

Abstract
Background
Alignment of sequence reads generated by next-generation sequencing is an integral part of most pipelines analyzing next-generation sequencing data. A number of tools designed to quickly align a large volume of sequences are already available. However, most existing tools lack explicit guarantees about their output. They also do not support searching genome assemblies, such as the human genome assembly GRCh38, that include primary and alternate sequences and placement information for alternate sequences to primary sequences in the assembly.
Findings
This paper describes SRPRISM (Single Read Paired Read Indel Substitution Minimizer), an alignment tool for aligning reads without splices. SRPRISM has features not available in most tools, such as (i) support for searching genome assemblies with alternate sequences, (ii) partial alignment of reads with a specified region of reads to be included in the alignment, (iii) choice of ranking schemes for alignments, and (iv) explicit criteria for search sensitivity. We compare the performance of SRPRISM to GEM, Kart, STAR, BWA-MEM, Bowtie2, Hobbes, and Yara using benchmark sets for paired and single reads of lengths 100 and 250 bp generated using DWGSIM. SRPRISM found the best results for most benchmark sets with error rate of up to ∼2.5% and GEM performed best for higher error rates. SRPRISM was also more sensitive than other tools even when sensitivity was reduced to improve run time performance.
Conclusions
We present SRPRISM as a flexible read mapping tool that provides explicit guarantees on results.
Keywords: NGS, short reads, alignment, SRPRISM, alternate loci
Background
Rapid development of DNA sequencing technology resulted in an increasingly large amount of sequence data being generated and submitted to public databases, creating a demand for efficient tools to align nucleotide reads to reference genomes. Several such tools have emerged, and they generally fall in two categories.
One group consists of BLAST-like programs that search for local alignments of reads and aim for high sensitivity. These tools are relatively slow, require computers with a large amount of RAM or clusters of several computers running in parallel, and do not guarantee full sensitivity. Such tools as SHRiMP [1], BFast [2], and nucleotide BLAST itself [3] fall in this category.
The other group consists of fast aligners that can align large volumes of reads. Aligners in this group have an additional advantage in that they do not require a lot of memory to work, which makes using them practical on medium- to high-level desktop computers. However, such aligners usually make some sacrifices to achieve good run time performance. Sensitivity is most commonly sacrificed, with some aligners having limits on the reference genome size, the ability to align with gaps, the number of alignments reported, or the alignment of paired reads. Examples of such tools are BWA [4], GEM [5], and Bowtie2 [6,7] based on Burrows-Wheeler transform; Ψ-RA aligner [8] based on suffix arrays; SNAP [9], SARUMAN [10], and SeqAlto [11] using k-mer indexing, and ZOOM [12] using spaced seeds. Kart [13] uses both Burrows-Wheeler transform and hash indexing. Some tools try to maintain sensitivity and achieve speed improvements using hardware-specific optimizations, such as SOAP3-dp [14], BarraCUDA [15], and CUSHAW [16]. Only a very small number of aligners, such as BatMis [17], RazerS 3 [18], Hobbes [19], Yara (formerly Masai) [20], and mrFAST [21], explicitly attempt to report all good alignments. STAR [22] is a popular splice-aware aligner with options that can be set to find unspliced alignments.
Comparison of read alignment tools has been done for software robustness [23], choice of parameters and algorithm features [24], effect of aligner on downstream processing [25,26], and properties of the reference genome to which reads are aligned [27]. A review of the read alignment problem for various applications and technologies was presented by Reinert et al. [28]. Comparisons of software tools use simulated reads, real-world read sets, or benchmarks developed using tools such as DWGSIM [29], Rabema [30], or Seal [31].
SRPRISM (Single Read Paired Read Indel Substitution Minimizer) is an aligner that aims to achieve low memory footprint, capability of being run on multiple platforms and commodity hardware, and options that provide a good balance between sensitivity and speed. SRPRISM has many capabilities present in most aligners—it can align both single and paired reads, supports alignments that have substitutions as well as gaps, and can process a large volume of reads efficiently. In addition, there are several important features that distinguish SRPRISM from other aligners, as described below. SRPRISM generates output in SAM format [32].
An important property of SRPRISM is that it is possible to list precisely the conditions that guarantee full sensitivity. Specifically, if certain limits on the read length and requested number of errors are satisfied and that read can be aligned to a reference with at most that many errors, then SRPRISM is guaranteed to report best scoring mappings for that read. Moreover, if the number of equivalent best mappings does not exceed a configurable upper bound on the number of mappings to be reported, then all such mappings are guaranteed to be reported.
Another distinguishing feature of SRPRISM is its support for genome assemblies, such as the current human assembly GRCh38, which uses the Genome Reference Consortium (GRC) [33] defined assembly model. This model accommodates sequence representations that can introduce allelic duplication, including alternate loci and patches. Alternate loci are extra-chromosomal assembly sequences that provide variant representations for chromosomal regions. Patches are operationally similar to alternate loci and provide a means for the public release of assembly updates without disruption to reference chromosome coordinates. The relationship of alternate loci and patches to the chromosomes is defined by alignments that are included as part of the assembly release. SRPRISM currently does not distinguish between alternate loci and patches and treats them as equivalent substitutions for defined chromosomal regions. SRPRISM does not support patches or alternate loci for other alternate loci. Consequently, all sequences in the genome assembly are divided into two disjoint classes: “primary sequences” and “alternate sequences.” The alignment between alternate and primary sequences is used by SRPRISM to adjust the mapping quality scores when reads align to chromosomal regions that have alternate sequence representations. This also finds mappings that partially cover both the alternate locus and the primary sequence at the junctions where the alternate locus joins the primary sequence. To our knowledge, SRPRISM, BWA-MEM [34] (an algorithm in BWA), and iBWA [35] are the only software that have support for assemblies with alternate loci. However, the alignment information between primary and alternate sequences in the reference needed by the tools differs. BWA-MEM requires base by base alignment information (commonly called “traceback”), but SRPRISM and iBWA only require end-points of alignments.
When aligning paired reads using SRPRISM, the desired range of insert sizes and strand configuration of mates can be specified explicitly. Alternatively, SRPRISM has the ability to discover these parameters automatically.
When reads are aligned globally, SRPRISM offers three schemes for ranking alignments: “minimum errors,” “bounded errors,” and “sum of errors.” In the minimum error mode, the maximum number of errors in alignment of either mate of a read is minimized with minimum number of errors in alignments of both mates as the second key. In bounded error mode, all alignments with number of errors at most the given bound are considered equally good. In sum of errors mode, alignments are ranked using the sum of errors seen in both mates of a paired alignment. Sum of errors limits total number of edits for paired reads and is the same as minimum errors for single reads. Minimum errors mode could be more appropriate when aligning to a very close genome where only a negligible number of differences per read are expected. Bounded errors could be useful for applications assessing number of similar copies for a region in an assembly or doing cross-species alignments.
In addition to aligning reads globally, SRPRISM offers a “partial” alignment mode. In the partial mode, SRPRISM looks for best partial read alignments (i.e., maximizing a certain score function) provided a user-designated trusted region of the read, called the “seeding region,” is fully aligned within the maximum number of errors allowed by the length of the specified seeding region. This gives an opportunity to align read sets with known defects, like low-quality tail regions.
SRPRISM has some limitations that must be considered when selecting it as an alignment tool. The maximum number of errors searched for by SRPRISM is limited to 15 errors in the aligned portion of the read. Second, SRPRISM can handle read lengths in the range 16–8,192 bp, but because of the limit on the number of errors, SRPRISM is best suited for aligning reads of length up to 250 bp, such as reads generated by Illumina sequencing technology, the dominant technology at this time [36].
An emerging alternative way to capture an organism’s genetic variation is by modeling genomes as graphs. Methods and tools to align sequences to graph genomes are described, e.g., in [37–44]. Comparison of SRPRISM to these aligners is outside of the scope of this paper.
In the following sections, we briefly describe the design of SRPRISM software. We also describe the testing methodology including details on benchmark sets and software settings used for comparison to GEM, Kart, STAR, BWA-MEM, Bowtie2, Hobbes, and Yara. We did not include iBWA for comparisons because BWA is the core aligner for both iBWA and BWA-MEM. Technical details, including pseudocode and software optimizations, are available in the supplementary material.
Algorithm
SRPRISM creates an index and stores it once per reference assembly. The index can then be used for multiple searches against that reference. We present the structure of the reference index, steps taken by the search procedure when reads are aligned using the index, properties of results reported, and additional information about the major steps including processing of alternate sequences in reference and key optimizations.
Reference index
SRPRISM indexes words on the positive strand of the reference genome and stores them in a database. An SRPRISM database consists of two main parts. The first part contains compressed sequence data encoded at two bits per nucleotide (A, C, G, and T encoded as integers 0, 1, 2, and 3, respectively) along with metadata that has information about ambiguities, individual sequence location in the database, correspondence between alternate and primary sequences, and a frequency map containing the base 2 logarithm of frequency lf(w) for each word w in the database. This part is always loaded in memory, requiring a relatively small amount of space (e.g., ∼800 MB for GRCh38).
The second part of an SRPRISM database contains the locations of occurrences of unambiguous words of length 16 in the genome, sorted in the order of integer values of those words in the 2-bit per base encoding. This represents the bulk of the database. The SRPRISM pattern of access for this part is strictly sequential; as a consequence, only a small part of the database has to be present in memory at any given time during the search. Additional information for the neighboring words is kept for the repetitive words (see supplementary material for details).
SRPRISM search: bird’s eye view
SRPRISM aligns both single reads and paired reads. For ease in describing the alignment procedure, we use the term “query” for user input that can be single reads or paired reads and the term “read” for single reads or individual mates of paired reads.
SRPRISM is designed for batch queries. The batch size can be specified explicitly by the user (in which case the user can also directly control which batches to process) or is otherwise inferred from the amount of available memory. The following subsections describe the processing of a single batch of queries. If there is more than one batch, they are all processed in the same way. Fig. 1 gives a high-level overview of the stages involved in the processing of one batch of queries by SRPRISM.
Figure 1:

Processing of a batch of queries by SRPRISM.
SRPRISM operates by selecting and sorting a subset of 16-bp words from all reads. This sorted subset is matched against the sorted set of words in the database. These initial matches are then extended via an alignment procedure that is greedy in nature but guarantees optimal results up to the number of errors requested by exploring the search space needed for the guarantee. For queries with paired reads, if a paired mapping is found for the query, no unpaired mappings are reported for the reads in that query. Four search modes are supported:
(i) Minimum error mode: For alignments of a query with single read or unpaired alignments of reads in a paired query, the rank is the same as the number of errors in the alignment. For a query with paired reads, the rank of a paired mapping has its first key as the maximum of the number of errors in individual read alignments and its second key as the minimum of the number of errors in individual read alignments. We report alignments with minimum rank.
(ii) Bounded error mode: All alignments for each read with up to the user-specified number of errors are reported.
(iii) Sum of errors mode: For single reads, the rank is the same as the rank in the minimum error mode. For queries with paired mappings, the rank is the sum of the number of errors in the individual read alignments. We report alignments with minimum rank.
(iv) Partial mode: The individual read alignments are assigned a rank equal to the number of read bases in the alignment, excluding the unaligned portion of the read, minus the number of errors. The rank of a paired alignment is the sum of ranks of the individual read alignments. We report alignments with maximum rank.
SRPRISM operates as a global aligner in the minimum error, bounded error, and sum of errors modes. In the partial mode, non-global alignments are allowed.
What is reported
Let N be the user-defined maximum number of mappings to be reported per query (default 10, maximum 254). If the number of best-ranked mappings for a query to primary or an alternate exceeds N, then SRPRISM reports N mappings to the primary and each alternate exceeding the limit; all the best mappings are reported for the rest. When more than N mappings are available for a query to primary or an alternate sequence, mappings with a larger number of mismatches are preferred for reporting to those with gaps. For paired mappings, the number of mismatches for each mapping is taken to be the maximum in the individual read alignments in the mapping.
Let ℓ be the length of the seeding region SR as described in the next section. SRPRISM is guaranteed not to miss a mapping that has at most K errors in the seeding region, where K = min (K0, K1, 15), K0 is the user-specified maximum number of errors for reported alignments (default 5), and K1 is defined as follows:
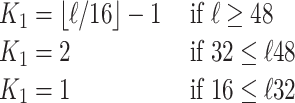 |
(1) |
Each reported mapping is assigned a mapping quality value in the following way. Let R be the number of mappings found for query q that will be reported. All mappings for q in R will have the same best rank and are assigned the same quality value
 |
(2) |
The computation of the number of mappings R includes adjustments needed for alternate loci, which are described in the section “Alignment to alternate sequences.” Q = 0 indicates that more mappings of the desired rank can be found in the database but are not being reported because only N mappings were requested.
SRPRISM supports a “heuristic” mode, where seeding is restricted to words with a frequency less than the user-specified threshold. In this mode, search is done exactly as in the sensitive mode. In the heuristic mode, if SRPRISM determines that it can find the complete set of best ranked mappings for a query, this is indicated in SAM format by a flag XA:i:1. Otherwise the flag is set to XA:i:0. This flag is supported for the minimum error and sum of error modes.
Query analysis and seed selection
In this subsection, we describe how a set of seeds 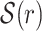 is selected for a given read r. For a set S of words from r, its combined frequency is defined as F(S) = ∑w ∈ Slf(w). The collection
is selected for a given read r. For a set S of words from r, its combined frequency is defined as F(S) = ∑w ∈ Slf(w). The collection 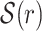 is selected differently depending on the length ℓ(r) of r as illustrated in Fig. 2.
is selected differently depending on the length ℓ(r) of r as illustrated in Fig. 2.
Figure 2:

Seed selection for reads of different lengths.
Let w be a word within the read r and s be a subsequence of w. We define 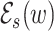 to be a set of all words that can be obtained from w by introducing one error in s (in the case of insertions/deletions, one letter from r is shifted in or out of w either from the left or from the right).
to be a set of all words that can be obtained from w by introducing one error in s (in the case of insertions/deletions, one letter from r is shifted in or out of w either from the left or from the right).
The seeding region SR for a query can be explicitly specified by the user. Otherwise, the entire read is considered to be the seeding region. All words in 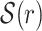 originate from an interval SA(r) ⊆ SR of the read r, called the “seeding area” of r. If SR is less than 48 bases, SA(r) is taken to be the whole of SR. Otherwise, SA(r) is defined in item 1 below, where ℓ is the length of SR and K is the number of errors.
originate from an interval SA(r) ⊆ SR of the read r, called the “seeding area” of r. If SR is less than 48 bases, SA(r) is taken to be the whole of SR. Otherwise, SA(r) is defined in item 1 below, where ℓ is the length of SR and K is the number of errors.
1. Case 16(K + 1) ≤ ℓ: let Wp be the set of K + 1 consecutive non-overlapping words starting at offset p of SR (see Fig. 2a), where 0 ≤ p ≤ ℓ − 16(K + 1). Then
 is taken to be one of Wp with the least combined frequency. In this case, SA(r) is the interval between the start of the first word and the end of the last word in
is taken to be one of Wp with the least combined frequency. In this case, SA(r) is the interval between the start of the first word and the end of the last word in 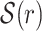 .
.2. Case K = 1 and 32 > ℓ ≥ 16: let s be the area of overlap of w1 and w2 (see Fig. 2b) and
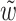 be one of w1 and w2 with the least frequency (w1 = w2 when ℓ = 16). Then
be one of w1 and w2 with the least frequency (w1 = w2 when ℓ = 16). Then 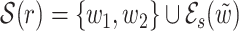 .
.3. Case K = 2 and 48 > ℓ ≥ 32: let W be one of two sets {w1, w2, w3} and {w1, w2, w4} (see Fig. 2c) with the least combined frequency (w3 = w4 when ℓ = 32). Let
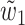 and
and  be two overlapping words in W and s be their area of overlap. Let also
be two overlapping words in W and s be their area of overlap. Let also  be one of
be one of 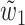 and
and  with the least frequency. Then
with the least frequency. Then 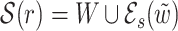 .
.
A deterministic tie-breaking procedure is used in cases where the frequency comparisons, as described above, result in a tie (see supplementary material for details).
The selection of seeds ensures that for any potential alignment of a read r to the database, satisfying conditions 1, there is at least one word in  that aligns exactly (see supplementary material for additional details).
that aligns exactly (see supplementary material for additional details).
It is possible that the same alignment can be found by extending several seeds in  . To avoid duplication of mappings, for each read, SRPRISM keeps track of which seeds it has seen already and will not keep the alignments that could have been found by previously seen seeds.
. To avoid duplication of mappings, for each read, SRPRISM keeps track of which seeds it has seen already and will not keep the alignments that could have been found by previously seen seeds.
Processing of queries with single reads
SRPRISM search proceeds by scanning sorted lists of 16-bp seeds from the reads and from the database. For any match found, the locations for the matching word are extracted from the database and alignments of corresponding reads to the database sequences at those locations are attempted. Successful alignments are stored in a temporary file, grouped by query, post-processed, and reported to the output stream.
Processing of queries with paired reads
For queries consisting of paired reads, alignments are first searched for each read independently, treating each read as a query with single read. If both reads in a query produce mappings, the number of mappings found for each read is within the bound for the number of mappings requested, and a paired alignment in the correct insert size range and with the correct strand configuration can be computed from the read alignments; then paired mappings for such queries are produced from the alignments of reads.
For all remaining queries where both reads produce at least one mapping, frequency information is used to decide which of the two reads in the query is less repetitive. The read that is less repetitive is selected as “master” and the other read is designated as “slave.” The single-read search is repeated for the master to find all alignments with up to the specified number of errors. For each candidate alignment found, an attempt is made to align the slave in–place within the insert size and strand configuration dictated neighborhood of the alignment location.
Alignments for reads in queries for which no paired alignments are found are reported as single alignments.
Alignment to alternate sequences
The relationship between each alternate sequence and corresponding primary sequences is available as a file in the assembly release. Specification has mapping information for the end points of alternate sequences to the coordinate space of the primary genome. If the precise mapping of an end point for an alternate sequence to its primary sequence is not known, such end points are marked as “fuzzy.”
SRPRISM extends alternate sequences over the non-fuzzy end points via segments of the primary sequences of configurable length as shown in Fig. 3. The length of the extension depends on read length and insert size and is determined so as to ensure that alignments overlapping the end point of an alternate sequence are correctly extended to the primary assembly. For any alternate sequence s in the database after such extension, we use notation s′ for the original non-extended alternate sequence (s and s′ are the same if both ends of the alternate sequence are marked as fuzzy).
Figure 3:

Metadata records for an alternate sequence and the corresponding primary sequence. Alternate sequence is extended in one or both directions with segments from the primary sequence of configurable length (shown in blue) to allow for correct identification of paired alignments. Extension is done for end points where the alignment of alternate to the primary is not fuzzy in the information provided by GRC. The dashed red sequence is the region of the primary sequence that is conceptually replaced by the solid red region from the alternate sequence when adjusting mapping quality scores.
We say that an alignment a of read r to an alternate locus sequence s is “proper” if the aligned portion of s overlaps with s′. A paired-end alignment is proper for s if at least one of the individual read alignments is proper.
The computation of quality values Q for the mappings corresponding to alternate sequences is described by equation (2), but R is defined differently.
For a query q and alternate sequence s corresponding to the primary sequence spri, let 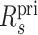 be the number of mappings of q to the primary portion of the database, not counting the mappings that overlap the region of spri replaced by s. Let Rs be the number of proper mappings of q to the sequence s. We then define
be the number of mappings of q to the primary portion of the database, not counting the mappings that overlap the region of spri replaced by s. Let Rs be the number of proper mappings of q to the sequence s. We then define 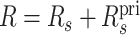 in equation (2) for the purpose of computing the quality of a mapping aligning q to s.
in equation (2) for the purpose of computing the quality of a mapping aligning q to s.
Key optimizations
SRPRISM implements a number of optimizations intended to improve the performance of the search.
For every 16-bp word that appears in the SRPRISM database more than hundred times, the database also contains information about its 16-bp neighbor words. This information is used by the aligner to quickly filter out 32-bp words that do not match the read sequences with up to two errors. This greatly reduces the number of initial matches designated for further extension without violating the guarantees (see supplementary material for details).
For each query, SRPRISM tracks the best alignment rank seen, which allows it to reduce the search space for subsequent alignments and to stop the search early when it can prove that all of the best alignments are already found.
In addition to the above, SRPRISM can be instructed not to use as seeds the 16-mers that appear more than a specified number of times in the reference database. This allows some sensitivity to be traded for performance. Our testing shows that limiting the word frequency to 4,096 can greatly improve SRPRISM run time performance while sacrificing very little in sensitivity. The number of queries that have XA:i set to value 0, indicating that the result is not guaranteed, is also typically a very small percentage of the total number of queries (data not shown).
Operation
SRPRISM is implemented in C++ on Linux OS. We recommend providing 4 GB memory to SRPRISM for alignment to reference genomes similar in size to the human genome. Steps needed for running SRPRISM are creating an index for the reference and searching the index with reads (see supplementary material Tables 1–3 for command lines). The reads can be provided as files in fasta or fastq format, or they can be accessed directly from the SRA.
Methods
Benchmark datasets generated, parameter settings used for different software compared, and the method used for evaluating alignments are presented in this section.
Datasets used for comparison
Paired query sets were created using DWGSIM version 0.1.12 (DWGSIM, RRID:SCR_002342) [29]. Reads of length 100 and 250 bp were generated using primary sequences in GRCh38 with insert size of 500 and 600 bp, respectively. Each set contains one hundred million paired queries. Error rates considered for both read lengths were in steps of 0.5% from 0.5% to 4%, excluding 3.5%. This gives a total of 14 different benchmark sets. Table S1 in the supplementary material has command lines for creating benchmark sets.
Benchmark sets with single queries consisted of the first read of the paired benchmark sets generated above.
For comparing the running time, we generated one million queries at the same read lengths, insert size, and error rates.
Test runs
SRPRISM performance was compared with GEM version 3.6.1 (GEM, RRID:SCR_005339) [5], Kart version 2.5.2 [13], STAR version 2.7.1a (STAR, RRID:SCR_015899) [22], BWA-MEM version 0.7.12 (BWA, RRID:SCR_010910) [4], Bowtie2 Beta version 2.0.0-beta6 (Bowtie, RRID:SCR_005476) [6], Hobbes (version 3.0.0) [19], and Yara (version 0.9.3) [20]. Index databases were created for each of the tested programs for GRCh38 primary assembly. All runs for all methods were single threaded as SRPRISM currently does not have a multi-threaded mode of operation.
Two sets of SRPRISM runs were performed: (i) runs with full sensitivity and (ii) runs where seeds were limited to k-mers occurring at most 4,096 times in the reference database. We refer to these runs as “sensitive” and “fast” runs correspondingly. In both cases SRPRISM was instructed to use 4 GB of RAM. Only the sum of the error ranking scheme was used for comparison.
GEM runs were performed using mapping mode fast and sensitive and are referred to as “fast” and “sensitive,” respectively. Kart was run with the option to report multiple mappings. We tested BWA-MEM with two sets of runs: (i) with most parameters set to their default values, which matches the common use; and (ii) with parameters set to closely match SRPRISM. These runs are referred to as “default” and “custom,” respectively. Bowtie2 runs were done with–very-sensitive mode and requesting up to 10 mappings per query to estimate the best sensitivity. STAR options for minimum and maximum intron length were set to 2 and 1 bp, respectively, to find unspliced alignments.
The database index for Hobbes was created with the recommended qgram length of 11. All programs that have options to specify insert size were given a range of 10–990 bp. Tables S2–S4 in the supplementary material provide the command lines for each aligner that were used for aligning paired reads, single reads, and creating the index.
Methods that gave the best result on at least one benchmark were GEM, BWA-MEM, and SRPRISM. We performed run time performance comparison using both settings for each of these three methods.
Evaluation of results
For each query q in each benchmark set S, we find the target number of errors for q in S as the minimum sum of errors for any valid alignment inferred from any of the methods tested. Every alignment reported is valid for single queries. Additional requirements for a paired alignment inferred from read alignments for a paired query reported by a method to be valid are that the paired alignment be in proper forward-reverse orientation and within the specified insert size. We use the number of queries that did not find the target number of errors as the criterion for comparing the sensitivity of different methods.
For sensitive SRPRISM runs, we further investigated all queries for which (i) there was a valid result reported by any method, (ii) the read in the single query or both reads in the paired query had at most the number of errors that SRPRISM guarantees to find (5 errors for 100 bp and 14 for 250 bp reads), and (iii) SRPRISM did not report a valid result at the target number of errors.
For each read r in each benchmark set S, the benchmark specifies position P in the genome from where r is generated and the number of errors E introduced. The second measure of evaluation defines the position p of an alignment for r in S to be at an “acceptable position” if and only if p differs from P by at most E. The deviation from P by E positions is to account for potentially equally good alignments at the same location in the genome. We use the number of reads that did not find an alignment at an acceptable position as a criterion for comparing the correctness of alignments reported by different methods. This criterion is also independent of the scoring scheme used by each software package.
All runs for run time performance tests were performed on a 2.2-GHz Intel Xeon E5-2660 CPU, with 128 GB of RAM. Each run was performed 3 times, and the final time was taken as a minimum total user and system time over 3 runs.
Results and Discussion
Quality of results
For each benchmark set and each method tested, Table 1 reports the number of queries for which a valid result at the target number of errors was not found. The sensitive mode of GEM performed well for 100-bp paired and single queries at all error rates except at 0.5%. The sensitive mode of GEM also performed well at high error rates for 250-bp paired and single queries. Both modes of SRPRISM performed well for paired and single 250-bp queries for error rates up to 2%. The sensitive mode of SRPRISM also performed best at 0.5% error rate for 100-bp paired and single queries. BWA-MEM in custom mode narrowly outperformed GEM in sensitive mode at high error rate of 4% for 250-bp single queries. Hobbes and Yara performed well only for single queries at low error rates.
Table 1:
Number of queries out of 100,000,000 in each benchmark set for which a mapping at the target number of errors is not reported
| Method | 0.5% | 1% | 1.5% | 2% | 2.5% | 3% | 4% |
|---|---|---|---|---|---|---|---|
| 100-bp paired sets | |||||||
| GEM fast | 208,716 | 452,079 | 687,939 | 915,017 | 1,152,014 | 1,420,600 | 2,144,373 |
| GEM sensitive | 24,808 | 54,032 | 92,371 | 142,406 | 204,455 | 281,705 | 523,960 |
| BWA-MEM default | 225,610 | 614,532 | 1,230,792 | 2,097,907 | 3,239,775 | 4,681,048 | 8,563,643 |
| BWA-MEM custom | 2,359,073 | 2,384,496 | 2,419,891 | 2,466,556 | 2,527,910 | 2,607,907 | 2,835,866 |
| SRPRISM fast | 616,704 | 830,673 | 1,785,953 | 4,346,647 | 9,318,663 | 16,987,001 | 38,310,302 |
| SRPRISM sensitive | 13,781 | 161,295 | 978,406 | 3,371,025 | 8,198,853 | 15,783,121 | 37,192,459 |
| (check count) | (2) | (2) | (13) | (17) | (18) | (27) | (32) |
| Hobbes | 13,791 | 161,310 | 978,421 | 3,371,041 | 8,198,870 | 15,783,133 | 37,192,468 |
| Yara | 2,028,722 | 2,320,285 | 3,218,186 | 5,616,159 | 10,368,429 | 17,790,749 | 38,676,640 |
| Kart | 3,099,642 | 4,906,886 | 7,970,113 | 12,270,554 | 17,617,775 | 23,734,421 | 37,223,055 |
| STAR | 3,936,831 | 5,998,191 | 8,153,960 | 10,520,656 | 13,461,104 | 17,566,471 | 30,942,844 |
| Bowtie2 | 566,714 | 824,542 | 1,216,097 | 1,659,296 | 2,180,881 | 2,783,325 | 4,344,807 |
| 250-bp paired sets | |||||||
| GEM fast | 191,675 | 309,943 | 396,546 | 493,401 | 620,435 | 783,967 | 1,250,672 |
| GEM sensitive | 17,437 | 38,105 | 73,760 | 128,479 | 205,774 | 309,672 | 620,641 |
| BWA-MEM default | 175,984 | 512,561 | 1,071,537 | 1,864,593 | 2,917,694 | 4,234,163 | 7,718,972 |
| BWA-MEM custom | 4,163,355 | 4,166,258 | 4,180,166 | 4,198,419 | 4,216,046 | 4,241,512 | 4,301,237 |
| SRPRISM fast | 7,243 | 10,171 | 20,220 | 82,247 | 485,610 | 2,158,312 | 15,963,870 |
| SRPRISM sensitive | 6 | 103 | 2,896 | 52,640 | 435,848 | 2,082,873 | 15,842,838 |
| (check count) | (0) | (1) | (0) | (2) | (4) | (7) | (6) |
| Hobbes | 712,059 | 9,957,606 | 34,283,316 | 62,109,460 | 81,094,426 | 90,103,730 | 94,380,319 |
| Yara | 1,118,568 | 1,139,838 | 1,182,725 | 1,584,865 | 3,609,820 | 9,519,961 | 37,597,089 |
| Kart | 3,291,334 | 6,529,283 | 12,026,850 | 19,338,546 | 27,932,293 | 37,124,040 | 55,155,698 |
| STAR | 3,705,691 | 7,288,696 | 20,709,476 | 46,098,586 | 70,500,124 | 85,216,290 | 93,852,773 |
| Bowtie2 | 511,367 | 1,029,144 | 1,575,666 | 1,944,531 | 2,258,863 | 2,568,040 | 3,311,748 |
| 100-bp single sets | |||||||
| GEM fast | 697,818 | 1,238,906 | 1,799,896 | 2,431,653 | 3,193,987 | 4,126,182 | 6,625,844 |
| GEM sensitive | 18,479 | 41,393 | 68,604 | 99,672 | 137,949 | 190,986 | 364,521 |
| BWA-MEM default | 131,829 | 353,621 | 708,158 | 1,211,188 | 1,883,318 | 2,750,676 | 5,143,324 |
| BWA-MEM custom | 41,331 | 89,456 | 161,560 | 259,535 | 388,985 | 554,852 | 1,017,491 |
| SRPRISM fast | 354,827 | 482,556 | 970,542 | 2,261,776 | 4,824,603 | 8,930,551 | 21,561,640 |
| SRPRISM sensitive | 6,871 | 79,825 | 487,266 | 1,686,967 | 4,163,766 | 8,201,636 | 20,791,635 |
| (check count) | (5) | (3) | (6) | (9) | (13) | (14) | (13) |
| Hobbes | 6,879 | 79,836 | 487,278 | 1,686,978 | 4,163,781 | 8,201,647 | 20,791,648 |
| Yara | 7,045 | 80,320 | 488,388 | 1,688,898 | 4,166,532 | 8,205,171 | 20,796,339 |
| Kart | 2,316,648 | 3,458,734 | 5,354,805 | 8,018,703 | 11,393,479 | 15,370,874 | 24,641,561 |
| STAR | 2,933,574 | 3,858,421 | 4,892,676 | 6,021,576 | 7,255,494 | 8,607,920 | 11,708,200 |
| Bowtie2 | 434,413 | 1,162,865 | 2,147,595 | 3,088,132 | 4,031,885 | 5,019,518 | 7,278,163 |
| 250-bp single sets | |||||||
| GEM fast | 470,499 | 613,415 | 711,556 | 829,955 | 984,829 | 1,185,478 | 1,753,151 |
| GEM sensitive | 12,230 | 21,540 | 35,493 | 58,151 | 92,135 | 141,456 | 294,110 |
| BWA-MEM default | 96,944 | 274,979 | 563,872 | 973,153 | 1,515,439 | 2,195,157 | 4,019,429 |
| BWA-MEM custom | 89,619 | 98,956 | 113,424 | 131,055 | 153,179 | 180,745 | 252,940 |
| SRPRISM fast | 6,119 | 8,583 | 14,575 | 46,442 | 249,263 | 1,092,460 | 8,346,408 |
| SRPRISM sensitive | 2 | 50 | 1,505 | 26,337 | 217,830 | 1,046,440 | 8,270,701 |
| (check count) | (0) | (0) | (1) | (2) | (2) | (1) | (2) |
| Hobbes | 355,895 | 5,112,610 | 19,046,975 | 39,121,004 | 58,784,299 | 73,835,948 | 89,223,792 |
| Yara | 17 | 671 | 20,079 | 229,846 | 1,278,929 | 4,380,059 | 20,794,991 |
| Kart | 1,974,474 | 3,801,390 | 6,906,889 | 11,136,617 | 16,332,521 | 22,220,722 | 35,305,069 |
| STAR | 2,093,088 | 3,075,609 | 4,234,139 | 6,327,695 | 10,836,887 | 19,020,755 | 44,485,933 |
| Bowtie2 | 628,816 | 1,231,834 | 1,747,783 | 2,077,950 | 2,361,942 | 2,636,580 | 3,246,201 |
For each benchmark set, cell in bold italic has the best result (lowest number) and cell in bold has the second best result among all methods tested. For SRPRISM sensitive mode, numbers in rows labeled “(check count)” give the number of queries where a mapping at the target number of errors is expected to be found but was not. All such cases were found to be due to an error in the software that reported the target number of errors. Every read for which SRPRISM sensitive mode found the target number of errors but SRPRISM fast mode did not had the XA:i flag set to 0 in the alignment of SRPRISM fast mode to indicate that an exhaustive search was not done on that read in the fast mode.
For all queries where SRPRISM sensitive mode did not report the best result, we verified that either at least one read in the query had more errors than what SRPRISM guarantees to find or the valid alignments giving the target number of errors under-reported the number of errors. There were 131 paired queries and 71 single queries across all benchmark sets where under-reporting of errors led to SRPRISM sensitive mode not finding the best result. Alignments at the target number of errors for these queries were generated by Kart or BWA-MEM. In the case of Kart, it seems to us to be incorrect reporting of flags or alignment information in the SAM output format. For BWA-MEM, all such alignments had an ambiguous letter in the genome that was not counted as an error.
Hobbes reported a large number of alignments. Sometimes the same alignment was reported tens of times. Yara showed very good performance on single queries but did not perform well on paired queries because it did not find paired alignments within the insert size using the alignments of single reads. The sensitivity of Kart, STAR, and Bowtie2 was poor on our benchmark sets.
For each software package and each benchmark set, Table 2 reports the number of reads for which an alignment was not reported at an acceptable position. These results show that SRPRISM performed best for up to ∼1.5–2% error rate for 100-bp single and paired reads and up to ∼2.5% error rate for 250-bp single and paired reads. GEM performed best at higher error rates.
Table 2:
Number of reads out of 100,000,000 queries in each benchmark set that do not have an alignment reported at an acceptable position
| Method | 0.5% | 1% | 1.5% | 2% | 2.5% | 3% | 4% |
|---|---|---|---|---|---|---|---|
| 100-bp paired sets | |||||||
| GEM fast | 2,158,326 | 2,447,764 | 2,738,941 | 3,037,229 | 3,364,602 | 3,747,831 | 4,765,773 |
| GEM sensitive | 1,901,673 | 1,981,195 | 2,059,397 | 2,130,620 | 2,195,788 | 2,259,858 | 2,403,473 |
| BWA-MEM default | 3,503,900 | 3,694,196 | 3,926,933 | 4,184,533 | 4,471,295 | 4,794,740 | 5,609,957 |
| BWA-MEM custom | 8,860,867 | 8,933,255 | 9,010,309 | 9,085,946 | 9,167,742 | 9,258,394 | 9,456,612 |
| SRPRISM fast | 1,006,006 | 1,356,965 | 2,447,345 | 5,183,959 | 10,475,460 | 18,866,496 | 44,464,395 |
| SRPRISM sensitive | 344,312 | 594,231 | 1,512,225 | 4,041,830 | 9,141,276 | 17,379,822 | 42,880,979 |
| Hobbes | 32,770 | 327,910 | 1,968,851 | 6,775,367 | 16,468,195 | 31,682,305 | 74,583,521 |
| Yara | 5,597,985 | 5,911,015 | 6,829,531 | 9,289,853 | 14,250,013 | 22,277,779 | 47,149,270 |
| Kart | 3,380,032 | 4,844,934 | 6,992,011 | 9,898,739 | 13,621,352 | 18,161,419 | 29,745,827 |
| STAR | 3,825,268 | 4,385,741 | 4,946,686 | 5,705,656 | 6,970,971 | 9,220,807 | 18,954,444 |
| Bowtie2 | 2,569,020 | 2,985,751 | 3,603,612 | 4,276,062 | 5,056,422 | 5,968,209 | 8,402,287 |
| 250-bp paired sets | |||||||
| GEM fast | 712,535 | 858,612 | 975,720 | 1,087,934 | 1,209,649 | 1,341,859 | 1,670,558 |
| GEM sensitive | 501,022 | 527,993 | 552,836 | 577,930 | 600,248 | 616,809 | 644,306 |
| BWA-MEM default | 1,722,764 | 1,820,361 | 1,904,660 | 1,983,740 | 2,067,370 | 2,154,285 | 2,353,065 |
| BWA-MEM custom | 12,392,113 | 12,433,803 | 12,477,092 | 12,521,397 | 12,557,453 | 12,594,864 | 12,656,512 |
| SRPRISM fast | 48,843 | 81,640 | 121,191 | 215,034 | 651,481 | 2,370,391 | 16,983,951 |
| SRPRISM sensitive | 40,196 | 68,277 | 97,944 | 176,190 | 588,588 | 2,278,047 | 16,833,181 |
| Hobbes | 1,427,639 | 19,925,780 | 68,589,403 | 124,242,516 | 162,204,242 | 180,214,260 | 188,761,386 |
| Yara | 3,307,303 | 3,348,270 | 3,396,860 | 3,817,185 | 5,880,648 | 12,011,464 | 44,494,919 |
| Kart | 1,587,623 | 3,198,421 | 5,618,063 | 8,748,124 | 12,535,330 | 16,891,570 | 27,148,917 |
| STAR | 2,400,076 | 3,663,337 | 12,851,873 | 36,141,759 | 67,514,161 | 98,592,297 | 148,388,393 |
| Bowtie2 | 1,338,406 | 2,281,072 | 3,199,528 | 3,728,392 | 4,079,316 | 4,333,211 | 4,748,836 |
| 100-bp single sets | |||||||
| GEM fast | 2,159,515 | 2,555,599 | 3,050,646 | 3,657,962 | 4,409,461 | 5,331,848 | 7,781,124 |
| GEM sensitive | 1,893,918 | 1,960,117 | 2,025,016 | 2,091,019 | 2,162,444 | 2,242,231 | 2,443,015 |
| BWA-MEM default | 2,951,614 | 3,119,119 | 3,325,630 | 3,577,959 | 3,891,307 | 4,285,172 | 5,394,488 |
| BWA-MEM custom | 3,029,335 | 3,145,431 | 3,274,838 | 3,423,952 | 3,605,689 | 3,822,017 | 4,393,113 |
| SRPRISM fast | 999,076 | 1,258,584 | 1,856,469 | 3,242,205 | 5,879,422 | 10,030,932 | 22,671,681 |
| SRPRISM sensitive | 693,373 | 865,651 | 1,360,105 | 2,637,064 | 5,177,257 | 9,255,935 | 21,860,106 |
| Hobbes | 9,448 | 82,624 | 494,466 | 1,707,672 | 4,209,527 | 8,282,759 | 20,964,792 |
| Yara | 4,346,841 | 4,480,521 | 4,930,710 | 6,135,782 | 8,568,196 | 12,488,626 | 24,599,222 |
| Kart | 2,267,023 | 3,333,555 | 4,791,532 | 6,685,044 | 9,033,451 | 11,812,503 | 18,570,389 |
| STAR | 2,432,233 | 2,692,112 | 2,924,912 | 3,187,834 | 3,524,174 | 3,955,793 | 5,204,898 |
| Bowtie2 | 1,979,322 | 2,714,956 | 3,658,181 | 4,546,995 | 5,426,656 | 6,339,945 | 8,431,806 |
| 250-bp single sets | |||||||
| GEM fast | 778,802 | 883,079 | 994,676 | 1,125,128 | 1,274,892 | 1,451,466 | 1,899,922 |
| GEM sensitive | 670,168 | 686,437 | 700,617 | 715,160 | 727,770 | 741,177 | 765,982 |
| BWA-MEM default | 1,456,796 | 1,532,430 | 1,600,115 | 1,661,964 | 1,729,293 | 1,795,530 | 1,951,414 |
| BWA-MEM custom | 1,559,406 | 1,617,696 | 1,671,388 | 1,718,129 | 1,766,689 | 1,812,703 | 1,910,370 |
| SRPRISM fast | 124,027 | 168,441 | 212,881 | 281,998 | 519,088 | 1,394,821 | 8,688,136 |
| SRPRISM sensitive | 116,631 | 157,842 | 196,829 | 258,090 | 483,006 | 1,343,260 | 8,605,573 |
| Hobbes | 357,877 | 5,120,961 | 19,070,195 | 39,156,258 | 58,821,287 | 73,866,934 | 89,237,145 |
| Yara | 2,514,572 | 2,540,678 | 2,580,642 | 2,807,726 | 3,852,121 | 6,891,634 | 22,897,410 |
| Kart | 1,006,947 | 1,889,442 | 3,175,251 | 4,831,006 | 6,850,275 | 9,200,480 | 14,925,505 |
| STAR | 1,139,441 | 1,166,238 | 1,252,693 | 1,682,787 | 3,100,911 | 6,344,045 | 19,878,712 |
| Bowtie2 | 1,115,272 | 1,675,553 | 2,119,728 | 2,395,088 | 2,611,429 | 2,800,029 | 3,151,637 |
For each benchmark set, cell in bold italic has the best result (lowest number) and cell in bold has the second best result among all methods tested.
Running time
Figs 4 and 5 present the running times of the programs GEM, BWA-MEM, and SRPRISM for paired benchmark sets with reads of length 100 and 250 bp, respectively. Figs 6 and 7 present the running times of the same methods for the benchmark sets for single queries of length 100 and 250 bp, respectively.
Figure 4:

Run time performance for 100-bp paired benchmark sets.
Figure 5:

Run time performance for 250-bp paired benchmark sets.
Figure 6:

Run time performance for 100-bp single benchmark sets.
Figure 7:

Run time performance for 250-bp single benchmark sets.
GEM run time performance was most uniform across different error rates while run time for other methods tended to increase with respect to the error rate. GEM in fast mode was the fastest of all methods. However, for 100-bp single and paired query sets, GEM in sensitive mode was the slowest.
Conclusions
We designed SRPRISM for reliable alignment of large volumes of sequences to large genomic databases. Its main strengths are guaranteed sensitivity and features that include support for paired alignments, support for up to 15 errors (including gaps) in alignments, configurable number of reported mappings, and support for alternate loci in the reference assembly. It has a relatively low memory footprint, which makes it suitable for running on most modern hardware even when searching very large query sets against human genome–sized databases. It can also be configured for faster performance at the expense of some sensitivity, and the mappings that are not guaranteed are flagged as such.
We compared SRPRISM performance with GEM, Kart, STAR, Bowtie2, BWA-MEM, Hobbes, and Yara. We found that the fast mode of SRPRISM provides a good compromise between running speed and sensitivity and the sensitive mode of SRPRISM has reasonable speed for sets with low error rates. We also found that changing parameters for both GEM and BWA-MEM can improve sensitivity with a relatively modest increase in running time for BWA-MEM but significant increase in running time for GEM. We showed that Hobbes and Yara do not find all expected mappings and Kart, STAR, and Bowtie2 have poor sensitivity.
SRPRISM software in its current form has room for enhancements and optimizations. The features planned for the future versions include support for concurrency, additional scoring schemes for alignments, and improved processing of ambiguities in reference.
The data presented support SRPRISM being an efficient aligner that has a combination of unique features including explicit guarantees for the result set, support for alternate loci, global and partial alignments of reads, and equally efficient handling of both gaps and substitutions in alignments.
Availability of Source Code and Requirements
Project name: SRPRISM
Source code: https://github.com/ncbi/SRPRISM
Operating system: Linux/Unix
Programming language: C++
License: https://github.com/ncbi/SRPRISM/blob/master/LICENSE
Availability of Supporting Data and Materials
A README and a binary for SRPRISM are available at ftp://ftp.ncbi.nlm.nih.gov/pub/agarwala/srprism.
The files needed for generating index and alignments to GRCh38 human genome assembly are available at ftp://ftp.ncbi.nlm.nih.gov/pub/agarwala/srprism/GRCh38.
The files needed for generating index and doing a test run using GRCh38 human genome assembly files above are available at ftp://ftp.ncbi.nlm.nih.gov/pub/agarwala/srprism/testrun.
Snapshots of the code are also available in the GigaScience GigaDB repository [45].
Abbreviations
BLAST: Basic Local Alignment Search Tool; bp: base pairs; BWA: Burrows-Wheeler Aligner; GRC: Genome Reference Consortium; RAM: random access memory; SRPRISM: Single Read Paired Read Indel Substitution Minimizer; SNAP: Scalable Nucleotide Alignment Program; SRA: Sequence Read Archive.
Competing Interests
The authors declare that they have no competing interests.
Funding
This research was supported by the Intramural Research Program of the National Library of Medicine, National Institutes of Health.
Authors' Contributions
A.M. did the software development. R.A. did testing and assisted A.M. in some design decisions.
Supplementary Material
Jia-Ming Chang -- 2/11/2019 Reviewed
Jia-Ming Chang -- 9/1/2019 Reviewed
Paolo Ribeca, Ph.D. -- 5/17/2019 Reviewed
Paolo Ribeca, Ph.D. -- 3/12/2020 Reviewed
ACKNOWLEDGEMENTS
We thank Alejandro Schaffer, Deanna Church, and Valerie Schneider for their helpful suggestions for improving the exposition of the manuscript. We thank the referees for suggesting that we use simulated reads and sum of errors ranking instead of focusing our comparison on correctness only.
References
- 1. Rumble SM, Lacroute P, Dalca AV, et al.. SHRiMP: Accurate mapping of short color-space reads. PLoS Comput Biol. 2009;5(5):e1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Homer N, Merriman B, Nelson SF. BFAST: An alignment tool for large scale genome resequencing. PLoS One. 2009;4(11):e7767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Altschul SF, Madden TL, Schäffer AA, et al.. Gapped BLAST and PSI-BLAST - A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marco-Sola S, Sammeth M, Guigo R, et al.. The GEM mapper: fast, accurate and versatile alignment by filtration. Nat Methods. 2012;9(12):1185–8. [DOI] [PubMed] [Google Scholar]
- 6. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Langmead B, Trapnell C, Pop M, et al.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Oğuzhan Külekci M, Hon WK, Shah R, et al.. Ψ-RA: a parallel sparse index for genomic read alignment. BMC Genomics. 2011;12(Suppl 2):S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zaharia M, Bolosky WJ, Curtis K, et al.. Faster and more accurate sequence alignment with SNAP. arXiv. 2011:1111.5572v1. [Google Scholar]
- 10. Blom J, Jakobi T, Doppmeier D, et al.. Exact and complete short-read alignment to microbial genomes using Graphics Processing Unit programming. Bioinformatics. 2011;27(10):1351–8. [DOI] [PubMed] [Google Scholar]
- 11. Mu JC, Jiang H, Kiani H, et al.. Fast and accurate read alignment for resequencing. Bioinformatics. 2012;28(18):2366–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lin H, Zhang Z, Zhang MQ, et al.. ZOOM! Zillions of oligos mapped. Bioinformatics. 2008;24(21):2431–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin HN, Hsu WL. Kart: a divide-and-conquer algorithm for NGS read alignment. Bioinformatics. 2017;33(15):2281–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luo R, Wong T, Zhu J, et al.. SOAP3-dp: fast, accurate and sensitive GPU-based short read aligner. PLoS One. 2013;8(5):e65632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Klus P, Lam S, Lyberg D, et al.. BarraCUDA - a fast short read sequence aligner using graphics processing units. BMC Res Notes. 2012;5:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Y, Schmidt B, Maskell DL, et al.. CUSHAW: a CUDA compatible short read aligner to large genomes based on the Burrows-Wheeler transform. Bioinformatics. 2012;28:1830–7. [DOI] [PubMed] [Google Scholar]
- 17. Tennakoon C, Purbojati RW, Sung WK, et al.. BatMis: a fast algorithm for k-mismatch mapping. Bioinformatics. 2012;28(16):2122–8. [DOI] [PubMed] [Google Scholar]
- 18. Weese D, Holtgrewe M, Reinert K. RazerS 3: faster, fully sensitive read mapping. Bioinformatics. 2012;28(20):2592–9. [DOI] [PubMed] [Google Scholar]
- 19. Kim J, Li C, Xie X. Improving read mapping using additional prefix grams. BMC Bioinformatics. 2014;15:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Siragusa E, Weese D, Reinert K. Fast and accurate read mapping with approximate seeds and multiple backtracking. Nuceic Acids Res. 2013;41(7):e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alkan C, Kidd JM, Marques-Bonet T, et al.. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet. 2009;41(10):1061–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dobin A, Davis CA, Schlesinger F, et al.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Giannoulatou E, Park SH, Humphreys DT, et al.. Verification and validation of bioinformatics software without a gold standard: a case study of BWA and Bowtie. BMC Bioinformatics. 2014;15(Suppl 16):S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hatem A, Bozdağ D, Toland AE, et al.. Benchmarking short sequence mapping tools. BMC Bioinformatics. 2013;14:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bao R, Huang L, Andrade J, et al.. Review of current methods, applications, and data management for the bioinformatics analysis of whole exome sequencing. Cancer Inform. 2014;13(Suppl 2):67–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lindner R, Friedel CC. A comprehensive evaluation of alignment algorithms in the context of RNA-seq. PLoS One. 2012;7(12):e52403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yu X, Guda K, Willis J, et al.. How do alignment programs perform on sequencing data with varying qualities and from repetitive regions?. BioData Min. 2012;5(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reinert K, Langmead B, Weese D, et al.. Alignment of next-generation sequencing reads. Annu Rev Genomics Hum Genet. 2015;16:133–51. [DOI] [PubMed] [Google Scholar]
- 29. DWGSIM whole genome simulator https://github.com/nh13/DWGSIM/wiki. Accessed on March 20,2020. [Google Scholar]
- 30. Holtgrewe M, Emde AK, Weese D, et al.. A novel and well-defined benchmarking method for second generation read mapping. BMC Bioinformatics. 2011;12:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ruffalo M, LaFramboise T, Koyutürk M. Comparative analysis of algorithms for next-generation sequencing read alignment. Bioinformatics. 2011;27(20):2790–6. [DOI] [PubMed] [Google Scholar]
- 32. SAM Format https://samtools.github.io/hts-specs/SAMv1.pdf. [Google Scholar]
- 33. Genome Reference Consortium. http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/. [Google Scholar]
- 34. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. iBWA Alpha http://gmt.genome.wustl.edu/packages/ibwa/index.html. [Google Scholar]
- 36. Levy SE, Myers RM. Advancements in next-generation sequencing. Annu Rev Genomics Hum Genet. 2016;17:99–115. [DOI] [PubMed] [Google Scholar]
- 37. Rakocevic G, Semenyuk V, Lee W, et al.. Fast and accurate genomic analyses using genome graphs. Nat Genet. 2019;51:354–62. [DOI] [PubMed] [Google Scholar]
- 38. Garrison E, Sirén J, Novak AM, et al.. Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat Biotechnol. 2018;36(9):875–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Paten B, Novak AM, Eizenga JM, et al.. Genome graphs and the evolution of genome inference. Genome Res. 2017;27(5):665–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee C, Grasso C, Sharlow M. Multiple sequence alignment using partial order graphs. Bioinformatics. 2002;18(3):452–64. [DOI] [PubMed] [Google Scholar]
- 41. Kavya VNS, Tayal K, Srinivasan R, et al.. Sequence alignment on directed graphs. J Comput Biol. 2019;26(1):53–67. [DOI] [PubMed] [Google Scholar]
- 42. Jain C, Zhang H, Gao Y, et al.. On the complexity of sequence to graph alignment. J Comput Biol. 2020, doi: 10.1089/cmb.2019.0066. [DOI] [Google Scholar]
- 43. Rautiainen M, Mäkinen V, Marschall T. Bit-parallel sequence-to-graph alignment. Bioinformatics. 2019;35(19):3599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rautiainen M, Marschall T. GraphAligner: Rapid and versatile sequence-to-graph alignment. bioRxiv. 2019, doi: 10.1101/810812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Morgulis A, Agarwala R, Supporting data for “SRPRISM (Single Read Paired Read Indel Substitution Minimizer): An efficient aligner for assemblies with explicit guarantees”. GigaScience Database. 2020;. 10.5524/100709. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Jia-Ming Chang -- 2/11/2019 Reviewed
Jia-Ming Chang -- 9/1/2019 Reviewed
Paolo Ribeca, Ph.D. -- 5/17/2019 Reviewed
Paolo Ribeca, Ph.D. -- 3/12/2020 Reviewed