

Abstract
Most attention is given to seasonal influenza and respiratory syncytial virus outbreaks, but the cumulative burden caused by other respiratory viruses (RV) is not widely considered. The aim of the present study is to describe the circulation of RV in the general population during six consecutive seasons from 2006 to 2012 in Catalonia, Spain. Cell culture, immunofluorescence and PCR-based assays were used for the RV laboratory-confirmation and influenza subtyping. Phylogenetic and molecular characterizations of viral haemagglutinin, partial neuraminidase and matrix 2 proteins were performed from a representative sampling of influenza viruses. A total of 6315 nasopharyngeal samples were collected, of which 64% were laboratory-confirmed, mainly as influenza A viruses and rhinoviruses. Results show the significant burden of viral aetiological agents in acute respiratory infection, particularly in the youngest cases. The study of influenza strains reveals their continuous evolution through either progressive mutations or by segment reassortments. Moreover, the predominant influenza B lineage was different from that included in the recommended vaccine in half of the studied seasons, supporting the formulation and use of a quadrivalent influenza vaccine. Regarding neuraminidase inhibitors resistance, with the exception of the 2007/08 H275Y seasonal A(H1N1) strains, no other circulating influenza strains carrying known resistance genetic markers were found. Moreover, all circulating A(H1N1)pdm09 and A(H3N2) strains finally became genetically resistant to adamantanes. A wide knowledge of the seasonality patterns of the RV in the general population is well-appreciated, but it is a challenge due to the unpredictable circulation of RV, highlighting the value of local and global RV surveillance.
Keywords: Influenza viruses, molecular epidemiology, respiratory syncytial virus, respiratory viruses, surveillance, type/subtype/lineage
Introduction
Respiratory viruses (RV) cause significant morbidity and mortality in the human population. Most attention is given to the impact of seasonal outbreaks by human respiratory syncytial (HRSV) and influenza viruses, but the cumulative burden caused by more than 200 other known RV (picornaviruses, paramyxoviruses, coronaviruses and adenoviruses, among others) is not widely appreciated [1]. In the present study the circulation and seasonality of RV from 2006 to 2012 in Catalonia (Spain) are described.
Materials and methods
From week 40/2006 (2006/07 season) to week 20/2012 (2011/12 season), including the 2009, 2010 and 2011 inter-seasonal periods, demographic characteristics (gender and age) and nasopharyngeal samples were systematically collected for virological diagnosis from outpatients with influenza-like illness (ILI) (two first ILI consultations per week per physician), through the PIDIRAC (Daily information on Acute Respiratory Illness Plan of Catalonia) Sentinel Surveillance Network. ILI is defined as acute respiratory tract infection presenting with sudden onset of symptoms; and at least one of the following four systemic symptoms: fever or feverishness, malaise, headache, myalgia; and at least one of the following three respiratory symptoms: cough, sore throat, shortness of breath, according to the European Centre for Disease Prevention and Control's clinical criteria of ILI [2]. The PIDIRAC Sentinel Surveillance Network is based on a medical sentinel network at primary-care centres coordinated by the Public Health Agency of Catalonia, that covers all seven Health regions into which the Catalan territory is divided. Primary-care centres involved in the sampling varied from the 2006/07 season to the 2011/12, ranging from 27 in the former to 38 in the latter, and covered approximately 1% of the total population in Catalonia.
Two independent nested multiplex RT-PCR were used to detect human influenza A (FLUAV), B (FLUBV) and C (FLUCV) viruses, HRSV, human adenoviruses (HAdV), human parainfluenza viruses (HPIV) 1–4, human coronaviruses (HCoV) 229E and OC43, human enteroviruses (HEV) and human rhinoviruses (HRV) A, B and C [3], [4]. Subtyping (seasonal H1, H1pdm09 and H3) of influenza A viruses isolated on MDCK or MDCK-SIAT1 (Vircell, Granada, Spain) cell culture was performed by using the annual WHO influenza immunofluorescence assay, or directly from laboratory-confirmed clinical samples using a one-step multiplex real-time RT-PCR assay [5]. Influenza laboratory-confirmed samples collected from patients belonging to different age groups (0–4, 5–14, 15–65, > 65 years old), from different geographical sites and in different weeks were selected for a good representativeness of the phylogenetic and molecular characterizations of circulating influenza viruses in Catalonia throughout the period of study.
The coding sequences of complete domain HA1 of viral haemagglutinin (HA) protein, and the partial neuraminidase (NA) and matrix 2 (M2) proteins from FLUAV and FLUBV laboratory-confirmed specimens were sequenced, as well as, the coding region of the haemagglutinin-esterase (HE) protein from FLUCV laboratory-confirmed specimens, as previously described [6]. Updated amplification and sequencing protocols are available on request. Phylogenetic analyses of sequences from the present study together with sequences from clade reference strains downloaded from the GISAID Database (Global Initiative on Sharing Avian Influenza Data, available at: www.platform.gisaid.org) were carried out with MEGA v5.2 [7]. Sequences were aligned using the MUSCLE program, and the molecular evolutionary models of nucleotide substitutions were fitted to the multiple sequence alignments using evolutionary analyses conducted in MEGA v5.2 [7]. The phylogenetic trees were reconstructed using the neighbour-joining (NJ) distance method as implemented in MEGA v5.2 [7] with the evolutionary model with the lowest Bayesian Information criterion score. Reliability for the internal branch was assessed using the non-parametric bootstrap analysis with 1000 replicates. The amino acid substitutions of predicted influenza protein sequences were studied using MEGA v5.2 [7] relative to the homologous sequences of the corresponding recommended vaccine strains [8]. The potential N-linked glycosylation sites in HA1 amino acid sequences were tracked using the N-GlycoSite tool [9].
Statistical analyses were performed using SPSS v17 (SPSS Inc., Chicago, IL, USA). Numeric variables were compared using the non-parametric Mann–Whitney U-test for comparisons between more than two groups. Chi-squared test, Fisher test, and the OR and their 95% CI were calculated to assess associations between categorical variables. Values of p <0.05 were considered to be statistically significant.
The nucleotide sequences from the present study were submitted to the GISAID Database.
No ethical approval was required for this study.
Results
A total of 6315 nasopharyngeal samples were collected from 3135 (49.7%) male and 3173 (50.3%) female patients. Eighteen samples had missing data by lacking either gender information (seven samples) or age (11 samples). The mean age (± SD) of the patients was 21.8 ± 22.0 years (age range from a few months to 95 years; median: 12.0 years; interquartile range (IQR) 3.0–37.0 years; mode 1.0 year). Age was not normally distributed (one-sample Kolmogorov–Smirnov test, p <0.001), and the non-parametric Mann–Whitney U-test was therefore used for comparison between two groups. Statistical age differences between male patients (mean 19.8 ± 20.9 years; median 10.0; IQR 3.0–33.0) and female patients (mean 23.7 ± 22.8 years; median 14.0; IQR 4.0–40.0) were significant (p <0.001). Table 1 and Supplementary material Table S1 summarize RV detection rates by age groups and by season, respectively. The largest proportion of samples (55%) was collected from children under 15 years old. The 64% of received samples were laboratory-confirmed for single (83%) or multiple (17%) detections, mainly by FLUAV and HRV, and followed by HAdV, FLUBV and HRSV. FLUAV or HRV were detected in 74% of multiple detections. In addition, HAdV, HCoV, HPIV-2, HPIV-4 and HEV were found together with other RVs in rates over 40% of detections (see Supplementary material, Table S1). Among RV laboratory-confirmed cases there were no general differences (p 0.906) between male (49.6%) and female (50.4%) patients, but higher percentages were particularly reported for HRSV (p 0.015; OR 1.302; 95% CI 1.053–1.611) and HEV (p 0.020; OR 1.391; 95% CI 1.053–1.837) in male patients. The highest detection percentages were reported in the youngest patient group, and HAdV, HEV, HPIV-1, HPIV-3 and HRSV (Table 1) circulated preferentially in patients younger than 5 years old. Multiple RV detections were also more frequently detected in the youngest patients (p <0.001). Age differences (Table 1) were not statistically significant between patients infected by the different FLUAV subtypes (seasonal H1N1 versus H3N2: p 0.123; seasonal H1N1 versus H1N1pdm09: p 0.176; and H1N1pdm09 versus H3N2: p 0.503).
Table 1.
Detection rates (%) of all respiratory viruses among the received respiratory samples by age patient groups
| Age group |
Age (years) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0–4 years |
5–14 years |
15–65 years |
>65 years |
Total (%) | Mean ± SD | Median [IQR] | |||||
| 11 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | ||||
| Samples received | 1881 (30%) | 1601 (25%) | 2483 (39%) | 339 (5%) | 63042 | 21.8 ± 22.0 | 12.0 [3.0–37.0] | ||||
| Positive samples | 1378 | 1099 | 1432 | 155 | 4064 (64%) | 19.1 ± 20.6 | 10.0 [3.0–32.0] | ||||
| 34% | 73% | 27% | 69% | 35% | 58% | 4% | 46% | ||||
| Single detection | 1043 (76%) | 938 (85%) | 1269 (89%) | 137 (88%) | 3387 (83%) | 20.0 ± 20.8 | 11.0 [3.0–33.0] | ||||
| Multiple detection | 335 (24%) | 161 (15%) | 163 (11%) | 18 (12%) | 677 (17%) | 14.1 ± 18.7 | 5.0 [2.0–19.0] | ||||
| Single or multiple respiratory virus infection | |||||||||||
| FLUAV | 394 | 528 | 634 | 43 | 1599 (25%) | 20.0 ± 19.2 | 12.0 [5.0–33.0] | ||||
| 25% | 21% | 33% | 33% | 40% | 26% | 3% | 13% | ||||
| Seasonal H1 | 42 | 33 | 57 | 1 | 133 (2%) | 18.1 ± 17.6 | 9.0 [3.0–30.0] | ||||
| 32% | 2% | 25% | 2% | 43% | 2% | 1% | <1% | ||||
| H1pdm09 | 113 | 272 | 246 | 1 | 632 (10%) | 18.2 ± 16.3 | 12.0 [6.0–29.0] | ||||
| 18% | 6% | 43% | 17% | 39% | 10% | <1% | <1% | ||||
| H3 | 218 | 206 | 302 | 40 | 766 (12%) | 21.9 ± 21.5 | 12.0 [4.0–36.0] | ||||
| 28% | 12% | 27% | 13% | 39% | 12% | 5% | 12% | ||||
| Unsubtyped | 21 | 17 | 29 | 1 | 68 (1%) | 19.4 ± 18.6 | 11.5 [3.0–33.0] | ||||
| 31% | 1% | 25% | 1% | 43% | 1% | 1% | <1% | ||||
| FLUBV | 103 | 255 | 147 | 20 | 525 (8%) | 17.4 ± 18.7 | 9.0 [5.0–25.0] | ||||
| 20% | 5% | 49% | 16% | 28% | 6% | 4% | 6% | ||||
| FLUCV | 9 | 3 | 7 | 0 | 19 (<1%) | 16.6 ± 18.8 | 10.0 [1.0–35.0] | ||||
| 47% | <1% | 16% | <1% | 37% | <1% | 0% | <1% | ||||
| HAdV | 343 | 117 | 110 | 11 | 581 (9%) | 10.9 ± 16.6 | 3.0 [1.0–11.0] | ||||
| 59% | 18% | 20% | 7% | 19% | 4% | 2% | 3% | ||||
| HCoV | 57 | 27 | 100 | 15 | 199 (3%) | 28.2 ± 24.5 | 25.0 [3.0–48.0] | ||||
| 29% | 3% | 14% | 2% | 50% | 4% | 8% | 4% | ||||
| HEV | 110 | 45 | 49 | 5 | 209 (3%) | 13.2 ± 18.1 | 4.0 [2.0–18.0] | ||||
| 53% | 6% | 22% | 3% | 23% | 2% | 2% | 1% | ||||
| HPIV-1 | 45 | 12 | 22 | 2 | 81 (1%) | 14.7 ± 19.2 | 4.0 [2.0–24.0] | ||||
| 56% | 2% | 15% | 1% | 27% | 1% | 2% | 1% | ||||
| HPIV-2 | 48 | 37 | 49 | 5 | 139 (2%) | 19.0 ± 20.0 | 11.0 [3.0–34.0] | ||||
| 35% | 3% | 27% | 2% | 35% | 2% | 4% | 1% | ||||
| HPIV-3 | 55 | 24 | 25 | 5 | 109 (2%) | 15.9 ± 21.6 | 4.0 [2.0–23.0] | ||||
| 50% | 3% | 22% | 1% | 23% | 1% | 5% | 1% | ||||
| HPIV-4 | 30 | 17 | 26 | 3 | 76 (1%) | 18.9 ± 20.7 | 7.5 [2.0–37.0] | ||||
| 39% | 2% | 22% | 1% | 34% | 1% | 4% | 1% | ||||
| HRSV | 243 | 42 | 71 | 8 | 364 (6%) | 11.3 ± 18.2 | 3.00 [1.0–11.0] | ||||
| 67% | 13% | 12% | 3% | 20% | 3% | 2% | 2% | ||||
| HRV | 307 | 158 | 359 | 58 | 882 (14%) | 22.8 ± 23.1 | 13.0 [3.0–39.0] | ||||
| 35% | 16% | 18% | 10% | 41% | 14% | 7% | 17% | ||||
Abbreviations: FLUCV, human influenza C virus; HAdV, human adenoviruses; HCoV, human coronaviruses; HEV, human enteroviruses; HPIV 1–4, human parainfluenza viruses 1, 2, 3 and 4; HRV, human rhinoviruses; IQR, interquartile range.
1. Detection rate per age group (% of row); 2. Detection rate per total received samples within each age group (% of column).
Missing data: 11 cases.
The seasonal HRSV outbreaks usually started early every season, before the seasonal influenza circulation. When FLUAV and FLUBV were co-detected during a season, FLUAV was first, with the predominance of a particular FLUAV subtype, and was followed by FLUBV later (Fig. 1 ). The only exception was the 2009 influenza pandemic. A(H1N1)pdm09, which was first noted in June 2009 and which circulated showing a biphasic pattern. A first peak was detected during weeks 24–35 (summer months), and a second peak during weeks 41–49 (autumn months), before HRSV circulation and outside the usual months of influenza outbreaks (from December to March). During the first two 2009 pandemic peaks, other FLUAV subtypes (seasonal H1 and H3) and FLUBV remained almost undetected despite the large sampling done to strengthen the A(H1N1)pdm09 surveillance. During these six consecutive seasons other RV than influenza viruses were mainly detected during the cold months, often just before and after the seasonal influenza epidemics (Fig. 2 ), with scarce circulation during the inter-seasonal periods. Differences between the RV detection rates (see Supplementary material, Table S1) were observed just before and after the 2009 pandemic (p <0.05). The detection rates of HCoV, HRV, HPIV 1–4 and HEV increased after the pandemic (OR <1), in comparison to FLUAV, FLUBV, HAdV and HRSV (OR >1), which decreased. Age differences of FLUAV, HAdV and HCoV between the periods before and after the 2009 pandemics (see Supplementary material, Table S1) were also shown.
Fig. 1.

Weekly distribution of laboratory-confirmation rates (%) for human influenza and respiratory syncytial viruses from week 40/2006 (2006/07 season) to week 20/2012 (2011/12 season).
Fig. 2.
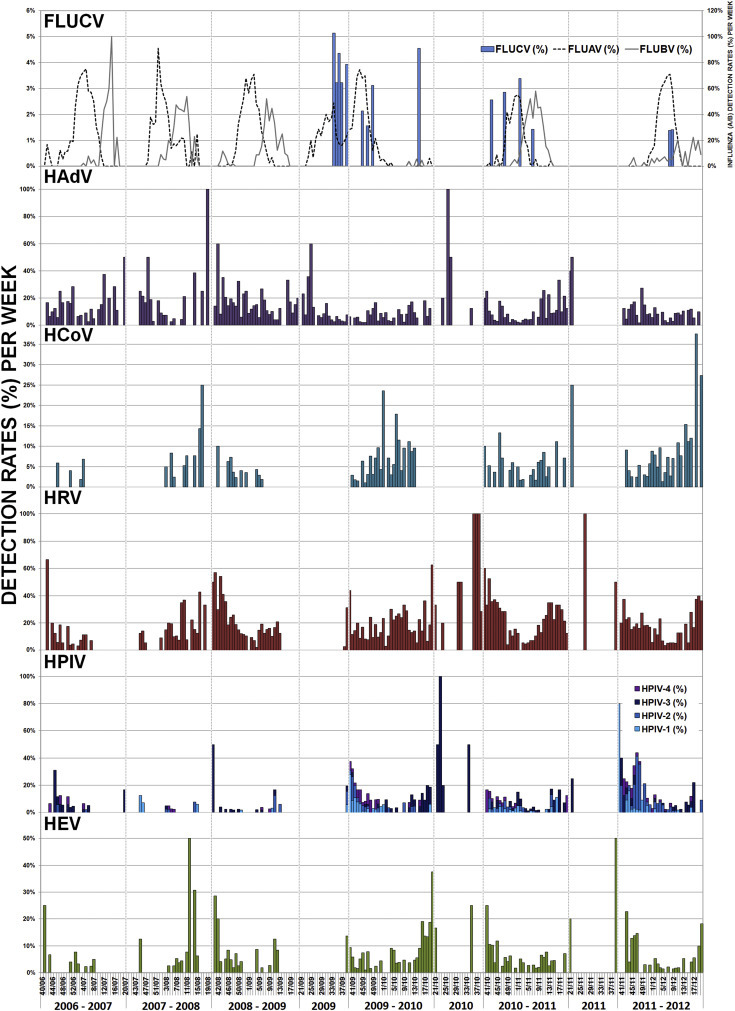
Weekly distribution of laboratory-confirmation rates (%) for human influenza C virus (FLUCV) viruses, human adenoviruses (HAdV), human parainfluenza viruses (HPIV) 1, 2, 3 and 4, human coronaviruses (HCoV), human enteroviruses (HEV) and human rhinoviruses (HRV), from week 40/2006 (2006/07 season) to week 20/2012 (2011/12 season).
Phylogenetic analyses (HA1 and NA) of 28/29 seasonal A(H1N1) strains (Table 2 ; and Supplementary material, Fig. S1) showed that most of them fell within subclade 2B, represented by the 2008/09 vaccine strain (A/Brisbane/59/2007). Two out of the 28 strains (7%) isolated during the 2007/08 season carried the mutation H275Y in NA, in addition to D354G and the compensatory mutations R222Q, V234M and D344N (see Supplementary material, Table S2) [10].
Table 2.
Summary of molecular characterization of influenza A and B viruses based on haemagglutinin sequences by season (the numbers of samples studied are shown in brackets), including the recommended vaccine strains to use
| 2006/07 | 2007/08 | 2008/09 | 2009/10 | 2010/11 | 2011/12 | |
|---|---|---|---|---|---|---|
| Influenza A virus—seasonal A(H1N1) | ||||||
| Recommended vaccine strain | A/New Caledonia/20/99 | A/Solomon Islands/3/2006 | A/Brisbane/59/2007 | A/Brisbane/59/2007 | – | – |
| Characterized strains | Tessaloniki/24-like (1) | Tessaloniki/24-like (1) | – | – | – | – |
| 29 1 | Solomon Islands/3-like (3) | |||||
| Brisbane/59-like (24) | ||||||
| Influenza A virus—A(H1N1)pdm09 | ||||||
| Recommended vaccine strain | – | – | – | A/California/7/2009 | A/California/7/2009 | A/California/7/2009 |
| Characterized strains | – | – | – | No-Nelson's Clade 7 (9) | No-Nelson's Clade 7 (2) | – |
| 123 1 | California/7-like (67) | California/7-like (1) | ||||
| Hong Kong/3934-like (1) | Christchurch/16-like (2) | |||||
| St. Petersburg/27-like (8) | ||||||
| St. Petersburg/100-like (1) | ||||||
| Astrakan/1-like (32) | ||||||
| Influenza A virus—A(H3N2) | ||||||
| Recommended vaccine strain | A/Wisconsin/67/2005 | A/Wisconsin/67/2005 | A/Brisbane/10/2007 | A/Brisbane/10/2007 | A/Perth/16/2009 | A/Perth/16/2009 |
| Characterized strains | California/7-like (1) | – | California/7-like (1) | – | Perth/10-like (2) | Perth/10-like (1) |
| 117 1 | Wisconsin/7-like (2) | Brisbane/10-like (19) | Iowa/19-like (1) | Iowa/19-like (18) 2 | ||
| Brisbane/10-like (12) | Iraq/7-like (1) | Stockholm/18-like (5) | ||||
| Stockholm/18-like (3) | England/259-like (26)3 | |||||
| Victoria/361-like (25) | ||||||
| Influenza B virus | ||||||
| Recommended vaccine strain | B/Malaysia/2506/2004 | B/Malaysia/2506/2004 | B/Florida/4/2006 | B/Brisbane/60/2008 | B/Brisbane/60/2008 | B/Brisbane/60/2008 |
| (Lineage) | (B/Victoria) | (B/Victoria) | (B/Yamagata) | (B/Victoria) | (B/Victoria) | (B/Victoria) |
| B/Victoria-lineage characterized strains | Malaysia/2506-like (5) | Brisbane/60-like (17) | – | Malaysia/2506-like (2) 4 | ||
| 54 1 | Brisbane/60-like (27) | Brisbane/60-like (3) | ||||
| B/Yamagata-lineage characterized strains | Florida/4-like (2) | Shangai/361-like (4) | – | – | Bangladesh/3333-like (1) | Brisbane/3-like (1) |
| 72 1 | Egypt/144-like (2) | Florida/4-like (13) | Florida/01-like (8) | Stockholm/12-like (39) | ||
| Bangladesh/3333-like (2) | ||||||
In case of different phylogenetic variants, the most frequent is marked in bold letters.
The partial NA sequences for phylogenetic and molecular characterization could not be obtained from some of these strains.
One intra-clade reassortant strain (Iowa/19 HA; England/259 NA).
One intra-clade reassortant strain (England/259 HA; Stockholm/18 NA).
Intra-clade reassortant (Brisbane/60 HA; Malaysia/2506 NA).
Phylogenetic analyses (HA1 and NA) of 111/117 seasonal A(H3N2) strains (Table 2; and Supplementary material, Fig. S2) revealed a high genetic diversity among HA and NA sequences. The majority of 2006/07 and 2008/09 strains belonged to clade represented by 2008/10 vaccine strain (A/Brisbane/10/2007). In the following seasons (2010/11 and 2011/12) HA sequences fell within up to six different genetic subgroups within Victoria/208 clade, and none was genetically close to the 2010–2012 vaccine strain.
Phylogenetic analyses (HA1 and NA) of 121/123 A(H1N1)pdm09 strains (Table 2; and Supplementary material, Fig. S3), showed that 116 strains were carrying the genetic features (S203T in HA1, and V106I and N248D in NA) of strains belonging to the clade 7 described by Nelson et al. [11]. Strains collected during the 2009/10 season remained genetically close to those first described at the beginning of the pandemic. Most of 2010/11 strains genetically evolved and fell within four different genetic subgroups based on HA sequences. In addition, at least ten strains without key genetic features of Nelson's clade 7 were detected during the first two pandemic seasons, of which the latest strains (2010/11 season) showed genetic drift from the early 2009 isolates.
Phylogenetic analysis of HA1 sequences of 126 FLUBV strains revealed the co-circulation of B/Victoria (54) and B/Yamagata-lineage (72) strains. An alternance in the predominant lineage, which was different from what was included in the recommended vaccine composition, was shown in three out of the six studied seasons (Table 2). But this alternance did not seem to affect the FLUBV detection rates (see Supplementary material, Table S1). In fact, the highest FLUBV detection rate (19%) was reported during the 2010/11 season, when the predominant circulating lineage was well-matched with the lineage included in the vaccine (Table 2).
Phylogenetic analyses (HA1 and NA) of 51/54 B/Victoria strains (Table 2; and Supplementary material, Fig. S4) and of 67/72 B/Yamagata strains (Table 2; and Supplementary material, Fig. S5) revealed the circulation of strains during the 2006–2008 seasons that were genetically close to both recommended 2006–2009 vaccine strains. However, the majority of B/Victoria strains detected since the 2008/09 season fell within clade represented by the recommended B/Victoria vaccine strain that has been used until now. Most of B/Yamagata strains collected since the 2010/11 season fell within the two genetic subgroups (Florida/01 and Stockholm/12) described within Bangladesh/3333 clade, and none within the 2012/13 vaccine strain (B/Wisconsin/01/2010) clade. In addition, one 2011/12 strain fell within the Brisbane/3 clade together with other 2007/08 strains, but with additional mutations that defined a new genetic subgroup represented by B/Estonia/55669/2011.
The HA and NA phylogenetic analyses revealed a few circulating intra-clade reassortants for B/Victoria lineage during the 2010/11 season (see Supplementary material, Fig. S4), and for A(H3N2) subtype during the 2011/12 season (see Supplementary material, Fig. S2).
Phylogenetic analysis of 19 FLUCV strains (see Supplementary material, Fig. S6) detected during the 2009/10 and 2011/12 seasons, revealed that strains belonged to the C/Kanagawa/1/76-related and to C/Sao Paulo/378/82-related lineages [5], remaining genetically similar.
Regarding NA mutations related to neuraminidase inhibitors (NAIs) resistance, known genetic markers were not found in the influenza strains studied, with the only exception being the H275Y mutation [10] in some 2007/08 seasonal A(H1N1) strains, as described above. Some mutations within the enzyme active site or its surroundings were found in the characterized strains (see Supplementary material, Table S2), which might be associated with decreased or reduced susceptibility to NAIs [10], [12], but are not yet characterized.
In M2 sequences, the predominance of the genetic adamantanes-resistant A(H3N2) strains during the 2006/07 season (13/15, 87%) and later (100%) by acquiring the S31N mutation [12] were observed. All characterized A(H1N1)pdm09 strains were also carrying S31N mutation as described at the beginning of the pandemic. Double mutations, S31N/V27A and S31N/V27F, in one 2011/12 A(H3N2) strain and in one 2009/10 A(H1N1)pdm09 strain were also found, respectively. No mutations in M2 protein sequences related to antiviral resistance were found in seasonal A(H1N1) strains.
Discussion
Our results show the significant burden of viral aetiological agents in acute respiratory infections, particularly in the youngest patient group, as well as the decline in RV detection rates as the age increases. In the adult population, viral respiratory infection might be underestimated because it is usually mild and self-limiting. Gender did not seem to be related to an increased infection susceptibility, except in HRSV or HEV. Overall, the most frequently detected RV were FLUAV, HRV, HAdV, FLUBV and HRSV, although HRSV and influenza viruses mostly circulated as seasonal outbreaks, and not continously throughout the year, such as HAdV and HRV.
Differences in age distribution among RV were found. Statistical differences between the age of patients infected by the several FLUAV subtypes were not found, although the means and IQR suggest that patients infected by seasonal A(H1N1) or A(H1N1)pdm09 cases were younger than those infected by A(H3N2). Variations in the pattern of age-specific positive proportions between different subtypes were previously described [13], [14]. More A(H1N1)pdm09 susceptibility in younger patients was attributed to the little or no pre-existing immunity to the virus among children and young adults [15]. HRV or HCoV were commonly detected in all age groups, which might be explained by their high genetic diversity or the incomplete cross-reactive immune response, leading to continuous re-infections throughout life.
Differences in the RV detection rates and in the ages of infected patients after the 2009 pandemic were observed as noted by other authors [16], [17], although these have not been reported in other studies [18]. This might be a result of specific and non-specific cross-reactive immunity against other RV following the A(H1N1)pdm09 infection [17]. But a consequence of the larger sampling cannot be discarded.
Continuous evolution through either progressive amino acid substitutions (with changes on potential N-glycosylation sites) or by segment reassortments [19], [20] can affect (a) the antigenicity and tropism features by changes in the protective antigenic epitopes or in the receptor binding site of HA protein [21], [22], [23], [24], [25], or (b) the susceptibility to the available antivirals through changes in the NA and M2 proteins [10], [12]. Drifted-strains with substantial antigenic changes, driven by the host immune response acting as an evolutionary selective pressure, lead to the annual vaccine composition update [8].
A(H3N2) strains, which circulated at varying levels throughout the study period, despite the wide community protection acquired by the natural infection and the seasonal vaccination since its appearance in 1968, belonged to several genetic subgroups, showing a great genetic heterogenity. A(H1N1)pdm09 has been genetically evolving into several phylogenetic groups since 2009, but remains antigenically similar [8]. However, close attention should be paid to future antigenic drift events in response to an increased natural or vaccine-induced immunity. Regarding FLUBV and vaccine composition, the predominant lineage did not match the recommended vaccine lineage in half of the studied seasons. The inaccurate prediction of the predominant FLUBV lineage in trivalent influenza vaccines supported the formulation of a quadrivalent influenza vaccine [26], [27], [28] to enhance protection. However, FLUBV lineage alternance has not been reported so far, with a high predominance of B/Yamagata lineage since the 2011/12 season [29].
Some intra-clade reassortant strains were found. It is well-known that viral segment reassortment is a powerful genetic mechanism for influenza evolution. As these genetic events are uncommon, these findings highlight the importance of studying at least the envelope HA and NA sequences to monitor their emergence and spread, as well as, the value of local surveillance to detect these minor viral populations.
Throughout these six consecutive seasons the picture of the available antiviral drugs to fight against influenza infection changed considerably, and our results also showed the trends reported worldwide [8], [30]. During the 2006/07 season both adamantane and NAIs were the two antiviral family drugs available. In Catalonia, the first genetic oseltamivir-resistant seasonal A(H1N1) variants, which carried H275Y mutation in NA associated with antiviral resistance [10], were detected in a percentage of 7% during the 2007/08 season. These strains also carried the compensatory mutations, that favoured the global spread of H275Y variants despite the absence of drug selective pressure [10], [30]. According to WHO data, an average of approximately 24% of characterized strains in Europe were shown to possess high-level oseltamivir-resistance, ranging from no detection in some countries to 68% in Norway [30]. In addition, circulating A(H1N1)pdm09 and A(H3N2) strains are also resistant to adamantanes, since they carry the S31N mutation in M2 protein [12], [30], remaining susceptible to NAIs. Indeed, adamantanes cannot now be considered suitable for seasonal influenza treatment. In the present study, with the exception of H275Y seasonal A(H1N1) strains that did not circulate since the 2009 pandemics, no other circulating influenza strains carrying genetic markers related to NAIs resistance were found [10]. Changes within the enzyme active site or its surroundings were found in NA sequences, but further phenotyping studies should be performed. There is public health concern that the antiviral resistance genetic markers could become fixed in the viral genome, as detected in a low percentage (<1%) among circulating viruses [31]. The rapid global spread of oseltamivir-resistant seasonal A(H1N1) influenza viruses without drug pressure should serve as a reminder for close local and global surveillance.
A wide knowledge of the seasonal patterns of RV in the general population contributes to a better diagnosis and management of respiratory infections, but it is considered a challenge because of the unpredictable nature of RV circulation. Indeed, continuous local and global surveillance of influenza and other RV must be carried out to monitor their prevalence, their genetic diversity and the emergence of antiviral resistance.
Transparency declaration
The authors have no conflicts to declare.
Acknowledgements
The authors would like to thank the Working Group of Influenza Surveillance Network in Catalonia. This work was partially supported by Fondo de Investigación Sanitaria (grants FIS PI08/0118 and FIS PI11/01864) from the Spanish Ministry of Health; Agència d’Avaluació de Tecnologia i Recerca Mèdiques (AATRM) (grant 402/02/2008); the Spanish Network for the Research in Infectious Diseases (REIPI 06/0008); and the European Regional Development Fund (ERDF).
Editor: L. Kaiser
Footnotes
Additional Supporting Information may be found in the online version of this article at http://dx.doi.org/10.1016/j.cmi.2016.02.007.
Appendix A. Supplementary materials
The following supplementary materials are available for this article:
Number and percentage of detections (single and multiple) per respiratory virus and by season.
Molecular characterization of predicted positions in NA protein (in the corresponding N2, N1 and B/Yam numbering) associated with decreased and reduced susceptibility to neuraminidase inhibitors.
Fig. S1.
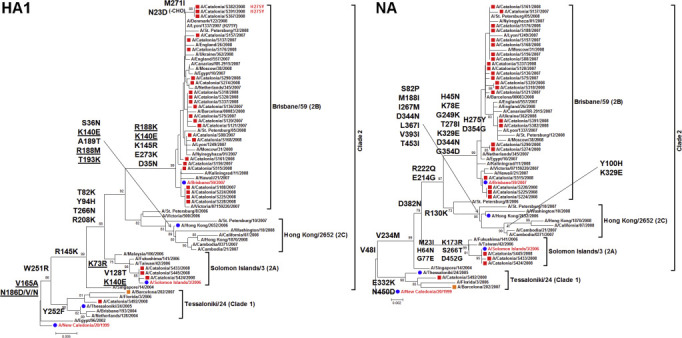
Phylogenetic reconstruction of complete HA1 (from nucleotide position 52 to 1029 of the coding HA sequence) and partial NA (from nucleotide position 46 to 1398 of the coding the NA sequence) sequences of influenza seasonal A(H1N1) viruses constructed using neighbour-joining method rooted to A/New Caledonia/20/1999 (2006/07 vaccine strain).
Fig. S2.

Phylogenetic reconstruction of complete HA1 (from nucleotide position 49 to 1035 of the coding HA sequence) and partial NA (from nucleotide position 27 to 1388 of the coding NA sequence) sequences of influenza seasonal A(H3N2) viruses constructed using neighbour-joining method rooted to A/Wisconsin/67/2005 (2006–2008 vaccine strain).
Fig. S3.

Phylogenetic reconstruction of complete HA1 (from nucleotide position 52 to 1032 of the coding HA sequence) and partial NA (from nucleotide position 160 to 1377 of the coding NA sequence) sequences of influenza A(H1N1)pdm09 viruses constructed using neighbour-joining method rooted to A/California/07/2009 (2009–2016 vaccine strain).
Fig. S4.

Phylogenetic reconstruction of complete HA1 (from nucleotide position 46 to 1086 of the coding HA sequence) and complete NA (from nucleotide position 1 to 1398 of the coding NA sequence) sequences of B/Victoria viruses constructed using neighbour-joining method, and rooted to B/Malaysia/2506/2004 (2006–2008 vaccine strain).
Fig. S5.

Phylogenetic reconstruction of complete HA1 (from nucleotide position 46 to 1083 of the coding HA sequence) and complete NA (from nucleotide position 1 to 1398 of the coding NA sequence) sequences of B/Yamagata viruses constructed using neighbour-joining method, and rooted to B/Shangai/361/2002 (2005/06 vaccine strain).
Fig. S6.

Phylogenetic reconstruction of complete HE sequences (excluding signal peptide, from nucleotide position 43 to 1968 of the the coding HE sequence) of FLUCV viruses constructed using neighbour-joining method, and rooted to strains represented by C/Taylor/1233/47.
References
- 1.Ruuskanen O., Lahti E., Jennings L.C., Murdoch D.R. Viral pneumonia. Lancet. 2011;377:1264–1275. doi: 10.1016/S0140-6736(10)61459-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Influenza surveillance: Influenza case definitions. European Centre for Disease Prevention and Control. Available at: http://ecdc.Europa.Eu/en/healthtopics/influenza/surveillance/pages/influenza_case_definitions.aspx.
- 3.Coiras M.T., Perez-Brena P., Garcia M.L., Casas I. Simultaneous detection of influenza A, B, and C viruses, respiratory syncytial virus, and adenoviruses in clinical samples by multiplex reverse transcription nested-PCR assay. J Med Virol. 2003;69:132–144. doi: 10.1002/jmv.10255. [DOI] [PubMed] [Google Scholar]
- 4.Coiras M.T., Aguilar J.C., Garcia M.L., Casas I., Perez-Brena P. Simultaneous detection of fourteen respiratory viruses in clinical specimens by two multiplex reverse transcription nested-PCR assays. J Med Virol. 2004;72:484–495. doi: 10.1002/jmv.20008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antón A., López-Iglesias A.A., Tórtola T., Ruiz-Camps I., Abrisqueta P., Llopart L. Selection and viral load kinetics of an oseltamivir-resistant pandemic influenza A (H1N1) virus in an immunocompromised patient during treatment with neuraminidase inhibitors. Diagn Microbiol Infect Dis. 2010;68(3):214–219. doi: 10.1016/j.diagmicrobio.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 6.Antón A., Marcos M.A., Codoñer F.M., de Molina P., Martínez A., Cardeñosa N. Influenza C virus surveillance during the first influenza A (H1N1) 2009 pandemic wave in Catalonia, Spain. Diagn Microbiol Infect Dis. 2011;69(4):419–427. doi: 10.1016/j.diagmicrobio.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 7.Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. Mega5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.World Health Organization (WHO). Global influenza programme. Influenza. Available at: http://www.Who.Int/influenza/en/.
- 9.Zhang M., Gaschen B., Blay W., Foley B., Haigwood N., Kuiken C. Tracking global patterns of N-linked glycosylation site variation in highly variable viral glycoproteins: HIV, SIV, and HCV envelopes and influenza hemagglutinin. Glycobiology. 2004;14(12):1229–1246. doi: 10.1093/glycob/cwh106. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen H.T., Fry A.M., Gubareva L.V. Neuraminidase inhibitor resistance in influenza viruses and laboratory testing methods. Antivir Ther. 2012;17:159–173. doi: 10.3851/IMP2067. [DOI] [PubMed] [Google Scholar]
- 11.Nelson M., Spiro D., Wentworth D., Beck E., Fan J., Ghedin E. The early diversification of influenza A/H1N1pdm. PLoS Curr. 2009;1:RRN1126. doi: 10.1371/currents.RRN1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pozo F., Lina B., Andrade H.R., Enouf V., Kossyvakis A., Broberg E. Guidance for clinical and public health laboratories testing for influenza virus antiviral drug susceptibility in Europe. J Clin Virol. 2013 May;57(1):5–12. doi: 10.1016/j.jcv.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 13.Khiabanian H., Farrell G.M., St George K., Rabadan R. Differences in patient age distribution between influenza A subtypes. PLoS One. 2009;4:e6832. doi: 10.1371/journal.pone.0006832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang L., Chan K.H., Suen L.K., Chan K.P., Wang X., Cao P. Age-specific epidemic waves of influenza and respiratory syncytial virus in a subtropical city. Sci Rep. 2015;5:10390. doi: 10.1038/srep10390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller E, Hoschler K, Hardelid P, Stanford E, Andrews N, Zambon M. Incidence of 2009 pandemic influenza A H1N1 infection in England: a cross-sectional serological study. Lancet 375:1100–08. [DOI] [PubMed]
- 16.Casalegno J.S., Ottmann M., Bouscambert-Duchamp M., Valette M., Morfin F., Lina B. Impact of the 2009 influenza A(H1N1) pandemic wave on the pattern of hibernal respiratory virus epidemics, France, 2009. Euro Surveill. 2010;15 [PubMed] [Google Scholar]
- 17.Meningher T., Hindiyeh M., Regev L., Sherbany H., Mendelson E., Mandelboim M. Relationships between A(H1N1)pdm09 influenza infection and infections with other respiratory viruses. Influenza Other Respir Virus. 2014;8:422–430. doi: 10.1111/irv.12249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Navarro-Marí J.M., Pérez-Ruiz M., Galán Montemayor J.C., Marcos Maeso M.A., Reina J., de Oña Navarro M. Circulation of other respiratory viruses and viral co-infection during the 2009 pandemic influenza. Enferm Infecc Microbiol Clin. 2012;30(Suppl 4):25–31. doi: 10.1016/S0213-005X(12)70101-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hay A.J., Gregory V., Douglas A.R., Lin Y.P. The evolution of human influenza viruses. Philos Trans R Soc Lond B Biol Sci. 2001;356:1861–1870. doi: 10.1098/rstb.2001.0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palese P., Shaw M. Orthomyxoviridae: the viruses and their replication. In: Knipe D.M., Howley P.M., editors. 5th ed. vol. 2. Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 1647–1689. (Fields virology). [Google Scholar]
- 21.Wang Q., Cheng F., Lu M., Tian X., Ma J. Crystal structure of unliganded influenza B virus hemagglutinin. J Virol. 2008;82:3011–3020. doi: 10.1128/JVI.02477-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshida R., Igarashi M., Ozaki H., Kishida N., Tomabechi D., Kida H. Cross-protective potential of a novel monoclonal antibody directed against antigenic site B of the hemagglutinin of influenza A viruses. PLoS Pathog. 2009;5(3):e1000350. doi: 10.1371/journal.ppat.1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soundararajan V., Zheng S., Patel N., Warnock K., Raman R., Wilson I.A. Networks link antigenic and receptor-binding sites of influenza hemagglutinin: mechanistic insight into fitter strain propagation. Sci Rep. 2011;1:200. doi: 10.1038/srep00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu R., Ekiert D.C., Krause J.C., Hai R., Crowe J.E., Jr., Wilson I.A. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science. 2010;328:357–360. doi: 10.1126/science.1186430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang C.C., Chen J.R., Tseng Y.C., Hsu C.H., Hung Y.F., Chen S.W. Glycans on influenza hemagglutinin affect receptor binding and immune response. Proc Natl Acad Sci U S A. 2009;106(43):18137–18142. doi: 10.1073/pnas.0909696106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ambrose C.S., Levin M.J. The rationale for quadrivalent influenza vaccines. Hum Vaccin Immunother. 2012;8:81–88. doi: 10.4161/hv.8.1.17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belshe R.B. The need for quadrivalent vaccine against seasonal influenza. Vaccine. 2010;28(Suppl. 4):D45–D53. doi: 10.1016/j.vaccine.2010.08.028. [DOI] [PubMed] [Google Scholar]
- 28.Reed C., Meltzer M.I., Finelli L., Fiore A. Public health impact of including two lineages of influenza B in a quadrivalent seasonal influenza vaccine. Vaccine. 2012;30:1993–1998. doi: 10.1016/j.vaccine.2011.12.098. [DOI] [PubMed] [Google Scholar]
- 29.Sistema de vigilancia de la gripe en españa. Red nacional de vigilancia epidemiológica. Instituto de Salud Carlos III. Ministerio de Economía y Competitividad. Gobierno de españa. Available at: http://vgripe.Isciii.Es/.
- 30.WHO Collaborating Centre for Reference and Research on Influenza (London, United Kingdom). Medical Research Council's National Institute for Medical Research (MIMR). Francis Crick Institute. Annual reports. Available at: http://www.Crick.Ac.Uk/research/worldwide-influenza-centre/annual-and-interim-reports.
- 31.Okomo-Adhiambo M., Fry A.M., Su S., Nguyen H.T., Elal A.A., Negron E. Oseltamivir-resistant influenza A(H1N1)pdm09 viruses, United States, 2013–14. Emerg Infect Dis. 2015;21(1):136–141. doi: 10.3201/eid2101.141006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Number and percentage of detections (single and multiple) per respiratory virus and by season.
Molecular characterization of predicted positions in NA protein (in the corresponding N2, N1 and B/Yam numbering) associated with decreased and reduced susceptibility to neuraminidase inhibitors.