

Abstract
Porcine reproductive and respiratory syndrome virus (PRRSV) is an enveloped RNA virus of the Arteriviridae family, genomically related to the coronaviruses. PRRSV is the causative agent of both severe and persistent respiratory disease and reproductive failure in pigs worldwide. The PRRSV virion contains a core made of the 123 amino acid nucleocapsid (N) protein, a product of the ORF7 gene. We have determined the crystal structure of the capsid-forming domain of N. The structure was solved to 2.6 Å resolution by SAD methods using the anomalous signal from sulfur. The N protein exists in the crystal as a tight dimer forming a four-stranded β sheet floor superposed by two long α helices and flanked by two N- and two C-terminal α helices. The structure of N represents a new class of viral capsid-forming domains, distinctly different from those of other known enveloped viruses, but reminiscent of the coat protein of bacteriophage MS2.
Introduction
Respiratory disease is a common and often serious problem in humans and animals and may be caused by a wide variety of pathological agents, including coronaviruses, myxoviruses, paramyxoviruses, and picornaviruses (Strauss and Strauss, 2002). Porcine reproductive and respiratory syndrome virus (PRRSV)—also known by the name of the European strain, Lelystad virus—is a ubiquitous and highly infectious virus of pigs, causing respiratory disease, weight loss, and spontaneous abortion in infected animals and leading to great economic loss in the U.S. and Europe (Wenswoort, 1993). Measures to regulate the disease have been complicated by the high heterogeneity of the virus (Meng, 2000) and its pattern of persistent, subclinical infection with occasional epidemic outbreaks Blaha 2000, Zimmermann et al. 1997. Vaccines have been developed, but have not proven to be highly effective Meng 2000, Mengeling et al. 2003. PRRSV infects exclusively alveolar macrophages, leading to apoptosis in secondary cells and immune system suppression Molitor et al. 1997, Murtaugh et al. 2002 and is often found in combination with other pathogens, such as Streptococcus suis and Haemophilus parasuis (Zimmermann et al., 1997).
PRRSV is a member of the Arteriviridae, a recently recognized viral family genomically related to the coronaviruses Meulenberg et al. 1993, Plagemann and Moennig 1992, Snijder and Meulenberg 1998. Other arteriviruses include lactate dehydrogenase-elevating virus (LDV) of mice, equine arteritis virus (EAV), and simian hemorrhagic fever virus (SHFV). Like the coronaviruses, arteriviruses have a positive-sense single-stranded (+) RNA genome, which is expressed through a set of subgenomic mRNA transcripts, each used for the translation of one open reading frame (ORF). In the 15 kb PRRSV genome, the first and largest ORF codes for the nonstructural proteins, including two proteases and the RNA-dependent RNA polymerase. ORFs 2–5 encode glycosylated membrane proteins (GP2 to GP5), ORF6 encodes a nonglycosylated membrane protein (M), and ORF7 encodes the nucleocapsid (N) protein Dea et al. 2000, Meulenberg et al. 1995b. The N protein is the most abundant protein expressed in infected cells, comprising 40% of the proteins in the virion, and is the most immunogenic protein of the virus (Meulenberg et al., 1995a). The N protein forms a spherical, possibly icosahedral nucleocapsid core of 20–30 nm diameter, which is surrounded by a lipid envelope containing the viral membrane proteins, yielding a relatively smooth spherical virion of about 60 nm diameter (Dea et al., 2000). The N-terminal half of the 123 residue N protein contains a large number of Lys, Gln, and Asn residues (Figure 1) and is presumed to be involved in RNA binding by analogy with similar regions in other enveloped RNA viruses. The C-terminal half constitutes the shell-forming domain of the protein, and the 11 C-terminal residues have been reported previously to be important for capsid formation (Wootton et al., 2001).
Figure 1.

Sequence Alignment of the PRRSV N Protein
Alignment of the N sequence from the North American strain VR-2332 of PRRSV with the corresponding sequences from the European (Lelystad) strain and the related arteriviruses LDV, EAV, and SHFV. Completely conserved residues are indicated by solid boxes, while open boxes indicate residues that are partially conserved, according to the BLOSUM matrix. The bullet indicates the start of the NΔ57 fragment, and the secondary structure elements are represented by spirals (α helices) and arrows (β strands) above the sequence. This figure was generated with ESPript (Gouet et al., 1999).
We previously reported the cloning, expression, and crystallization of NΔ57, the capsid-forming C-terminal domain of N (Doan and Dokland, 2003). Here, we report the structure of NΔ57 solved to 2.6 Å resolution by single-wavelength anomalous diffraction (SAD) using the anomalous signal from the sulfur atoms in the native protein. The structure consists of a dimer with a four-stranded β sheet floor surrounded and superposed by α helices. It represents a novel fold and a new class of viral capsid-forming domains.
Results and Discussion
Structure Determination
NΔ57, the C-terminal 65 amino acids of the N protein from the North American strain VR-2332 of PRRSV, tagged with a polyhistidine-tag at the amino terminus, was cloned and expressed in Escherichia coli and purified as previously described (Doan and Dokland, 2003). Hexagonal rod-shaped crystals were produced by vapor diffusion against 8% PEG 3,350. These crystals diffracted anisotropically to about 2.2 Å in one direction with synchrotron radiation, but only to 2.6 Å in the worst direction. The crystals belong to space group P3221 with unit cell dimensions of a = 44.67 Å and c = 125.6 Å, giving a solvent content of 43% and a Matthews coefficient of 2.2 with a dimer in the asymmetric unit.
Earlier attempts at solving the structure by MIR or MAD methods had not been successful, as we were unable to produce suitable heavy atom derivatives and the native sequence contained no Met residues that could be used for Se-Met phasing. Finally, using single-wavelength anomalous diffraction from the two sulfur atoms in the sequence (S-SAD) has allowed us to solve the structure from the native protein. S-SAD for ab initio phasing of native data has recently been shown to be a useful technique Dauter 2002, Micossi et al. 2002, and the fact that we could solve the structure by S-SAD phasing using only relatively low-resolution data demonstrates the general applicability of this method even for data of modest quality.
For this method, a highly redundant native data set was collected at 1.74 Å wavelength at beamline BM7A at DESY, Hamburg, and processed with the HKL package (Otwinowski and Minor, 1997) (Table 1) . Starting phases were calculated with the program SOLVE (Terwilliger, 2002) using data to 2.8 Å resolution. Four S sites were identified, consistent with the presence of two Cys residues per N monomer (Figure 2A) . Density modification and autotracing was carried out with RESOLVE (Terwilliger, 2003) and resulted in a partial backbone trace of 78 (out of 144) residues (Figure 2A). Although about a third of the initial trace turned out to be incorrect, a 2-fold noncrystallographic symmetry (NCS) axis could be identified by manual inspection of the map, using this trace and the positions of the Cys residues known from the S sites. This information was used for NCS averaging and solvent flattening with the program DM (Cowtan and Main, 1998). The resulting map was sufficiently good to allow most of the sequence to be built, except for the polyhistidine tag and two amino acids at the N terminus and four residues at the C terminus (Figures 2B and 2C). Refinement with REFMAC5 (Murshudov et al., 1997) yielded a final Rcryst= 0.21 and Rfree= 0.26 (Table 1).
Table 1.
Crystallographic Data
| Data collection statistics | |
|---|---|
| X-ray source | EMBL BW7A |
| Wavelength | 1.74 Å |
| Oscillation angle | 0.5° |
| Number of frames | 720 |
| Resolution (Å) | 20.0–2.6 (2.68–2.6) |
| Number of unique reflections | 4813 (389) |
| Completeness (%) | 99.0 (100.0) |
| Redundancy | 19.9 |
| Rmergea | 0.041 (0.220) |
| <I>/σ(I) | 24.6 (12.0) |
| Refinement statistics | |
| Resolution range (Å) | 10.0–2.6 (2.71–2.6) |
| Number of reflections | 4456 (493) |
| Rcrystb | 0.210 (0.247) |
| Rfreeb | 0.267 (0.278) |
| Total number of atoms (Z > 1) | 919 |
| Number of solvent atoms (Z > 1) | 25 |
| Mean bond length deviation (Å) | 0.015 |
| Mean bond angle deviation (°) | 1.77 |
| Ramachandran statistics (% residues) | |
| in most favored region | 99.0 |
| in allowed region | 1.0 |
Numbers in parentheses apply to the highest resolution shell.
Rmerge = ΣhΣi|Ih,i − <Ih>|/ΣhΣi|Ih,i|.
Rcryst = _Σ||Fobs| − |Fcalc||/Σ|Fobs|. Rfree is the same, calculated from a random 5% subset of the reflections that were not included in the refinement.
Figure 2.

Structure Determination of N
(A) Stereo view of the initial backbone trace from RESOLVE (blue) superimposed on the final Cα backbone of the NΔ57 dimer (red). The S atoms found by SOLVE and used for phasing are indicated (yellow).
(B) Stereo view showing a detail of the 2Fo-Fc electron density map around β2, residues Arg97 to Phe104, rendered at 1 σ cutoff level Esnouf 1999, Kraulis 1991, Merrit and Bacon 1997.
(C) Stereo view of the Cα backbone of the NΔ57 dimer. The A and B subunits are colored blue and red, respectively. The residues are numbered according to the full-length N sequence.
(D) Backbone trace of NΔ57 in the “side” view. The Cys residues and their sulfur atoms (yellow) are indicated in ball-and-stick representation, as are the Arg 116 and Asp 94 residues, which are involved in interdimeric interactions.
Structure of N
NΔ57 exists as a dimer in the crystal Figure 2, Figure 3 . The NΔ57 monomer contains two antiparallel β strands flanked on both sides by α helices Figure 1, Figure 2, Figure 3. The NΔ57 dimer is organized such that the four β strands together form a continuous, flat antiparallel β sheet floor, superposed by the two long α2 helices and flanked on both sides by the N- and C-terminal helices α1 and α3 (Figure 3A). A DALI search of the protein data bank (Holm and Sander, 1995) did not identity any other significantly similar folds. The top hit (chorismate mutase, PDB code 1ECM) gave a Z score of 3.0 and had a dimeric structure, but the similarity did not extend beyond the two long α2 helices. However, we were struck by the similarity of the overall organization of the N protein to that of the coat protein of the RNA bacteriophage MS2 (Valegård et al., 1990) and the peptide recognition domain of the human histocompatibility antigen (HLA) (Saper et al., 1991). Both these proteins have a dimeric arrangement of two long α helices superposing a flat β sheet (Figure 3B). However, the similarity is only superficial, as the topology of these proteins is different from N, and MS2 and HLA have more extensive, four-stranded β sheets.
Figure 3.
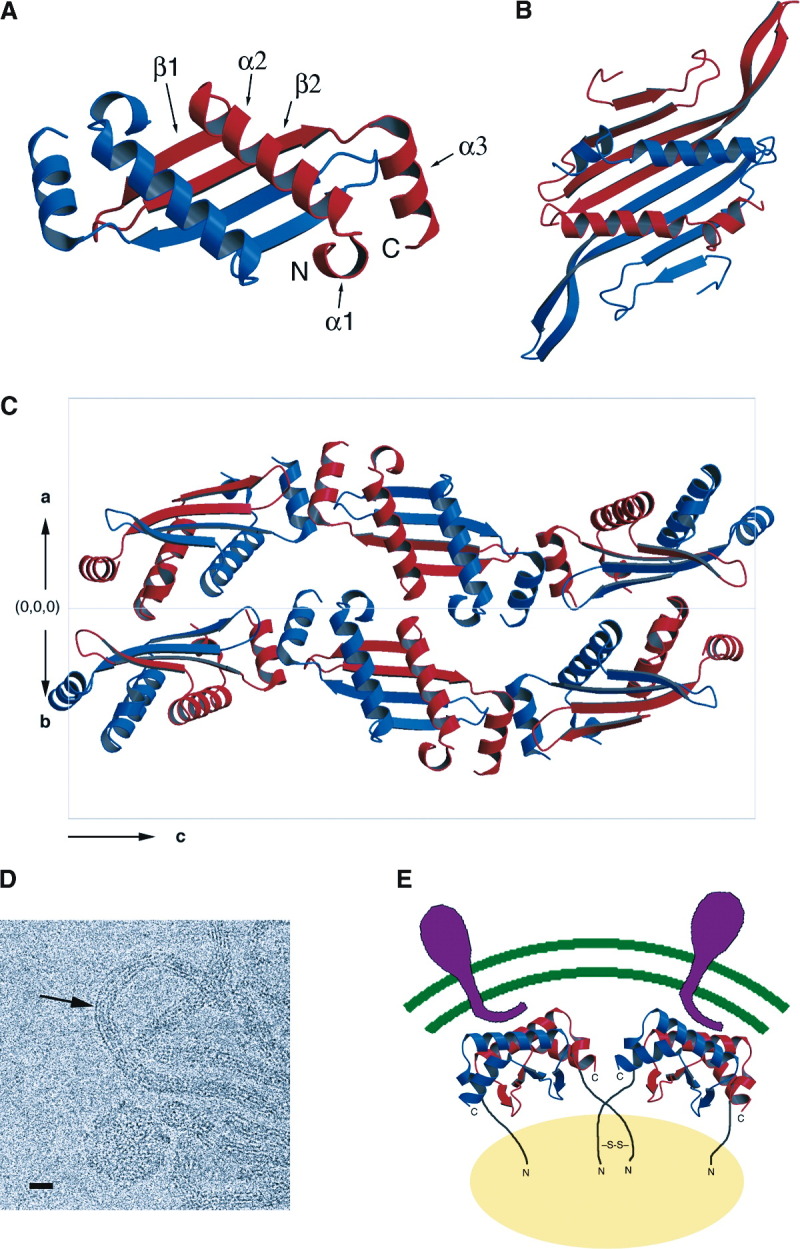
Crystal Structure of N
(A) Ribbon diagram of the NΔ57 dimer structure, showing the A (blue) and B (red) subunits, in the “top” view, thought to represent the outside of the nucleocapsid. Secondary structure elements are indicated on the B subunit, according to the labeling in Figure 1.
(B) Ribbon diagram of the coat protein dimer of bacteriophage MS2 (Valegård et al., 1990).
(C) The arrangement of NΔ57 dimers into ribbons in the unit cell, viewed down the a+b vector, perpendicular to c. The origin of the unit cell (0,0,0) and the a, b, and c axes are indicated.
(D) Cryoelectron micrograph of in vitro assembled full-length N protein (bar, 200 Å). The spacing between filaments in the ribbon (arrow) is equal to the length of the a and b crystal axes (44.67 Å).
(E) Model for shell formation in the PRRSV nucleocapsid. The N dimers interact with the RNA core (orange), forming dimeric interactions via their N-terminal domains. The α2 helices of N interact with the envelope proteins (magenta) in the lipid bilayer (green), shown schematically (not to scale). Ribbon drawings were made with MOLSCRIPT and RASTER3D Kraulis 1991, Merrit and Bacon 1997.
Considering the tight and symmetric nature of the N dimers, they almost certainly also represent the building block for the shell. Furthermore, antigenic analysis suggested an adjacent location for residues 51–67 and 80–90, and dimer formation puts these residues close together (Meulenberg et al., 1998). It was previously reported that N is incorporated into virions as disulphide-linked dimers Mardassi et al. 1996, Wootton and Yoo 2003. Indeed, the two Cys residues Cys75 and Cys90 from separate chains were positioned opposite one another in the structure (Figure 2D). However, the distance (5.3 Å) was too great for a disulphide bond to form. Furthermore, the Lelystad strain of PRRSV lacks Cys90, and the NΔ57 crystals were grown under reducing conditions, so disulphide formation is clearly not a requirement for dimerization. Disulphide formation during nucleocapsid assembly may instead involve the conserved Cys24 in the RNA binding domain and may primarily play a role in stabilizing the nucleocapsid structure (Wootton and Yoo, 2003). The N-terminal half of the protein can dimerize independently of the capsid-forming domain and in the absence of disulphide bonding (Wootton and Yoo, 2003). Such dimerization could lead to crosslinking of the N dimers, providing a possible mechanism for particle assembly (Figure 3E). Wootton et al. (2002) reported phosphorylation on Ser residues in both the RNA binding domain and the capsid-forming domain. The proposed phosphorylated Ser residues are located near the surface of the protein, on α2 (Ser 70, Ser 77, and Ser 78), in the β turn (Ser 93 and Ser 95), and in β2 (Ser 99). However, no phosphorylation occurred upon expression in bacteria, and no electron density corresponding to phosphate groups was observed in these positions in the NΔ57 map. Therefore, phosphorylation in these residues is unlikely to play a role in dimerization.
Nucleocapsid Formation
In the crystal, the N dimers are arranged in parallel, undulating ribbons located on the 3-fold screw axes (Figure 3C). The end-to-end contacts between the dimers in the ribbon are accommodated by interactions between Val 112, Ile 115, and Arg 116 in α3 and residues Ser 93 and Asp 94 in the β loop Figure 2, Figure 3, Figure 4 . Lateral interactions between ribbons, on the other hand, are scarce and mainly accommodated through a hydrogen bond between Ser70 in α2 and Glu103 in β2. Indeed, the protein was found to have a propensity for the formation of fibrous material during purification, and these properties also explain the typical morphology of the crystals, which grew as long needles and thin rods.
Figure 4.
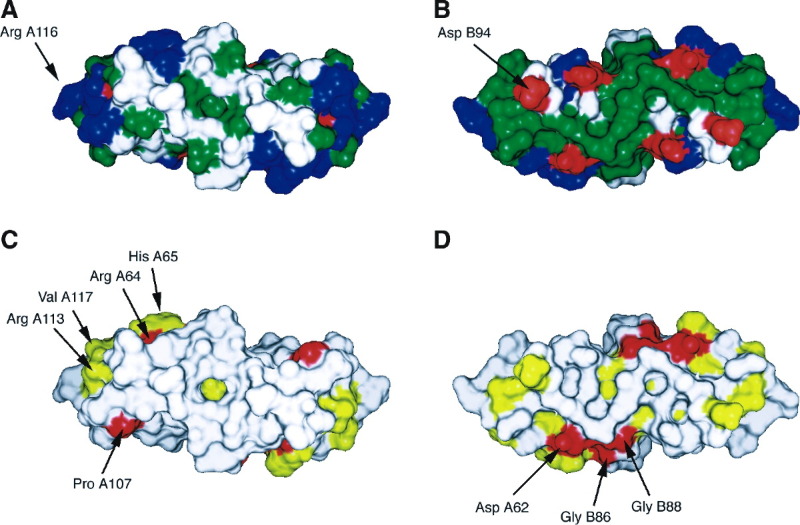
Molecular Surface of the NΔ57 Dimer
(A and C) Top views, as in Figure 3A; (B and D) “Bottom” view, from the side of the β sheet. (A and B) Showing hydrophobic (green), positively charged (blue), and negatively charged (red) residues. (C and D) Showing sequence conservation between PRRSV, Lelystad virus, LDV, EAV, and SHFV. Completely conserved residues are indicated in red, while residues that are partially conserved according to the alignment in Figure 1 are shown in yellow. The figure was generated with CHIMERA (Huang et al., 1996).
The C-terminal 11 amino acids were previously shown to be essential for binding of N-specific conformation-dependent monoclonal antibodies and important for N oligomerization (Wootton et al., 2001). Thus, the end-to-end interactions between α3 and the β loop seen in the crystal structure could be representative of the side-by-side interactions of subunits in the nucleocapsid shell. Full-length N protein expressed in E. coli can be assembled in vitro into ribbons similar to those found in the crystals, as well as shells of various sizes (Figure 3D), suggesting that the crystal arrangement may be relevant to the organization of N protein in the nucleocapsid shells. Of course, in the crystal the dimers rotate through a 3-fold screw axis, which is incompatible with the formation of a spherical shell; hence, the interactions cannot be exactly the same.
We suggest that the β sheet floor of the N dimer comprises the internal surface of the shell (Figure 3E). The floor is rather hydrophobic in character, consistent with an internal location (Figure 4B). This arrangement would place the N termini on the interior side of the shell, allowing the preceding RNA binding domains to interact with the genome (Figure 3E). This arrangement is similar to that of MS2 (Figure 3B), where the dimers are organized through 3-fold interactions on a T=3 lattice (Valegård et al., 1990). We imagine a similar arrangement in PRRSV, perhaps effected through dimerization in the RNA binding domains and most likely involving interactions with the RNA genome (Wootton and Yoo, 2003) (Figure 3E). Such an arrangement could easily be extended to larger shells. Even if the shell lacks global icosahedral symmetry, such interactions could form the basis for a nucleocapsid organized by local symmetry alone. Indeed, viral capsid proteins often show considerable flexibility to form a wide range of interactions while retaining the same tertiary structure (Dokland, 2000). In this arrangement, α2 would be on the surface of the nucleocapsid and would most likely interact with the cytoplasmic part of the envelope membrane proteins (Figure 3E), perhaps in a fashion similar to the interaction of HLA with its target peptide (Garboczi et al., 1996).
Evolutionary Relationships between Viral Nucleocapsid Proteins
The European (Lelystad) and American strains represent two distinct subgroups of PRRSV Dea et al. 2000, Le Gall et al. 1998. The greatest sequence difference in the N protein is in the RNA binding domain. The same pattern is observed when the PRRSV N protein is compared to the equivalent proteins from the related species LDV, EAV, and SHFV Figure 1, Figure 4. This could be due to virus-specific RNA differences or, more likely, due to relaxation of the structural constraints in this domain, as long as there is an overall positive charge. The most highly conserved residues include the Pro and Gly residues between the β strands and α helices, presumably essential to the correct folding of the protein. The conserved residues in α2 and the β sheet tend to be internal and are involved in dimer interactions. In contrast, there is rather little conservation of the outer surface of the dimer (Figures 4C and 4D). There is a patch of conserved residues, including several charged amino acids, made up by the terminal helices α1 and α3 (Figure 4C), perhaps suggesting a role in forming interdimer interactions in the nucleocapsid shell.
Only a few types of structural motifs have so far been found in viral structures. The most common type of viral capsid domain of nonenveloped (+) RNA viruses is the eight-stranded β barrel motif also common in DNA viruses (Strauss and Strauss, 2002). Interestingly, the structural context of the fold is quite different in all these viruses, with little conservation of the subunit interfaces, suggesting that the viruses have evolved by divergent evolution from a common ancestor rather than based on any particular advantages of this fold in particular. The main known exception to this motif is the RNA bacteriophage MS2 and its relatives, which have a fold more similar to that of the HLA proteins Saper et al. 1991, Valegård et al. 1990.
Nucleocapsid proteins of enveloped (+) RNA viruses, however, appear to belong to a different lineage. Superficially, the virions of toga-, flavi- and arterivirus virions have a similar architecture. However, based on differences in genomic organization as well as homologies in nonstructural proteins, these three groups are considered representatives of three evolutionarily separate superfamilies (Strauss and Strauss, 2002). The nucleocapsid protein of Sindbis virus, a togavirus, has a chymotrypsin-like fold (Choi et al., 1991). Like the PRRSV N protein, the Sindbis virus nucleocapsid protein has a positively charged and flexible amino-terminal half. However, the PRRSV protein is only half the size of that of Sindbis virus and has a different fold, supporting the notion that the arteriviruses are evolutionarily separate from the togaviruses. Interestingly, coronaviruses, which are clearly related to arteriviruses based on their similar genomic organization, are structurally quite different and have a much larger capsid protein that forms a helical nucleocapsid and most likely has a different fold (Strauss and Strauss, 2002). Although the nucleocapsid proteins of flaviviruses are comparable in size to that of PRRSV, their structures are also most likely different, as the charge distribution in the sequence differs completely. These differences may not tell the whole story, however, since structural similarities between the glycoproteins of Semliki Forest virus, a togavirus (Lescar et al., 2001), and tick-borne encephalitis virus, a flavivirus (Rey et al., 1995), have suggested a closer evolutionary relationship between these two groups of viruses. Thus, structurally, the enveloped RNA viruses are a much more diverse group than the nonenveloped viruses, displaying a mixed evolutionary heritage. Evolution of these viruses may be based on a “mix and match” principle, whereby different structural motifs are borrowed and used in different contexts. Our results shed some light on these structural relationships. Knowledge of the structures of other arterivirus proteins, such as the envelope glycoproteins, will be helpful in further unraveling the evolutionary relationships between the arteriviruses and other viral families.
Experimental Procedures
Cloning, Purification, and Crystallization
The C-terminal 65 amino acids of the N protein from PRRSV strain VR-2332 was cloned into the pET14b vector (Novagen) with a 6×His tag immediately N-terminal to the protein sequence. The clone was expressed in E. coli strain BL21(DE3) (Novagen) and purified by affinity and ion exchange chromatography, as previously described (Doan and Dokland, 2003). The N protein was crystallized by vapor diffusion against 8% PEG 3,350 (pH 6.5) at 15°C and grew as hexagonal rods with typical dimensions of 200 μm length and 25 μm width after 1–3 weeks.
Structure Determination
Diffraction data were collected under cryogenic conditions on a 165 mm marCCD detector (Mar USA, Inc.) at DESY, Hamburg, beamline BW7A, using an X-ray wavelength of 1.74 Å. A total of 720 frames were collected with an oscillation angle of 0.5° to provide a highly redundant data set for SAD phasing (Table 1). The data were processed using the HKL package (Otwinowski and Minor, 1997). Local scaling, location of sulfur sites, and initial SAD phasing was done with the program SOLVE using data out to 2.8 Å resolution (Terwilliger, 2002). Four S sites with an occupancy of >0.5 were identified, giving a Z score of 10.8 and an average figure of merit of 0.26. Density modification and autotracing with RESOLVE (Terwilliger, 2003) resulted in an improved map with a mean figure of merit of 0.50 and a partial trace of the protein backbone (Figure 2A). Using this partial trace and the known positions of the S atoms, an approximate axis of 2-fold NCS could be identified by manual inspection of the map using the program O (Jones et al., 1991). This information was used for NCS averaging, histogram matching, and solvent flattening with the program DM (Cowtan and Main, 1998), yielding mean figure of merit of 0.79. This resulting map was sufficiently good to allow most of the sequence to be built manually, using O (Jones et al., 1991). Subsequent maximum-likelihood refinement, including 20 cycles of TLS refinement, was done with REFMAC5 (Murshudov et al., 1997), yielding a final Rcryst= 0.210 and Rfree= 0.267 (Table 1), after placement of 25 water molecules in the map.
Acknowledgements
We are indebted to Jimmy Kwang from whom the idea of working on the PRRSV proteins originated and who supplied the original GST fusion clone of N. We are grateful to Julian Lescar for invaluable assistance with the initial model building and suggestions on how to improve the map. Thanks to Paul Tucker at EMBL Hamburg for assistance in collecting the S-SAD data, to Sarah Butcher and Pasi Laurinmaki for cryo-EM, and to Tommy Wang for his earlier contributions to the data analysis.
Published: November 4, 2003
Accession Numbers
The final coordinates have been submitted to the Protein Data Bank with accession code 1P65.
References
- Blaha T. The “colorful” epidemiology of PRRS. Vet. Res. 2000;31:77–83. doi: 10.1051/vetres:2000109. [DOI] [PubMed] [Google Scholar]
- Choi H.-K., Tong L., Minor W., Dumas P., Boege U., Rossmann M.G., Wengler G. Structure of Sindbis virus core protein reveals a chymotrypsin-like serine protease and the organization of the virion. Nature. 1991;354:37–43. doi: 10.1038/354037a0. [DOI] [PubMed] [Google Scholar]
- Cowtan K., Main P. Miscellaneous algorithms for density modification. Acta Crystallogr. D Biol. Crystallogr. 1998;54:487–493. doi: 10.1107/s0907444997011980. [DOI] [PubMed] [Google Scholar]
- Dauter Z. New approaches to high-throughput phasing. Curr. Opin. Struct. Biol. 2002;12:674–678. doi: 10.1016/s0959-440x(02)00372-x. [DOI] [PubMed] [Google Scholar]
- Dea S., Gagnon C.A., Mardassi H., Pirzadeh B., Rogan D. Current knowledge on the structural proteins of porcine reproductive and respiratory syndrome (PRRS) virus: comparison of the North American and European isolates. Arch. Virol. 2000;145:659–688. doi: 10.1007/s007050050662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan D., Dokland T. Cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis of the structural domain of the nucleocapsid N protein from porcine reproductive and respiratory syndrome virus (PRRSV) Acta Crystallogr. D Biol. Crystallogr. 2003;59:1504–1506. doi: 10.1107/s0907444903012721. [DOI] [PubMed] [Google Scholar]
- Dokland T. Freedom and restraint: themes in virus capsid assembly. Structure. 2000;8:R157–R167. doi: 10.1016/s0969-2126(00)00181-7. [DOI] [PubMed] [Google Scholar]
- Esnouf R.M. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr. D Biol. Crystallogr. 1999;55:938–940. doi: 10.1107/s0907444998017363. [DOI] [PubMed] [Google Scholar]
- Garboczi D.N., Ghosh P., Utz U., Fan Q.R., Biddison W.E., Wiley D.C. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature. 1996;384:134–141. doi: 10.1038/384134a0. [DOI] [PubMed] [Google Scholar]
- Gouet P., Courcelle E., Stuart D.I., Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- Holm L., Sander C. Dali: a network tool for protein structure comparison. Trends Biochem. Sci. 1995;20:345–347. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- Huang C.C., Couch G.S., Pettersen E.F., Ferrin T.E. Chimera: an extensible molecular modeling application constructed using standard components. Pac. Symp. Biocomput. 1996;1:724. [Google Scholar]
- Jones T.A., Zou J.-Y., Cowan S.W., Kjeldgaard M. Improved methods for the building of protein models in electron density and the location of errors in these models. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. Molscript: a program to produce both detailed and schematic plots of protein structure. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- Le Gall A., Legeay O., Bourhy H., Arnauld C., Albina E., Jestin A. Molecular variation in the nucleoprotein gene (ORF7) of the porcine reproductive and respiratory syndrome virus (PRRSV) Virus Res. 1998;54:9–21. doi: 10.1016/s0168-1702(97)00146-9. [DOI] [PubMed] [Google Scholar]
- Lescar J., Roussel A., Wien M.W., Navaza J., Fuller S.D., Wengler G., Wengler G., Rey F.A. The Fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell. 2001;105:137–148. doi: 10.1016/s0092-8674(01)00303-8. [DOI] [PubMed] [Google Scholar]
- Mardassi H., Massie B., Dea S. Intracellular synthesis, processing and transport of proteins encoded by ORFs 5 to 7 of porcine reproductive and respiratory syndrome virus. Virology. 1996;221:98–112. doi: 10.1006/viro.1996.0356. [DOI] [PubMed] [Google Scholar]
- Meng X.J. Heterogeneity of porcine reproductive and respiratory syndrome virus: implications for current vaccine efficacy and future vaccine development. Vet. Microbiol. 2000;12:309–329. doi: 10.1016/S0378-1135(00)00196-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengeling W.L., Lager K.M., Vorwald A.C., Koehler D.F. Comparative safety and efficacy of attenuated single-strain and multi-strain vaccines for porcine reproductive and respiratory syndrome. Vet. Microbiol. 2003;93:25–38. doi: 10.1016/s0378-1135(02)00426-1. [DOI] [PubMed] [Google Scholar]
- Merrit E.A., Bacon D.J. Raster3D photorealistic molecular graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- Meulenberg J.J., Hulst M.M., de Meijer E.J., Moonen P.L., den Besten A., de Kluyver E.P., Wenswoort G., Moormann R.J. Lelystad virus, the causative agent of porcine epidemic abortion and respiratory syndrome (PEARS), is related to LDV and EAV. Virology. 1993;192:62–72. doi: 10.1006/viro.1993.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meulenberg J.J., Bende R.J., Pol J.M., Wenswoort G., Moormann R.J. Nucleocapsid protein N of Lelystad virus: expression by recombinant baculovirus, immunological properties, and suitability for detection of serum antibodies. Clin. Diagn. Lab. Immunol. 1995;2:652–656. doi: 10.1128/cdli.2.6.652-656.1995. a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meulenberg J.J., Petersen-den Besten A., De Kluyverer E.P., Moormann R.J., Schaaper W.M., Wenswoort G. Characterization of proteins encoded by ORFs 2 to 7 of Lelystad virus. Virology. 1995;206:155–163. doi: 10.1016/S0042-6822(95)80030-1. b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meulenberg J.J., van Nieuwstadt A.P., van Essen-Zandbergen A., Bos-de Ruijter J.N.A., Langeveld J.P.M., Meloen R.H. Localization and fine mapping of antigenic sites on the nucleocapsid protein N of porcine reproductive and respiratory syndrome virus with monoclonal antibodies. Virology. 1998;252:106–114. doi: 10.1006/viro.1998.9436. [DOI] [PubMed] [Google Scholar]
- Micossi E., Hunter W.N., Leonard G.A. De novo phasing of two crystal forms of tryparedoxin II using the anomalous scattering from S atoms: a combination of small signal and medium resolution reveals this to be general tool for solving protein structures. Acta Crystallogr. D Biol. Crystallogr. 2002;58:21–28. doi: 10.1107/s0907444901016808. [DOI] [PubMed] [Google Scholar]
- Molitor T.W., Bautista E.M., Choi C.S. Immunity to PRRS: double-edged sword. Vet. Microbiol. 1997;55:265–276. doi: 10.1016/S0378-1135(96)01327-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Murtaugh M.P., Xiao Z., Zuckermann F. Immunological responses of swine to porcine reproductive and respiratory syndrome virus infection. Viral Immunol. 2002;15:533–547. doi: 10.1089/088282402320914485. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z., Minor W. In: Methods in Enzymology, Volume 276, Macromolecular Crystallography, Part A. Carter C.W. Jr, Sweet R.M., editors. Academic Press; San Diego: 1997. Processing of X-ray diffraction data collected in oscillation mode. 307–326.pp. [DOI] [PubMed] [Google Scholar]
- Plagemann P.G.W., Moennig V. Lactate dehydrogenase-elevating virus, equine arteritis virus, and simian hemorrhagic fever virus, a new group of positive-strand RNA viruses. Adv. Virus Res. 1992;41:99–192. doi: 10.1016/S0065-3527(08)60036-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey F.A., Heinz F.X., Mandl C., Kunz C., Harrison S.C. The envelope glycoprotein from tick-borne encephalitis virus at 2 Å resolution. Nature. 1995;375:291–298. doi: 10.1038/375291a0. [DOI] [PubMed] [Google Scholar]
- Saper M.A., Bjorkman P.J., Wiley D.C. Refined structure of the human histocompatibility antigen HLA-A2 at 2.6 Å resolution. J. Mol. Biol. 1991;219:277–319. doi: 10.1016/0022-2836(91)90567-p. [DOI] [PubMed] [Google Scholar]
- Snijder E.J., Meulenberg J.J.M. The molecular biology or arteriviruses. J. Gen. Virol. 1998;79:961–979. doi: 10.1099/0022-1317-79-5-961. [DOI] [PubMed] [Google Scholar]
- Strauss J.H., Strauss E.G. Viruses and Human Disease. Academic Press; San Diego: 2002. [Google Scholar]
- Terwilliger T.C. Automated structure solution, density modification and model building. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1937–1940. doi: 10.1107/s0907444902016438. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr. D Biol. Crystallogr. 2003;59:38–44. doi: 10.1107/S0907444902018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valegård K., Liljas L., Fridborg K., Unge T. The three-dimensional structure of the bacterial virus MS2. Nature. 1990;345:36–41. doi: 10.1038/345036a0. [DOI] [PubMed] [Google Scholar]
- Wenswoort G. Lelystad virus and the porcine epidemic abortion and respiratory syndrome. Vet. Res. 1993;24:117–124. [PubMed] [Google Scholar]
- Wootton S., Yoo D. Homo-oligomerization of the porcine reproductive and respiratory syndrome virus nucleocapsid protein and the role of disulfide linkages. J. Virol. 2003;77:4546–4557. doi: 10.1128/JVI.77.8.4546-4557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootton S., Koljesar G., Yang L., Yoon K.J., Yoo D. Antigenic importance of the carboxy-terminal beta-strand of the porcine reproductive and respiratory syndrome virus nucleocapsid protein. Clin. Diagn. Lab. Immunol. 2001;8:598–603. doi: 10.1128/CDLI.8.3.598-603.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootton S.K., Rowland R.R., Yoo D. Phosphorylation of the porcine reproductive and respiratory syndrome virus nucleocapsid protein. J. Virol. 2002;76:10569–10576. doi: 10.1128/JVI.76.20.10569-10576.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann J.J., Yoon K.J., Wills R.W., Swenson S.L. General overview of PRRSV: a perspective from the United States. Vet. Microbiol. 1997;55:187–196. doi: 10.1016/s0378-1135(96)01330-2. [DOI] [PubMed] [Google Scholar]