

Abstract
Styrene–maleic acid copolymers have received significant attention because of their ability to interact with lipid bilayers and form styrene–maleic acid copolymer lipid nanoparticles (SMALPs). However, these SMALPs are limited in their chemical diversity, with only phenyl and carboxylic acid functional groups, resulting in limitations because of sensitivity to low pH and high concentrations of divalent metals. To address this limitation, various nucleophiles were reacted with the anhydride unit of well-defined styrene–maleic anhydride copolymers in order to assess the potential for a new lipid disk nanoparticle-forming species. These styrene–maleic anhydride copolymer derivatives (SMADs) can form styrene–maleic acid derivative lipid nanoparticles (SMADLPs) when they interact with lipid molecules. Polymers were synthesized, purified, characterized by Fourier-transform infrared spectroscopy, gel permeation chromatography, and nuclear magnetic resonance and then used to make disk-like SMADLPs, whose sizes were measured by dynamic light scattering (DLS). The SMADs form lipid nanoparticles, observable by DLS and transmission electron microscopy, and were used to reconstitute a spin-labeled transmembrane protein, KCNE1. The polymer method reported here is facile and scalable and results in functional and robust polymers capable of forming lipid nanodisks that are stable against a wide pH range and 100 mM magnesium.
Graphical Abstract
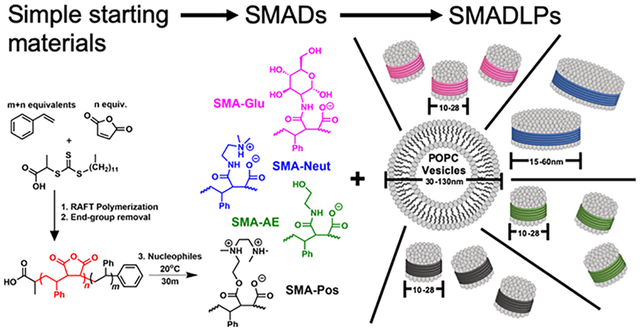
INTRODUCTION
Membrane proteins are ubiquitous and critical drug targets because of their roles in cell–cell signaling and recognition, immune response, ion transport, and other regulatory processes.1–3 However, they are challenging to characterize because of the innate heterogeneity of the lipid bilayer environment and surrounding water. To glean accurate structural information, membrane proteins must be incorporated into native-like membrane-mimetic environments, which enable retention of the protein’s native conformation.4
Many types of membrane mimetics have been developed for isolation and characterization of membrane proteins. Membrane protein isolation typically requires disruption of the cell membrane and solubilizing its contents using micelle-forming detergents.5 However, micelles have high curvature and a different lateral pressure profile than the native bilayer, and thus, characterization data obtained from protein–detergent micelle samples carry the question of whether the data are biologically relevant.
In membrane protein characterization, a few classes of membrane mimetics have been developed, such as micelles, bicelles, liposomes, and nanodisks, each with their own applications.6 The name “bicelle” is a portmanteau combining the words “bilayer” and “micelle”. To form a bicelle, bilayer-forming long-chain lipids are mixed with a short-chain detergent, such as CHAPSO or DHPC, to form a two-phased nanoparticle. The bilayer lipids form a bilayer which surrounds the protein of interest, and the detergents form a ring around the bilayer edge to shield the hydrophobic lipid tails from interaction with water. However, the select group of bicelle-forming detergents all fall on the hard-surfactant side of the spectrum and run the risk of denaturing the protein of interest.7,8
Liposomes are aggregates of lipid molecules in aqueous solution, which form a large spherical bilayer having an enclosed aqueous phase. This property can be useful in characterizing the activity of membrane transport proteins. Methods of preparing liposomes also quite often retain the native mimetic complex of protein and closely associated lipids. However, their large size confers anisotropy, making them unsuitable for solution nuclear magnetic resonance (NMR) applications.9 In addition, it is difficult to concentrate proteins into liposomes, which diminishes signal in biophysical experiments.10
Lipid disks, also called nanodisks, have recently attracted attention as novel membrane mimetic systems. In this architecture, bilayer-forming lipids are solubilized as nanoparticles by a belt of amphipathic macromolecules. The first generation of nanodisks utilized membrane scaffold protein or similar shorter peptides as the solubilizing unit.11–13 However, these peptides themselves can contribute background noise in certain membrane protein spectroscopic studies. Additionally, detergents must still be used to stabilize the protein of interest before addition of nanodisk-forming peptides.14 Amphipathic block copolymers are known to have interesting and useful phase behaviors, including the ability to form a variety of nanoparticles and architectures.15 Recently, such copolymers have been developed for nanodisk applications. When mixed with a lipid suspension, the hydrophobic portions of these polymers interact with the lipid acyl chains, inserting themselves into the bilayer. The hydrophilic portions make the lipid–protein–copolymer assembly soluble, disrupting the larger bilayer source, often without the assistance of detergents.16
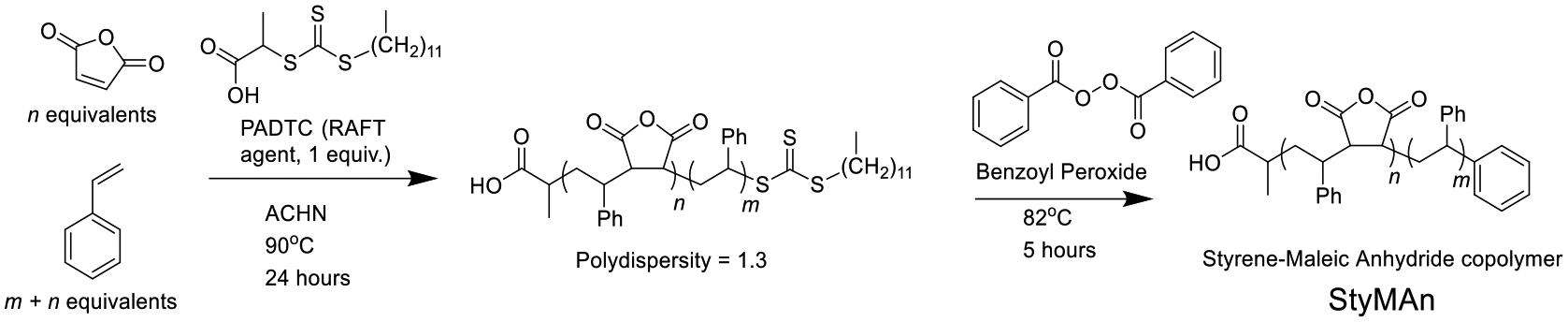
The most common polymeric surfactant used to make nanodisks is the synthetic copolymer styrene–maleic acid (SMA). When a lipid sample, such as a liposome, interacts with SMA, SMA lipid nanoparticles (SMALPs) are formed. The transformation is usually complete at or above a 1:1 weight ratio of polymer/lipid.17 We have previously reported the synthesis of SMA suitable for use in SMALPs using reversible addition-fragmentation chain transfer (RAFT) radical polymerization (Scheme 1).18 RAFT polymerization of styrene and maleic anhydride affords SMA that generates SMALPs with tunable sizes between 10 and 50 nm based on the ratio of monomers used, on multigram scale, with a cost of dollars per gram of material.
Scheme 1. Synthesis of a Styrene–Maleic Acid Copolymer with Styrene Tail.
There are, however, limits to the utility of SMA; the most common problems encountered are due to the diacids in the copolymer. Protonation of the acids or binding to divalent metal cations such as calcium or magnesium causes SMA to become too hydrophobic to maintain the lipid disk structure. The presence of magnesium at a concentration above 10 mM or pH less than 6 often causes precipitation of the polymer, resulting in SMALP disassembly.19 This sensitivity to magnesium is unfortunate as many membrane proteins with enzymatic function, such as ATPases, require magnesium as a cofactor. Other groups have successfully overcome these problems by derivatizing cheaper commercially available materials (on their own, these materials are unable to form lipid disks), at times using somewhat expensive reagents, or long protocols with multiple reaction and precipitation steps for the derivatization.20–22
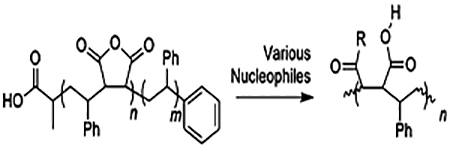
To overcome these limitations, nucleophilic ring opening of RAFT-synthesized styrene–maleic anhydride (SMAn) copolymers is explored in this article. This allows a wide range of chemical spaces to be explored. The rationale for the choice of nucleophile is summarized in Table 1. All the nucleophiles could be obtained for less than $2.00/g.
Table 1.
SMA Derivatives for New SMALP Formulations
 | ||
|---|---|---|
| Product Name | R-group | Rationale |
| SMA-Glu |  |
Mimic some lipid head groups Mimic some mild detergents High number of H-bond donors |
| SMA-AE | 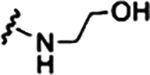 |
Recently reported using commercially available polymer23 |
| SMA-Neut |  |
Net neutral charge at neutral pH, charged over entire pH range, recently reported using commercially available polymer |
| SMA-Pos | 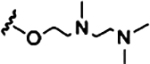 |
Net positive charge at neutral and acidic pH |
All of the synthesized styrene–maleic anhydride copolymer derivatives (SMADs) are hypothesized to be milder surfactants than the parent SMA copolymer. Reacting SMA with glucosamine can mimic to some extent the alkyl glucosides which have proved useful in membrane protein work.24 SMA-Neut and SMA-Pos should be milder than SMA because of their zwitterionic character, mimicking surfactants such as CHAPSO and DHPC, which have been used in bicelle research.7 Finally, a different formulation of SMA-AE has been previously reported as useful in converting cheap yet impotent commercial SMA into a lipid-disk-forming material.23 SMA-AE has fewer charged groups than SMA and is therefore also expected to behave as a milder surfactant.
This work expands the biochemist’s toolkit for studying membrane proteins by synthesizing styrene–maleic anhydride copolymers by RAFT and then took advantage of the high reactivity of the anhydride functional group to derivatize the polymers with various inexpensive, commercially available nucleophiles under ambient conditions in only 30 min, with only one or two precipitation steps. The products, the new R-group added, and rationale for each choice are listed in Table 1. Conversion of the anhydride group was determined by infrared spectroscopy. The resulting polymers were then mixed with 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) liposomes in order to assess their potential as new lipid disk polymers.
EXPERIMENTAL SECTION
Typical Synthesis of a One-Pot Block Copolymer of Poly(styrene-alt-maleic anhydride-b-styrene) (SMAn).
All materials were obtained from commercial suppliers and used as received unless otherwise specified. The synthesis of poly(styrene-alt-maleic anhydride-b-styrene) was carried out as an adaptation of a previously described procedure.18 Briefly, styrene (3.6608 g, 35.1 mM), maleic anhydride (0.98 g, 10 mM), and 2-(dodecylthiocarbonothioylthio)propionic acid (0.1404 g, 0.4 mM) were combined in a 20 mL vial and dissolved in 4.64 g of 1,4-dioxane. 1,1-Azobis(cyclohexanecarbonitrile) (0.0195 g, 0.008 mM) was added and dissolved. The contents of the vial were transferred to a 50 mL round-bottom flask with a magnetic stir bar, and a small aliquot of solution was set aside. The flask was capped with a rubber stopper and bubbled with nitrogen for 15 min. The solution was heated to 90 °C for 20 h. Once comparison by NMR with the previously set aside aliquot showed sufficient conversion (~80%), the polymer was purified by three sequential precipitations from tetrahydrofuran (THF) into a large excess of cold hexanes, yielding 4 g of yellow powder.
End-Group Removal of Poly(styrene-alt-maleic anhydride-b-styrene).
The polymer (ca. 4 g) was dissolved in dioxane and combined with 2.4 g of benzoyl peroxide (9.9 mM) in a 50 mL round-bottom flask. The flask was sealed with a rubber stopper and bubbled with nitrogen for 15 min. The escape needle was left in the flask, and the flask was heated to 82 °C for 5 h. Upon completion, the polymer was precipitated twice from THF into a large excess of cold hexanes, yielding a white to off-white powder. This material was stored in a desiccator at room temperature to prevent hydrolysis of the anhydride moiety by atmospheric moisture.
Synthesis of 2–1 SMA-Glu.
To a 20 mL glass vial was added 400 mg of 2–1 SMAn, which had ca. 3 mmol/g of anhydride, for a total of 1.2 mmol anhydride. The polymer was dissolved in minimal dimethylformamide (DMF). Then, to the vial were added simultaneously triethylamine (TEA) (502.5 μL, 3.6 mmol) and d-glucosamine (HCl salt) (0.3887 g, 1.8 mmol). The solution was stirred at room temperature for 30 min. Longer stirring periods will result in an immobile cross-linked matrix. Then, the polymer was precipitated by adding this solution dropwise to rapidly stirring diethyl ether (40 mL) in an ice bath. The ether was decanted, and the polymer was washed with 20 mL of ice-cold ethyl acetate and mixed well. The ethyl acetate was decanted, and the polymer was dried in a vacuum oven at low temperature (ca. 50 °C) for 30 min to remove all volatiles. Then, the sample was suspended in 0.1 M HCl (ca. 10 mL), well mixed in a 15 mL centrifuge tube, and spun into a pellet at 7k rpm for 5 min. HCl was decanted, and the material was rinsed again with 10 mL of HCl, spun down, and HCl was decanted. Finally, the polymer was freeze-dried, yielding 100 mg of off-white powder. Conversion of the anhydride was confirmed by infrared spectroscopy.
Synthesis of 2–1 SMA-AE.
To a 20 mL glass vial was added 400 mg of 2–1 SMAn, which had ca. 3 mmol/g of anhydride, for a total of 1.2 mmol anhydride. The polymer was dissolved in minimal DMF. Then, to the vial were added simultaneously TEA (251.2 μL, 1.8 mmol) and aminoethanol (109.0 μL, 1.8 mmol). The solution was stirred at room temperature for 30 min. Then, the polymer was precipitated by adding this solution dropwise to rapidly stirring diethyl ether (40 mL) in an ice bath. The ether was decanted, and the polymer was washed with 20 mL of ice-cold ethyl acetate and mixed well. The ethyl acetate was decanted, and the polymer was dried in a vacuum oven at low temperature (ca. 50 °C) for 30 min to remove all volatiles. Then, the sample was suspended in 0.1 M HCl (ca. 10 mL), well mixed in a 15 mL centrifuge tube, and spun into a pellet at 7k rpm for 5 min. HCl was decanted, and the material was rinsed again with 10 mL of HCl, spun down, and HCl was decanted. Finally, the polymer was freeze-dried, yielding 190 mg of off-white powder. Conversion of the anhydride was confirmed by infrared spectroscopy.
Synthesis of 2–1 SMA-Pos.
To a 20 mL glass vial was added 400 mg of 2–1 SMAn, which had ca. 3 mmol/g of anhydride, for a total of 1.2 mmol anhydride. The polymer was dissolved in minimal DMF. Then, to the vial was added 2-{[2-(dimethylamino)ethyl]-methylamino}ethanol (289.7 μL, 1.8 mmol). The solution was stirred at room temperature for 30 min. Then, the polymer was precipitated by adding this solution dropwise to rapidly stirring diethyl ether (40 mL) in an ice bath. The ether was decanted, and the polymer was washed with 20 mL of ice-cold ethyl acetate and crushed with a metal spatula while being submerged. This was repeated twice for a total of 60 mL ethyl acetate used in washing. The ethyl acetate was decanted, and the polymer was dried in a vacuum oven at low temperature (ca. 50 °C) for 30 min to remove all volatiles, followed by freeze-drying, yielding 245 mg of off-white powder. Conversion of the anhydride was confirmed by infrared spectroscopy.
Synthesis of 2–1 SMA-Neut.
To a 20 mL glass vial was added 400 mg of 2–1 SMAn, which had ca. 3 mmol/g of anhydride, for a total of 1.2 mmol anhydride. The polymer was dissolved in minimal DMF. Then, to the vial was added N,N-dimethylethylenediamine (196.9 μL, 1.8 mmol). The solution was stirred at room temperature for 30 min. Then, the polymer was precipitated by adding this solution dropwise to rapidly stirring diethyl ether (40 mL) in an ice bath. The ether was decanted, and the polymer was washed with 20 mL of ice-cold ethyl acetate and crushed with a metal spatula while submerged. This was repeated twice for a total of 60 mL ethyl acetate used in washing. The ethyl acetate was decanted, and the polymer was dried in a vacuum oven at low temperature (ca. 50 °C) for 30 min to remove all volatiles, followed by freeze-drying, yielding 220 mg of off-white powder. Conversion of the anhydride was confirmed by infrared spectroscopy.
Synthesis of 2–1M/3–1M SMA.
The anhydride moieties of the polymer were hydrolyzed to their succinic acid counterparts as previously described18 with slight modifications. Briefly, equal mass of polymer and THF (1 g) was combined in a vial and briefly heated to 95 °C while swirling to dissolve all the polymer. Then, a 4X molar excess of NaOH was added, and the mixture was heated at 50 °C for 24 h. After this period, there are two layers, so another 10 mL of distilled water was added, and the mixture was heated for another 24 h. At the end of this period, the solution was found to be stable at room temperature. THF and excess base were removed by dialysis in 3.5 kDa cutoff tubing using two 1 L portions of ultrapure water. The polymer was collected in a 50 mL conical tube, frozen in a −80 °C refrigerator overnight, and then lyophilized, yielding a white to off-white powder.
Determination of Polymer Molecular Weight Parameters via Size Exclusion Chromatography.
Approximately 5 mg of polymer was weighed out and dissolved in either 1.5 mL of THF with 0.025% butylated hydroxy toluene or 1.5 mL of DMF with 0.01% LiBr–THF for the parent anhydride and DMF for the derivatives and SMA. To the resulting mixture was added two drops of toluene as the flow rate marker. The solution was filtered through a 0.22 μm filter. Size exclusion chromatography was performed using an Agilent 1260 gel permeation chromatography system equipped with an autosampler, a guard and 2X PLgel MIXED B columns, and a refractive index detector. The eluent was THF or DMF running at 1 mL/min at 25 °C. The system was calibrated with poly(methyl methacrylate) standards in the range of 617,000–1010, and the parent anhydride copolymer was corrected to polystyrene using the standard Mark–Houwink parameters KMMA = 12.8, αMMA = 0.69, KSty = 11.4, αSty = 0.716. For the derivatives, no mass corrections could be done; no correctors exist for the derivatized anhydride units.
Determination of pKa.
In most cases, approximately 15 mg of polymer was weighed out and dissolved in 15 mL of distilled water with vigorous stirring. Dissolution was promoted by the addition of 50 μL of 2 M NaOH. The undissolved material was pelleted by centrifugation at 10,000 rpm for 10 min. The supernatant was transferred to a vial. A pH probe was submerged into solution, and pH was recorded with the addition of small volumes of 0.3–0.6 M HCl. Because of the labile nature of esters in aqueous base, SMA-Pos was weighed out and dissolved in distilled water with vigorous stirring to promote dissolution of all visible materials. The SMA-Pos sample was then brought to low pH by the addition of a small volume of 6 M HCl. Then, the pH was recorded with the addition of small volumes of 2 M NaOH.
The pKa was determined by plotting the first derivative of the pH. The first derivative can be easily obtained using eq 1.
| (1) |
where ΔpHn is the change in pH at volume n; pHn is the pH at volume n; and pHn–1 is the pH at the volume preceding volume n. The pKa is equal to the pH at the volume corresponding to local minima in the derivative plot. Very low (under 2.5) and very high (over 11) values were not recorded as pKa values because of the high degree of dilution of these systems for which the ionization of water obscures observable changes in buffer capacity contributed by the polymers.
Infrared Spectroscopy of SMA Derivatives.
Infrared spectra were collected on a PerkinElmer Spectrum One FT-IR spectrometer. All samples were interrogated as dry solids. Spectra were normalized by setting the lowest transmittance value to zero and the highest transmittance near 4000 cm−1 to 100%.
Preparation of POPC Vesicles.
A previously published procedure was used to prepare the POPC vesicles.18 POPC was chosen because the phosphatidylcholine head group is the most abundant head group in eukaryotic cell membranes.25,26 Powdered lipid was dissolved and suspended in a buffer containing 100 mM NaCl and 20 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES) at a pH 7.0 to a final concentration of 25 mM. The lipid slurry was vortexed vigorously to mix completely and vesicles were spontaneously formed, resulting in a homogeneous milky solution after 10 freeze/sonication cycles (<30 °C). Vesicle solutions were then frozen with liquid nitrogen and placed in a freezer overnight (−20 °C). Dynamic light scattering (DLS) was used to confirm the size of the vesicles the next day.
Formation of Styrene–Maleic Acid Derivative Lipid Nanoparticles.
SMADs were dissolved in buffer (20 mM HEPES, 100 mM NaCl pH 7) at an approximate concentration of 2.5% (m/v) and sonicated at 30–40 °C for several hours. The insoluble material was allowed to settle to the bottom for 24 h. The styrene–maleic acid derivative lipid nanoparticles (SMADLPs) were formed by adding the top layer of the polymer solution dropwise to the POPC vesicles at a volume ratio of 1/1.5 lipid to polymer. Samples were then equilibrated via two freeze/sonication cycles. Samples were allowed to mix and equilibrate overnight at room temperature. The insoluble material was allowed to settle for 24 h before making the DLS measurement.
DLS Measurement.
DLS measurements were performed on a ZetaSizer Nano series (Malvern Instruments) at 25 °C in disposable 40 μL micro cuvettes. Data were collected for 20 s and averaged for 10 scans.
Transmission Electron Microscopy.
One drop of either liposome control or SMADLP sample was adsorbed to 200 mesh copper carbon-coated grids for 10 s for full absorbance. The grids were stained with two drops of 1.5% ammonium molybdate. Images were recorded using Joel-1200.
pH Stability of SMADLPs.
SMADLPs were formed by mixing together SMAD copolymers (2.5% w/v) and POPC vesicles (2% w/v) at a 7.5:1 weight ratio (6:1 by volume), followed by two freeze–sonication cycles, rotating at room temperature overnight, and then waiting at least 24 h for the insoluble material to settle to the bottom. Then, the turbidity of solutions of varying pH was measured in a 96-well plate. The supernatant SMADLP solution (10–20 μL) was combined with 120 μL of 0.1 M NaCl with 0.1 M buffer–pH 3 HEPES, pH 5 acetate, pH 7 HEPES, and pH 9 CHES. The optical density at 620 nm was used to measure turbidity,27 and all points were run in triplicates.
Mg2+ Tolerance of SMADLPs.
SMADLPs and SMALPs were prepared as described in the previous section. In a 96-well plate, 20 μL of supernatant SMADLP solution was combined with Mg2+ containing buffer for a final volume of 150 μL at the desired [Mg2+] up to 100 mM. The optical density at 620 nm was used to measure turbidity, and all points were run in triplicates and baselined against a solution of SMADLPs at 0 mM Mg2+.
Preparation and Reconstitution of KCNE1 into Liposomes and Nanodisks for the CW-EPR Spectroscopic Study.
The KCNE1 mutant T58C was overexpressed in BL21 Escherichia coli cells grown in TB minimal media with 50 μg/mL of chloramphenicol and 50 μg/mL of ampicillin. The cell cultures were incubated at 250 rpm and 37 C until an OD600 of 0.6 was reached; the cells were induced with 1 mM isopropyl-1-thio-D-galactopyranoside. Purification of the KCNE1 protein was carried out using a previously described protocol.28 The pure protein was eluted in 0.5% dodecylphosphocho-line (DPC) detergent and concentrated using a Microcon YM-3 (molecular weight cutoff, 3000) filter (Amicon). The protein concentration was determined from A280 using a molecular extinction coefficient of 1.2 mg/mL protein per OD280 on a NanoDrop 2000c (Thermo Scientific). Protein purity was confirmed with sodium dodecyl sulfate polyacrylamide gel electrophoresis.
The protein sample was reduced with 2.5 mM dithiothreitol, with gentle agitation at room temperature for 24 h. The MTSL spin label was added in 10X molar excess to KCNE1 solution and incubated at room temperature 30 min, followed by agitation at 37·°C for 3 h and then agitation at room temperature for the remaining 24 h. The sample was then buffer-exchanged into 50 mM phosphate, 0.5% DPC, pH 7.0. After buffer exchange, samples were bound to nickel resin in a gravity column that was washed with 300 mL of 50 mM phosphate, 0.05% DPC, pH 7.0 to remove excess spin label. Spin-labeled KCNE1 was eluted in 50 mM phosphate, 250 mM imidazole, 0.5% DPC, pH 7.0.
Liposomes were prepared using the thin-film method. POPC and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG) were measured in a 3:1 molar ratio with a final concentration of 100 mM. The powder lipid was dissolved in minimal chloroform, and then the chloroform was evaporated off with nitrogen. The flask was rotated during evaporation to form a thin film of lipids on the surface of the flask. The lipids were desiccated overnight, dissolved in 50 mM phosphate pH 7.0, and then underwent three freeze–thaw cycles to form liposomes.
The concentrated spin-labeled KCNE1 protein was mixed with the liposomes to a 1:400 protein to lipid molar ratio. The sample underwent three freeze–thaw cycles to ensure incorporation of the protein into the liposome. The sample then underwent dialysis for 48 h in 4 L of dialysis buffer (10 mM imidazole and 0.1 mM ethylenediaminetetraacetic acid at pH 7.0) with buffer changed twice daily. The completion of detergent removal was determined when the KCNE1–liposome sample became turbid and viscous when compared to predialysis.
KCNE1 inPOPC/POPG vesicles was reconstituted into nanodisks upon the addition of an SMAD similar to previous studies.29 Briefly, about 500 μL of protein in the multilamellar vesicles was mixed with 2.5% SMAD solution (20 mM HEPES, 100 mM NaCl, pH 7) over 5 min and brought to a final weight ratio of 2:1 (polymer/lipid solution). The mixture was equilibrated at 4 °C for at least 3 h to allow for nanodisks to spontaneously form.
RESULTS AND DISCUSSION
Gel Permeation Chromatography Analysis of SMA Derivatives.
RAFT polymerization was used to synthesize the parent SMAn polymer. Gel permeation chromatography (GPC) analysis of the parent 2–1M SMAn shows that it is of the correct size and well-controlled with a dispersity less than 2.0 (Table 2 and Figure 1a). Small differences between the theoretical and experimental molecular weights are likely due to the use of poly(methyl methacrylate) standards to calibrate the size exclusion chromatography system. As for the derivatives, the calculated molecular weights are for the most part only slightly higher than expected (Table 2). SMA-Neut is very close or even slightly lower than the expected molecular weight. The mass of the other derivatives may be inflated by the aggregation behavior and differing Mark–Houwink parameters.
Table 2.
GPC Analysis of SMA Derivatives
| polymer | Mn (kDa) | PD |
|---|---|---|
| 2–1M SMA-Glu | 42.1 | 6.93 |
| 2–1M SMA-Neut | 6.9 | 1.46 |
| 2–1M SMA-AE | 18.3 | 1.72 |
| 2–1M SMA-Pos | 11.1 | 1.43 |
| 3–1M SMA-Pos | 21.9 | 1.33 |
| 3–1M SMA | 17.8 | 1.51 |
| 2–1M SMAn | 6.8 | 1.29 |
Figure 1.
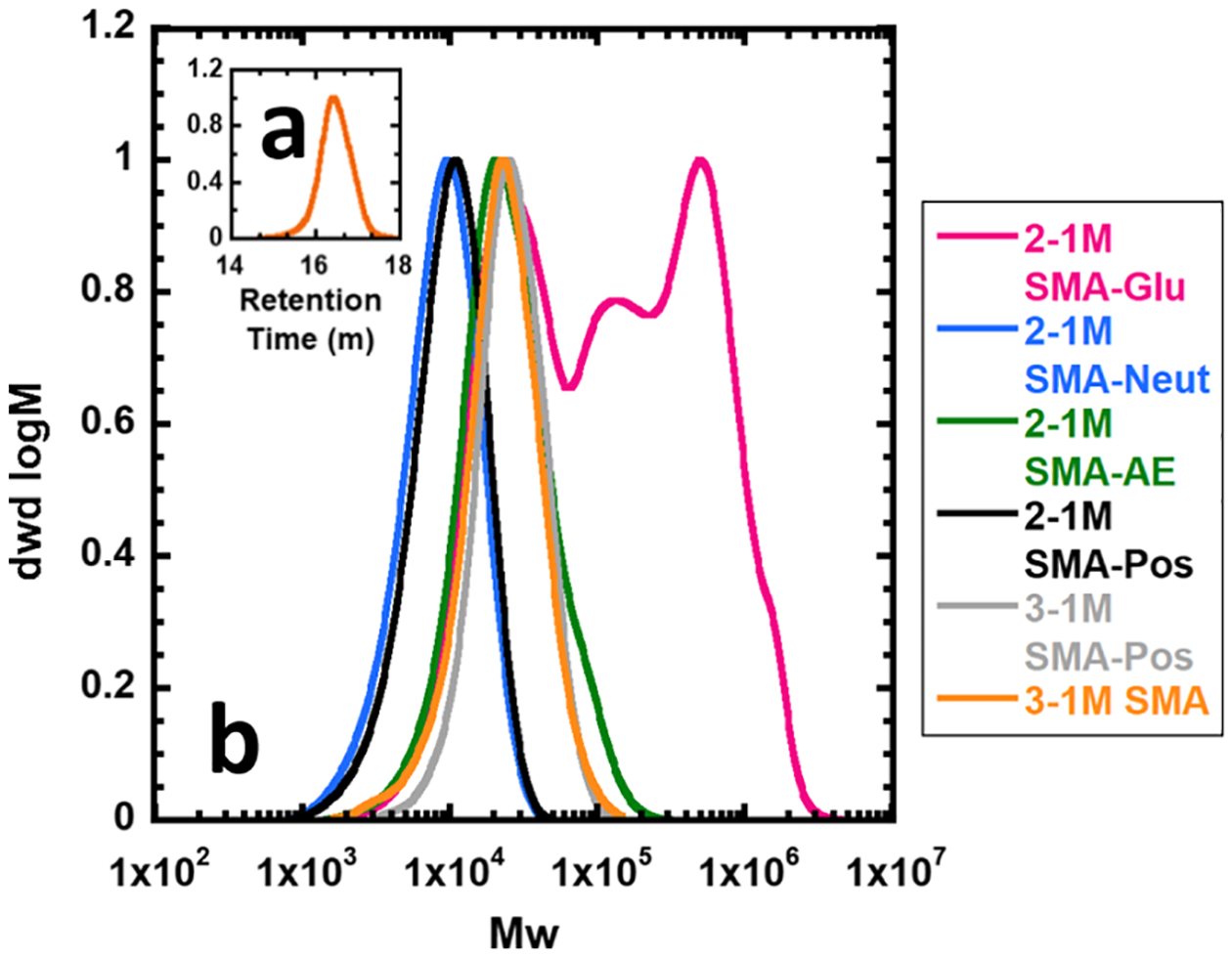
GPC traces of SMA materials. (a) (inset) 2–1M styrene–maleic anhydride starting material in THF. (b) SMA derivatives in DMF. Maximum intensity normalized to 1.
SMA-Glu was observed to have a very high dispersity, with multiple observable peaks (Figure 1b). The lowest molecular weight peak appears at the expected size (ca. 11 kDa); however, several larger peaks appear. This indicates that there is a large degree of branching and/or aggregation behavior. Indeed, if the SMA-Glu reaction is left stirring for longer than 30 min, it will completely solidify. This may be due to a combination of nucleophilic substitution of the hydroxyl groups of glucosamine with anhydride and esterification of those same groups with carboxyl groups, catalyzed by TEA. More control could potentially be achieved by refluxing in aqueous base to selectively cleave the ester bonds that appear to be cross-linking the system (Figure 1).
GPC analysis of hydrolyzed SMA (Figure 1b) was also performed, but the comparison is complicated because dissolution of SMA in DMF requires neutralizing the acidic groups with HCl, resulting in a mostly noncharged polymer. In any case, the low dispersity values for SMA-Neut, SMA-AE, and SMA-Pos indicate complete functionalization, which is supported by the IR data.
pKa Analysis of SMA Derivatives.
Many of the polymers displayed pKa values outside of the expected ranges for their functional groups. This is likely due to the differences between the local environments of these functional groups in a polymeric versus small-molecule form. There may be field effects for both amines and carboxylic acids, which favor the less ionic state (carboxylic acid or amine) and cause a shift in the pKa of the polymer compared to the small-molecule analogues. A protonated amine in proximity to a neutral amine or carboxylic acid will prevent more hydronium cations from approaching, decreasing the pKa. Nearby hydrogen bond acceptors could help stabilize the protonated states of carboxylic acids and amines, increasing pKa. Moreover, a large concentration of negative charge such as with carboxylic acids strung together could cause an increase in pKa by attracting more hydronium and by the stabilization of the protonated state resulting from a decrease in local charge density; this phenomenon is known as the polyelectrolyte effect.30 As the protonation state changes, the magnitude of the polyelectrolyte effect also changes. This leads to a gradual change in apparent pKa over the course of the titration and generally smoothens the otherwise sharp pH transitions that occur outside the buffer region (Table 3).
Table 3.
pKa Analysis of Derivatives
| polymer | pKa1 | pKa2 | macroscopic behavior |
|---|---|---|---|
| SMA-Glu | 7.4 | no precipitated observed | |
| SMA-Neut | 6.4 | 10.4 | no precipitated observed |
| SMA-AE | 9.5 | above pH 2.3, the undissolved material swells and forms a gel-like phase. Below pH 2.3, the transparent solution becomes cloudy, and the nebulous material settles to bottom within 24 h. | |
| SMA-Pos | 6.3 | 9.7 | cloudy solution quickly clarifies when moving from pH 5.63 to pH 6.30. |
The protonatable functional groups are also expected to be forced to the outside of a coiling polymer in which the styrene units are forced inside because of the hydrophobic effect. This agrees with the computed structure of dissolved SMA.16 This forced proximity may be what allows the carboxylic acids in some cases to have far higher pKa values than expected. From a thermodynamic standpoint, this would decrease the entropic penalty of intramolecular ring formation via hydrogen bonding; the coiled chain forces the functional groups into a favorable geometry for hydrogen bonding.
The hydrolyzed SMA parent material itself displays some interesting features. Although it does display one pKa in the expected range of 4.5, there is also a reproducible shoulder in the ΔpH curve at pH 9.6, which is suspected to be a weak pKa2 where the first carboxyl group of each pair is protonated. This is consistent with the literature reporting pKa values of 6 and 10.19
SMA-Neut displayed pKa values in the expected range, corresponding to an amine(10.4) and a carboxylic acid (6.4). While the latter is on the high side for a carboxylic acid, it is consistent with prior work that has shown that the standard SMA copolymer exhibits a high pKa for its carboxylic acids.19 This is likely due to the proximity of carboxylic acids on the polymer chain and stabilization of a protonated carboxylic acid by the nearby amide carbonyl oxygen.
SMA-Pos showed only two pKa values, both close to those of SMA-Neut. These two polymers have analogous structures, but the pKa of 9.7 may be due to the amine nitrogen atoms sharing a proton, decreasing their pKa value relative to the typical amine pKa (ca. 10.5). A higher pKa may exist but may be too high for this simple method to detect.
SMA-Glu exhibits a single, wide, and very high pKa of 7.4 for its carboxylic acids. We propose that this is due to the numerous hydrogen bonding hydroxyl groups on the pendant glucose units, which cooperatively stabilize the protonated carboxylic acid. No pH-induced precipitation was observed for this polymer, which suggests that the glucose units shelter the protonated acid from the hydrophobic effect enough to prevent aggregation and precipitation. In addition, it is possible that any macromolecules which were capable of precipitating were dissolved and lost in one of the purification steps, which involved suspension in 0.1 M HCl and centrifugation into a pellet. It is noteworthy that two carbonyl stretches were observed for this polymer in the IR, leading us to conclude that some ester bonds may also have formed alongside the expected amide bonds, leaving some amines unreacted (Figure 3a). This is further supported by the GPC trace, which shows a peak with two shoulders that seem to indicate the presence of two-chain and three-chain species that could only form from slower esterification reactions. However, the difference in peak intensity by IR is too high to make these free amines significant contributors to pKa.
Figure 3.
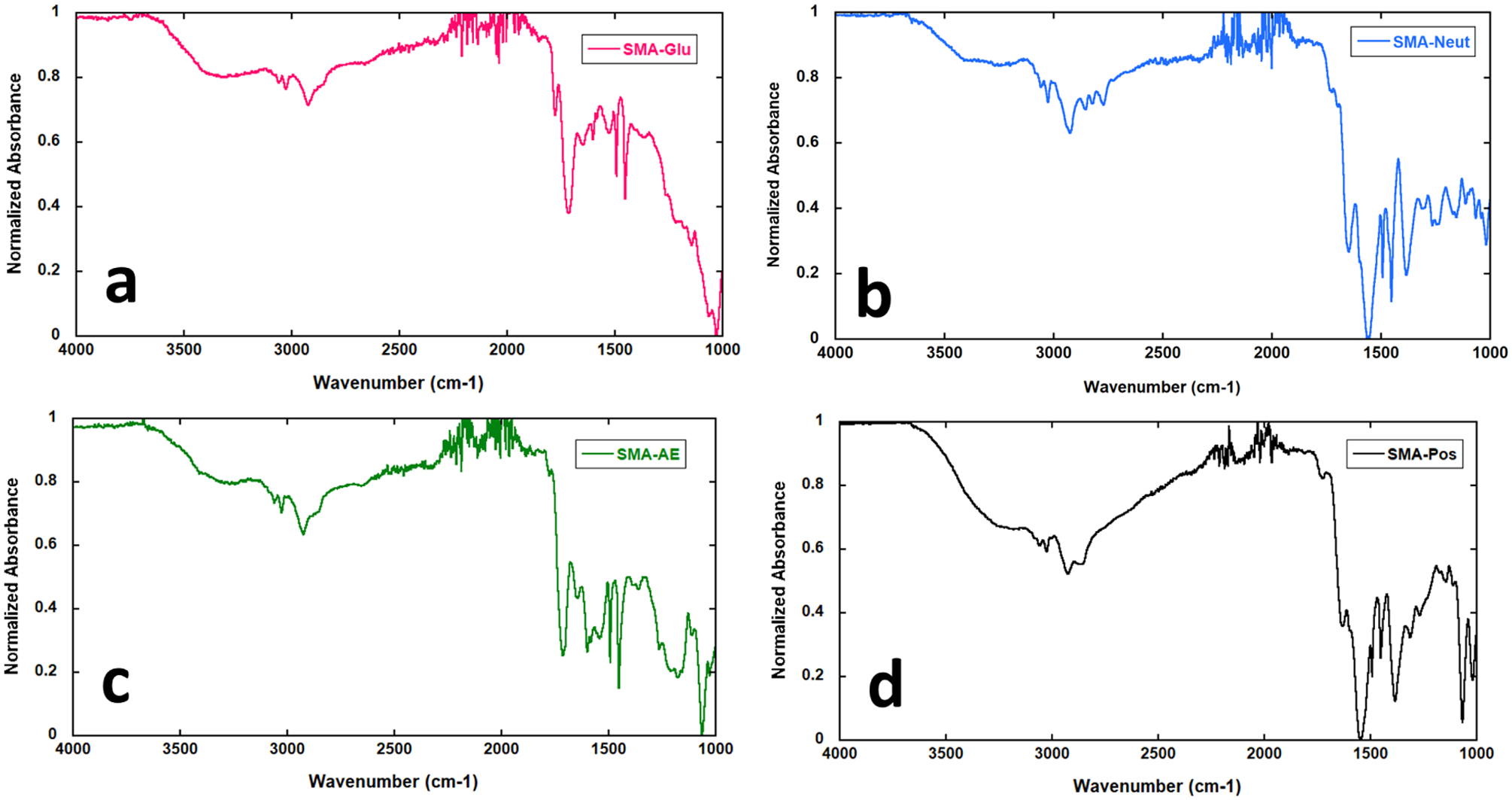
Infrared spectra of SMA derivatives. (a) 2–1M SMA-Glu; (b) 2–1M SMA-Neut; (c) 2–1M SMA-AE; and (d) 2–1M SMA-Pos.
Finally, SMA-AE seems to form a buffer region everywhere but the region expected for carboxylic acid, all the way from 10.5 to 8.5. This observation was reproduced even when the polymer was fully dissolved in distilled water (ca. 25 mg/L solubility). The presence of the amide and the hydroxyl of the new functional group must provide an extremely favorable geometry for a stabilizing hydrogen bond network. No buffer region could be observed below 2.5, but the polymer does precipitate below around 2.3, suggesting a change in protonation state, but unfortunately not one we can easily measure because of the contribution of bulk water. To our knowledge, this is the first reported pKa for an SMA of this type.
Infrared Spectroscopy of SMA Derivatives.
In all cases, it appears that complete conversion of the anhydride to the nucleophile-functionalized derivative has occurred (Figure 3). While it is possible that the weak peaks at 1778 and 1779 cm−1 (SMA-Glu and SMA-AE) correspond to unreacted anhydride, it is more likely that they correspond to contributions from intra- or intermolecular ester bond formation with the less reactive hydroxyl groups of the nucleophiles used. This seems especially plausible when considering how for SMA-Glu, the peak at 1779 cm−1 is more intense than the 1778 cm−1 peak in SMA-AE. This would be a result of the presence of five hydroxyl groups in glucosamine as opposed to just one in aminoethanol, resulting in more ester bonds formed when glucosamine is used.
As expected, SMA-Neut and SMA-Pos both display low wavenumber peaks for their carboxylic acids, which are deprotonated as a result of their preparation and/or the presence of basic amine groups in the polymers. They also display weak peaks in the typical carboxylic acid range, which we attribute to the protonated forms of carboxylic acids in the polymers, which may be present in a lower proportion.
Solution NMR Spectroscopy of sMa Derivatives.
Generally, NMR spectra of polymers can be difficult to assign and interpret, especially for polymers almost 10 kDa in size and with complex internal structure such as the alternation and block-like structure in SMA. Assignment is difficult because the size and complex structure of these derivatives cause peaks to appear as multiplets and also broaden out, frustrating meaningful peak integration. Generally, this broadening is most substantially closer to the backbone, which helped inform some of the peak assignments; sharper peaks were assumed to be further from the backbone where fast isotropic motion could more easily occur. Nevertheless, Tables S5–S8 give proposed assignments of NMR spectra of the SMA-AE, SMA-Glu, SMA-Neut, and SMA-Pos derivatives. NMR spectra are given in Figure 4.
Figure 4.

Solution NMR spectra of SMA derivatives. (a) 2–1M SMA-Glu; (b) 2–1M SMA-Neut; (c) 2–1M SMA-AE; and (d) 2–1M SMA-Pos.
Formation and Stability of Lipid Disks.
The performance of SMA polymers and their derivatives in forming nanoscale disk-like particles is a critical parameter to evaluate the impact of these derivatives. Vesicles of 25 mM POPC were titrated with ca. 2.5% (w/v) solutions of polymer up to a volume ratio of 1:1.5 (Figure 5a–e), which is a typical lipid/polymer ratio used in SMALP-like systems. These SMADLPs typically resulted in narrow particle diameter distributions shifted by a factor of 4–8, as measured by DLS. All polymers formed nanoparticles (SMADLPs) of sizes in good agreement with our previous work,18,29,31 where 2–1M SMA formed SMADLPs with an average particle diameter of 28 nm and 3–1M SMA formed SMADLPs with a particle diameter of 10 nm.18 The SMADLPs in this work are also likely to be formed as disk-like aggregates of lipid molecules, wrapped by the synthetic polymer, where the styrene units interact with lipid hydrophobic chains, and the polar maleic acid derivative is exposed to the aqueous phase.
Figure 5.
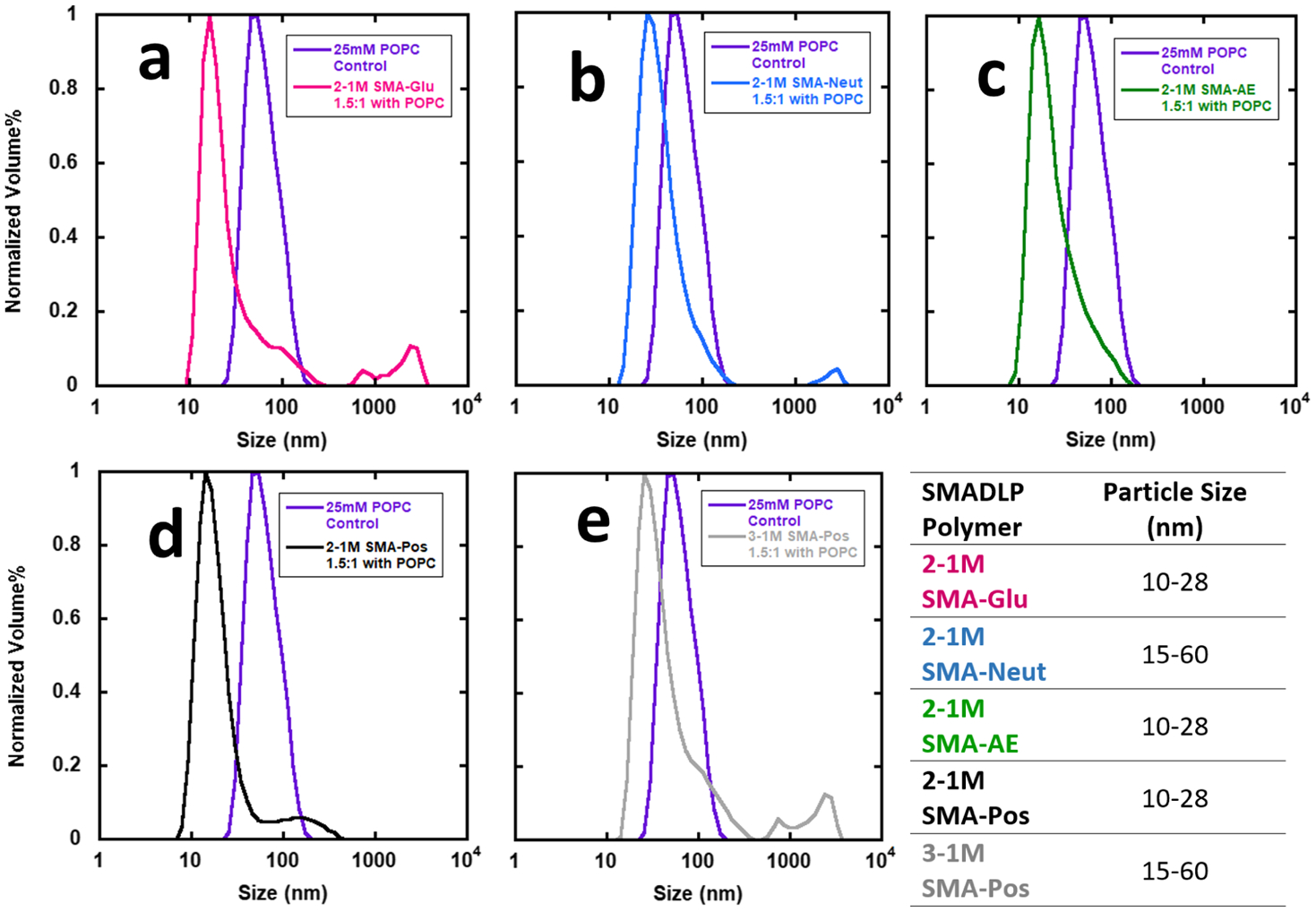
DLS volume plots of nanoparticles formed by SMA derivatives titrated in a 1.5:1 polymer/lipid ratio (v/v). (a) 2–1M SMA-Glu; (b) 2–1M SMA-Neut; (c) 2–1M SMA-AE; (d) 2–1M SMA-Pos; and (e) 3–1M SMA-Pos.
All five of the copolymers used were capable of forming SMADLPs, although their level of performance varies. 2–1M SMA-Glu, 2–1M SMA-Pos, and 3–1M SMA-Pos (5a,d,e) display high-diameter shoulders that appear to be unreacted POPC vesicles, and this is most pronounced for the SMA-Pos samples. This indicates that these materials may not be effective at solubilizing vesicles. SMA-Glu, SMA-Neut, and 3–1M SMA-Pos (5a,b,e) also display some very large particles near or above 1000 nm in diameter. These may be aggregates of many nanodisks, which can be seen in some of the transmission electron microscopy (TEM) images (Figure S1c). The samples were allowed to sediment for 24 h before data collection, so these particles must not have significantly higher density than the bulk solution, leading us to conclude that they are composed largely of lipids.
Finally, the SMADs used generate SMADLPs in two distinct size ranges. The polymers in the smaller size category (SMA-Glu, SMA-AE, 2–1M SMA-Pos) have characteristics associated with hard surfactants, whereas those in the larger size category (SMA-Neut and 3–1M SMA-Pos) have characteristics associated with soft surfactants. As discussed in the Introduction, the softness of a surfactant refers to its tendency to not disrupt native protein structure, with the caveat that a soft surfactant is not effective at solubilizing membranes. Having a zwitterionic or uncharged polar head group makes a surfactant softer than a head group with net charge. Despite the difference in scale, the same relationship seems to apply for SMADLPs because the zwitterionic SMA-Neut produced particles of larger size than the polymers with overall net charge. Having a longer acyl chain also makes for a softer surfactant. As seen in Figure 5e, 3–1M SMA-Pos produced significantly larger particles than 2–1M SMA-Pos (5d). The only chemical difference between these two polymers is that 3–1M SMA has a longer styrene tail; the alternating region should be of almost exactly the same structure between these two copolymers. The styrene tail may be acting to soften the surfactant character of 3–1M SMA-Pos, which hampered its ability to form nanoparticles.
The copolymer–lipid nanoparticles generated from the SMADs in this study are very robust against both pH and magnesium. SMALPs are known to be sensitive to pH values below 6 and sensitive to even modest concentrations of magnesium at or below 10 mM.32 Figure 6a shows the overall robust pH behavior of the SMADLPs. SMA-Pos is highly sensitive to pH 5 but tolerates pH 3 and 9. This behavior is predicted by the pKa experiment, where the cloudy SMA-Pos solution clarified as the pH was raised past 5.63. SMA-Glu nanoparticles begin to become cloudy at pH 3 but only slightly and tolerate the other pH values as well. SMA-AE and SMA-Neut are completely insensitive to all pH values tested. These results are in contrast to previously reported maleimide derivatives, which can be sensitive to high pH.
Figure 6.
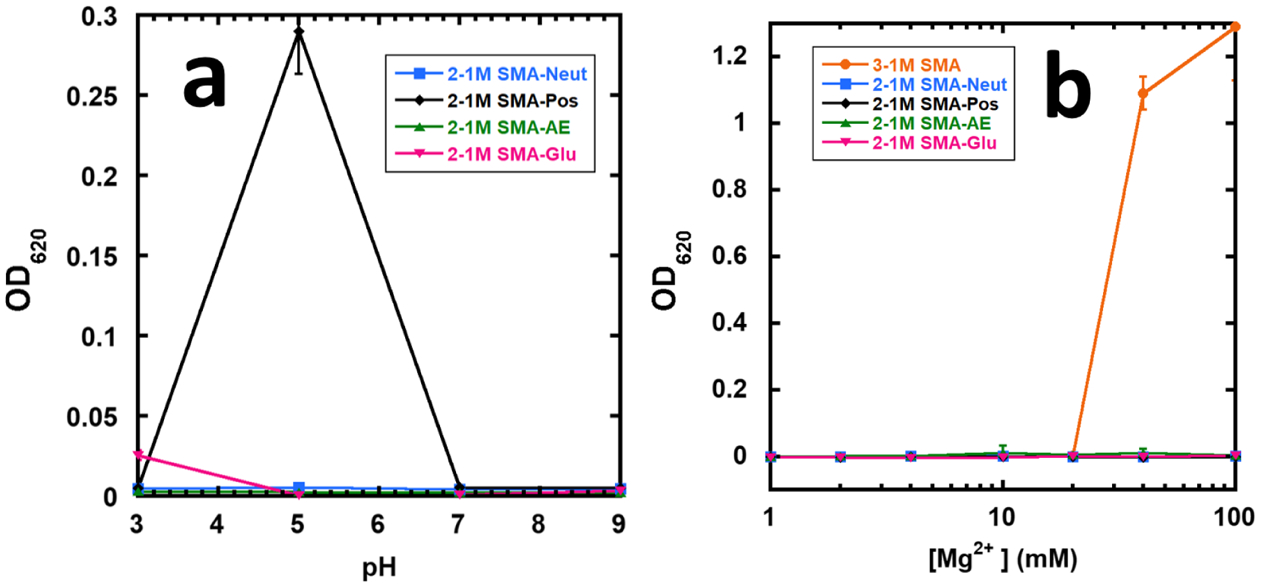
Stability of SMADLPs against (a) pH and (b) magnesium ions. SMADLPs and SMALPS were formed by combining SMAD or SMA and POPC at approximately a 7.5:1 weight ratio.
Figure 6b shows that the SMADLPs are completely insensitive to Mg2+. In our experiment, the synthesized SMA seems to be less sensitive to Mg than usual, but this is accounted for by the high polymer/lipid ratio used in this experiment (7.5:1), which was used to limit the size of the pellets seen in the SMADLP samples. In any case, a sharp increase in turbidity is observed for SMA from 20 to 40 mM, while the SMADLPs had no observable response to even 100 mM magnesium (Figure 6b), which is due to the loss of the strong chelating character of the diacids present in SMA.
Incorporation of KCNE1–58 into Nanodisks.
KCNE1 is a 129 amino acid, single transmembrane protein that assists in the proper functioning of several voltage-gated potassium ion channels.25,28,33 Mutations of genes that code for KCNE1 protein cause congenital deafness, congenital long QT syndrome, ventricular tachyarrhythmia, syncope, and sudden cardiac death.28,34,35
This study serves as a guide to help determine which SMADLP system to use based on the overall charge of the membrane protein of interest to reconstitute. In this study, human KCNE1 with a spin label in the transmembrane domain (introduced via a mutant cysteine residue, T58C) was incorporated into two different nanodisks; each SMADLP system contained polymer belts SMA-Glu and SMA-AE. CW-EPR lineshape analysis was used to compare T58C reconstituted in the two nanodisk systems to T58C reconstituted into POPC/POPG vesicles. The lineshape of the control is verified by comparing it to the literature that has already characterized this transmembrane protein in both lipid bilayers and SMALPs.36 Despite the ability for any SMA derivative to spontaneously form nanodisks when mixed with vesicles, a recent study has transformed our understanding of protein functionality when reconstituted into nanodisks; using a variety of biophysical techniques, this study confirmed protein dynamics and functionality are preserved when overall protein charge matches that of the polymer belt.37 Human KCNE1, a negatively charged protein at pH 7, was reconstituted into nanodisks with SMA-Glu and SMA-AE, both negatively charged polymers, to preserve a more native environment.
The lineshape of the reconstituted mutant KCNE1 into SMA-AE (Figure 7c) nanodisks shows significant amplification of the slow/rigid component of the spin label much similar to that of previous studies that use traditional SMA to reconstitute transmembrane proteins into nanodisks.29,36 Interestingly, reconstitution of the mutant KCNE1 into SMA-Glu nanodisks (Figure 7d) only slightly magnifies the slow/rigid component of the spin label and closely resembles the lineshape of the control (Figure 7b).
Figure 7.
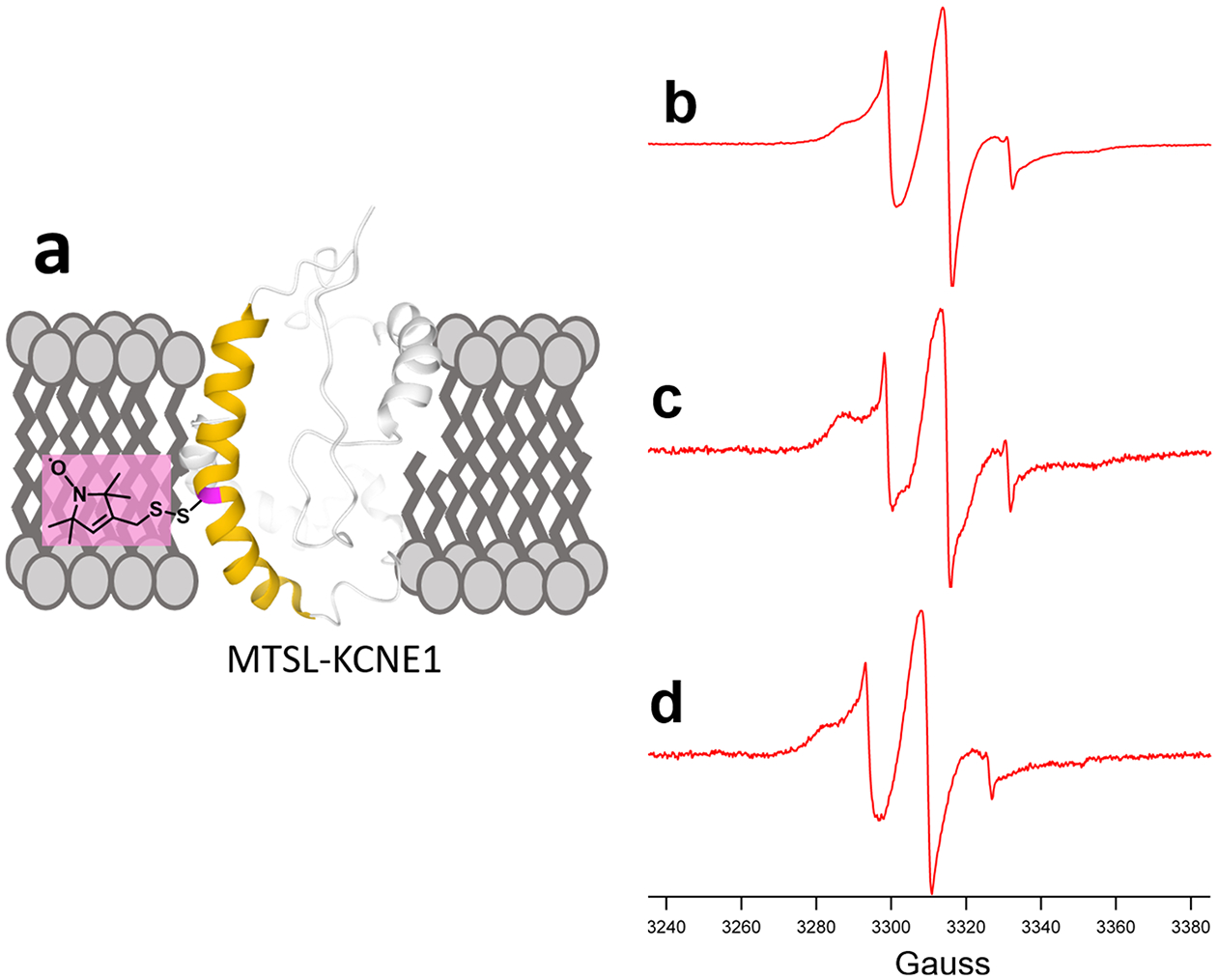
CW-EPR spectra of (a) MTSL spin-labeled KCNE1 T58C reconstituted in (b) POPC/POPG liposomes, (c) SMA-AE nanodisks, and (d) SMA-Glu nanodisks.
These differences in lineshapes are due to the relationship of the charge density of the polymer belt to the charge density of the membrane protein. Using SMA-AE (7c), the CW-EPR lineshape is most similar to lineshapes of similar mutants reconstituted into nanodisks with traditional SMA because of similarity in high charge density between the two polymers. SMA-Glu has a lower charge density than SMA-AE or SMA and displays a lineshape most similar to the control.
Human KCNE1 mutant T58C has a relatively low charge density because of the similar number of negatively charged residues (13) and positively charged residues (16, including 3 histidines), resulting in a calculated charge between −1 and 0 at pH 7. Not only has this study shown to preserve protein dynamics of KCNE1 using negatively charged polymer belts, but CW-EPR lineshape analysis reveals differences in protein dynamics based on the charge density of the polymer belt. Analysis of the lineshapes shows that SMA-Glu provides the more suitable membrane mimetic system for KCNE1.
CONCLUSIONS
Multiple derivatives of SMA were synthesized by taking advantage of the high reactivity of the maleic anhydride functionality of the parent polymer. The GPC data support preservation of the low dispersity of SMA-AE, SMA-Pos, and SMA-Neut. The infrared spectra show efficient conversion of the anhydride groups to give a functional polymer, suggesting complete functionalization for each derivative. It is noteworthy that essentially complete conversion of the anhydride occurs in each system, despite the rapid and mild conditions used in the functionalization step. Several SMA derivative polymers display pKa values far outside the range expected of carboxylic acids, especially SMA-Glu, which shows a weak buffering region near neutral pH. We attribute this behavior to the unique macromolecular structure of the copolymers. The NMR data are unique for each polymer, and despite the complexity, each proton in the intended products can be assigned, indicating successful reaction. All of the copolymers were capable of reacting with POPC vesicles to form SMADLPs of varying sizes, and there is some correlation between the net polymer charge and the sizes of particles formed. All the SMADLPs display robust physical characteristics, functioning at a wide pH range and up to 100 mM magnesium. Finally, we observed reconstitution of the transmembrane protein KCNE1 into SMADLPs by CW-EPR, finding that minimal perturbation of protein dynamics occurred when using SMA-Glu, which has the lowest charge density of the negatively charged polymers. This work highlights the potential of the styrene–maleic anhydride polymers toward the facile development of functional surfactants and will encourage the syntheses of materials with a wider range of uses than the current SMA standard.
Supplementary Material
Figure 2.
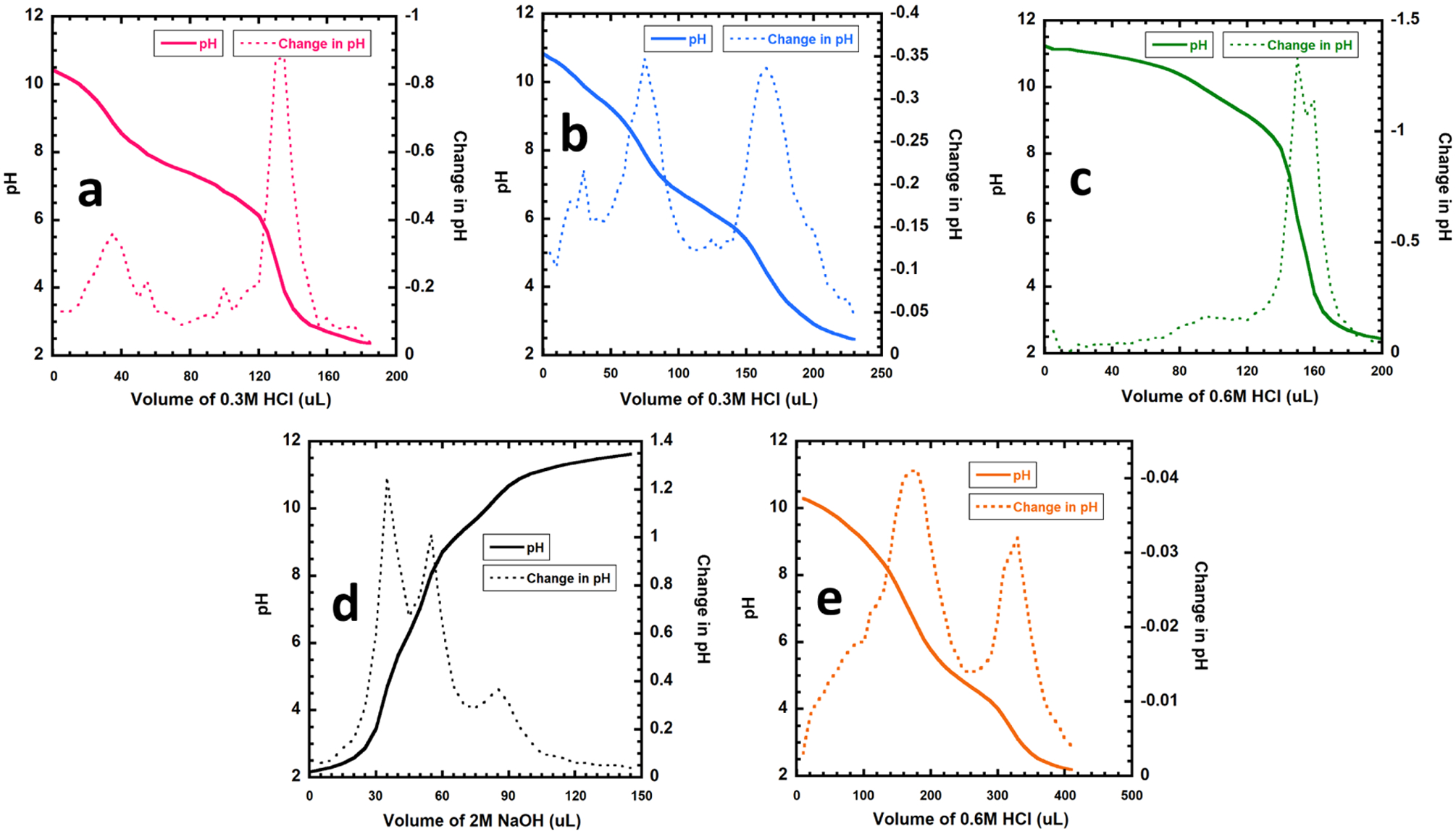
Titration curves of SMA derivatives. (a) 2–1M SMA-Glu; (b) 2–1M SMA-Neut; (c) 2–1M SMA-AE; (d) 2–1M SMA-Pos; and (e) 2–1M SMA for reference.
ACKNOWLEDGMENTS
This work was generously supported by NIGMS/NIH Maximizing Investigators’ Research Awards (MIRA) R35GM126935 (GAL) and R35GM128595 (RCP) and NSF grant CHE-1807131 (GAL), the Ohio Board of Regents, and Miami University. G.A.L. would also like to acknowledge support from the John W. Steube Professorship. D.K would also like to acknowledge support from the Robert H. and Nancy J. Blayney Professorship. D.K. and R.C.P. would also like to acknowledge support from The Army Research Office (Award# 74112-CH-II).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biomac.0c00041.
Peak assignments of the Fourier-transform infrared spectra and solution NMR spectra of the synthesized SMADs and representative TEM images of SMADLPs made from POPC liposomes (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.biomac.0c00041
The authors declare no competing financial interest.
Contributor Information
Kevin M. Burridge, Department of Chemistry and Biochemistry, Miami University of Oxford Ohio, Oxford, Ohio 45056, United States
Benjamin D. Harding, Department of Chemistry and Biochemistry, Miami University of Oxford Ohio, Oxford, Ohio 45056, United States
Indra D. Sahu, Department of Chemistry and Biochemistry, Miami University of Oxford Ohio, Oxford, Ohio 45056, United States; Natural Science Division, Campbellsville University, Campbellsville KY 42718, United States
Madison M. Kearns, Department of Chemistry and Biochemistry, Miami University of Oxford Ohio, Oxford, Ohio 45056, United States
Rebecca B. Stowe, Department of Chemistry and Biochemistry, Miami University of Oxford Ohio, Oxford, Ohio 45056, United States
Madison T. Dolan, Department of Chemistry and Biochemistry, Miami University of Oxford Ohio, Oxford, Ohio 45056, United States
Richard E. Edelmann, Center for Advanced Microscopy & Imaging, Miami University, Oxford, Ohio 45056, United States
Carole Dabney-Smith, Department of Chemistry and Biochemistry, Miami University of Oxford Ohio, Oxford, Ohio 45056, United States.
Richard C. Page, Department of Chemistry and Biochemistry, Miami University of Oxford Ohio, Oxford, Ohio 45056, United States
Dominik Konkolewicz, Department of Chemistry and Biochemistry, Miami University of Oxford Ohio, Oxford, Ohio 45056, United States.
Gary A. Lorigan, Department of Chemistry and Biochemistry, Miami University of Oxford Ohio, Oxford, Ohio 45056, United States.
REFERENCES
- (1).Bull SC; Doig AJ Properties of Protein Drug Target Classes. PLoS One 2015, 10, No. e0117955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Yin H; Flynn AD Drugging Membrane Protein Interactions. Annu. Rev. Biomed. Eng 2016, 18, 51–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Overington JP; Al-Lazikani B; Hopkins AL How Many Drug Targets Are There? Nat. Rev. Drug Discovery 2006, 5, 993–996. [DOI] [PubMed] [Google Scholar]
- (4).Sachs JN; Engelman DM Introduction to the Membrane Protein Reviews: The Interplay of Structure, Dynamics, and Environment in Membrane Protein Function. Annu. Rev. Biochem 2006, 75, 707–712. [DOI] [PubMed] [Google Scholar]
- (5).Tate CG Practical Considerations of Membrane Protein Instability during Purification and Crystallisation. Methods Mol. Biol 2010, 601, 187–203. [DOI] [PubMed] [Google Scholar]
- (6).Hardy D; Desuzinges Mandon E; Rothnie AJ; Jawhari A The Yin and Yang of Solubilization and Stabilization for Wild-Type and Full-Length Membrane Protein. Methods 2018, 147, 118–125. [DOI] [PubMed] [Google Scholar]
- (7).Ujwal R; Bowie JU Crystallizing Membrane Proteins Using Lipidic Bicelles. Methods 2011, 55, 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Dürr UHN; Soong R; Ramamoorthy A When Detergent Meets Bilayer: Birth and Coming of Age of Lipid Bicelles. Prog. Nucl. Magn. Reson. Spectrosc 2013, 69, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Raschle T; Hiller S; Etzkorn M; Wagner G Nonmicellar Systems for Solution NMR Spectroscopy of Membrane Proteins. Curr. Opin. Struct. Biol 2010, 20, 471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Shen H-H; Lithgow T; Martin L Reconstitution of Membrane Proteins into Model Membranes: Seeking Better Ways to Retain Protein Activities. Int. J. Mol. Sci 2013, 14, 1589–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bayburt TH; Grinkova YV; Sligar SG Self-Assembly of Discoidal Phospholipid Bilayer Nanoparticles with Membrane Scaffold Proteins. Nano Lett. 2002, 2, 853–856. [Google Scholar]
- (12).Larsen AN; Sørensen KK; Johansen NT; Martel A; Kirkensgaard JJK; Jensen KJ; Arleth L; Midtgaard SR Dimeric Peptides with Three Different Linkers Self-Assemble with Phospholipids to Form Peptide Nanodiscs That Stabilize Membrane Proteins. Soft Matter 2016, 12, 5937–5949. [DOI] [PubMed] [Google Scholar]
- (13).Kondo H; Ikeda K; Nakano M Formation of Size-Controlled, Denaturation-Resistant Lipid Nanodiscs by an Amphi-philic Self-Polymerizing Peptide. Colloids Surf., B 2016, 146, 423–430. [DOI] [PubMed] [Google Scholar]
- (14).Bayburt TH; Sligar SG Membrane Protein Assembly into Nanodiscs. FEBS Lett. 2010, 584, 1721–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Burridge KM; Wright TA; Page RC; Konkolewicz D Photochemistry for Well-Defined Polymers in Aqueous Media: From Fundamentals to Polymer Nanoparticles to Bioconjugates. Macromol Rapid Commun. 2018, 39, 1800093–1800114. [DOI] [PubMed] [Google Scholar]
- (16).Xue M; Cheng L; Faustino I; Guo W; Marrink SJ Molecular Mechanism of Lipid Nanodisk Formation by Styrene-Maleic Acid Copolymers. Biophys. J 2018, 115, 494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zhang R; Sahu ID; Bali AP; Dabney-Smith C; Lorigan GA Characterization of the Structure of Lipodisq Nanoparticles in the Presence of KCNE1 by Dynamic Light Scattering and Transmission Electron Microscopy. Chem. Phys. Lipids 2017, 203, 19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Craig AF; Clark EE; Sahu ID; Zhang R; Frantz ND; Al-Abdul-Wahid MS; Dabney-Smith C; Konkolewicz D; Lorigan GA Tuning the Size of Styrene–maleic Acid Copolymer-Lipid Nanoparticles (SMALPs) Using RAFT Polymerization for Biophysical Studies. Biochim. Biophys. Acta, Biomembr 2016, 1858, 2931–2939. [DOI] [PubMed] [Google Scholar]
- (19).Dörr JM; Scheidelaar S; Koorengevel MC; Dominguez JJ; Schäfer M; van Walree CA; Killian JA The Styrene–maleic acid copolymer: a versatile tool in membrane research. Eur. Biophys. J 2016, 45, 3–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Fiori MC; Jiang Y; Altenberg GA; Liang H Polymer-Encased Nanodiscs with Improved Buffer Compatibility. Sci. Rep 2017, 7, 7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ravula T; Hardin NZ; Ramadugu SK; Cox SJ; Ramamoorthy A Formation of PH-Resistant Monodispersed Polymer-Lipid Nanodiscs. Angew. Chem., Int. Ed 2018, 57, 1342–1345. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- (22).Ravula T; Hardin NZ; Ramadugu SK; Ramamoorthy A PH Tunable and Divalent Metal Ion Tolerant Polymer Lipid Nanodiscs. Langmuir 2017, 33, 10655–10662. [DOI] [PubMed] [Google Scholar]
- (23).Ravula T; Ramadugu SK; Di Mauro G; Ramamoorthy A Bioinspired, Size-Tunable Self-Assembly of Polymer-Lipid Bilayer Nanodiscs. Angew. Chem 2017, 129, 11624–11628. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- (24).Newstead S; Ferrandon S; Iwata S Rationalizing α-helical membrane protein crystallization. Protein Sci. 2008, 17, 466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Coey AT; Sahu ID; Gunasekera TS; Troxel KR; Hawn JM; Swartz MS; Wickenheiser MR; Reid R.-j; Welch RC; Vanoye CG; Kang C; Sanders CR; Lorigan GA Reconstitution of KCNE1 into Lipid Bilayers: Comparing the Structural, Dynamic, and Activity Differences in Micelle and Vesicle Environments. Biochemistry 2011, 50, 10851–10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Gennis RB Biomembranes : Molecular Structure and Function; Springer: New York, 1989. [Google Scholar]
- (27).Hall SCL; Tognoloni C; Charlton J; Bragginton ÉC; Rothnie AJ; Sridhar P; Wheatley M; Knowles TJ; Arnold T; Edler KJ; Dafforn TR An Acid-Compatible Co-Polymer for the Solubilization of Membranes and Proteins into Lipid Bilayer-Containing Nanoparticles. Nanoscale 2018, 10, 10609–10619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Tian C; Vanoye CG; Kang C; Welch RC; Kim HJ; George AL; Sanders CR Preparation, Functional Characterization, and NMR Studies of Human KCNE1, a Voltage-Gated Potassium Channel Accessory Subunit Associated with Deafness and Long QT Syndrome. Biochemistry 2007, 46, 11459–11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Harding BD; Dixit G; Burridge KM; Sahu ID; Dabney-Smith C; Edelmann RE; Konkolewicz D; Lorigan GA Characterizing the Structure of Styrene–maleic Acid Copolymer-Lipid Nanoparticles (SMALPs) Using RAFT Polymerization for Membrane Protein Spectroscopic Studies. Chem. Phys. Lipids 2019, 218, 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Koper GJM; Borkovec M Proton Binding by Linear, Branched, and Hyperbranched Polyelectrolytes. Polymer 2010, 51, 5649–5662. [Google Scholar]
- (31).Bali AP; Sahu ID; Craig AF; Clark EE; Burridge KM; Dolan MT; Dabney-Smith C; Konkolewicz D; Lorigan GA Structural Characterization of Styrene–maleic Acid Copolymer-Lipid Nanoparticles (SMALPs) Using EPR Spectroscopy. Chem. Phys. Lipids 2019, 220, 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Dörr JM; Scheidelaar S; Koorengevel MC; Dominguez JJ; Schäfer M; van Walree CA; Killian JA The Styrene–maleic acid copolymer: a versatile tool in membrane research. Eur. Biophys. J 2016, 45, 3–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kang Q; Zhao Z-A; You S-L Highly Enantioselective Friedel–Crafts Reaction of Indoles with Imines by a Chiral Phosphoric Acid. J. Am. Chem. Soc 2007, 129, 1484–1485. [DOI] [PubMed] [Google Scholar]
- (34).Wang Z; Fermini B; Nattel S Rapid and Slow Components of Delayed Rectifier Current in Human Atrial Myocytes. Cardiovasc. Res 1994, 28, 1540–1546. [DOI] [PubMed] [Google Scholar]
- (35).Harmer SC; Tinker A The Role of Abnormal Trafficking of KCNE1 in Long QT Syndrome 5. Biochem. Soc. Trans 2007, 35, 1074–1076. [DOI] [PubMed] [Google Scholar]
- (36).Sahu ID; Zhang R; Dunagan MM; Craig AF; Lorigan GA Characterization of KCNE1 inside Lipodisq Nanoparticles for EPR Spectroscopic Studies of Membrane Proteins. J. Phys. Chem. B 2017, 121, 5312–5321. [DOI] [PubMed] [Google Scholar]
- (37).Ravula T; Hardin NZ; Bai J; Im S-C; Waskell L; Ramamoorthy A Effect of Polymer Charge on Functional Reconstitution of Membrane Proteins in Polymer Nanodiscs. Chem. Commun 2018, 54, 9615–9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.