroos E. The balance between acetylation and deacetylation controls smad7 stability. J Biol Chem. 2005;280:21797–803. [DOI] [PubMed] [Google Scholar]
- 80.Kinugasa F, Noto T, Matsuoka H, Urano Y, Sudo Y, Takakura S, et al. Prevention of renal interstitial fibrosis via histone deacetylase inhibition in rats with unilateral ureteral obstruction. Transpl Immunol. 2010;23:18–23. [DOI] [PubMed] [Google Scholar]
- 81.Marumo T, Hishikawa K, Yoshikawa M, Hirahashi J, Kawachi S, Fujita T. Histone deacetylase modulates the proinflammatory and -fibrotic changes in tubulointerstitial injury. Am J Physiol Renal Physiol. 2010;298:F133–41. [DOI] [PubMed] [Google Scholar]
- 82.Brilli Skvarca L, Han HI, Espiritu EB, Missinato MA, Rochon ER, McDaniels MD, et al. Enhancing regeneration after acute kidney injury by promoting cellular dedifferentiation in zebrafish. Dis Model Mech. 2019;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Reddy P, Sun Y, Toubai T, Duran-Struuck R, Clouthier SG, Weisiger E, et al. Histone deacetylase inhibition modulates indoleamine 2,3-dioxygenase-dependent dc functions and regulates experimental graft-versus-host disease in mice. J Clin Invest. 2008;118:2562–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu WP, Tsai YG, Lin TY, Wu MJ, Lin CY. The attenuation of renal fibrosis by histone deacetylase inhibitors is associated with the plasticity of foxp3(+)il-17(+) t cells. BMC Nephrol. 2017;18:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ellmeier W, Seiser C. Histone deacetylase function in cd4(+) t cells. Nat Rev Immunol. 2018;18:617–34. [DOI] [PubMed] [Google Scholar]
- 86.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in g2/m mediates kidney fibrosis after injury. Nat Med. 2010;16:535–43, 1p following 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tang J, Yan Y, Zhao TC, Bayliss G, Yan H, Zhuang S. Class i histone deacetylase activity is required for proliferation of renal epithelial cells. Am J Physiol Renal Physiol. 2013;305:F244–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tang J, Yan Y, Zhao TC, Gong R, Bayliss G, Yan H, et al. Class i hdac activity is required for renal protection and regeneration after acute kidney injury. Am J Physiol Renal Physiol. 2014;307:F303–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rubin EH, Agrawal NG, Friedman EJ, Scott P, Mazina KE, Sun L, et al. A study to determine the effects of food and multiple dosing on the pharmacokinetics of vorinostat given orally to patients with advanced cancer. Clin Cancer Res. 2006;12:7039–45. [DOI] [PubMed] [Google Scholar]
- 90.Fakih MG, Pendyala L, Fetterly G, Toth K, Zwiebel JA, Espinoza-Delgado I, et al. A phase i, pharmacokinetic and pharmacodynamic study on vorinostat in combination with 5-fluorouracil, leucovorin, and oxaliplatin in patients with refractory colorectal cancer. Clin Cancer Res. 2009;15:3189–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lassen U, Molife LR, Sorensen M, Engelholm SA, Vidal L, Sinha R, et al. A phase i study of the safety and pharmacokinetics of the histone deacetylase inhibitor belinostat administered in combination with carboplatin and/or paclitaxel in patients with solid tumours. Br J Cancer. 2010;103:12–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bailey H, McPherson JP, Bailey EB, Werner TL, Gupta S, Batten J, et al. A phase i study to determine the pharmacokinetics and urinary excretion of belinostat and metabolites in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2016;78:1059–71. [DOI] [PubMed] [Google Scholar]
- 93.Klimek VM, Fircanis S, Maslak P, Guernah I, Baum M, Wu N, et al. Tolerability, pharmacodynamics, and pharmacokinetics studies of depsipeptide (romidepsin) in patients with acute myelogenous leukemia or advanced myelodysplastic syndromes. Clin Cancer Res. 2008;14:826–32. [DOI] [PubMed] [Google Scholar]
- 94.Byrd JC, Marcucci G, Parthun MR, Xiao JJ, Klisovic RB, Moran M, et al. A phase 1 and pharmacodynamic study of depsipeptide (fk228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood. 2005;105:959–67. [DOI] [PubMed] [Google Scholar]
- 95.Zaccara G, Messori A, Moroni F. Clinical pharmacokinetics of valproic acid--1988. Clin Pharmacokinet. 1988;15:367–89. [DOI] [PubMed] [Google Scholar]
- 96.McGuire BM, Zupanets IA, Lowe ME, Xiao X, Syplyviy VA, Monteleone J, et al. Pharmacology and safety of glycerol phenylbutyrate in healthy adults and adults with cirrhosis. Hepatology. 2010;51:2077–85. [DOI] [PMC free article] [PubMed] [Google Scholar]