

Abstract
Rationale:
Angiogenesis promotes neurological recovery after stroke and is associated with longer survival of stroke patients. Cerebral angiogenesis is tightly controlled by certain microRNAs (miRs), such as the miR-15a/16–1 cluster, among others. However, the function of the miR-15a/16–1 cluster in endothelium on post-ischemic cerebral angiogenesis is not known.
Objective:
To investigate the functional significance and molecular mechanism of endothelial miR-15a/16–1 cluster on angiogenesis in the ischemic brain.
Methods and Results:
Endothelial cell -selective miR-15a/16–1 conditional knockout (EC-miR-15a/16–1 cKO) mice and WT littermate controls were subjected to 1h middle cerebral artery occlusion (MCAO) followed by 28d reperfusion. Deletion of miR-15a/16–1 cluster in endothelium attenuates post-stroke brain infarction and atrophy, and improves the long-term sensorimotor and cognitive recovery against ischemic stroke. Endothelium-targeted deletion of the miR-15a/16–1 cluster also enhances post-stroke angiogenesis by promoting vascular remodeling and stimulating the generation of newly formed functional vessels, and increases the ipsilateral cerebral blood flow. Endothelial cell-selective deletion of the miR-15a/16–1 cluster up-regulated the protein expression of pro-angiogenic factors vascular endothelial growth factor (VEGFA), fibroblast growth factor 2 (FGF2), and their receptors VEGFR2 and FGFR1 after ischemic stroke. Consistently, lentiviral knockdown of the miR-15a/16–1 cluster in primary mouse or human brain microvascular endothelial cell cultures enhanced in vitro angiogenesis and up-regulated pro-angiogenic proteins expression after oxygen-glucose deprivation (OGD), whereas lentiviral overexpression of the miR-15a/16–1 cluster suppressed in vitro angiogenesis and down-regulated pro-angiogenic proteins expression. Mechanistically, miR-15a/16–1 translationally represses pro-angiogenic factors VEGFA, FGF2, and their receptors VEGFR2 and FGFR1, respectively, by directly binding to the complementary sequences within three prime untranslated regions (3’-UTRs) of those mRNAs.
Conclusions:
Endothelial miR-15a/16–1 cluster is a negative regulator for post-ischemic cerebral angiogenesis and long-term neurological recovery. Inhibition of miR-15a/16–1 function in cerebrovascular endothelium may be a legitimate therapeutic approach for stroke recovery.
Keywords: Cerebrovascular endothelial cell, microRNAs, miR-15a/16–1, ischemic stroke, angiogenesis
Subject Terms: Angiogenesis, Basic Science Research, Gene Expression and Regulation, Ischemic Stroke, Vascular Biology
Angiogenesis refers to the growth of new blood vessels from pre-existing vessels. Post-stroke cerebral angiogenesis contributes to improved neurological recovery by promoting brain tissue repair, vascular remodeling, and plasticity. Cerebral angiogenesis is tightly controlled by certain miRs, such as the miR-15a/16–1 cluster. However, the functional significance of the miR-15a/16–1 cluster in endothelium on post-ischemic cerebral angiogenesis is yet to be defined. This study uncovered a novel role of the endothelial-specific miR-15a/16–1 cluster in regulating post-stoke angiogenesis in mouse brains following transient cerebral ischemia. We demonstrated for the first time that endothelium-targeted deletion of the miR-15a/16–1 cluster improves post-stroke sensorimotor and cognitive recovery and enhances post-stroke cerebral angiogenesis by promoting the generation of newly formed functional vessels. MiR-15a/16–1 inhibits post-stroke cerebral angiogenesis by repressing the protein expression of key pro-angiogenic factors VEGFA, FGF2, and their receptors VEGFR2 and FGFR1, respectively, through directly binding to the complementary sequences of three prime untranslated regions (3’-UTR) in their mRNAs. The study provides a solid theoretical basis for the future development of miR-15a/16–1-based and endothelium-targeted restorative therapeutics for ischemic stroke.
Graphical Abstract
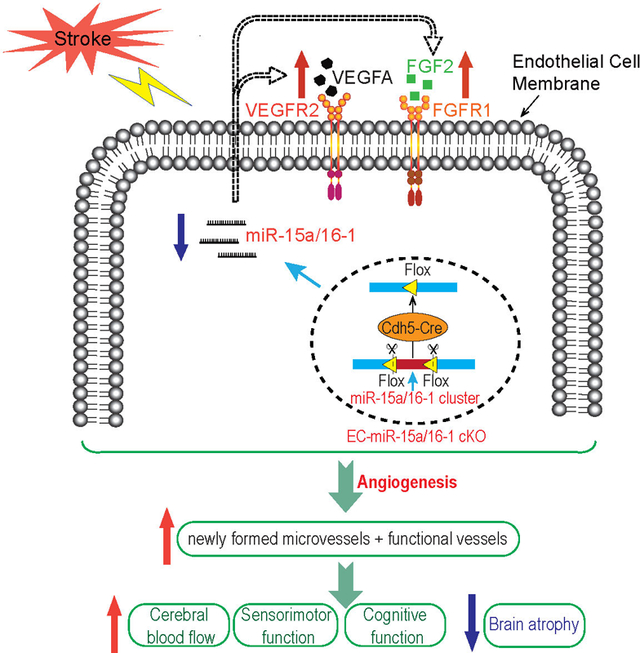
INTRODUCTION
Ischemic stroke accounts for ~87% of all strokes in the United States. Although clinical treatments by means of the tissue plasminogen activator (tPA)-mediated thrombolysis and endovascular thrombectomy have significantly improved options within the acute time window of ischemic attack1, therapeutic approaches for improving long-term post-stroke recovery remains limited2. Angiogenesis is a biological process referring to the growth of new blood vessels from pre-existing vessels3. Although angiogenesis is strictly controlled under physiological conditions in adult brains, studies from human and experimental stroke models indicate that neovascularization is present in the adult brains after cerebral ischemia4, 5. Cumulative evidence has indicated that post-ischemic angiogenesis plays a crucial role in the recovery of blood flow and neuronal metabolism after stroke6–8. Moreover, the increased microvessel density in the penumbral areas in stroke patients has been associated with longer post-stroke survival9, suggesting that the promotion of post-ischemic angiogenesis may become a promising therapeutic strategy for the restorative treatment of ischemic stroke.
MicroRNAs (miRs) function as a novel class of endogenous non-coding small (~21–25 nucleotide) RNA molecules that negatively modulate gene translation by hybridizing to three prime untranslated regions (3’-UTR) of one or more messenger RNAs (mRNAs) in a sequence-specific manner10. MiRs play an essential role in almost all cellular processes, including cell proliferation, differentiation, metabolism, and immune responses, under both physiological as well as pathological conditions11, 12. MiR-based therapeutics involve a wide range of mechanisms, including anti-oxidative stress, anti-inflammation, anti-apoptosis, anti-neurodegeneration, blood-brain barrier (BBB) protection, pro-angiogenesis, neuronal and axonal regeneration, among other tissue remodeling13. As an example, a miR-107 mimic reduced ischemic brain infarction and increased the number of capillaries in the penumbral area presumably by enhancing endothelial VEGF165/164 levels to promote angiogenesis14. However, due to the wide and potent tissue distribution of miRs, delineating cell-specific targets and their effects has been challenging, particularly in complex tissue environments such as the brain.
MiR-15a and miR-16–1 are two highly conserved miRs located in a cluster 250 base pairs (bp) apart in human chromosome 13q14 and 54 bp apart in mouse chromosome 1415. They can form a structural and functional cluster (the miR-15a/16–1 cluster) as they can similarly bind to their common mRNA targets16, 17. Dysregulated miR-15a and miR-16–1 levels in stroke patients have been proposed as useful diagnostic and prognostic biomarkers18. Previous research reported that vascular endothelial cell-selective miR-15a/16–1 transgenic overexpression could significantly suppress hindlimb ischemia-induced cell-autonomous angiogenesis in peripheral circulation19, suggesting that miR15a/16–1 may be an attractive target to inhibit for improved angiogenesis in tissue recovery. However, whether suppression of the miR-15a/16–1 cluster can promote vascular regeneration in brain and aid in long term functional restoration after stroke is unknown.
Endothelial cells (ECs) play a prominent role in vascular remodeling and angiogenesis20, but the role of miR-15a/16–1 and potential mechanism in ECs remains unknown. In this study, we generated an endothelium-targeted miR-15a/16–1 conditional knockout (EC-miR-15a/16–1 cKO) mouse model to investigate the long-term effects and potential molecular mechanisms of endothelial miR-15a/16–1 cluster on cerebral ischemia-induced angiogenesis. The results suggest that EC-targeted deletion of the miR-15a/16–1 cluster enhances angiogenesis in the penumbral areas and improves long-term neurological outcomes after cerebral ischemia. Moreover, we demonstrate that VEGFA, VEGF receptor 2 (VEGFR2), FGF2 and FGF receptor 1 (FGFR1) are direct downstream targets of miR-15a/16–1 translational repression. Loss-of-miR-15a/16–1 function in vascular endothelium contributes to post-stroke angiogenesis and overall long-term neurological recovery.
METHODS
All data that support the findings of this study are available within the article and its online supplementary files, or from the corresponding author upon reasonable request.
C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, Marine, USA). Endothelium-targeted miR-15a/16–1 conditional knockout (EC-miR-15a/16–1 cKO) mice were generated on C57BL/6J background, by crossing miR-15a/16–1flox/flox mice (a generous gift from Dr. Riccardo Dallla-Favera at Columbia University)17 with VE-Cadherin Cre mice21. Generally, EC-miR-15a/16–1 cKO mice were viable and fertile with normal appearance, behavior, growth, and litter size. All procedures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All stroke experiments follow the Stroke Therapy Academic Industry Roundtable (STAIR) guidelines22. Animals were randomly assigned to various experimental groups using a lottery box. All stroke outcomes assessments were performed in a blinded manner. All efforts were made to minimize animal suffering and the number of experimental animals. For detailed material and methods, please refer to the Major Resources Table and Supplemental Materials.
RESULTS
Pathological expression of cerebrovascular miR-15a/16–1 and pro-angiogenic factors after ischemic insults.
We first measured miR-15a and miR-16–1 expression in primary mBMEC cultures subjected to 16 h OGD and 4–24 h reoxygenation (Reox). qPCR data revealed that OGD alone or in combination with Reox increased with statistical significance the expression of miR-15a (Online Figure IA) and miR-16–1 (Online Figure IB) at comparable levels. We then analyzed the expression of miR-15a and miR-16–1 in microvessels isolated from ischemic mouse brains following MCAO. MiR-15a expression remained elevated with statistical significance over 28 d after MCAO compared with sham group, peaking 3–14 d following ischemia (Online Figure IC). Similarly, miR-16–1 expression was statistically significantly up-regulated over 14 d following MCAO compared to sham controls (Online Figure ID), but peaked earlier (3 d following ischemia) than miR-15a. To compare the timecourse of miR-15a/16–1 expression to induction of angiogenesis, we analyzed the mRNA expression of several proangiogenic factors in isolated cerebral microvessels following MCAO. Stroke induced a statistically significant increase of VEGFA expression 1–3 d following MCAO. A statistically significant decline occurred at 7 d following stroke compared with 3 d group, but VEGFA expression remained statistically significantly higher than the sham group, and gradually increased until 28 d after stroke (Online Figure IE). Additionally, qPCR data revealed a statistically significant decreased expression of VEGFR2, FGF2, and FGFR1 (Online Figure IF–H), with lowest expression levels at 7 d following MCAO. Interestingly, miR-15a and miR-16–1 exhibited the highest expression levels around 7 d following MCAO, suggesting, although not proving, a negative correlation between the miR-15a/16–1 cluster and these angiogenic factors after ischemic insults, similar to that observed in myeloma cells by Li et al.23
Generation and identification of EC-targeted miR-15a/16–1 conditional knockout mice.
To determine the functional role of endothelial-specific miR-15a/16–1 cluster in regulating post-stroke angiogenesis in vivo, we generated the EC-miR-15a/16–1 cKO mice (Fig. 1A, lane 5), by cross-mating miR-15a/16–1flox/flox mice17 with VE-Cadherin Cre mice21. These mice had both a VE-Cadherin Cre band (933 bp) and a miR-15a/16–1flox/flox band (650 bp). The flox/flox mice (lane 1 and 4) that only had a miR-15a/16–1flox/flox band (650 bp) were used as the wild type littermate controls (EC-miR-15a/16–1 WT). Mice that showed a WT band (558 bp) were either VE-Cadherin Cre TG mice or miR-15a/16–1flox heterozygous (Ht) mice, which were not included in this study. As shown (Fig. 1B), qPCR products of miR-15a and miR-16–1 were almost undetectable from the cerebral microvessels from EC-miR-15a/16–1 cKO mice. Of note, there were no statistically significant changes in vascular miR-497 expression in the cKO mice (Fig. 1B), indicating the specificity of endothelial miR-15a/16–1 deletion. We also examined the histological structure of brain microvasculature in the cerebral cortex and striatum using CD31 immunostaining (Fig. 1C). In non-ischemic brains, no statistically significant difference was observed in the microvasculature profiles between EC-miR-15a/16–1 cKO and WT mice, including surface area (Fig. 1D), vascular length (Fig. 1E), branch points (Fig. 1F), and capillary number (Fig. 1G). We further examined the microvasculature in hindlimb gastrocnemius muscles in sham-operated mice using CD31 immunostaining (Online Figure II), and found no statistically significant difference between two genotypes regarding branch points (Online Figure IIB), capillary number (Online Figure IIC) and vascular length (Online Figure IID). These results suggest that endothelium-targeted deletion of the miR-15a/16–1 cluster does not affect in a statistically significant manner the anatomical structure of cerebral or peripheral microvessels under physiological conditions, and EC-miR-15a/16–1 cKO mice are suitable for studying the role of endothelial-specific miR-15a/16–1 cluster in cerebral ischemia in vivo.
Fig. 1. Generation and identification of EC-miR-15a/16–1 cKO mice.
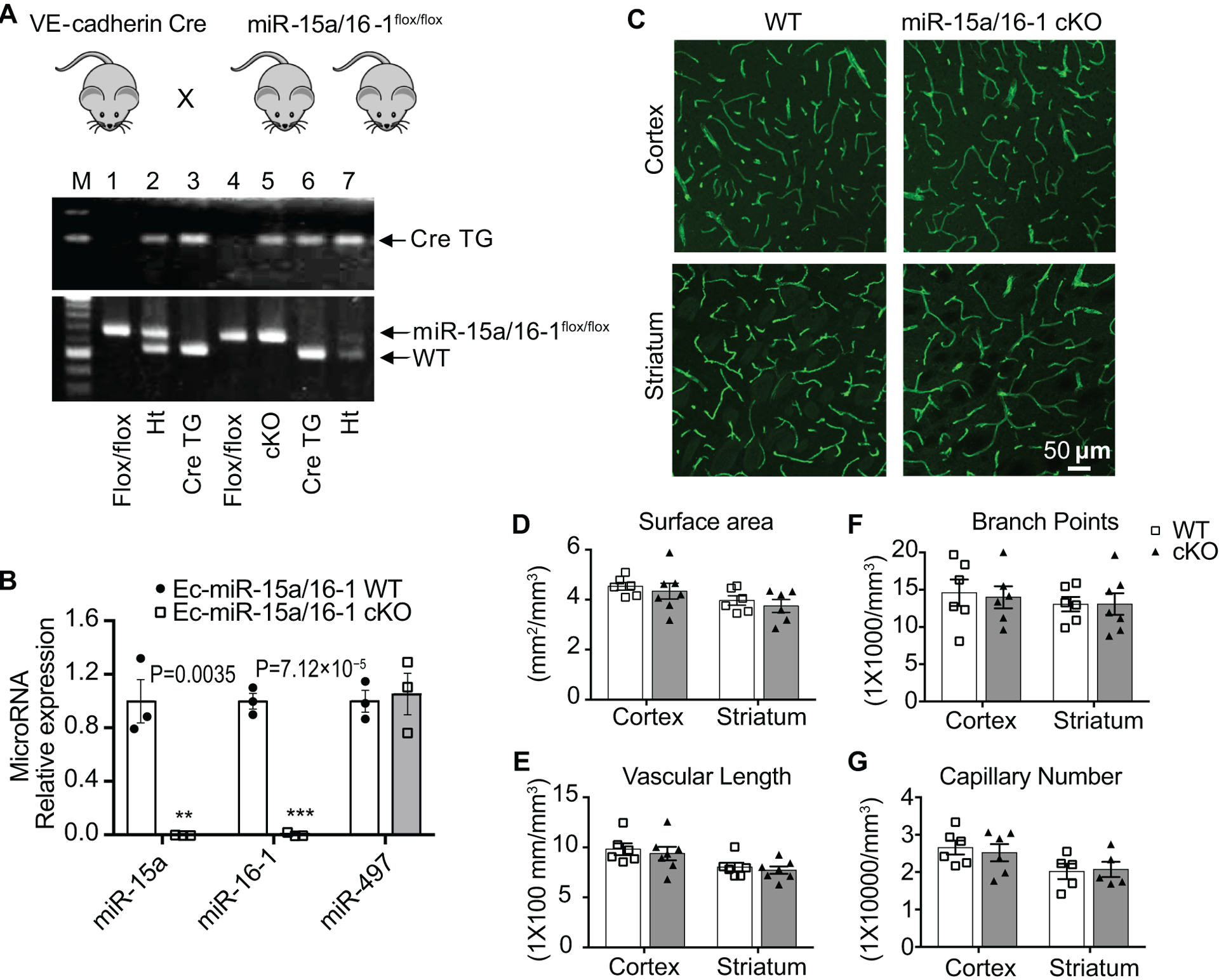
A, PCR genotyping showed that EC-miR-15a/16–1 cKO (lane 5) and WT littermate control (lane 1 and 4) mice were generated by crossing VE-Cadherin-Cre transgenic mice with miR-15a/16–1flox/flox mice. Heterozygotes (Ht) which contain both miR-15a/16–1 WT and miR-15a/16–1flox bands were shown as lane 2 and 7. B, Total RNA was extracted from the isolated cerebral microvasculature of EC-miR-15a/16–1 WT and cKO mice. qPCR data revealed almost no expression of miR-15a and miR-16–1 in the isolated cerebral microvasculature from EC-miR-15a/16–1 cKO mice compared with WT controls. n = 3; **p < 0.01, ***p < 0.001 versus EC-miR-15a/16–1 WT group; statistical analyses were performed by two-tailed Student’s t-test. C-G, Cerebral microvasculature was examined and quantified in the cerebral cortex and striatum of EC-miR-15a/16–1 cKO and WT mice by immunostaining for CD31. Representative CD31 fluorescent images (C), the quantification of vascular surface area (D), vascular length (E), branch points (F) and capillary number (G) showing no statistically significant difference in the microvascular anatomy between WT and EC-miR-15a/16–1 cKO animals. n = 5–7 for each group; statistical analyses were performed by two-tailed Student’s t-test.
Endothelium-targeted miR-15a/16–1 deletion improves sensorimotor and cognitive functions, and attenuates brain infarction and atrophy after cerebral ischemia.
We investigated whether EC-targeted miR-15a/16–1 deletion in mice affects long-term neurological outcomes after stroke. We applied a battery of behavioral tests to comprehensively evaluate the sensorimotor functions before and up to 28 d after MCAO (Online Figure III and Fig. 2A–D). Compared to sham groups, both WT and EC-miR-15a/16–1 cKO groups exhibited significant sensorimotor functional deficits in all the behavioral tests (foot fault, rota-rod, and adhesive tape removal tests), and these sensorimotor deficits were most prominent in the first week after MCAO. Interesting, the EC-miR-15a/16–1 cKO mice showed statistically significant less sensorimotor deficits and better neurological recovery than WT controls in all the three behavioral tests at most time points, as evidenced by statistically significant fewer foot faults (3–28 d after MCAO), longer time on the rota-rod (3–21 d after MCAO), and quicker response time to touch (3–14 d after MCAO) and to remove (3–10 d after MCAO) the adhesive tape from the contralateral forepaw (Fig. 2A–D).
Fig. 2. Endothelium-targeted miR-15a/16–1 deletion improves sensorimotor and cognitive outcomes, and attenuates brain infarction and atrophy after cerebral ischemia.
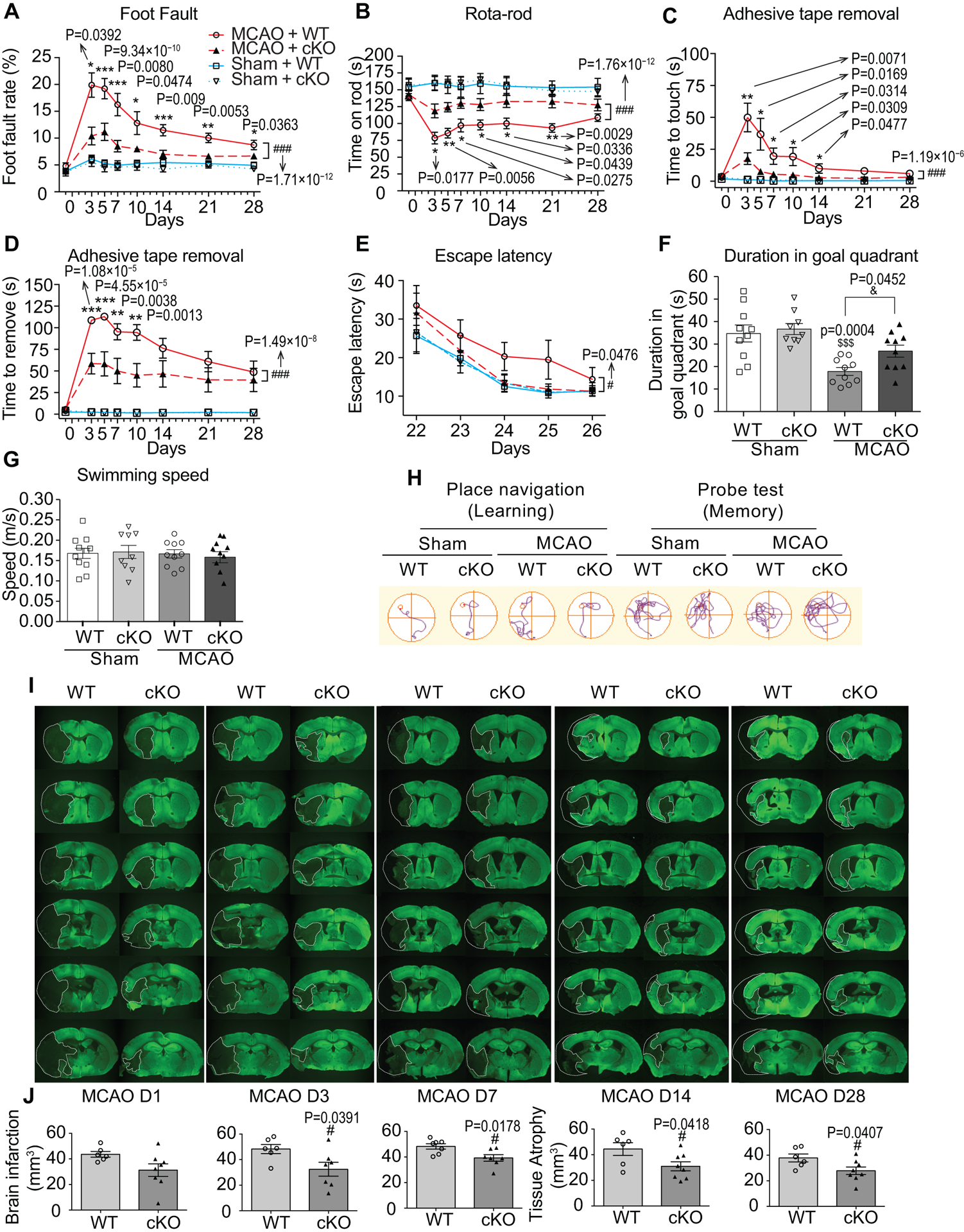
A-D, Sensorimotor deficits were assessed 1 d before and up to 28 d after MCAO by foot fault test (A), rota-rod test (B), and adhesive tape removal test (C, time to touch the tape and D, time to remove the tape). EC-miR-15a/16–1 cKO mice exhibited statistically significantly less sensorimotor deficits and better neurological recovery than WT controls after MCAO. n = 13–14 for MCAO + WT and MCAO + cKO groups; n = 10 for Sham + WT and Sham + cKO groups. *p < 0.05, **p < 0.01, ***p < 0.001 between MCAO + WT and MCAO + cKO groups by one-way ANOVA followed by Tukey’s multiple comparison tests (individual time point); ###p < 0.001 between MCAO + WT and MCAO + cKO groups by two-way ANOVA followed by Bonferroni’s multiple comparison tests (bracket); Kruskal-Wallis test followed by Dunn’s post hoc analysis was performed for Foot Fault test at 3 d time point in A. E-H, Long-term cognitive functions were assessed by the Morris water maze, showing that EC-miR-15a/16–1 cKO mice exhibited better learning and memory abilities than WT controls after ischemic stroke. E, The time for the animals to locate the submerged platform (escape latency) was measured at 22–26 d after MCAO. F, Spatial memory was evaluated at 27 d after MCAO by measuring the time spent in the goal quadrant when the platform was removed. G, Gross locomotor functions was measured by average swim speed. H, Representative swim path for different experimental groups. n = 10 for MCAO + WT and MCAO + cKO groups; n = 9–10 for Sham + WT and Sham + cKO groups. #p < 0.05 between MCAO + WT and MCAO + cKO groups by two-way ANOVA followed by Bonferroni’s multiple comparison tests (bracket). $$$p < 0.001 versus Sham + WT group, &p < 0.05 as indicated by one-way ANOVA followed by Tukey’s multiple comparison tests. I, J, brain infarction and atrophy were also measured and quantified by using MAP2 (green) immunostaining at 1–28 d reperfusion following MCAO. Dashed lines outline the brain infarction or atrophy. Representative MAP2 immunofluorescent images (I), and quantitative analysis (J) showing that EC-miR-15a/16–1 deletion statistically robustly down-regulated brain infarction at 3–7 d after MCAO, and reduced brain atrophy at 14–28 d following MCAO compared to WT controls. n = 6–8 for each group; #p < 0.05 versus MCAO + WT group; statistical analyses were performed by two-tailed Student’s t-test.
We performed the Morris water maze test to investigate whether EC-targeted miR-15a/16–1 deletion affects long-term spatial cognitive functions in mice after MCAO. Under sham conditions, the latency to find the platform (escape latency) improved over time, reflecting a normal spatial learning process that was identical between genotypes (Fig. 2E). Both EC-miR-15a/16–1 cKO and WT mice exhibited a similar trend in improvement over time in learning behavior after stroke (Fig. 2E,F), but compared to ischemic WT mice, long-term learning and memory behavior were improved in EC-miR-15a-16–1 cKO mice after MCAO, as shown by the statistically significant decreased escape latency during the learning phase (bracket) and increased time spent in the goal quadrant during the memory phase (Fig. 2E,F,H). Swimming speed between EC-miR-15a-16–1 cKO and WT mice was comparable (Fig. 2G), eliminating speed as a confounding factor in assessing time-based outcomes. No statistically significant difference was observed between WT and EC-miR-15a/16–1 cKO mice in body weight (Online Figure IV) and survival rate (Online Figure V) after stroke. Taken together, these data suggest that EC-targeted miR-15a/16–1 deletion enhances the overall long-term neurological recovery after stroke. By measuring the loss of microtubule-associated protein 2 (MAP2) in neurons at 1–28 d after MCAO, we explored whether the EC-targeted miR-15a/16–1 deletion affects histological outcomes after stroke. As shown in Figures 2I–J, compared with WT, EC-targeted miR-15a/16–1 deletion statistically significantly reduced brain infarct or atrophy volume at 3 to 28 d after MCAO, indicating long-term reduction of ischemic infarct at both early and late stages of recovery. Taken together, these data indicated that genetic deletion of miR-15a/16–1 in endothelium improves stroke outcome and recovery at both the histological and neurobehavioral levels.
Endothelium-targeted miR-15a/16–1 deletion enhances revascularization and newly formed microvessels in the penumbral area of ischemic brains.
The improvement in stroke outcomes in EC-miR-15a/16–1 cKO mice led to the hypothesis that EC-directed miR-15a/16–1 deletion may improve cerebral blood flow (CBF) during stroke recovery. To this end, we measured the spatiotemporal changes of ischemic cortical CBF using the laser speckle imaging. No statistically significant difference was found in the relative CBF values between WT and EC-miR-15a/16–1 cKO mice prior to, during, or 15 min, 24 h, or 7 d reperfusion after MCAO (Online Figure VI,VII). However, EC-miR-15a/16–1 cKO mice exhibited considerably improved CBF recovery at 14 d and 21 d reperfusion after MCAO, compared to WT controls (Online Figure VII). CBF recovery returned to comparable levels in both genotypes at 28 d post-ischemic reperfusion. These data were consistent with better long-term neurological recovery in EC-miR-15a/16–1 cKO mice after stroke above-described and demonstrate for the first time that EC-targeted deletion of miR-15a/16–1 potentiates revascularization of damaged ischemic tissue.
Previous studies suggested that an early rise in cerebral blood volume (CBV) in the ischemic hemisphere may be a result of improved collateral flow, whereas the late phase increase in CBV is attributed to a surge of angiogenesis7, 24. Thus, we hypothesized that the improvement in CBF recovery in EC-miR-15a/16–1 cKO mice after stroke may be associated with enhanced post-ischemic vascular remodeling. We chose the penumbral region to investigate revascularization since activated angiogenesis in this region might correlate with prolonged survival in ischemic stroke patients9, 25. As shown (Fig. 3A), compared to sham groups, brain microvasculature in both genotypes were dramatically decreased 1 d after MCAO. Cerebral revascularization progressively developed during the 28 d recovery period in both genotypes but was more prominent in the EC-miR-15a/16–1 cKO mice. Further quantitative analysis revealed that vascular surface area (Fig. 3B), vascular length (Fig. 3C), branch points (Fig. 3D), and capillary number (Fig. 3E) were generally higher in EC-miR-15a/16–1 cKO mouse brains than WT controls (bracket). Specifically, cerebral microvasculature in ischemic brains of EC-miR-15a/16–1 cKO mice exhibited statistically significant increased surface area, vascular length and branch points than WT controls at multiple time points throughout the recovery period following MCAO (asterisks, Fig. 3B,C). We further identified newly formed microvessels by BrdU/CD31 double-immunostaining. As illustrated (Fig. 3F), more newly formed microvessels (BrdU+ cells (red) co-labeled with CD31+ microvessels (green) were present in EC-miR-15a/16–1 cKO ischemic brains than WT controls 28 d after MCAO. Quantitative analysis further confirmed increased post-stroke cerebral angiogenesis in EC-miR-15a/16–1 cKO mice, as presented by statistically significant increased BrdU+/CD31+ double positive cells (Fig. 3H). No BrdU+ signals were detected in the sham animals.
Fig. 3. Endothelium-targeted miR-15a/16–1 deletion enhances revascularization and newly formed microvessels in the penumbral regions of mouse brains after cerebral ischemia.
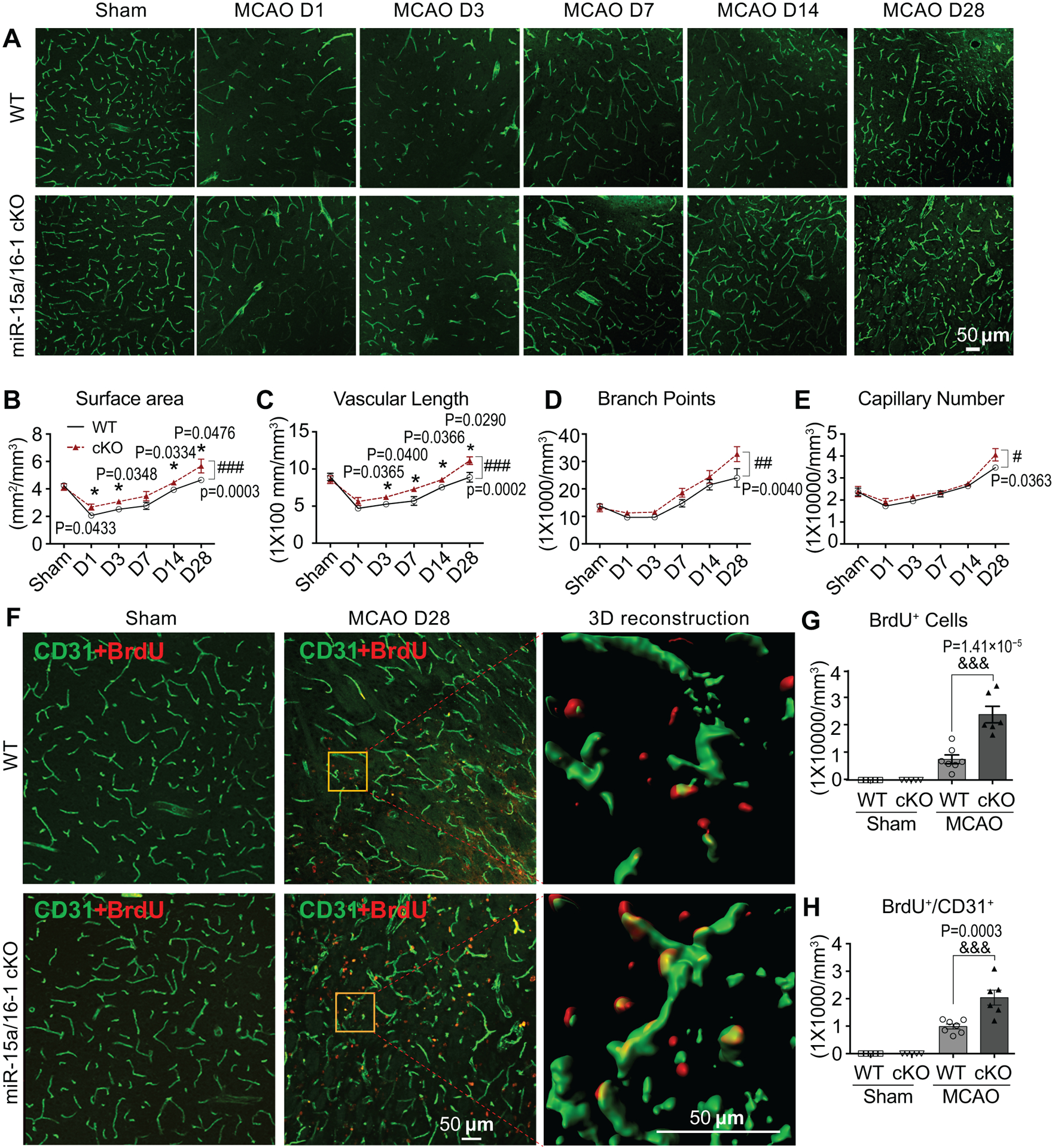
A-E, Cerebral vasculature was detected and quantified by CD31 immunostaining in the penumbral regions at 1–28 d reperfusion after MCAO. Representative CD31 immunostaining images (A), quantitative analysis of surface area (B), vascular length (C), branch points (D), and capillary number (E) showing faster enhancement of revascularization in penumbral regions of EC-miR-15a/16–1 cKO mice than WT controls during 1–28 d recovery period following MCAO. n = 5–7 for each group. *p < 0.05 versus WT mice by two-tailed student t-test (individual time point) and #p < 0.05, ##p < 0.01, ###p < 0.001 versus WT mice by two-way ANOVA followed by Bonferroni’s multiple comparison tests (bracket). F-H, CD31 and BrdU (red) double-immunostaining was used to determine the newly formed microvessels in the penumbral regions of the brains at 28 d reperfusion after MCAO. Yellow boxes indicated areas were enlarged and 3D reconstructed in the 3rd column of F. Representative images (F), quantification of the BrdU+ cells (G) and BrdU+/CD31+ signals (H) demonstrated more newly formed microvessels (angiogenesis) in the EC-miR-15a/16–1 cKO mice than WT controls in the brain penumbral regions following MCAO. Scale bar, 50 μm. n = 5–7 for each group; &&&p < 0.001 as indicated; statistical analyses were performed by one-way ANOVA followed by Tukey’s multiple comparison tests.
Endothelium-targeted miR-15a/16–1 deletion enhances newly formed functional vessels in the penumbra after cerebral ischemia.
BrdU/Biotinylated Tomato lectin double-immunostaining was used to identify newly formed functional (perfused) vessels26. In sham brain of either genotype, no newly formed functional vessels were detected (Fig. 4A–C). Twenty-eight days following MCAO, ischemic brains in both genotypes contained BrdU+ cells (red) along with functional vessels (green) in the penumbral regions, but more BrdU+ cells were present in the EC-miR-15a/16–1 cKO mice (Fig. 4A,B). Furthermore, BrdU+/Tomato lectin+ double positive cells were statistically significantly increased in the EC-miR-15a/16–1 cKO ischemic penumbra compared to WT (Fig. 4A,C), indicating that endothelium-targeted miR-15a/16–1 deletion might have increased the number of newly formed functional vessels in the penumbral areas of ischemic brains. Quantitative analyses indeed confirmed the increases of functional vessels in EC-miR-15a/16–1 cKO mice than WT controls following stroke, as illustrated by statistically significant increased surface area (Fig. 4D), branch points (Fig. 4F) and capillary number (Fig. 4G) in the penumbral areas 28 d after reperfusion.
Fig.4. Endothelium-targeted miR-15a/16–1 deletion enhances functional vessels in the penumbral areas after cerebral ischemia.
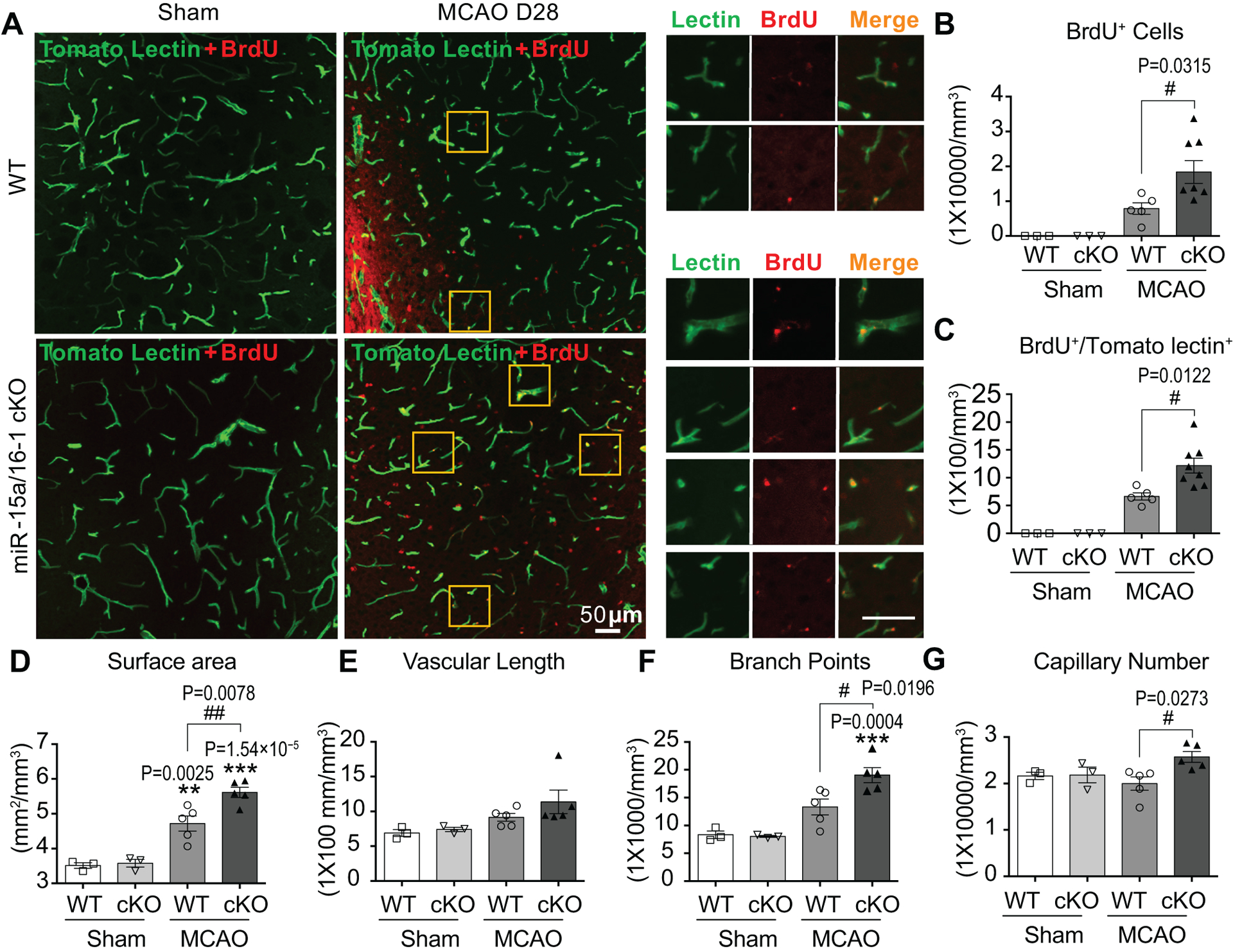
A-G, Tomato lectin (green) and BrdU (red) double-immunofluorescent staining were utilized to detect functional microvessels in the penumbral regions at 28 d reperfusion after MCAO. BrdU+/lectin+ signals exhibit yellow color, and yellow boxes indicated magnified areas with splitted tomato lectin+ and BrdU+ immunofluorescent signals. Representative images (A) and quantification of the BrdU+ cells (B), BrdU+/lectin+ signals (C), surface area (D), vascular length (E), branch points (F), and capillary number (G) showed more functional microvessels and angiogenesis in EC-miR-15a/16–1 cKO mice than WT controls in the penumbral regions following MCAO. Scale bar, 50 μm. n = 3 for Sham + WT and Sham + cKO groups; n=5 for MCAO + WT group; n = 5–8 for MCAO + cKO group. **p < 0.01, ***p < 0.001 versus sham group of each genotype; #p < 0.05, ##p < 0.01 as indicated; statistical analyses were performed by one-way ANOVA followed by Tukey’s multiple comparison tests.
Knockdown of the miR-15a/16–1 cluster promotes in vitro angiogenesis in cultures of mouse and human brain microvascular endothelial cells.
We further examined the effects of miR-15a on pro-angiogenic activities using capillary tube formation assay27, scratch assay28 and BrdU cell proliferation assay29 in mBMECs. To achieve loss- or gain-of-miR-15a functions in mBMEC cultures, we generated lentiviruses carrying small hairpin miR-15a (miRZip 15a, Online Figure VIIIB), pre-miR-15a (Lenti miR-15a, Online Figure VIIID), or their non-functional lentiviral controls (miRZip GFP, Online Figure VIIIA; or Lenti GFP, Online Figure VIIIC). Representative fluorescent images confirmed the successful infection of these lentiviruses into mBMECs (Fig. 5A,B). TaqMan® miRNA assay indicated successful down-regulation or up-regulation of miR-15a levels in mBMECs by miRZip 15a (Fig. 5C) and Lenti miR-15a (Fig. 5D) lentivirus, respectively. No difference was observed regarding miR-497 after lentiviral infection. Capillary tube formation assay revealed that loss-of-miR-15a function in mBMECs by miRZip 15a statistically significantly increased the tube formation, showing elevated branch points (Fig. 5E,G) and tube length (Fig. 5E,H). On the contrary, miR-15a overexpression by Lenti miR-15a abolished these pro-angiogenic effects (Fig. 5F,I,J). Moreover, the in vitro scratch assay demonstrated that loss-of-miR-15a function led to statistically significant increased endothelial cell migration (Fig. 5K,M), whereas miR-15a overexpression reversed this effect (Fig. 5L,N). Infection of the lentiviral GFP controls into mBMECs did not affect to statistical significance endothelial migration when compared to non-infected cells. In vitro angiogenesis was also measured by BrdU incorporation, since cell proliferation actively participates in angiogenesis. Compared to controls, lentiviral loss-of-miR-15a function statistically significantly up-regulated (Fig. 5O) cell proliferation in mBMECs, while gain-of-miR-15a function led to statistically significant down-regulation (Fig. 5P). Moreover, to explore if these effects can be translated to human cells, in vitro angiogenesis was also examined in primary human brain microvascular endothelia cells (hBMECs). Interestingly, lentivirus-mediated loss- or gain-of-miR-15a/16–1 function in primary hBMECs also statistically significantly increases or decreases BrdU cell proliferation (Online Figure IX), migration (Online Figure X), and tube formation (Online Figure XI), respectively. Collectively, these results demonstrate that the miR-15a/16–1 cluster inhibits endogenous angiogenic activities in mBMECs and hBMECs.
Fig. 5. Lentivirus-mediated loss- or gain-of-miR-15a/16–1 function increases or decreases in vitro angiogenesis, respectively.
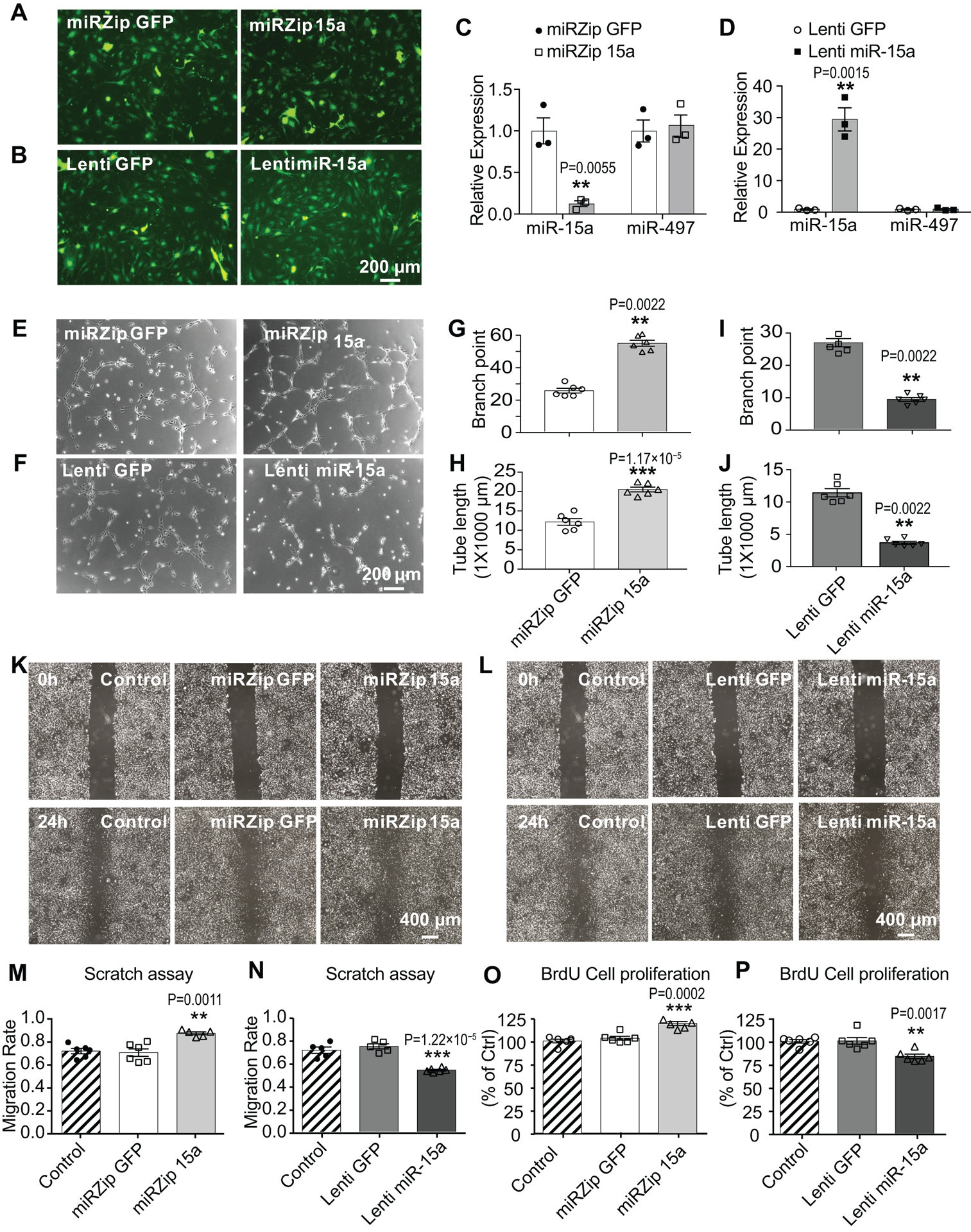
A-B, mBMECs were infected with lentivirus (1–2 MOI) containing small hairpin miR-15a (miRZip 15a, A) or pre-miR-15a (Lenti miR-15a, B) for 48–72h. C-D, qPCR data showed statistically significantly reduced miR-15a levels in mBMECs after infection with miRZip 15a (C), while elevated miR-15a levels after infection with Lenti miR-15a (D), compared to their GFP controls. No statistically significant change was detected for miR-497 expression. n = 3; **p < 0.01, ***p < 0.001 versus miRZip GFP or Lenti GFP groups; statistical analyses were performed by two-tailed Student’s t-test. E-J, Capillary tube formation assay indicated that, loss-of-miR-15a function by miRZip 15a statistically significantly increased tubular-like structure (E), branch points (G) and total tube length (H). On the contrary, gain-of-miR-15a function by lentivirus dramatically reversed these effects (F,I,J). n = 6; **p < 0.01, ***p < 0.001 versus miRZip GFP or Lenti GFP groups; statistical analyses were performed by Mann-Whitney test for G,I,J, and by two-tailed Student’s t-test for H. K-N, In vitro scratch assay in mBMEC cultures after lentiviral infections. Representative images (K) and quantitative analysis (M) showed that loss-of-miR-15a function by miRZip 15a statistically significantly increases the endothelial migration compared to miRZip GFP group or non-transduction control, whereas gain-of-miR-15a function (L,N) by Lenti miR-15a statistically significantly reverses this effect. n = 5–6; **p < 0.01, ***p < 0.001 versus miRZip GFP or Lenti GFP groups; statistical analyses were performed by one-way ANOVA followed by Tukey’s multiple comparison tests. O-P, BrdU incorporation assays showed that, lentivirus-mediated loss-of-miR-15a function in mBMECs statistically significantly up-regulated (O) while gain-of-miR-15a function statistically significantly down-regulated (P) cell proliferation, compared to lentiviral GFP groups or non-transduction controls. n = 6; **p < 0.01, ***p < 0.001 versus miRZip GFP or Lenti GFP groups; statistical analyses were performed by one-way ANOVA followed by Tukey’s multiple comparison tests.
Endothelium-targeted miR-15a/16–1 deletion enhances the expression of pro-angiogenic factors in the ischemic mouse brain.
In post-stroke brains, several angiogenic factors contribute to the pro-angiogenic responses, including VEGF, FGF, platelet-derived growth factor (PDGF), transforming growth factor beta (TGF-β), and others30. We focused on VEGFA, FGF2 and their receptors VEGFR2 and FGFR1 since we previously found that endothelial FGF2 and VEGF play a central role in peripheral angiogenesis after hindlimb ischemia19. EC-miR-15a/16–1 cKO exhibited overall higher cortical VEGFA, VEGFR2, FGF2 and FGFR1 mRNA levels than WT group in the tested time points after MCAO (Fig. 6A–D, bracket comparisons). Moreover, compared with WT, EC-miR-15a/16–1 cKO statistically significantly enhanced total cortical VEGFA protein expression at 1, 7, 14 and 28 d after MCAO (Fig. 6E,F). VEGFR2 protein level was statistically significantly increased at 1 to 7 d after MCAO (Fig. 6E,G) in EC-miR-15a/16–1 cKO mice. Similarly, statistically significant increased FGF2 protein level was detected at 7–28 d (Fig. 6E,H), and statistically significant increased FGFR1 protein level was detected at 3–28 d after MCAO (Fig. 6E,I). We also examined the mRNA levels of PDGFA, PDGFB, and TGF-β by qPCR, but no statistically significant difference was observed between two genotypes in the ischemic mouse brains (Online Figure XIII). Moreover, to confirm the expression of these pro-angiogenic factors in the brain microvasculature, we further isolated microvessels from the ischemic brains at 14 d after MCAO. Similar to our findings above, qPCR data showed statistically significant up-regulated mRNA expression of FGF2 and FGFR1, and an enhanced trend for VEGFA and VEGFR2 (Online Figure XIVA–D) in the brain microvessels of EC-miR-15a/16–1 cKO mice in comparison with WT controls. No statistically significant changes were observed in the mRNA expression of PDGFA, PDGFB, and TGF-β in two genotypes (Online Figure XIVE–G).
Fig. 6. Endothelium-targeted miR-15a/16–1 deletion enhances the expression of pro-angiogenic factors in ischemic mouse brains.
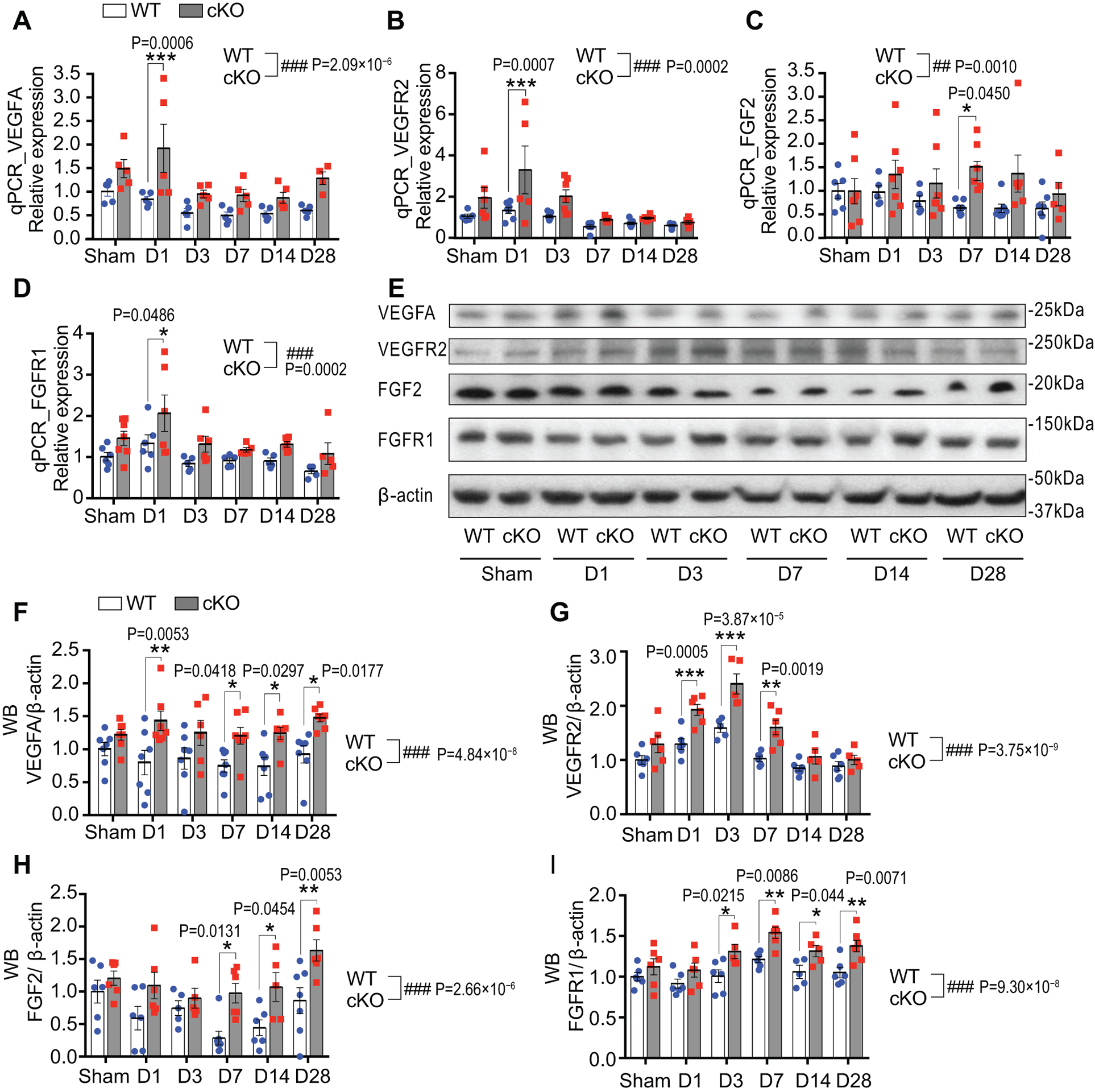
Total RNAs and proteins were isolated from the ipsilateral cortex of mouse brains at 1–28 d reperfusion after MCAO. qPCR and Western Blotting (WB) were carried out to detect the expression of pro-angiogenic factors. A-D, qPCR data showed elevated mRNA expression of VEGFA (A), VEGFR2 (B), FGF2 (C) and FGFR1 (D) in the cerebral cortex of EC-miR-15a/16–1 cKO mice than WT controls at two or more reperfusion time points after 1h MCAO. Accordingly, representative WB images (E) and quantitative analysis (F-I) indicated enhanced protein levels of VEGFA (F), VEGFR2 (G), FGF2 (H) and FGFR1 (I) in the cerebral cortex of EC-miR-15a/16–1 cKO mice than WT controls at three or more reperfusion time points after 1h MCAO. n = 5–7/group for all qPCR and WB experiments; *p < 0.05, **p < 0.01, ***p < 0.001 versus WT controls at each time point; ##p < 0.01, ###p < 0.001 for the overall difference between WT and EC-miR-15a/16–1 cKO groups; statistical analyses were performed by two-way ANOVA followed by Bonferroni’s multiple comparison tests.
Taken together, these results suggest that endothelium-targeted deletion of the miR-15a/16–1 cluster promotes post-stroke angiogenesis and potentiates the expression of pro-angiogenic factors, including VEGFA, VEGFR2, FGF2, and FGFR1.
Knockdown of the miR-15a/16–1 cluster enhances pro-angiogenic factors expression in mBMEC and hBMEC cultures after OGD and reoxygenation.
To further corroborate the role of miR-15a/16–1 in regulating endothelial expression of pro-angiogenic factors, we used mBMEC and hBMEC cultures subjected to 4 h of OGD and followed by up to 48 h of reoxygenation; this model does not result in statistically significant cell death, mimicking the penumbral regions described above, yet is still associated with an upregulation of miR-15a/16–131. Knockdown of miR-15a/16–1 expression by miRZip 15a statistically significantly up-regulated VEGFR2 and FGFR1 mRNA expression after 4h OGD and 4, 24, or 48h Reoxygenation (O4hR4h, O4hR24 or O4hR48h), compared to miRZip GFP control (Fig. 7B,D). VEGFA and FGF2 mRNA levels increased to statistical significance by miRZip 15a at O4hR4h time point compared to miRZip GFP group (Fig. 7 A,C). Consistent with qPCR findings, we found statistically significant enhanced protein levels of pro-angiogenic factors at longer reoxygenation periods (O4hR24h or/and O4hR48h) (Fig. 7E–I). On the contrary to the effects of gene knockdown, lentiviral overexpression of miR-15a in mBMECs by Lenti miR-15a statistically significantly down-regulated VEGFA, VEGFR2, and FGFR1 mRNA expression at longer Reox time points (O4hR24 or/and O4hR48) (Fig. 7J,K,M) and down-regulated FGF2 mRNA expression at a shorter Reox time point (O4hR4h) (Fig. 7L), compared with lentiviral GFP controls. Accordingly, statistically significant reduced protein levels of VEGFA, VEGFR2, FGF2, and FGFR1 (Fig. 7N–R) were observed in Lenti miR-15a treated cultures under OGD and Reox. In order to confirm these effects occur in human cells, qPCR (Online Figure XVA–D) and western blotting (Online Figure XVI) data from hBMECs illustrated that lentivirus-mediated knockdown of miR-15a/16–1 cluster in hBMEC cultures statistically significantly enhanced the mRNA and protein expression of VEGFA, FGF2, and their receptors VEGFR2 and FGFR1 after OGD and Reox, without statistically significantly altering the expression of PDGFA, PDGFB, and TGF-β (Online Figure XVE–G). Furthermore, statistically significant enhanced cell proliferation triggered by the loss-of-miR-15a/16–1 function in hBMECs can be abolished by co-treatments with VEGFA and FGF2 siRNAs (Online Figure XVII), indicating that VEGFA and FGF2 signaling pathways are direct downstream targets of miR-15a/16–1. Collectively, these in vitro data clearly illustrated the vital role of brain endothelium-originated miR-15a/16–1 in the regulation of post-ischemia angiogenesis at both the cellular and molecular levels.
Fig. 7. Knockdown or overexpression of miR-15a/16–1 cluster enhances or suppresses the pro-angiogenic factors expression in mBMECs cultures after OGD and reoxygenation, respectively.
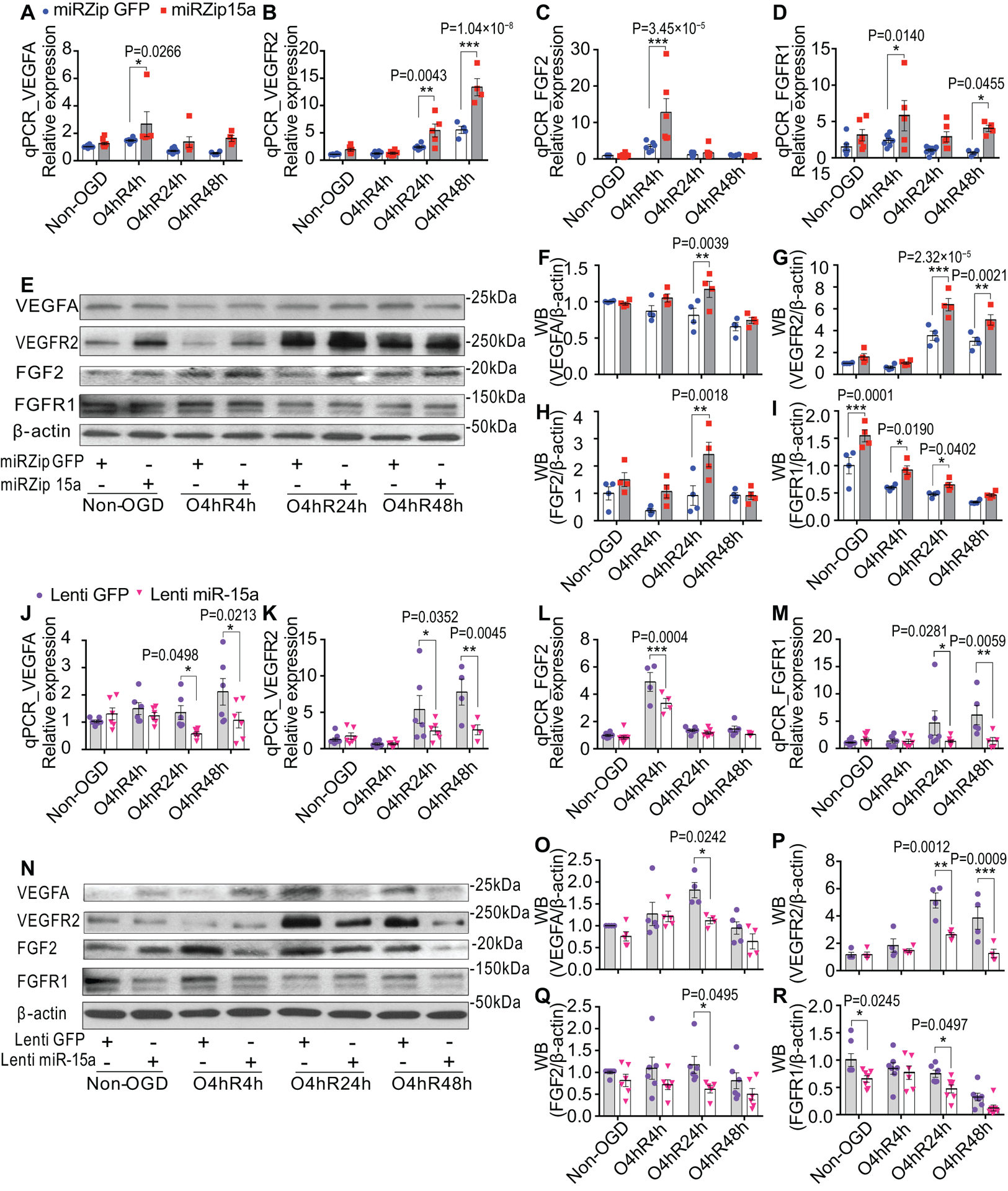
Primary mBMECs were infected with lentiviruses and then cells were subjected to OGD 4h followed by reoxygenation 4h (O4hR4h), 24h (O4hR24h) and 48h (O4hR48h). A-D, qPCR data showed that loss-of-miR-15a function by miRzip15a lentivirus statistically significantly up-regulated mRNA expressions of VEGFA (A), VEGFR2 (B), FGF2 (C) and FGFR1 (D) at multiple time points of reoxygenation after OGD, compared to lentiviral GFP group (miRZip GFP). Accordingly, representative WB images (E) and quantitative analysis (F-I) indicated that enhanced protein levels of VEGFA (F), VEGFR2 (G), FGF2 (H) and FGFR1 (I) were also observed in miRZip 15a treated group after OGD and reoxygenation, compared to miRZip GFP. However, gain-of-miR-15a function by Lenti miR-15a dramatically reversed these effects. J-M, qPCR data showed that gain-of-miR-15a function by Lenti miR-15a statistically significantly down-regulated mRNA expressions of VEGFA (J), VEGFR2 (K), FGF2 (L) and FGFR1 (M) at multiple time points of reoxygenation after OGD, compared to lentiviral GFP group (Lenti GFP). Accordingly, representative WB images (N) and quantitative analysis (O-R) indicated that statistically significantly reduced protein levels of VEGFA (O), VEGFR2 (P), FGF2 (Q) and FGFR1 (R) were also observed in Lenti miR-15a treated group after OGD and reoxygenation, compared to Lenti GFP. n = 4–6/group for all qPCR and WB experiments. *p < 0.05, **p < 0.01, ***p < 0.001 versus lentiviral GFP controls at each time point; statistical analyses were performed by two-way ANOVA followed by Bonferroni’s multiple comparison tests.
MiR-15a/16–1 translationally represses mouse VEGFA, VEGFR2, FGF2 and FGFR1 in mouse brain microvascular endothelial cells.
To elucidate the molecular mechanism underlying endothelial miR-15a/16–1 in regulation of VEGFA, VEGFR2, FGF2 and FGFR1 expression, we performed both bioinformatics analysis and dual-luciferase assays. Bioinformatics analysis indicated one conserved miR-15a binding site within the 3’-UTR of mouse VEGFA and VEGFR2 mRNAs (Fig. 8A,D) and two conserved miR-15a binding sites within the 3’-UTR of mouse FGF2 and FGFR1 mRNAs (Fig. 8G,J). These results indicate that miR-15a might translationally repress these angiogenic factors through direct interaction with the putative binding sites located on their 3’-UTR regions. Thus, we constructed firefly/renilla dual-luciferase reporter plasmids in which a cytomegalovirus (CMV) driven-luciferase cDNA is fused to wild-type mouse VEGFA, VEGFR2, FGF2, and FGFR1 3’-UTR (WT Seq.), or to mutants with the miR-15a/16–1 binding site deleted 3’-UTRs (Dele Seq.). Detailed structures and sequences of plasmids can be found in Online Figure XVIII. Mouse brain microvascular endothelial cells (bEnd3) were infected with miRZip 15a or lenti miR-15a to knockdown or overexpress miR-15a/16–1 levels 72 h prior to co-transducing with above-described dual-luciferase reporter plasmids. Lentiviral knockdown of miR-15a in bEnd3 cells statistically significantly increased luciferase activity of reporter vectors containing the 3’-UTR mRNAs of mouse VEGFA, VEGFR2, FGF2 and FGFR1 (Fig. 8B,E,H,K). In contrast, lentiviral overexpression of miR-15a statistically significantly decreased the luciferase activity of reporter vectors containing the 3’-UTR mRNAs of these pro-angiogenic factors (Fig. 8C,F,I,L). Importantly, neither knocking down nor overexpressing miR-15a in bEnd3 cells has any statistically significant effects on the activities of luciferase reporter vectors containing a miR-15a binding site-deleted sequences (Dele Seq) from VEGFA, VEGFR2, FGF2 or FGFR1 3’-UTR. Collectively, these results confirmed that miR-15a specifically represses mouse VEGFA, VEGFR2, FGF2, and FGFR1 mRNA translation by directly binding to the complementary sequences located in their 3’-UTR mRNA regions.
Fig. 8. MiR-15a/16–1 translationally suppress mouse VEGFA, VEGFR2, FGF2 and FGFR1 in mouse brain microvascular endothelial cells.
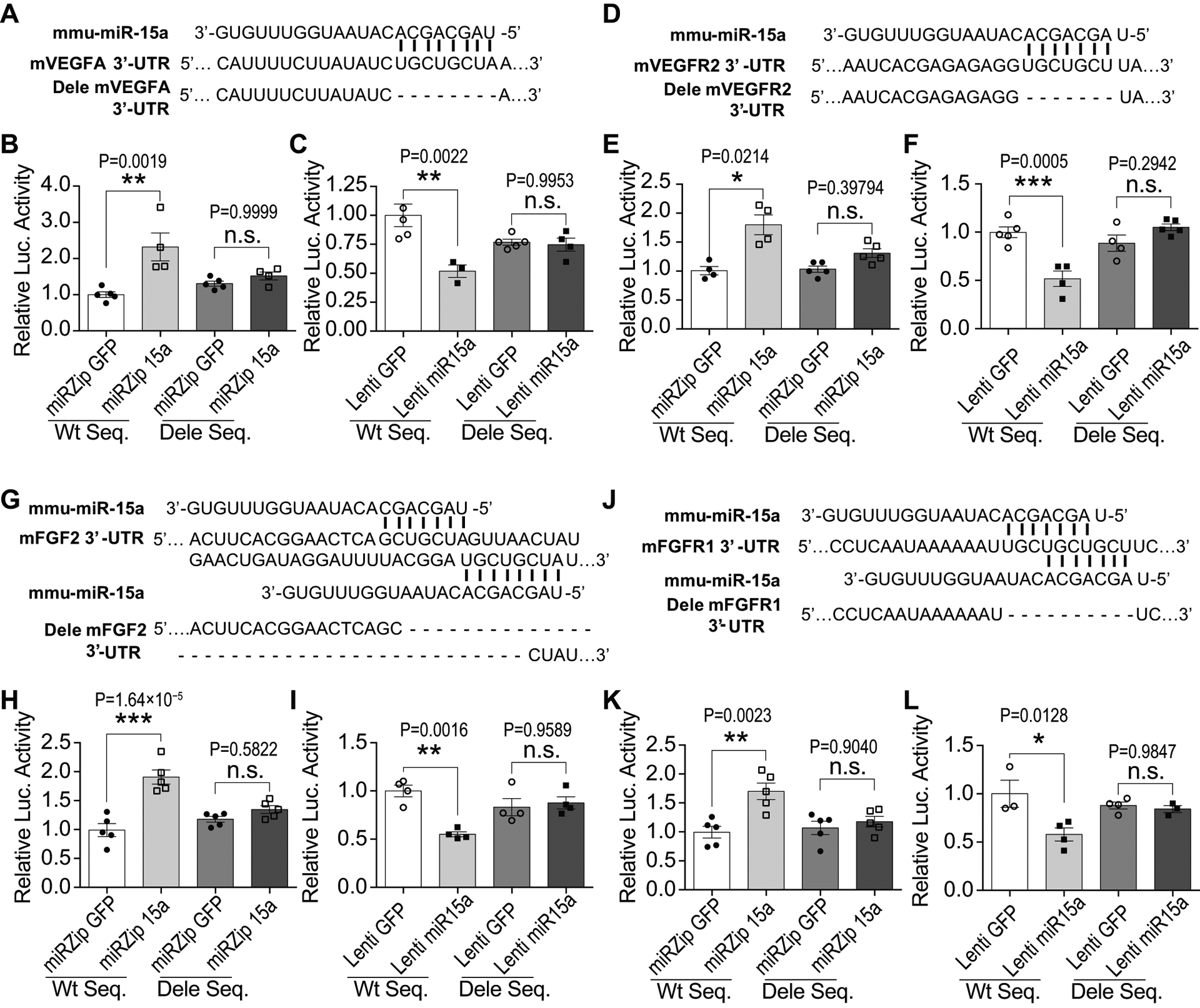
A,D,G,J, The partial sequence of mature mouse miR-15a (mmu-miR-15a) and its binding sites in the 3’-UTR region of the mouse VEGFA (A), VEGFR2 (D), FGF2 (G) and FGFR1 (J). MiR-15a reporter vectors containing CMV-driven expression of luciferase cDNA fused to mouse VEGFA (mVEGFA), mVEGFR2, mFGF2 and mFGFR1 wild-type 3’-UTR (WT Seq) or to miR-15a/16–1 binding site deleted 3’-UTR (the dash line nucleotides are deleted, Dele Seq) were constructed and transfected into bEnd3 cells. Meanwhile, bEnd3 cells were co-transduced with miRZip 15a or Lenti miR-15a for 72h prior to performing luciferase reporter activity assays. B,E,H,K, Quantitative data showed that loss-of-miR-15a function by miRZip 15a in bEnd3 cells statistically significantly increased luciferase activity of the reporter vector containing wild-type 3’-UTR sequence from mVEGFA (B), mVEGFR2 (E), mFGF2 (H) and mFGFR1 (K), in comparison with miRZip GFP. C,F,I,L, Gain-of-miR-15a function by Lenti miR-15a statistically significantly reduced luciferase activity of the reporter vector containing wild-type 3’-UTR sequence from mVEGFA (C), mVEGFR2 (F), mFGF2 (I) and mFGFR1 (L), in comparison with Lenti GFP. However, loss- or gain-of-miR-15a functions in bEnd3 cells had no effects on the luciferase activity of the reporter vector containing miR-15a binding site deleted 3’-UTR sequence (Dele Seq) from mouse VEGFA, VEGFR2, FGF2 or FGFR1. n = 3–5/group for all assays; *p < 0.05, **p < 0.01, ***p < 0.001 versus lentiviral GFP controls; n.s., no statistical significance; statistical analyses were performed by one-way ANOVA followed by Tukey’s multiple comparison tests for C, F, H, I, K, L; and statistical analyses were performed by Kruskal-Wallis test followed by Dunn’s multiple comparison tests for B and E.
DISCUSSION
Angiogenesis is a crucial restorative or protective mechanism in response to cerebral ischemia9, 32. Occurring in the ischemic boundary zone (IBZ) as early as 12h, angiogenesis may last for more than 21 days following experimental cerebral ischemia4, 5, 33. Post-stroke angiogenesis contributes to improved neurological recovery by promoting tissue repair, vascular remodeling, and plasticity, in both stroke patients and animal stroke models4, 9, 34, 35. For example, omega-3 polyunsaturated fatty acids (n–3 PUFAs) can induce a robust improvement in revascularization and angiogenesis, which ultimately lead to long-term histological and behavioral protection against ischemic stroke in young mice34. Consistent with these studies, our findings also confirmed the existence of vascular remodeling throughout a prolonged recovery time window (1–28 d following MCAO), and described a role for endothelium-targeted deletion of the miR-15a/16–1 cluster promoted the revascularization and angiogenesis in the peri-infarct brain areas following ischemic stroke.
MiRs actively participate in various pathophysiological cascades of ischemic stroke, including angiogenesis and neurogenesis36. Several miRs have important roles in the regulation of post-stroke angiogenesis, such as miR-10714 and miR-15037. Here, our study systematically demonstrated that genetic deletion of the miR-15a/16–1 cluster in endothelium induced a potent pro-angiogenic effect following reperfusion of the ischemic brain, which finally led to the improvements of long-term neurological recovery after ischemic stroke. Previously, our group described that EC-targeted miR-15a/16–1 overexpression statistically significantly suppressed angiogenesis in mouse hindlimb ischemia19; permanent hindlimb ischemia is a fairly simple tissue system, and neovascularization in peripheral circulation may be quite different from ischemic brain injury. Others have shown up-regulated VEGF expression in the infarcted hemisphere as early as three hours after ischemic insult5, which persisted over 7 d in animal models of experimental stroke38. VEGF immunoreactivity was also elevated in cerebrovascular endothelial cells within the ischemic penumbral areas39, 40. Consistent with these observations, we detected statistically significant increased expression of VEGFA in cerebral microvessels up to 28 d following reperfusion, most notably at 3 d after transient MCAO; however, at 7 d following reperfusion, we detected a dramatic decline in the VEGFA expression. This discrepancy may be attributed to the use of whole brain tissues or serum in the previous studies, while we employed isolated brain microvessels. Interestingly, the lowest expressions of VEGFR, FGF2, and FGFR1 were observed at 7 d following stroke, while miR-15a/16–1 cluster exhibited highest expression around the same time point revealing a negative correlation between miR-15a/16–1 and pro-angiogenic factors. Data from EC-miR-15a/16–1 cKO mouse further indicated that endothelial miR-15a/16–1 modulates long-term sensorimotor and cognitive functions after ischemic stroke, and also remarkedly attenuated brain infarct volume in the acute phase and brain atrophy in the chronic phase of stroke. Moreover, EC-targeted deletion of the miR-15a/16–1 cluster considerably up-regulated the CBF recovery at 14 d and 21 d after stroke. As angiogenesis is associated with an increase in CBF and cerebral blood volume (CBV)24, our data strongly suggest that EC-targeted deletion of the miR-15a/16–1 cluster statistically significantly enhances revascularization and generation of newly formed microvessels and functional vessels in the penumbral areas after ischemic stroke.
Endothelial cells are the primary constituents of newly-generating cerebral microvessels, and their functions are required for angiogenesis, including morphogenesis, migration, proliferation, and others29. Here we also examined the influence of the miR-15a/16–1 cluster on in vitro angiogenic activities. We found that loss-of-miR-15a function by lentivirus statistically significantly enhanced the in vitro angiogenesis in primary mBMEC or hBMEC cultures, as revealed by the elevated branch points and tube length in the capillary tube formation assay, boosted endothelial cell migration in the scratch assay, and increased cell proliferation in the BrdU cell proliferation assay. As an additional proof, gain-of-miR-15a function by lentivirus completely reversed those pro-angiogenic activities. These findings on in vitro angiogenesis are consistent with our in vivo results.
Various angiogenic factors are able to regulate angiogenesis in the brain6–8. VEGF plays a central role in the post-stroke angiogenesis6. VEGFA-VEGFR2 signaling pathway triggers most of the mechanisms activating angiogenesis41. VEGFA can bind to its receptor (VEGFR2) on the surface of vascular endothelial cells to activate multiple downstream signals to stimulate angiogenesis, including endothelial cell proliferation, invasion, migration and survival6. FGF2 is also a potent angiogenic inducer by binding to its receptors FGFR1 or FGFR242. Our mechanistic study reveals that EC-targeted deletion of the miR-15a/16–1 cluster robustly increased levels of VEGFA, FGF2, and their receptors VEGFR2 and FGFR1 in the mouse cerebral cortex and microvessels in a long-term pattern after stroke. Consistently, our in vitro study demonstrates that loss-of-miR-15a/16–1 function also statistically significantly enhanced these pro-angiogenic factors in primary mBMECs or hBMECs following in vitro ischemia, while gain-of-miR-15a/16–1 function caused the opposite effects. Thus, we suggest that the up-regulated VEGFA-VEGFR2 and FGF2-FGFR1 signaling pathways resulting from EC-targeted deletion of the miR-15a/16–1 cluster may augment post-stroke angiogenesis in brain and functions as a restorative neurovascular response against ischemic brain injury. Nevertheless, our current data cannot exclude the possibility that other pro-angiogenic mechanisms may be also involved in the miR-15a/16–1-mediated post-stroke angiogenesis, which warrants further comprehensive examinations.
This study focused on investigating endothelial miR-15a/16–1 regulation of angiogenesis and the long-term neurological recovery after ischemic stroke. However, we also observed early-stage neurovascular protection at 3–7 d after stroke in EC-miR-15a/16–1 cKO mice, which accompanied elevated cerebral VEGFA levels (Fig. 2A–D,J and Fig 6A,F), implying the involvement of several protective mechanisms. Besides proangiogenic property, VEGF-VEGFR signaling can increase endothelial nitric oxide synthase (eNOS) expression, enhance nitric oxide (NO) production to promote endothelial cell survival and vascular protection43, 44. Also, VEGFA exhibits neuroprotective effects after ischemia, including neuronal survival in vitro45, and local application of VEGFA protein to the surface of the reperfused brain also reduced the infarction after transient ischemia46. Moreover, we previously showed that miR-15a/16–1 was enriched and up-regulated in mBMEC cultures after OGD and caused apoptosis via inhibiting bcl-2 expression31. We also demonstrated that mBMECs can secrete miR-15a to the culture medium via exosomes after OGD (Online Figure XIX). Thus, it is possible that the elevated brain endothelial miR-15a/16–1 can be secreted and affect the surrounding neurovascular cells upon cerebral ischemia in vivo. Furthermore, we previously demonstrated that genetic deletion of miR-15a/16–1 statistically significantly reduced acute (3 d) ischemic brain injury in mice correlated with anti-inflammatory effects15. Thus, VEGFA-mediated neurovascular protection and miR-15a/16–1 inhibition-triggered anti-apoptotic and anti-inflammatory effects in endothelium and other neural cells may synergistically contribute to EC-miR-15a/16–1 cKO-mediated neurovascular protection during the acute stage of ischemic stroke.
In conclusion, the present study demonstrates that endothelium-targeted deletion of the miR-15a/16–1 cluster promotes cerebral angiogenesis after experimental ischemic stroke, and may be applicable to human systems. Endothelial miR-15a/16–1 appears to be a promising pharmacological target to improve post-stroke neurological recovery through enhancement of cerebral angiogenesis. Thus, miR-15a/16–1 inhibition may shed light on developing novel restorative therapeutics for ischemic stroke.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Cerebral ischemia results in severe cerebral blood flow (CBF) reduction and ultimately leads to neuronal cell death and brain infarction.
Post-ischemic cerebral angiogenesis can improve regional CBF supply and promote neurological recovery, contributing to prolonged survival of stroke patients.
MicroRNAs (miRs) function as a novel class of endogenous non-coding small RNA molecules and play an essential role in regulating almost all cellular processes, including angiogenesis.
What New Information Does This Article Contribute?
Endothelium-targeted deletion of the miR-15a/16–1 cluster significantly enhances cerebral angiogenesis and improves long-term neurological function in a murine model of stroke.
The miR-15a/16–1 cluster down-regulates angiogenesis by translationally repressing pro-angiogenic factors vascular endothelial growth factor (VEGFA), fibroblast growth factor 2 (FGF2), and their receptors VEGFR2 and FGFR1 after ischemic stroke.
ACKNOWLEDGMENTS
We thank Patricia Strickler for the administrative support.
SOURCES OF FUNDING
This work was supported by the National Institutes of Health Grants: NS091175 (K.J. Yin and J. Chen), NS094930, NS086820 (K.J. Yin); and American Heart Association Grant: 20POST35210900 (P. Sun); JC is also supported by the Senior Research Career Scientist Award and Merit Review grants BX002495 and BX003377 from the Department of Veterans Affairs.
Nonstandard Abbreviations and Acronyms:
- MCAO
Middle cerebral artery occlusion
- VEGFA
Vascular endothelial growth factor
- FGF2
Fibroblast growth factor 2
- VEGFR2
Vascular endothelial growth factor receptor 2
- FGFR1
Fibroblast growth factor receptor 1
- EC
Endothelial cell
- mBMEC
Mouse brain microvascular endothelial cell
- OGD
Oxygen-glucose deprivation
- tPA
Tissue plasminogen activator
- miRs
MicroRNAs
- 3’-UTR
Three prime untranslated regions
- MAP2
Microtubule-associated protein 2
- CBF
Cerebral blood flow
- CBV
Cerebral blood volume
- BrdU
5-bromo-2’-deoxyuridine
- PDGF
Platelet-derived growth factor
- TGFβ
Transforming growth factor beta
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Nogueira RG, Jadhav AP, Haussen DC, et al. Thrombectomy 6 to 24 Hours after Stroke with a Mismatch between Deficit and Infarct. N Engl J Med 2018;378:11–21. [DOI] [PubMed] [Google Scholar]
- 2.George PM and Steinberg GK. Novel Stroke Therapeutics: Unraveling Stroke Pathophysiology and Its Impact on Clinical Treatments. Neuron. 2015;87:297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carmeliet P Angiogenesis in life, disease and medicine. Nature. 2005;438:932–6. [DOI] [PubMed] [Google Scholar]
- 4.Hayashi T, Noshita N, Sugawara T and Chan PH. Temporal profile of angiogenesis and expression of related genes in the brain after ischemia. J Cereb Blood Flow Metab. 2003;23:166–80. [DOI] [PubMed] [Google Scholar]
- 5.Marti HJ, Bernaudin M, Bellail A, Schoch H, Euler M, Petit E and Risau W. Hypoxia-induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am J Pathol. 2000;156:965–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beck H and Plate KH. Angiogenesis after cerebral ischemia. Acta Neuropathol. 2009;117:481–96. [DOI] [PubMed] [Google Scholar]
- 7.Arai K, Jin G, Navaratna D and Lo EH. Brain angiogenesis in developmental and pathological processes: neurovascular injury and angiogenic recovery after stroke. FEBS J. 2009;276:4644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang ZG and Chopp M. Neurorestorative therapies for stroke: underlying mechanisms and translation to the clinic. Lancet Neurol. 2009;8:491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krupinski J, Kaluza J, Kumar P, Kumar S and Wang JM. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke. 1994;25:1794–8. [DOI] [PubMed] [Google Scholar]
- 10.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. [DOI] [PubMed] [Google Scholar]
- 11.Bi Y, Liu G and Yang R. MicroRNAs: novel regulators during the immune response. J Cell Physiol. 2009;218:467–72. [DOI] [PubMed] [Google Scholar]
- 12.Gauthier BR and Wollheim CB. MicroRNAs: ‘ribo-regulators’ of glucose homeostasis. Nat Med. 2006;12:36–8. [DOI] [PubMed] [Google Scholar]
- 13.Sun P, Liu DZ, Jickling GC, Sharp FR and Yin KJ. MicroRNA-based therapeutics in central nervous system injuries. J Cereb Blood Flow Metab. 2018;38:1125–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, Mao L, Gao Y, Baral S, Zhou Y and Hu B. MicroRNA-107 contributes to post-stroke angiogenesis by targeting Dicer-1. Sci Rep. 2015;5:13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang X, Tang X, Sun P, Shi Y, Liu K, Hassan SH, Stetler RA, Chen J and Yin KJ. MicroRNA-15a/16–1 Antagomir Ameliorates Ischemic Brain Injury in Experimental Stroke. Stroke. 2017;48:1941–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calin GA, Cimmino A, Fabbri M, et al. MiR-15a and miR-16–1 cluster functions in human leukemia. Proc Natl Acad Sci U S A. 2008;105:5166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, Ambesi-Impiombato A, Califano A, Migliazza A, Bhagat G and Dalla-Favera R. The DLEU2/miR-15a/16–1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17:28–40. [DOI] [PubMed] [Google Scholar]
- 18.Tan KS, Armugam A, Sepramaniam S, Lim KY, Setyowati KD, Wang CW and Jeyaseelan K. Expression profile of MicroRNAs in young stroke patients. PLoS One. 2009;4:e7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yin KJ, Olsen K, Hamblin M, Zhang J, Schwendeman SP and Chen YE. Vascular endothelial cell-specific microRNA-15a inhibits angiogenesis in hindlimb ischemia. J Biol Chem. 2012;287:27055–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamalice L, Le Boeuf F and Huot J. Endothelial cell migration during angiogenesis. Circ Res. 2007;100:782–94. [DOI] [PubMed] [Google Scholar]
- 21.Alva JA, Zovein AC, Monvoisin A, Murphy T, Salazar A, Harvey NL, Carmeliet P and Iruela-Arispe ML. VE-Cadherin-Cre-recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Dev Dyn. 2006;235:759–67. [DOI] [PubMed] [Google Scholar]
- 22.Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI, Lo EH and Group S. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Zhang B, Li W, Wang L, Yan Z, Li H, Yao Y, Yao R, Xu K and Li Z. MiR-15a/16 regulates the growth of myeloma cells, angiogenesis and antitumor immunity by inhibiting Bcl-2, VEGFA and IL-17 expression in multiple myeloma. Leuk Res. 2016;49:73–9. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, Wang Y, Akamatsu Y, Lee CC, Stetler RA, Lawton MT and Yang GY. Vascular remodeling after ischemic stroke: mechanisms and therapeutic potentials. Prog Neurobiol. 2014;115:138–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krupinski J, Kaluza J, Kumar P, Wang M and Kumar S. Prognostic value of blood vessel density in ischaemic stroke. Lancet. 1993;342:742. [DOI] [PubMed] [Google Scholar]
- 26.Robertson RT, Levine ST, Haynes SM, Gutierrez P, Baratta JL, Tan Z and Longmuir KJ. Use of labeled tomato lectin for imaging vasculature structures. Histochem Cell Biol. 2015;143:225–34. [DOI] [PubMed] [Google Scholar]
- 27.Arnaoutova I and Kleinman HK. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat Protoc. 2010;5:628–35. [DOI] [PubMed] [Google Scholar]
- 28.Liang CC, Park AY and Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–33. [DOI] [PubMed] [Google Scholar]
- 29.Goodwin AM. In vitro assays of angiogenesis for assessment of angiogenic and anti-angiogenic agents. Microvasc Res. 2007;74:172–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yin KJ, Hamblin M and Chen YE. Angiogenesis-regulating microRNAs and Ischemic Stroke. Curr Vasc Pharmacol. 2015;13:352–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yin KJ, Deng Z, Hamblin M, Xiang Y, Huang H, Zhang J, Jiang X, Wang Y and Chen YE. Peroxisome proliferator-activated receptor delta regulation of miR-15a in ischemia-induced cerebral vascular endothelial injury. J Neurosci. 2010;30:6398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin CY, Chang C, Cheung WM, Lin MH, Chen JJ, Hsu CY, Chen JH and Lin TN. Dynamic changes in vascular permeability, cerebral blood volume, vascular density, and size after transient focal cerebral ischemia in rats: evaluation with contrast-enhanced magnetic resonance imaging. J Cereb Blood Flow Metab. 2008;28:1491–501. [DOI] [PubMed] [Google Scholar]
- 33.Beck H, Acker T, Wiessner C, Allegrini PR and Plate KH. Expression of angiopoietin-1, angiopoietin-2, and tie receptors after middle cerebral artery occlusion in the rat. Am J Pathol. 2000;157:1473–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J, Shi Y, Zhang L, Zhang F, Hu X, Zhang W, Leak RK, Gao Y, Chen L and Chen J. Omega-3 polyunsaturated fatty acids enhance cerebral angiogenesis and provide long-term protection after stroke. Neurobiol Dis. 2014;68:91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoffmann CJ, Harms U, Rex A, Szulzewsky F, Wolf SA, Grittner U, Lattig-Tunnemann G, Sendtner M, Kettenmann H, Dirnagl U, Endres M and Harms C. Vascular signal transducer and activator of transcription-3 promotes angiogenesis and neuroplasticity long-term after stroke. Circulation. 2015;131:1772–82. [DOI] [PubMed] [Google Scholar]
- 36.Li G, Morris-Blanco KC, Lopez MS, Yang T, Zhao H, Vemuganti R and Luo Y. Impact of microRNAs on ischemic stroke: From pre- to post-disease. Prog Neurobiol. 2018;163–164:59–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He QW, Li Q, Jin HJ, Zhi F, Suraj B, Zhu YY, Xia YP, Mao L, Chen XL and Hu B. MiR-150 Regulates Poststroke Cerebral Angiogenesis via Vascular Endothelial Growth Factor in Rats. CNS Neurosci Ther. 2016;22:507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hayashi T, Abe K, Suzuki H and Itoyama Y. Rapid induction of vascular endothelial growth factor gene expression after transient middle cerebral artery occlusion in rats. Stroke. 1997;28:2039–44. [DOI] [PubMed] [Google Scholar]
- 39.Lennmyr F, Ata KA, Funa K, Olsson Y and Terent A. Expression of vascular endothelial growth factor (VEGF) and its receptors (Flt-1 and Flk-1) following permanent and transient occlusion of the middle cerebral artery in the rat. J Neuropathol Exp Neurol. 1998;57:874–82. [DOI] [PubMed] [Google Scholar]
- 40.Margaritescu O, Pirici D and Margaritescu C. VEGF expression in human brain tissue after acute ischemic stroke. Rom J Morphol Embryol. 2011;52:1283–92. [PubMed] [Google Scholar]
- 41.Pauty J, Usuba R, Cheng IG, Hespel L, Takahashi H, Kato K, Kobayashi M, Nakajima H, Lee E, Yger F, Soncin F and Matsunaga YT. A Vascular Endothelial Growth Factor-Dependent Sprouting Angiogenesis Assay Based on an In Vitro Human Blood Vessel Model for the Study of Anti-Angiogenic Drugs. EBioMedicine. 2018;27:225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beenken A and Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8:235–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Papapetropoulos A, Garcia-Cardena G, Madri JA and Sessa WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest. 1997;100:3131–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zachary I Signaling mechanisms mediating vascular protective actions of vascular endothelial growth factor. Am J Physiol Cell Physiol. 2001;280:C1375–86. [DOI] [PubMed] [Google Scholar]
- 45.Jin KL, Mao XO and Greenberg DA. Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proc Natl Acad Sci U S A. 2000;97:10242–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hayashi T, Abe K and Itoyama Y. Reduction of ischemic damage by application of vascular endothelial growth factor in rat brain after transient ischemia. J Cereb Blood Flow Metab. 1998;18:887–95. [DOI] [PubMed] [Google Scholar]
- 47.Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI, Lo EH and Group S. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alkayed NJ, Harukuni I, Kimes AS, London ED, Traystman RJ and Hurn PD. Gender-linked brain injury in experimental stroke. Stroke. 1998;29:159–65; discussion 166. [DOI] [PubMed] [Google Scholar]
- 49.Sohrabji F, Park MJ and Mahnke AH. Sex differences in stroke therapies. J Neurosci Res. 2017;95:681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Girijala RL, Sohrabji F and Bush RL. Sex differences in stroke: Review of current knowledge and evidence. Vasc Med. 2017;22:135–145. [DOI] [PubMed] [Google Scholar]
- 51.Bushnell CD, Chaturvedi S, Gage KR, Herson PS, Hurn PD, Jimenez MC, Kittner SJ, Madsen TE, McCullough LD, McDermott M, Reeves MJ and Rundek T. Sex differences in stroke: Challenges and opportunities. J Cereb Blood Flow Metab. 2018;38:2179–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koellhoffer EC and McCullough LD. The effects of estrogen in ischemic stroke. Transl Stroke Res. 2013;4:390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi Y, Jiang X, Zhang L, Pu H, Hu X, Zhang W, Cai W, Gao Y, Leak RK, Keep RF, Bennett MV and Chen J. Endothelium-targeted overexpression of heat shock protein 27 ameliorates blood-brain barrier disruption after ischemic brain injury. Proc Natl Acad Sci U S A. 2017;114:E1243–E1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shi Y, Zhang L, Pu H, Mao L, Hu X, Jiang X, Xu N, Stetler RA, Zhang F, Liu X, Leak RK, Keep RF, Ji X and Chen J. Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat Commun. 2016;7:10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang X, Tang X, Sun P, Shi Y, Liu K, Hassan SH, Stetler RA, Chen J and Yin KJ. MicroRNA-15a/16–1 Antagomir Ameliorates Ischemic Brain Injury in Experimental Stroke. Stroke. 2017;48:1941–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang X, Tang X, Liu K, Hamblin MH and Yin KJ. Long Noncoding RNA Malat1 Regulates Cerebrovascular Pathologies in Ischemic Stroke. J Neurosci. 2017;37:1797–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tang X, Liu K, Hamblin MH, Xu Y and Yin KJ. Genetic Deletion of Kruppel-Like Factor 11 Aggravates Ischemic Brain Injury. Mol Neurobiol. 2018;55:2911–2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang J, Yuan L, Zhang X, Hamblin MH, Zhu T, Meng F, Li Y, Chen YE and Yin KJ. Altered long non-coding RNA transcriptomic profiles in brain microvascular endothelium after cerebral ischemia. Exp Neurol. 2016;277:162–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yin KJ, Fan Y, Hamblin M, Zhang J, Zhu T, Li S, Hawse JR, Subramaniam M, Song CZ, Urrutia R, Lin JD and Chen YE. KLF11 mediates PPARgamma cerebrovascular protection in ischaemic stroke. Brain. 2013;136:1274–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yin KJ, Hsu CY, Hu XY, Chen H, Chen SW, Xu J and Lee JM. Protein phosphatase 2A regulates bim expression via the Akt/FKHRL1 signaling pathway in amyloid-beta peptide-induced cerebrovascular endothelial cell death. J Neurosci. 2006;26:2290–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yin KJ, Deng Z, Hamblin M, Xiang Y, Huang H, Zhang J, Jiang X, Wang Y and Chen YE. Peroxisome proliferator-activated receptor delta regulation of miR-15a in ischemia-induced cerebral vascular endothelial injury. J Neurosci. 2010;30:6398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yin KJ, Deng Z, Huang H, Hamblin M, Xie C, Zhang J and Chen YE. miR-497 regulates neuronal death in mouse brain after transient focal cerebral ischemia. Neurobiol Dis. 2010;38:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Correction to: MicroRNA-17–92 Cluster in Exosomes Enhance Neuroplasticity and Functional Recovery After Stroke in Rats. Stroke. 2017;48:e137. [DOI] [PubMed] [Google Scholar]
- 64.Liu X, Liu J, Zhao S, Zhang H, Cai W, Cai M, Ji X, Leak RK, Gao Y, Chen J and Hu X. Interleukin-4 Is Essential for Microglia/Macrophage M2 Polarization and Long-Term Recovery After Cerebral Ischemia. Stroke. 2016;47:498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang X, Suenaga J, Pu H, Wei Z, Smith AD, Hu X, Shi Y and Chen J. Post-stroke administration of omega-3 polyunsaturated fatty acids promotes neurovascular restoration after ischemic stroke in mice: Efficacy declines with aging. Neurobiol Dis. 2018. [DOI] [PubMed] [Google Scholar]
- 66.Wang J, Shi Y, Zhang L, Zhang F, Hu X, Zhang W, Leak RK, Gao Y, Chen L and Chen J. Omega-3 polyunsaturated fatty acids enhance cerebral angiogenesis and provide long-term protection after stroke. Neurobiol Dis. 2014;68:91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sun P, Bu F, Min JW, Munshi Y, Howe MD, Liu L, Koellhoffer EC, Qi L, McCullough LD and Li J. Inhibition of calcium/calmodulin-dependent protein kinase kinase (CaMKK) exacerbates impairment of endothelial cell and blood-brain barrier after stroke. Eur J Neurosci. 2019;49:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sun P, Sole M and Unzeta M. Involvement of SSAO/VAP-1 in oxygen-glucose deprivation-mediated damage using the endothelial hSSAO/VAP-1-expressing cells as experimental model of cerebral ischemia. Cerebrovasc Dis. 2014;37:171–80. [DOI] [PubMed] [Google Scholar]
- 69.Sun P, Hernandez-Guillamon M, Campos-Martorell M, Simats A, Montaner J, Unzeta M and Sole M. Simvastatin blocks soluble SSAO/VAP-1 release in experimental models of cerebral ischemia: Possible benefits for stroke-induced inflammation control. Biochim Biophys Acta Mol Basis Dis. 2018;1864:542–553. [DOI] [PubMed] [Google Scholar]
- 70.Yin KJ, Olsen K, Hamblin M, Zhang J, Schwendeman SP and Chen YE. Vascular endothelial cell-specific microRNA-15a inhibits angiogenesis in hindlimb ischemia. J Biol Chem. 2012;287:27055–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arnaoutova I and Kleinman HK. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat Protoc. 2010;5:628–35. [DOI] [PubMed] [Google Scholar]
- 72.Smits M, Mir SE, Nilsson RJ, van der Stoop PM, Niers JM, Marquez VE, Cloos J, Breakefield XO, Krichevsky AM, Noske DP, Tannous BA and Wurdinger T. Down-regulation of miR-101 in endothelial cells promotes blood vessel formation through reduced repression of EZH2. PLoS One. 2011;6:e16282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liang CC, Park AY and Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.